


 下载:
下载:

-
近年来,锂电池 (LIBs) 因其高能量密度、高电压和良好的循环性能等优点,在交通运输和大规模能源存储行业广泛应用。据估计,LIBs的产量将以59%的增长速率从2012年的1.07×104 t增加到2025年的4.64×105 t[1],这导致大量的废旧LIBs亟待处理处置。然而这些废旧LIBs含有高值Li (5%~7%) 、Ni (5%~10%) 、Mn (5%~11%) 、Co (5%~25%) 等[2],其金属含量比自然界矿石中金属含量还高,可以作为二次能源物质加以利用。同时,随着LIBs需求量不断增加,可利用金属资源越来越稀缺[3],LIBs原材料价格不断上涨,而与开采和提炼原材料金属相比,回收废旧LIBs中的金属可以节省大量的能源。此外,废旧LIBs若不被有效处理,其含有的重金属可能会对环境产生不利影响[4]。因此,从废旧LIBs中回收有价金属不仅是保护环境的迫切需求,还是缓解资源短缺、节约生产成本的有效途径。
目前,从废旧LIBs中浸出有价金属是金属回收的一个重要环节,其浸出方法包括化学浸出和生物浸出[5]。化学浸出一般利用强酸或碱将固体废物中金属离子浸出,但该方法消耗大量的化学试剂且需要热量促进反应进行[6]。与化学浸出相比,生物浸出法因能量投入低、成本低廉、操作简单、浸出过程绿色环保等潜在优势而受到越来越多的关注[7-8];而且,微生物可以不断产生H2SO4和细胞的接触机制使浸出具有更好性能[9]。生物浸出废旧LIBs的方法可分为一步生物浸出法及两步生物浸出法。在一步生物浸出方法中,微生物与废旧LIBs一起接种在微生物培养基中,其中微生物培养和生长以及金属浸出同时发生[10]。这种浸出方式较为简单,但由于LIBs中存在的金属离子对微生物生长和活性产生不利影响,生物浸出效率可能会受到限制[11],Co、Li的浸出率均分别低于60%和80%[12-14]。为了解决上述问题,有学者开始采用两步生物浸出法浸出废旧LIBs,即微生物在培养基中生长达到活性较强、生长速度较快的对数增长期,然后添加废旧LIBs开始浸出,以抵御金属离子对微生物生长的不利影响,避免微生物的延滞期延长[15-17],有效地减少废旧LIBs对微生物的毒性作用,Co、Li的浸出率可以分别达到70%及90%以上[8, 18-19]。因此,运用两步生物浸出废旧LIBs具有一定的优势。然而,对于两步生物浸出,浸出微生物活性较强、生长速度较快的对数增长期会维持一定的时间,可分为对数增长期的前、中、后期,而不同时期的微生物活性有一定差异,但在微生物对数增长期的不同时期加入废旧LIBs对金属浸出率的影响鲜少报道。因此,选择微生物活性最强、生长速度最快的时期投加废旧LIBs以抵御加入固体废弃物对微生物生长的不良影响,也许可以简单有效地提高金属浸出率,优化两步生物浸出。
此外,选择活性高的浸出微生物是进一步提高金属浸出率的重要因素。目前,已发现40余种微生物可用于生物浸出[20],而在生物浸出废旧LIBs的研究中应用最广泛的是氧化亚铁硫杆菌(Acidithiobacillus ferrooxidans)、氧化亚铁钩端螺旋菌(Leptospirillum ferrooxidans)和氧化硫硫杆菌 (Acidithiobacillu thiooxidans)[19],这些细菌具有嗜酸、以CO2为碳源,利用铁/硫作为能源物质的特点。此外,也有利用真菌浸出废旧LIBs[10],但利用真菌进行浸出LIBs所需时间比嗜酸菌长且需要外加碳源,能耗较高[7]。因此,选择嗜酸菌浸出LIBs更有利。同时,XIN等[9]研究发现,具有铁硫氧化能力的生物浸出体系有较高的浸出效率。因此,本研究选取具有铁硫氧化能力且应用较广泛的A. ferrooxidans作为浸出微生物。
本研究采用两步生物浸出法对废旧LIBs进行浸出,并探究在A. ferrooxidans对数增长期的前、中和后期向浸出体系中投加废旧LIBs对金属浸出率的影响和作用机制,以期通过优化两步生物浸出,获得更高的金属浸出率,为生物浸出废旧LIBs的工业化提供参考。
-
浸出原料。本实验使用的废旧LIBs由广东深圳某废旧电池回收厂提供,该废旧LIBs已经过放电及初步破碎处理。
实验菌种。本实验选用A. ferrooxidans DX作为浸出微生物,该菌由中南大学教育部生物冶金实验室提供。
实验药品。硫酸 (H2SO4) 、硫酸铵 ((NH4)2SO4) 、氯化钾 (KCl) 、磷酸氢二钾 (K2HPO4) 、硝酸钙 (Ca(NO3)2) 、硫粉 (S0) 、七水合硫酸亚铁 (FeSO4·7H2O) 。所有化学试剂均为分析纯,实验用水为超纯水。
实验仪器。电热恒温鼓风干燥箱 (DHG-9240A,上海一恒科学仪器有限公司) ;恒温振荡器 (HZQ-X300C,上海一恒科学仪器有限公司) ;真空干燥箱 (DZF-6050,上海一恒科学仪器有限公司) ;荧光相差显微镜 (CX43,日本奥林巴斯公司) ;数字pH计 (STARTER 2100,美国奥豪斯仪器有限公司) ;紫外可见分光光度计 (UV-2100,北京瑞利分析仪器有限公司) ;原子吸收分光光度计 (AAS,AA-6880,日本岛津公司) ;扫描电子显微镜-能量色散x射线光谱 (SEM-EDS,MIRA LMS,捷克泰思肯电镜公司) ;X射线衍射 (XRD,Smartlab 9KW,日本理学公司) ;X射线电子能谱 (XPS,Scientific K-Alpha,美国赛默飞世尔科技有限公司) 。
-
1) 材料的预处理及表征。将LIBs碎片过200目筛,获得LIBs粉末。然后用蒸馏水洗涤[21],以去除黏附的电解质和粘合剂。随后在电热恒温鼓风干燥箱中烘干[22],并对样品的形貌、物相和元素组成进行分析。 同时,采用王水微波消解LIBs粉末,并用AAS测定目标金属质量浓度。
2) 浸出微生物的培养。在250 mL锥形烧瓶中,将10 mL初始浓度为108 cells·mL−1 的A. ferrooxidans菌液添加到90 mL的无铁9k培养基[23],培养基初始pH为1.85,并加入44.7 g·L−1 FeSO4·7H2O和5 g·L−1 S0[8, 24]作为能源物质。最后,将锥形瓶置于30 ℃和170 r·min−1的恒温振荡器中培养,定期测定pH、细菌数、Fe2+和Fe3+质量浓度以观察浸出菌生长状况,以确定A. ferrooxidans对数增长期的前、中和后期。
3) 生物浸出实验。在上述的培养条件下,对A. ferrooxidans进行培养,当培养至对数增长期的前、中、后期,分别向体系加入10 g·L−1废旧LIBs开始浸出,从而构建了A、B和C浸出体系,以探究废旧LIBs在对数期的投加时间对金属浸出的影响。浸出过程中,测定pH、细菌数、Fe2+和Fe3+质量浓度及目标金属质量浓度,以上实验重复不少于2次。浸出结束后,从浸出体系中离心收集固体物质,并用蒸馏水洗涤[21],放入40 ℃真空干燥箱干燥24 h获得浸出渣。通过分析浸出前后废旧LIBs的表面形貌、物相及元素价态变化,揭示A. ferrooxidans浸出废旧LIBs的机理。
-
采用荧光相差显微镜通过血细胞计数法测定细菌数;利用数字pH计测定pH;运用紫外分光光度计采用5-磺基水杨酸法[25]和邻菲罗啉法[26]分别测定浸出液中Fe3+和Fe2+质量浓度;采用AAS测定目标金属质量浓度;采用SEM-EDS、XRD和XPS分别分析浸出前后材料的形貌与元素分布、物相和价态变化。
-
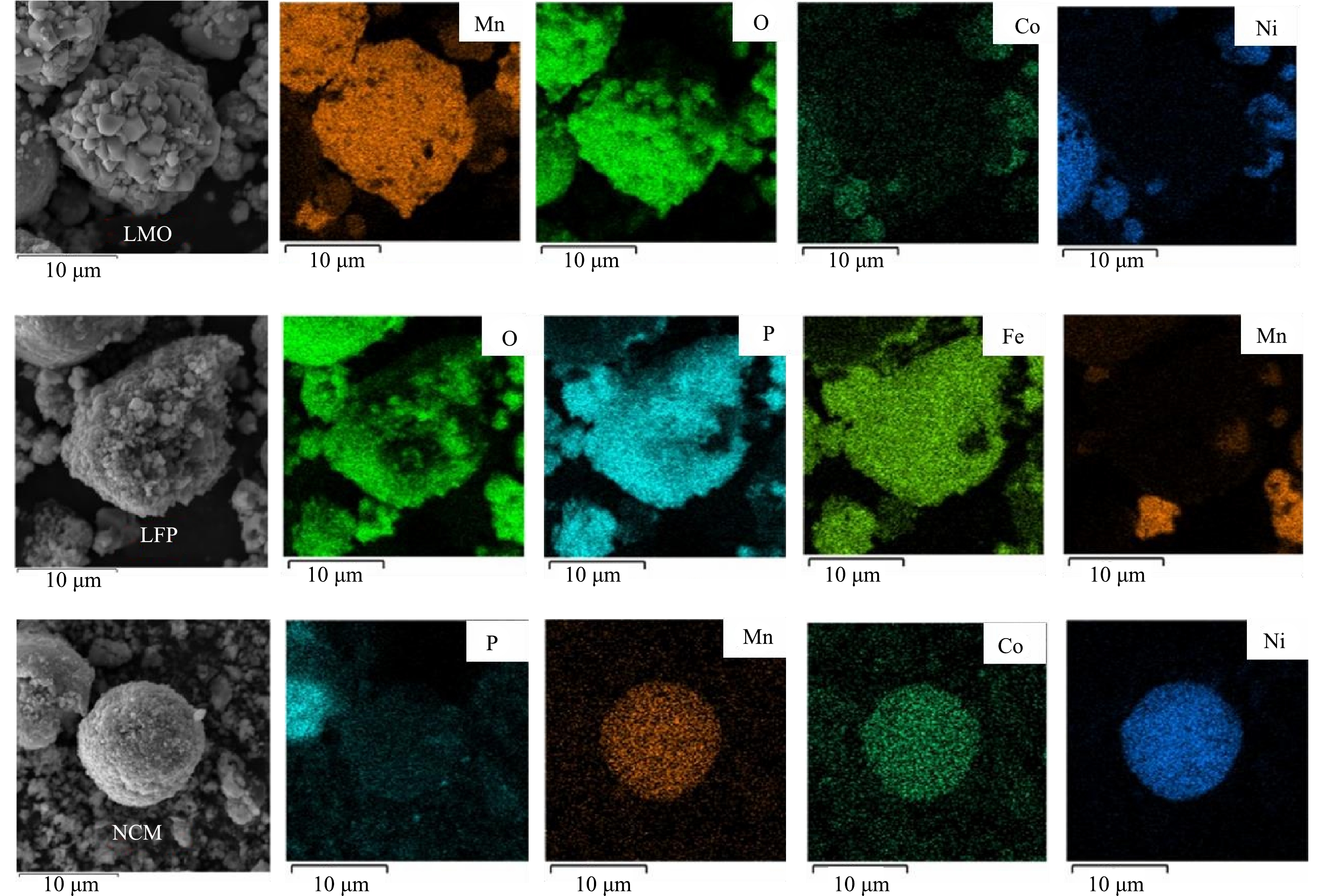
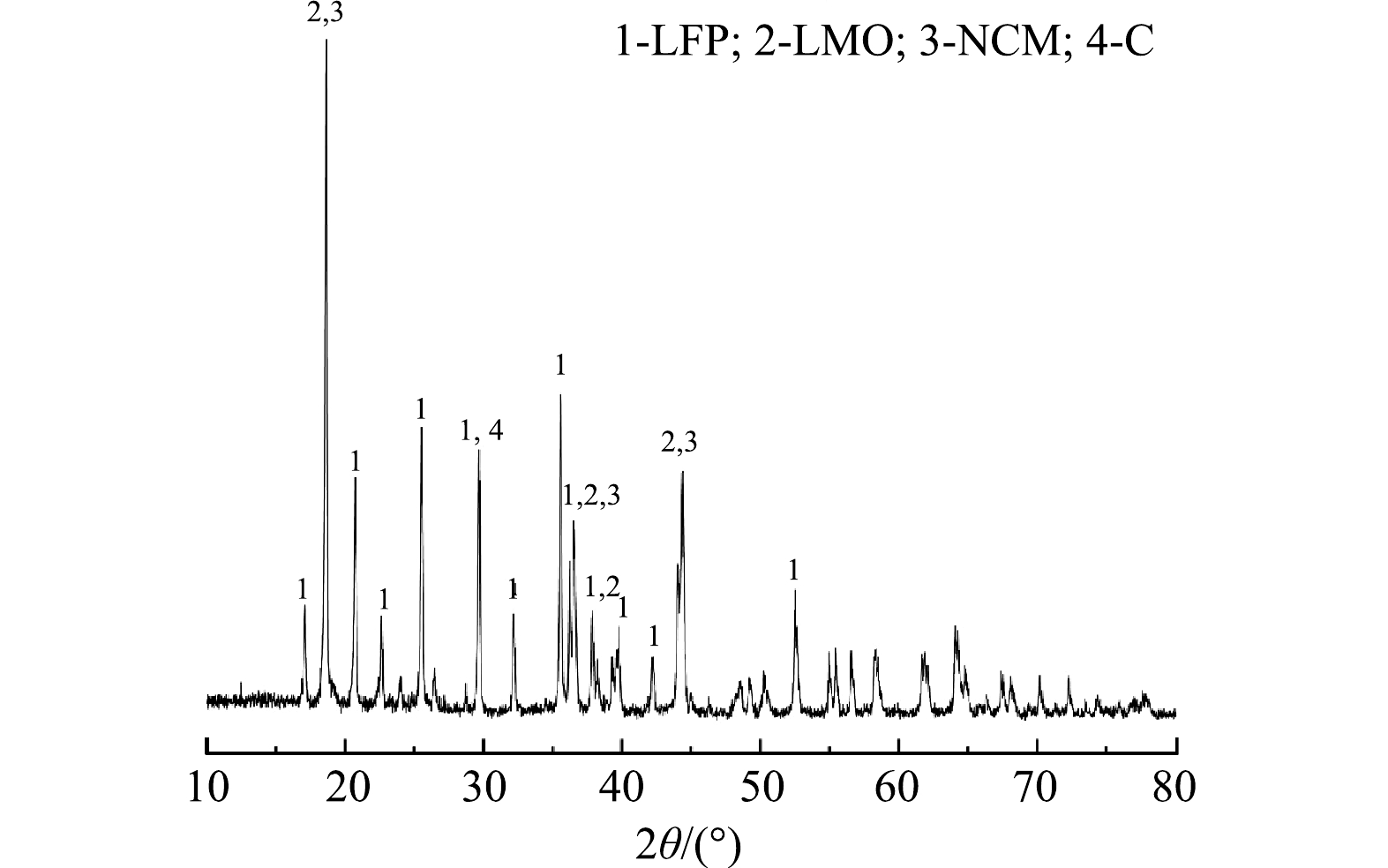
1) 原料分析。利用XRD对浸出材料的物相组成进行分析,如图1所示。结果表明,废旧LIBs含有LiNixCoyMn1–x–yO2 (NCM) 、LiFePO4 (LFP) 、LiMn2O4 (LMO) 多种阴极材料以及阳极材料石墨 (C) 。同时,利用SEM-EDS对原料的形貌及元素分布 (图2) 进一步分析,可以观察到,原料中有多种形貌:尖晶石型的LMO[27]、橄榄石型的LFP[28]和球状的NCM[29]。因此可了解到,该材料由C、LMO、LFP、NCM等组成。因此,本研究以Li、Ni、Co和Mn作为浸出实验的目标金属。化学分析结果表明,废旧LIBs中含有2.95% Co、8.53% Mn、10.59% Ni、3.66% Li以及17.04% Fe,证实该种废旧LIBs具有较高的回收价值。
2) A. ferrooxidans的生长特性。为了了解微生物在浸出过程中发挥的作用及更好地选择投加废旧LIBs的时间,分析了浸出菌的生长特性,结果如图3所示。A. ferrooxidans将Fe2+氧化成Fe3+过程中可获得生长所需的能源能量,导致Fe2+和H+消耗 (式(1)) 。因而在0~1 d,pH呈上升趋势,而Fe2+质量浓度不断降低[30]。随着A. ferrooxidans利用S0产生生物硫酸 (式(2)) 及氧化形成的Fe3+不断水解、与9k培养基中的K2HPO4及(NH4)2SO4生成K+/NH4+Fe3(SO4)2(OH)6沉淀,促进H+的产生 (式(3)、式(4)) [31],pH开始下降。同时,Fe3+也呈先增加后减少趋势。而从图3可知,细菌数量呈现“S”型变化趋势。因此,2~6 d为A. ferrooxidans的对数增长期,并分别以2、4和6 d作为对数增长期的前、中、后期。然而,目前废旧LIBs浸出多数在对数期加入到浸出体系,但究竟在对数期哪个阶段投加LIBs获得更好的浸出效率,需要进一步的探究。
3) 浸出废旧LIBs的过程分析。在对数增长期不同时期投加废旧LIBs后,浸出过程中可能发生的反应由式(1)~式(7)表示,浸出体系中pH、细菌数、Fe3+以及Fe2+质量浓度的变化如图4所示 (以加入废旧LIBs作为浸出实验的0点,下同) 。当废旧LIBs投加到浸出体系后,pH在0~0.25 d急剧增加 (图4(a)) 。这可能是因为LIBs的碱性[8]及金属的溶出会导致H+消耗。但pH在1~8 d不断下降。这是因为, A. ferrooxidans能通过氧化S0促进生物酸产生 (式(2)) 。同时, Fe3+会发生水解、或形成K+/NH4+Fe3(SO4)2(OH)6促进H+释放 (式(3)、式(4)) ,进一步加速pH下降。对于不同时期投加废旧LIBs,体系中pH的变化趋势不同。在2~8 d,C体系中保持最低pH (2.25~1.61) ,明显低于A体系 (2.23~1.95) 和B体系 (2.23~1.7) 。因此,采用两步法浸出废旧LIBs时,在对数期后期加入废旧LIBs,浸出体系可以产生更多的生物酸。
废旧LIBs浸出过程中细菌数量的变化如图4(b)所示。当废旧LIBs加入后,细菌浓度在0~0.25 d呈下降趋势。这可能是因为pH迅速上升以及释放的重金属和电池中的电解质对A. ferrooxidans的生长产生不利影响[32]。而在0.25~8 d,细菌数量逐渐增加,表明A. ferrooxidans对不利的生长环境具有适应性,可以继续保持自身活性,产生生物酸。对于不同时期投加废旧LIBs,C体系中的A. ferrooxidans适应能力大于A和B体系。而且,浸出过程中C体系中细菌量显著高于A和B体系。因此,在微生物对数期后期添加LIBs,细菌对浸出体系的适应性更好,可以保持较高的细菌浓度。
废旧LIBs生物浸出过程中Fe2+质量浓度的变化如图4(c)所示。当加入废旧LIBs后,在生物酸的攻击作用下,LIBs中LFP会发生溶解,释放Fe2+,导致Fe2+质量浓度逐渐增加 (式(5),式(6)) 。对于不同时期加入废旧LIBs,体系中Fe2+的产生速度不同。B体系中Fe2+的产生速度快于A和C体系,但是C体系可以实现更多Fe2+产生,且浸出的Fe2+可以将废旧LIBs中难溶解的Ni(Ⅲ)、Mn(Ⅳ)、Co(Ⅲ)还原成易溶解的Ni(Ⅱ)、Mn(Ⅱ)、Co(Ⅱ)[33] (式(7)) ,同时也可作为A. ferrooxidans生长的能源物质 (式(2)) ,促进浸出菌生长。因此,在对数期后期加入LIBs,A. ferrooxidans可以促进LIBs更多的Fe2+释放,有望促进金属浸出。
对于Fe3+,在0~4 d,Fe3+质量浓度呈显著下降趋势,这与Fe3+的水解、沉淀释放H+有关,且该反应有利于浸出体系维持较低pH,为废旧LIBs中金属的溶出提供H+。在4~8 d,Fe3+质量浓度趋于稳定,这说明在此过程中pH的下降主要是由于浸出微生物的产酸作用。
综上所述,与前期和中期投加LIBs的体系相比,在对数增长期后期加入废旧LIBs,浸出体系中细菌数量最高、Fe2+和生物酸的产生量最多,为LIBs中金属的溶出创造了有利的条件。
4) 不同投加时间对浸出效果的影响。图5显示了在A. ferrooxidans对数增长期的不同时期投加废旧LIBs的金属浸出率。从图5中可知,Mn、Li较容易浸出,而Ni、Co较难浸出。但是在对数期不同时期投加废旧LIBs,对金属浸出率有明显影响。其中,对Mn的影响最小,其次是易溶解金属Li,影响较大的是浸出率较低的Ni和Co。对于具有酸溶性的Li,生物浸出8 d后,B和C体系均能完全浸出,而A体系浸出率仅为88.56%。这可能是由于A. ferrooxidans在对数期前期耐受性较差、细菌量较低和产酸能力较弱,pH下降不明显 (图4) ,导致Li未能完全浸出。而对于浸出率较低的Ni和Co,生物浸出8 d后,C体系中Ni和Co的浸出率高于A和B体系。这是由于生物浸出过程中,C体系中细菌量最高,可以促进产生更多的H+释放,促进更多的Fe2+产生,而Fe2+和H+可以同时攻击废旧LIBs,导致金属释放[7]。此外,高细菌量能分泌更多的胞外聚合物 (EPS) 黏附在废旧LIBs上,并吸附Fe3+、Fe2+,在细胞-EPS-LIBs界面形成高质量浓度的Fe3+、Fe2+,对浸出固体有更强的侵蚀作用[34],更好地利用Fe2+还原浸出。综上所述,运用微生物两步浸出废旧LIBs过程中,在对数增长期不同时期加入废旧LIBs对金属浸出率有明显影响,且后期加入废旧LIBs可以实现高效的金属浸出率。
-
1) 形貌分析。利用SEM对生物浸出过程中固体形貌变化进行分析,深入了解浸出微生物对浸出固体的作用效果 (图6) 。在微生物对数期不同时期添加LIBs,浸出体系中LIBs的形貌具有明显的差异。生物浸出4 d后,A、C体系均无LMO形貌,C体系浸出渣中的橄榄石LFP结构消失,而A体系的浸出渣中仍能观察到块状的LFP (图6(a)、图6(d)) ,说明在微生物对数期后期加入废旧LIBs,A. ferrooxidans能更快地浸出LFP,释放Fe2+,这可作为还原剂及细菌生长的能源物质,提高浸出效率。生物浸出8 d后,2个体系的生物浸出渣均不存在LFP形貌的物质,表明LFP均完全浸出 (图6(b)、图6(e)) 。但2个体系浸出渣中NCM的结构有明显差别 (图6(c)、图6(f)) 。在A体系中,NCM球状结构的内部较光滑,无明显裂缝,仅有细小孔洞,而C体系浸出渣中的NCM内部变得多孔且粗糙、有明显的腐蚀痕迹,这表明C体系中微生物对NCM结构有更强的破坏能力。这是因为,在微生物对数期后期加入LIBs,A. ferrooxidans对浸出体系的适应性更强,产生更多的生物酸,攻击LIBs,导致其结构破坏。总之,A. ferrooxidans对废旧LIBs中阴极材料浸出的难易程度不同,LMO最容易被浸出,其次到LFP,而球状的NCM结构最为稳定,难以浸出;对于不同时期加入废旧LIBs,后期加入废旧LIBs时,A. ferrooxidans能较快浸出LFP中Fe2+,且对材料结构破坏更明显,可以促进LIBs中金属的溶出。
2) 物相分析。通过XRD分析对数增长期的不同时期加入废旧LIBs在浸出过程中固体物相的变化,探究不同时期投加废旧LIBs的浸出效果 (图7) 。观察发现,浸出原料以LFP、LMO、NCM为主,以及部分C,而浸出渣以K+/NH4+Fe3(SO4)2(OH)6、S0、C、FePO4为主,以及部分NCM。与浸出原料相比,LFP和LMO的特征峰均消失,其中LMO特征峰的消失均仅需4 d。这说明,A. ferrooxidans率先浸出LMO,且在不同时期加入废旧LIBs对LMO的浸出影响不大。然而对于对数增长期不同时期加入废旧LIBs,浸出渣的物相有明显差异。生物浸出4 d后,A体系的浸出渣可观察到LFP的特征峰,而C体系的浸出渣中未发现该谱峰。这说明,在微生物对数期后期向浸出体系加入废旧LIBs时,微生物能在较短时间内完全浸出LFP,产生Fe2+,从而为LIBs中金属的还原提供电子以及细菌生长提供能源物质;但生物浸出8 d后,A和C体系的浸出渣中仍然存在NCM的特征峰,而且C体系的NCM的特征峰强度低于A体系。该结果表明,A. ferrooxidans作用下未能实现NCM完全浸出,但在微生物对数期后期加入废旧LIBs,A. ferrooxidans对NCM浸出效果较好。综上所述,A. ferrooxidans会优先促进废旧LIBs中LMO浸出,其次到LFP,NCM最难浸出。而在对数期不同时期添加废旧LIBs,会影响A. ferrooxidans对LFP和NCM的浸出,在后期添加LIBs时,A. ferrooxidans对LFP和NCM的浸出作用更大,可以促进更多的金属离子释放。
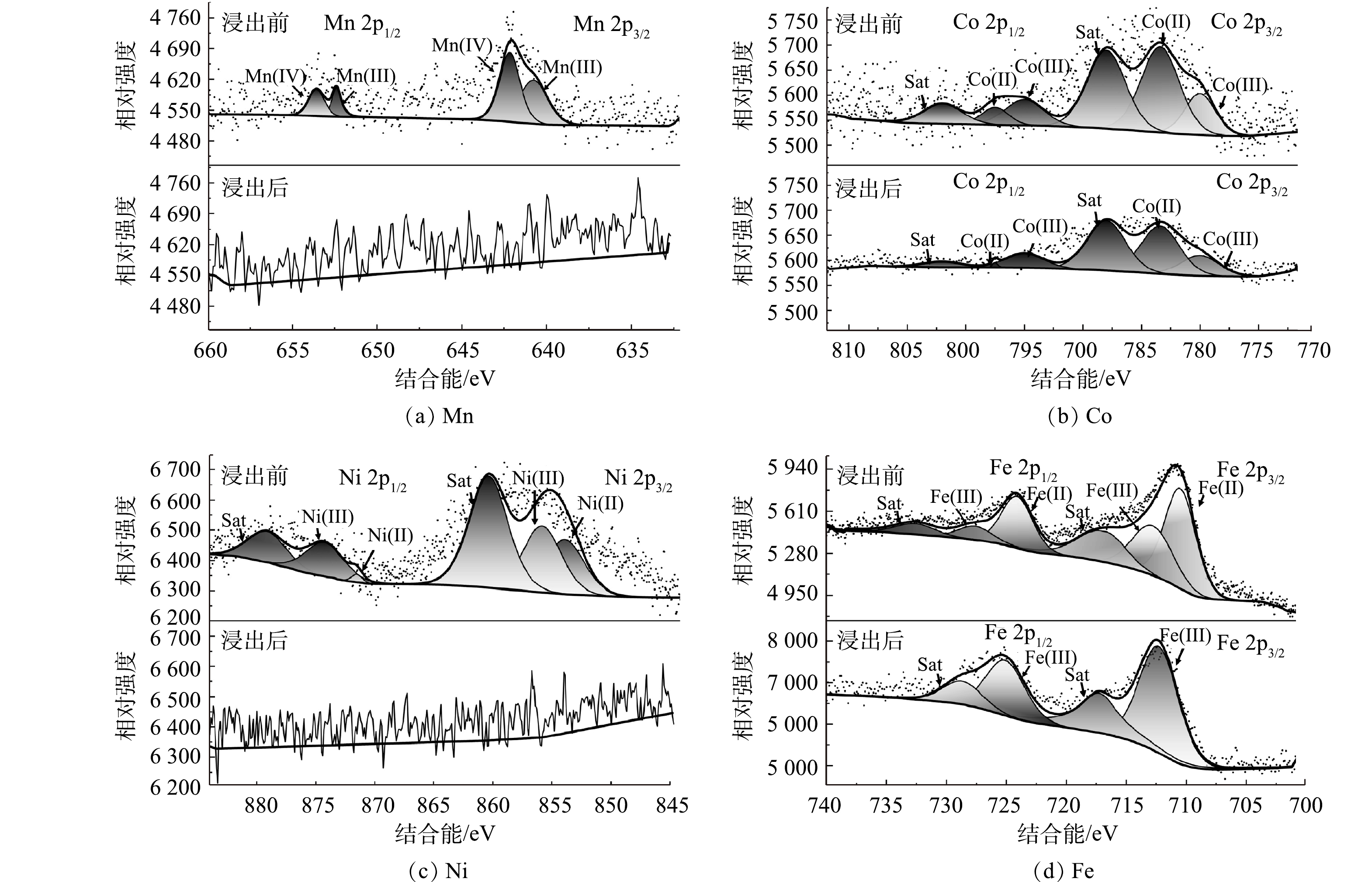
3) 金属价态分析。为了揭示A. ferrooxidans浸出废旧LIBs的机理,利用XPS分析C体系浸出前后固体金属价态变化,如图8所示。废旧LIBs中含有Mn(Ⅳ)、Mn(Ⅲ) [35]、Ni(Ⅲ)、Ni(Ⅱ) [36-37]、Co(III)、Co(II) [38]、Fe(Ⅱ) [39]和Fe(Ⅲ) [40],但是Mn(Ⅳ)、Ni(Ⅲ)和Co(III)是难以浸出的[33]。浸出完成后,Mn 2p的XPS谱图光谱嘈杂,浸出渣无明显谱峰,这表明浸出渣中Mn的表面浓度低,Mn基本浸出完成。这可能是因为,浸出体系中产生的Fe2+释放电子将Mn(Ⅳ)还原为Mn(Ⅲ),促进Mn容易被浸出。对于Ni 2p,浸出渣无明显谱峰,与AAS、SEM、XRD结果不一致,这可能是因为XPS是一种表面分析技术,分析深度约5~10 nm[41],而Ni表面覆盖率低导致谱峰不明显,这说明需要进一步破坏材料结构才能继续浸出Ni;对于Co 2p,与Ni 2p相比,有明显谱峰,这说明材料表面的Co未能完全浸出,Co相比于Ni更难浸出,同时,Co(Ⅱ)与Co(Ⅲ)面积比值增加,说明浸出过程中部分Co(Ⅲ)还原成Co(Ⅱ)[39];对于Fe 2p,浸出8 d后,浸出渣以Fe(Ⅲ)为主,表明大部分Fe(Ⅱ)氧化为Fe(Ⅲ),而该过程可以为Ni、Co和Mn的还原提供电子。因此,A. ferrooxidans能浸出废旧LIBs的LFP,释放的Fe(Ⅱ)可以还原高价态难溶解金属,从而有效提高金属浸出率。
-
1) 在A. ferrooxidans对数期的前、中和后期加入废旧LIBs,会影响金属浸出率。在对数期后期加入废旧LIBs,A. ferrooxidans有较强的产酸能力,较高的Fe2+产生量,从而获得最高的浸出率。对于不同金属,Mn和Li较容易浸出;Ni和Co较难浸出,且不同时期加入废旧LIBs对Ni和Co浸出率影响较大。
2) A. ferrooxidans均率先浸出废旧LIBs中的LMO,且可完全浸出LFP,而球状的NCM结构较为稳定,未能完全浸出;在对数增长期后期加入废旧LIBs,微生物对废旧LIBs的结构破坏更彻底,从而有利于金属的溶出。
3) A. ferrooxidans浸出废旧LIBs过程中产生的Fe2+可将LIBs中难溶解的Co(Ⅲ)、Ni(Ⅲ)、Mn(Ⅳ)还原为易溶的Co(Ⅱ)、Ni(Ⅱ)、Mn(Ⅱ),从而促进金属的浸出。
Acidithiobacillus ferrooxidans对数期的不同时期添加废旧锂电池对两步生物浸出工艺性能的影响
Effects of two-step bioleaching process on spent lithium ion batteries leaching during different stages of logarithmic growth of Acidithiobacillus ferrooxidans
-
摘要: 为了提高废旧锂电池 (LIBs) 的生物浸出效率,采用了氧化亚铁硫杆菌 (Acidithiobacillus ferrooxidans,简称A. ferrooxidans) 两步浸出废旧LIBs,考察在A. ferrooxidans对数期的前、中和后期向浸出体系中添加废旧LIBs对金属浸出效率的影响。结果表明,在对数期后期加入LIBs,A. ferrooxidans实现了100% Mn、76.82% Co、84.42% Ni和100% Li的浸出,比前和中期投加LIBs提高了4.51%~17.85% Co和16.38%~20.42% Ni。机理分析表明,A. ferrooxidans在对数期后期具有较强的产酸能力,比对数期前期和中期产生更多生物酸攻击废旧LIBs,导致金属释放。而释放的Fe2+可为A. ferrooxidans的生长提供能源物质,同时提供电子将LIBs中难溶解的Co(Ⅲ)、Ni(Ⅲ)和Mn(Ⅳ)还原为易被生物酸浸出的Co(Ⅱ)、Ni(Ⅱ)、Mn(Ⅱ),从而促进金属浸出。采用两步法生物浸出废旧LIBs时,为获得较高的生物浸出效率,需要在对数期的后期加入LIBs。本研究结果可为生物浸出废旧LIBs的工业化提供参考。Abstract: To improve the bioleaching efficiency of spent lithium batteries (LIBs), this study investigated Acidithiobacillus ferrooxidans (A. ferrooxidans) two-step leaching of spent LIBs during early, middle and late stages of logarithmic growth. The results showed that A. ferrooxidans could achieve a leaching efficiency of 100% Mn, 76.82% Co, 84.42% Ni and 100% Li by adding LIBs during late logarithmic phase and improve 4.51% to 17.85% Co and 16.38% to 20.42% Ni compared with early and mid-logarithmic growth. Mechanism analysis showed that A. ferrooxidans has a high capacity for acid production during the late logarithmic phase to attack spent LIBs, leading to an increased release of metal ions, as compared to early and mid-logarithmic growth. The released of Fe2+ could provide energy for the growth of A. ferrooxidans and release electrons to reduce the insoluble Co(Ⅲ), Ni(Ⅲ) and Mn(Ⅳ) in LIBs to Co(Ⅱ), Ni(Ⅱ) and Mn(Ⅱ), which were easily bioleached by this microorganism. In conclusion, to reach high leaching efficiencies, spent LIBs should be exposed to A. ferrooxidans during its the late logarithmic phase, when leaching was carried out by a two-step bioleaching method and the results of this study may provide a reference for the industrialisation of bioleaching of spent LIBs.
-
化工、医药等行业产生的难降解有机物会排入污水厂。然而,当前以生物处理为核心的传统污水厂无法有效降解这些难降解有机物,如何采用新工艺或者改良当前的污水工艺以应对新型污染物的挑战,已经成为了环境工作者的研究热点之一[1]。高级氧化法可产生具有强氧化能力的羟基自由基(·OH)、硫酸根自由基(SO4·−)等活性氧物种,使难降解有机物氧化成低毒或无毒的小分子物质[2-3],亦可改善其可生化性,故被认为是处理这类污染物的有效方法之一。
各类高级氧化法中,芬顿反应(式(1))因其条件温和、操作简便和价格低廉等优势而被广泛研究[4]。然而,在中性/碱性条件下,Fe2+会以氢氧化物的形式沉淀,失去活性,故该反应体系需要控制pH在3.0~4.0,以保证有高浓度的溶解态亚铁离子(Fe2+)持续驱动自由基的生成[5-6]。调节pH大幅提高了芬顿工艺的运行费用,阻碍了其在含有缓冲溶液的工业废水中的推广使用[7-8]。络合剂能使高浓度Fe2+在中性/碱性条件下以金属络合物(Fe2+-EDTA)形式存在,Fe2+-EDTA在中性/碱性条件下依然具有激活H2O2(式(2))并氧化降解核酸链的能力[9]。在中性条件下,添加NTA(氨三乙酸),TA(对苯二甲酸)等络合剂的芬顿体系可快速降解新诺明(pH=7.0)[10]。然而,络合剂与亚铁离子配比至少为1∶1,这表明将向待处置污水中引入大量的有机物,且络合剂与芬顿反应处置后的Fe3+形成的Fe3+-EDTA提高了体系内去除Fe3+的难度。CHEN等[11]向芬顿体系中投加还原剂(羟胺、NH2OH、HA)以降低铁离子用量,式(1)中失活的Fe3+可被羟胺重新转化成具有活性的Fe2+(式(3)~式(4)),故该强化体系能以痕量铁(2 mg∙L−1)持续驱动芬顿反应的进行与·OH的持续生成[11-12];相似的方法也用于持续驱动Fe2+/过一硫酸根体系和Fe2+/过二硫酸根体系[13-14]。
Fe2++H2O2→Fe3++OH−+⋅OH (1) Fe2+−EDTA+H2O2→Fe3+−EDTA+OH−+⋅OH (2) Fe3++NH2OH→Fe2++NH2O⋅+H+ (3) Fe3++NH2O⋅→Fe2++NHO+H+ (4) 在后续研究中发现,体系内的羟胺并不会生成对水体健康有害的NO3−和NO2−,而是转化成N2O(式(5)),因此,其不会有二次污染。考虑到羟胺的强还原性,羟胺加入到Fe2+-EDTA/H2O2体系内或许可以实现Fe2+-EDTA的再生,最终实现中性/碱性条件下活性氧自由基(·OH)的持续生成。
2NHO→N2O+H2O (5) 基于以上研究结果,本研究拟通过在芬顿体系中投加羟胺与EDTA,以伊文思蓝为·OH的指示剂,考察强化后的芬顿体系持续产生·OH的能力。伊文思蓝可与H2O2、过硫酸盐等过氧化物共存,且其与·OH可快速反应,因此,本研究中伊文思蓝的降解率可反映芬顿体系中·OH的生成情况。本文分析了溶液pH、铁离子浓度、H2O2浓度、EDTA浓度和伊文思蓝浓度对降解过程的影响,优化了体系对伊文思蓝降解的最佳条件,并通过电子自旋共振(ESR)技术和苯基甲基亚砜(PMSO)推测了体系中伊文思蓝的主要降解路径,从而确定该方法的稳定性,以期为芬顿反应在特定应用场景中的应用提供参考。
1. 材料与方法
1.1 实验主要试剂
实验用30%过氧化氢(H2O2)购自上海沃凯生物技术有限公司、硫酸亚铁(FeSO4)、盐酸羟胺(HONH2HCl, HA)、乙二胺四乙酸二钠(C10H14N2Na2O8, EDTA)、99.7%乙醇(C2H5OH)、叔丁醇(C4H10O)、甲醇(CH3OH)、乙腈(CH3CN)、伊文思蓝(C34H24N6Na4O14S4)、氢氧化钠(NaOH)、硫酸(H2SO4)等均购自上海麦克林生化科技,叔丁醇、甲醇、乙腈为色谱级纯,伊文思蓝为生物技术级纯,其余均为分析纯。
1.2 实验主要仪器
紫外可见分光光度计(UV-3000,上海元析仪器有限公司)、pH计(FE28,梅特勒-托利多仪器(上海)有限公司)、磁力搅拌器(MS-H-Pro+,大龙兴创实验仪器(北京)股份公司)、超纯水机(Milli-Q,厦门锐思捷水纯化技术有限公司)、电热鼓风干燥箱(GFL-45,天津市莱玻特仪器设备有限公司)、高效液相色谱仪(HPLC-20AD,Shimadzu)、顺磁共振波谱仪(EMXnano,BRUKER)。
1.3 实验方法
所有实验均于150 mL锥形瓶中进行,室温条件下将反应装置置于磁力搅拌器以350 r∙min−1的速度搅拌,先加入Fe2+、羟胺、EDTA、伊文思蓝药品后使用稀硫酸与氢氧化钠调节pH,再加入H2O2开始计时并定时取样,每次取样品2 mL加入5 mL离心管内,并加入1.5 mL 1 mol∙L−1乙醇淬灭、加入1 mL 10 mmol∙L−1 H2KPO4-HK2PO4 磷酸缓冲液调节样品pH为7,加盖后待测;未及时测定的样品则密封后置于4 ℃冰柜中冷藏保存;实验过程中始终监测系统pH并及时进行调节,保证系统pH始终处于±0.2范围内波动。
1.4 分析方法
以超纯水为参比,在波长为611 nm的条件下测定伊文思蓝原始样品及经芬顿/羟胺/EDTA系统处理后样品吸光值,根据朗伯-比尔定律计算样品去除率。使用高效液相色谱(HPLC)检测PMSO(苯基甲基亚砜)与PMSO2(苯基甲基砜),将样品过0.22 μm滤膜后装入1.5 mL玻璃小瓶,按顺序加入HPLC进样板,以1‰乙酸为水相、色谱级纯乙腈为有机相进行测定,设定流速1.0 mL∙min−1,水相80%,有机相20%,停留时间15 min,PMSO测定波长230 nm、PMSO2测定波长254 nm的方法进行测定,测定结果以吸收峰的面积相关去除率进行分析[15]。羟基自由基(·OH)的检测使用电子自旋共振法进行定性分析,以二甲基吡咯氮氧化物(DMPO)为捕获剂,取样后以10∶1加入捕获剂,混匀后置于顺磁共振波谱仪进行测定,若图像中存在比例约为1∶2∶2∶1的4个吸收峰,则表示体系中有·OH存在。
2. 结果与讨论
2.1 HA/Fe2+-EDTA/H2O2系统中伊文思蓝的降解
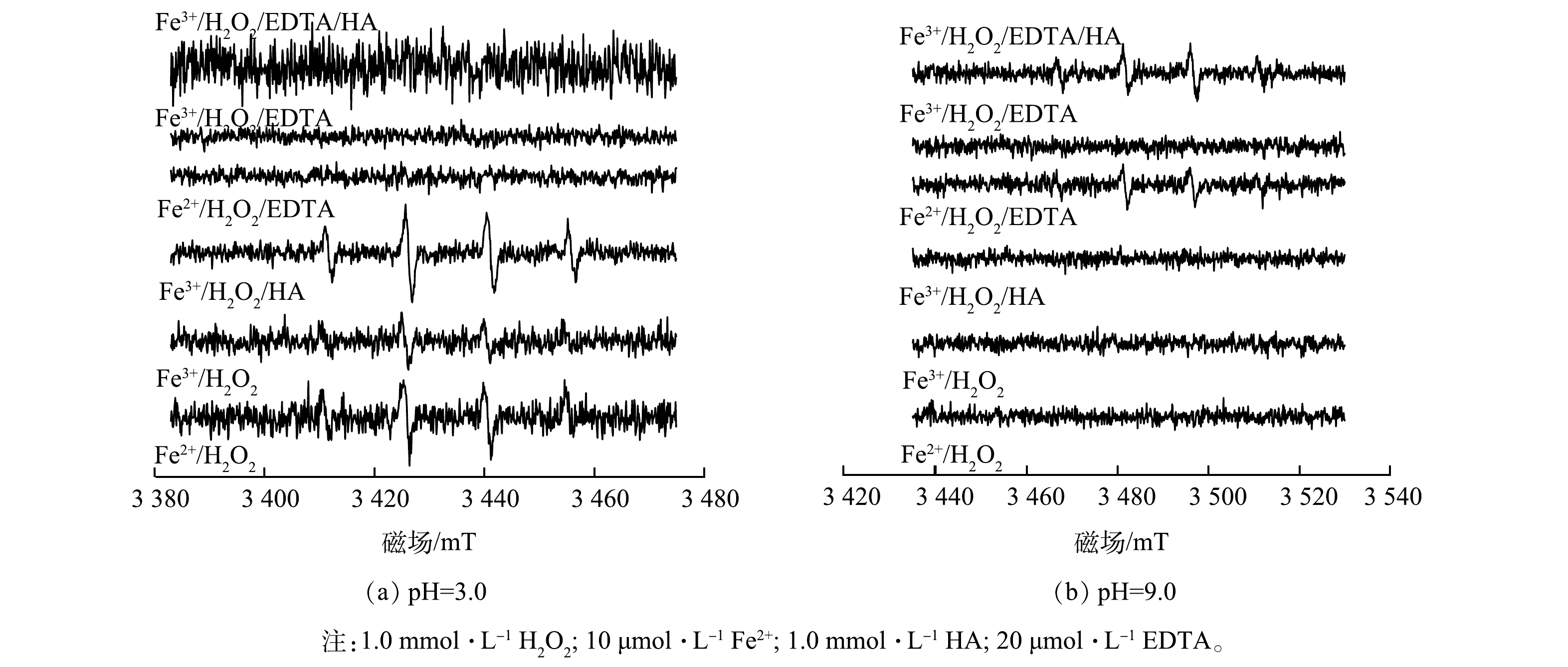
如图1(a)所示,当pH为3.0时,随着时间推移,HA/芬顿体系中伊文思蓝的降解率不断下降,30 min时达到86.5%;随着pH降至4.0或5.0,其降解率分别提升至93.8%和92.8%。CHEN等[11]的研究表明,这主要是因为羟胺的投加可以促进Fe3+向Fe2+转化,溶解态Fe2+的不断再生保证了芬顿体系中活性氧物质的持续生成。但是,随着pH的进一步提高(6.0~9.0),体系对伊文思蓝的降解效果骤然下降,仅维持在约10%。这与传统芬顿的适用pH类似,在中性或者碱性条件下,Fe3+与Fe2+主要以氢氧化物形式存在,故无法有效地被羟胺还原或激发H2O2。
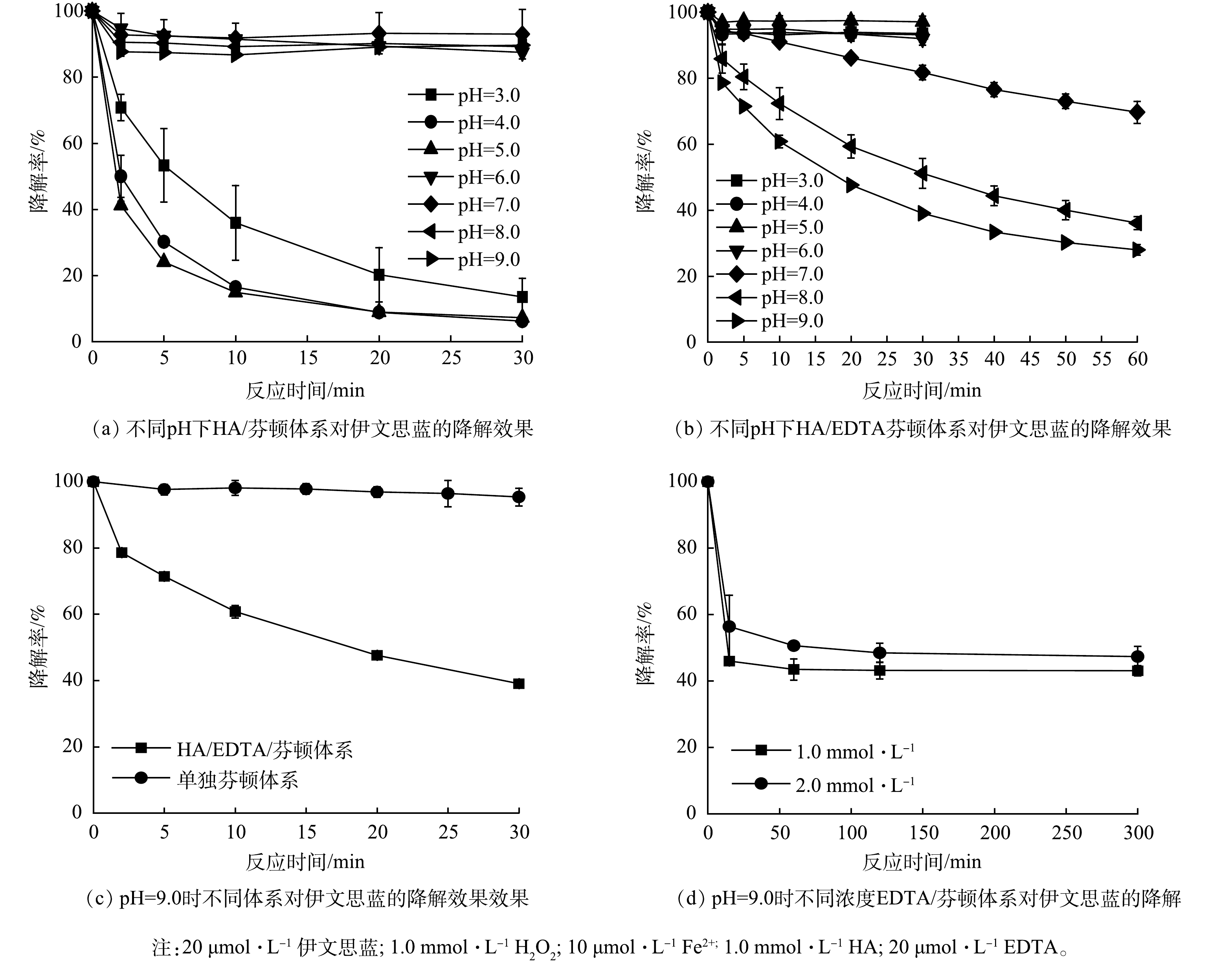 图 1 不同pH条件下HA/芬顿体系及HA/EDTA/芬顿体系对伊文思蓝的降解效果Figure 1. Effect of solution pH on the degradation of Evans Blue by HA/Fenton process and HA/EDTA/Fenton process
图 1 不同pH条件下HA/芬顿体系及HA/EDTA/芬顿体系对伊文思蓝的降解效果Figure 1. Effect of solution pH on the degradation of Evans Blue by HA/Fenton process and HA/EDTA/Fenton process图1(b)为在不同pH条件下HA/EDTA/芬顿体系对伊文思蓝的降解效果。酸性条件下的HA/EDTA/芬顿体系对伊文思蓝几乎没有降解。Fe3+-EDTA/Fe2+-EDTA氧化还原对的电位会因铁离子被络合而偏离Fe3+/Fe2+的氧化还原电位[16]。羟胺的还原性受pH影响,其在酸性条件下还原性较弱,碱性条件下还原性增强。随着pH提高,羟胺可缓慢将Fe3+-EDTA转化成Fe2+-EDTA,进一步用于H2O2的活化(反应2)。在羟胺较强的还原性保证了溶液内溶解氧维持在较低水平的前提下,较高浓度H2O2的竞争使Fe2+-EDTA会更倾向于与H2O2而不是氧气发生反应。在pH为9.0时,体系反应60 min可实现伊文思蓝72.1%的降解效率。值得一提的是,体系内铁离子浓度仅10 μmol∙L−1,而伊文思蓝的降解浓度(14.4 μmol∙L−1)高于铁离子浓度。因此,EDTA与羟胺的共同作用可有效避免碱性条件下铁离子易沉淀的问题,在提高羟胺还原能力的同时,实现Fe3+-EDTA/Fe2+-EDTA循环的持续驱动,而不仅仅是初始的亚铁离子起到了活化作用。
如图1(c)所示,当pH为9.0时,在传统芬顿体系中伊文思蓝基本没有降解(30 min内约5%降解)。而在相同条件下,HA/EDTA/芬顿体系对伊文思蓝的降解率在30 min时达到61.0%。如图1(d)所示,在pH为9.0不添加HA的情况下,Fe2+-EDTA投加量为原体系的100倍与200倍时(分别为1.0 mmol∙L−1和2.0 mmol∙L−1),降解率仅约50%。这主要是因为过量的EDTA与目标污染物竞争·OH导致降解率的下降。因此,HA/EDTA/芬顿体系不仅可降低铁离子及EDTA的用量,还可强化自由基定向氧化伊文思蓝。另外,由于体系在pH=9.0处的优异表现,本研究选取了9.0为后续反应的最佳pH。
2.2 芬顿/羟胺/EDTA系统中活性物质探究
为明确体系中存在的活性物质种类,以DMPO为捕获剂,利用顺磁共振波谱仪检测各体系中的自由基,以便定性判断系统中是否有·OH或其他活性物质存在。如图2所示,当出现比例约为1∶2∶2∶1的吸收峰波形时,代表体系中有DMPO-·OH的存在,即该体系有·OH生成[17-18]。由图2(a)可见,当反应条件为酸性(pH=3.0)时,可检测出·OH的系统有Fe2+/H2O2系统,即酸性条件下的传统芬顿体系;由于酸性条件下有利于Fe3+向Fe2+的转化,故Fe3+/H2O2系统能在一定程度上进行,即图中出现不明显的特征吸收峰;Fe3+/H2O2/HA系统,即HA的存在更有利于铁循环的优化,使反应得以持续进行;其余组别则未出现明显特征吸收峰。
由图2(b)可见,当反应条件为碱性(pH=9.0)时,Fe2+/H2O2系统没有任何波形出现,这与“芬顿反应无法在碱性条件下运行”的传统认知相符;随着EDTA的加入(Fe2+/H2O2/EDTA),明显的DMPO-·OH峰出现,这说明Fe2+-EDTA可有效活化H2O2;相比之下,Fe3+/H2O2/EDTA体系中则没有检测到自由基信号,这说明Fe3+-EDTA体系缺乏活性;而在羟胺和EDTA共存的体系中(Fe3+/H2O2/HA/EDTA),谱图中出现明显的1:2:2:1的·OH的特征峰。综上所述,EDTA的引入可让Fe2+在碱性条件下保持活性,而羟胺的引入则可强化Fe3+-EDTA/Fe2+-EDTA循环,从而实现Fe2+-EDTA源源不断地再生,从而活化H2O2并产生·OH。
为进一步验证·OH在降解过程中的作用,采用淬灭实验来反推降解体系中可能存在的活性氧物种。·OH与叔丁醇和甲醇的反应动力学常数均较大(3.8×108 L∙mol−1∙s−1与3.0×108 L∙mol−1∙s−1),被认为是一种理想的·OH淬灭剂。如图3(a)所示,HA/EDTA/芬顿体系对伊文思蓝的降解率为72.1%,当在体系内分别投加10 mmol∙L−1的叔丁醇和10 mmol∙L−1甲醇后,伊文思蓝的降解率降至62.3%和47.1%;随着叔丁醇与甲醇浓度分别提高到100 mmol∙L−1,降解率进一步降低至43.3%和28.9%。为了更直观地表达·OH在伊文思蓝降解中的作用,本研究对体系进一步做了一级动力学拟合,叔丁醇的加入使得反应动力学常数(k1)由0.050 3 min−1降至0.037 6 min−1(10 mmol∙L−1)和0.021 5 min−1(100 mmol∙L−1);甲醇的加入则使得伊文思蓝的降解动力学常数变为0.025 3 min−1(10 mmol∙L−1)和0.014 5 min−1(100 mmol∙L−1)。结合ESR的测试结果,叔丁醇和甲醇对伊文思蓝的降解抑制作用表明,在HA/EDTA/芬顿体系降解伊文思蓝的过程中,·OH起到主要作用。
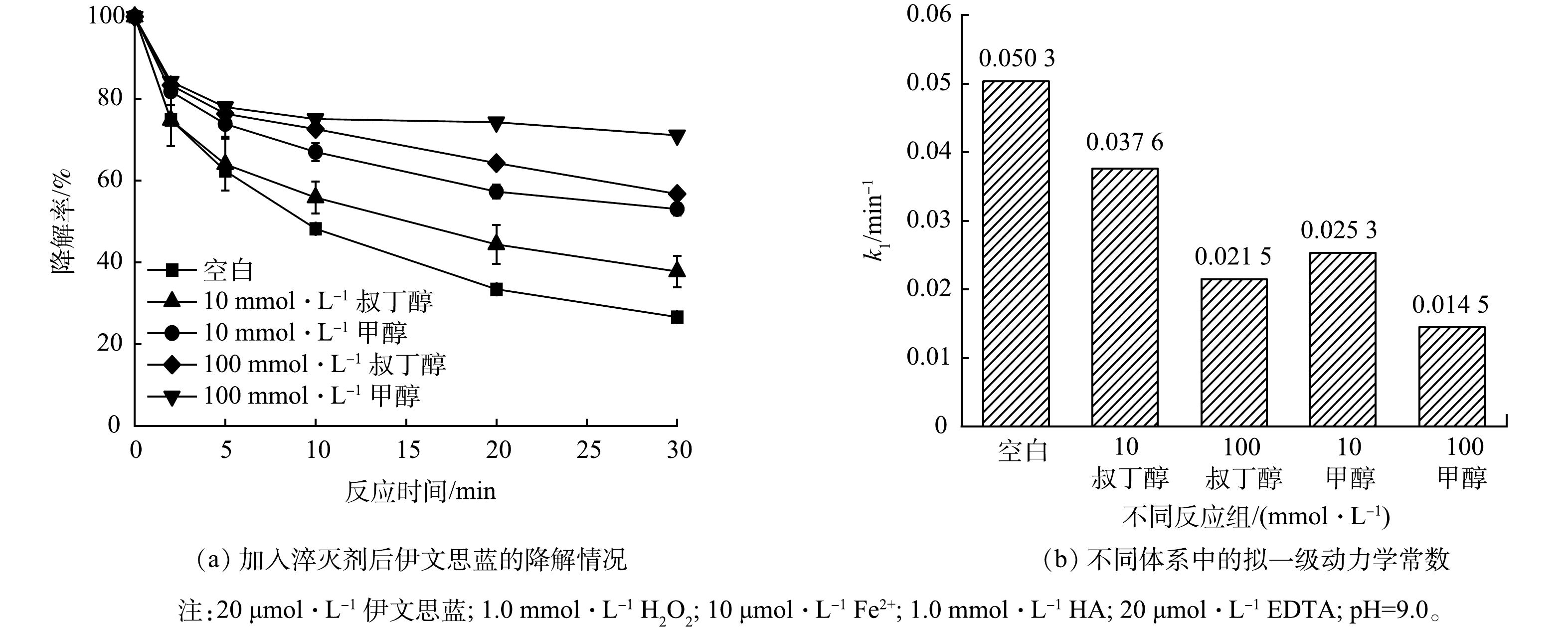 图 3 在HA/EDTA/芬顿体系中投加淬灭剂后伊文思蓝的降解情况及各条件下的拟一级动力学常数Figure 3. Effect of quenching agent on the degradation of Evans Blue by HA/EDTA/Fenton process and first-order kinetic constant of Evans Blue degradation by HA/EDTA/Fenton process
图 3 在HA/EDTA/芬顿体系中投加淬灭剂后伊文思蓝的降解情况及各条件下的拟一级动力学常数Figure 3. Effect of quenching agent on the degradation of Evans Blue by HA/EDTA/Fenton process and first-order kinetic constant of Evans Blue degradation by HA/EDTA/Fenton process近年来,在芬顿/类芬顿反应研究中,高价铁(FeIV)被认为是芬顿/类芬顿体系中除了·OH以外的另一类核心活性氧物质(式(6))[19]。尤其是在碱性条件下,高价铁被认为是主要活性物质[20]。为确认在新体系中高价铁的作用,采用PMSO为HA/EDTA/芬顿体系的探针物质,PMSO在高价铁作用下会转化成PMSO2,而在·OH的作用下则会转化成苯亚磺酸(PhSO2)[21]。因此,通过鉴定PMSO2是否存在,即可确认高价铁在该过程中的作用。
H2O2+Fe2+→H2O+FeⅣO2+ (6) PMSO标样和PMSO2标样浓度均为100 μmol∙L−1。在高效液相色谱图中,PMSO的出峰位置约在3.6 min,而PMSO2的出峰位置则处于7.0 min。如图4(b)所示,在HA/EDTA/芬顿体系中,随着时间的推移,PMSO的峰强逐渐降低,而代表PMSO2的峰形则始终保持在较低的强度上。这表明PMSO2并不是PMSO在新体系中的主要产物,由此推断即使是在偏碱性的条件下,新体系的活性物质依然以·OH为主。
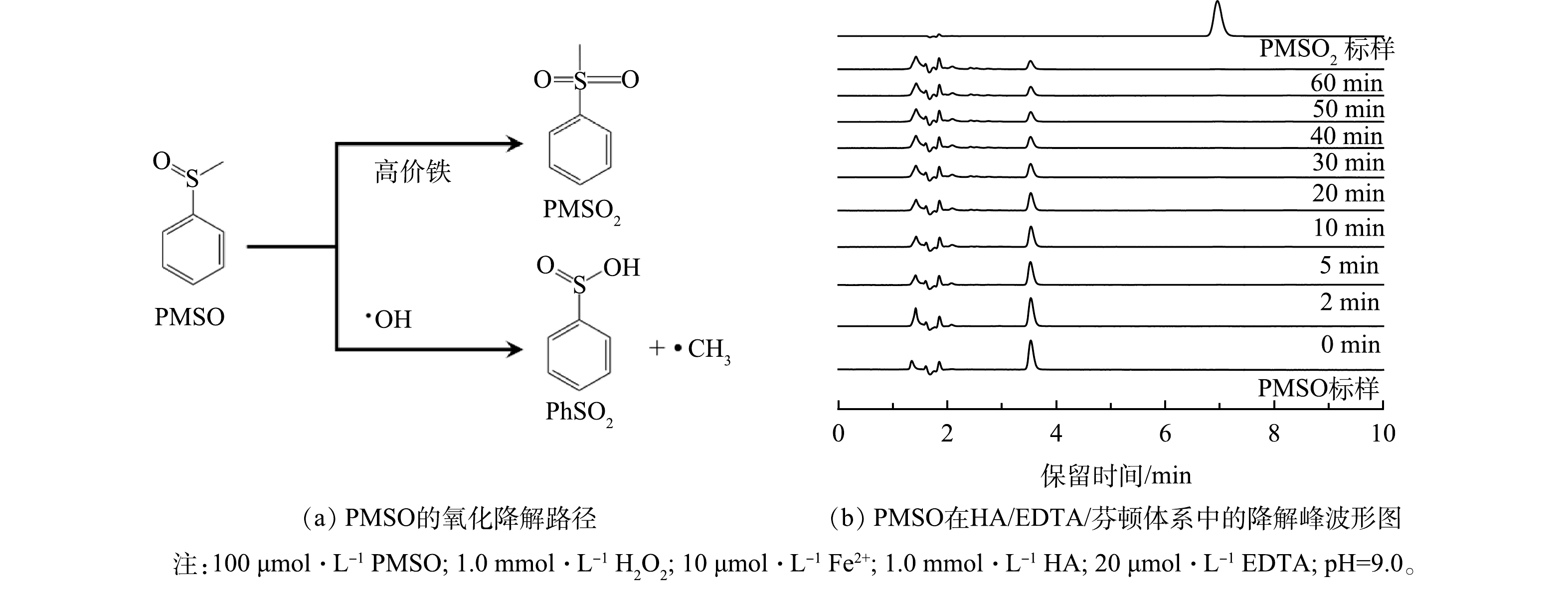 图 4 PMSO的氧化降解路径其在HA/EDTA/芬顿体系中富有不同时间的高效液相色谱图谱Figure 4. Degradation pathway of PMSO by different reactive oxygen species and degradation products analysis of PMSO in HA/EDTA/Fenton process
图 4 PMSO的氧化降解路径其在HA/EDTA/芬顿体系中富有不同时间的高效液相色谱图谱Figure 4. Degradation pathway of PMSO by different reactive oxygen species and degradation products analysis of PMSO in HA/EDTA/Fenton process2.3 芬顿/羟胺/EDTA系统降解伊文思蓝影响因素
作为Fe3+-EDTA/Fe2+-EDTA循环对的成分,铁离子与EDTA缺一不可,因此本工作考察了铁离子与EDTA对伊文思蓝降解的影响。如图5(a)~(b)所示,铁投加量和EDTA投加量对伊文思蓝降解有明显的正向促进作用。为更好地比较不同投加量药剂的影响,比较了不同铁投加量和EDTA投加量条件下伊文思蓝降解的拟一级动力学常数。如图5(c)所示,在低浓度条件下(Fe2+ <20 μmol∙L−1),铁离子浓度的增加提高了伊文思蓝降解的一级动力学常数;但是当铁离子浓度高于EDTA(20 μmol∙L−1)浓度时,一级动力学常数不再提高。这主要是因为铁离子只有和EDTA络合时才可以发挥作用,因此,当Fe2+<20 μmol∙L−1时,Fe3+-EDTA/Fe2+-EDTA循环才可以驱动H2O2生成·OH;当Fe2+浓度更高时,额外的铁离子则将以氢氧化物形式存在,无法进一步促进体系运行。类似的,当EDTA投加量低于10 μmol∙L−1时,EDTA对伊文思蓝降解有明显的线性提高,随着EDTA投加量提高至10 μmol∙L−1以上,体系降解速度则难以进一步提升。综合经济、效率等因素考虑,可认为铁离子和EDTA的最佳投加摩尔比为1:1。值得一提的是,10 μmol∙L−1的铁离子质量浓度仅为0.56 mg∙L−1,满足污水的排放标准,这表明处理后的污水可直接排放,而无需担心铁离子的超标问题。
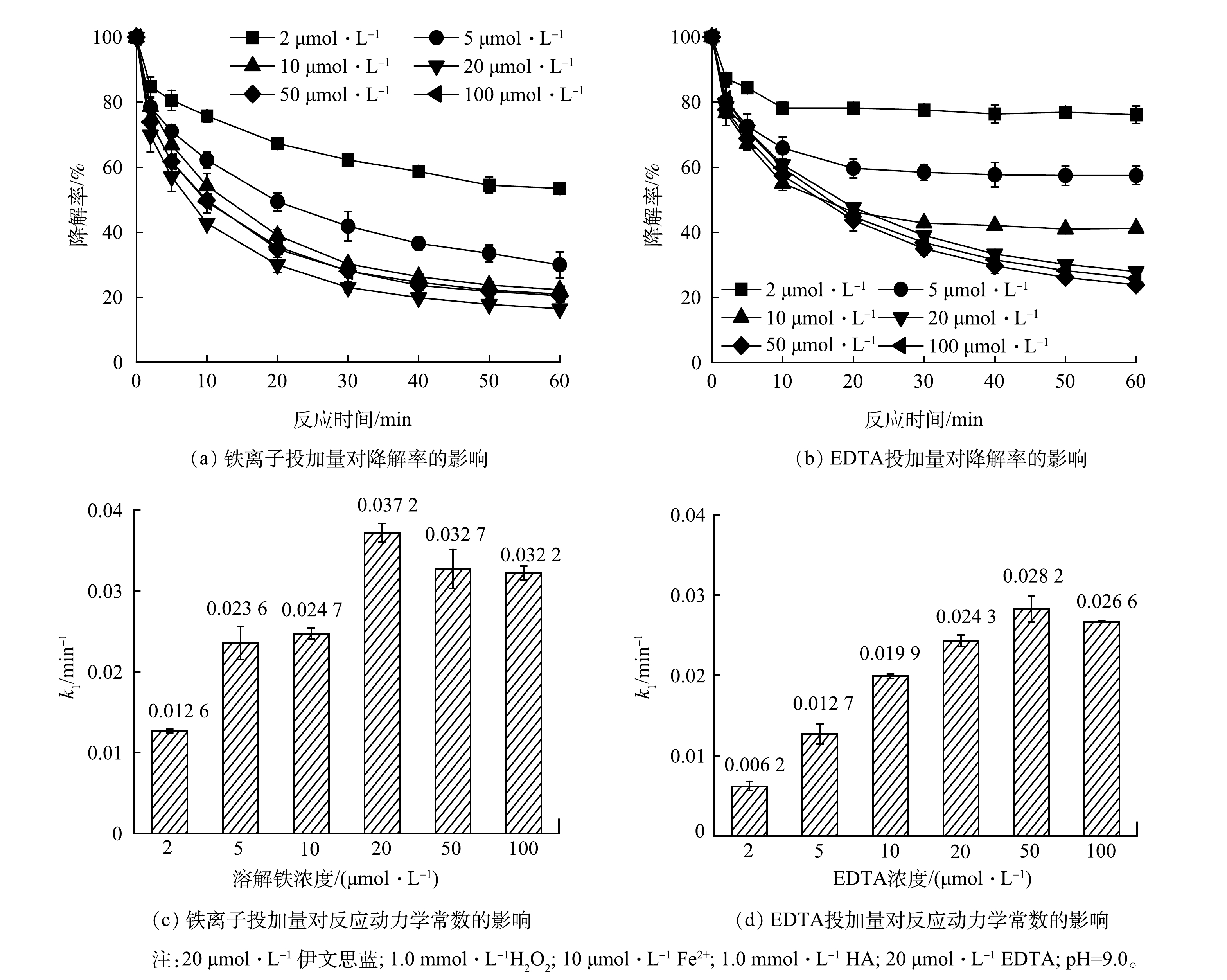 图 5 铁离子/EDTA投加量对HA/EDTA/芬顿体系降解伊文思蓝的影响Figure 5. Effect of iron concentration/EDTA concentration on the degradation of Evans Blue by HA/EDTA/Fenton process
图 5 铁离子/EDTA投加量对HA/EDTA/芬顿体系降解伊文思蓝的影响Figure 5. Effect of iron concentration/EDTA concentration on the degradation of Evans Blue by HA/EDTA/Fenton process作为体系的电子供体(HA)和·OH前驱体(H2O2),高投加量同样对体系有明显的提升作用(图6(a)~(b))。然而,对H2O2而言,当浓度达到0.5 mmol∙L−1以上时,降解速率的提升的速度明显减缓,60 min的处理效果相似。因此,在一定范围内,提高芬顿/羟胺/EDTA系统中H2O2浓度有助于反应的快速发生,提高处理效率,而当底物浓度过量时,即使继续提升其浓度,但受到其他因素的限制,体系对污染物的处理效率也无法继续提升。使用反应动力学进行拟合,随H2O2浓度的提升,系统的动力学常数k1也随之提升(图6(c)),观察发现H2O2浓度为0~0.5 mmol∙L−1时,与动力学常数之间近似线性关系,对其进行拟合所得R2 = 0.965 4,拟合效果较好;而0.5 mmol∙L−1后这种线性关系便不再显著。当H2O2浓度超过0.5 mmol∙L−1时,其浓度增加对于反应体系效率提升较低,反而可能由于温度等因素的影响导致体系效率产生一定波动,故确定系统中最适H2O2浓度为0.5 mmol∙L−1。
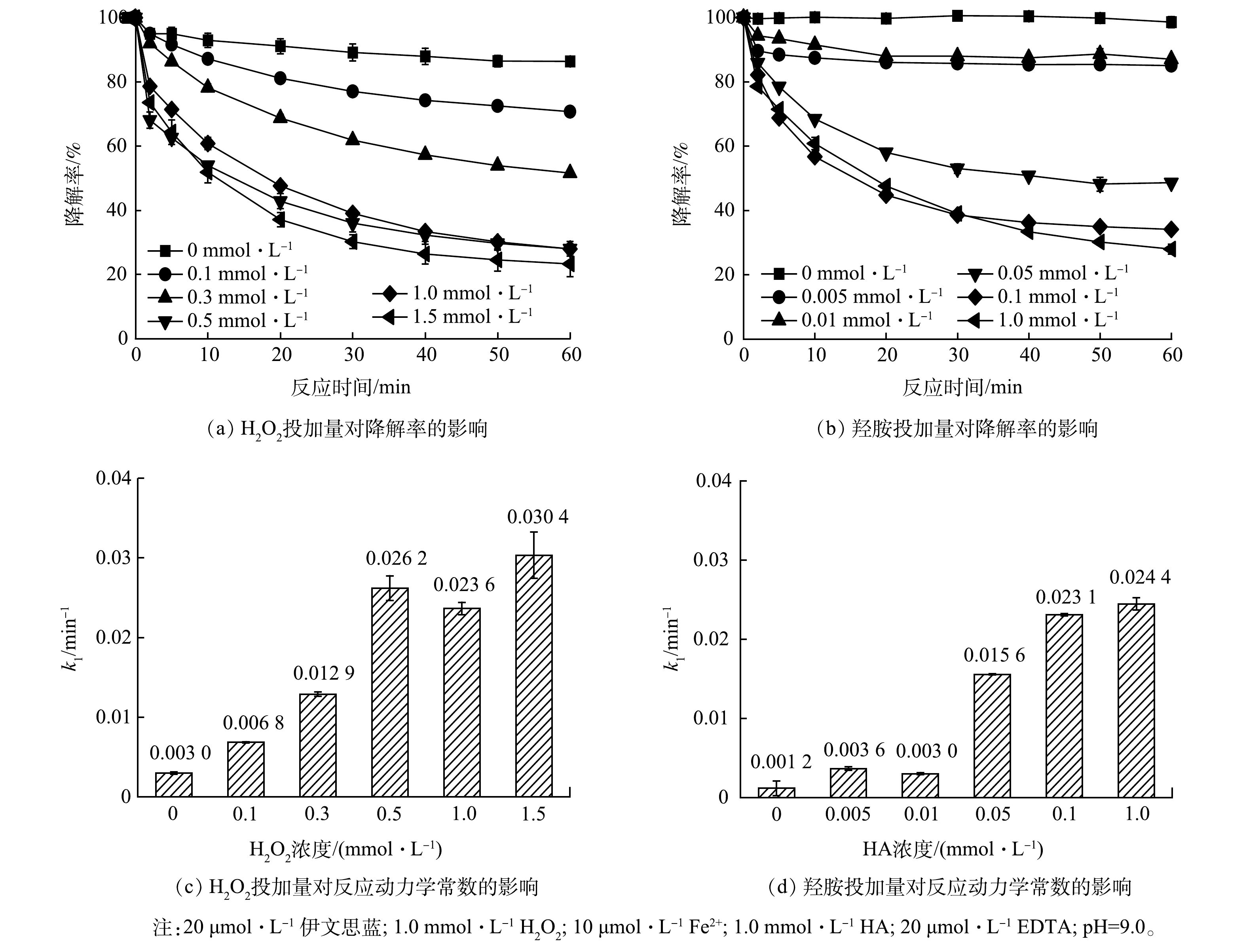 图 6 H2O2/羟胺投加量对HA/EDTA/芬顿体系降解伊文思蓝的影响Figure 6. Effect of H2O2/HA concentration on the degradation of Evans Blue by HA/EDTA/Fenton process
图 6 H2O2/羟胺投加量对HA/EDTA/芬顿体系降解伊文思蓝的影响Figure 6. Effect of H2O2/HA concentration on the degradation of Evans Blue by HA/EDTA/Fenton process羟胺在体系中能够起到还原Fe3+、促进铁循环的作用。当不加入羟胺时,该体系很快便会停止,故伊文思蓝降解率很低。在加入羟胺后,随加入量的提升,体系的降解效果会逐渐增强。使用反应动力学进行拟合,随羟胺浓度的提升,系统的一级动力学常数k1也随之提升(图6(d)),观察发现0~0.1 mmol∙L−1内羟胺浓度与动力学常数之间近似线性关系,对其进行拟合所得R2=0.973 6,拟合效果较好;而0.1 mmol∙L−1后这种线性关系便不再显著,故认为系统中最适羟胺浓度为0.1 mmol∙L−1。
3. 结论
1) EDTA的投加可使芬顿反应在碱性条件下(pH=7~9)运行,而羟胺可以使Fe3+-EDTA持续转化成具有活性的Fe2+-EDTA,从而实现碱性条件下·OH的连续生成与有机污染物的持续降解。当pH为9.0、羟胺浓度为1.0 mmol∙L−1、H2O2浓度为1.0 mmol∙L−1、铁离子浓度为10 μmol∙L−1、EDTA浓度为20 μmol∙L−1时,伊文思蓝在60 min内的降解率可达到72.1%。
2)在HA/EDTA/芬顿体系中主要的活性物种为·OH;PMSO的降解路径证明在碱性体系中高价铁(FeIV)几乎不起任何作用。
3)对于HA/EDTA/芬顿体系降解伊文思蓝(20 μmol∙L−1),铁离子和EDTA的最佳投料比为1:1,H2O2的最优投加量为0.5 mmol∙L−1,HA的最优投加量为0.1 mmol∙L−1。
4)羟胺的引入可缓解EDTA/芬顿体系中铁离子与络合剂的消耗性使用问题,在降低EDTA与铁离子的投加量的同时,避免了体系中络合剂的二次污染问题与铁离子的后续去除问题。
-
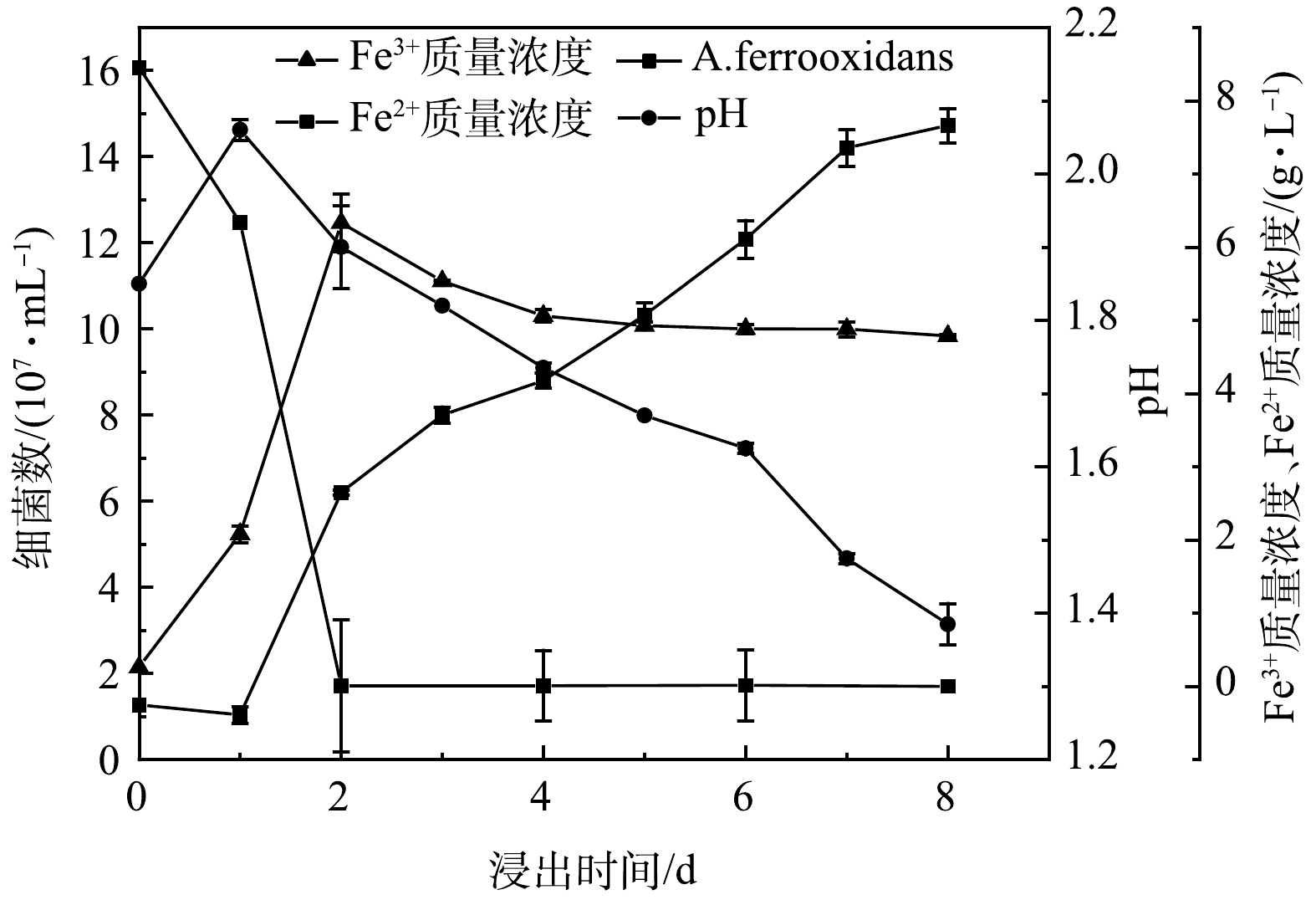
图 3 A. ferrooxidans的生长曲线及pH、Fe2+、Fe3+变化
Figure 3. A. ferrooxidans growth profile and changes in pH, Fe2+, and Fe3+ concentration
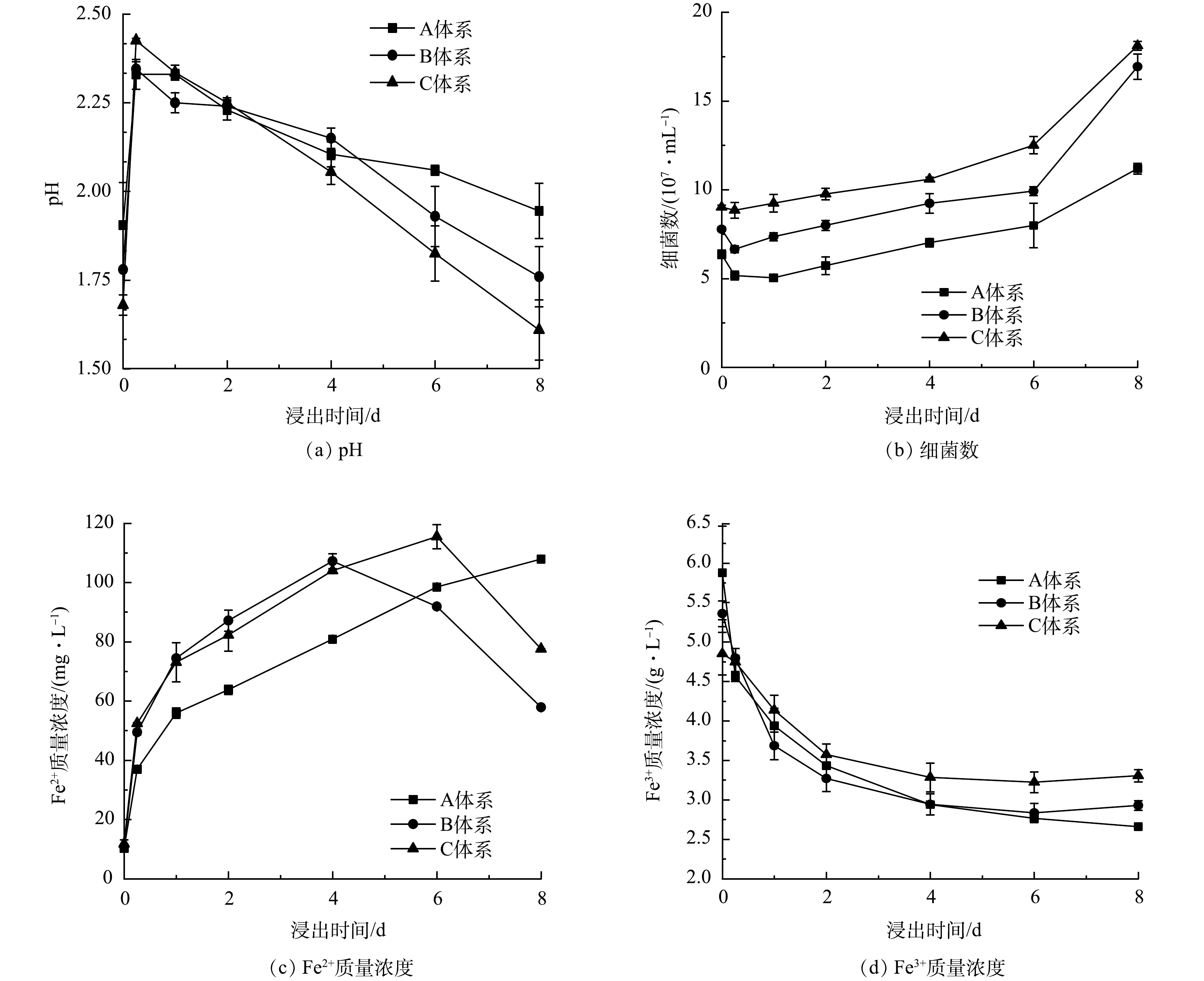
图 4 A.ferrooxidans对数增长期的不同时期加入废旧LIBs对浸出参数的影响
Figure 4. The effects of adding spent LIBs at different stages of the A.ferrooxidans logarithmic growth period on leaching parameters
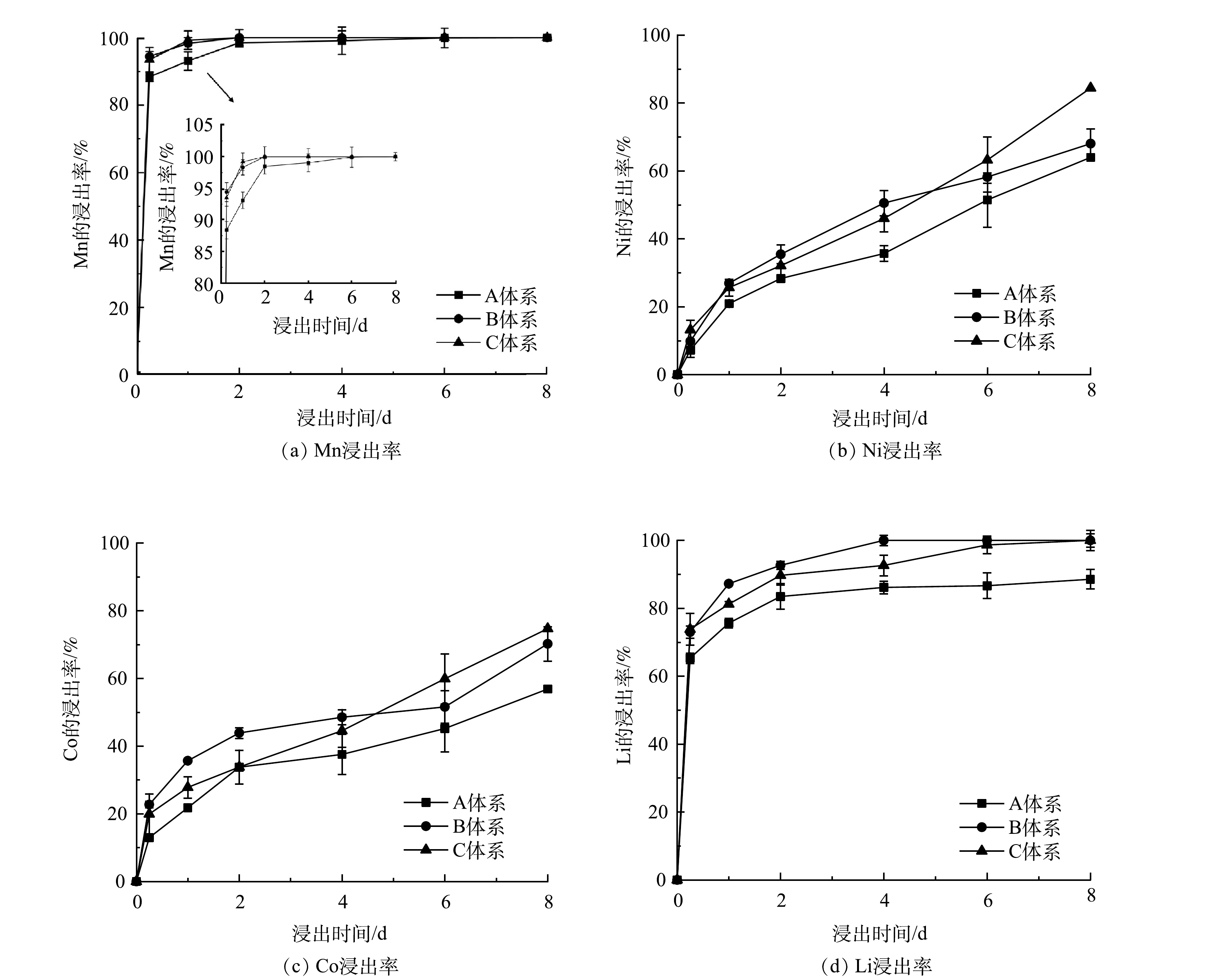
图 5 废旧LIBs的投加时间对金属浸出率影响
Figure 5. The influence of the adding time of spent LIBs on the metal leaching efficiency
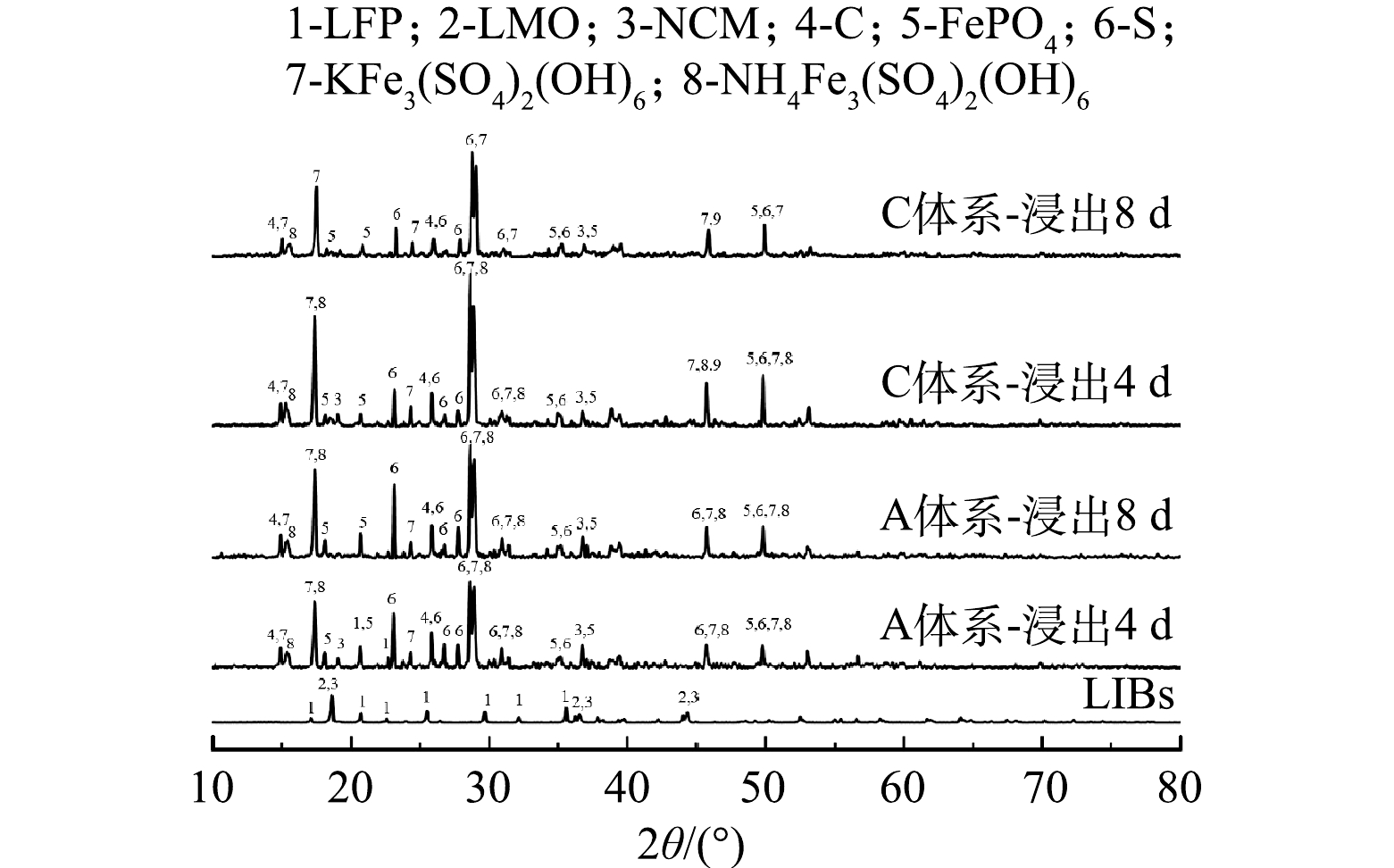
图 7 生物浸出前后废旧LIBs的物相变化
Figure 7. Phase changes of spent LIBs before and after bioleaching
-
[1] ZHAO S Q, HE W Z, LI G M. Recycling technology and principle of spent Lithium-Ion Battery[M]. Cham: Springer International Publishing. 2019: 1-26. [2] ROY J J, CAO B, MADHAVI S. A review on the recycling of spent lithium-ion batteries (LIBs) by the bioleaching approach[J]. Chemosphere, 2021, 282: 130944. doi: 10.1016/j.chemosphere.2021.130944 [3] SETHURAJAN M, GAYDARDZHIEV S. Bioprocessing of spent lithium ion batteries for critical metals recovery – A review[J]. Resources, Conservation and Recycling, 2021, 165: 105225. doi: 10.1016/j.resconrec.2020.105225 [4] WANG X, GAUSTAD G, BABBITT C W, et al. Economic and environmental characterization of an evolving Li-ion battery waste stream[J]. Journal of Environmental Management, 2014, 135: 126-134. doi: 10.1016/j.jenvman.2014.01.021 [5] WANG M X, TIAN Y H, LIU W, et al. A moving urban mine: The spent batteries of electric passenger vehicles[J]. Journal of Cleaner Production, 2020, 265: 121769. doi: 10.1016/j.jclepro.2020.121769 [6] SRIVASTAVA N, SINGH S K, GUPTA H, et al. Electrochemical performance of Li-rich NMC cathode material using ionic liquid based blend polymer electrolyte for rechargeable Li-ion batteries[J]. Journal of Alloys and Compounds, 2020, 843: 155615. doi: 10.1016/j.jallcom.2020.155615 [7] LIAO X J, YE M Y, LIANG J L, et al. Feasibility of reduced iron species for promoting Li and Co recovery from spent LiCoO2 batteries using a mixed-culture bioleaching process[J]. Science of The Total Environment, 2022, 830: 154577. doi: 10.1016/j.scitotenv.2022.154577 [8] ROY J J, MADHAVI S, CAO B. Metal extraction from spent lithium-ion batteries (LIBs) at high pulp density by environmentally friendly bioleaching process[J]. Journal of Cleaner Production, 2021, 280: 124242. doi: 10.1016/j.jclepro.2020.124242 [9] XIN Y Y, GUO X M, CHEN S, et al. Bioleaching of valuable metals Li, Co, Ni and Mn from spent electric vehicle Li-ion batteries for the purpose of recovery[J]. Journal of Cleaner Production, 2016, 116: 249-58. doi: 10.1016/j.jclepro.2016.01.001 [10] BISWAL B K, JADHAV U U, MADHAIYAN M, et al. Biological Leaching and Chemical Precipitation Methods for Recovery of Co and Li from Spent Lithium-Ion Batteries[J]. ACS Sustainable Chemistry & Engineering, 2018, 6(9): 12343-12352. [11] MONBALLIU A, CARDON N, TRI NGUYEN M, et al. Tolerance of Chemoorganotrophic Bioleaching Microorganisms to Heavy Metal and Alkaline Stresses[J]. Bioinorganic Chemistry and Applications, 2015, 2015: 861874. [12] MISHRA D, KIM D-J, RALPH D E, et al. Bioleaching of metals from spent lithium ion secondary batteries using Acidithiobacillus ferrooxidans[J]. Waste Management, 2008, 28(2): 333-338. doi: 10.1016/j.wasman.2007.01.010 [13] ZENG G S, DENG X R, LUO S L, et al. A copper-catalyzed bioleaching process for enhancement of cobalt dissolution from spent lithium-ion batteries[J]. Journal of Hazardous Materials, 2012, 199-200: 164-169. doi: 10.1016/j.jhazmat.2011.10.063 [14] ZENG G S, LUO S L, DENG X R, et al. Influence of silver ions on bioleaching of cobalt from spent lithium batteries[J]. Minerals Engineering, 2013, 49: 40-44. doi: 10.1016/j.mineng.2013.04.021 [15] BRYAN C G, WATKIN E L, MCCREDDEN T J, et al. The use of pyrite as a source of lixiviant in the bioleaching of electronic waste[J]. Hydrometallurgy, 2015, 152: 33-43. doi: 10.1016/j.hydromet.2014.12.004 [16] NATARAJAN G, TING Y-P. Gold biorecovery from e-waste: An improved strategy through spent medium leaching with pH modification[J]. Chemosphere, 2015, 136: 232-238. doi: 10.1016/j.chemosphere.2015.05.046 [17] ZHU N W, XIANG Y, ZHANG T, et al. Bioleaching of metal concentrates of waste printed circuit boards by mixed culture of acidophilic bacteria[J]. Journal of Hazardous Materials, 2011, 192(2): 614-9. doi: 10.1016/j.jhazmat.2011.05.062 [18] GHASSA S, FARZANEGAN A, GHARABAGHI M, et al. Novel bioleaching of waste lithium ion batteries by mixed moderate thermophilic microorganisms, using iron scrap as energy source and reducing agent[J]. Hydrometallurgy, 2020, 197: 105465. doi: 10.1016/j.hydromet.2020.105465 [19] JEGAN ROY J, SRINIVASAN M, CAO B. Bioleaching as an Eco-Friendly Approach for Metal Recovery from Spent NMC-Based Lithium-Ion Batteries at a High Pulp Density[J]. ACS Sustainable Chemistry & Engineering, 2021, 9(8): 3060-3069. [20] MA L Y, WANG X J, FENG X, et al. Co-culture microorganisms with different initial proportions reveal the mechanism of chalcopyrite bioleaching coupling with microbial community succession[J]. Bioresource Technology, 2017, 223: 121-130. doi: 10.1016/j.biortech.2016.10.056 [21] NASERI T, BAHALOO-HOREH N, MOUSAVI S M. Bacterial leaching as a green approach for typical metals recovery from end-of-life coin cells batteries[J]. Journal of Cleaner Production, 2019, 220: 483-92. doi: 10.1016/j.jclepro.2019.02.177 [22] HOREH N B, MOUSAVI S M, SHOJAOSADATI S A. Bioleaching of valuable metals from spent lithium-ion mobile phone batteries using Aspergillus niger[J]. Journal of Power Sources, 2016, 320: 257-266. doi: 10.1016/j.jpowsour.2016.04.104 [23] 聂红燕, 朱能武, 杨婷婷, 等. 嗜酸性细菌对废旧线路板浸出的吸附行为及动力学[J]. 环境科学学报, 2015, 35(5): 1471-1476. doi: 10.13671/j.hjkxxb.2014.0913 [24] BOXALL N J, CHENG K Y, BRUCKARD W, et al. Application of indirect non-contact bioleaching for extracting metals from waste lithium-ion batteries[J]. Journal of Hazardous Materials, 2018, 360: 504-511. doi: 10.1016/j.jhazmat.2018.08.024 [25] LIU L Z, NIE Z Y, YANG Y, et al. In situ characterization of change in superficial organic components of thermoacidophilic archaeon Acidianus manzaensis YN-25[J]. Research in Microbiology, 2018, 169(10): 590-597. doi: 10.1016/j.resmic.2018.08.003 [26] 陈志, 王敏, 葛淑萍. 工科基础化学实验汇编[J]. 重庆:重庆大学出版社, 2018: 125. [27] SEONG M J, YIM T. Critical role of corrosion inhibitors modified by silyl ether functional groups on electrochemical performances of lithium manganese oxides[J]. Journal of Energy Chemistry, 2020, 51: 425-433. doi: 10.1016/j.jechem.2020.02.029 [28] MAHANDRA H, GHAHREMAN A. A sustainable process for selective recovery of lithium as lithium phosphate from spent LiFePO4 batteries[J]. Resources, Conservation and Recycling, 2021, 175: 105883. doi: 10.1016/j.resconrec.2021.105883 [29] LIU P, XIAO L, TANG Y, et al. Resynthesis and electrochemical performance of LiNi0.5Co0.2Mn0.3O2 from spent cathode material of lithium-ion batteries[J]. Vacuum, 2018, 156: 317-324. doi: 10.1016/j.vacuum.2018.08.002 [30] 王鹤茹, 杨琳琳, 王蕊, 等. 3种填料A. ferrooxidans挂膜效果及对模拟酸性矿山废水中Fe~(2+)生物矿化能力比较[J]. 环境科学学报, 2022, 42(5): 160-168. [31] 邱冠周, 柳建设, 王淀佐, 等. 氧化亚铁硫杆菌生长过程铁的行为[J]. 中南工业大学学报, 1998(3): 25-27. [32] JANG H-C, VALIX M. Overcoming the bacteriostatic effects of heavy metals on Acidithiobacillus thiooxidans for direct bioleaching of saprolitic Ni laterite ores[J]. Hydrometallurgy, 2017, 168: 21-25. doi: 10.1016/j.hydromet.2016.08.016 [33] HE L P, SUN S Y, SONG X F, et al. Leaching process for recovering valuable metals from the LiNi1/3Co1/3Mn1/3O2 cathode of lithium-ion batteries[J]. Waste Management, 2017, 64: 171-181. doi: 10.1016/j.wasman.2017.02.011 [34] WANG J, TIAN B Y, BAO Y H, et al. Functional exploration of extracellular polymeric substances (EPS) in the bioleaching of obsolete electric vehicle LiNixCoyMn1-x-yO2 Li-ion batteries[J]. Journal of Hazardous Materials, 2018, 354: 250-7. doi: 10.1016/j.jhazmat.2018.05.009 [35] GRISSA R, MARTINEZ H, COTTE S, et al. Thorough XPS analyses on overlithiated manganese spinel cycled around the 3V plateau[J]. Applied Surface Science, 2017, 411: 449-456. doi: 10.1016/j.apsusc.2017.03.205 [36] GAUTHIER N, COURRèGES C, DEMEAUX J, et al. Impact of the cycling temperature on electrode/electrolyte interfaces within Li4Ti5O12 vs LiMn2O4 cells[J]. Journal of Power Sources, 2020, 448: 227573. doi: 10.1016/j.jpowsour.2019.227573 [37] LIU J, GAO S, SI Z, et al. Lanthanum Oxyfluoride modifications boost the electrochemical performance of Nickel-rich cathode[J]. Applied Surface Science, 2022, 599: 153928. doi: 10.1016/j.apsusc.2022.153928 [38] HAN H, LEE H, LIM J, et al. Hopping conduction in (Ni, Co, Mn)O4 prepared by different synthetic routes: Conventional and spark plasma sintering[J]. Ceramics International, 2017, 43(18): 16070-16075. doi: 10.1016/j.ceramint.2017.08.105 [39] CHEN B P, LIU M, CAO S, et al. Regeneration and performance of LiFePO4 with Li2CO3 and FePO4 as raw materials recovered from spent LiFePO4 batteries[J]. Materials Chemistry and Physics, 2022, 279: 125750. doi: 10.1016/j.matchemphys.2022.125750 [40] XU Z D, DAI Y, HUA D, et al. Creative Method for Efficiently Leaching Ni, Co, Mn, and Li in a Mixture of LiFePO4 and LiMO2 Using Only Fe(III)[J]. ACS Sustainable Chemistry & Engineering, 2021, 9(11): 3979-3984. [41] HU W Q, ZHANG C H, JIANG H, et al. Improving the electrochemistry performance of layer LiNi0.5Mn0.3Co0.2O2 material at 4.5V cutoff potential using lithium metaborate[J]. Electrochimica Acta, 2017, 243: 105-111. doi: 10.1016/j.electacta.2017.05.075 期刊类型引用(1)
1. 王乔,李海英,汪小琪,王晓东,薛海瑞. 废旧锂离子电池正极材料有价金属回收研究进展. 化学通报. 2024(04): 441-448 .  百度学术
百度学术
其他类型引用(0)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 4480
- HTML全文浏览数: 4480
- PDF下载数: 149
- 施引文献: 1
