



















-
作为重要的化工原材料,三氯乙烯(trichloroethylene)应用于金属脱脂、干洗、化工产品原料以及药品生产加工等过程[1]。尽管企业严格控制TCE的使用和处置,但仍存在由于使用管理和废物排放不当或容器泄露等事件引起土壤和地下水的污染。TCE具有粘度低、水溶性差和密度大等特性[2],能够轻易地穿透土壤并迁移至地下水中,在土壤和地下水中长期赋存,进而对环境和人类健康造成持续危害[3]。
近年来,表面活性剂增溶-化学氧化联合修复技术被广泛应用于受有机物污染的地下水治理中,其原理主要是将表面活性剂注入污染区域,通过表面活性剂的增溶作用,使有机污染物溶解到水相中,之后在原位或抽出至地面上通过化学氧化去除污染物[4]。当表面活性剂在水溶液中的浓度大于其临界胶束浓度(critical micellar concentration)时,表面活性剂会形成胶束包裹污染物,发挥其增溶作用[5]。表面活性剂主要分为阴离子、阳离子和非离子型[6],在实际应用中,非离子表面活性剂吐温-80(Tween-80)使用频率较高,因此,本研究选择TW-80作为表面活性剂的代表。目前在污染地下水化学氧化修复技术中常用的氧化剂主要有过一硫酸盐(peroxymonosulfate)、过二硫酸盐(persulfate)和过氧化氢(hydrogen peroxide)等。其中,PMS凭借其不对称结构和高于PDS和H2O2过氧化物键能而展现出更强的氧化能力[7]。在热、紫外、过渡金属或碱等活化方式下,PMS能够生成硫酸根自由基(SO4–·)、羟基自由基(HO·)和超氧自由基(O2–·),进而氧化降解氯代烃和多环芳烃等有机污染物[8]。铁作为廉价的过渡金属常被用作活化剂[9],其中Fe(Ⅱ)能快速活化PMS,产生大量的自由基。但随着反应的进行,Fe(Ⅱ)不断被消耗并转化为Fe(Ⅲ),且以氢氧化物的形式沉淀析出[10],导致反应体系催化性能下降。为了进一步提高体系氧化效率,需要将Fe(Ⅲ)及时还原为Fe(Ⅱ),促进反应过程中Fe(Ⅱ)/Fe(Ⅲ)循环。因此,本研究使用硫化亚铁(ferrous sulfide)作为催化PMS的手段,其中FeS不仅能够提供Fe(Ⅱ),而且可以将反应中生成的Fe(Ⅲ)还原为Fe(Ⅱ),反应过程如式(1)~(4)所示[11]。
目前为止,关于FeS活化PMS降解含表面活性剂水溶液中TCE的研究尚鲜有报道。本研究选择PMS作为氧化剂、FeS作为活化剂、TCE作为污染物、TW-80作为表面活性剂,研究了PMS/FeS体系对含有TW-80水溶液中TCE的降解效果,考察了PMS和FeS投加量、溶液初始pH和无机阴离子对PMS/FeS体系中TCE降解效果的影响,探究了PMS/FeS体系在反应过程中产生的自由基种类以及TCE降解的机制,并在实际地下水中验证PMS/FeS体系对TCE的降解效果,以期为该技术在实际工程中的应用提供参考。
-
实验试剂包括过硫酸氢钾、FeS、TCE、TW-80、异丙醇(IPA)、叔丁醇(TBA)、氯化钠、硝酸钠、磷酸钠、碳酸钠、硫酸、氢氧化钠、5,5-二甲基-1-氧化吡咯啉(DMPO)、四氯乙烯(perchlorethylene)、1,1,1-三氯乙烷(trichloroethane)、四氯化碳(tetrachloromethane)、1,2-二氯乙烷(dichloroethane)、三氯甲烷(trichloromethane)和二氯甲烷(dichloromethane),以上试剂均为分析纯。实际地下水取自上海市松江区地表以下20 m的深井。
实验仪器包括气相色谱分析仪(7890A,安捷伦科技有限公司)、电子顺磁共振仪(EMX-8/2.7C,德国Burker)、离子色谱仪(ICS-1000,美国Dionex)、紫外可见光分光光度计(DR-6000,美国HACH)和pH测定仪(AL204,瑞士Metter-Toledo集团)。
-
向250 mL定制玻璃反应器中加入预先配制好的13 g·L−1 TW-80溶液。将一定体积的超纯水配制的TCE储备液加入到反应器中,加超纯水至250 mL,使TW-80和TCE分别稀释至1.3 g·L−1和0.15 mmol·L−1,再依次加入FeS和PMS后开始计时,反应过程中使混合液充分搅拌,在预定时间点取样分析。分别添加不同浓度的氯化钠和碳酸氢钠探究Cl–和HCO3–对降解的影响。实验中用0.1 mol·L−1硫酸或氢氧化钠调节溶液pH。每组实验均进行3次,取其平均值。
除探究某物质浓度对实验结果的影响和实际地下水实验外,其余条件下PMS、FeS、TW-80和TCE的初始浓度均分别设定为0.8 mmol·L−1、0.6 g·L−1、1.3 g·L−1和0.15 mmol·L−1。
淬灭实验选择叔丁醇(淬灭HO·)、异丙醇(淬灭HO·和SO4–·)和三氯甲烷(淬灭O2–·)作为淬灭剂。测定TCE降解过程中氯离子的释放时,在不同时间点采集5 mL样品,加入1 mL甲醇终止反应,放置12 h挥发掉残余的TCE,采用离子色谱仪分析氯离子浓度。除实际地下水实验外,其他实验均使用超纯水。
-
采用正己烷萃取法测定TCE浓度。取0.2 mL反应样品注入到预先装有2.8 mL正己烷的棕色瓶中,旋涡振荡5 min后再静置5 min,用滴管吸取上层有机相至进样瓶中密封,使用气相色谱分析仪分析。溶液中Fe(Ⅱ)和总铁浓度采用邻菲啰啉分光光度法测定[12];PMS浓度采用碘化钾分光光度法测定[13];TW-80浓度采用硫氰酸钴铵显色-分光光度法测定[14];氯离子和硫酸根离子浓度通过离子色谱仪测定。
-
图1反映了不同反应体系对TCE的降解效果。其中PMS、FeS、Fe(Ⅱ)、TW-80和TCE的初始浓度分别设定为0.8 mmol·L−1、0.6 g·L−1、0.6 g·L−1、1.3 g·L−1和0.15 mmol·L−1。空白组中由于挥发导致TCE在60 min内减少了5.7%。当只加入PMS时,TCE降解率仅为14.7%,表明在没有活化剂加入的条件下,PMS的氧化性能没有得到充分发挥。但当体系中加入Fe(Ⅱ)或FeS后,TCE的降解率由14.7%分别显著提升到44.7%和90.0%。这表明Fe(Ⅱ)和FeS均能有效活化PMS,产生活性自由基可降解TW-80和TCE(式(5)和式(6))。与Fe(Ⅱ)相比,FeS活化性能更为显著。当加入Fe(Ⅱ)后,Fe(Ⅱ)在5 min内被快速消耗,在之后的反应过程中因Fe(Ⅱ)转化为Fe(Ⅲ)并以氢氧化物的形式沉淀析出,活化PMS效果急剧下降,最终导致TCE降解停滞;反应60 min后,PMS/Fe(Ⅱ)体系中Fe(Ⅱ)和溶解性总铁浓度分别为0.02 mmol·L−1和1.86 mmol·L−1。而在TW-80共存的PMS/FeS体系中,因FeS的还原作用,体系中Fe(Ⅱ)与Fe(Ⅲ)不断循环,TCE降解率在60 min内提升至90.0%[15]。当无TW-80存在时,TCE在10 min内降解99.4%,表面活性剂的加入略微抑制了TCE的降解。由于TW-80的增溶作用,TCE分子被其包裹,反应中产生的活性自由基在攻击TCE分子前需先攻击TW-80分子,因此,导致TCE降解效率有所下降[16]。
-
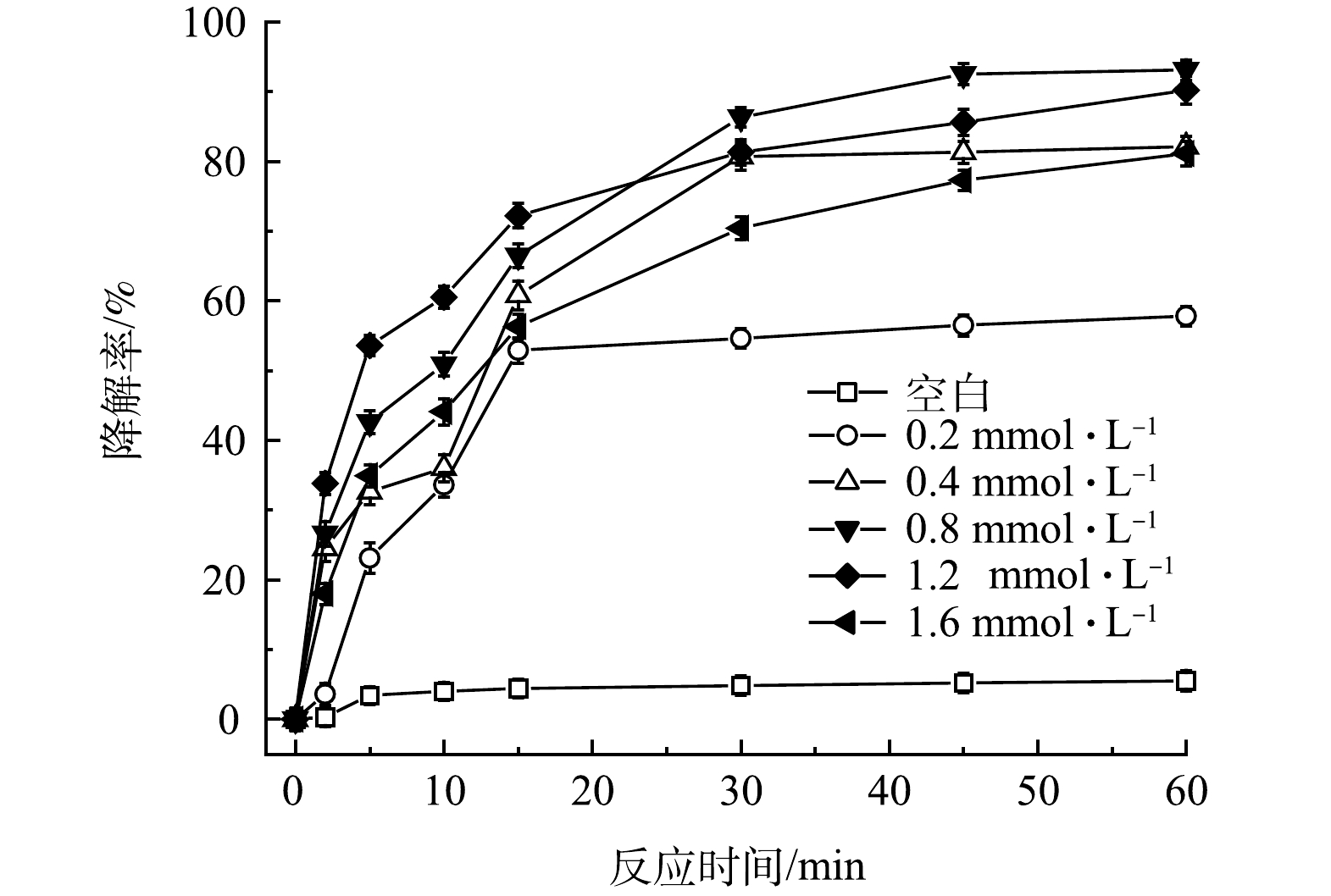
控制FeS、TW-80和TCE的初始浓度分别为0.6 g·L−1、1.3 g·L−1和0.15 mmol·L−1,考察PMS投加量对PMS/FeS体系降解TCE的影响。如图2所示,当不加入PMS时,整个反应过程中TCE降解率仅为5.5%。随着提高PMS投加量,TCE降解率显著升高。当PMS浓度为0.2、0.4和0.8 mmol·L−1时,反应60 min时TCE降解率分别为57.8%、82.1%和93.1%;但进一步增加PMS浓度至1.2 mmol·L−1和1.6 mmol·L−1时,TCE降解率由93.1%分别下降至90.2%和81.1%,表明提高PMS浓度并不会一直促进TCE的降解。当体系中PMS浓度较高时,产生的活性自由基会与多余的PMS发生反应,生成SO5–·(式(7)和式(8)),而SO5–·的氧化能力(E0 = 1.10 eV)低于SO4−·(E0 = 2.50 - 3.10 eV)和HO·(E0 = 2.76 eV)。此外,如式(9)和式(10)所示,PMS浓度较高时,体系中产生的高浓度SO4−·和SO5–·也会发生自我消耗[17]。此外,由图3可见,在TCE的降解过程中,PMS也随之消耗,在60 min内其分解率高达95%。以上结果表明PMS被充分利用。
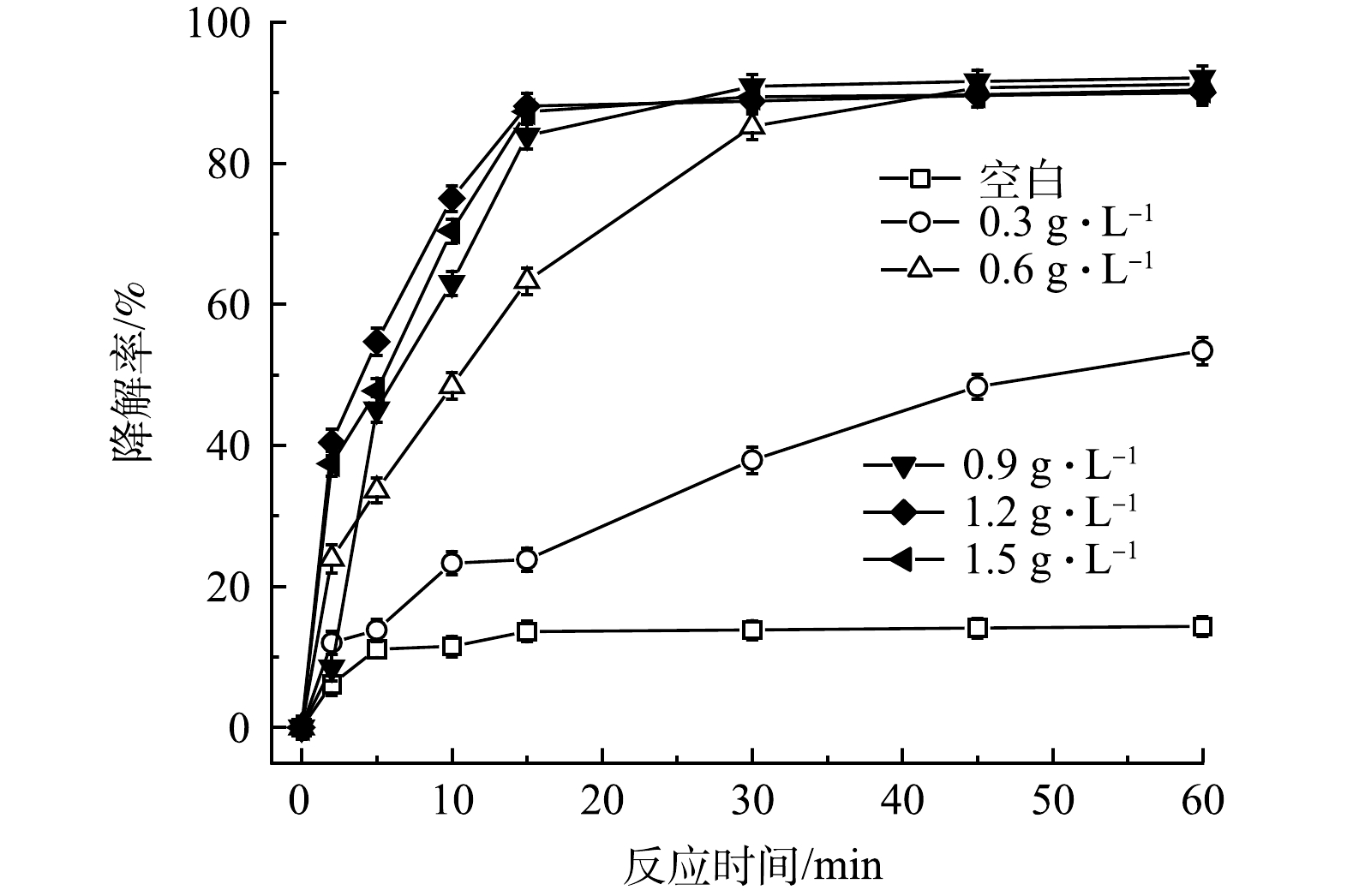
在相同实验条件下,考察了FeS投加量对PMS/FeS体系降解TCE的影响。如图4所示,当不加FeS时,TCE仅降解了14.3%。当FeS为0.3 g·L−1和0.6 g·L−1时,TCE降解率由14.3%分别上升至53.4%和91.2%。随着FeS投加量的增加,PMS得到有效活化,TCE降解率也随之提高。值得注意的是,当FeS投加量增加到0.9、1.2和1.5 g·L−1时,TCE降解率分别为92.1%、90.0%和90.1%,与加入0.6 g·L−1的FeS剂量对比,TCE的降解效果改变甚微,表明在FeS剂量为0.6 g·L−1条件下,PMS被充分活化和消耗,继续增加FeS剂量不会有效提高TCE降解率。
溶解性铁浓度在反应过程中的变化情况如图5所示。随着反应的进行,体系中Fe(Ⅱ)和溶解性总铁含量不断上升,在60 min内从初始点0分别增加至0.35 mmol·L−1和0.90 mmol·L−1,特别是在15 min后Fe(Ⅱ)浓度增加较大。而Fe(Ⅲ)浓度的变化不同于两者,Fe(Ⅲ)含量(溶解性总铁含量减去Fe(Ⅱ)含量)在0、2、5、10、15、30、45和60 min时刻分别为0、0.24、0.42、0.54、0.65、0.61、0.55和0.55 mmol·L−1。可以看出,Fe(Ⅲ)含量在前15 min大幅度增加,之后略微减少,最后保持稳定,由此可以间接判断在反应后期部分Fe(Ⅲ)转变为Fe(Ⅱ),使其显著优于PMS/Fe(Ⅱ)体系。同时,反应结束后体系中Fe(Ⅱ)浓度为0.35 mmol·L−1(19.6 mg L−1),根据《地下水质量标准》(GB/T 14848-2017),铁离子含量超过IV类水质标准规定的2 mg L−1。为避免造成二次污染,可采用曝气充氧的方法将水中Fe(Ⅱ)氧化为高价态离子,形成氢氧化铁胶体,再经过滤处理后达到降低地下水中铁离子含量的目的。
-
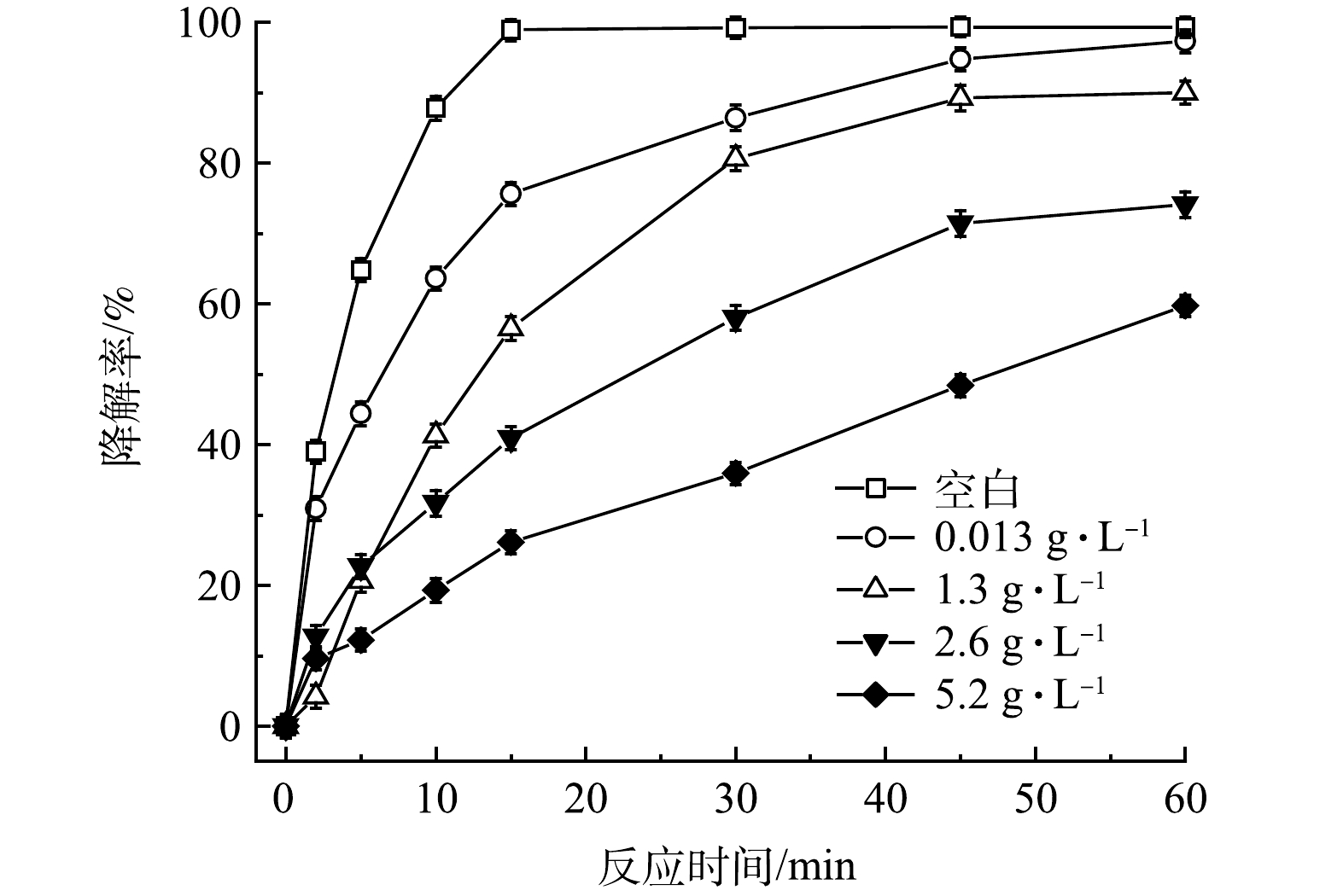
TW-80的临界胶束浓度(CMC)为13 mg·L−1,考虑到在污染场地实际修复中常使用较高浓度的表面活性剂[18],故控制PMS、FeS和TCE的初始浓度分别为0.8 mmol·L−1、0.6 g·L−1和0.15 mmol·L−1,设置不同浓度的TW-80(0、0.013、1.3、2.6和5.2 g·L−1)以探究其对TCE降解效果的影响。如图6所示,当添加0.013 g·L−1(1 CMC)的TW-80时,TCE的降解率由99.3%(空白)下降至97.3%,TCE的降解率仅下降了2%,由此推断0.013 g·L−1以内的TW-80对TCE降解的影响可忽略不计。当TW-80浓度增加至1.3、2.6和5.2 g·L−1时,TCE降解率分别下降至90.0%、74.1%和59.7%。由上述实验结果可知,当表面活性剂TW-80浓度超过0.013 g·L−1时,其会在水溶液中形成胶束包裹TCE分子,反应过程中产生的SO4–·和HO·需先攻击胶束,之后再攻击TCE分子,因此,会消耗一部分SO4–·和HO·,进而影响TCE的降解[19]。
为进一步证实TW-80会消耗体系中的自由基,包括HO·、SO4–·和O2–·,实验选择叔丁醇(TBA)作为HO·的淬灭剂,异丙醇(IPA)作为SO4–·和HO·的淬灭剂,三氯甲烷(trichloromethane)作为O2–·的淬灭剂[20],设置PMS、FeS、TW-80和TCE的初始浓度分别为0.8 mmol·L−1、0.6 g·L−1、1.3 g·L−1和0.15 mmol·L−1,在不添加淬灭剂以及分别添加100 mmol·L−1 TBA、100 mmol·L−1 IPA或50 mmol·L−1 CF时,探究TW-80在不同条件下的分解情况,结果如图7所示。当不添加淬灭剂时,TW-80在60 min内分解了51.5%;当分别添加TBA、IPA和CF后,TW-80分解率从51.5%分别降至44.6%、16.2%和37.7%。结果表明,HO·和SO4–·会直接攻击TW-80使其分解,造成了自由基的消耗,而O2–·主要通过链式反应参与HO·和SO4–·自由基的生成,促进TW-80的分解,且相比于HO·和O2–·,SO4–·对TW-80的分解起主要作用。
-
1)溶液初始pH。溶液初始pH对PMS/FeS体系降解TCE有很大影响,因此在初始实验条件下,探究pH其对TCE降解的影响。反应60 min后溶液pH变化及TCE降解率如表1所示。当溶液初始pH为3、5、7和9时,60 min内TCE降解率分别为91.5%、90.0%、89.5%和90.7%,无明显变化;当pH为11时,TCE降解率有所下降,为79.8%。该结果表明,PMS/FeS体系在降解含TW-80的TCE时对溶液pH有较宽的适用范围,反应结束后溶液pH均在3以下,这与PMS在水溶液中呈酸性有关。在酸性条件下,PMS能够产生更多的自由基,攻击TW-80和TCE分子,有利于TCE的降解[21]。
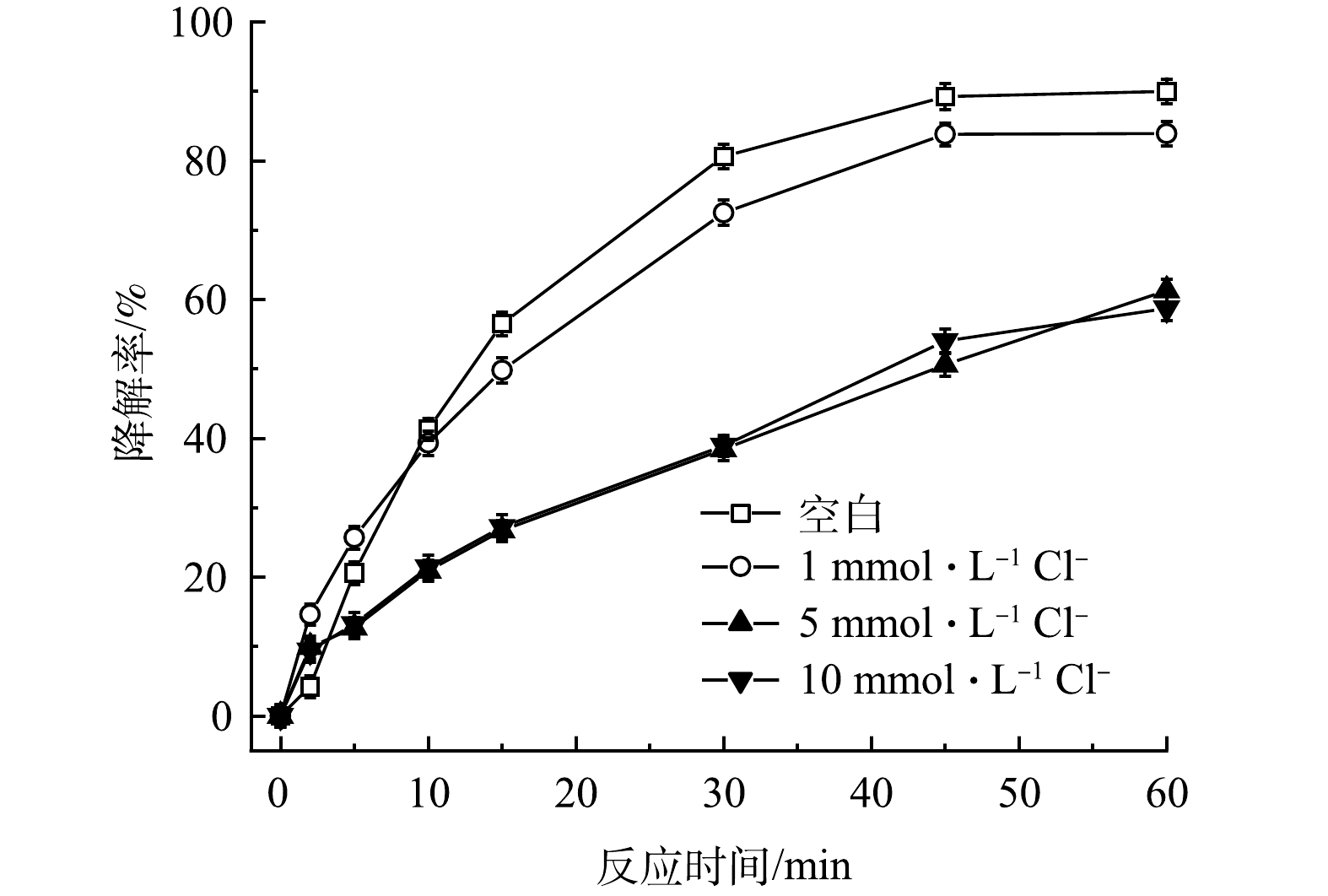
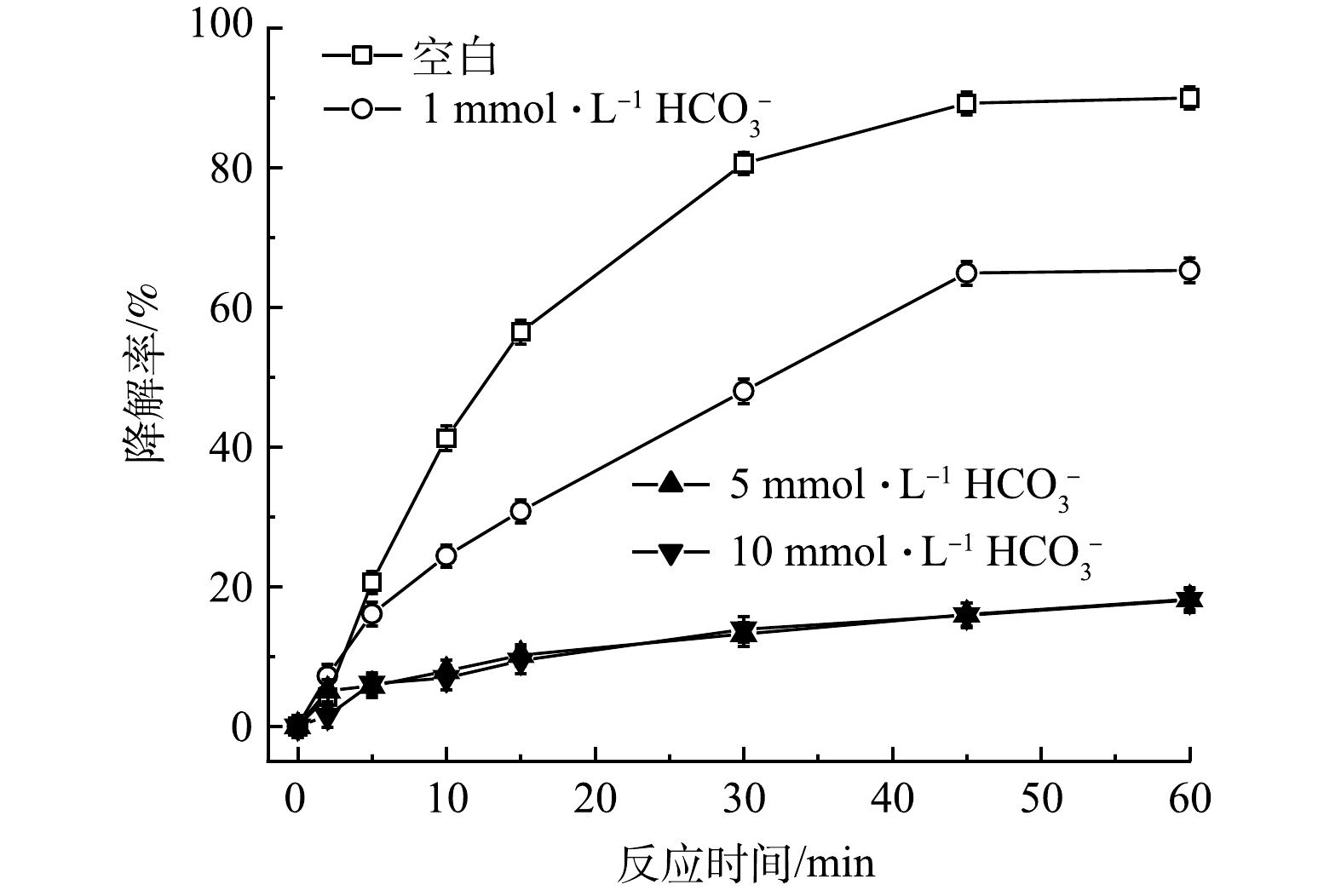
2)无机阴离子。在实际地下水中存在各种无机阴离子,其中Cl–和HCO3–对高级氧化影响较大。通过添加不同浓度的Cl–和HCO3–,探究其对TCE降解效果的影响。如图8所示,与空白相比,当加入1 mmol·L−1的Cl–后,TCE降解率从90.0%降至83.9%,随着Cl–浓度增加,对TCE降解的抑制作用也越来越强,在加入5和10 mmol·L−1 Cl–条件下,TCE降解率从90.0%分别降至61.3%和58.7%。造成这种现象可能有两方面的原因,首先,由式(11)可知,PMS会与Cl–发生反应,生成Cl2并消耗PMS,使反应过程中有效参与TCE降解的氧化剂减少[22];其次,HO·和SO4–·也会与Cl–反应生成Cl·和ClOH–(式(12)和式(13)),造成自由基的消耗[23]。如图9所示,HCO3–对TCE降解的抑制作用比Cl–更强。与空白相比,当加入1、5和10 mmol·L−1 HCO3–后,TCE降解率从90.0%分别降至65.3%、18.2%和18.1%。一方面,HCO3–的加入会使溶液pH升高,且HCO3–具有一定缓冲能力;另一方面,HCO3–也会淬灭HO·和SO4–·并生成氧化性较弱且选择性较强的CO3–·自由基(式(14)和式(15)),造成体系中HO·和SO4–·的消耗。所以,当PMS/FeS体系内存在高浓度HCO3–时,TCE的降解会受到较强的抑制[24]。
-
1)自由基淬灭实验。为了解PMS/FeS体系中产生的主导自由基种类,开展了自由基淬灭实验。选择叔丁醇(TBA)作为HO·的淬灭剂,异丙醇(IPA)作为SO4–·和HO·的淬灭剂,三氯甲烷(CF)作为O2–·的淬灭剂[20]。结果如图10所示。不加淬灭剂时,60 min内TCE降解90.0%,当加入10和100 mmol·L−1的TBA后,TCE降解率分别降至84.4%和69.3%,表明HO·是PMS/FeS体系中降解TCE的主导自由基之一;考虑到CF在水中溶解度较低,因此加入的CF最高浓度为50 mmol·L−1。当加入10和50 mmol·L−1的CF后,TCE降解率分别降至79.3%和67.4%,表明O2–·也是影响TCE降解的主导自由基之一;当加入10和100 mmol·L−1的IPA后,TCE降解率分别降至31.1%和24.7%,表明SO4–·也是PMS/FeS体系中降解TCE的主导自由基之一,且SO4–·的贡献程度比HO·和O2–·大。另外,这一结果也与SO4–·对TW-80分解的作用程度比HO·和O2–·大相一致。
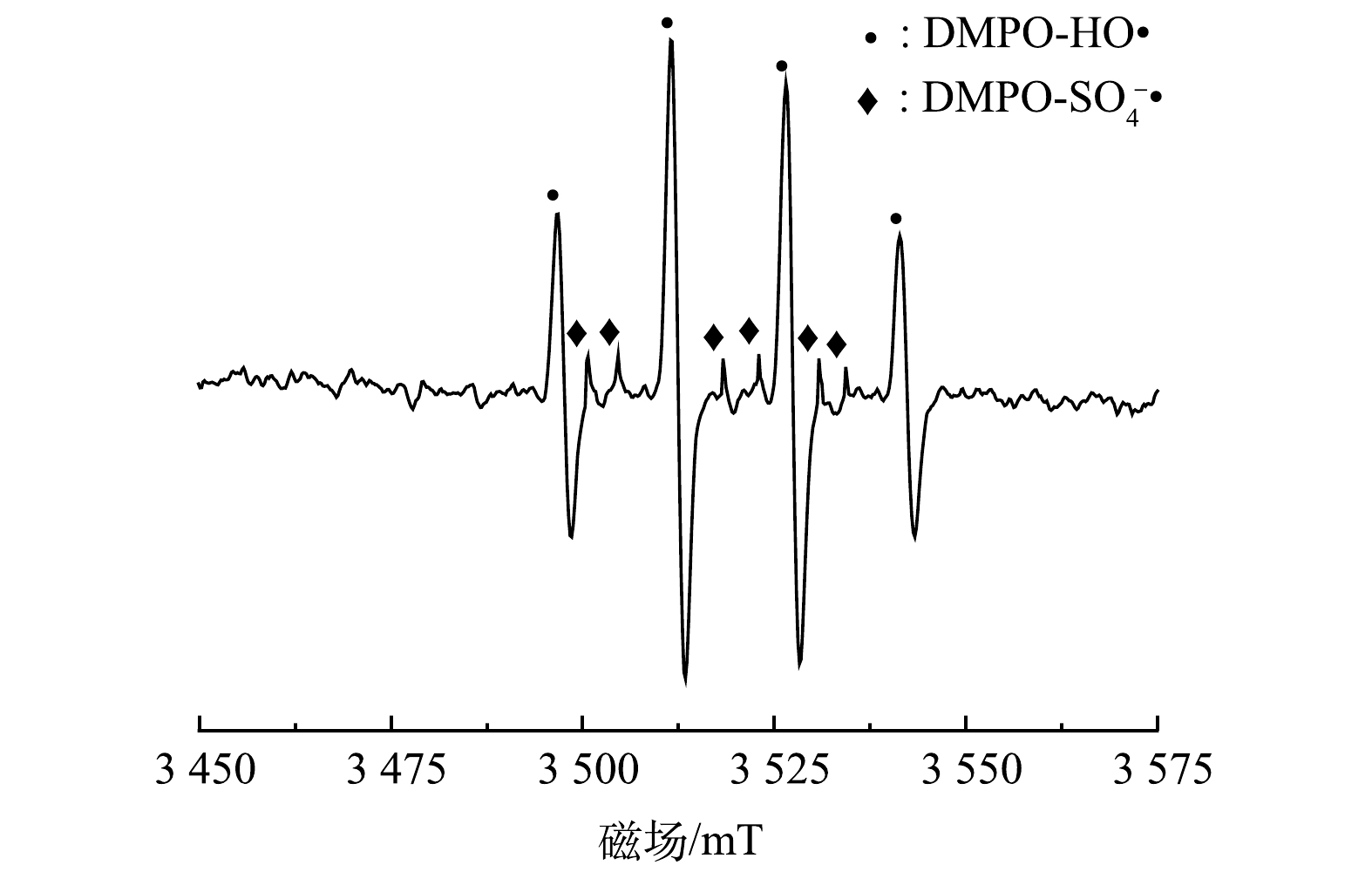
2)电子顺磁共振实验。为了进一步验证PMS/FeS体系中产生的自由基,选择DMPO作为捕捉剂,利用电子顺磁共振仪(EPR)探究体系中存在的自由基[25]。反应15 min后,如图11所示,在EPR谱图中检测到DMPO-HO·和DMPO-SO4–·加合物的特征峰。这证明HO·和SO4–·是PMS/FeS体系中的主要自由基[26]。
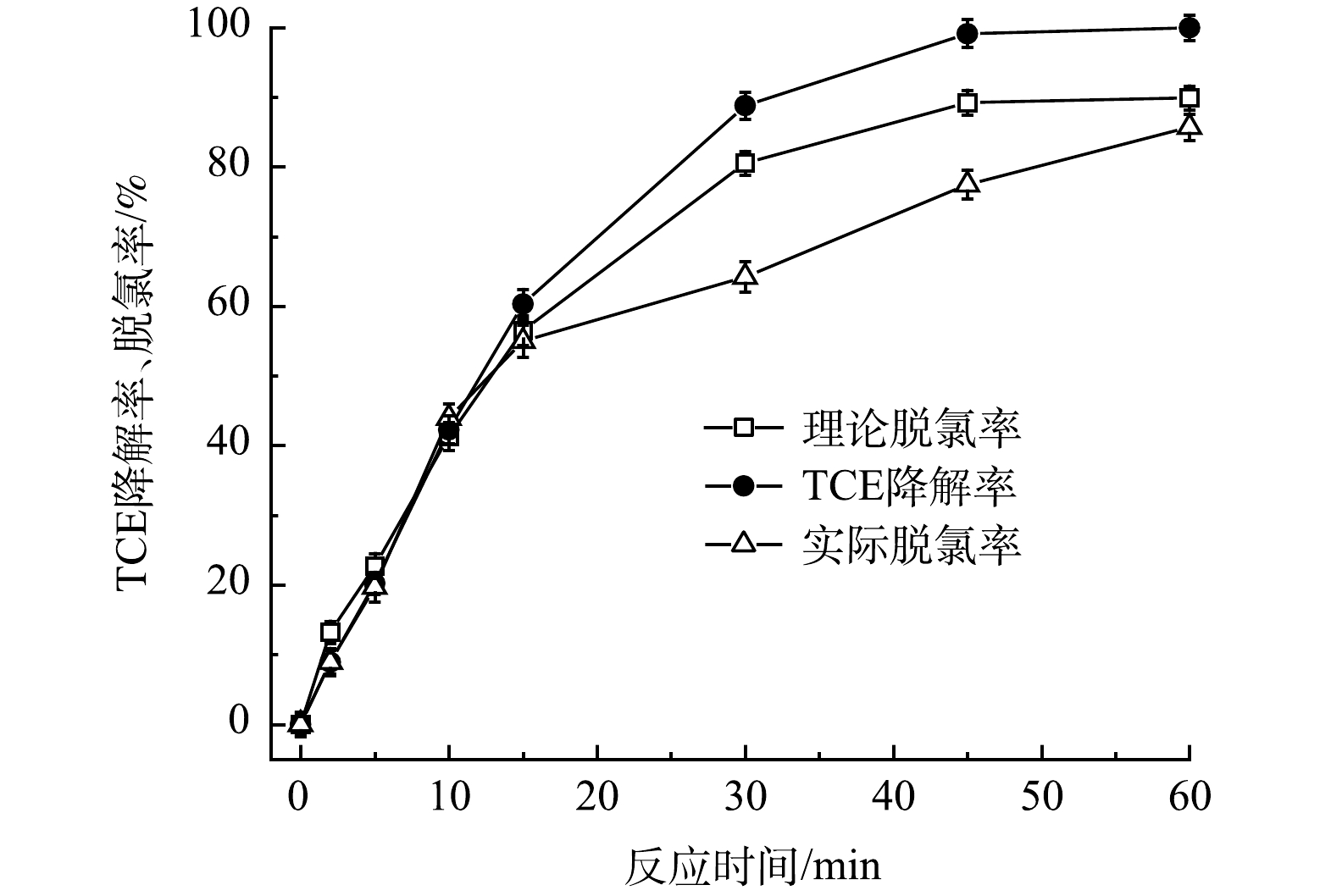
3) TCE脱氯及S形态的转化。为探究含TW-80的TCE降解过程中脱氯情况,利用IC进一步测定了TCE降解过程中Cl–浓度变化,结果如图12所示。随着TCE的降解,Cl–释放量逐渐升高,60 min后达到66.6%,考虑到反应过程中5.7%的TCE挥发,因此实际Cl–的释放率为72.3%。鉴于在此药剂投加量下TCE的去除率为90%,扣除5.7%的TCE挥发量后理论Cl–释放量为84.3%,可以计算出在TCE降解过程中超过85.8%的TCE被完全脱氯。
在反应过程中FeS中的硫元素会转化为SO42−,SO42−浓度(去除PMS自身分解产生的SO42−)随时间的变化如图13所示。在60 min内,SO42−由0增加至127.3 mg·L−1,FeS利用率达78.2%。表明在PMS/FeS体系中FeS充分发挥其还原作用,促进了体系中Fe(Ⅱ)/Fe(Ⅲ)循环和TCE的降解。
-
在实际污染场地中除TCE外,还常有其他氯代烃污染物检出,如四氯乙烯(PCE)、1,1,1-三氯乙烷(TCA)、四氯化碳(CT)、1,2-二氯乙烷(DCA)、三氯甲烷(CF)和二氯甲烷(DCM)等。利用PMS/FeS体系降解含TW-80的多种氯代烃实验,PCE、TCA、CT、DCA、CF和DCM浓度均设置为0.15 mmol·L−1。结果如图14所示。在60 min内超过63.9%的PCE和94.4%的CT被降解,但TCA、DCA、CF和DCM去除率分别仅为6.3%、3.5%、3.2%和1.7%,表明PCE能够被PMS/FeS体系产生的自由基氧化。对于有机物CT,体系中产生的还原性O2–·自由基有助于CT的降解。此外,根据相关研究报道,表面活性剂在降解过程中会产生醇类等分解产物。HO·和SO4−·作为强氧化性自由基,可能会与醇类物质反应生成具有还原性的醇自由基。因此,CT作为高氯代有机物,能够被高效降解的主要原因可能是由于其被相关醇自由基和体系中生成的O2–·还原所致[27]。遗憾的是,TCA、DCA、CF和DCM降解率均未超过10%,表明PMS/FeS体系中产生的自由基无法促进上述烷烃的去除。
-
为验证PMS/FeS体系在实际地下水中(含1.3 g·L−1 TW-80)降解TCE的效果,进一步开展了不同投加剂量及预先调节溶液pH实验研究。实际地下水中TOC、Cl–、HCO3–、NO3–、CO32–、Ca2+、Ma2+质量浓度分别为16.44、134、91.2、0.408、172、59和55 mg·L−1,pH为7.49。各种实验条件下反应前后溶液pH变化如表2所示。当不调节溶液pH时,在0.8 mmol·L−1 PMS和0.6 g·L−1 FeS投加量下,如图15所示,TCE降解率仅为6.8%;将PMS和FeS剂量增加至2倍和4倍时,TCE的降解率由6.8%分别增加到13.0%和26.7%。在实际地下水中的实验结果与在超纯水中的结果相差较大。其主要原因是实际地下水pH呈弱碱性,且存在高浓度的无机阴离子(Cl–和HCO3–/CO32–)[28]。因此,增加药剂量无法达到较好的降解效果,且增加修复药剂成本。为了提高TCE降解率和节省修复成本,使用H2SO4将溶液初始pH预先调节为3,令人意外的是,在0.8 mmol·L−1 PMS和0.6 g L−1 FeS投加量下,TCE降解率达到63.8%,将PMS和FeS剂量同时增加至2倍和4倍,TCE降解率提高至70.2%和82.8%,相比未调节pH的实验组,TCE降解率显著增加。虽然反应后溶液pH在3以下,但可以采用生石灰等廉价碱性物质调节至中性。显然,对于在实际工程中调节pH的应用,可以考虑通过注射井,在污染区域上游将表面活性剂注入,在表面活性剂的增溶和迁移作用下,通过下游提升井,利用水泵将表面活性剂和被增溶的有机污染物从污染区域下游抽出至地表进行处理,在地表上可预先调节溶液pH,经处理达标后可回灌至地下或通过市政管道纳管排放。
-
1)相比PMS/Fe(Ⅱ)体系,PMS/FeS体系对含TW-80的TCE降解效果更好,当药剂投加量PMS为0.8 mmol·L−1、FeS为0.6 g·L−1时,60 min内TCE降解率为90.0%,FeS的还原作用使反应过程中Fe(Ⅲ)及时还原成Fe(Ⅱ),促进Fe(Ⅱ)/Fe(Ⅲ)的循环。
2) TW-80会抑制TCE的降解,且TW-80浓度越高,抑制作用越强,PMS/FeS体系产生的自由基会攻击TW-80,且SO4–·对TW-80分解起主要作用;在超纯水实验中PMS/FeS体系适应的pH范围较宽,当pH在3~9时,TCE的降解效果均较好,当pH=11时,TCE降解受到抑制;HCO3–和Cl–均会影响TCE的降解,其中HCO3–对TCE降解的抑制作用更强。
3) SO4–·、HO·和O2–·是PMS/FeS体系降解TCE的主导自由基,其中SO4–·对降解的贡献最大。
4) PMS/FeS体系对PCE和CT同样具有较好的降解效果,但对其他氯代烷烃的降解效果不佳。
5)与超纯水相比,PMS/FeS处理实际地下水中的TCE效果不佳;预先调节溶液pH=3后,TCE降解率显著提高,表明PMS/FeS体系在修复实际地下水工程中具有较好的应用潜力。
硫化亚铁活化过一硫酸盐降解含表面活性剂水溶液中三氯乙烯的效果与机制
Degradation of trichloroethene in water solution containing surfactant by peroxymonosulfate activated by ferrous sulfide
-
摘要: 采用硫化亚铁(FeS)活化过一硫酸盐(PMS)降解含表面活性剂吐温-80(TW-80)水溶液中的三氯乙烯(TCE),考察了PMS和FeS投加量、TW-80浓度、溶液初始pH、无机阴离子(Cl–和HCO3–)对TCE降解的影响,确定了PMS/FeS体系中的主导自由基及TCE降解机理,验证了PMS/FeS体系处理实际地下水中含TW-80的TCE效果。结果表明:增加PMS或FeS投加量有利于TCE的降解,但当其投加剂量分别超过0.8 mmol·L–1和0.6 g·L–1时,TCE降解反而受到抑制,且TCE的降解率随TW-80浓度的增加而下降;PMS/FeS体系对pH有较宽的适用范围,在pH=11时受到抑制,Cl–和HCO3–对TCE降解有抑制作用;通过自由基淬灭实验和电子顺磁共振实验确定了SO4–·、HO·和O2–·是PMS/FeS体系中的主导自由基;相比于其他氯代烃,该体系对四氯乙烯和四氯化碳也有较好的降解效果;实际地下水的处理结果证实PMS/FeS体系在实际应用中具有潜力和优势。Abstract: Ferrous sulfide (FeS) was used to activate peroxymonosulfate (PMS) for trichloroethylene (TCE) degradation in solution containing Tween-80 (TW-80). The effects of PMS and FeS doses, the initial solution pH, inorganic anions (Cl– and HCO3–) on TCE degradation were investigated. The main reactive radicals generated in PMS/FeS system were determined and TCE degradation mechanisms were revealed. The degradation performance of TCE in actual groundwater containing TW-80 was verified by PMS/FeS process. The experimental results showed that the increase of PMS or FeS dosage was favorable to TCE removal, while TCE degradation was inhibited at PMS dose over 0.8 mmol·L–1 or FeS dose over 0.6 g·L–1. In addition, TCE degradation rate decreased with the increase of TW-80 concentration. PMS/FeS system performed well in a wide pH range, but its effect was seriously inhibited at pH = 11. Both Cl– and HCO3– had inhibitive effects on TCE removal. SO4–·, HO· and O2–· were identified as the main reactive radicals in PMS/FeS system by radical scavenging tests and electron paramagnetic resonance detection. Compared with other chlorinated hydrocarbons, PMS/FeS had a better performance on perchloroethylene and carbon tetrachloride degradation. Finally, the test results using actual groundwater demonstrated that PMS/FeS system had a great application potential in actual groundwater remediation.
-
Key words:
- trichloroethylene /
- surfactants /
- peroxymonosulfate /
- ferrous sulfide /
- groundwater remediation
-
六溴环十二烷(HBCD)是一种高含溴量的添加型阻燃剂,其产量仅次于多溴联苯醚(decaBDE)和四溴双酚A(TBBPA),被广泛用于建筑、纺织材料、电子设备、塑料制品等产品中[1]。基于其广泛污染及具有富集性、远距离迁移性、生物毒性等特点,HBCD作为持久性有机污染物,于2013年被列入斯德哥尔摩公约受控名单[2]。
HBCD具有手性中心,理论而言共含有16种同分异构体,常见的有α-HBCD、β-HBCD和γ-HBCD,以及其相应的(+)和(−)对映体。不同立体构型的HBCD在环境中的富集、代谢和毒性行为均存在差异[3]。一般而言,在土壤[4]和水体[5]等非生物介质中γ-HBCD含量较高,在生物介质中α-HBCD则占主导。尽管β-HBCD在环境介质中的检出浓度一般不是最高的,但有研究表明,其累积能力和毒性作用不容忽视。对斜生栅藻[6]和玉米[7]的研究发现,β-HBCD在其体内的累积动力学速率和最高吸收量均高于α-和γ-HBCD。奥斯卡鱼肠对β-和γ-HBCD的吸收速率也高于对α-HBCD的吸收[8]。在生物毒性方面,Palace等[9]发现,与α-HBCD相比,β和γ-HBCD会显著增高鱼体内脱碘酶的活性,从而降低鱼对碘的吸收能力。对人肝细胞L02和人肝癌细胞HepG2的研究表明β-HBCD的毒性影响高于γ-和α-HBCD[10]。可见,在特定环境下β-HBCD的富集能力及生物毒性高于其他异构体。同时,在生物体内亦有研究表明,γ-HBCD会发生向β-HBCD的异构体转化,导致β-构型浓度增加[11],加剧其环境生态风险。因此,在研究HBCD的环境行为及影响时,对β-HBCD的考察需要引起重视。而目前关于β-HBCD的毒性研究较为匮乏,亟待开展更多工作。
有关HBCD植物毒性方面的考察相对较少,植物是生态系统中的第一营养级,植物对污染物的吸收及其迁移转化、环境影响和归趋均有重要作用,研究HBCD对植物的毒性作用具有深远意义。Zhang等[12]研究了商品HBCD混合物对拟南芥的基因表达和蛋白功能的毒性作用,发现机体能量产生-转化和氨基酸转运-代谢方面均受到不同程度的负面影响。Huang等[13]在体外研究α-和γ-HBCD对玉米细胞色素P450(CYP)酶的影响表明,(−)/(+)γ-HBCD会诱导CYP酶活性增加,α-HBCD则表现为抑制,且(−)α-HBCD对CYP酶活性的抑制高于(+)α-HBCD。本团队前期研究发现HBCD可诱导玉米体内羟基自由基产生,造成一定程度的DNA损伤,其中β-HBCD的毒性高于γ-HBCD[7];在对映体水平上也分别考察了α-和γ-HBCD对玉米的生长、形态改变、抗氧化酶和DNA损伤方面的对映体选择性影响[14-15]。然而遗憾的是上述研究均未开展对β-HBCD对映体的相关探讨,不同光旋纯活性的β-HBCD是否会对植物产生选择性毒性还需要进一步判断。
本研究选择玉米为受试植物,采用不同浓度梯度的β-HBCD及其对映体对玉米开展水培暴露研究,通过玉米体内抗氧化物质活性及含量变化,从抗氧化酶系统和非酶抗氧化系统两方面说明机体对β-HBCD选择性毒性的响应,结合细胞内典型活性氧(ROS)物质超氧阴离子(
O⋅−2 1. 实验部分 (Experimental section)
1.1 试剂与仪器
主要试剂:β-HBCD标准品(50 μg·mL−1)购于AccuStandard公司。流动相甲醇为色谱纯,其余试剂均为分析纯。
主要仪器:高效液相色谱(HPLC,岛津LC-20AD)、紫外-可见分光光度计(岛津UV-2550)、高速冷冻离心机(Multifuge X1R,Thermo公司)、恒温培养箱(A1000,Conviron公司)、多功能酶标仪(SPARK,德国TECAN公司)、电子天平(精度±0.0001g,上海双杰有限公司)。
1.2 对映体的分离与制备
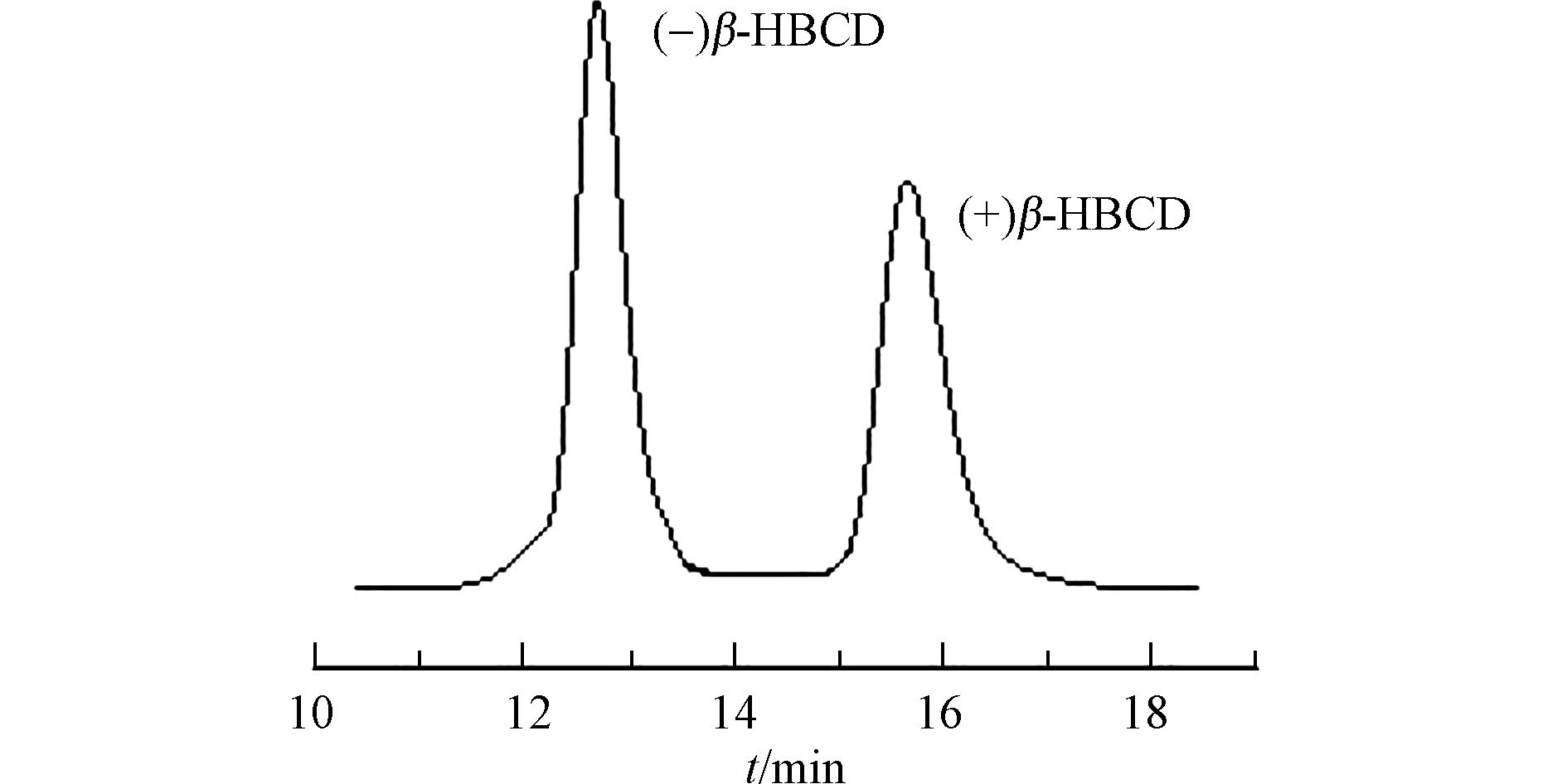
选择β-PM手性柱(200 mm×4 mm,5 μm,德国MN公司),采用高效液相色谱法(HPLC)优化得到最佳色谱分离条件:流动相为95%甲醇:5%水,流速0.4 mL·min−1,柱温25 ℃,检测器波长220 nm。在最佳分离条件下制备单一对映体,根据紫外检测器出峰信号和保留时间手动收集相应的流出液,并采用外标法进行定量。根据Zhang等[16]的研究,流出检测器的对映体依次为(−)β-HBCD、(+)β-HBCD对映体。分离后的对映体纯度均在98%以上,最终获得的(+)和(−)β-HBCD浓度分别为1.81 μg·mL−1和1.63 μg·mL−1。分离谱图如图1所示。
1.3 植物的暴露与培养
选取均匀且饱满的玉米种子(购于中国农业科学研究院),去离子水清洗后,用10%H2O2的消毒溶液浸泡10 min,用去离子清洗干净后,将玉米种子均匀平铺在覆盖有湿滤纸的托盘上,置于(27±0.5)℃的恒温培养箱中于暗处发芽,保持相对湿度在60%—80%。配置不同浓度梯度(0.5、1.0、2.0、3.0、5.0 μg·L−1)的(+)β-HBCD、(rac)β-HBCD和(−)β-HBCD暴露液。挑选培养4 d后长势均一的玉米幼苗转移至盛有150 mL HBCD溶液的黑色玻璃水培罐中进行暴露,每盆放置5棵幼苗,每24 h更换1次暴露液,更换暴露液前检测溶液中对映体外消旋化的比率(< 9%),确定对映体转化的影响可忽略不计。恒温培养箱每天设置14 h光照时间,相对湿度为60%—80%,每天改变玉米幼苗的位置以减少光照和温度的空间差异。暴露3 d后进行毒性指标测定。实验中,每个处理设置3个平行,每个实验重复3次,设置去离子水和甲醇空白对照,两组对照结果一致,说明溶剂甲醇未对植物生长产生影响。
1.4 酶提取与测定
将玉米幼苗用去离子水清洗并用滤纸吸干,分别称取0.5 g玉米根部与地上部,置于研钵中,在冰浴下加入6 mL磷酸缓冲液(0.05 mol·L−1、pH =7.8)研磨成匀浆,于4 ℃,5000 r·min−1离心15 min,提取上清液,即得到样品粗酶液。
采用Bradford的方法[17],以100 μg·mL−1牛血清蛋白为标准溶液,0.005%的考马斯亮蓝G-250溶液为染料,在595 nm下绘制标准曲线,检测样品中的蛋白水平。超氧化物歧化酶(SOD)的测定参照Beauchamp等[18]的方法并略作改动。采用氮蓝四唑(NBT)光还原法进行检测,将粗酶液与显色液(50 mmol·L−1磷酸缓冲液(pH =7.8)、130 mmol·L−1甲硫氨酸、750 μmol·L−1氮蓝四唑、100 μmol·L−1 EDTA-Na2及20 μmol·L−1核黄素)混匀,将1支对照管避光放置作为空白,其它于4000 lx光照下反应15—30 min,在560 nm处测定各管吸光值。抗坏血酸过氧化物酶(APX)的测定参照Nakano[19]的方法,0.5 mL上清液混于1 mL反应体系(包含50 mmol·L−1磷酸钾,0.5 mmol·L−1抗坏血酸,0.1 mmol·L−1过氧化氢以及0.1 mmol·L−1 EDTA),在290 nm处测定各管吸光值以确定抗坏血酸活性。
1.5 花青素的测定
称取幼苗叶片0.10 g,剪成1—2 mm碎片,置于5 mL 2%的盐酸甲醇溶液中,于37 ℃恒温箱中放置3 h,直到叶片用肉眼观察已完全变白,取出过滤获得上清液,于530 nm下测花青素的含量,由于可溶性糖和叶绿素的干扰,需要在620 nm下测可溶性糖和650 nm下叶绿素的光密度值。计算公式为:
OD花青素=(OD530−OD620)−0.1×(OD650−OD620) (1) 花青素含量(nmol⋅g−1)=[(OD花青素/ε)×(V/m)×106] (2) 其中,ε为花青素的摩尔消光系数值为4.62×106;V为所取上清液体积(mL);m为所取样品质量(g);nmol·g−1为每克样品中的花青素含量。
1.6 超氧阴离子的检测
植物样品的提取和粗酶液的提取方法一致,获得的上清液采用NBT法[20]检测。取0.5 mL样品、0.5 mL 65 mol·L−1的磷酸缓冲液和1.5 mL 1.0 mol·L−1的盐酸羟胺混匀,于25 ℃保温1 h,加入2 mL 17 mmol·L−1对氨基苯磺酸和2 mL 7 mmol·L−11-萘胺,摇匀后在25 ℃保温20 min,在波长530 nm下测吸光度。
1.7 根系活力检测
根系活力采用TTC法[21]并略作改动。在0.1 mL 1%的2,3,5-三苯基氯化四氮唑溶液中加入少许Na2S2O4粉末,无水甲醇定容至10 mL,即得到三苯甲月替(TTF)标准溶液,以空白作参比,在485 nm下测定吸光度值绘制标准曲线。称取0.3 g根样品于小烧杯中,加入5 mL 1% TTC和5 mL 0.1 mol·L−1磷酸缓冲液(pH =7.0)摇匀,使溶液完全浸没根,37 ℃无光保温2 h,加入2 mL 1 mol·L−1硫酸停止反应。取出根,用滤纸吸干反应液并切段置于10 mL甲醇中,于40 ℃恒温箱中放置,使根尖切段完全变白为止(4—6 h),在485 nm下测定吸光度值,通过标准曲线计算出四氮唑还原量。
1.8 数据分析
所有数据均为3次重复实验所得结果,实验数据以平均值±标准偏差(Mean±SD)来表示。采用Origin 8.0对数据进行回归分析,SPSS 17.0对数据进行显著性检验。
2. 结果与讨论 (Results and discussion)
2.1 β-HBCD对玉米抗氧化酶活性的影响
SOD是生物体抗氧化系统的第一道防线,可催化
O⋅−2 随着β-HBCD暴露浓度的增加,玉米体内SOD和APX活性(除(−)β-HBCD处理外)均呈现显著的先增加后减少的趋势(图2),表明β-HBCD诱发了玉米机体的氧化应激效应。图2a显示玉米根部SOD活性在(+)、(rac)和(−)β-HBCD最低暴露浓度下(0.5 μg·L−1)即显著增加到最大值(P < 0.05),分别为对照组的1.85倍、1.71倍和1.63倍,但三者之间未呈现出显著性差异,可能是因为低浓度的污染物对玉米SOD活性的影响较小;在最高浓度5.0 μg·L−1时,SOD活性降到最低,但仍比对照组增加了58.7%、56.0%和26.8%,表明β-HBCD对玉米产生了氧化胁迫,且不同立体构型产生的毒性为(+)β-HBCD > (rac)β-HBCD > (−)β-HBCD。在(+)和(rac)β-HBCD暴露下,玉米地上部SOD活性分别在3.0 μg·L−1与2.0 μg·L−1时达到最大(图2b),较对照组分别增加了15.6%和9.5%,表明根部受到的影响更大。(−)β-HBCD处理的地上部SOD活性与对照组相比无显著差异,可见(−)β-HBCD对玉米的影响较小。
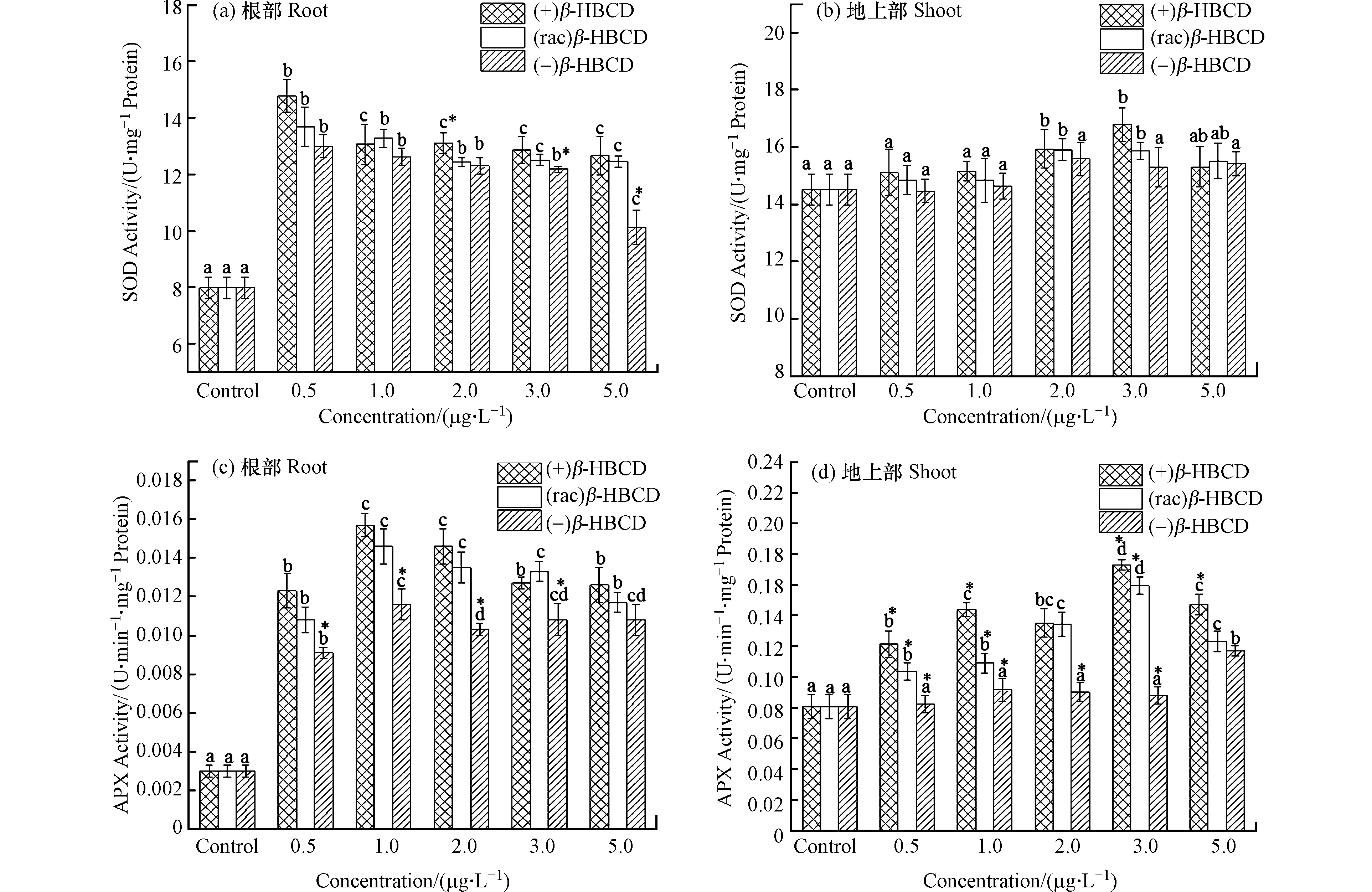 图 2 β-HBCD对玉米体内SOD和APX活性的影响Figure 2. Effects of β-HBCD on SOD and APX activity in maize(不同字母表示不同浓度之间存在显著差异(P < 0.05),* 表示不同对映体之间存在显著差异(P < 0.05))(Different letters represent statistically significant difference between different concentrations at P << 0.05,while * represents a significant difference between the enantiomers at P < 0.05)
图 2 β-HBCD对玉米体内SOD和APX活性的影响Figure 2. Effects of β-HBCD on SOD and APX activity in maize(不同字母表示不同浓度之间存在显著差异(P < 0.05),* 表示不同对映体之间存在显著差异(P < 0.05))(Different letters represent statistically significant difference between different concentrations at P << 0.05,while * represents a significant difference between the enantiomers at P < 0.05)β-HBCD及其对映体均诱导了玉米根部和地上部APX活性增加(图2c和d)。玉米根部在暴露浓度为1.0 μg·L−1时APX活性最高,(+)、(rac)和(−)β-HBCD处理分别是对照组的5.26倍、4.91倍和3.90倍,随后与浓度呈现负相关关系。地上部分,APX活性在(+)和(rac)β-HBCD浓度为3.0 μg·L−1时达到最大值,较对照组分别增加了114.6%和97.6%,随后逐渐降低;(−)β-HBCD处理的玉米地上部APX活性在低浓度下无显著差异,仅在最大暴露浓度(5.0 μg·L−1)时显著增加到对照组的1.45倍。可见,APX活性变化结果与SOD类似,均表现出了(+)β-HBCD的影响最大,其次是(rac)β-HBCD,(−)β-HBCD影响最小。Fang等[23]在甲霜灵对映体对烟草幼苗的影响研究中发现:R-对映体比S-对映体对SOD活性的促进作用更高,对幼苗的影响更大,说明同种物质不同立体构型对生物体产生的毒性作用亦是存在差异的。
外源污染物会影响植物体内的活性氧(ROS)平衡,ROS的大量生成,可以激发植物自身的抗氧化防御系统,显著诱导酶活性增加以消除过量的ROS[24]。本研究中β-HBCD诱导了玉米体内ROS的产生,使细胞抗氧化酶SOD和APX活性增加以清除玉米体内的
O⋅−2 2.2 β-HBCD对玉米体内花青素含量的影响
植物在受到氧化胁迫时,为保护自身生理发育及新陈代谢的正常进行,形成了一套复杂完善的抗氧化保护系统,包括抗氧化酶系统和非酶系统,花青素是非酶系统中一种重要的抗氧化剂,属于生物类黄酮物质,主要存在于植物叶片中,和酚类化合物的作用机理相似,花青素可通过酚羟基与自由基反应生成较稳定的半醌式自由基,从而终止自由基链式反应[27]。
随着β-HBCD暴露浓度的增加,玉米地上部花青素含量与抗氧化酶类似,亦呈现先增加后减少的趋势(图3)。1.0 μg·L−1的(+)、(rac)和(−)β-HBCD处理下玉米叶片中花青素含量最高,分别达到对照组的3.55倍、3.22倍和2.28倍;在最高浓度5.0 μg·L−1时,花青素含量最低,(+)、(rac)和(−)β-HBCD处理组分别比对照组增加了77.6%、94.2%和43.9%。研究表明,花青素具有清除自由基、促进激活抗氧化酶系统的能力,是植物抗氧化的基础[27],本研究中植物体内抗氧化酶系统和非酶系统随β-HBCD暴露的变化趋势一致,说明其在植物防御外源氧化胁迫过程中同样起到非常重要的作用。不同对映体处理组相比,与抗氧化酶系统的SOD和APX活性检测结果一致,均表现出对玉米的毒性作用大小顺序为(+)β-HBCD > (rac)β-HBCD > (−)β-HBCD。
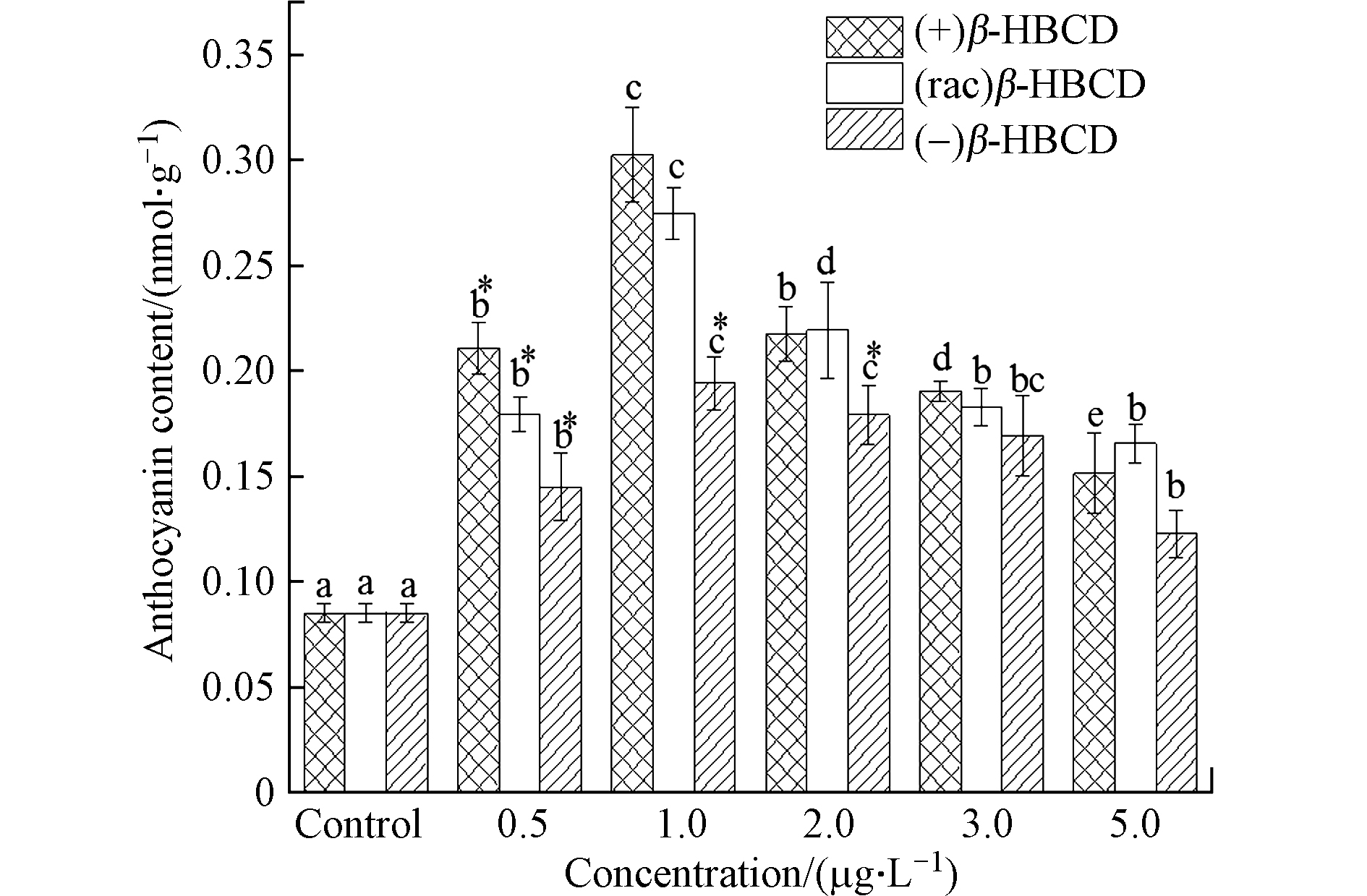 图 3 β-HBCD对花青素含量的影响Figure 3. Effect of β-HBCD on anthocyanidin content in maize(不同字母表示不同浓度之间存在显著差异(P < 0.05),* 表示不同对映体之间存在显著差异(P < 0.05))(Different letters represent statistically significant difference between different concentrations at P < 0.05,while * represents a significant difference between the enantiomers at P < 0.05)
图 3 β-HBCD对花青素含量的影响Figure 3. Effect of β-HBCD on anthocyanidin content in maize(不同字母表示不同浓度之间存在显著差异(P < 0.05),* 表示不同对映体之间存在显著差异(P < 0.05))(Different letters represent statistically significant difference between different concentrations at P < 0.05,while * represents a significant difference between the enantiomers at P < 0.05)2.3 β-HBCD对玉米超氧阴离子含量的影响
β-HBCD的暴露诱导了玉米体内SOD、APX酶活性和花青素含量的增加,那么这种应激响应是否对机体内部的ROS进行了有效的控制呢?本研究进一步考察了玉米组织中的
O⋅−2 结果显示(图4),玉米体内的
O⋅−2 O⋅−2 O⋅−2 O⋅−2 O⋅−2 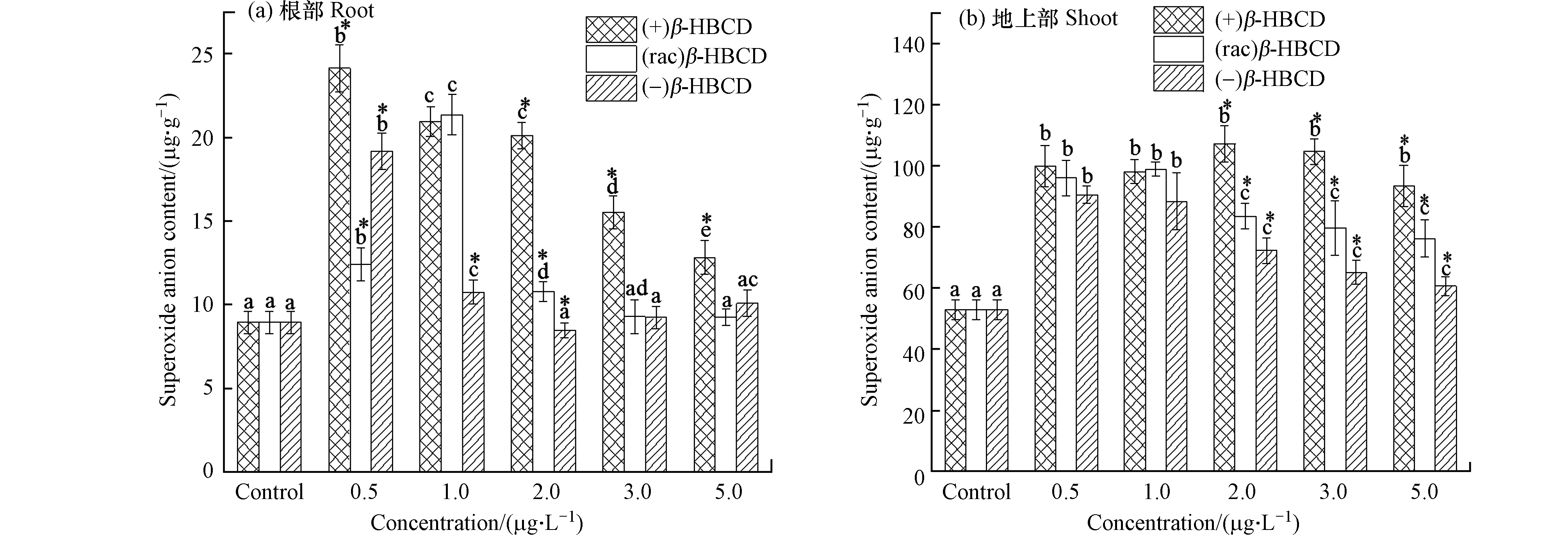 图 4 β-HBCD对映体对玉米体内
图 4 β-HBCD对映体对玉米体内O⋅−2 Figure 4. Effects of β-HBCD enantiomers onO⋅−2 (不同字母表示不同浓度之间存在显著差异(P < 0.05),* 表示不同对映体之间存在显著差异(P < 0.05))(Different letters represent statistically significant difference between different concentrations at P < 0.05,while * represents a significant difference between the enantiomers at P < 0.05)在外界胁迫下,生物体内更多的分子氧经过单电子还原反应转变为
O⋅−2 O⋅−2 O⋅−2 O⋅−2 2.4 β-HBCD对玉米根系活力的影响
根系是植物获得营养和水分,维持稳定较大生物量的关键部位,根系活力的高低直接影响作物的正常生长并间接干扰叶片的光合效率[29-30]。本研究中玉米根系是直接接触暴露液的部位,根部
O⋅−2 O⋅−2 由图5可以看出,经(+)和(rac)β-HBCD胁迫后,幼苗根系活力在0.5 μg·L−1时与空白对照组无显著差异,在1.0 μg·L−1时显著下降,于最大浓度(5.0 μg·L−1)时降到最低,分别较对照组减少了65.1%和39.7%;(−)β-HBCD胁迫下,幼苗根系活力仅在最大浓度胁迫下,较空白对照组显著下降了28.5%,其余处理均与空白无显著差异。显然,尽管植物机体存在庞大的抗氧化保护系统,但β-HBCD及其对映体仍降低了玉米幼苗的根系活力,影响了其健康状态,且不同对映体对幼苗根系活力的抑制作用为(+)β-HBCD > (rac)β-HBCD > (−)β-HBCD。植物根系活力反映根系整体的代谢强度,包括根系呼吸、氧化、还原和合成能力等[31],植物处于逆境生理环境时,其呼吸代谢关键酶表达量会发生改变,进而影响植物的呼吸过程[30]。本研究中β-HBCD很可能是通过诱导ROS的变化影响了玉米根部代谢关键酶的表达或活性,从而导致其代谢强度的改变,根系活力发生变化。这与Zhang等[32]关于碱环境胁迫下大米机体ROS累积导致根系损伤的作用结果一致。不同的β-HBCD对映体对幼苗根系活力的抑制作用存在显著差异,可能与其空间立体构型有关。有文献报道,不同立体构型的HBCD与细胞内代谢相关酶的结合位点具有差异性,其中(−)β-HBCD能够选择性进入玉米代谢相关酶-细胞色素氧化酶(CYP)和谷胱甘肽转移酶(GST)亚型酶CYP71C3v2、GST31的活性位点并与其键合,进而被转化成不同的代谢产物,从而降低了其在植物体内的浓度[25],这可能是(−)β-构型产生的毒性效应较小的主要原因。
 图 5 β-HBCD对玉米根系活力的影响Figure 5. Effects of β-HBCD on root activity in maize(不同字母表示不同浓度之间存在显著差异(P < 0.05),* 表示不同对映体之间存在显著差异(P < 0.05))(Different letters represent statistically significant difference between different concentrations at P < 0.05,while * represents a significant difference between the enantiomers at P < 0.05)
图 5 β-HBCD对玉米根系活力的影响Figure 5. Effects of β-HBCD on root activity in maize(不同字母表示不同浓度之间存在显著差异(P < 0.05),* 表示不同对映体之间存在显著差异(P < 0.05))(Different letters represent statistically significant difference between different concentrations at P < 0.05,while * represents a significant difference between the enantiomers at P < 0.05)3. 结论 (Conclusion)
手性化合物β-HBCD对玉米的生长代谢具有显著的对映体选择性影响,毒性大小为(+)β-HBCD>(rac)β-HBCD>(−)β-HBCD,主要源于植物抗氧化酶系统和非酶抗氧化系统未能有效控制玉米体内
O⋅−2 -
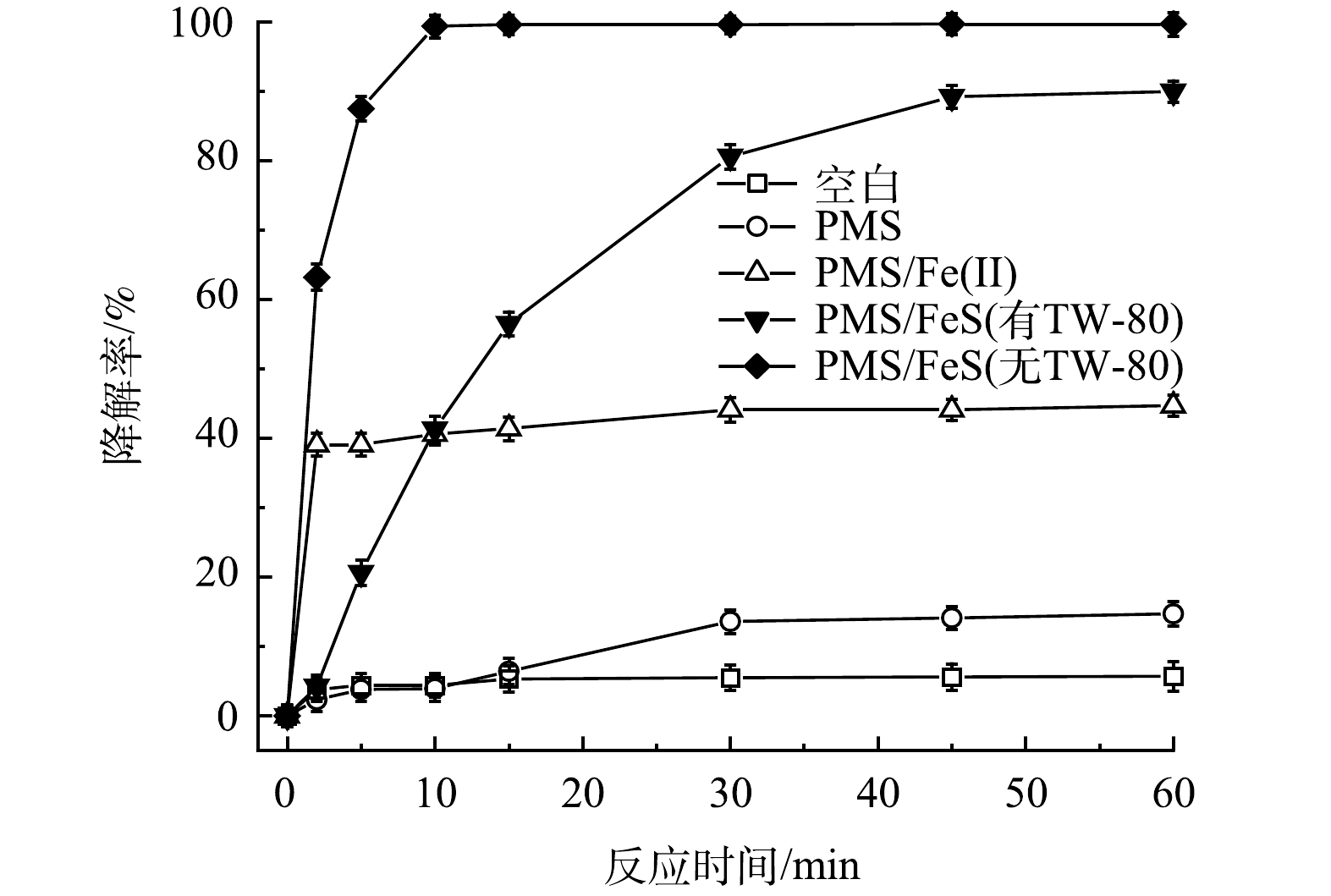
图 1 TCE在不同体系中的降解效果比较
Figure 1. Comparison of TCE degradation performance in different systems
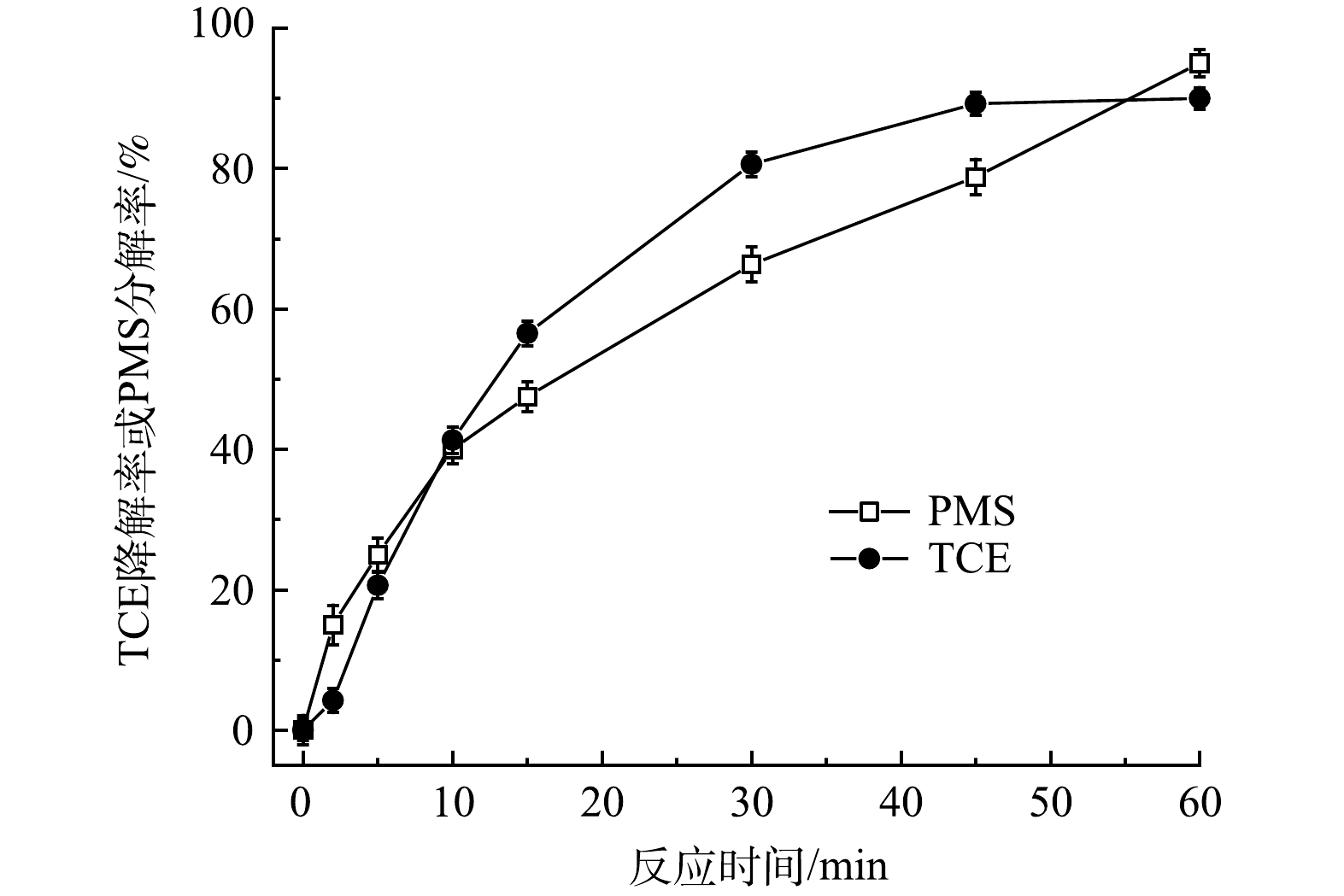
图 3 PMS/FeS体系中TCE降解率与PMS分解率
Figure 3. TCE degradation and PMS decomposition in PMS/FeS system

图 5 PMS/FeS体系中Fe(II)、Fe(III)及溶解性总铁浓度变化
Figure 5. Changes of Fe(II), Fe(III), and total soluble iron concentrations in PMS/FeS system
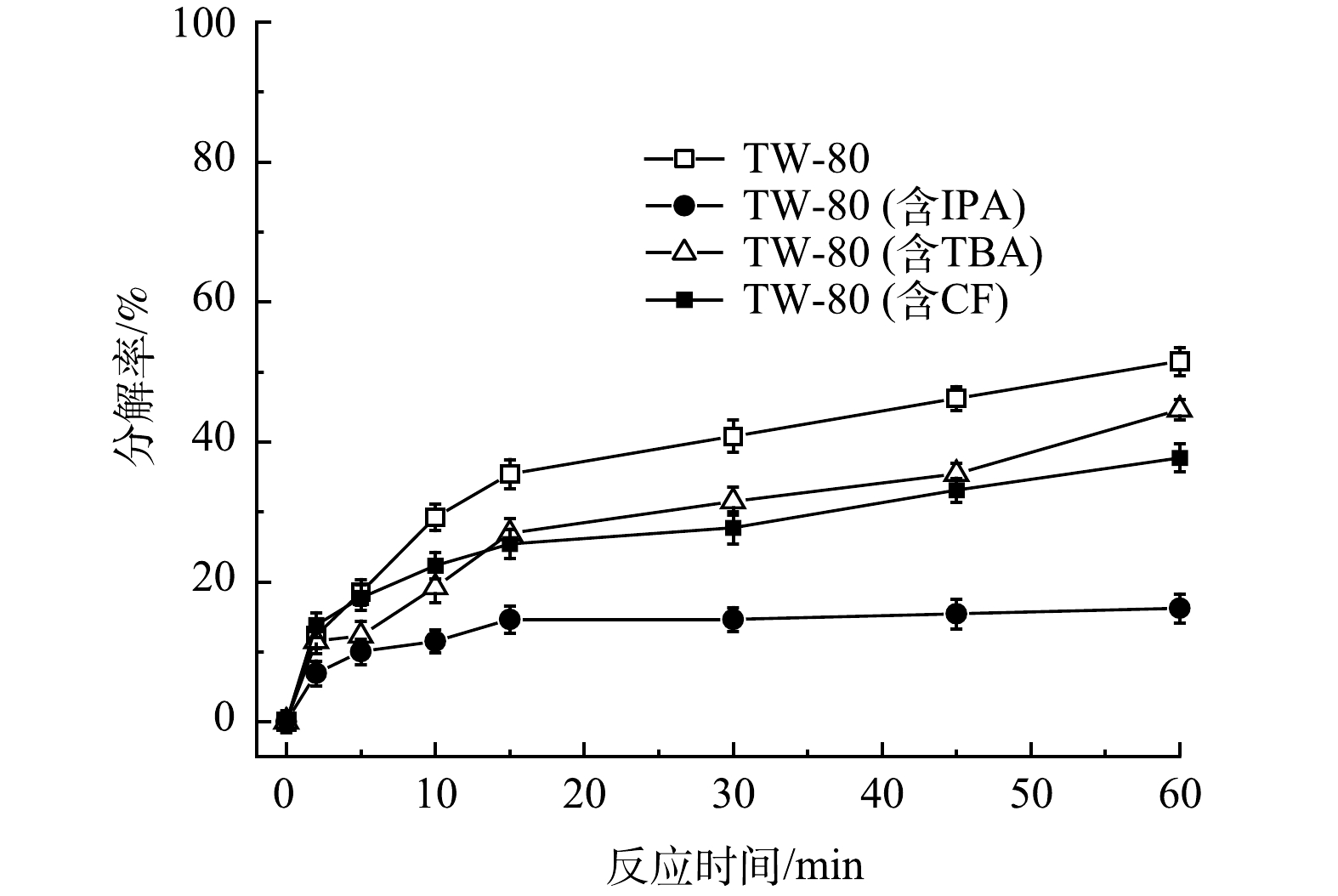
图 7 吐温-80在不同自由基淬灭条件下的分解
Figure 7. Decomposition of TW-80 under different radical scavenging conditions
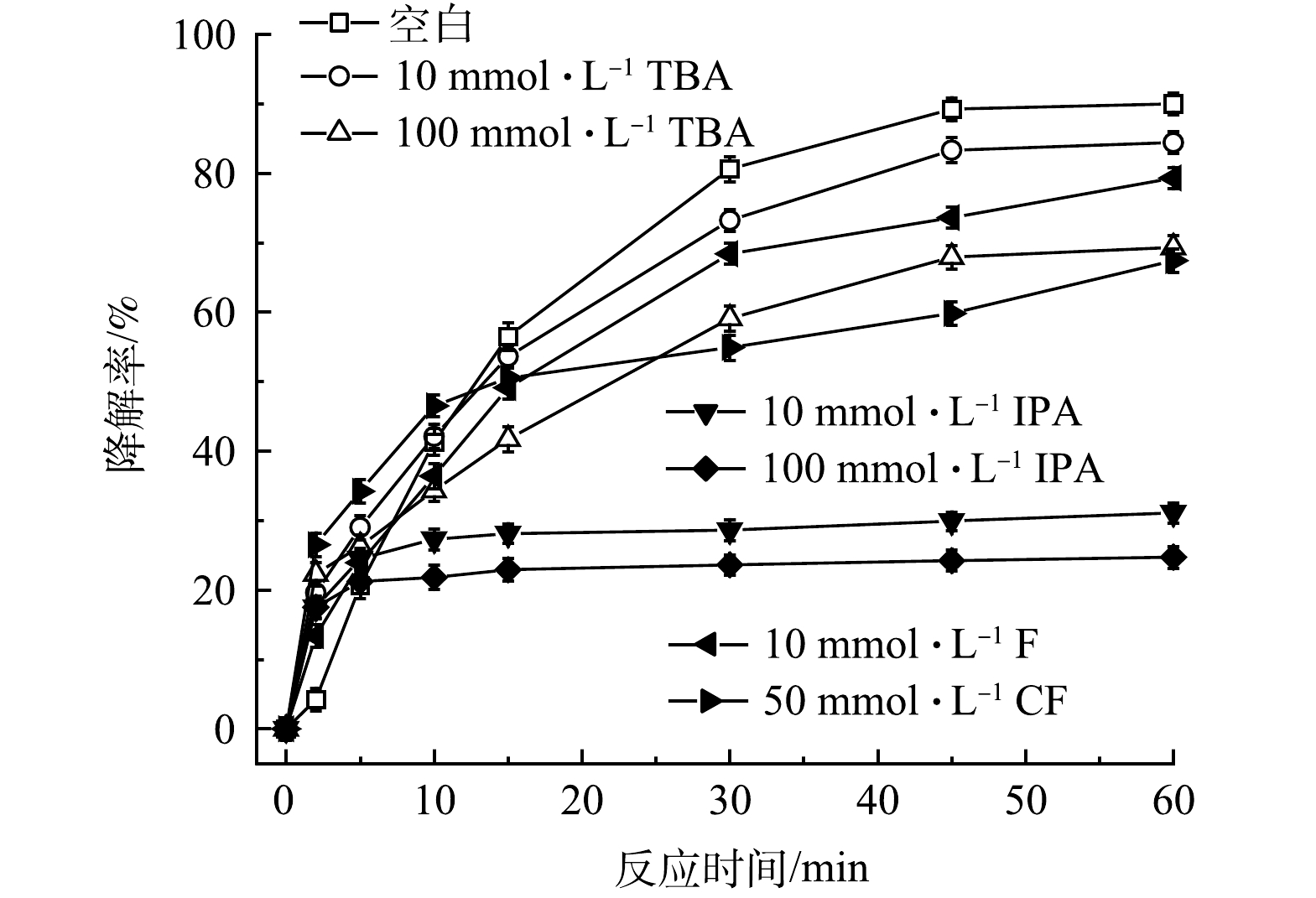
图 10 自由基淬灭剂对TCE降解效果的影响
Figure 10. Effect of radical scavengers on TCE degradation performance
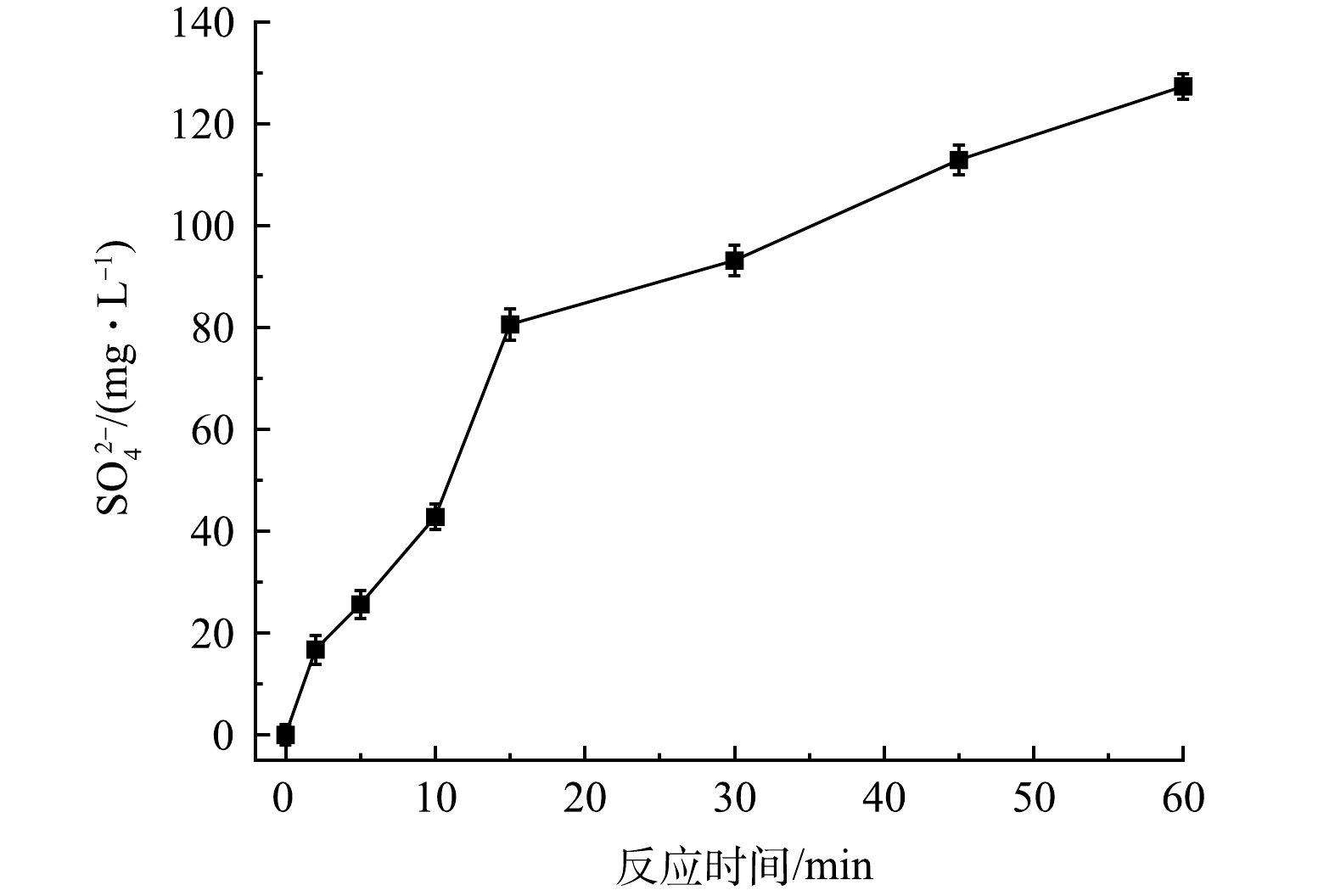
图 13 PMS/FeS体系中SO42–浓度变化
Figure 13. Variation of SO42– concentration in PMS/FeS system
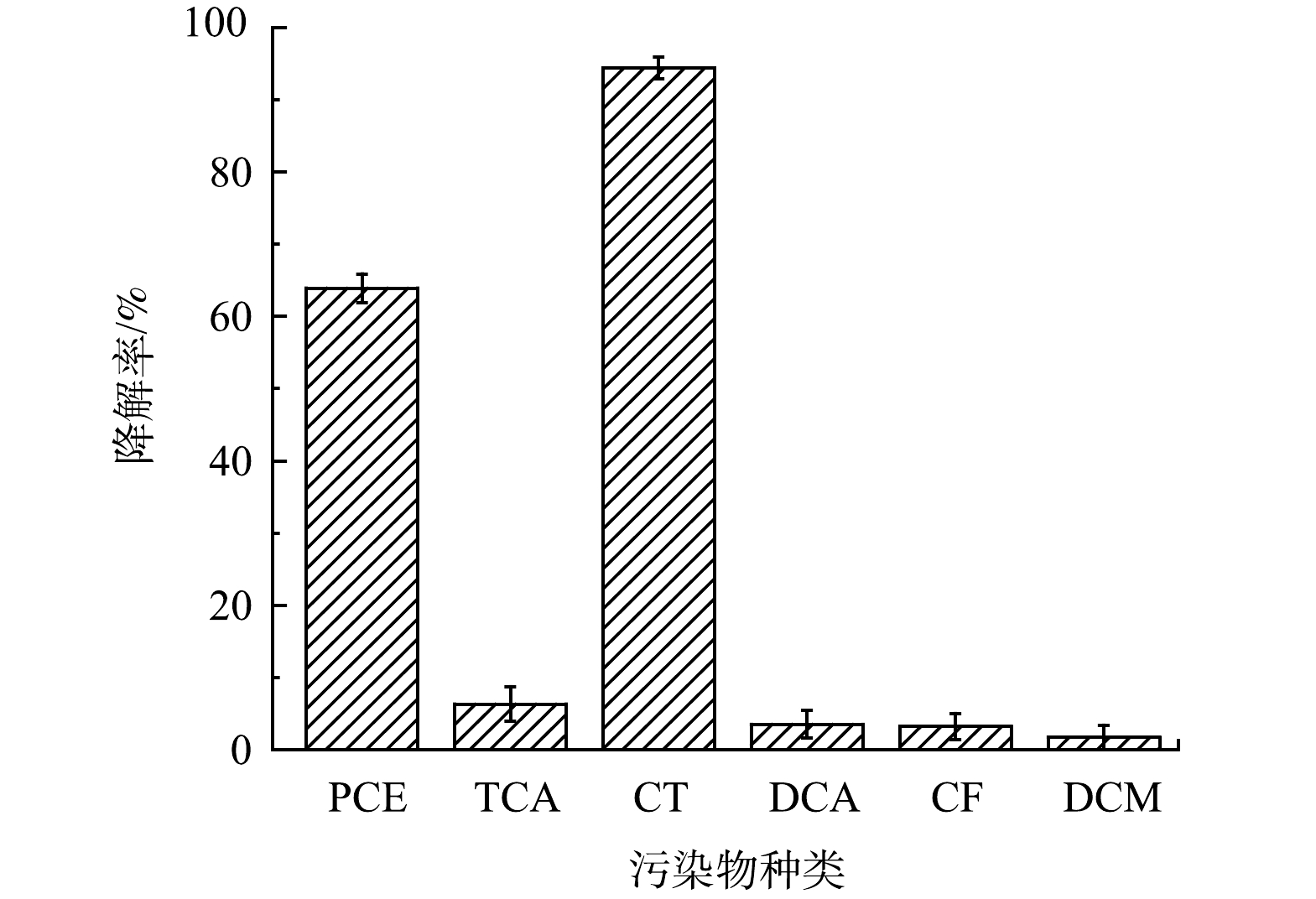
图 14 PMS/FeS体系中不同氯代烃的降解效果
Figure 14. The degradation of different chlorinated hydrocarbons in PMS/FeS system
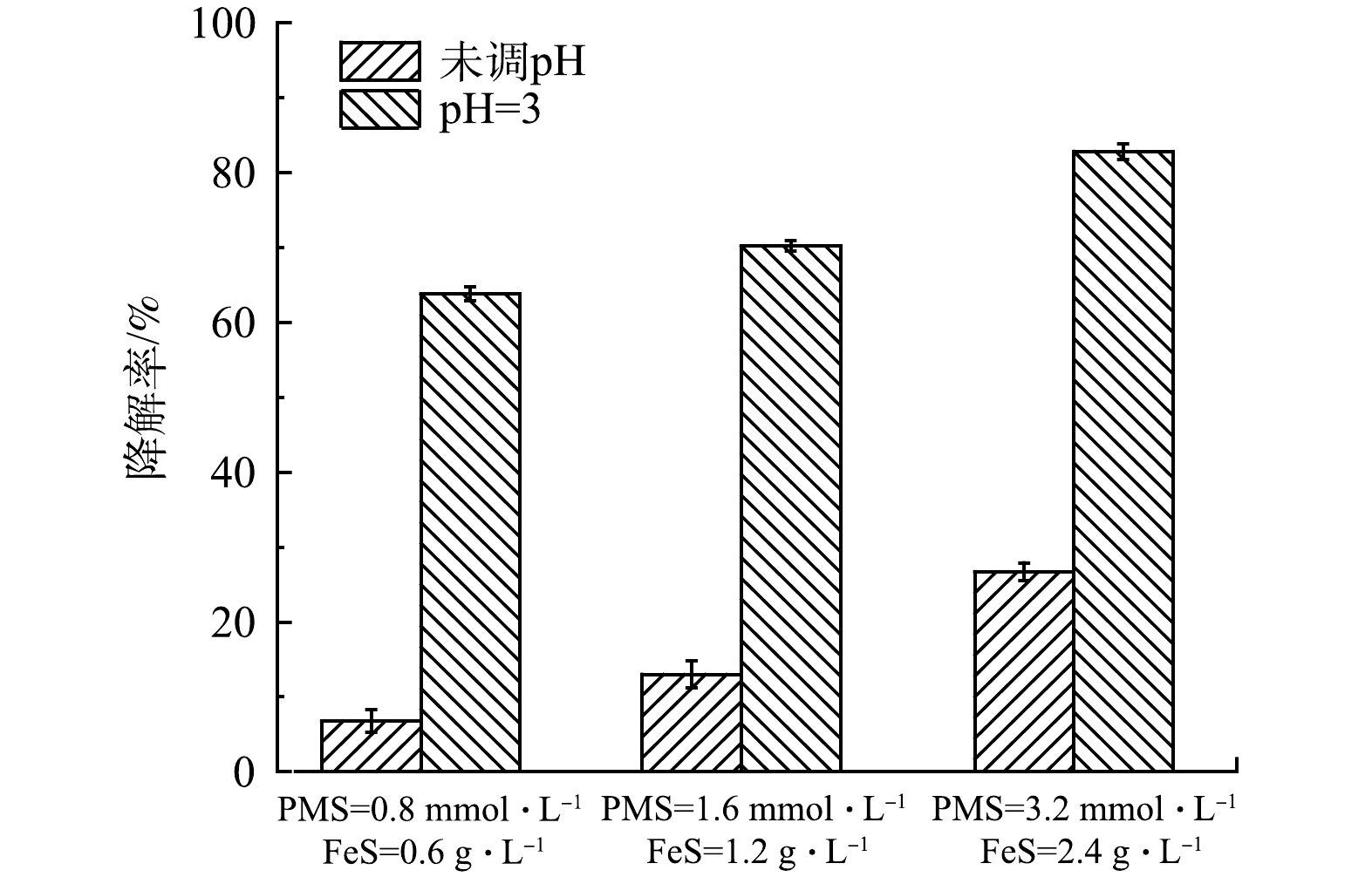
图 15 PMS/FeS体系对实际地下水中TCE的降解效果
Figure 15. TCE degradation using actual groundwater by PMS/FeS system
表 1 不同溶液初始pH对TCE降解率的影响
Table 1. Effect of different initial solution pH on TCE degradation
反应前pH 反应后pH TCE降解率/% 3.06 2.69 91.5 5.02 2.76 90.0 7.01 2.86 89.5 9.05 2.88 90.7 11.03 2.93 79.8  下载: 导出CSV
下载: 导出CSV
表 2 实际地下水试验中反应前后溶液pH变化
Table 2. Change of pH values before and after reactions in actual groundwater tests
PMS(mmol·L−1) FeS(g·L−1) 反应前pH 反应后pH 0.8 0.6 7.49(未调节) 6.45 1.6 1.2 7.48(未调节) 5.97 3.2 2.4 7.49(未调节) 3.22 0.8 0.6 3.04(调节后) 2.82 1.6 1.2 3.05(调节后) 2.73 3.2 2.4 3.02(调节后) 2.54
下载: 导出CSV
-
[1] CHOI K, LEE W. Reductive dichlorination of carbon tetrachloride in acidic soil manipulated with iron(II) and bisulfide ion[J]. Journal of Hazardous Materials, 2009, 172: 623-630. doi: 10.1016/j.jhazmat.2009.07.041 [2] ALONSO-DE-LINAJE V, MANGAYAYAM M C, TOBLER D J, et al. Sorption of chlorinated hydrocarbons from synthetic and natural groundwater by organo-hydrotalcites: Towards their applications as remediation nanoparticles[J]. Chemosphere, 2019, 236: 124369. doi: 10.1016/j.chemosphere.2019.124369 [3] WU X L, GU X G, LU S G, et al. Accelerated degradation of tetrachloroethylene by Fe(II) activated persulfate process with hydroxylamine for enhancing Fe(II) regeneration[J]. Journal of Chemical Technology and Biotechnology, 2016, 91: 1280-1289. doi: 10.1002/jctb.4718 [4] MAO X, JIANG R, XIAO W, et al. Use of surfactants for the remediation of contaminated soils: A review[J]. Journal of Hazardous Materials, 2015, 285: 419-435. doi: 10.1016/j.jhazmat.2014.12.009 [5] WEST C C, HARWELL J H. Surfactants and subsurface remediation[J]. Environmental Science and Technology, 1992, 26: 2324-2330. doi: 10.1021/es00036a002 [6] 李思明, 刘汉桥, 魏国侠. 表面活性剂应用于浮选中的研究进展[J]. 天津城建大学学报, 2022, 28(3): 192-197. [7] RASTOGI A, AL-ABED S R, DIONYSIOU D D. Sulfate radical-based ferrous-peroxymonosulfate oxidative system for PCBs degradation in aqueous and sediment systems[J]. Applied Catalysis B-Environmental, 2009, 85: 171-179. doi: 10.1016/j.apcatb.2008.07.010 [8] ANTONIOU M G, DELACRUZ A A, DIONYSIOU D D. Degradation of microcystin-LR using sulfate radicals generated through photolysis, thermolysis and e(-) transfer mechanisms[J]. Applied Catalysis B-Environmental, 2010, 96: 290-298. doi: 10.1016/j.apcatb.2010.02.013 [9] 王克全. 零价铁活化过硫酸盐降解水中萘普生的研究[J]. 四川环境, 2022, 41(1): 1-7. [10] STANISTAW W, HOLGER V L, KLAUDIUSZ G, et al. Chemistry of persulfates in water and wastewater treatment: A review[J]. Chemical Engineering Journal, 2017, 330: 44-62. doi: 10.1016/j.cej.2017.07.132 [11] WEI D W, OSSEO-ASARE K. Particulate pyrite formation by the Fe3+ HS- reaction in aqueous solutions: effects of solution composition[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 1996, 118: 51-61. [12] HARVEY A E, SMART J A, AMIS E S. Simultaneous spectrophotometric determination of iron(II) and total iron with 1, 10-phenanthroline[J]. Analytical Chemistry, 1955, 27(1): 26-29. doi: 10.1021/ac60097a009 [13] LIANG C J, HUANG C F, MOHANTY N, et al. A rapid spectrophotometric determination of persulfate anion in ISCO[J]. Chemosphere, 2008, 73(9): 1540-1543. doi: 10.1016/j.chemosphere.2008.08.043 [14] LIN J T, CORNELL D G, MICICH T J. Cobaltothiocyanate colorimetric analysis for homologous polyoxyethylated alkyl amides[J]. Journal of the American Oil Chemists' Society, 1986, 63(12): 1575-1579. doi: 10.1007/BF02553089 [15] OH S Y, KANG S G, KIM D W, et al. Degradation of 2, 4-dinitrotoluene by persulfate activated with iron sulfides[J]. Chemical Engineering Journal, 2011, 127: 641-646. [16] FOUNTAIN J C, STARR R C, MIDDLETON T, et al. A controlled field test of surfactant-enhanced aquifer remediation[J]. Groundwater, 1996, 34(5): 910-916. doi: 10.1111/j.1745-6584.1996.tb02085.x [17] TIAN X K, TIAN C, NIE Y L, et al. Controlled synthesis of dandelion-like NiCo2O4 microspheres and their catalytic performance for peroxymonosulfate activation in humic acid degradation[J]. Chemical Engineering Journal, 2018, 331: 144-151. doi: 10.1016/j.cej.2017.08.115 [18] BESHA A T, BEKELE D N, NAIDU R, et al. Recent advances in surfactant-enhanced In-Situ Chemical Oxidation for the remediation of non-aqueous phase liquid contaminated soils and aquifers[J]. Environmental Technology & Innovation, 2017, 9: 303-322. [19] MATURI K, REDDY K R, CAMESELLE C. Surfactant-enhanced electrokinetic remediation of mixed contamination in low permeability soil[J]. Separation Science and Technology, 2009, 44: 2385-2409. doi: 10.1080/01496390902983745 [20] JIANG W L, SPURGEON S R, ZHU Z H, et al. Chemical imaging and diffusion of hydrogen and lithium in lithium aluminate[J]. Journal of Nuclear Materials, 2018, 511: 1-10. doi: 10.1016/j.jnucmat.2018.08.057 [21] ZHOU Y, WANG X L, ZHU C Y, et al. New insight into the mechanism of peroxymonosulfate activation by sulfur-containing minerals: Role of sulfur conversion in sulfate radical generation[J]. Water Research, 2018, 142: 208-216. doi: 10.1016/j.watres.2018.06.002 [22] AO X, LIU W. Degradation of sulfamethoxazole by medium pressure UV and oxidants: Peroxymonosulfate, persulfate, and hydrogen peroxide[J]. Chemical Engineering Journal, 2017, 313: 629-637. doi: 10.1016/j.cej.2016.12.089 [23] WANG Z, AL L, HUANG Y, et al. Degradation of azo dye with activated peroxygens: When zero-valent iron meets chloride[J]. Rsc Advances, 2017, 7: 30941-30948. doi: 10.1039/C7RA03872K [24] WU X L, GU X G, LU S G, et al. Degradation of trichloroethylene in aqueous solution by persulfate activated with citric acid chelated ferrous ion-ScienceDirect[J]. Chemical Engineering Journal, 2014, 255: 585-592. doi: 10.1016/j.cej.2014.06.085 [25] LIANG C J, SU H W. Identification of sulfate and hydroxyl radicals in thermally activated persulfate[J]. Industrial and Engineering Chemistry Research, 2009, 48(11): 5558-5562. doi: 10.1021/ie9002848 [26] MADDEN K P, TANIGUCHI H. The role of DMPO-hydrated electron spin adduct in DMPO-OH spin trapping[J]. Free Radical Biology and Medicine, 2001, 30(12): 1374-1380. doi: 10.1016/S0891-5849(01)00540-8 [27] LIU L, LI Y N, LI W, et al. The efficient degradation of sulfisoxazole by singlet oxygen (1O2) derived from activated peroxymonosulfate (PMS) with Co3O4-SnO2/RSBC[J]. Environmental Research, 2020, 187: 109665. doi: 10.1016/j.envres.2020.109665 [28] 刘敏, 崔然, 褚秀玲. 地下水水质分析及污染治理[J]. 能源与节能, 2022, 5: 153-155. doi: 10.3969/j.issn.2095-0802.2022.10.042 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4556
- HTML全文浏览数: 4556
- PDF下载数: 184
- 施引文献: 0
