



-
甲醛作为主要室内空气污染物[1],其来源主要为装修过程中使用的含有粘合剂的人造板材、劣质油漆和地毯等的缓慢释放[2]。控制室内低浓度甲醛的途径有2种 [3]:一是源头控制,如使用环保材料以降低室内空气中甲醛浓度;二是末端治理,即去除空气中游离的甲醛气体。催化氧化法被认为是最为有效的吸附处理技术,包括常温催化氧化和光催化氧化技术,具有脱除效率高、无二次污染等优点,可将HCHO直接转化为CO2和H2O[4],其核心在于研制出能在低温潮湿环境下高效催化氧化甲醛的催化剂。
二氧化钛(TiO2)为最常用的半导体材料,具有较高的太阳光敏感性、化学稳定性和低毒性等优点[5],且TiO2的高能带态密度能实现高效的光电流转换,使得其比ZnO、SnO2、ZrO2、CdS和g-C3N4等半导体材料具有更高的氧化活性[6],可在紫外光激发下生成强氧化性的羟基自由基(·OH)和超氧阴离子自由基(·O2−)等[7-8]。然而,TiO2仅能吸收紫外光,无法吸收更多的可见光,且较低的电子转移率和较高的光致电子与空穴复合率亦会严重影响整体量子产率。通常采用2种解决策略:一是将TiO2辐射吸收范围扩展到可见光区域;二是抑制激发电子和正空穴的复合[9]。为此需要对TiO2光催化剂进行改性,如贵金属沉积[10],半导体耦合[11]等。TiO2与半导体Bi2O3耦合[12]亦可将吸收光谱扩展到可见光区域,显著增强光催化氧化活性 [13]。掺杂的Bi离子以化合物形式部分取代了一些钛原子形成的锐钛矿TiO2呈现出更大的可见光偏移,且Bi和Y共掺杂致使光生空穴和电子的复合速率降低,有利于促进催化剂氧化性能[14]。但由于Bi2O3-TiO2受光照条件限制,且Bi2O3和TiO2亦可作为光生电子和空穴的复合中心而降低其氧化性能。
锰基催化剂为一种高效、深度的常温催化氧化催化剂。隐钾锰矿型和水钠锰矿型氧化锰中存在多种价态的锰,易相互转化,使得氧化锰具有较高的催化氧化活性[15],但锰基催化剂亦会出现结晶性差、易团聚、易失活等现象,会直接影响催化剂稳定性和再生活性[16]。结构型、电子型CeO2助催化剂有较强的储氧能力[17],可在高空速条件下为氧化反应提供充足的氧,并借助Ce4+/Ce3+离子偶的Redox循环,有效改善活性位点间的电子传递。易变价的Ce又导致晶界处存在各种非化学计量缺陷,因在混合氧化物存在下易形成更多的晶格缺陷,增加活性位点。TANG等[18-19]通过改进的共沉淀方法制备了一系列MnOx-CeO2催化剂,在100 ℃反应温度下实现了100%的HCHO降解。这是通过氧转移机制有效激活分子氧,MnOx-CeO2固溶体的形成致使氧化锰具有更高的氧化状态,且表面更丰富的晶格氧物种对低温催化氧化甲醛发挥着至关重要的作用。ZHANG等[20]采用溶胶-凝胶柠檬酸法制备了一系列高效的MnOx-Co3O4-CeO2三效催化剂,当三者摩尔比为16∶19∶1,反应温度为100 ℃时,催化剂展示出对HCHO的最佳去除效率,但MnOx-CeO2存在稳定性差,一段时间后可能由于大量吸附空气中水蒸气致使HCHO去除效率大幅降低,且显示室温下氧化锰表现为HCHO的氧化物,而非催化剂[21]。
本研究以常温催化氧化性能的MnCeOx氧化物为载体,起到吸附和常温催化作用,以具有可见光催化氧化性能的Bi3+-TiO2为活性组分,采用不同制备方法研制Bi3+-TiO2与MnCeOx相耦合的可见光光热协同催化氧化催化剂,以实现光生电子和活性氧物种(O2−, O−, ·OH )的转移与氧化,同时利用MnOx-CeO2的黑色组分吸光促使Bi3+-TiO2光催化性能提升或升高温度进而提升其常温催化氧化性能,并探索制备方法、负载量、组分配比以及结构形貌等对催化剂性能的影响,研究考察Bi3+-TiO2/MnCeOx耦合情况下的催化氧化性能及可能机理,以期为低浓度HCHO治理提供参考。
-
1) MnCeOx活性粉末的制备。取55.0 mL硝酸锰溶液(质量分数为50%)和250 mL蒸馏水于烧杯中,依次投加25.5 g硝酸铈和16.0 g柠檬酸至上述溶液中,搅拌至完全溶解,再将上述溶液置于60 ℃水浴锅蒸煮直至形成溶胶状,自然陈化24 h,将所得样品置于100 ℃烘箱干燥直至形成粉末状,再经550 ℃,7 h煅烧制得MnCeOx活性粉末备用。
2) Bi3+-TiO2/MnCeOx催化剂的制备。研究采用浸渍法、水热法和物理混合等3种方法将Bi3+-TiO2(Bi2O3负载至MnCeOx上形成复合催化剂,最佳负载量为7.0%(质量分数)。浸渍法是先取7.1 mL钛酸四丁酯、8.5 mL乙醇和丙三醇混合溶液(体积比1:1)混合搅拌形成溶液A,另取8.5 mL乙醇和丙三醇混合溶液并加入适量乙酸、碳酸铵和硝酸铋搅拌制成溶液B;再将溶液B缓慢滴加至溶液A中形成混合溶液,并向其中投加2.0 g MnCeOx活性粉末形成悬浊液C,持续搅拌10 min后置于80 ℃烘箱中干燥直至形成粉末状;研磨成细粉状并经450 ℃,8.5 h焙烧制得10.0%(质量分数)Bi3+-TiO2/MnCeOx催化剂。不同Bi3+-TiO2负载量(质量分数1.0%、2.0%、5.0%、20.0%)的Bi3+-TiO2/MnCeOx催化剂亦可采用上述方法制得。在10.0%Bi3+-TiO2/MnCeOx基础上,探索了不同焙烧温度(350、550、和650 ℃)对催化剂性能的影响。同时,将乙醇和丙三醇混合溶液替换为纯乙醇,亦采用上述方法制备10.0%Bi3+-TiO2/MnCeOx(EtOH)催化剂(550 ℃)。水热法则是将上述所得悬浊液C置于具有聚四氟乙烯内胆的高压反应釜中110 ℃陈化48 h,再经多次洗涤(水和乙醇)、干燥和550 ℃焙烧所得。混合法是将分别制得的Bi3+-TiO2光催化剂粉末和MnCeOx活性粉末按照适当比例通过简单物理混合而成。与此同时,将浸渍法所制的10.0%Bi3+-TiO2/MnCeOx(550 ℃)进行NaOH改性处理。具体方法如下:取适量Bi3+-TiO2/MnCeOx粉末与0.1 mol·L−1 NaOH水溶液混合均匀,置于反应釜110 ℃陈化,用蒸馏水洗涤至中性,在HCl溶液中浸泡2 h,经蒸馏水洗涤、干燥制得Bi3+-TiO2/MnCeOx(NaOH)催化剂。
-
本实验为模拟室内环境条件下的光催化耦合常温催化HCHO性能评价,在尺寸为60 cm×60 cm×60 cm 密闭、含盖的玻璃反应器中进行,以反应器底部正上方10 cm处悬挂的36W LED灯为可见光光源,并通过反应器内HCHO的质量浓度变化来确定催化剂活性。首先将质量分数为38%的甲醛溶液滴加至培养皿中,并直接置入玻璃反应器内待其充分挥发,并将装填量为1.0 g催化剂均匀涂抹于Φ=5 cm培养皿中,并置于反应器内LED灯正下方,迅速取出装有甲醛溶液的培养皿并采用凡士林密封玻璃盖,连续测量反应器内HCHO质量浓度,始终保持其初始质量浓度为 (1.05±0.05) mg·m−3。HCHO质量浓度直接由甲醛分析仪(PPM-400st)测量得出,每次连续测定3次并取平均值。甲醛转化率按如式 (1) 计算。
式中:φ表示为催化氧化HCHO转化率;Ct表示每反应12 h后反应器内的气态甲醛质量浓度,mg·m−3;C0表示反应器内起始甲醛质量浓度,mg·m−3。
-
为研究考察催化剂微观结构在催化氧化反应过程的作用,用AXSD8衍射仪对催化剂进行了XRD检测,选用Cu靶射线管,扫描速率为4°·min−1,2θ衍射角范围为10°~80°;催化剂的比表面积、孔径大小及其分布情况采用以N2吸附的Autosorb-iQ-AG-MP表面积分析仪检测。表面形貌采用日本Hitachi公司生产的SU1510扫描电镜检测分析。材料纳米尺度的结构、晶格面以及晶格间距由日本JEOL公司生产的200 kV场发射透射电子显微镜(JEM-2100F)检测分析。样品傅里叶变换红外(FTIR)光谱分析是在美国Thermo Scientific Nicolet 6700 FTIR光谱仪上进行,催化剂粉末与KBr混合压片后置于DRIFT池中,分辨率为4 cm−1,扫描范围为4 000~400 cm−1,每个光谱均需扣除背景值。样品紫外-可见吸收光谱由日本岛津仪器公司生产的UV-3600 Plus紫外-可见-近红外分光光度计检测分析。
-
1) 结构分析。为考察不同负载量的Bi3+-TiO2对催化剂比表面积及孔容孔径产生的影响,对部分样品进行了BET分析,如表1和图1所示。Bi3+-TiO2/MnCeOx催化剂比表面积随TiO2负载量的增加先降低后增加,而孔体积和孔径持续降低,其中10.0%Bi3+-TiO2/MnCeOx催化剂展示出最低比表面积(54.0 m2·g−1),却显示出最佳的催化氧化活性。这表明催化剂比表面积与催化剂氧化活性并非正相关。负载有1.0和10.0%TiO2的催化剂显示出II型气体等温吸附线,且相对压力P/P0在0.8~1.0具有H3型滞后环[22],高压区吸附量上升较快。这说明孔径尺寸较大、分布相对较宽且以大孔居多[23],这有利于催化剂对气态甲醛吸附及反应产物的排出,而当TiO2负载量达到20.0%,催化剂显示出Ⅴ型气体吸附等温线,亦伴有滞后环,以小孔径为主(平均孔径3.88 nm)。这表明MnCeOx活性氧化物孔道被TiO2大量占据,致使孔容孔径降低,这不利于气态甲醛在孔道内扩散,且孔道内的Bi3+-TiO2因无法接收光源致使催化剂无法充分发挥其光催化性能,故TiO2负载量不是越多越好。
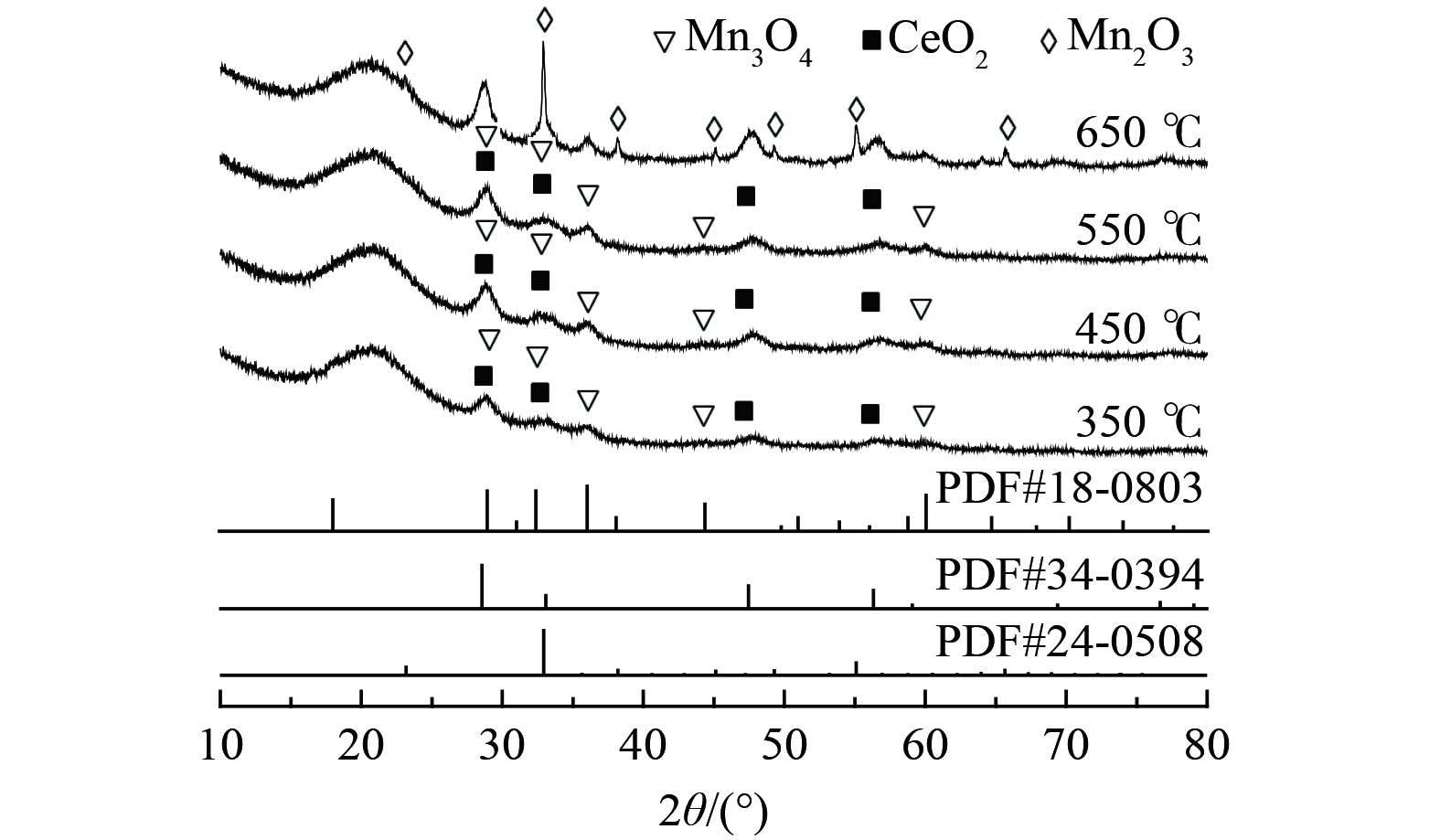
对经不同煅烧温度处理的Bi3+-TiO2/MnCeOx催化剂晶型结构进行了XRD分析,如图2所示。随着煅烧温度的升高,样品中氧化物晶体结晶度不断增强,其晶体粒径逐渐增大。2θ为28.9、35.9、44.3和59.9°归属为Mn3O4特征衍射峰(PDF #18-0803),位于28.8、47.8和56.6°处特征衍射峰为CeO2晶体(PDF #34-0394) [24],其所展示的氧化锰和氧化铈晶体衍射峰强度较低,衍射峰较宽,Mn3O4和CeO2衍射角分别向小角度(0.1°)和大角度(0.3°)偏移。考虑到Mn4+的离子半径(0.25 nm)小于Ce4+的半径(0.31 nm),锰离子易被引入到氧化铈晶格中形成固溶体。这表明氧化铈抑制了Mn3O4晶体粒径的增大,且氧化锰易进入CeO2晶格中形成缺陷[19]。多价态的Mn3O4(+2和+3)提高了氧的流动性,两者间的协同作用有利于氧空位数量的增加,进而提升催化剂常温催化氧化活性。在以上结果中均未发现TiO2特征衍射峰,可能仍为无定形态或粒径较小、高度分散于MnCeOx表面而低于XRD检出限。随着煅烧温度提升至650 ℃时, 2θ为23.1、32.9、38.2、45.1、49.3、55.1和65.7°归属为Mn2O3的特征衍射峰(PDF #24-0508);晶面分别为(211)、(222)、(400)、(323)、(413)、(044)和(622);衍射峰逐渐由宽峰演变为尖峰,Mn2O3晶体粒径逐渐增大。这表明高温致使氧化物晶型及晶体粒径发生显著变化。
利用SEM和TEM对催化剂的表观结构形貌进行了分析,如图3所示。MnCeOx粉末以无规则形态存在,表面结构蓬松,富含大量孔隙,属介孔材料,但其内部富有大量类似球状颗粒物,为MnCeOx的氧化物,其晶格间距0.27和0.31 nm分别归属为Mn2O3 (2 2 2)和CeO2 (1 1 1)。而Bi3+-TiO2/MnCeOx底部为密实结构的MnCeOx氧化物,部分展示出长条状结构,其表面富含类似球状TiO2。MnCeOx和球状TiO2通过长条状结构或直接担载紧密结合,而催化剂表面未被TiO2完全覆盖,其边缘富含MnCeOx氧化物,晶格间距0.25、0.27和0.31 nm分别归属为Mn3O4 (2 1 1)、Mn2O3 (2 2 2)和CeO2 (1 1 1),且晶格条纹中均存在大量凹陷状,即晶格缺陷。与此同时,实验采用纯乙醇代替乙醇和丙三醇混合溶液制备了Bi3+-TiO2/MnCeOx(EtOH)催化剂,其SEM如图3(e)所示。催化剂底部为具有密实结构的MnCeOx氧化物,其上负载有短棒状的TiO2,且相互搭接分布于MnCeOx氧化物上。同时,对Bi3+-TiO2/MnCeOx使用NaOH改性处理,结果表明(图3(f)),其表观结构遭受不同程度的侵蚀,且表面呈现出大量纳米片状结构。
为考察Mn、Ce、O、Ti和Bi元素在催化剂中分布情况,对Bi3+-TiO2/MnCeOx催化剂进行了TEM-EDS分析,如图4所示。Mn、Ce、O、Ti和Bi5种元素分布与催化剂形态完全一致,其元素含量依次为O、Mn、Ce、Ti和Bi。这与所制催化剂实际结果相一致,氧化锰与氧化铈均匀紧密结合,形成混、复合氧化物,彼此间发挥协同常温催化作用[25]。与此同时,掺Bi的TiO2亦均匀分布与MnCeOx上,且Bi原子部分取代Ti原子[26]。两者紧密结合,发挥协同光催化性能。由于负载量低,Bi3+-TiO2未将MnCeOx表面全部覆盖,这有利于Bi3+-TiO2/MnCeOx催化剂发挥光催化耦合常温催化氧化HCHO性能。
2) UV-vis DRS图谱。图5为450 ℃下制备的Bi3+-TiO2/MnCeOx催化剂的UV-vis DRS图。相较于P25,Bi3+-TiO2催化剂显示出明显的可见光吸收,吸收光发生红移。通过计算其带隙能分别为3.1 eV和2.8 eV,因此,Bi3+-TiO2光催化氧化HCHO性能明显优于P25,这与实际测试结果相一致。而Bi3+-TiO2/MnCeOx催化剂显示出在200~800 nm波长范围内全波段吸收,且吸收度随着Bi3+-TiO2负载量的增加先增加后降低。其中,10.0%Bi3+-TiO2/MnCeOx催化剂最佳,优于Bi3+-TiO2及MnCeOx催化剂,而展示出最佳的氧化活性。这与实际结果相一致。10.0% Bi3+-TiO2/MnCeOx复合催化剂在紫外可见光区域全波段吸收可能与载体MnCeOx氧化物为黑色组成有关,可充分吸收紫外和可见光。尤其在可见光区域吸收明显增强,产生热能,亦可致使氧化锰价态的变化[27],进而有利于催化氧化活性的提升。因此,以MnCeOx为载体,Bi3+-TiO2为活性组分可进一步优化催化剂催化氧化甲醛性能。
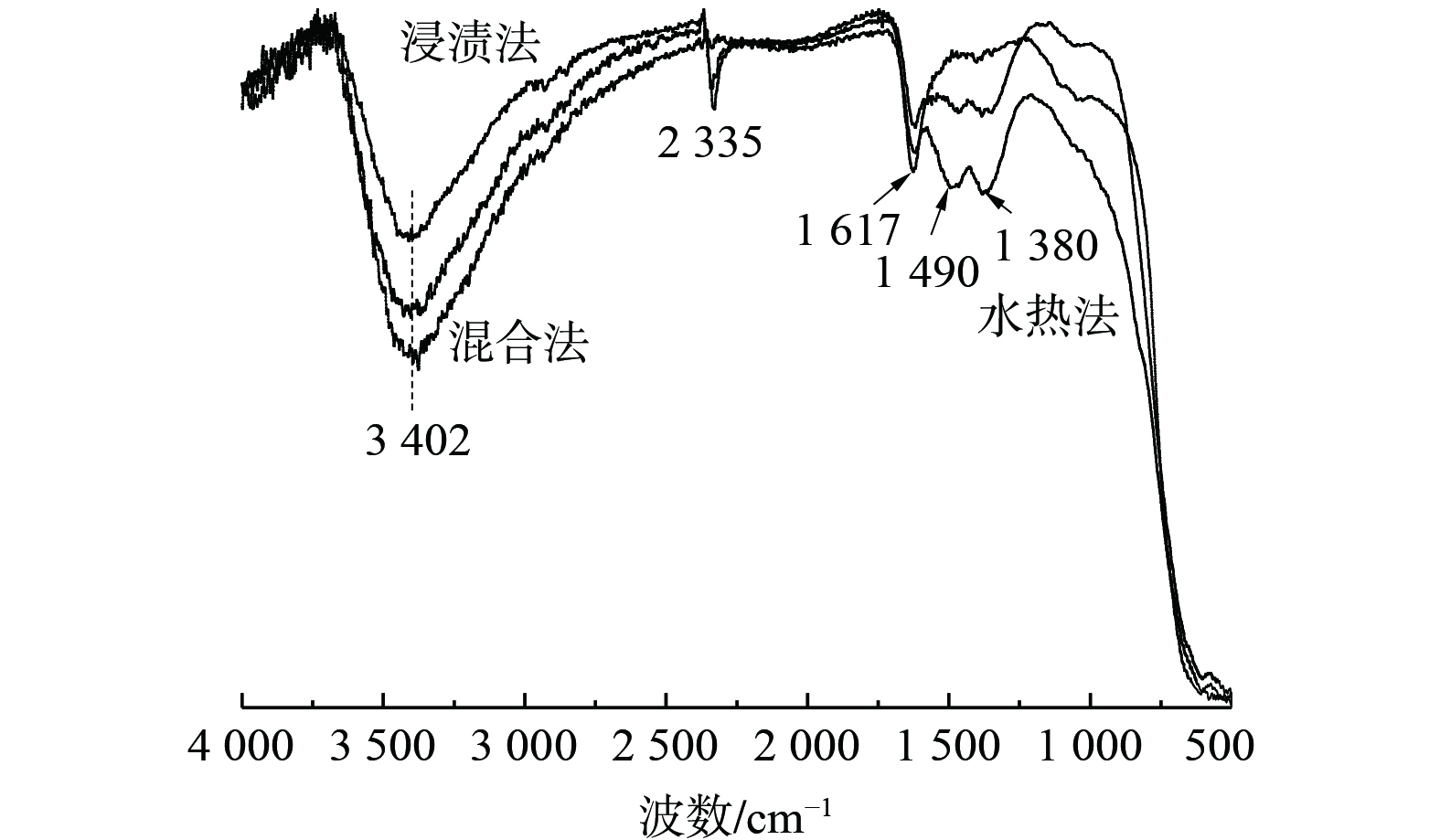
3) IR光谱。图6为3种采用不同制备方法所制的Bi3+-TiO2/MnCeOx催化剂的傅里叶红外光谱图。波数为3 402 cm−1宽峰和1 617 cm−1处特征峰均归属为催化剂上羟基自由基或含水物种的对称伸缩振动νs(—OH)峰,这些羟基通过氢键参与对气态HCHO吸附[28]。在2 335 cm−1处的特征峰可归属为C—O(νs (HCO3−))的拉伸或弯曲振动,而在1 380 cm−1处特征峰被归属为甲酸盐的对称伸缩振动νs(COO−)[29],1 490 cm−1处特征峰归属为参与催化氧化反应生成的二氧甲基(DOM)的δ(CH−)振动峰[30]。结果表明,3种方法所制催化剂吸水性能依次为混合法>水热法>浸渍法,其与催化氧化活性成反比。这表明过多水蒸气和HCHO之间存在明显的竞争吸附关系[31],而适量羟基通过氢键参与对气态HCHO吸附有利于催化剂氧化性能的提升。通过采用水热法和浸渍法所制催化剂的IR光谱图发现:有DOM和甲酸的存在,且均为催化氧化HCHO中间产物[32],还有碳酸氢的存在。这表明催化氧化HCHO反应最终形成CO2和H2O,而采用混合法所制催化剂未发现上述情况。
-
为研究探索Bi3+-TiO2/MnCeOx催化剂光催化耦合常温催化氧化HCHO性能,在同一测试条件下(36W LED灯,温度:20±4 ℃) 进行了一组对比实验,结果如图7所示。空白对照实验(图7(a))显示玻璃反应器内甲醛浓度略有下降,反应48 h后由1.052 mg·m−3降至0.923 mg·m−3。这是由于昼夜温差及吸附导致,但未见显著性下降趋势,亦说明玻璃反应器密封性良好,完全满足实验测试要求。为对比分析,对P25,Bi3+-TiO2和MnCeOx三者还进行了活性测试。结果表明:三者均展示出良好的吸附及催化氧化性能,反应48 h后甲醛降解率分别为78.9%、88.0%和85.5%。其中,Bi3+-TiO2优于P25和MnCeOx,其性能提升可能与Bi3+-TiO2展示出一定的可见光吸收特性有关。同时,对10.0% Bi3+-TiO2/MnCeOx催化剂进行避光性能测试。结果表明,在无光的条件下催化剂性能较差。这可能与Bi3+-TiO2的光催化性能受光照条件限制有关。催化剂在48 h内吸附及催化降解率均展示出逐渐降低的趋势,这是由于吸附气态低浓度甲醛是催化反应限速步骤,且空气中水蒸气与甲醛间存在竞争吸附所致[33]。为进一步提升催化剂的性能,将光催化剂Bi3+-TiO2和具有常温催化氧化性能的MnCeOx相结合(图7(b)),制备了不同Bi3+-TiO2负载量的Bi3+-TiO2/MnCeOx催化剂。结果显示,450 ℃焙烧所得Bi3+-TiO2/MnCeOx催化氧化HCHO性能较Bi3+-TiO2略有提升,其催化氧化性能随着Bi3+-TiO2负载量的增加先提升后降低。一般来言,活性组分越多所提供的活性位越多,其比表面积越大,越有利于反应的进行,但当活性组分负载量超过分散阈值后,多余晶相会覆盖MnCeOx活性位,并堵塞起表面孔道[34],这不利于气态甲醛及反应产物在孔道内扩散,会阻碍氧化反应的进行致使催化剂氧化活性降低。这与BET及孔径分布分析结果相一致。其中,10% Bi3+-TiO2/MnCeOx的性能最佳,其反应48 h后甲醛质量浓度为0.122 mg·m−3,但仍高于《民用建筑工程室内环境污染控制规范》(GB50325-2001)所规定的甲醛限值(0.08 mg·m−3),性能仍需优化。
煅烧温度能影响金属氧化物晶型、粒径及孔道结构,进而影响催化剂氧化性能[35]。为进一步优化催化剂性能,考察了煅烧温度对催化剂氧化性能的影响,如图8所示。随着煅烧温度的增加,催化氧化HCHO降解率呈现出先增加后降低的趋势。经550 ℃和650 ℃煅烧所制催化剂明显优于350 ℃和450 ℃。其中,经550 ℃煅烧所制10.0%Bi3+-TiO2/MnCeOx催化剂HCHO降解率(48 h)最佳,HCHO去除率达93.4%,其质量浓度由1.066 mg·m−3 降至0.070 mg·m−3,性能较Bi3+-TiO2和MnCeOx有显著提升,低于室内HCHO浓度控制限值。随着煅烧温度的提升,二氧化钛逐渐由无定形态转变为锐钛矿型,氧化锰由Mn3O4逐渐转变为Mn2O3,且晶体粒径逐渐增大。其晶型转变、粒径增大且大量的晶格缺陷是催化剂氧化性能提升的重要原因,MnCeOx氧空位及流动性和掺Bi3+的锐钛矿TiO2利于催化剂常温催化及光催化氧化作用的发挥,故后续实验均以经550 ℃焙烧所制催化剂为研究对象。
还考察了制备方法(水热法,混合法和浸渍法)以及改性处理(NaOH和EtOH)对Bi3+-TiO2/MnCeOx催化剂氧化性能的影响,结果如图9所示。经浸渍法所制催化剂(550 ℃)表现较佳,其氧化效率较混合法和水热法分别提升了11%和38%,实现了Bi3+-TiO2与MnCeOx间的协同耦合作用。促使催化剂性能提升的原因可能与Bi3+-TiO2/MnCeOx催化剂展示出良好的抗水蒸气性能有关。这一结果与红外光谱分析结果相吻合。与此同时,由于纳米结构的TiO2可通过影响孔结构的排列及比表面积对复合材料的光催化性能产生显著影响[36-37]。采用浸渍法制备的以乙醇替代乙醇和丙三醇混合溶液的Bi3+-TiO2/MnCeOx(EtOH)也显示出较佳的氧化性能(92.5%),其TiO2表观结构由球状转变为短棒状,且相互搭接分布于MnCeOx氧化物上,其结构未发生明显变化。而采用浸渍法所制催化剂经NaOH改性处理后的Bi3+-TiO2/MnCeOx(NaOH)样品的48 h HCHO去除率为84.1%,其氧化性能显著降低。这与催化剂表观结构遭受不同程度的侵蚀有关,其表面存有大量纳米片状结构,可能为Na2O晶体,其结构可由SEM表征结果证实。
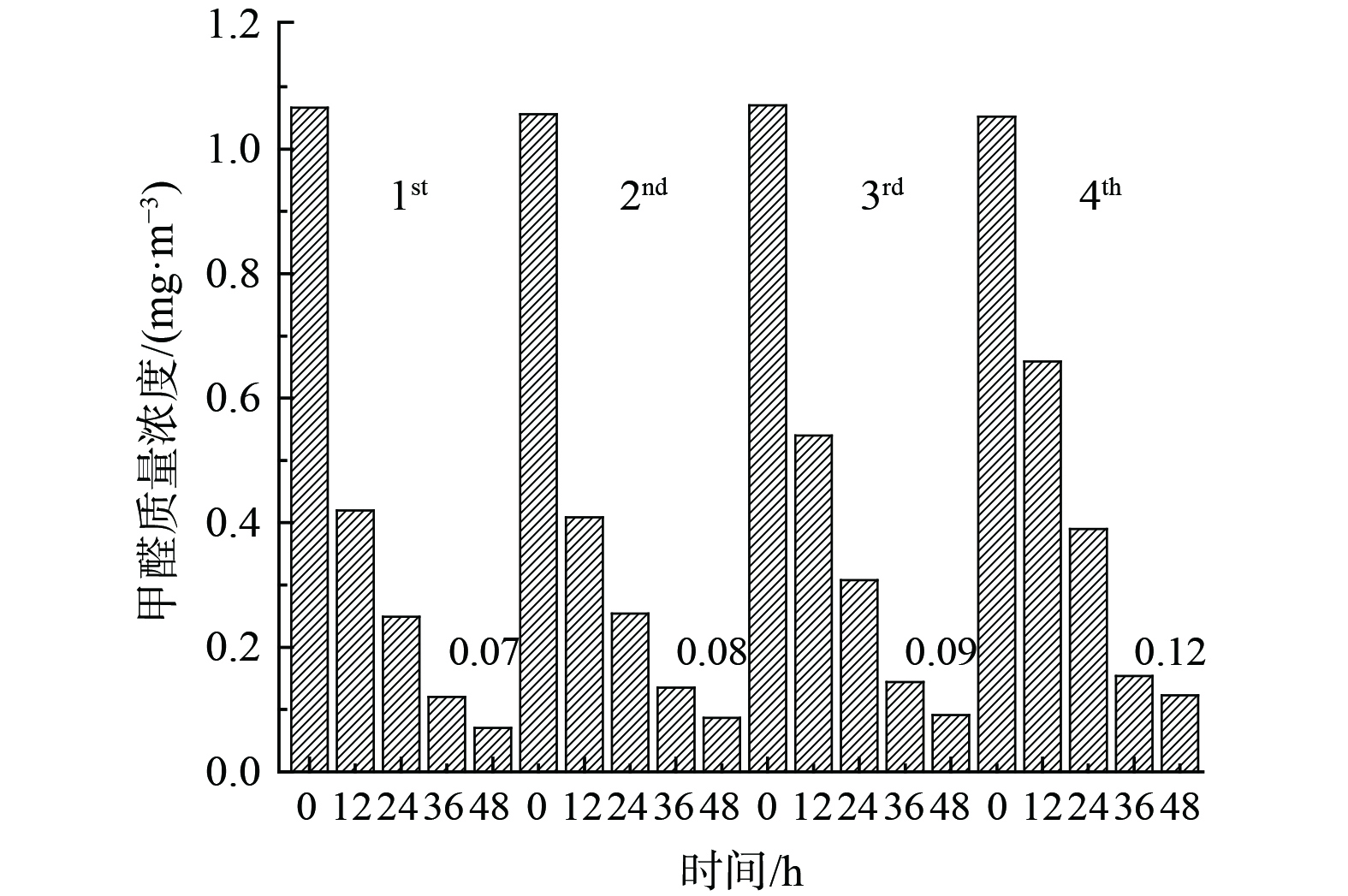
催化剂可吸附空气中大量水蒸气而导致催化剂氧化性能(竞争吸附)及稳定性能下降。为考察Bi3+-TiO2/MnCeOx催化剂的稳定性,对经550 ℃煅烧处理所制的10.0%Bi3+-TiO2/MnCeOx催化剂进行4次平行测定 (图10) 发现,催化剂稳定性良好,未见显著性氧化活性下降趋势,仅表现为小幅度降低,其48 h内HCHO质量浓度由0.07 mg·m−3升至0.12 mg·m−3。分析其主要原因可能与催化剂吸收空气中水分有关,HCHO和水蒸气间存在竞争性吸附致使催化剂氧化性能降低。这表明Bi3+-TiO2/MnCeOx催化剂显示出良好的抗水蒸气性能。
-
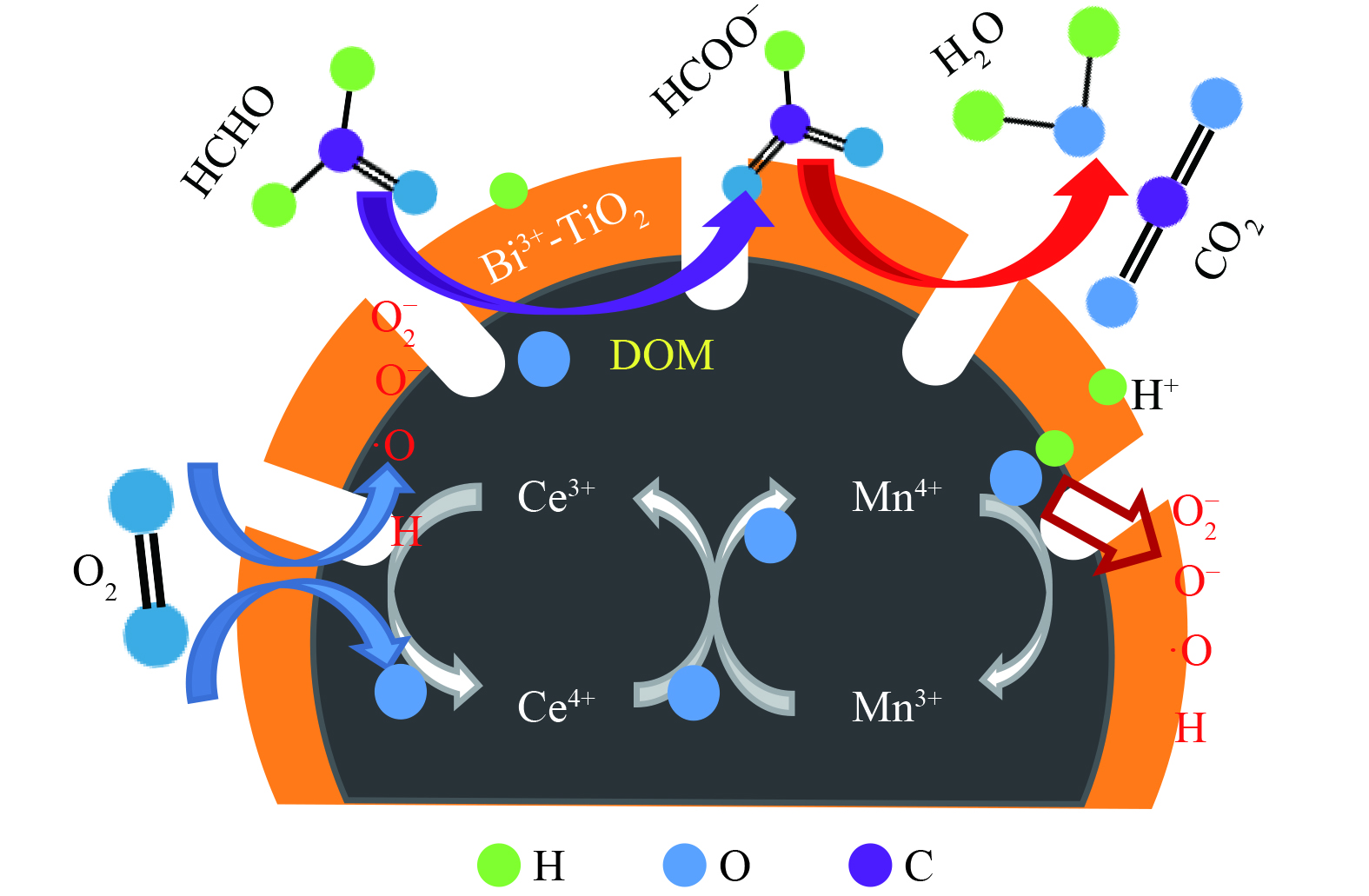
基于上述研究结果,推测Bi3+-TiO2/MnCeOx去除HCHO的催化氧化机理如图11所示。Bi3+-TiO2/MnCeOx表面羟基通过氢键参与对气态HCHO吸附,形成吸附态。在Bi3+-TiO2(光生电子e−和空穴h+)的光催化氧化和MnCeOx (Oads和Olatt)的常温催化氧化的共同作用下,O2在催化剂表面解离,立即形成活性氧物种或表面羟基等(O2−, O− , ·OH)。吸附的HCHO与活性氧或表面羟基反应形成DOM和甲酸盐,进而在Bi3+-TiO2/MnCeOx晶格氧的作用下进一步氧化为CO2和H2O[38]。
-
1) 将具备常温催化氧化性能的MnCeOx与光催化性能的Bi3+-TiO2相结合,采用浸渍法制备了一系列Bi3+-TiO2/MnCeOx催化剂。采用浸渍法所制的10.0%Bi3+-TiO2/MnCeOx(550 ℃)性能最佳,其48 h可将气态HCHO降低至0.07 mg·m−3,低于室内HCHO控制标准,且稳定性良好。
2) 基于具备常温催化氧化性能的MnCeOx氧化物为载体,其氧化锰和氧化铈间的协同作用及晶格缺陷有利于氧空位和氧的流动性的增加,进而提升其常温氧化性能。同时,以具备可见光催化氧化能力的Bi3+-TiO2为活性组分,其表观结构形态、晶型、粒径、孔道、掺杂、可见光吸收以及抗水性为Bi3+-TiO2/MnCeOx提升其氧化性能提供必要条件。两者间的多重协同耦合作用是核心,研究可为光催化耦合常温催化氧化新材料的制备提供参考。
光催化耦合常温催化氧化HCHO的Bi3+-TiO2/MnCeOx催化剂性能
Catalytic oxidation of HCHO over Bi3+-TiO2/MnCeOx catalyst based on photocatalytic coupled catalytic oxidation at ambient temperature
-
摘要: 常用净化室内挥发性有机物(VOCs)的方法主要有吸附、低温等离子体、光催化氧化、常温催化氧化等,而鲜有基于光催化耦合常温催化催化剂的报道。以具备常温催化氧化性能的MnCeOx为载体,以具有可见光催化性能的Bi3+-TiO2为活性组分,考察负载量、煅烧温度、制备方法以及结构形态等对Bi3+-TiO2/MnCeOx催化氧化甲醛(HCHO)性能的影响,并利用XRD、BET、SEM、TEM、UV-vis DRS和IR等技术对催化剂进行微观表征与分析。结果表明,催化剂表观结构形态、晶型、粒径、孔道、掺杂、可见光吸收及抗水性为Bi3+-TiO2/MnCeOx氧化性能提升提供了必要条件,两者间的多重协同耦合作用是核心,其中采用浸渍法所制的负载量10.0%Bi3+-TiO2/MnCeOx(550 ℃)表现最佳,48 h催化降解率高达93.4%,其HCHO浓度低于室内控制标准(GB50325-2001),且稳定性良好。本研究结果可为室内HCHO高效控制耦合光催化和常温催化技术提供参考。Abstract: The common methods for removing indoor volatile organic compounds (VOCs) mainly included adsorption, low-temperature plasma, photocatalytic oxidation, catalytic oxidation at ambient temperature, etc. However, there are few research reports based on the combination of photocatalytic oxidation and catalytic oxidation at ambient temperature. In this paper, MnCeOx were used as supports to catalytic oxidation of formaldehyde at ambient temperature, and Bi3+-TiO2 as the active component were utilized to photocatalytic performance under visible light. The effects of supporting, calcination temperature, preparation method and structure on the performance of formaldehyde (HCHO) oxidation were investigated, and their physicochemical properties were characterized by XRD, BET, SEM, TEM, UV-vis DRS and IR. The results exihibited that the apparent structural morphology, crystal type, particle size, pore, doping, absorption under visible light and water resistance provided the necessary conditions, and the potentially synergistic multiple effect of Bi3+-TiO2 and MnCeOx was the critical factor for improving the performance of Bi3+-TiO2/MnCeOx. Among them, 10.0% Bi3+-TiO2/MnCeOx (550 ℃) prepared by the impregnation method exhibited the highest activity and stability, and the degradation rate was as high as 93.4% at 48 h. Finally, the concentration of HCHO was lower than the indoor control standard (GB50325-2001). The synergistic effects are responsible for the enhanced indoor HCHO removing and it provides some reference for the research.
-
新污染物(emerging contaminants, ECs)是指新近发现或被关注,对生态环境或人体健康存在风险,尚未纳入管理或者现有管理措施不足以有效防控其风险的污染物[1]. 主要包括但不限于药物及个人护理品、抗生素抗性基因、内分泌干扰物、消毒副产物、纳米材料以及包括多氯联苯、有机氯农药、多环芳烃和全氟化合物在内的持久性有机污染物. 这类化学物质稳定性高、亲水性强,它们可以在地下水、饮用水、地表水,甚至是污水处理厂的废水中检测到,其中大部分物质最终会进入并滞留在沿海水域,影响水产品的质量和安全,因其对水质环境的长期不良影响而受到科学界和公众的广泛关注[2 − 3]. 因此,迫切需要研究ECs在水生环境中的迁移转化过程机制. 为了调查和探究ECs在沿海水域的迁移转化并评估其生态环境风险,有必要提高对其降解过程的认识.
研究表明光化学降解是水体中有机污染物的主要转化途径[4 − 6],ECs的光化学降解包括直接、间接和自敏化光解3种方式[7]. 在直接光降解过程中,目标化合物对光子的吸收导致键断裂或重排以形成新的稳定产物;在自敏化光降解过程中,ECs吸收光子跃迁至激发态,同时将能量转移给基态3O2或H2O,产生活性氧自由基,进而引发自身降解;在间接光降解中,具有光活性的化合物吸收阳光,产生活性反应中间体(reactive intermediates,RIs),这些物质在ECs的光降解过程中起着重要作用[8 − 9]. 溶解有机物(dissolved organic matter, DOM)是一种具有优异光化学活性的天然光敏剂,在ECs的间接光解过程中发挥着重要作用:一方面DOM可以通过屏蔽阳光、清除RIs和淬灭目标污染物的激发态来减弱有机污染物的光降解;另一方面主要通过产生RIs促进光降解[10]. DOM在特定有机污染物光降解中的具体作用主要取决于DOM的类型、来源和组成[11 − 12]. 另外,环境因素如pH、光照强度等也会影响DOM的微观形态和光化学性质. 不同DOM的分子结构和特征性质存在内在差异,进而对ECs光降解表现出不同的作用.
然而,不同ECs在自然水体中的光降解行为并不一致,环境条件的动态变化也在影响该行为. 因此,研究DOM组分及环境因素对ECs在自然水体中的间接光降解作用,对预测其光化学命运至关重要,也将有助于更好地了解其他ECs的环境命运. 本文介绍了DOM诱导ECs间接光降解的降解途径和作用,讨论了DOM来源、类型、组分和其他环境条件对光降解效率的影响,以全面了解该行为机制.
1. DOM的性质和来源及光化学过程(Properties, sources and photochemical processes of DOM)
1.1 DOM的性质和来源
DOM是一类由多种活性有机物(如多糖、蛋白质和木质素)组成的复杂且不均匀的混合物[13]. 它由各种官能团组成,包括醛、氨基、羧基、酯、羟基、酮、苯酚和其他官能团. DOM中的芳香酮类结构具有较强的得电子能力;酚类结构具有较强的给电子能力. 这两种结构可使DOM作为电子传递体,通过电子转移与水体中的其他物质发生氧化还原反应[14]. 研究表明,芳香酮和酚类结构在DOM中的比例对其光敏化过程起决定作用[15]. 按荧光结构划分,DOM分为类腐殖质和类蛋白质[16]. 在水生环境中主要存在两种腐殖酸组分:腐殖酸和富里酸. 腐殖酸和富里酸在分子量、化学成分、化学性质、芳香性和光学性质方面各有差异[17]. 由于有机物种的多样性和丰富的化学结构,DOM被认为是地球上最具有化学活性的有机物质之一[18].
DOM中能够吸收光的基团称为生色团,在DOM的光化学活性中起主导作用. 由于这部分结构的光吸收随着可见光(VIS)和紫外(UV)光谱中波长的降低而呈指数增长,其光吸收在UV区域最高,在光谱的红色区域下降到接近零的水平[19]. DOM在受到光辐照后通常会发生两个过程,即光漂白和光矿化. 光漂白是指DOM吸收光后发色基团吸光能力及芳香性降低的现象;而光矿化则是指随着光化学反应的发生,DOM中连接到芳香结构上的羧基发生脱羧,分解为简单矿物质后形成无机小分子化合物(如CO和CO2)的过程[20]. 由于DOM具有特殊的荧光特性,在受到光激发时会发射出荧光,通常采用三维荧光光谱与平行因子相结合的方式来分析其组成、来源和性质[21].
水体环境中的DOM可通过两个主要来源引入:内源或外源[22]. 内源主要是指水生生物,包括藻类、浮游生物和大型植物[23]. 在海洋中,浮游植物是DOM的主要来源[22];而在淡水湖泊中,植物凋谢物的分解被视为水生DOM的重要来源. 外源是指从外部水体环境中引入的外来DOM,引入途径主要包括土壤、雨水径流、大气(如雨水、沙尘暴)、地下水和人类活动等[24]. 由于营养状况、周围环境和水力特性等因素的不确定性,水体环境中DOM的来源及其比例变化很大,从而导致了其复杂性和异质性[25 − 26].
鉴于DOM在水生环境中的整体作用,深入了解其性质、反应性和环境影响在环境和生态化学领域具有重要意义.
1.2 DOM光致生成自由基的过程
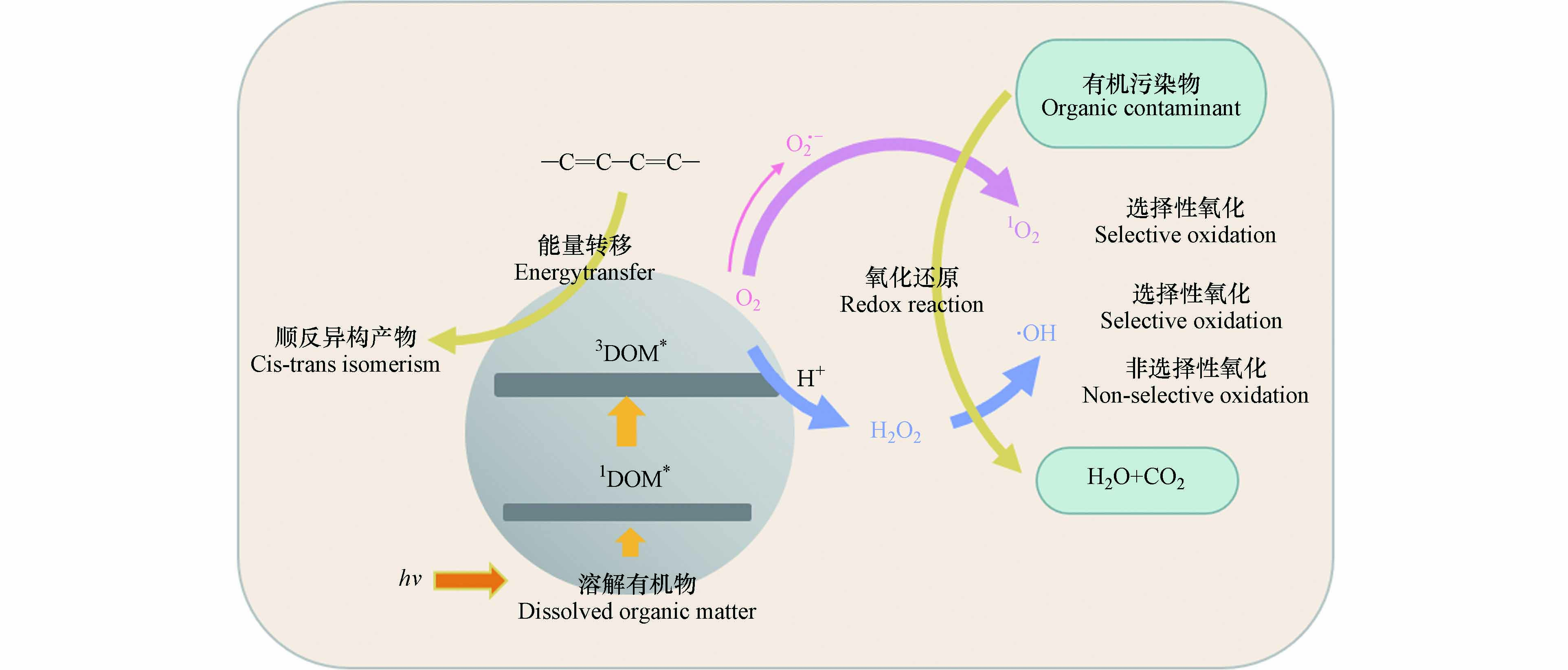
DOM是连接生命形态碳和无机碳的关键纽带,参与各种生物地球化学循环过程[27]. 尤其是DOM中的高分子量成分作为“太阳屏障”,对水生生态环境非常重要[18]. 吸光反应是DOM在水体中的重要光化学过程[28]. 由于DOM分子具有很强的光反应性,尤其是含有苯环、羧基、羟基等发色团,使其能够在水生环境中吸收光后诱导形成RIs,如羟基自由基(·OH)、单线态氧(1O2)、DOM的激发三重态(3DOM*)、过氧化氢(H2O2)等[29]. 这些光生反应物种由于其高度不稳定性和化学反应性而具有瞬态性质,可以对ECs的光降解产生不同程度的影响. 其主要反应途径如图1所示.
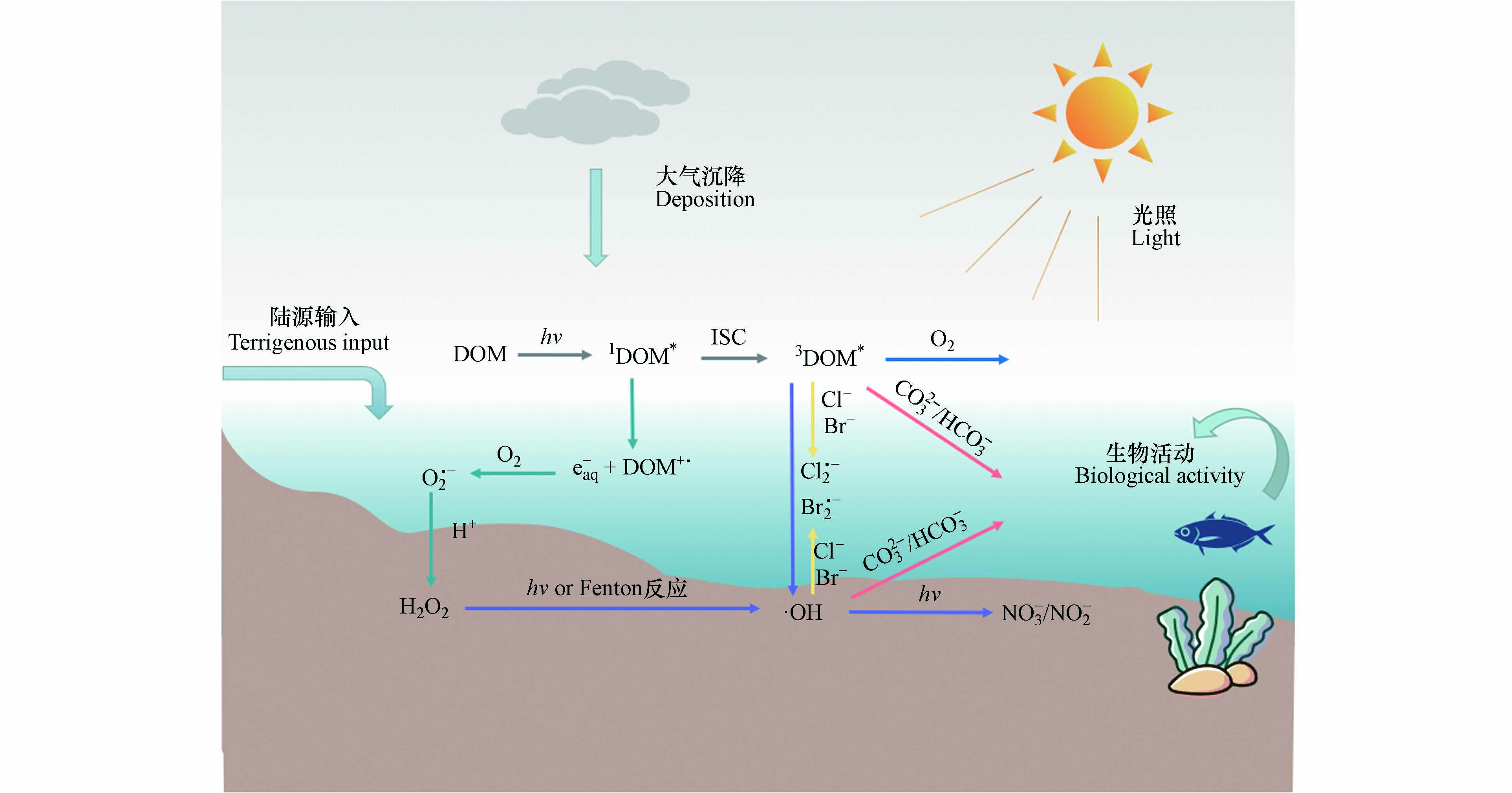 图 1 水体中DOM生成活性物种的反应途径Figure 1. Reaction pathway of DOM producing active species in water
图 1 水体中DOM生成活性物种的反应途径Figure 1. Reaction pathway of DOM producing active species in waterDOM通过辐射吸收,从电子基态(S0)光化学激发到激发单线态(1DOM*),通过荧光发射和无辐射跃迁释放能量回到基态,或在有利条件下通过系统间交叉(ISC,Intersystem Crossing)进化到三线态,从而形成3DOM*[30]. 3DOM*是水体中重要的过渡物种,可以通过能量、电子或氢转移与ECs反应,在后两种情况下,三重态通常表现为氧化剂. 在含氧水环境中,3DOM*可以将能量转移到基态分子氧以产生1O2[31]. 1O2在有机微污染物的转化中起着重要作用[32]. 3DOM*还能够通过水的氧化产生羟基化物种·OH[33]. 另外,1DOM*存在时间非常短,它会向水中释放电子形成水合电子(e-),从而在含氧水环境中产生超氧自由基(O2·-),与H+结合后转化为H2O2[34]. H2O2作为·OH的前驱体,通过直接光解或与过渡金属离子发生芬顿反应(Fenton)生成·OH[35]. ·OH还可以通过水体中NO3-/NO2-的光解产生,这也是·OH的重要来源[36]. ·OH是地表水中能产生的反应性最强的瞬态物种,它们对许多持久污染物都有很高的二阶动力学常数,因此涉及·OH的反应速率往往受到传质(扩散)现象的限制,但是它们通常可以与许多难降解的污染物反应[35]. 水体中的卤素离子也是有机污染物进行光化学转化的重要参与者. 3DOM*可以氧化卤素离子(如Cl-、Br-),从而生成卤素自由基(Cl·-、Br·-). 有研究表明,卤素自由基在富含卤化物的河口和沿海水域对污染物的光化学降解具有重要影响[37].
2. 水体中ECs的光降解过程(Photodegradation process of ECs in water)
ECs通常水溶性较低,易被有机物质吸附,在水体中主要涉及一系列生物、物理和化学过程,包括吸附、生物降解和光化学降解[38 − 40]. 其中光化学降解是天然水环境中ECs的主要转化过程之一,尤其是在表层水体中[41]. 光化学降解一般分为直接光降解、间接光降解和自敏化光降解. 大多数ECs对光高度敏感,会对光子进行吸收,当光子具有足够的能量时ECs分子断裂进行直接分解;间接光降解是由水中的光敏剂产生RIs引发的;自敏化光降解是指ECs在吸收光子活化至激发态的同时,将能量转移给基态3O2或H2O,导致活性氧自由基的产生,进而引发自身降解[27,42 − 43]. ECs在水体中的光降解途径及影响因素如图2所示.
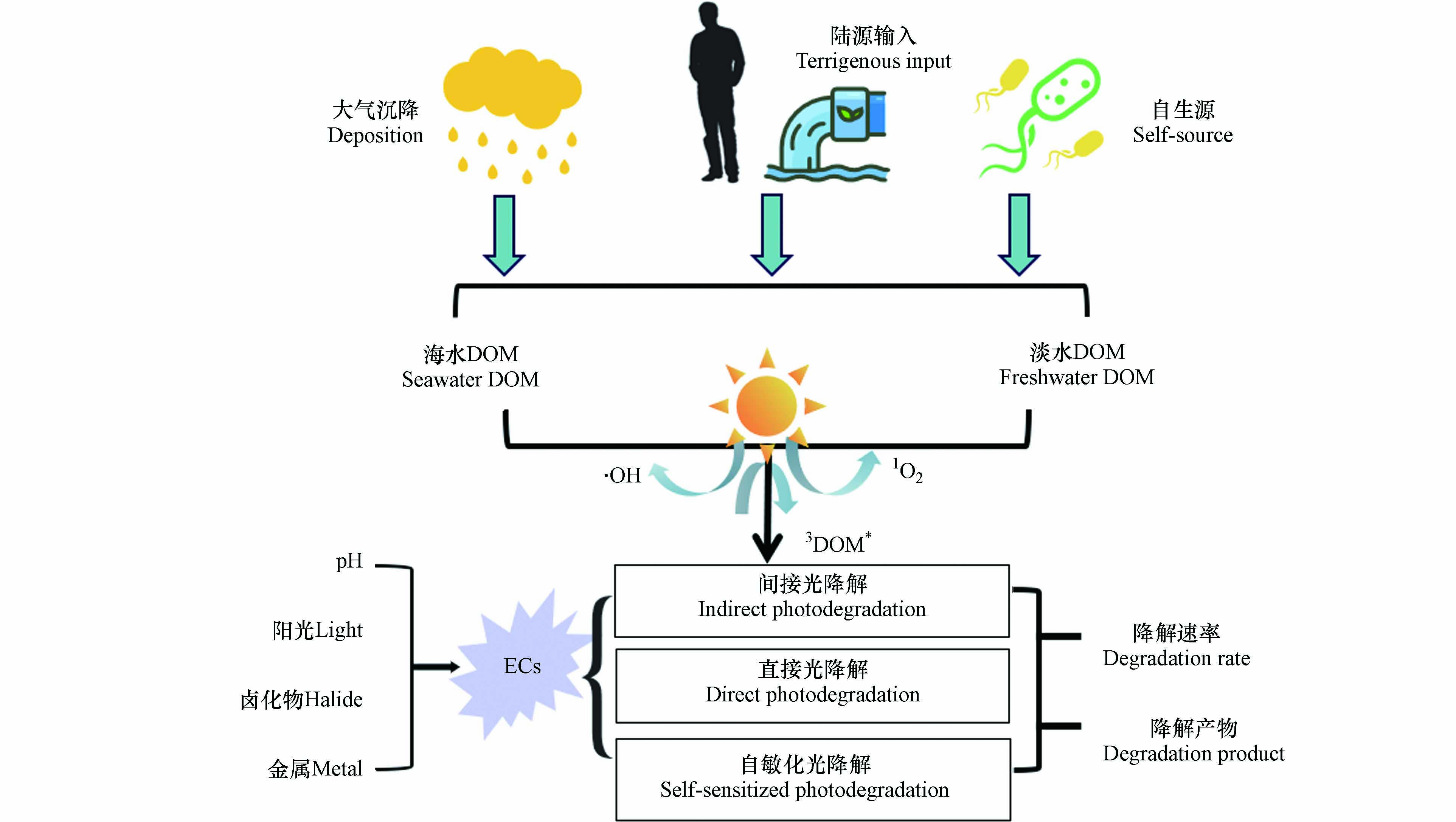 图 2 ECs在水体中的光降解途径及影响因素Figure 2. Photodegradation pathways and influencing factors of ECs in water
图 2 ECs在水体中的光降解途径及影响因素Figure 2. Photodegradation pathways and influencing factors of ECs in water直接光降解和间接光降解的区别主要在于吸收光子的对象不同. 直接光降解是由ECs自身吸收光子引发的,而在间接光降解中吸收光子的对象则是光敏剂. 当ECs吸光能力较强时,DOM对其光降解的影响较弱. 例如,Zeng等[44]研究发现,相对于直接光降解速率,光敏剂的存在对氟乐灵的光降解速率没有影响. Carena等[45]研究发现,即使将苯达松溶解在含有光敏剂的天然水样中,苯达松的光降解速率与在纯水中相比并无显著差异. 该类物质在太阳光谱中具有很强的吸收作用,因此直接光降解是其在环境相关条件下的主要光降解途径. 对于这类物质来说,光敏剂如DOM的存在可能不会改变其光降解速率.
ECs的光降解过程与其化学结构密切相关. 在间接光降解过程中,光敏剂诱导ECs的光降解效率会受到ECs自身结构的影响. 先前研究发现敌草隆直接光降解速率非常缓慢,当加入DOM时光降解速率变快[46]. 由于敌草隆芳环上存在吸电子取代基,光敏剂DOM在阳光诱导下产生·OH,带有亲电性和非选择性,导致其与富电子的芳香族有机化合物的反应性最强[44]. Bertoldi等[47]研究发现与短波长辐射相比,在长波长辐射下腐殖酸对双酚A的光降解作用更弱. 由于双酚A分子结构中含有不饱和苯环,而不饱和烃、芳烃及其类似化合物对短波紫外光有很强的吸收,对波长大于290 nm的UVA和UVB几乎没有吸收作用[48]. 因此,双酚A在短波长辐射下光降解作用显著,而在长波长辐射下的光降解作用并不明显. Bertoldi等[47]还发现雌三醇(E3)比17β-雌二醇(E2)更易受到腐殖酸的诱导作用. 与E2相比,E3具有更多数量的羟基自由基,而腐殖酸结构中含有大量的羧基,它是由两个平行p轨道的横向重叠形成,有利于π-π相互作用[49]. 由此可见,ECs的自身结构会决定化学性质,从而影响间接光降解行为.
在自然水体中,DOM是有效的光敏剂,可以在辐照下生成RIs从而诱导ECs进行间接光降解[39,50]. 例如Ozaki等[51]将DOM浓度设置为与自然水体相似的浓度,发现TCS的光降解比在纯水中显著增强;Carena等[52]研究发现在DOM存在下,除草剂丙腈的间接光降解速率显著提高;任文华等[53]研究发现不同的DOM对丁吡吗啉降解存在不同程度的促进作用. 光降解效率增强的一个可能原因是在间接光降解过程中,DOM吸收光进行光解反应生成RIs,然后与化合物进行反应一起消耗,提高ECs降解效率并缩短残留ECs的半衰期. 相反,郑晓东等[54]研究三氯生光降解行为时发现DOM的存在降低其光降解速率;刘师宇等[55]向阿特拉津溶液中加入不同浓度的DOM后发现,DOM的存在对阿特拉津的光降解起到抑制作用,且浓度越高,抑制作用越显著. 这归因于DOM的抑制作用:DOM与ECs竞争光吸收进行自身光降解从而对其具有光屏蔽作用,另外DOM可以与RIs直接反应清除部分RIs,这也是抑制ECs光降解的一个重要原因[56]. 上述研究表明光敏化过程是由DOM诱导的. 因此,可以合理假设水体中的DOM是诱导ECs间接光降解的主要驱动因素.
3. DOM诱导ECs间接光降解机制(Indirect photodegradation mechanism of ECs induced by DOM)
DOM诱导的ECs光降解是一种间接光降解,而间接光降解主要取决于氧化剂种类. 例如,DOM在光照下产生的·OH、1O2、H2O2、和3DOM*等[29,57]. DOM通常对ECs光降解过程产生两种影响:促进或抑制作用. DOM的促进作用主要表现在DOM吸光后产生RIs与ECs反应并促进其光降解;抑制作用主要包括两种机制:①DOM吸收阳光进行光降解,从而与有机污染物发生竞争作用;②清除RIs和淬灭目标污染物的激发态减弱有机污染物的光降解. 除影响ECs光降解速率外,DOM还可以改变其光降解产物,如Mansour等[58]研究发现二甲戊灵在没有DOM存在的情况下发生脱烷基化,在DOM存在的情况下发生硝基还原. DOM产生的·OH、1O2及3DOM*均可以作为氧化剂与供电子体发生氧化还原反应. 其中·OH为非选择性氧化剂;1O2为选择性氧化剂,易与烯烃、硫化物及富电子酚类物质发生氧化还原反应. 3DOM*在遇到共轭二烯结构时会发生能量转移,使含有该结构的ECs生成自身顺反异构产物;在遇到芳香胺、富电子酚类等富电子物质时发生氧化还原反应[27]. ECs在DOM水体中降解的大致路径如图3所示. 同时,DOM对ECs光降解的影响机制具有很强的特异性,这种特异性的产生可能有4个原因,即DOM类型不同,来源不同,组分不同,且DOM的性质及功能还会受到环境因素的影响. DOM对不同ECs光降解的影响如表1所示.
表 1 DOM对不同污染物光降解的影响Table 1. Effects of DOM on photodegradation of different pollutants污染物Pollutants CAS号CAS number 主要光降解途径Main photodegradation pathways DOM来源DOM source 促进/抑制作用Promotion/inhibition DOM主要作用机制Main mechanism of DOM 参考文献Reference 氟乐灵 1582-09-8 直接光降解 SRHA/SRFA/SRNOM — — [44] 苯达松 25057-89-0 直接 湖水/稻田 — — [45] 二甲戊灵 40487-42-1 直接 湖水 — — [58] 敌草隆 330-54-1 间接 SRFA 促进 光致产生·OH [46] 雌三醇 50-27-1 间接 SRFA/SRNOM 促进 π-π相互作用 [47] 17-β雌二醇 50-28-2 间接 滇池底泥 促进 光致产生·OH,1O2,3DOM* [59] 三氯生 3380-34-5 间接 腐殖酸(购自日本) 促进 光致产生·OH,1O2,3DOM* [51] NLHA/NLFA 抑制 光屏蔽;动态猝灭 [54] 丙腈 107-12-0 间接 蒽醌-2-磺酸钠 促进 光致产生3DOM* [52] 丁吡吗啉 868390-90-3 间接 腐殖酸(购自中国天津) 促进 氢键、离子交换等作用力 [53] 阿特拉津 1912-24-9 间接 腐殖酸(购自中国上海) 抑制 竞争光吸收 [55] 17α-乙炔基雌二醇 57-63-6 间接 河水腐殖酸 促进 光致产生·OH [60] 河水富里酸 促进 光致产生3DOM* [60] 二苯甲酮-1 131-56-6 间接 海水DOM/SRFA/SRNOM 促进 光致产生·OH,1O2,3DOM* [61] 二苯甲酮-3 131-57-7 间接 淡水DOM/海水DOM 促进 光致产生·OH,1O2,3DOM* [4] 2-(2-羟基-5-苯甲基)苯并三唑 2440-22-4 间接 海水DOM 促进 光致产生3DOM* [12] 磺胺嘧啶 68-35-9 间接 SRHA/SRFA/SRNOM/JKHA 促进 光致产生3DOM* [3] 布洛芬 15687-27-1 间接 SRHA/SRFA/SRNOM/JKHA 促进 光致产生·OH,1O2 [62] 对乙酰氨基酚 103-90-2 间接 SRHA/SRFA/SRNOM/JKHA 促进 光致产生·OH,1O2,3DOM* [63] 注:蒽醌-2-磺酸钠为DOM替代物,SRHA为苏万尼河腐殖酸,SRFA为苏万尼河富里酸,SRNOM为苏万尼河天然有机物,NLHA为Nordic湖腐植酸,NLFA为Nordic湖富里酸,JKHA为J&K科技有限公司腐殖酸,上述均为商品化DOM. Note:anthraquinone-2-sulfonate is a DOM substitute, SRHA is Suwannee River humic acid, SRFA is Suwannee River fulvic acid, SRNOM is Suwannee River natural organic matter, NLHA is Nordic Lake humic acid, NLFA is Nordic Lake fulvic acid, JKHA is humic acid from J&K Scientific Ltd. All of the above are commercial DOM. | Show Table DownLoad:
CSV
DownLoad:
CSV
3.1 不同类型DOM对光降解的影响
一般来说,DOM主要分为腐殖酸和富里酸[64]. 腐殖酸(humic acid,HA)又称胡敏酸,是一种天然有机高分子化合物,也是腐殖质的主要组成部分. 富里酸(fulvic acid,FA)又称黄腐酸,既溶于酸也溶于碱,是土壤腐殖质的组成成分之一[65]. HA和FA都是腐殖质中易溶于水的部分,主要由碳、氢、氧、氮、硫等元素构成[66]. 与HA相比,FA碳氢比值较低,分子结构方面芳香核的聚合度较小,官能团中酚羟基和甲氧基的数目比较多,含有更多含氧官能团和脂肪结构,且腐殖化程度更深[67]. 此外,FA的酸性官能团(—COOH)含量也高于HA,因此其在水溶液中具有较强的酸性[68]. HA和FA分子结构和特征性质的内在差异往往导致其对ECs的间接光降解作用表现不一致.
研究表明,DOM结构对光活性有重要影响[69 − 70]. 任东等[57]在研究E2光降解时发现,与HA相比FA的促进作用更强. 这主要与HA和FA的含氧官能团含量和分子极性大小有关. 含氧官能团如酚、醌、酮等组分结构与·OH、1O2等活性氧物种的产生密切相关. 杜超等[71]研究发现,含氧官能团含量会影响3DOM*的电子转移和能量传递过程,且含量高的组分在光照下产生RIs的能力更高. Fang等[72]研究发现,醌类结构在DOM生成·OH和1O2过程中起主要作用. 含氧官能团的含量制约着腐殖酸的可溶性、亲疏水性等,是光诱导DOM生成RIs的重要影响因素. 大分子作为较小组分的超分子聚集体,其特点是电荷转移相互作用有利于内部转化,而牺牲了光物理(例如荧光)和光化学过程[73 − 74]. 腐殖质的低分子量组分具有更高的形成三重态的能力[75 − 76]. 从这个角度来看,与HA相比,FA具有更高的生成3DOM*的能力. Ren等[60]研究发现,与HA相比,FA表现出较弱的光吸收,但在促进17α-乙炔基雌二醇(EE2)光降解方面,FA的促进作用比HA更强. 其猝灭实验表明·OH是HA溶液中EE2光降解的主要贡献者,而3DOM*在FA溶液中主导EE2的降解. 这种机制差异主要归因于以下两个原因:①FA中酮羰基和羟基的数量高于HA;②HA中的分子内相互作用可以强烈抑制3DOM*的形成[77 − 79]. 由于HA生成的3DOM*产率受到抑制,导致·OH在ECs光降解过程中起主导作用. 综上来看,与HA相比,FA具有更强的光敏能力、更低的反应物种猝灭效应和较弱的光衰减. 因此FA在ECs光降解中的促进作用强于HA.
3.2 海水和淡水DOM对光降解的影响
目前研究的淡水DOM主要购自腐殖酸协会,如SRHA、SRFA、SRNOM及JKHA等[43,46],而海水DOM主要取自海水水域,通过电渗析/反渗透、固相萃取等方式获得[61,80]. 由于水域不同,海水DOM与淡水DOM在参与ECs间接光降解过程中通常会表现出不同的作用.
Wang等[61]在研究沿海海水DOM和淡水河流DOM对二苯甲酮-1(BP-1)光降解影响时发现,BP-1的光降解在不同DOM水体中出现了不同结果. 研究发现与沿海海水DOM相比,淡水DOM的光吸收率较高,导致吸光后产生的RIs稳态浓度较高,DOM光致产生活性中间体的稳态浓度决定了DOM对有机污染物的反应性[81]. 而且淡水DOM通常具有更高的发色团和荧光团含量,这与稳定状态下的挥发性有机物浓度有关[82]. Li等[4]研究也发现二苯甲酮-3在海水中的光降解速率相对低于在淡水中的光降解速率. 不同的是,在海水中的间接光降解主要归因于3DOM*,而在淡水中,3DOM*和·OH是其间接光降解的主要原因. 对于不同来源的DOM,组成成分不同,相应的3DOM*也具有不同的激发态还原电势[82]. 说明沿海海水和淡水DOM的光反应性对污染物的光降解具有内在差异,且淡水DOM对ECs的光降解促进作用更明显.
由于人为排放,例如海水养殖活动的影响,来自养殖海域的DOM与来自原始海水的DOM对ECs光降解表现出不同程度的行为. 人为投放的营养饵料和高强度动物活动是海水养殖的特定DOM来源[83]. 之前有研究表明,海水DOM对磺胺类抗生素的光降解具有显著的促进作用,且受海水养殖影响较大DOM的促进作用强于其他海域DOM[84 − 85]. Chen等[12]研究也发现,与来自原始海水DOM相比,受海水养殖活动影响较大的DOM表现出更高的吸光度,且对2-(2-羟基-5-苯甲基)苯并三唑的光降解促进作用更显著. 这表明养殖活动会增强DOM的光敏化降解能力. 光漂白是从天然水中去除DOM的主要过程,可以降低海洋中DOM的紫外线吸收率和分子量[86 − 87]. 与来自沿海水域的DOM相比,受海水养殖影响较大海域的DOM光漂白较少,类腐殖质物质比例较高,芳香结构和羰基结构比例较高,通常具有较高的光吸收率、3DOM*的形成量子产率和3DOM*稳态浓度,从而会对ECs的光降解产生更高的光敏效应[84]. 说明养殖区海水DOM具有更强的光敏化降解能力,并且主要是通过3DOM*加速光降解过程.
综上,光漂白过程弱、富含发色团和芳香结构的DOM光敏化降解能力更强. 由于来源不同,DOM经历的光漂白过程及所含的发色团和芳香结构含量不同,其产生RIs的能力不同,从而导致来自淡水、海水及特殊海水水域的DOM在诱导ECs间接光降解过程中表现出不同的作用.
3.3 不同荧光组分DOM对光降解的影响
激发发射矩阵光谱(EEM)与平行因子分析(PARAFAC)相结合是表征DOM的常用分析技术[88 − 89]. 该方法可以根据不同的光源、特性以及迁移和转换方式,将DOM划分为具有特殊光学性质的不同组分[90]:首先利用PARAFAC得到荧光组分图,根据最大激发发射波长得到荧光峰的位置,通过与前人所研究的荧光峰位置对比及荧光参数分析,从而得到DOM组分性质及来源的相关信息[91]. DOM组分一般分为类色氨酸、类络氨酸、类腐殖质等荧光组分[92]. 由于DOM组分千差万别,ECs与不同组分的DOM发生相互作用时,效果也不一致.
白莹等[62]研究发现,布洛芬主要进行间接光降解且光降解速率与DOM的组成成分密切相关. Bai等[63]在研究DOM各组分对对乙酰氨基酚光降解影响时发现,对乙酰氨基酚与所有组分的间接光降解速率常数均呈显著正相关,且外源DOM的相关性强于自源DOM. 为进一步分析探究,Bai等[3]在研究DOM组分对磺胺类抗生素光降解影响时,将DOM分为4个组分C1、C2、C3和C4,通过与前人研究结果对照发现C1、C2、C3为外源DOM,C4为自源DOM,且各组分的分子量和芳香度顺序为C3>C1>C2>C4,与组分荧光强度的衰减和光降解速率的相关系数一致. 说明这4种组分在光降解过程中起重要作用,且外源性荧光组分起主要作用. 与源自微生物合成的自源DOM相比,外源DOM可能具有更多芳香环和电供体基团,因此往往具有更高的芳香性[93 − 95]. DOM中含羰基和芳香结构的部分为主要发色团,RIs的稳态浓度与DOM的芳香性呈显著正相关[96]. 这是由于高分子量DOM的量子产率下降,而DOM的高芳香性超过了其量子产率,能够产生RIs或表现出较低的猝灭速率[97]. 与上述研究相符,Batista等[96]报道了芳香性与·OH稳态浓度及·OH生成速率之间的密切关系. Timko等[11]观察到芳香性与1O2和3DOM*的形成速率呈正相关. Zhou等[98]的研究表明,DOM(E2/E3)的吸光度比与1O2的表观量子产率之间存在正相关,并且DOM的芳香结构是产生3DOM*和1O2的主要部分. RIs的稳态浓度随DOM的分子量、光学性质和组成而变化. 由于DOM组分不同,其产生的RIs活性效率不同. 芳香度更高的组分对RIs的生成贡献更大,从而对ECs间接光降解过程促进作用更明显. 因此,我们应该了解DOM组分的变化,以评估DOM对ECs间接光降解的影响.
3.4 环境因素的影响
除了DOM的类型、来源及组分差异外,DOM诱导的ECs光降解还受到环境因素的影响,例如pH大小、光照强度、离子强度及金属元素等.
3.4.1 光源及光照强度
吸光是光化学反应的前提条件,DOM中的发色团可以吸收290—500 nm的太阳光,且光吸收强度随着波长增长呈现近似指数的下降趋势[81,99]. 当光源强度不同,产生的光子能量自然不同,从而会影响DOM对ECs的光降解效率. Peng等[100]研究发现,在UV-vis辐照下(λ>200 nm),DOM抑制了普萘洛尔的光降解,然而在模拟太阳辐照下(λ>290 nm),DOM促进其光降解. 研究表明在不同光源照射下,DOM对普萘洛尔光降解的双重作用十分典型. 一方面由于DOM具有很强的光敏性,能进行光解产生大量的RIs;另一方面当DOM自身发生光解时,会与污染物竞争光吸收从而产生光屏蔽作用. 在波长较小的UV-vis辐照下,DOM吸光强度变大,此时可能以竞争光吸收的抑制作用为主. 另外,光源强度还会影响DOM产生活性物种的能力. 如刘砚弘等[101]研究发现,汞灯照射下DOM产生活性物种的能力显著高于氙灯照射条件下,说明光源强度越大,DOM产生的活性物种越多.
3.4.2 卤化物浓度
上述研究中提到DOM可以产生RIs,其中3DOM*能够氧化卤化物离子(如Cl-,Br-),从而生成卤素自由基(Reactive halogen species,RHs),如Cl·、Br·、Cl·-、Br·-等. 这些自由基可以促进海水中许多ECs的光解. 例如,Zhao等[102]研究发现,磺胺嘧啶的光解随着上游至下游河口水域卤化物浓度的增加而增强. Pinto等[103]研究表明,在低浓度的DOM溶液和盐水中,毒死蜱主要进行直接光降解;而在高浓度的DOM溶液和盐水中,其主要进行间接光降解. 由于3DOM*的寿命变化和3DOM*与其他卤化物离子生成RHs,从而增强ECs的间接光降解速率.
但是,3DOM*与卤化物离子反应产生的RHs也可能导致3DOM*的猝灭. Hou等[104]研究发现,向DOM溶液中添加卤化物,卡马西平光解速率明显降低. 因此,即使在RHs主导的河口水域中有些污染物出现了光降解增强的现象,可能也会有部分污染物光降解受到抑制. 3DOM*诱导产生的RHS反应无法补偿3DOM*因猝灭效应对降解产生抑制,这可能会减缓ECs在卤化物浓度相对较高的河口水域中的光降解. 此外,卤化物离子的离子强度效应也会影响3DOM*衰变,从而影响3DOM*的稳态浓度[105].
3.4.3 pH
pH值的变化会严重影响ECs的电离形式并改变其光化学反应性[69],pH值在污染物的光降解中起着重要作用. 例如Bai等[63]研究发现,在碱性条件下APAP的光降解速率显著增强. 在pH为碱性时,APAP中的羟基转变为酚阴离子,由于酚阴离子在芳香环上的电子密度较高,因此其与3DOM*的反应比酚羟基更容易[106]. Jin等[107]研究发现,氧四环素直接光解速率常数随pH的增加而增加. Ge等[69]研究发现,沙拉沙星和加替沙星的光解速率常数随着pH值的增加先增大后减小. pH决定了ECs的离子存在形式. 可电离污染物最佳离子形式占主导地位,光吸收率也随之增加;而当污染物以最稳定形式存在时,光化学活性降低. 因此,ECs的不同解离状态具有不同的光化学反应性,光降解速率随pH的变化与其在给定pH值下的解离状态密切相关.
另外,pH还会改变DOM的性质和微观形态,从而影响DOM产生RIs的浓度[108]. 于莉莉等[109]研究发现,DOM吸光系数随pH值的增大而增大. 由于在碱性条件下DOM结构发生膨胀,导致显色基团暴露增多,从而其吸光系数变大[110]. Zhang等[111]研究发现,氧四环素的光解速率常数与含DOM水溶液的酸碱度呈正相关. DOM中含有较多的含氧官能团和芳香官能团,活性氧在光照下介导形成苯氧自由基,含氧官能团和芳香物质与四环素络合. 在碱性条件下,四环素更容易受到活性氧的攻击,提高DOM与四环素在光照下的反应效率从而加速四环素的光降解[112]. Gao等[113]研究发现,随着离子强度的增加,DOM吸光度始终在较高pH下增加更明显. 离子强度可以影响DOM的光化学特性,增强DOM诱导的间接光降解作用,而这种增强效应主要依赖于酸碱度[111,113].
3.4.4 金属离子
在水环境中,DOM能够与痕量金属(如Cu2+、Fe3+、Mn2+和Zn2+)结合后产生不同络合强度的稳定配合物[114 − 115]. 据研究称,光激发的金属DOM络合物可以进行配体到金属的电荷转移,从而产生·HO2和O2·-,进一步反应生成·OH[116 − 117]. 这一过程可能会显著影响金属离子和DOM共存体系ECs的间接光降解. Wang等[118]研究发现,Fe3+和丙酮酸形成的络合物是·OH的来源. Liu等[119]研究发现,Fe(Ⅲ)黄腐酸络合物通过在盐水中产生更多·OH,促进了双酚A的光氯化. 庄晓虹等[120]研究发现与只含有DOM的体系相比,DOM和铁(Ⅲ)的络合体系对壬基酚的光降解表现出更显著的促进作用. 在辐照下Fe(Ⅲ)产生自由基时更具有光活性[121]. Fe(Ⅲ)-DOM络合物很容易被光解,引入的氧气促进了这一过程. Fe3+首先被还原为Fe2+,同时生成H2O2,这两种产物进行类芬顿反应,从而生成·OH促进有机物的光降解[122 − 123].
除生成·OH外,向DOM溶液中添加金属离子还会导致DOM的荧光猝灭[115,124]. Wan等[125]研究发现,金属离子和DOM的络合能力与3DOM*的猝灭呈正相关. Liu等[126]研究发现,对于具有较强给电子基团的ECs,Cu(Ⅱ)-DOM络合物显著抑制了3DOM*诱导的有机污染物光降解. 这可能因为Cu2+的络合作用降低了3DOM*形成速率,导致3DOM*稳态浓度的降低. 通过金属离子的静态猝灭效应,1DOM*的形成受到抑制,从而形成3DOM*以及水生环境中其他RIs的进一步减少. 同时,金属离子还可以通过形成偶合络合物直接淬灭ECs的激发三重态,然后进行电子转移和能量转移或无辐射跃迁到基态,产生动态猝灭[127]. 因此,共存的金属离子对DOM诱导的ECs光降解有明显的影响. 研究这一因素对于系统地了解金属离子配位在DOM中如何影响ECs光降解的机制是必要的.
综上,DOM的光敏化降解能力会受到光源、卤化物、pH及金属离子等因素的影响:(1)光源会影响DOM诱导ECS的间接光降解过程. 在短波长辐射下,DOM的光吸收强度更强,在ECs光降解过程中产生的光屏蔽作用更明显. 在强光源辐射下,DOM产生的光活性物种更多,从而对ECs光降解的促进作用更明显. (2)卤化物离子通常在DOM诱导ECs间接光降解过程中表现出双重作用. 3DOM*可以诱导卤素离子产生卤素自由基促进ECs的光降解,但在该过程中3DOM*也可能发生猝灭从而抑制ECs光降解. (3)pH可以改变DOM的性质和微观形态,影响DOM产生的RIs浓度,从而影响DOM诱导ECs的间接光降解过程. (4)金属离子能够与DOM络合,在电荷转移过程中,DOM能够产生·HO2和O2·-,同时又会发生荧光猝灭,对诱导ECs的间接光降解过程起到双重作用.
4. 结论与展望(Conclusion and prospects)
DOM介导的光化学过程对水生系统的生物地球化学有着深远的影响. 目前对ECs在DOM水体中的光转化行为研究常在实验室开展,且选取的DOM溶液大多为实验室配制. 虽然研究的目标物质化学性质各有不同,但大多研究的结论也不尽相同. 另外,许多环境因素包括盐度、pH、离子强度、硝酸盐和碳酸氢盐等也被列入研究范围. 随着分析技术和手段不断进步,对ECs在水环境中光解动力学和机理的研究也逐渐深入.
目前的研究多集中于DOM对单种ECs光降解过程的影响,但是水体环境所含成分较为复杂,且DOM成分复杂、功能多样,同时自然水体中往往是多种DOM与多种ECs共存的情况;另外,部分ECs的代谢物比母体化合物具有更高的活性. 基于此,今后可重点关注以下几个方面:
(1)探究DOM的化学成分与ECs相互作用的机制. 注意与实际相结合,对多种DOM-ECs混合体系中的相互作用规律进行探究,明晰其作用过程和机理,建立相应数据库.
(2)探究ECs在间接光降解后可能形成的降解产物及产物转化途径. 应特别关注母体化合物及其代谢物的环境归宿,分析DOM作用后ECs性质的变化及毒性,为预测ECs的环境行为及评估其生态环境风险作出理论支持.
(3)探究水质因素和环境因素等可变因素综合作用对ECs光降解过程的影响. 河口是受海洋和陆地共同影响的动态系统,在环境条件(如pH、离子强度和卤化物浓度)是动态的河口地区,阐明ECs的光化学转化行为具有重要意义.
-
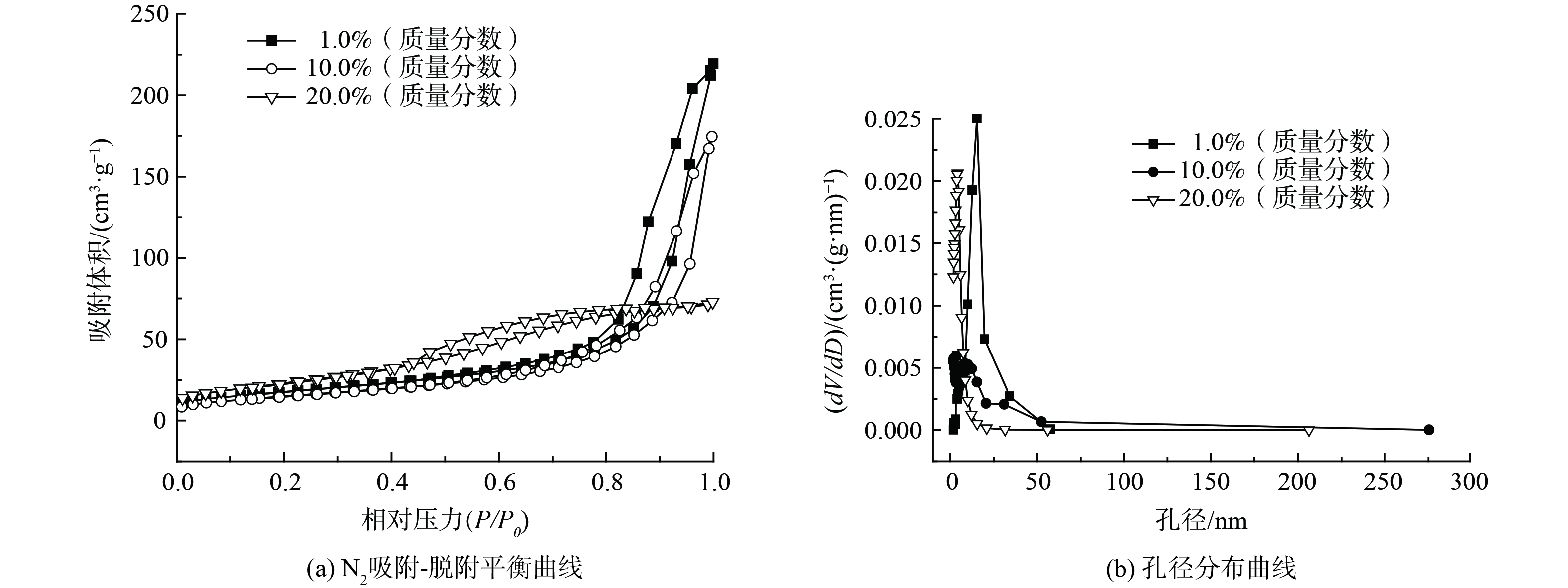
图 1 Bi3+-TiO2/MnCeOx的吸附等温线及孔径分布
Figure 1. Adsorption isotherms and pore size distribution of Bi3+-TiO2/MnCeOx
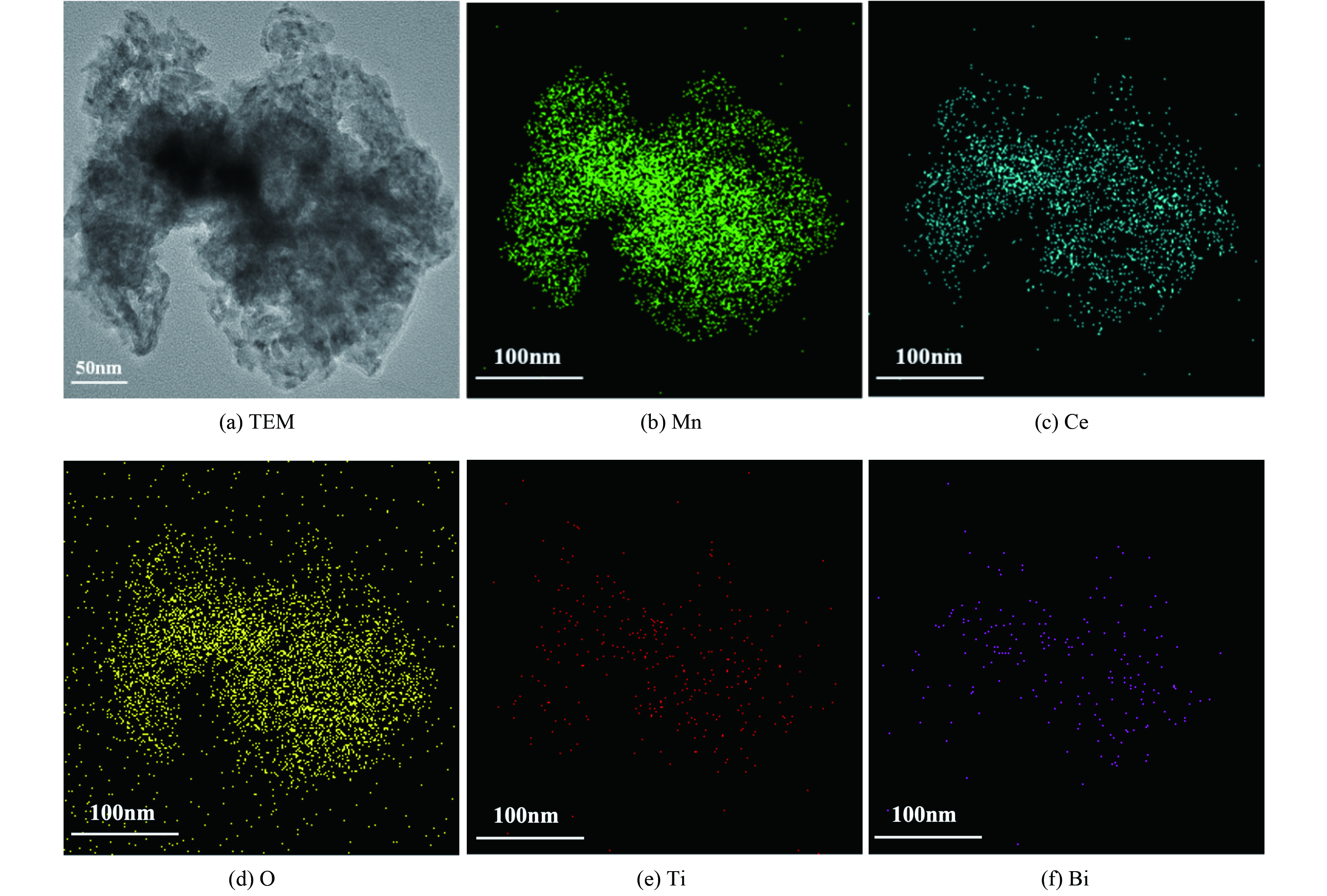
图 4 TEM及Mn, Ce, O, Ti和Bi元素的EDS映射图
Figure 4. TEM and EDS map of Mn, Ce, O, Ti and Bi elements
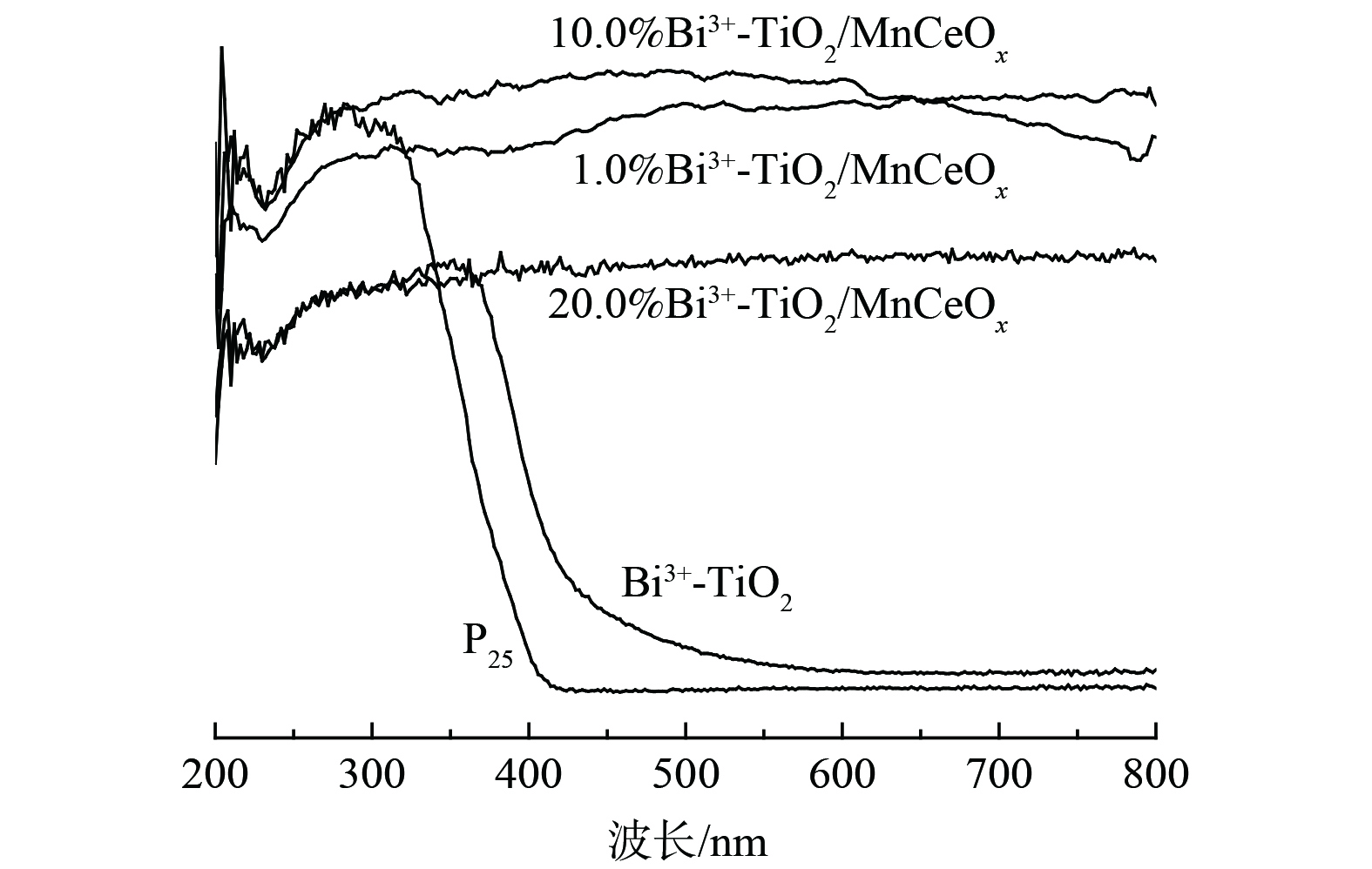
图 5 Bi3+-TiO2/MnCeOx催化剂的UV-vis DRS图谱
Figure 5. UV-vis DRS spectrum of Bi3+-TiO2/MnCeOx catalysts
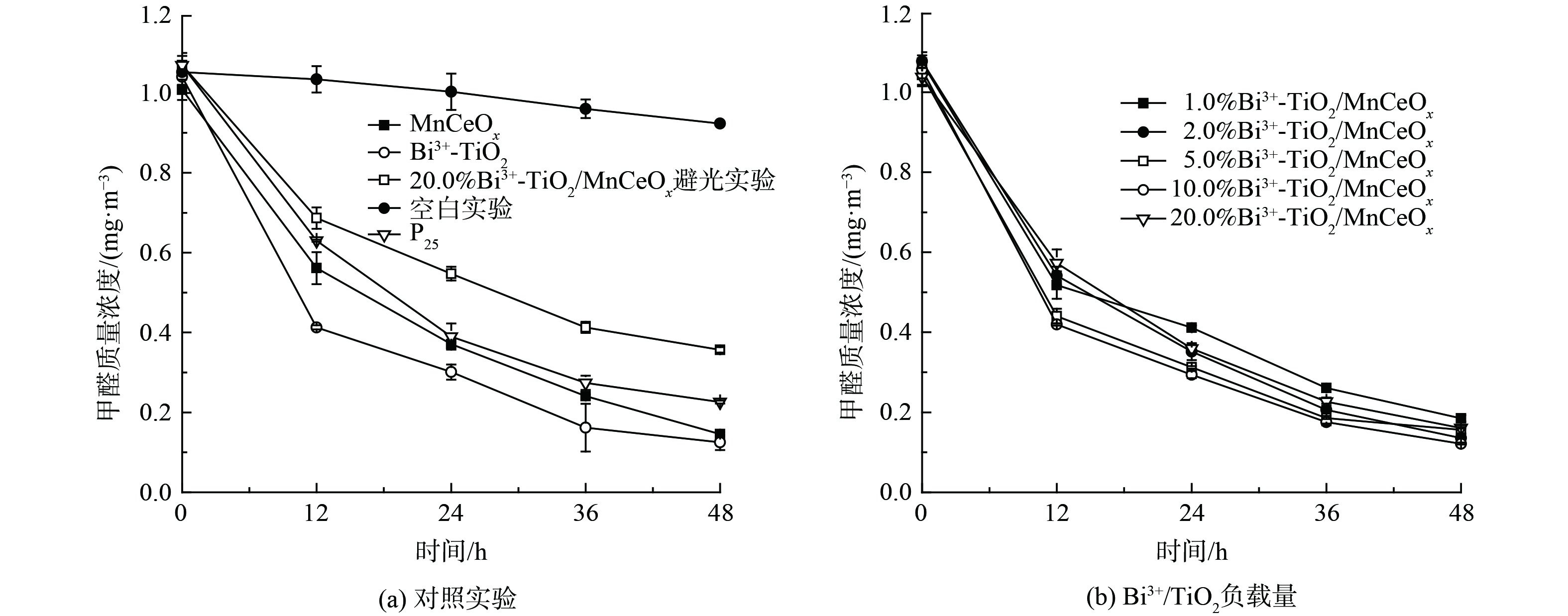
图 7 Bi3+-TiO2/MnCeOx催化剂催化氧化HCHO
Figure 7. Catalytic oxidation activity of HCHO over Bi3+-TiO2/MnCeOx catalysts
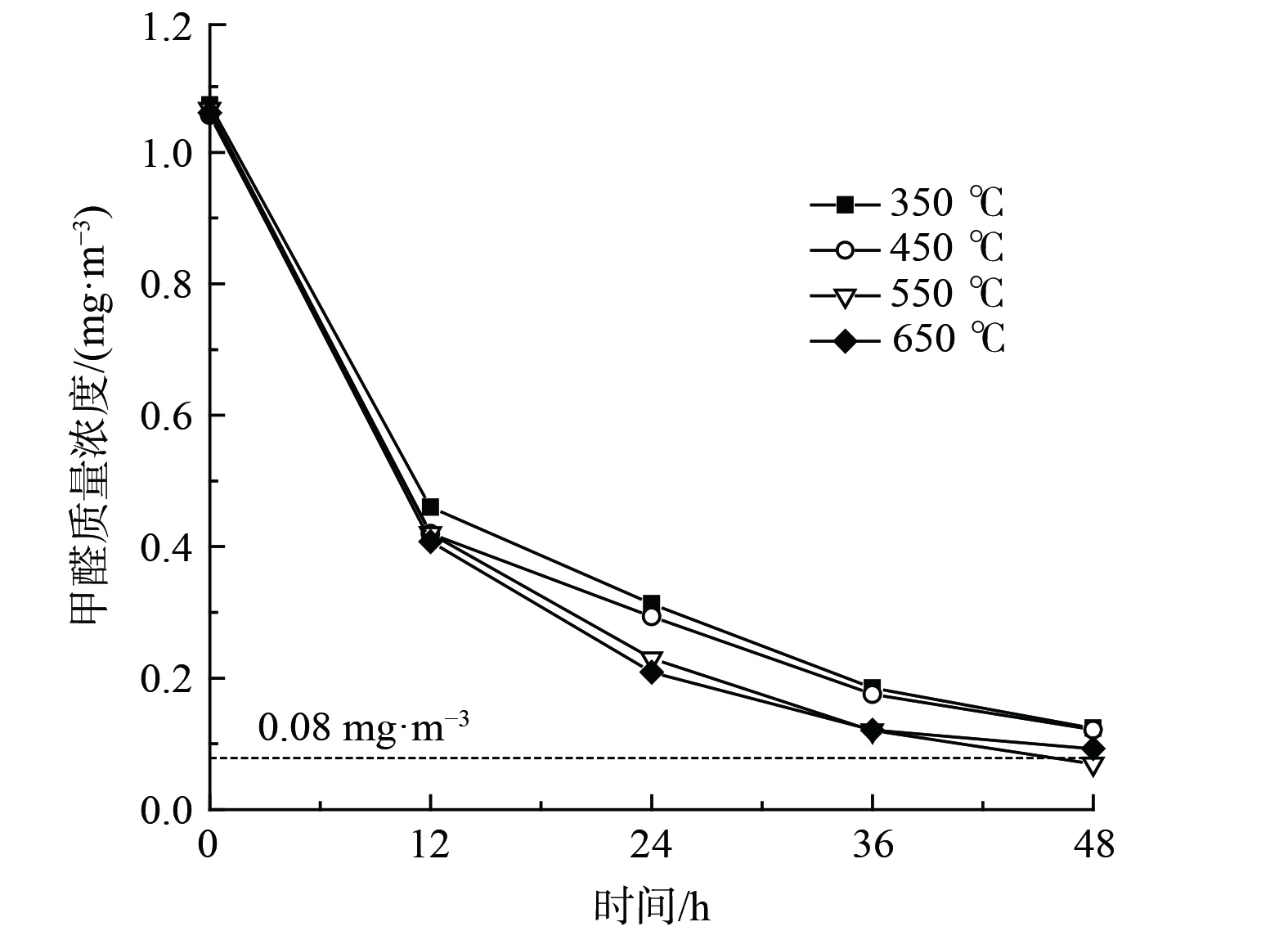
图 8 煅烧温度对Bi3+-TiO2/MnCeOx氧化性能的影响
Figure 8. The effect of calcination temperature on oxidation performance of Bi3+-TiO2/MnCeOx
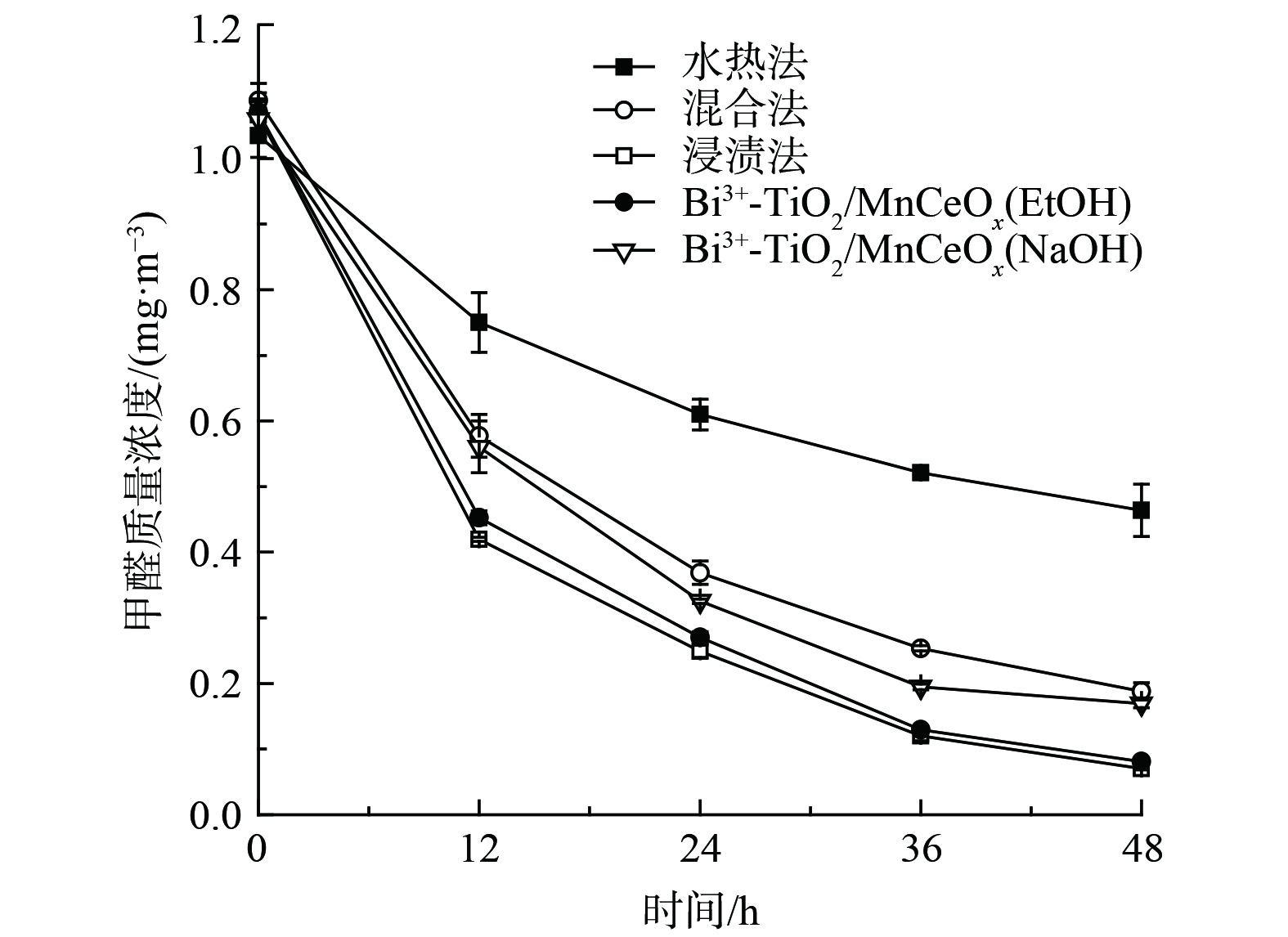
图 9 制备方法及改性处理对Bi3+-TiO2/MnCeOx氧化性能的影响
Figure 9. The effect of preparation method and modification on oxidation performance of Bi3+-TiO2/MnCeOx
表 1 部分样品比表面积及孔容孔径
Table 1. Physical properties of these catalysts in the orthogonal experiment
样品名称 比表面积/(m2·g−1) 孔体积/(cm3·g−1) 平均孔径/nm Bi3+-TiO2 27.3 0.07 6.73 MnCeOx 87.0 0.21 38.9 1.0% Bi3+-TiO2/MnCeOx 63.4 0.334 15.3 10.0% Bi3+-TiO2/MnCeOx 54.0 0.235 13.3 20.0%Bi3+-TiO2/MnCeOx 84.4 0.112 3.88
下载: 导出CSV
-
[1] SALTHAMMER T, MENTESE S, MARUTZKY R. Formaldehyde in the indoor environment[J]. Chemical Reviews, 2010, 110(4): 2536-2572. doi: 10.1021/cr800399g [2] 李省吾, 吴晓航, 黄荣珠, 林建标. 室内空气中甲醛污染去除技术进展研究[J]. 广东化工, 2022, 49(8): 148-149. doi: 10.3969/j.issn.1007-1865.2022.08.047 [3] 徐倩, 曲振平. 介孔氧化硅材料负载Au、Ag催化剂及其甲醛催化氧化性能研究[D]. 大连: 大连理工大学, 2021. [4] 关圣楠, 张琦. 锰基催化剂催化氧化甲醛脱除的研究[D]. 合肥: 中国科学技术大学, 2021. [5] ZHANG G X, SUN Z M, DUAN Y W, et al. Synthesis of nano-TiO2/diatomite composite and its photocatalytic degradation of gaseous formaldehyde[J]. Applied Surface Science, 2017, 412: 105-112. doi: 10.1016/j.apsusc.2017.03.198 [6] LI X, QIAN X R, AN X H, et al. Preparation of a novel composite comprising biochar skeleton and “chrysanthemum” g-C3N4 for enhanced visible light photocatalytic degradation of formaldehyde[J]. Applied Surface Science, 2019, 487: 1262-1270. doi: 10.1016/j.apsusc.2019.05.195 [7] ZHANG G K, XIONG Q, WEI X, et al. Synthesis of bicrystalline TiO2 supported sepiolite fibers and their photocatalytic activity for degradation of gaseous formaldehyde[J]. Applied Clay Science, 2014, 102: 231-237. doi: 10.1016/j.clay.2014.10.001 [8] MALAYERI M, HAGHIGHAT F, LEE C S. Modeling of volatile organic compounds degradation by photocatalytic oxidation reactor in indoor air: A review[J]. Building and Environment, 2019, 154: 309-323. doi: 10.1016/j.buildenv.2019.02.023 [9] YANG Y, LI X J, CHEN J T, et al. Effect of doping mode on the photocatalytic activities of Mo/TiO2[J]. Journal of Photochemistry and Photobiology A:Chemistry, 2004, 163(3): 517-522. doi: 10.1016/j.jphotochem.2004.02.008 [10] PAN X Y, XU Y J. Defect-mediated growth of noble-metal (Ag, Pt, and Pd) nanoparticles on TiO2 with oxygen vacancies for photocatalytic redox reactions under visible light[J]. The Journal of Physical Chemistry C, 2013, 117(35): 17996-18005. doi: 10.1021/jp4064802 [11] LI J, ZHANG M, LI Q Y, et al. Enhanced visible light activity on direct contact Z-scheme g-C3N4-TiO2 photocatalyst[J]. Applied Surface Science, 2017, 391: 184-193. doi: 10.1016/j.apsusc.2016.06.145 [12] HUANG Q, WANG P, FAN Y Z, et al. Synthesis and photocatalytic activity of N-doped BixTi1-xO2 photocatalysts under energy saving lamp illumination[J]. Indoor and Built Environment, 2017, 26(6): 785-795. doi: 10.1177/1420326X16641177 [13] HUANG Y F, WEI Y L, WANG J, et al. Controllable fabrication of Bi2O3/TiO2 heterojunction with excellent visible-light responsive photocatalytic performance[J]. Applied Surface Science, 2017, 423: 119-130. doi: 10.1016/j.apsusc.2017.06.158 [14] HAMDI A, FERRARIA A M, BOTELHO DO REGO A M, et al. Bi–Y doped and co-doped TiO2 nanoparticles: Characterization and photocatalytic activity under visible light irradiation[J]. Journal of Molecular Catalysis A:Chemical, 2013, 380: 34-42. doi: 10.1016/j.molcata.2013.09.005 [15] TIAN H, HE J H, ZHANG X D, et al. Facile synthesis of Porous manganese oxide K-OMS-2 materials and their catalytic activity for formaldehyde oxidation[J]. Microporous and Mesoporous Materials, 2011, 138(1/2/3): 118-122. [16] 何小云, 葛笑, 宋留名, 等. 室温下MnOx/HZSM-5催化氧化甲醛的性能和机理分析[J]. 材料工程, 2021, 49(1): 144-152. [17] YANG P, YANG S S, SHI Z N, et al. Deep oxidation of chlorinated VOCs over CeO2-based transition metal mixed oxide catalysts[J]. Applied Catalysis B:Environmental, 2015, 162: 227-235. doi: 10.1016/j.apcatb.2014.06.048 [18] TANG X F, LI Y G, HUANG X M, et al. MnOx-CeO2 mixed oxide catalysts for complete oxidation of formaldehyde: Effect of preparation method and calcinations temperature[J]. Applied Catalysis B:Environmental, 2006, 62(3/4): 265-273. [19] TANG X F, CHEN J L, HUANG X M, et al. Pt/MnOx CeO2 catalysts for the complete oxidation of formaldehyde at ambient temperature[J]. Applied Catalysis B:Environmental, 2008, 81(1/2): 115-121. [20] ZHANG Y, CHEN M X, ZHANG Z X, et al. Simultaneously catalytic decomposition of formaldehyde and ozone over manganese cerium oxides at room temperature: Promotional effect of relative humidity on the MnCeOx solid solution[J]. Catalysis Today, 2019, 327: 323-333. doi: 10.1016/j.cattod.2018.04.027 [21] SEKINE Y, NISHIMURA A. Removal of formaldehyde from indoor air by passive type air-cleaning materials[J]. Atmospheric Environment, 2001, 35(11): 2001-2007. doi: 10.1016/S1352-2310(00)00465-9 [22] 陶涛, 肖瑶, 胡宇欣, 等. MnCeOx/凹凸棒土催化剂的制备、表征及常温催化氧化性能[J]. 功能材料, 2020, 12(51): 12001-12008. [23] WANG T, YAN X Q, ZHAO S S, et al. Preparation, characterization and photocatalytic activity of three-dimensionally ordered mesoporous /macroporous TiO2 microspheres[J]. Journal of Molecular Catalysis, 2014, 28(4): 359-366. [24] 黄琼, 白梦天, 任超, 等. Mn基金属氧化物催化剂常温催化氧化甲醛[J]. 中国环境科学, 2018, 38(1): 103-111. doi: 10.3969/j.issn.1000-6923.2018.01.013 [25] LI H J, QI G S, TAN A, et al. Low-temperature oxidation of ethanol over a Mn0.6Ce0.4O2 mixed oxide[J]. Applied Catalysis B:Environmental, 2011, 103(1/2): 54-61. [26] HUANG Q, YE J, SI H, et al. Differences of characteristics and performance with Bi3+ and Bi2O3 doping over TiO2 for photocatalytic oxidation under visible light[J]. Catalysis Letters, 2020, 150: 1098-1110. doi: 10.1007/s10562-019-03017-w [27] 朱立才, 袁中直, 李伟善. 现场紫外-可见吸收光谱研究电解二氧化锰的还原过程[J]. 电化学, 2004, 10(2): 168-174. doi: 10.13208/j.electrochem.2004.02.008 [28] CHEN B B, SHI C, CROKER M, et al. Catalytic removal of formaldehyde at room temperature over supported gold catalysts[J]. Applied Catalysis B: Environmental, 2013, 132–133: 245-255. [29] 庞光龙. MnOx基催化剂上甲醛室温催化氧化反应的研究 [D]. 北京: 北京理工大学, 2015. [30] 朱杰, 孙月吟, 顾名扬, 等. TiO2负载MnFeOx催化剂的制备及常温催化氧化甲醛性能研究[J]. 功能材料, 2022, 53(4): 4011-4019. [31] LIU F, RONG S P, ZHANG P Y, et al. One-step synthesis of nanocarbon-decorated MnO2 with superior activity for indoor formaldehyde removal at room temperature[J]. Applied Catalysis B:Environmental, 2018, 235: 158-167. doi: 10.1016/j.apcatb.2018.04.078 [32] SUN D, WAGEH S, Al-GHAMDI A A, et al. Pt/C@MnO2 Composite hierarchical hollow microspheres for catalytic formaldehyde decomposition at room temperature[J]. Applied Surface Science, 2019, 466: 301-308. doi: 10.1016/j.apsusc.2018.10.044 [33] 李妍, 张舸, 蒋贞. 锐钛矿型TiO2吸附甲醛影响因素的模拟与实验研究[J]. 现代化工, 2019, 39(4): 207-210. [34] 段宝庆, 丁雅萍, 陈英文等. 铈基催化剂上CVOCs催化研究进展[J]. 现代化工, 2017, 37(12): 24-27. doi: 10.16606/j.cnki.issn0253-4320.2017.12.006 [35] 张萍, 许丽, 王莉. 水热法合成二氧化钛纳米管的晶型与形貌控制的研究[J]. 当代化工, 2018, 47(5): 893-896. doi: 10.13840/j.cnki.cn21-1457/tq.2018.05.005 [36] YUAN Z Y, SU B L. Titanium oxide nanotubes, nanofibers and nanowires[J]. Colloids and Surfaces A:Physicochemical and Engineering Aspects, 2004, 241(3): 173-183. [37] 赵红花, 马树平. 负载型 TiO2光催化降解含酚废水的研究[J]. 兰州理工大学学报, 2007, 33(1): 74-78. [38] 朱杰. MnFeOx氧化物催化剂常温催化氧化低浓度HCHO性能研究[D]. 南京: 南京信息工程大学, 2022. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5635
- HTML全文浏览数: 5635
- PDF下载数: 150
- 施引文献: 0
