


-
对农业废水中有机砷的去除是近年来的研究热点之一[1]。阿散酸p-ASA(p-arsanilic acid),作为最常见的畜禽有机砷添加剂之一,常用于促进肉鸡肉鸭生长和抑制禽体内寄生虫病。然而,诸如p-ASA此类苯胂酸在动物食用后并非停留在其体内,大部分会经过动物的排泻行为释放到环境中,在外界环境的生物和化学作用下,p-ASA会转化为强毒性和强流动性的无机砷(As(V)和As(III))[2],从而导致一系列砷污染的环境问题。其中,p-ASA通过直接排放或是淋溶作用进入附近地表水和地下水中,造成水体环境介质被破坏,且由于苯胂酸类物质具有微生物毒性,不易在水环境中自然降解,就会进一步因食物链富集作用进入人体造成更大的危害。
为解决砷污染的源头问题,亟待探索高效去除有机砷的方法。目前,解决有机砷污染主流的2种方法是吸附及氧化降解[3],尽管氧化法对有机砷具有高去除率的特点,然而存在无法妥善解决降解产物,即无机砷的释放问题[4],而吸附法兼具使用成本低、可重复利用性好、生产方便高效、工艺简单、中间产物向环境释放风险低等优点,是一种应用前景较好的有机砷去除技术[5]。有机砷的吸附剂主要包括铁基材料、高分子吸附剂、碳材料以及金属有机框架(MOFs)[6]等材料。其中,已有较多研究表明,铁基材料(如MIL-100(Fe)和MIL-101(Fe)等铁基MOFs)对有机砷化合物具有良好的吸附性能[7-8]。GAO等[5]考察了苯胂酸在铁基金属-有机骨架(MIL-88A)上的吸附行为及机理,发现配位(Fe-O-As)和氢键在吸附过程中起了主要作用。而锆基材料作为一种阴离子污染物吸附剂,也被加以探究[9]。WANG等[10]率先采用UiO-66对As(V)进行吸附,发现Zr—O键与目标阴离子发生配位作用的过程是UiO-66吸附容量大的主要原因。此外,赵娜等[11]发现当Zn2+被引入Fe3O4水溶液体系时,Zn2+与As(V)发生络合反应并形成金属-配体络合物,其与Fe3O4的结合力增强,对As(V)的去除产生协同作用,去除率从66%增强至高达99%。综上所述,若能将3种金属有机结合,或能进一步促进MOFs对有机砷的吸附作用。
因此,本研究选择铁、锌、锆3种吸附性能优良的金属为原料,通过水热法制备得到多元金属氧化物吸附剂(ZnFeZrOx),采用FT-IR、XRD、XPS等分析手段对ZnFeZrOx的结构和面貌进行表征,拟研究其对有机砷中的典型代表—阿散酸(p-ASA)的吸附去除效率,并探究相关的吸附机理,本研究以期为有机砷废水的处理提供参考。
-
本研究中主要试剂为:硝酸铁(Fe(NO3)3·9H2O)、硝酸锌(Zn(NO3)2·6H2O)、硝酸锆(Zr(NO3)4·5H2O)购自国药集团化学试剂有限公司;硝酸锆(Zr(NO3)4·5H2O)购自上海麦克林生化科技有限公司;氯化氢(HCl)购自上海沪试实验室器材股份有限公司;氢氧化钠(NaOH)、十六烷基三甲基溴化铵(CTAB)、无水乙醇购自天津福晨化学试剂厂;对氨基苯胂酸(p-ASA)纯度98%,氯化钠(NaCl)、氯化镁(MgCl2)、氯化钙(CaCl2)、硫酸钠(Na2SO4)、硝酸钠(NaNO3)、磷酸二氢钾(KH2PO4)购自上海阿拉丁生化科技股份有限公司。以上试剂均为分析纯。
-
在30 mL的0.2 mol·L−1 Zn(NO3)2 溶液中投加0.2 g CTAB,然后加入30 mL 0.4 mol·L−1 Fe(NO3)3溶液,再加入30 mL 0.1 mol·L−1 Zr(NO3)4。在常温度下进行磁力搅拌30 min,用0.6 mol·L−1 NaOH溶液将pH调节到12。再将其进行30 min的超声水浴预处理,而后通过500 mL高压釜在130 ℃下反应24 h。反应结束后冷却至室温,抽滤分离后水洗沉淀物,以减少产品中的杂质。最后,通过真空干燥箱(70 ℃,7 h以上)干燥所得吸附剂。
-
1)吸附特征及影响因素。称取50 mL 50 mg·L−1的p-ASA溶液,投加一定量自制ZnFeZrOx吸附剂粉末(平均粒径为42 nm),并分别探究吸附剂投加量、pH、吸附时间、离子强度、共存离子的干扰等对吸附效果的影响,在一定温度和摇床转速条件下反应一定时间,达到吸附平衡后,通过紫外可见分光光度计在252 nm处测定上清液中p-ASA吸光度,计算p-ASA的去除率和ZnFeZrOx的平衡吸附容量以测定吸附剂对p-ASA的吸附效果。其中,事先测得的p-ASA标准曲线为y= 0.062 7 x+0.016 3 (R2=0.999 3),其检测限为0.553 mg·L−1。
吸附剂投加量对吸附效果的影响:在pH=4的条件下,改变ZnFeZrOx吸附剂粉末投加量(投加量分别为0.02、 0.05、0.08、0.11、0.14 g),吸附15 h后测定不同含量的样品对p-ASA的吸附效果。pH对吸附效果的影响:分别调节反应体系的pH至1~12,投加0.11 g吸附剂,吸附15 h后测定不同pH对p-ASA的吸附效果。离子强度对吸附效果的影响:配置一系列以NaCl作为背景电解质成分的p-ASA溶液(NaCl浓度分别0、0.1、1、10、100 mmol·L−1),调节体系pH=4,其余条件不变,测定不同离子强度对p-ASA吸附效果的影响。共存离子干扰对吸附效果的影响:选取不同浓度(0、1、10 mmol·L−1)的Cl−1、NO3−、SO42−、CO32−作为共存离子,保持其余条件不变,反应结束后,测定其对p-ASA吸附效果的影响。
2)吸附等温线。改变温度和p-ASA溶液浓度,其余的条件相同,进行吸附平衡实验,采用Langmuir和Freundlich吸附等温线模型对ZnFeZrOx吸附p-ASA的过程进行研究。
3)吸附动力学。在其他实验条件相同时,间隔一定时间进行取样,测定该时刻样品中p-ASA的去除率及ZnFeZrOx的吸附容量,采用Lagergren准一级和准二级动力学模型对ZnFeZrOx吸附p-ASA的过程进行吸附性能考察。
-
X射线衍射分析(XRD)采用X射线衍射仪(MiniFlex600,Rigaku Corporation)对本实验吸附前后的ZnFeZrOx进行分析,扫描范围为10°~80°。傅里叶红外光谱分析(FT-IR)采用傅立叶红外光谱仪(AVAT-AR 360型)对吸附前后的ZnFeZrOx进行研究。样品粉末与KBr混合,混合比为1:100,测量范围为4 000~400 cm−1。采用 X射线光电子能谱分析(XPS) (Thermo Scientific ESCALAB 250 X)测定吸附前后的ZnFeZrOx的表面组成、化学状态以及元素成分。
-
称取8份已达饱和吸附量的ZnFeZrOx各0.05 g于锥形瓶中,分别加入10 mL质量浓度为1 mol·L−1的NaHCO3、NaOH、Na2CO3和NaCl溶液,每2份为1组做平行实验。通过恒温培养振荡器进行解吸实验,设置温度30 ℃,转速150 r·min−1,振荡12 h。反应结束后,采用紫外可见分光光度法测定p-ASA的浓度,计算解吸率和吸附容量。并探究解吸剂浓度对解吸效果的影响。称取8份已达饱和吸附量的ZnFeZrOx各0.05 g于锥形瓶中,分别加入质量浓度分别为0.1、0.5、1、2 mol·L−1最优解吸剂各10 mL,每2份为一组做平行实验。随后放入恒温培养振荡器中,以上述解吸条件进行同样操作,并计算解吸率。
-
配置固体浓度为50 g·L−1的猪粪浸出液备用,测得pH为7.2,取部分浸出液调节pH至4。同时配置质量浓度为50 mg·L−1的p-ASA溶液,分别调节pH为4和7.2。称取pH=7.2的浸出液、pH=4的浸出液、pH=7.2的p-ASA溶液、pH=4的p-ASA溶液各50 mL,并分别投加0.11 g吸附剂,吸附15 h后通过紫外可见分光光度计测定不同pH下猪粪浸出液样品及p-ASA溶液的吸附效果。
-
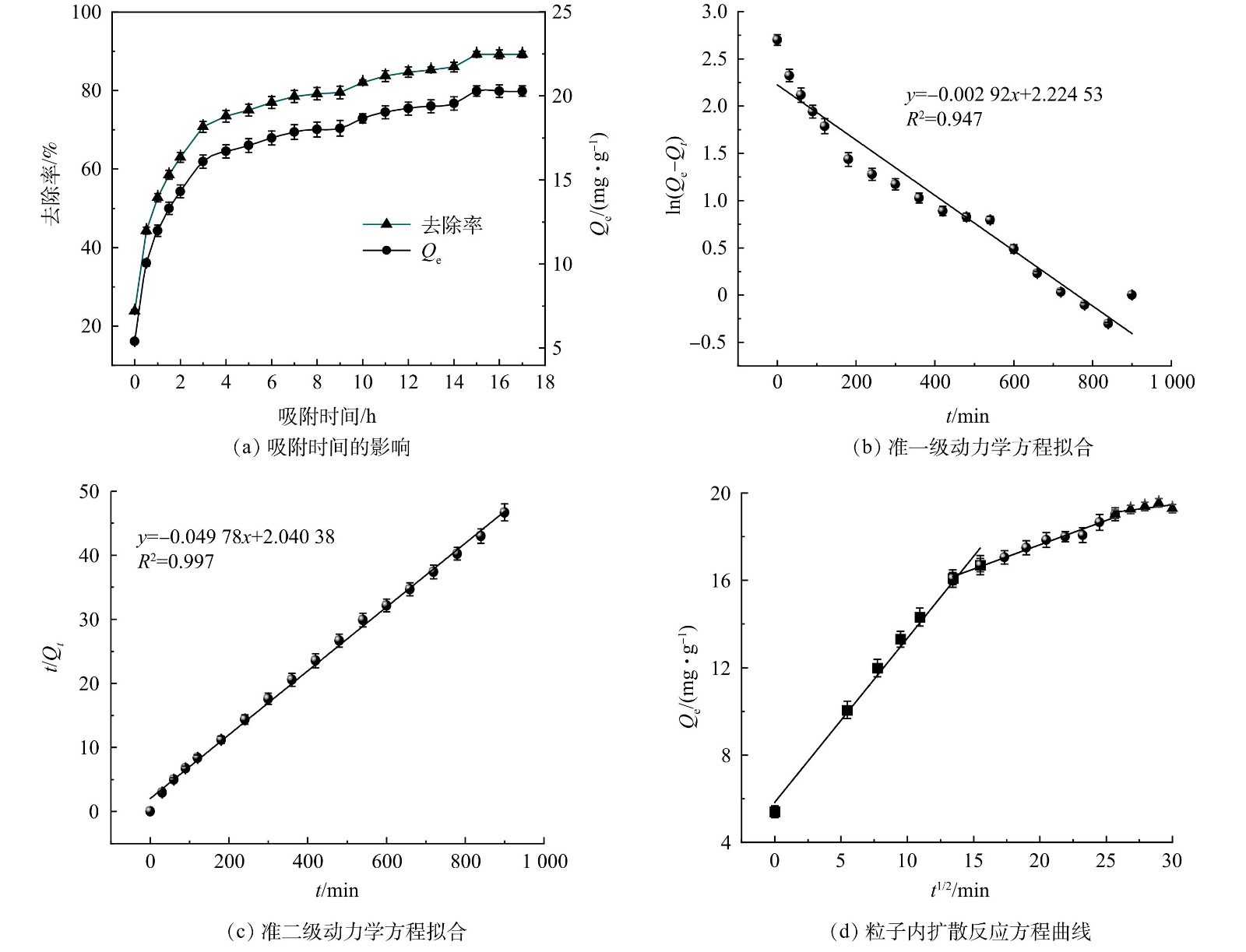
1) ZnFeZrOx投加量对吸附p-ASA的影响。投加量对ZnFeZrOx吸附p-ASA的影响如图1(a)所示。从图1(a)可以看出,随着投加量的增加,ZnFeZrOx吸附p-ASA的去除率呈现先升高再降低的特征,当投加量由0.02 g增至0.11 g时,去除率由25.15%增至75.73%;当投加量增至0.14 g时,去除率降至71.23%。原因可能是当ZnFeZrOx吸附剂的投加量增加,会使吸附剂与p-ASA结合的活性位点增加,此时去除率升高;而当体系中吸附剂量进一步增加,造成溶液中p-ASA的相对浓度降低,此外,还可能会发生吸附剂之间互相团聚的现象,导致吸附剂内部空隙堵塞,比表面积降低,进而导致去除率略有所下降[12]。从整体上看,随ZnFeZrOx投加量的增加,平衡吸附容量Qe由31.43 mg·g−1降至12.72 mg·g−1。p-ASA的吸附平衡容量随ZnFeZrOx投加量的升高而降低。这可能是由于当体系中p-ASA的初始浓度和体积不变,随着吸附剂的增加,单位质量所吸附的p-ASA减少,造成吸附平衡容量的下降,这表明p-ASA的吸附过程是一个扩散主导的过程,包括吸附剂表面扩散和颗粒内扩散过程[13-15]。综上所述,选取0.11 g作为ZnFeZrOx对p-ASA进行吸附的最佳投加量。
2) pH对ZnFeZrOx吸附p-ASA的影响。由图1(b)所示,当pH 在1~4时,吸附剂对p-ASA的去除率较高,均维持在 93% 以上,在pH=4时去除率最高达到95.15%;当pH>4 时,去除率逐步下降。以pH=4为转折点的去除率变化趋势与其他研究[14]结果一致。ZnFeZrOx吸附p-ASA的平衡吸附容量和p-ASA去除率的变化趋势趋于一致。在pH=4 时,Qe=21.63 mg·g−1。由此可见,该吸附受pH影响严重,可能因为该吸附是以物理吸附为主,配位体交换和表面配位作用为辅[14]。p-ASA的pKa为1.9、4.1和9.2,分别对应于p-ASA上对位-NH3+和2个胂酸基团(As-OH)的去质子化[16]。在pH<4 时,由于溶液中[H+]高,静电吸引力强,而p-ASA为阴离子污染物,因此,此时去除率高;当pH>4时,溶液pH升高会促进p-ASA被离子化而溶于水中,从而不易被吸附[17];此时溶液中OH−的增多可增加ZnFeZrOx表面上的直接竞争吸附行为,导致去除率下降[18]。随着OH−被大量吸附,ZnFeZrOx表面的正电荷不断减少,而负电荷不断增多,进一步增加其表面静电斥力,静电作用被削弱,导致p-ASA去除率降低。在低pH条件下,吸附剂表面以OH2为主,可产生更多的配位体交换位点,也起到一定吸附作用[19]。此外,Fe离子与p-ASA可能发生表面配位作用形成Fe-O-As基团[20],溶液pH的增大可能导致Fe离子从ZnFeZrOx中溶解,也会引起p-ASA去除率的降低。实验中虽然在pH=1时,平衡吸附容量和去除率最大,然而考虑到实际应用时满足相关条件的实施难度和成本问题以及强酸条件的潜在威胁,选取pH=4作为后续实验的最佳pH。
3)离子强度对ZnFeZrOx吸附p-ASA的影响。如图1(c)所示,随着离子强度的增大,吸附行为受影响,p-ASA去除率由95.15%降至74.95%。这可能是因为Cl−与p-ASA共同竞争ZnFeZrOx上的吸附位点,说明ZnFeZrOx吸附p-ASA的过程是外层络合吸附和静电吸附[12, 21]。
4)共存离子干扰对ZnFeZrOx吸附p-ASA的影响。在天然水体中往往存在多种离子,常见的阴离子有SO42−、CO32−、NO3−、PO43−等,在吸附除砷的过程中,他们可能同样与p-ASA间存在竞争机制。由图1(d)可知,不同共存离子对ZnFeZrOx吸附p-ASA的干扰程度不同,PO43−的干扰最强,NO3−次之,SO42−和Cl−的干扰作用较弱。当PO43−浓度由0 mmol·L−1增强到1 mmol·L−1和10 mmol·L−1时,ZnFeZrOx对p-ASA的去除率分别由95.15%降至15.26%和8.91%。这说明磷酸根与胂酸根之间存在极强的竞争机制,在高磷水体中,吸附剂ZnFeZrOx对p-ASA的吸附存在明显的空间位阻效应[22]。
5)等温吸附特征研究及p-ASA初始浓度的影响。由图2(a)~(b)可知,当p-ASA加入初始质量浓度为10 mg·L−1时,p-ASA的去除率为76.16%。当初始浓度增至160 mg·L−1,去除率降至41.9%。总体来看,去除率与p-ASA初始浓度呈负相关,这可能是因为在p-ASA初始浓度较高时,ZnFeZrOx吸附位点有限,吸附趋于饱和后,吸附难以继续进行,从而造成去除率下降。
以Ce为横坐标、Ce/Qe为纵坐标和以lnCe为横坐标、lnQe为纵坐标进行作图,拟合结果如图2(b)和图2(c)所示。利用Langmuir以及Freundlich公式对拟合后的数据进行计算,得到Langmuir和Freundlich模型的相关参数如表1和表2所示。比较图2(c)和图2(d)可以明显看出,Langmuir吸附等温线模型的拟合程度明显低于Freundlich模型,结合表1可知,在3种温度下,Langmuir模型的R2均远小于0.9,且计算所得理论Qe远大于实际实验值(20.285 mg·g−1)。这表明Langmuir模型不适合用于描述该吸附过程。但由表2可以看出,Freundlich吸附等温线模型对该吸附过程拟合所得R2均大于0.98,表明拟合程度良好,说明p-ASA通过内置配位层与ZnFeZrOx中的ZnFe2O4、Fe3O4和Fe等结合。此外,由表2中KL值可见,该值随温度升高而略微增大,表明升温能促进吸附反应的进行。吸附强度系数n>1 (1/n<1),表明吸附容易发生,说明p-ASA容易被ZnFeZrOx吸附。
-
对所得数据进行吸附动力学分析,结果如图3(a)所示。从图3(a)可以看出,ZnFeZrOx对的吸附过程可分为3个阶段。在0~3 h内,吸附去除率和平衡吸附量快速升高,表明该过程为快速吸附过程,此阶Qe段时,吸附剂表面吸附位点空缺较多且吸附剂表面与溶液之间浓度差较大,溶液中p-ASA分子与吸附剂碰撞,p-ASA分子被吸附剂吸附,物理吸附起主导作用,吸附速率快。在3~15 h内,去除率和Qe缓慢增加,表明该过程为慢速吸附过程。此时ZnFeZrOx表面吸附位点已被逐渐占用,溶液中p-ASA分子在接触过程中碰到空缺位点的概率减小,溶液浓度梯度也减小,从而导致吸附速率减缓。此外,ZnFeZrOx可能与吸附在表面的p-ASA分子发生电子转移、离子交换、配位体交换等反应,化学吸附为主导过程,去除速率减缓但去除率却保持上升。反应至15 h后,去除率和平衡吸附容量基本保持不变,此时吸附反应达到平衡。在15 h时去除率达到最高,为89.25%;在17 h时Qe达到最高,为20.28 mg·g−1。选取15 h为后续实验的最佳反应时间。
并分别以ln(Qe−Qt)对t、t/Qt对t、Qt对t1/2作图,对该反应的吸附动力学进行研究,结果如图3(b)、图3(c)、图3(d)和表3所示。如图3(b)~(c)所示,与准一级方程相比,准二级动力学方程曲线的拟合程度更好。结合表3可知,准二级动力学方程的R2值更接近于1 (0.997)。准一级动力学方程计算的理论Qe' (9.250 mg·g−1)与实验所得平衡吸附量Qe相比有很大差异,而准二级动力学方程的Qe' (20.008 mg·g−1)与实验Qe (20.285 mg·g−1)接近。说明准二级动力学方程更适用于ZnFeZrOx对p-ASA的吸附动力学过程,因此ZnFeZrOx对p-ASA吸附主要是化学吸附。由图3(d)可见,p-ASA在ZnFeZrOx上的吸附可被分为3个过程:液膜扩散过程、颗粒内扩散过程和吸附表面反应过程。3个过程的曲线斜率依次减小,说明吸附过程的速度依次减缓,第3阶段边界层对吸附效果的影响较大。另外,3个过程在图3(d)上都显示出较好的线性关系,但折线不通过原点,说明粒子内扩散反应影响了吸附速率。
-
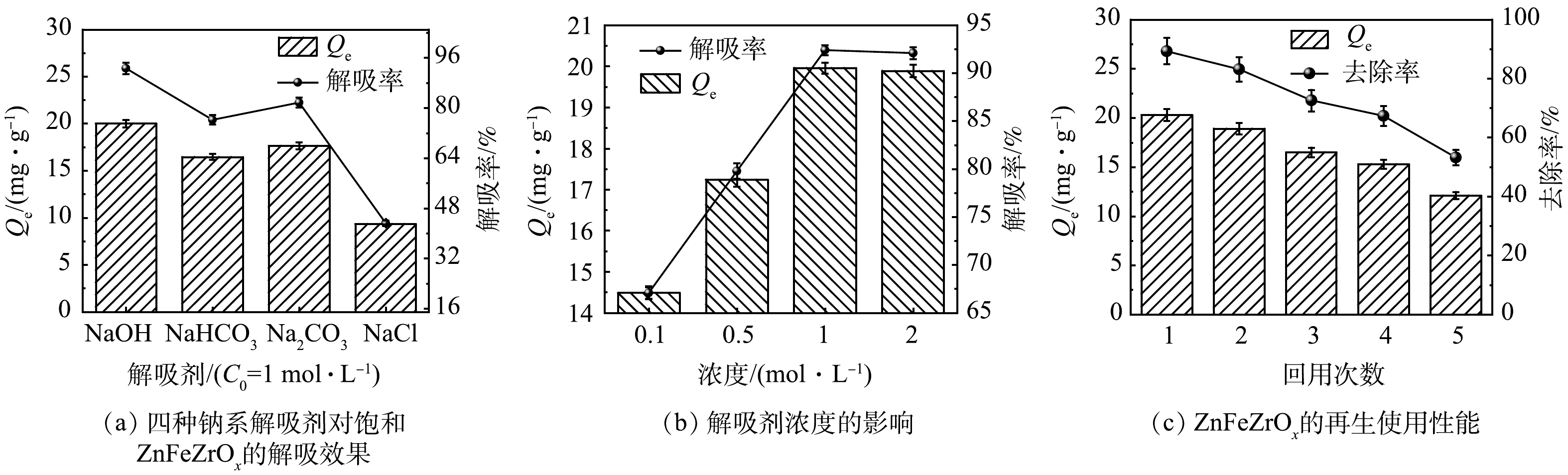
1)解吸剂的选择。4种钠系解吸剂对饱和ZnFeZrOx的解吸效果如图4(a)所示。其中,NaOH表现出最优解吸效果,在第1次解吸实验后解吸率为92.60%,吸附容量为20.00 mg·g−1。Na2CO3溶液和NaHCO3溶液的解吸效果相对较好,而NaCl的解吸效果则相对较低。因此,后续实验选取NaOH作为最佳解吸剂。
2)解吸剂浓度的影响。如图4(b)可知,随着NaOH浓度的增大,解吸率和ZnFeZrOx吸附容量先增大后保持几乎不变。当NaOH溶液为0.1 mol·L−1和0.5 mol·L−1时,解吸率相对较低;当NaOH浓度为1 mol·L−1时,解吸率为92.60%,吸附容量为20.00 mg·g−1;继续将NaOH浓度增加至2 mol·L−1后,对解吸率无进一步提高。因此,选取1 mol·L−1为NaOH的最佳解吸浓度。
3) ZnFeZrOx的再生使用性能。如图4(c)所示,饱和ZnFeZrOx经过5次循环再生后,吸附容量由20.3 mg·g−1降至12.1 mg·g−1,去除率相应由89.32%降至53.24%。在循环再生到第3次时,去除率为72.60%。表明ZnFeZrOx再生3次后仍可对水体中大部分p-ASA进行吸附。此外,以上结果间接表明1 mol·L−1 NaOH可以成功将p-ASA从吸附剂表明解吸下来,且ZnFeZrOx再生效果较好。
-
1) XRD分析。图5(a)为ZnFeZrOx吸附p-ASA前后的XRD对比图。如图5(a)所示,吸附前后ZnFeZrOx的XRD图几乎未有发生变化,表明吸附剂在吸附过程中能维持较稳定的化学形态,间接表明ZnFeZrOx的化学稳定性。
2) FI-IR分析。由图5(b)可见,在3 100~3 700 cm−1处形成一个宽频带区域[23],可能是由于H2O分子以及吸附剂中-OH官能团的吸收附带,并且比较吸附前和吸附后曲线观察到,-OH由3 550 cm−1 蓝移至 3 450 cm−1附近,说明吸附过程中-OH基团也参与吸附。1 642 cm−1为H2O和-OH的弯曲振动峰[24]。由吸附后的曲线可见,在837 cm−1处新出现一个峰。经查验为As-O的特征峰[16],可能是As-O-M(M代表Zr、Fe)的振动吸收[14],表明p-ASA通过配位体交换作用被吸附在三相金属氧化物吸附剂表面[25]。1 358 cm−1应为-OH结构伸缩振动的特征峰[26-27],与吸附前相比,该峰在吸附后明显减弱,表明p-ASA在吸附过后才能中替代了部分-OH并与ZnFeZrOx相连,证明-OH参与反应且p-ASA并非单纯沉积而可能是发生了配位反应。1 095 cm−1应为C-As的弹性振动峰,1 215 cm−1和1 560 cm−1分别为p-ASA上的C-N伸缩振动特征峰和-NH2的剪切振动峰[28-29]。
3) XPS分析。图6为ZnFeZrOx吸附p-ASA溶液的前后的XPS图谱。图6(a)~f)分别为元素As、C、O、Zn、Fe、Zr的高分辨图。在图6(a)中可以看出,在44.6 eV处出现新的吸收峰为As3d的特征峰。这表明吸附后的p-ASA中As元素主要以As(V)的形式存在,并已成功结合到ZnFeZrOx表面[27]。图6(b)中ZnFeZrOx样品吸附p-ASA前的C1s 谱图能够拟合出3个峰,分别为284.6 eV的C-C或C=C、288.5 eV的C-O以及289.0 eV的O-C=O。吸附后,原284.6 eV的峰蓝移至284.8 eV,且C-C与C=C二者占比由70.92%升至94.78%,这可能是因为p-ASA分子在吸附剂表面堆积。此外C-O和O-C=O的峰,在吸附p-ASA后,占比分别由17.02%和12.06%降至3.73%和1.49%,说明在p-ASA的吸附过程中,-COOH官能团发挥了重要作用[30]。
由图6(c)可见,吸附前样品中O信号较强,表明制备出的三相金属复合材料为含氧化合物[31]。O1s的3个原始峰为530.0、531.60、532.97 eV,分别代表由Fe、Zn、Zr和O的氧化物M-O(锌铁锆三种金属皆以M表示)、M-OH、C=O。在吸附p-ASA后,消耗了部分羧基,还形成了M-O-As、As-O、M-O-C等有机配体。此外,530.09 eV处的峰红移至529.89 eV,表明M-OH基团参与了p-ASA的吸附并在ZnFeZrOx表面形成M-O基团[24],这与FT-IR的分析结果一致[8]。由图6(d)、图6(e)、图6(f)可以看出,ZnFeZrOx中Zn主要为二价,Fe主要为二价,而ZnFeZrOx中的Zr则主要为正四价。
由图6(d)可见,吸附前,1 021.9 eV对应的是Zn2p3/2的Zn-O,1 045.0 eV对应于Zn2p1/2.吸附后,Zn-O和Zn2p1/2的特征峰分别红移至1 021.6 eV和1 044.8eV,表明Zn-O参与了p-ASA的配位。由图6(e)可见,吸附前,归属于Fe2p3/2的特征峰有709.6 eV和711.4 eV,而723.2 eV和725.6 eV则归属于Fe2p1/2[32]。吸附后,属于Fe2p3/2的峰移至709.7 eV和712.1 eV,归属于Fe2p1/2的峰移至723.3 eV和725.7 eV。吸附前后,Fe由二价转化为三价,表明Fe-OH是吸附阿散酸的活性位点[30]。由图6(f)可见,在吸附前XPS曲线上的182.4 eV对应Zr-O的结合能,而184.6 eV处对应Zr-Zr[33];吸附后,Zr-O的结合能偏移至178.7 eV,同时Zr-Zr的结合能偏移至181.1 eV,二者均向结合能低的方向偏移。表明Zr-O参与了p-ASA的配位,并在吸附剂吸附p-ASA时起到一定的作用。
-
牲畜养殖废水对ZnFeZrOx吸附p-ASA的影响。实际牲畜养殖废水往往组分复杂,具有高COD的特点[34]。图7反映了ZnFeZrOx对猪粪浸出液中p-ASA的吸附效果。如图7所示,在未调节pH的浸出液中(pH=7.2),ZnFeZrOx对p-ASA的去除率较低,仅有42.23%,而将其pH调节至4后,去除率却保持在较高的水平(84.92%),与对照组的结果接近,说明ZnFeZrOx在实际处理高碳源的牲畜养殖废水时受废水pH影响较大。
-
1)三相金属氧化物吸附剂ZnFeZrOx 对典型的有机砷污染物p-ASA具有良好的吸附性能,最大去除率达95.15%,最大吸附容量为595.23 mg·g−1。
2)三相金属氧化物材料ZnFeZrOx中,Fe、Zn、Zr主要以Fe3+、Zn2+及Zr4+的形式存在,Fe-OH为当中主要吸附活性点位,Zn-O和Zr-O起到一定的吸附作用。静态吸附实验结果表明,ZnFeZrOx吸附p-ASA的最佳条件为:吸附剂投加量为0.11 g,pH=4,吸附时间为15 h,在此条件下,吸附过程为吸热过程,其遵循准二级动力学方程动力学模型。
3)在温度30 ℃,转速150 r·min−1,解吸时间12 h的条件下,选用1 mol·L−1的NaOH溶液作为解吸剂,材料的最佳解吸率达92.60%,重复再生3次后,对p-ASA的去除率仍达到70%以上。
4)在调节牲畜养殖废水pH至4后,吸附剂对其的去除率仍能达到84.92%,具有实际应用前景。
多元金属氧化物对水中苯胂酸类污染物的吸附性能及机理
Adsorption performance and mechanism of phenylarsonic acid in water onto multi-component metal oxides
-
摘要: 砷污染一直是全球关注的环境问题。以典型苯胂酸—阿散酸(p-ASA)为目标污染物,通过水热法制备了锌-铁-锆复合金属氧化物(ZnFeZrOx)吸附剂,探究了ZnFeZrOx对p-ASA的吸附行为和吸附机理。结果表明:在pH=4,吸附温度为60 ℃,ZnFeZrOx投加量为2.2 g·L−1,p-ASA初始质量浓度为50 mg·L−1时,ZnFeZrOx对p-ASA的去除率可达95.15%。该吸附过程符合Freundlich等温吸附模型和准二级动力学模型,最大吸附容量为595.23 mg·g−1。而在吸附实际废水的实验中,ZnFeZrOx对牲畜养殖废水中p-ASA的去除率仍保持在较高的水平(84.92%)。表征结果表明,ZnFeZrOx在吸附过程中具有良好的化学稳定性,Fe-OH为主要吸附活性位点,Zn-O和Zr-O起到一定作用。ZnFeZrOx具有良好的再生性能,重复使用3次后,对p-ASA的去除率仍能达到70%以上。Abstract: Arsenic contamination has been a global environmental issue. In this study, the typical phenylarsonic acid, named p-ASA, was selected as the target pollutant to investigated the adsorption behaviour and mechanism of zinc-iron-zirconium metallic oxide (ZnFeZrOx) prepared by hydrothermal method. The results indicated that 95.15% of p-ASA was absorbed on the ZnFeZrOx under the optimal conditions: pH4, 60 ℃, ZnFeZrOx dosage of 2.2 g·L−1, and initial p-ASA concentration of 50 mg·L−1. The adsorption process was in accordance with the Freundlich isothermal adsorption model and quasi-secondary kinetic model, and the maximum adsorption capacity was 595.23 mg·g−1. In the experiment of adsorption of livestock breeding wastewater, the removal rate maintained at a high level (84.92%). In addition, ZnFeZrOx showed a good chemical stability during p-ASA adsorption, Fe-OH was the main adsorption active site, and Zn-O and Zr-O played a certain role. After three times of recycle, the removal efficiency of p-ASA still reached higher than 70%, manifesting that ZnFeZrOx had a good regeneration performance.
-
Key words:
- adsorption /
- p-arsanilic acid /
- ZnFeZrOx
-
对农业废水中有机砷的去除是近年来的研究热点之一[1]。阿散酸p-ASA(p-arsanilic acid),作为最常见的畜禽有机砷添加剂之一,常用于促进肉鸡肉鸭生长和抑制禽体内寄生虫病。然而,诸如p-ASA此类苯胂酸在动物食用后并非停留在其体内,大部分会经过动物的排泻行为释放到环境中,在外界环境的生物和化学作用下,p-ASA会转化为强毒性和强流动性的无机砷(As(V)和As(III))[2],从而导致一系列砷污染的环境问题。其中,p-ASA通过直接排放或是淋溶作用进入附近地表水和地下水中,造成水体环境介质被破坏,且由于苯胂酸类物质具有微生物毒性,不易在水环境中自然降解,就会进一步因食物链富集作用进入人体造成更大的危害。
为解决砷污染的源头问题,亟待探索高效去除有机砷的方法。目前,解决有机砷污染主流的2种方法是吸附及氧化降解[3],尽管氧化法对有机砷具有高去除率的特点,然而存在无法妥善解决降解产物,即无机砷的释放问题[4],而吸附法兼具使用成本低、可重复利用性好、生产方便高效、工艺简单、中间产物向环境释放风险低等优点,是一种应用前景较好的有机砷去除技术[5]。有机砷的吸附剂主要包括铁基材料、高分子吸附剂、碳材料以及金属有机框架(MOFs)[6]等材料。其中,已有较多研究表明,铁基材料(如MIL-100(Fe)和MIL-101(Fe)等铁基MOFs)对有机砷化合物具有良好的吸附性能[7-8]。GAO等[5]考察了苯胂酸在铁基金属-有机骨架(MIL-88A)上的吸附行为及机理,发现配位(Fe-O-As)和氢键在吸附过程中起了主要作用。而锆基材料作为一种阴离子污染物吸附剂,也被加以探究[9]。WANG等[10]率先采用UiO-66对As(V)进行吸附,发现Zr—O键与目标阴离子发生配位作用的过程是UiO-66吸附容量大的主要原因。此外,赵娜等[11]发现当Zn2+被引入Fe3O4水溶液体系时,Zn2+与As(V)发生络合反应并形成金属-配体络合物,其与Fe3O4的结合力增强,对As(V)的去除产生协同作用,去除率从66%增强至高达99%。综上所述,若能将3种金属有机结合,或能进一步促进MOFs对有机砷的吸附作用。
因此,本研究选择铁、锌、锆3种吸附性能优良的金属为原料,通过水热法制备得到多元金属氧化物吸附剂(ZnFeZrOx),采用FT-IR、XRD、XPS等分析手段对ZnFeZrOx的结构和面貌进行表征,拟研究其对有机砷中的典型代表—阿散酸(p-ASA)的吸附去除效率,并探究相关的吸附机理,本研究以期为有机砷废水的处理提供参考。
1. 材料与方法
1.1 实验材料
本研究中主要试剂为:硝酸铁(Fe(NO3)3·9H2O)、硝酸锌(Zn(NO3)2·6H2O)、硝酸锆(Zr(NO3)4·5H2O)购自国药集团化学试剂有限公司;硝酸锆(Zr(NO3)4·5H2O)购自上海麦克林生化科技有限公司;氯化氢(HCl)购自上海沪试实验室器材股份有限公司;氢氧化钠(NaOH)、十六烷基三甲基溴化铵(CTAB)、无水乙醇购自天津福晨化学试剂厂;对氨基苯胂酸(p-ASA)纯度98%,氯化钠(NaCl)、氯化镁(MgCl2)、氯化钙(CaCl2)、硫酸钠(Na2SO4)、硝酸钠(NaNO3)、磷酸二氢钾(KH2PO4)购自上海阿拉丁生化科技股份有限公司。以上试剂均为分析纯。
1.2 吸附剂的制备
在30 mL的0.2 mol·L−1 Zn(NO3)2 溶液中投加0.2 g CTAB,然后加入30 mL 0.4 mol·L−1 Fe(NO3)3溶液,再加入30 mL 0.1 mol·L−1 Zr(NO3)4。在常温度下进行磁力搅拌30 min,用0.6 mol·L−1 NaOH溶液将pH调节到12。再将其进行30 min的超声水浴预处理,而后通过500 mL高压釜在130 ℃下反应24 h。反应结束后冷却至室温,抽滤分离后水洗沉淀物,以减少产品中的杂质。最后,通过真空干燥箱(70 ℃,7 h以上)干燥所得吸附剂。
1.3 等温吸附特征及动力学特征实验
1)吸附特征及影响因素。称取50 mL 50 mg·L−1的p-ASA溶液,投加一定量自制ZnFeZrOx吸附剂粉末(平均粒径为42 nm),并分别探究吸附剂投加量、pH、吸附时间、离子强度、共存离子的干扰等对吸附效果的影响,在一定温度和摇床转速条件下反应一定时间,达到吸附平衡后,通过紫外可见分光光度计在252 nm处测定上清液中p-ASA吸光度,计算p-ASA的去除率和ZnFeZrOx的平衡吸附容量以测定吸附剂对p-ASA的吸附效果。其中,事先测得的p-ASA标准曲线为y= 0.062 7 x+0.016 3 (R2=0.999 3),其检测限为0.553 mg·L−1。
吸附剂投加量对吸附效果的影响:在pH=4的条件下,改变ZnFeZrOx吸附剂粉末投加量(投加量分别为0.02、 0.05、0.08、0.11、0.14 g),吸附15 h后测定不同含量的样品对p-ASA的吸附效果。pH对吸附效果的影响:分别调节反应体系的pH至1~12,投加0.11 g吸附剂,吸附15 h后测定不同pH对p-ASA的吸附效果。离子强度对吸附效果的影响:配置一系列以NaCl作为背景电解质成分的p-ASA溶液(NaCl浓度分别0、0.1、1、10、100 mmol·L−1),调节体系pH=4,其余条件不变,测定不同离子强度对p-ASA吸附效果的影响。共存离子干扰对吸附效果的影响:选取不同浓度(0、1、10 mmol·L−1)的Cl−1、NO3−、SO42−、CO32−作为共存离子,保持其余条件不变,反应结束后,测定其对p-ASA吸附效果的影响。
2)吸附等温线。改变温度和p-ASA溶液浓度,其余的条件相同,进行吸附平衡实验,采用Langmuir和Freundlich吸附等温线模型对ZnFeZrOx吸附p-ASA的过程进行研究。
3)吸附动力学。在其他实验条件相同时,间隔一定时间进行取样,测定该时刻样品中p-ASA的去除率及ZnFeZrOx的吸附容量,采用Lagergren准一级和准二级动力学模型对ZnFeZrOx吸附p-ASA的过程进行吸附性能考察。
1.4 表征分析
X射线衍射分析(XRD)采用X射线衍射仪(MiniFlex600,Rigaku Corporation)对本实验吸附前后的ZnFeZrOx进行分析,扫描范围为10°~80°。傅里叶红外光谱分析(FT-IR)采用傅立叶红外光谱仪(AVAT-AR 360型)对吸附前后的ZnFeZrOx进行研究。样品粉末与KBr混合,混合比为1:100,测量范围为4 000~400 cm−1。采用 X射线光电子能谱分析(XPS) (Thermo Scientific ESCALAB 250 X)测定吸附前后的ZnFeZrOx的表面组成、化学状态以及元素成分。
1.5 吸附剂解吸及回用性能实验
称取8份已达饱和吸附量的ZnFeZrOx各0.05 g于锥形瓶中,分别加入10 mL质量浓度为1 mol·L−1的NaHCO3、NaOH、Na2CO3和NaCl溶液,每2份为1组做平行实验。通过恒温培养振荡器进行解吸实验,设置温度30 ℃,转速150 r·min−1,振荡12 h。反应结束后,采用紫外可见分光光度法测定p-ASA的浓度,计算解吸率和吸附容量。并探究解吸剂浓度对解吸效果的影响。称取8份已达饱和吸附量的ZnFeZrOx各0.05 g于锥形瓶中,分别加入质量浓度分别为0.1、0.5、1、2 mol·L−1最优解吸剂各10 mL,每2份为一组做平行实验。随后放入恒温培养振荡器中,以上述解吸条件进行同样操作,并计算解吸率。
1.6 吸附剂的实际应用性能实验
配置固体浓度为50 g·L−1的猪粪浸出液备用,测得pH为7.2,取部分浸出液调节pH至4。同时配置质量浓度为50 mg·L−1的p-ASA溶液,分别调节pH为4和7.2。称取pH=7.2的浸出液、pH=4的浸出液、pH=7.2的p-ASA溶液、pH=4的p-ASA溶液各50 mL,并分别投加0.11 g吸附剂,吸附15 h后通过紫外可见分光光度计测定不同pH下猪粪浸出液样品及p-ASA溶液的吸附效果。
2. 结果与讨论
2.1 等温吸附特征及影响因素
1) ZnFeZrOx投加量对吸附p-ASA的影响。投加量对ZnFeZrOx吸附p-ASA的影响如图1(a)所示。从图1(a)可以看出,随着投加量的增加,ZnFeZrOx吸附p-ASA的去除率呈现先升高再降低的特征,当投加量由0.02 g增至0.11 g时,去除率由25.15%增至75.73%;当投加量增至0.14 g时,去除率降至71.23%。原因可能是当ZnFeZrOx吸附剂的投加量增加,会使吸附剂与p-ASA结合的活性位点增加,此时去除率升高;而当体系中吸附剂量进一步增加,造成溶液中p-ASA的相对浓度降低,此外,还可能会发生吸附剂之间互相团聚的现象,导致吸附剂内部空隙堵塞,比表面积降低,进而导致去除率略有所下降[12]。从整体上看,随ZnFeZrOx投加量的增加,平衡吸附容量Qe由31.43 mg·g−1降至12.72 mg·g−1。p-ASA的吸附平衡容量随ZnFeZrOx投加量的升高而降低。这可能是由于当体系中p-ASA的初始浓度和体积不变,随着吸附剂的增加,单位质量所吸附的p-ASA减少,造成吸附平衡容量的下降,这表明p-ASA的吸附过程是一个扩散主导的过程,包括吸附剂表面扩散和颗粒内扩散过程[13-15]。综上所述,选取0.11 g作为ZnFeZrOx对p-ASA进行吸附的最佳投加量。
 图 1 不同因素对ZnFeZrOx吸附p-ASA的影响Figure 1. Effect of different factors on adsorption of p-ASA onto ZnFeZrOx
图 1 不同因素对ZnFeZrOx吸附p-ASA的影响Figure 1. Effect of different factors on adsorption of p-ASA onto ZnFeZrOx2) pH对ZnFeZrOx吸附p-ASA的影响。由图1(b)所示,当pH 在1~4时,吸附剂对p-ASA的去除率较高,均维持在 93% 以上,在pH=4时去除率最高达到95.15%;当pH>4 时,去除率逐步下降。以pH=4为转折点的去除率变化趋势与其他研究[14]结果一致。ZnFeZrOx吸附p-ASA的平衡吸附容量和p-ASA去除率的变化趋势趋于一致。在pH=4 时,Qe=21.63 mg·g−1。由此可见,该吸附受pH影响严重,可能因为该吸附是以物理吸附为主,配位体交换和表面配位作用为辅[14]。p-ASA的pKa为1.9、4.1和9.2,分别对应于p-ASA上对位-NH3+和2个胂酸基团(As-OH)的去质子化[16]。在pH<4 时,由于溶液中[H+]高,静电吸引力强,而p-ASA为阴离子污染物,因此,此时去除率高;当pH>4时,溶液pH升高会促进p-ASA被离子化而溶于水中,从而不易被吸附[17];此时溶液中OH−的增多可增加ZnFeZrOx表面上的直接竞争吸附行为,导致去除率下降[18]。随着OH−被大量吸附,ZnFeZrOx表面的正电荷不断减少,而负电荷不断增多,进一步增加其表面静电斥力,静电作用被削弱,导致p-ASA去除率降低。在低pH条件下,吸附剂表面以OH2为主,可产生更多的配位体交换位点,也起到一定吸附作用[19]。此外,Fe离子与p-ASA可能发生表面配位作用形成Fe-O-As基团[20],溶液pH的增大可能导致Fe离子从ZnFeZrOx中溶解,也会引起p-ASA去除率的降低。实验中虽然在pH=1时,平衡吸附容量和去除率最大,然而考虑到实际应用时满足相关条件的实施难度和成本问题以及强酸条件的潜在威胁,选取pH=4作为后续实验的最佳pH。
3)离子强度对ZnFeZrOx吸附p-ASA的影响。如图1(c)所示,随着离子强度的增大,吸附行为受影响,p-ASA去除率由95.15%降至74.95%。这可能是因为Cl−与p-ASA共同竞争ZnFeZrOx上的吸附位点,说明ZnFeZrOx吸附p-ASA的过程是外层络合吸附和静电吸附[12, 21]。
4)共存离子干扰对ZnFeZrOx吸附p-ASA的影响。在天然水体中往往存在多种离子,常见的阴离子有SO42−、CO32−、NO3−、PO43−等,在吸附除砷的过程中,他们可能同样与p-ASA间存在竞争机制。由图1(d)可知,不同共存离子对ZnFeZrOx吸附p-ASA的干扰程度不同,PO43−的干扰最强,NO3−次之,SO42−和Cl−的干扰作用较弱。当PO43−浓度由0 mmol·L−1增强到1 mmol·L−1和10 mmol·L−1时,ZnFeZrOx对p-ASA的去除率分别由95.15%降至15.26%和8.91%。这说明磷酸根与胂酸根之间存在极强的竞争机制,在高磷水体中,吸附剂ZnFeZrOx对p-ASA的吸附存在明显的空间位阻效应[22]。
5)等温吸附特征研究及p-ASA初始浓度的影响。由图2(a)~(b)可知,当p-ASA加入初始质量浓度为10 mg·L−1时,p-ASA的去除率为76.16%。当初始浓度增至160 mg·L−1,去除率降至41.9%。总体来看,去除率与p-ASA初始浓度呈负相关,这可能是因为在p-ASA初始浓度较高时,ZnFeZrOx吸附位点有限,吸附趋于饱和后,吸附难以继续进行,从而造成去除率下降。
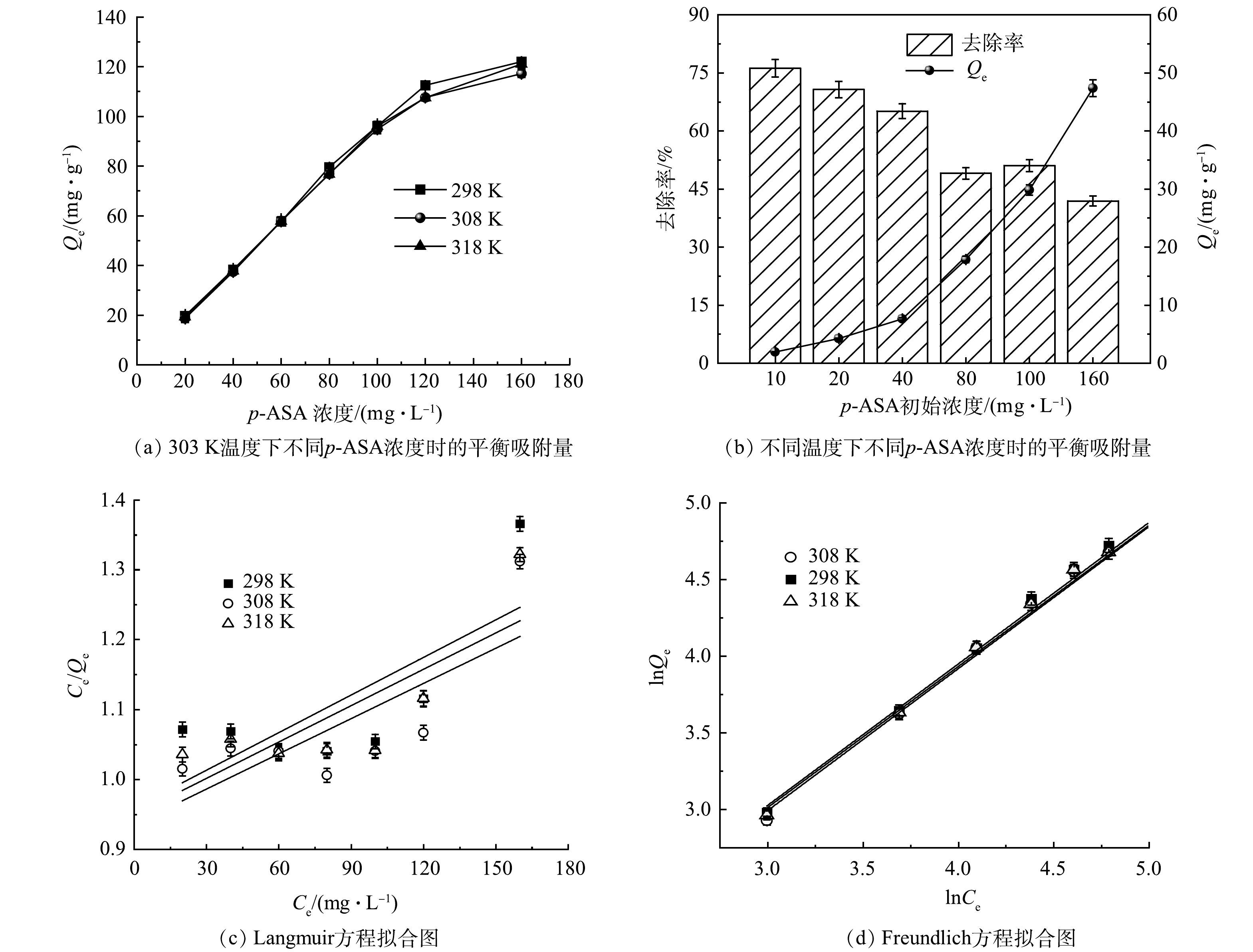 图 2 不同温度下ZnFeZrOx吸附p-ASA的吸附等温线及线性拟合Figure 2. Adsorption isotherms and linear fitting of p-ASA adsorbed by ZnFeZrOx at different temperatures
图 2 不同温度下ZnFeZrOx吸附p-ASA的吸附等温线及线性拟合Figure 2. Adsorption isotherms and linear fitting of p-ASA adsorbed by ZnFeZrOx at different temperatures以Ce为横坐标、Ce/Qe为纵坐标和以lnCe为横坐标、lnQe为纵坐标进行作图,拟合结果如图2(b)和图2(c)所示。利用Langmuir以及Freundlich公式对拟合后的数据进行计算,得到Langmuir和Freundlich模型的相关参数如表1和表2所示。比较图2(c)和图2(d)可以明显看出,Langmuir吸附等温线模型的拟合程度明显低于Freundlich模型,结合表1可知,在3种温度下,Langmuir模型的R2均远小于0.9,且计算所得理论Qe远大于实际实验值(20.285 mg·g−1)。这表明Langmuir模型不适合用于描述该吸附过程。但由表2可以看出,Freundlich吸附等温线模型对该吸附过程拟合所得R2均大于0.98,表明拟合程度良好,说明p-ASA通过内置配位层与ZnFeZrOx中的ZnFe2O4、Fe3O4和Fe等结合。此外,由表2中KL值可见,该值随温度升高而略微增大,表明升温能促进吸附反应的进行。吸附强度系数n>1 (1/n<1),表明吸附容易发生,说明p-ASA容易被ZnFeZrOx吸附。
表 1 ZnFeZrOx吸附p-ASA的Langmuir吸附等温式Table 1. The Langmuir adsorption isotherm equations for p-ASA onto ZnFeZrOx温度/K Qm/(mg·g−1) KL/(L·mg−1) Langmuir吸附等温式 R2 298 558.66 0.001 87 Ce/Qe=0.001 8 Ce+0.959 5 0.459 308 595.23 0.001 80 Ce/Qe=0.001 7 Ce+0.935 5 0.496 318 578.03 0.001 82 Ce/Qe=0.001 7 Ce+0.949 6 0.565 | Show Table DownLoad:
CSV
表 2 ZnFeZrOx吸附p-ASA的Freundlich吸附等温式Table 2. The Freundlich adsorption isotherm equations for p-ASA onto ZnFeZrOx
DownLoad:
CSV
表 2 ZnFeZrOx吸附p-ASA的Freundlich吸附等温式Table 2. The Freundlich adsorption isotherm equations for p-ASA onto ZnFeZrOx温度/K KF n Freundlich吸附等温式 R2 298 1.24 1.079 lnQe=0.926 6 lnCe+0.212 4 0.982 308 1.29 1.083 lnQe=0.923 3 lnCe+0.256 3 0.985 318 1.28 1.086 lnQe=0.921 2 lnCe+0.248 3 0.987 | Show TableDownLoad:
CSV
2.2 吸附动力学特征
对所得数据进行吸附动力学分析,结果如图3(a)所示。从图3(a)可以看出,ZnFeZrOx对的吸附过程可分为3个阶段。在0~3 h内,吸附去除率和平衡吸附量快速升高,表明该过程为快速吸附过程,此阶Qe段时,吸附剂表面吸附位点空缺较多且吸附剂表面与溶液之间浓度差较大,溶液中p-ASA分子与吸附剂碰撞,p-ASA分子被吸附剂吸附,物理吸附起主导作用,吸附速率快。在3~15 h内,去除率和Qe缓慢增加,表明该过程为慢速吸附过程。此时ZnFeZrOx表面吸附位点已被逐渐占用,溶液中p-ASA分子在接触过程中碰到空缺位点的概率减小,溶液浓度梯度也减小,从而导致吸附速率减缓。此外,ZnFeZrOx可能与吸附在表面的p-ASA分子发生电子转移、离子交换、配位体交换等反应,化学吸附为主导过程,去除速率减缓但去除率却保持上升。反应至15 h后,去除率和平衡吸附容量基本保持不变,此时吸附反应达到平衡。在15 h时去除率达到最高,为89.25%;在17 h时Qe达到最高,为20.28 mg·g−1。选取15 h为后续实验的最佳反应时间。
并分别以ln(Qe−Qt)对t、t/Qt对t、Qt对t1/2作图,对该反应的吸附动力学进行研究,结果如图3(b)、图3(c)、图3(d)和表3所示。如图3(b)~(c)所示,与准一级方程相比,准二级动力学方程曲线的拟合程度更好。结合表3可知,准二级动力学方程的R2值更接近于1 (0.997)。准一级动力学方程计算的理论Qe' (9.250 mg·g−1)与实验所得平衡吸附量Qe相比有很大差异,而准二级动力学方程的Qe' (20.008 mg·g−1)与实验Qe (20.285 mg·g−1)接近。说明准二级动力学方程更适用于ZnFeZrOx对p-ASA的吸附动力学过程,因此ZnFeZrOx对p-ASA吸附主要是化学吸附。由图3(d)可见,p-ASA在ZnFeZrOx上的吸附可被分为3个过程:液膜扩散过程、颗粒内扩散过程和吸附表面反应过程。3个过程的曲线斜率依次减小,说明吸附过程的速度依次减缓,第3阶段边界层对吸附效果的影响较大。另外,3个过程在图3(d)上都显示出较好的线性关系,但折线不通过原点,说明粒子内扩散反应影响了吸附速率。
表 3 ZnFeZrOx吸附p-ASA的动力学参数Table 3. Adsorption kinetic parameters of p-ASA onto ZnFeZrOx动力学方程 K1/min−1 K2/(g·(mg·min)−1) Qe'/(mg·g−1) R2 准一级 0.00292 — 9.250 0.947 准二级 — 0.00121 20.088 0.997 | Show TableDownLoad:
CSV
2.3 吸附剂再生性能分析
1)解吸剂的选择。4种钠系解吸剂对饱和ZnFeZrOx的解吸效果如图4(a)所示。其中,NaOH表现出最优解吸效果,在第1次解吸实验后解吸率为92.60%,吸附容量为20.00 mg·g−1。Na2CO3溶液和NaHCO3溶液的解吸效果相对较好,而NaCl的解吸效果则相对较低。因此,后续实验选取NaOH作为最佳解吸剂。
2)解吸剂浓度的影响。如图4(b)可知,随着NaOH浓度的增大,解吸率和ZnFeZrOx吸附容量先增大后保持几乎不变。当NaOH溶液为0.1 mol·L−1和0.5 mol·L−1时,解吸率相对较低;当NaOH浓度为1 mol·L−1时,解吸率为92.60%,吸附容量为20.00 mg·g−1;继续将NaOH浓度增加至2 mol·L−1后,对解吸率无进一步提高。因此,选取1 mol·L−1为NaOH的最佳解吸浓度。
3) ZnFeZrOx的再生使用性能。如图4(c)所示,饱和ZnFeZrOx经过5次循环再生后,吸附容量由20.3 mg·g−1降至12.1 mg·g−1,去除率相应由89.32%降至53.24%。在循环再生到第3次时,去除率为72.60%。表明ZnFeZrOx再生3次后仍可对水体中大部分p-ASA进行吸附。此外,以上结果间接表明1 mol·L−1 NaOH可以成功将p-ASA从吸附剂表明解吸下来,且ZnFeZrOx再生效果较好。
2.4 吸附前后材料的表征分析
1) XRD分析。图5(a)为ZnFeZrOx吸附p-ASA前后的XRD对比图。如图5(a)所示,吸附前后ZnFeZrOx的XRD图几乎未有发生变化,表明吸附剂在吸附过程中能维持较稳定的化学形态,间接表明ZnFeZrOx的化学稳定性。
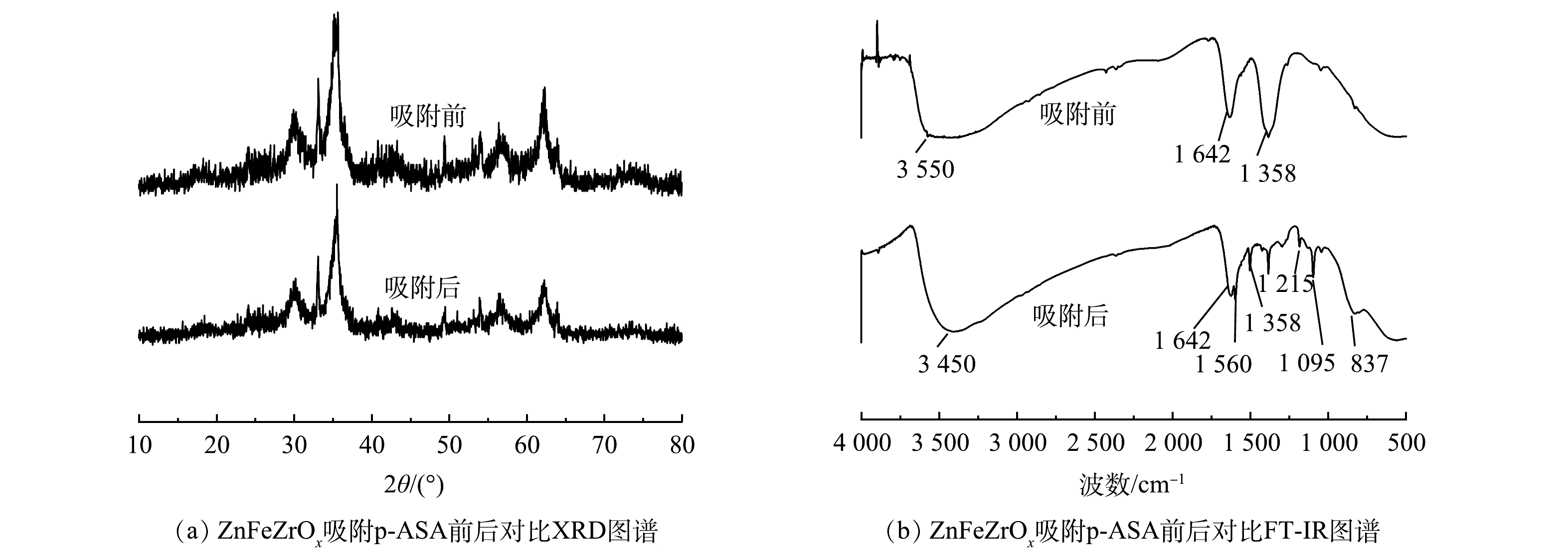 图 5 ZnFeZrOx吸附p-ASA前后对比XRD和FT-IR图谱Figure 5. Comparison of XRD and FT-IR spectra before and after adsorption of p-ASA onto ZnFeZrOx
图 5 ZnFeZrOx吸附p-ASA前后对比XRD和FT-IR图谱Figure 5. Comparison of XRD and FT-IR spectra before and after adsorption of p-ASA onto ZnFeZrOx2) FI-IR分析。由图5(b)可见,在3 100~3 700 cm−1处形成一个宽频带区域[23],可能是由于H2O分子以及吸附剂中-OH官能团的吸收附带,并且比较吸附前和吸附后曲线观察到,-OH由3 550 cm−1 蓝移至 3 450 cm−1附近,说明吸附过程中-OH基团也参与吸附。1 642 cm−1为H2O和-OH的弯曲振动峰[24]。由吸附后的曲线可见,在837 cm−1处新出现一个峰。经查验为As-O的特征峰[16],可能是As-O-M(M代表Zr、Fe)的振动吸收[14],表明p-ASA通过配位体交换作用被吸附在三相金属氧化物吸附剂表面[25]。1 358 cm−1应为-OH结构伸缩振动的特征峰[26-27],与吸附前相比,该峰在吸附后明显减弱,表明p-ASA在吸附过后才能中替代了部分-OH并与ZnFeZrOx相连,证明-OH参与反应且p-ASA并非单纯沉积而可能是发生了配位反应。1 095 cm−1应为C-As的弹性振动峰,1 215 cm−1和1 560 cm−1分别为p-ASA上的C-N伸缩振动特征峰和-NH2的剪切振动峰[28-29]。
3) XPS分析。图6为ZnFeZrOx吸附p-ASA溶液的前后的XPS图谱。图6(a)~f)分别为元素As、C、O、Zn、Fe、Zr的高分辨图。在图6(a)中可以看出,在44.6 eV处出现新的吸收峰为As3d的特征峰。这表明吸附后的p-ASA中As元素主要以As(V)的形式存在,并已成功结合到ZnFeZrOx表面[27]。图6(b)中ZnFeZrOx样品吸附p-ASA前的C1s 谱图能够拟合出3个峰,分别为284.6 eV的C-C或C=C、288.5 eV的C-O以及289.0 eV的O-C=O。吸附后,原284.6 eV的峰蓝移至284.8 eV,且C-C与C=C二者占比由70.92%升至94.78%,这可能是因为p-ASA分子在吸附剂表面堆积。此外C-O和O-C=O的峰,在吸附p-ASA后,占比分别由17.02%和12.06%降至3.73%和1.49%,说明在p-ASA的吸附过程中,-COOH官能团发挥了重要作用[30]。
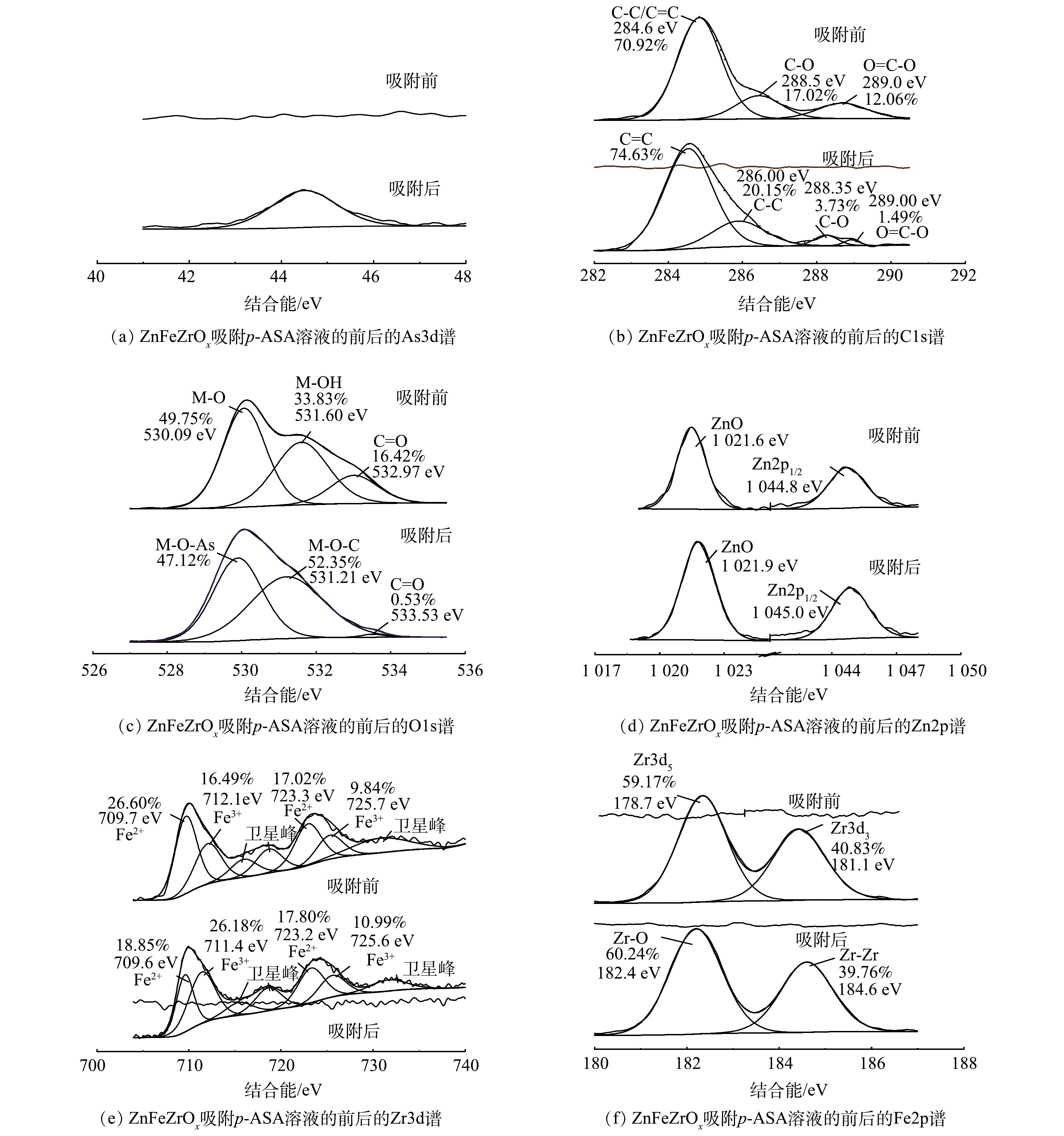 图 6 ZnFeZrOx吸附p-ASA前后的XPS图谱Figure 6. Comparison of XPS spectra before and after adsorption of p-ASA onto ZnFeZrOx
图 6 ZnFeZrOx吸附p-ASA前后的XPS图谱Figure 6. Comparison of XPS spectra before and after adsorption of p-ASA onto ZnFeZrOx由图6(c)可见,吸附前样品中O信号较强,表明制备出的三相金属复合材料为含氧化合物[31]。O1s的3个原始峰为530.0、531.60、532.97 eV,分别代表由Fe、Zn、Zr和O的氧化物M-O(锌铁锆三种金属皆以M表示)、M-OH、C=O。在吸附p-ASA后,消耗了部分羧基,还形成了M-O-As、As-O、M-O-C等有机配体。此外,530.09 eV处的峰红移至529.89 eV,表明M-OH基团参与了p-ASA的吸附并在ZnFeZrOx表面形成M-O基团[24],这与FT-IR的分析结果一致[8]。由图6(d)、图6(e)、图6(f)可以看出,ZnFeZrOx中Zn主要为二价,Fe主要为二价,而ZnFeZrOx中的Zr则主要为正四价。
由图6(d)可见,吸附前,1 021.9 eV对应的是Zn2p3/2的Zn-O,1 045.0 eV对应于Zn2p1/2.吸附后,Zn-O和Zn2p1/2的特征峰分别红移至1 021.6 eV和1 044.8eV,表明Zn-O参与了p-ASA的配位。由图6(e)可见,吸附前,归属于Fe2p3/2的特征峰有709.6 eV和711.4 eV,而723.2 eV和725.6 eV则归属于Fe2p1/2[32]。吸附后,属于Fe2p3/2的峰移至709.7 eV和712.1 eV,归属于Fe2p1/2的峰移至723.3 eV和725.7 eV。吸附前后,Fe由二价转化为三价,表明Fe-OH是吸附阿散酸的活性位点[30]。由图6(f)可见,在吸附前XPS曲线上的182.4 eV对应Zr-O的结合能,而184.6 eV处对应Zr-Zr[33];吸附后,Zr-O的结合能偏移至178.7 eV,同时Zr-Zr的结合能偏移至181.1 eV,二者均向结合能低的方向偏移。表明Zr-O参与了p-ASA的配位,并在吸附剂吸附p-ASA时起到一定的作用。
2.5 吸附剂在牲畜养殖废水中的应用
牲畜养殖废水对ZnFeZrOx吸附p-ASA的影响。实际牲畜养殖废水往往组分复杂,具有高COD的特点[34]。图7反映了ZnFeZrOx对猪粪浸出液中p-ASA的吸附效果。如图7所示,在未调节pH的浸出液中(pH=7.2),ZnFeZrOx对p-ASA的去除率较低,仅有42.23%,而将其pH调节至4后,去除率却保持在较高的水平(84.92%),与对照组的结果接近,说明ZnFeZrOx在实际处理高碳源的牲畜养殖废水时受废水pH影响较大。
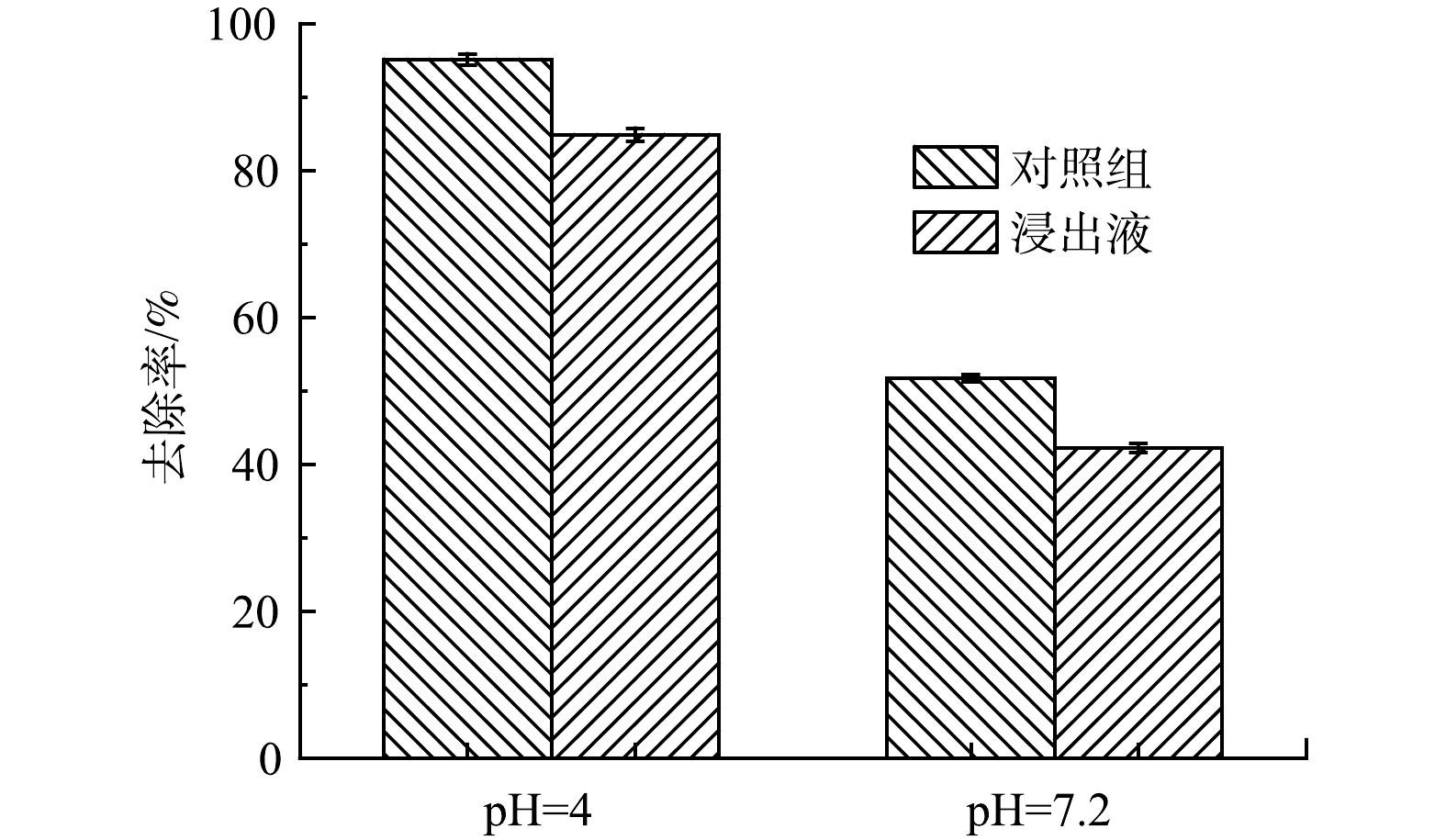 图 7 ZnFeZrOx在牲畜养殖废水中的应用Figure 7. Application of ZnFeZrOx in livestock breeding wastewater treatment
图 7 ZnFeZrOx在牲畜养殖废水中的应用Figure 7. Application of ZnFeZrOx in livestock breeding wastewater treatment3. 结论
1)三相金属氧化物吸附剂ZnFeZrOx 对典型的有机砷污染物p-ASA具有良好的吸附性能,最大去除率达95.15%,最大吸附容量为595.23 mg·g−1。
2)三相金属氧化物材料ZnFeZrOx中,Fe、Zn、Zr主要以Fe3+、Zn2+及Zr4+的形式存在,Fe-OH为当中主要吸附活性点位,Zn-O和Zr-O起到一定的吸附作用。静态吸附实验结果表明,ZnFeZrOx吸附p-ASA的最佳条件为:吸附剂投加量为0.11 g,pH=4,吸附时间为15 h,在此条件下,吸附过程为吸热过程,其遵循准二级动力学方程动力学模型。
3)在温度30 ℃,转速150 r·min−1,解吸时间12 h的条件下,选用1 mol·L−1的NaOH溶液作为解吸剂,材料的最佳解吸率达92.60%,重复再生3次后,对p-ASA的去除率仍达到70%以上。
4)在调节牲畜养殖废水pH至4后,吸附剂对其的去除率仍能达到84.92%,具有实际应用前景。
-
图 1 不同因素对ZnFeZrOx吸附p-ASA的影响
Figure 1. Effect of different factors on adsorption of p-ASA onto ZnFeZrOx
图 2 不同温度下ZnFeZrOx吸附p-ASA的吸附等温线及线性拟合
Figure 2. Adsorption isotherms and linear fitting of p-ASA adsorbed by ZnFeZrOx at different temperatures
图 5 ZnFeZrOx吸附p-ASA前后对比XRD和FT-IR图谱
Figure 5. Comparison of XRD and FT-IR spectra before and after adsorption of p-ASA onto ZnFeZrOx
图 6 ZnFeZrOx吸附p-ASA前后的XPS图谱
Figure 6. Comparison of XPS spectra before and after adsorption of p-ASA onto ZnFeZrOx
图 7 ZnFeZrOx在牲畜养殖废水中的应用
Figure 7. Application of ZnFeZrOx in livestock breeding wastewater treatment
表 1 ZnFeZrOx吸附p-ASA的Langmuir吸附等温式
Table 1. The Langmuir adsorption isotherm equations for p-ASA onto ZnFeZrOx
温度/K Qm/(mg·g−1) KL/(L·mg−1) Langmuir吸附等温式 R2 298 558.66 0.001 87 Ce/Qe=0.001 8 Ce+0.959 5 0.459 308 595.23 0.001 80 Ce/Qe=0.001 7 Ce+0.935 5 0.496 318 578.03 0.001 82 Ce/Qe=0.001 7 Ce+0.949 6 0.565
下载: 导出CSV
表 2 ZnFeZrOx吸附p-ASA的Freundlich吸附等温式
Table 2. The Freundlich adsorption isotherm equations for p-ASA onto ZnFeZrOx
温度/K KF n Freundlich吸附等温式 R2 298 1.24 1.079 lnQe=0.926 6 lnCe+0.212 4 0.982 308 1.29 1.083 lnQe=0.923 3 lnCe+0.256 3 0.985 318 1.28 1.086 lnQe=0.921 2 lnCe+0.248 3 0.987
下载: 导出CSV
表 3 ZnFeZrOx吸附p-ASA的动力学参数
Table 3. Adsorption kinetic parameters of p-ASA onto ZnFeZrOx
动力学方程 K1/min−1 K2/(g·(mg·min)−1) Qe'/(mg·g−1) R2 准一级 0.00292 — 9.250 0.947 准二级 — 0.00121 20.088 0.997
下载: 导出CSV
-
[1] MANGALGIRI KP, ADAK A, BLANEY L. Organoarsenicals in poultry litter: Detection, fate, and toxicity[J]. Environment International, 2015, 75: 68-80. doi: 10.1016/j.envint.2014.10.022 [2] SU S, CAO C, ZHAO Y, et al. Efficient transformation and elimination of roxarsone and its metabolites by a new alpha-FeOOH@GCA activating persulfate system under UV irradiation with subsequent As(V) recovery[J]. Applied Catalysis B:Environmental, 2019, 245: 207-219. doi: 10.1016/j.apcatb.2018.12.050 [3] LIANG L, XI F, TAN W, et al. Review of organic and inorganic pollutants removal by biochar and biochar-based composites[J]. Biochar, 2021, 3: 255-281. doi: 10.1007/s42773-021-00101-6 [4] YE C, DENG J, HUAI L, CAI A, et al. Multifunctional capacity of CoMnFe-LDH/LDO activated peroxymonosulfate for p-arsanilic acid removal and inorganic arsenic immobilization: performance and surface-bound radical mechanism[J]. Science of the Total Environment, 2022, 806: 150379. doi: 10.1016/j.scitotenv.2021.150379 [5] GAO MW, LI B, LIU J, et al. Adsorption behavior and mechanism of p-arsanilic acid on a Fe-based metal–organic framework[J]. Journal of Colloid and Interface Science, 2023, 629: 616-627. doi: 10.1016/j.jcis.2022.08.133 [6] WANG Z, FANG Y, YANG Y, et al. Synthesis of ε-MnO2@MIL-100(Fe) composite for p-arsanilic acid removal[J]. Journal of Environmental Chemical Engineering, 2022, 10(3): 107876. doi: 10.1016/j.jece.2022.107876 [7] FURUKAWA H, CORDOVA KE, O'KEEFFE M, et al. The chemistry and applications of metal-organic frameworks[J]. Science, 2013, 341: 1230444. doi: 10.1126/science.1230444 [8] LI Z, LIU X, JIN W, et al. Adsorption behavior of arsenicals on MIL101(Fe): The role of arsenic chemical structures[J]. Journal of Colloid and Interface Science, 2019, 554: 692-704. doi: 10.1016/j.jcis.2019.07.046 [9] LAKSHMANAN D, CLIFFORD D, SAMANTA G. Arsenic removal by coagulation with aluminum, iron, titanium, and zirconium[J]. American Water Works Association Journal, 2008, 100(2): 76-89. doi: 10.1002/j.1551-8833.2008.tb08144.x [10] WANG C, LIU X, CHEN J P, et al. Superior removal of arsenic from water with zirconium metal-organic framework UiO-66[J]. Scientific Reports, 2015, 5: 16613. doi: 10.1038/srep16613 [11] 赵娜, 王瑜, 杨卫春, 等. Zn2+和HCO3-对纳米磁性铁去除水中砷的影响[J]. 中国有色金属学报, 2011, 21(9): 2285-2290. [12] 彭云. 铁氧化物对有机胂类药物的吸附行为研究[D]. 南京: 南京师范大学, 2016. [13] 郭家骏. 污泥基生物炭的制备及其改性材料对水体中有机砷的吸附性能研究[D]. 长沙: 湖南农业大学, 2019. [14] LI B, ZHU X, HU K, et al. Defect creation in metal-organic frameworks for rapid and controllable decontamination of roxarsone from aqueous solution[J]. Journal of hazardous materials, 2016, 302: 57-64. doi: 10.1016/j.jhazmat.2015.09.040 [15] ZHU X, LI B, YANG J, et al. Effective adsorption and enhanced removal of organophosphorus pesticides from aqueous solution by Zr-based MOFs of UiO-67[J]. ACS applied materials and interfaces, 2014, 7(1): 223-231. [16] DEPALMA S, COWEN S, HOANG T, et al. Adsorption thermodynamics of p-arsanilic acid on iron (oxyhydr) oxides: in-situ ATR-FTIR studies[J]. Environmental Science and Technology, 2008, 42(6): 1922-1927. doi: 10.1021/es071752x [17] PENG Y, WEI W, ZHOU H, et al. Iron humate as a novel adsorbent for p-arsanilic acid removal from aqueous solution[J]. Journal of Dispersion Science and Technology, 2016, 37(11): 1590-1598. doi: 10.1080/01932691.2015.1120219 [18] HU Q, LIU Y, GU X, et al. Adsorption behavior and mechanism of different arsenic species on mesoporous MnFe2O4 magnetic nanoparticles[J]. Chemosphere, 2017, 181: 328-336. doi: 10.1016/j.chemosphere.2017.04.049 [19] CHEN W R, HUANG C H. Surface adsorption of organoarsenic roxarsone and arsanilic acid on iron and aluminum oxides[J]. Journal of Hazardous Materials, 2012, 227: 378-385. [20] 朱瑾. 铁锰氧化物/石墨烯复合材料对水中砷的吸附性能及机理研究[D]. 杭州: 浙江大学, 2016. [21] MOHAPATRA D, MISHRA D, CHAUDHURY G R, et al. Arsenic adsorption mechanism on clay minerals and its dependence on temperature[J]. Korean Journal of Chemical Engineering, 2007, 24(3): 426-430. doi: 10.1007/s11814-007-0073-z [22] 范芳. 铁锰铈三元复合氧化物的合成及重金属砷的吸附性能研究[D]. 西安: 西安工程大学, 2017. [23] ZHENG Y M, LIM S F, CHEN J P. Preparation and characterization of zirconium-based magnetic sorbent for arsenate removal[J]. Journal of Colloid and Interface Science, 2009, 338(1): 22-29. doi: 10.1016/j.jcis.2009.06.021 [24] 叶树芯. 铁锰复合氧化物新型固定化及其除砷特性研究[D]. 武汉: 华中农业大学, 2016. [25] JUN J W, TONG M, JUNG B K, et al. Effect of central metal ions of analogous metal–organic frameworks on adsorption of organoarsenic compounds from water: plausible mechanism of adsorption and water purification[J]. Chemistry-A European Journal, 2015, 21(1): 347-354. doi: 10.1002/chem.201404658 [26] 赵丹丹. 纳米锆颗粒吸附砷的应用基础研究[D]. 武汉: 华中科技大学, 2012. [27] 侯栋科, 彭兵, 柴立元, 等. 铁酸锌选择性还原的反应机理[J]. 中国有色金属学报, 2014, 24(10): 2634-2641. doi: 10.19476/j.ysxb.1004.0609.2014.10.027 [28] SARKER M, SONG J Y, JHUNG S H, et al. Adsorption of organic arsenic acids from water over functionalized metal-organic frameworks[J]. Journal of Hazardous Materials, 2017, 335: 162-169. doi: 10.1016/j.jhazmat.2017.04.044 [29] 唐秋莎, 张东生, 顾宁, 等. 聚乙烯亚胺在锰锌铁氧体纳米粒子表面定量吸附[J]. 东南大学学报(自然科学版), 2007, 37(5): 867-872. [30] WU Q, YE X, LV Y, et al. Lignin-based magnetic activated carbon for p-arsanilic acid removal: Applications and absorption mechanisms[J]. Chemosphere, 2020, 258: 127276. doi: 10.1016/j.chemosphere.2020.127276 [31] 张艳素. 铁锆复合氧化物去除砷氟的性能研究及机制探讨[D]. 北京: 北京林业大学, 2012. [32] WANG Y, JIANG B, WANG L L, et al. Hierarchically structured two-dimensional magnetic microporous biochar derived from hazelnut shell toward effective removal of p-arsanilic acid[J]. Applied Surface Science, 2020, 540: 148372. [33] 何兴羽. 锆基MOFs吸附去除水中砷, 锑离子和汞离子检测性能研究[D]. 南昌: 南昌航空大学, 2016. [34] TIAN C, ZHAO J, OU X W, et al. Enhanced adsorption of p-Arsanilic acid from water by amine-modified UiO-67 as examined using extended X-ray absorption fine structure, X-ray photoelectron spectroscopy, and density functional theory calculations[J]. Environmental Science and Technology, 2018, 52(6): 3466-3475. doi: 10.1021/acs.est.7b05761 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3088
- HTML全文浏览数: 3088
- PDF下载数: 117
- 施引文献: 0
