


 下载:
下载:



-
目前全球染料年产量超过700×104 t,染料品种已经超过10×104余种,常用染料有2 000种以上,而且每年人工合成的新型染料也层出不穷,各地的江河湖泊都受到不同程度的污染[1-2]。而且这些染料大多为酚类化合物、苯类化合物[3],其结构复杂、难以生物降解、对生态环境危害极大[4-5]。因此,处理有机废水中的染料大分子是当前必须解决的热点问题。目前常用于有机废水治理的方法主要有物理吸附法、化学法、生物处理法、膜分离技术等[6-7]。但是这些方法往往对系统条件要求苛刻,成本和能耗高,需要二次维护[8-9]。与上述处理方法相比,光催化技术具有操作简单、成本低廉、循环性好等优点[10-11]。
g-C3N4作为一种通过π-π共轭形成的可见光响应型催化剂,通过范德华力作用堆砌形成二维层状结构,与石墨烯的层状结构类似,其具有优异的光稳定性和热稳定性[12]、良好的生物相容性、合适的能带结构以及优异的光电转化性能,常用于光催化分解水制氢[13-15]、二氧化碳还原[16-17]、有机污染物降解[18-19]等。然而,g-C3N4也有不可避免的缺陷,如比表面积小导致对有机污染物的吸附性能差[20];光生电子-空穴对复合率高导致催化活性差等,严重限制了其对有机污染物的降解性能[21-22]。目前,有研究者通过制备三维多孔氮化碳改善了上述问题。LIU等[23]以三聚氰酸-三聚氰胺超分子和离子液体分别做为前体和模板,合成了三维多孔超薄g-C3N4纳米片。其中,3D多孔结构增大了g-C3N4的比表面积,暴露了更多的活性位点,超薄结构的纳米片降低了载流子传输距离,抑制了载流子的复合率。WANG等[24]在软模板P-123存在下对超分子前驱体进行水热处理,制备出由空心气泡组成的三维g-C3N4催化剂。硬模板法[25]在控制造孔孔径大小和孔径分布上具有明显优势,但脱除模板的过程中要用到强酸或强碱进行处理,容易使氮化碳的—N—、═NH和—NH2官能团发生质子化作用,最终破坏其缩聚结构,且模板脱除过程产生的废酸、废碱过多[26-27]。软模板法可选择大多数表面活性剂以及低沸点分子充当模板,但是实验过程中需要调控的因素很多,生成的孔不如硬模板法整齐,并且表面活性剂可能不随高温完全分解从而残留于样品表面[28]。与上述方法相比,NH4Cl作为气体模板辅助造孔,无需在后续过程中去除模板,实验操作步骤更加简便,且不容易改变g-C3N4的基本结构。此外,以NH4Cl为模板辅助制备多孔g-C3N4不仅有利于对污染物的吸附[29],还可在煅烧前驱体的过程中促进CN−的生成,进而抑制光生电子空穴对的复合,提高对有机污染物的降解性能。
本研究采用气体模板NH4Cl辅助制备出多孔g-C3N4,通过XRD、FT-IR、UV-vis和XPS等表征研究样品的化学结构和晶相结构,通过SEM和TEM表征样品的表面形貌和微观结构,通过光催化降解水中RhB评价样品的光催化性能和循环性能,利用荧光光谱仪、瞬态荧光光谱仪和电化学工作站研究光生电子与空穴的复合和迁移状况,最后通过自由基捕获实验和高分辨质谱仪测试分析其光催化降解机理,旨在为开发新型光催化剂和建立RhB的降解方法提供参考。
-
实验所用的药品均为分析级试剂,使用前无需纯化。三聚氰胺和RhB购自天津科密欧科技有限公司,NH4Cl购自上海麦克林生化科技有限公司,对苯醌购自上海麦克林生化科技有限公司,三乙醇胺购自天津市大茂化学试剂公司,无水乙醇和异丙醇购自天津富宇精细化工有限公司,去离子水由超纯水仪处理所得。
-
马弗炉(KSL-1400X-A1,合肥科晶材料技术有限公司),X射线衍射仪(D2 PHASER,德国Bruker公司),X射线光电子衍射仪(ESCALAB 250XI,美国Thermo Fisher Scientific Inc公司),场发射扫描电镜(SU5000,日本Hitachi公司),透射电子显微镜(FEI Tecnai G2 F20 S-TWIN,美国FEI公司),紫外-可见分光光度计(UV-2600,日本岛津公司),傅里叶红外光谱仪(INVENIO,德国Bruker公司),BET比表面积测试仪(Autosorb-IQ-II,陕西盖卓电子科技有限公司),荧光光谱仪(FS5,英国爱丁堡公司),电化学工作站(CHI600E,上海辰华仪器有限公司),氙灯光源(CEL-HXF300-T3,北京中教金源有限公司),总有机碳分析仪(vario TOC cube,Elemental仪器公司),高分辨质谱仪(Thermo Scientific Q Exactive,美国Thermo Scientific公司)。
-
1) g-C3N4的制备。以三聚氰胺为前驱体,通过高温煅烧法制备g-C3N4。取1.5 g三聚氰胺,用锡箔纸包裹后,置于有盖坩埚中,在马弗炉以550 ℃煅烧3 h。自然冷却所得黄色即为g-C3N4,研磨后进行光催化性能测试。
2) 多孔g-C3N4的制备。采用高温煅烧三聚氰胺和NH4Cl混合物的方法制备多孔g-C3N4。将1.5 g三聚氰胺分别与相应质量(0.675、0.75、0.825、0.9、0.975 g)的NH4Cl溶于50 mL去离子水中,磁力搅拌4 h,放入70 ℃烘箱中,蒸干水分。随后用锡箔纸包裹后,放入有盖的坩埚内,在马弗炉以550 ℃煅烧3 h,自然冷却所得黄色固体即为x%-PCN(x%表示前驱体中NH4Cl的质量分数),研磨后进行光催化性能测试。
-
1) 各项表征和测试。XRD测试:以Cu Kα为辐射源,扫描范围为2θ=5°~80°,扫描速度为2(°)·min−1。FT-IR测试:样品与KBr以1:100质量比混合研磨,压成半透明薄片测样,波数为400~4 000 cm−1。SEM测试:取少量样品贴于黑色导电胶上,真空喷金后测样。TEM测试:将样品溶于无水乙醇,超声混匀后滴在铜网上,装样测试。BET测试:样品以150 ℃脱气6 h。UV-vis测试:采用紫外-可见分光光度计,扫描波长为200~800 nm。PL测试:以波长为385 nm的激发光测量样品波长为390~640 nm的发射光谱。光电流-阻抗测试:采用三电极体系,0.1 g催化剂溶于10 mL溶剂(V水∶V无水乙醇=9∶1),超声后取上清液涂膜于FTO玻璃上,以此作为工作电极,参比电极为饱和甘汞电极,铂片电极为对电极,电解液为0.5 mol·L−1的Na2SO4溶液。高分辨质谱:取1.5 mL降解后RhB液体,甲醇为溶剂,EIS为离子源。
2) 催化剂降解性能测试。取0.1 g催化剂,加入100 mL质量浓度为30 mg·L−1的RhB溶液中,在黒暗环境下磁力搅拌20 min,以达到吸附-脱附平衡。随后用300 W氙灯模拟太阳光照射,每隔10 min取5 mL溶液,离心后取上清液,用UV-vis分光光度计测试吸光度。将使用过后的催化剂用水和乙醇洗至中性,烘干后按照上述步骤重复4次,以考察催化剂的稳定性。
-
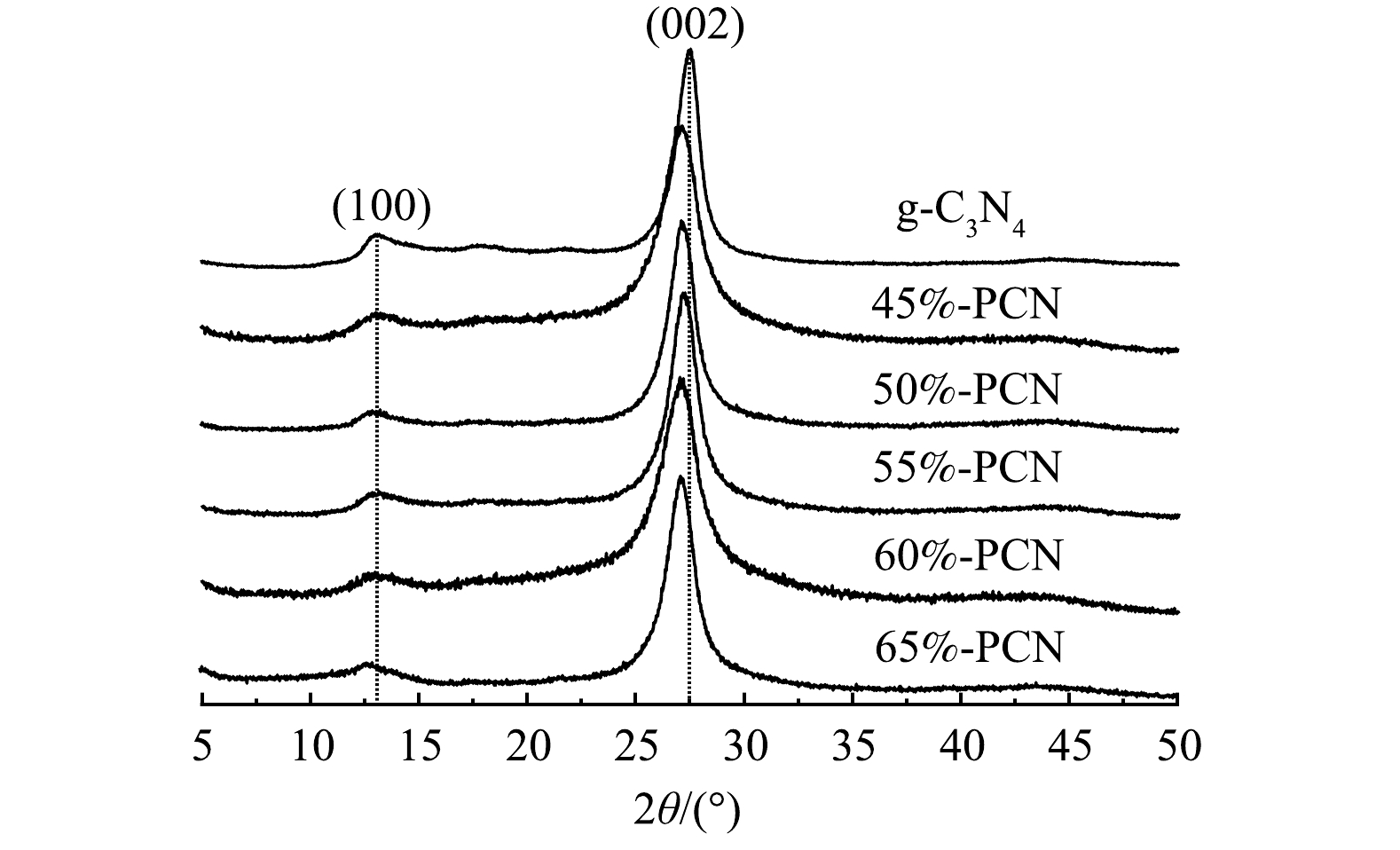
1) XRD分析。由图1可以看出,g-C3N4在13.5°和27.5°处有2个特征衍射峰,13.5°处的峰是面内三嗪环之间相互连接的特征峰,对应g-C3N4的(100)晶面,27.5°处的峰是环状芳香物层与层之间的堆积特征峰,对应g-C3N4的(002)晶面。与g-C3N4相比,PCN的2个特征峰均未发生明显改变,说明以气体模板法辅助制备PCN没有改变g-C3N4的晶相结构。此外,55%-PCN与g-C3N4相比,27.5°处的峰向左偏移至27.16°,根据布拉格公式(2dsinθ=nλ)[30]计算g-C3N4和55%-PCN的晶面层间距,分别为0.324 nm和0.329 nm,说明层间距变大。这可能是由于,三聚氰胺在高温下团聚脱氨的过程中,NH4Cl同时受热分解为NH3和HCl气体,导致生成的g-C3N4层与层之间的堆积作用变弱[31]。以上分析表明,气体模板的加入可以使g-C3N4片层与片层之间分离开来,从而进一步影响样品的性能。
2) 形貌分析。由图2(a)和图2(b)可以观察到g-C3N4整齐的片层堆叠,由图2(e)也可以观察到其表面光滑完整的大块结构。由图2(c)可以看出,55%-PCN的形貌与g-C3N4相比发生很大的改变,大片层破裂为小碎片,并且由图2(d)也可以看到样品表面产生很多孔径为50~100 nm的介孔。由图2(f)也可以看出孔的生成。
3) FT-IR分析。如图3所示,在g-C3N4的FT-IR光谱中,3 000~3 500 cm−1的宽峰为前驱体中未聚合的氨基(—NH2或═NH)的伸缩振动峰,1 200~1 640 cm−1对应于三嗪环间C—N和C═N键的特征峰,位于810 cm−1附近的吸收峰对应三嗪环的伸缩振动。与g-C3N4相比,PCN在1 200~1 600 cm−1和810 cm−1处的特征峰没有发生明显的变化,表明以NH4Cl为模板并未破坏g-C3N4的主体结构,在3 000~3 500 cm−1处的吸收峰变宽,可能是NH4Cl分解时的氨基与g-C3N4边缘位置未聚合的氨基或羟基结合,导致吸收范围变宽。值得注意的是,PCN在2 173 cm−1处出现1个明显的吸收峰,此为—C≡N的特征吸收峰[32]。并且随着前驱体中NH4Cl添加量的增多,峰强度越强,说明改性后的多孔结构有利于氰基的形成,使g-C3N4的面内形成更多的孔道结构。
4) UV-vis表征。如图4(a)所示,g-C3N4在波长小于470 nm处的蓝紫光和紫外光区吸收较强,可见光区的吸收较弱。55%-PCN在紫外光区和可见光区的吸收明显增强。此外,禁带宽度可利用UV-vis光谱数据、按照Kubelka-Munk函数由式(1)计算获得。
式中:α为吸光度;h代表普朗克常数;ν为频率;Eg为禁带宽度;A为常数。以(αhν)2为纵坐标,hν为横坐标作图,对所得曲线取切线,切线与横坐标的交点即为Eg,即所对应的样品的禁带宽度[33]。由图4(b)可以看出,55%-PCN的禁带宽度为2.73 eV,与g-C3N4禁带宽度(2.78 eV)相似。这说明改性后的PCN具有较强光吸收性能且带隙宽度变化不大。
5) XPS分析。由图5(a)可以看出,g-C3N4和55%-PCN的C1s、N1s、O1s峰均出现在285、400、520 eV左右,表明二者的元素组成一致。在图5(b)中,284.8、286.6、288.3 eV的3个峰分别是g-C3N4的C—C键、C—O键和N═C—N键的特征峰。这表明,以NH4Cl为模板,只在g-C3N4中产生了较多的孔道结构,没有影响到C的键合状态。在图5(c)中,g-C3N4和55%-PCN在398.7、399.5、401.1 eV处的3个峰,分别对应三嗪环内C═N—C键、环与环之间的H—N—(C)3键和末端氨基上的C—N—H键,表明N的键合状态也未受到气体模板的影响。由图5(d)可以看出,g-C3N4和55%-PCN的价带电位分别为+1.14 eV和+0.88 eV。采用文献中的方法[34]计算出g-C3N4和55%-PCN的导带电位,分别为−1.64 eV和−1.85 eV。这表明多孔结构不仅有利于提高g-C3N4的光吸收性能,还使导带上电子的还原电势更负。
6) BET分析。如图6(a)所示,二者的曲线趋势为Ⅳ型等温曲线,表明g-C3N4和55%-PCN均为中孔材料。另外,测试结果显示,55%-PCN的比表面积(28.548 m2·g−1)与g-C3N4(13.878 m2·g−1)相比有所提高,55%-PCN的孔体积(0.143 cm3·g−1)与g-C3N4(0.046 cm3·g−1)相比也有所提高。这表明PCN的表面性能得到了改善。由图6(b)可以看出:g-C3N4的孔径集中分布于5 nm左右;55%-PCN的孔径分布比较宽泛,增加了10 nm左右和10~80 nm孔道结构。这表明以NH4Cl为模板可增加g-C3N4的孔道结构,有利于提高催化剂对有机污染物的吸附性能。
-
1) 光致发光(PL)光谱和瞬态荧光光谱分析。PL峰强度越强表明光生电子空穴对的复合率越高。由图7(a)可以看出,55%-PCN的发射峰强度较g-C3N4明显降低,表明光生电子空穴对的复合率受到一定程度的限制,有利于光催化性能的提升。图7(b)是g-C3N4和55%-PCN的瞬态荧光光谱图。当荧光强度衰减到最大值的1/e时所用的时间为荧光寿命,即光生电子的平均寿命,其寿命越长,越有利于光催化性能的提升。可以看出,55%-PCN的平均寿命为1.42 ns,接近g-C3N4的7倍(0.21 ns),表明55%-PCN中载流子具有较长的寿命来参与光催化反应。氰基是一种常见的吸电子基团,可以更好地使55%-PCN表面的光生电子和空穴分离[32],提升载流子的寿命,最终影响其光催化性能。
2) 光电性能。虽然通过PCN的制备可以抑制光生电子空穴对的复合,但光生电子-空穴对参与到光催化反应的数量尚不清楚。采用瞬态光电流强度和电化学阻抗谱研究光生电子的迁移效率和迁移阻力,结果如图8所示。在图8(a)中,光电流强度越大,表示催化剂中的激发电子向导电玻璃表面的迁移效率越高,越有利于光催化性能的提升,PCN-55%的光电流强度最大,是g-C3N4的2.5倍,表明其电子迁移效率最高。电化学阻抗谱的圆弧半径越小,电阻越小,电荷转移效率更高效。由图8(b)同样可以看出,55%-PCN的电子迁移阻力最小,可提高光生电子向催化剂表面的迁移效率。综合以上分析,55%-PCN样品中的电子迁移阻力较小,有利于激发电子的迁移。
3) 光催化降解罗丹明B性能。在图9(a)中,−20~0 min表示催化剂在黑暗环境下对RhB的吸附过程,0~50 min表示光照下光催化降解RhB的过程。可以看出,不加催化剂时,RhB在光照50 min后的自去除率只有7.4%,g-C3N4的暗吸附率为2.8%,而45%-PCN、50%-PCN、55%-PCN、60%-PCN和65%-PCN的暗吸附率分别为5.2%、7.7%、8.9%、10.3%、11.5%。这表明,PCN中的孔道随气体模板NH4Cl的增加而增多,同时对RhB的吸附性能也越强。55%-PCN在50 min内即可将RhB完全降解,而在相同时间内g-C3N4的降解率只有50%左右。
为了更直观地比较样品在光照下降解RhB溶液的光催化性能,可根据拟合伪一级动力模型[35]计算反应的表观速率常数。光催化降解RhB的伪一级动力学速率常数和降解率关系如式(2)所示。
式中:k为表观速率常数,min−1;C0和C分别为RhB的初始质量浓度和光照时间t时的质量浓度,mg·L−1。由于0~40 min的降解数据更符合伪一级动力学方程,故选择此范围数据,根据文献中的方法[32]作图。以ln(C0/C)为纵坐标,t为横坐标做图,斜率为k,计算所得的k值标记于图9(b)中。g-C3N4的k值为1.23×10−2 min−1,55%-PCN的k值最大(6.07×10−2 min−1),是g-C3N4的5倍。
根据有机碳含量计算g-C3N4和55%-PCN降解RhB的TOC去除率,g-C3N4为85.7%,55%-PCN为95.8%(图9(c)和图9(d)),所以RhB有机物大分子基本被全部分解为无机物小分子。55%-PCN与g-C3N4相比,去除率升高,说明其可以更好地将RhB大分子吸附并降解。
-
以上研究表明,55%-PCN具有对RhB优异的吸附性能和降解性能,但催化剂的稳定性是限制其能否工业化应用的又一关键指标。由图10(a)可以看出,55%-PCN在相同循环时间内重复使用4次的降解率分别为97.4%、95.4%、93.6%、92.7%,对水中RhB的降解率未发生明显变化。由图10(b)可以看出,55%-PCN使用前后的XRD图谱出峰位置不变,说明反应前后样品的结构和化学组成也没有发生改变。以上均表明55%-PCN具有稳定的光催化活性。
-
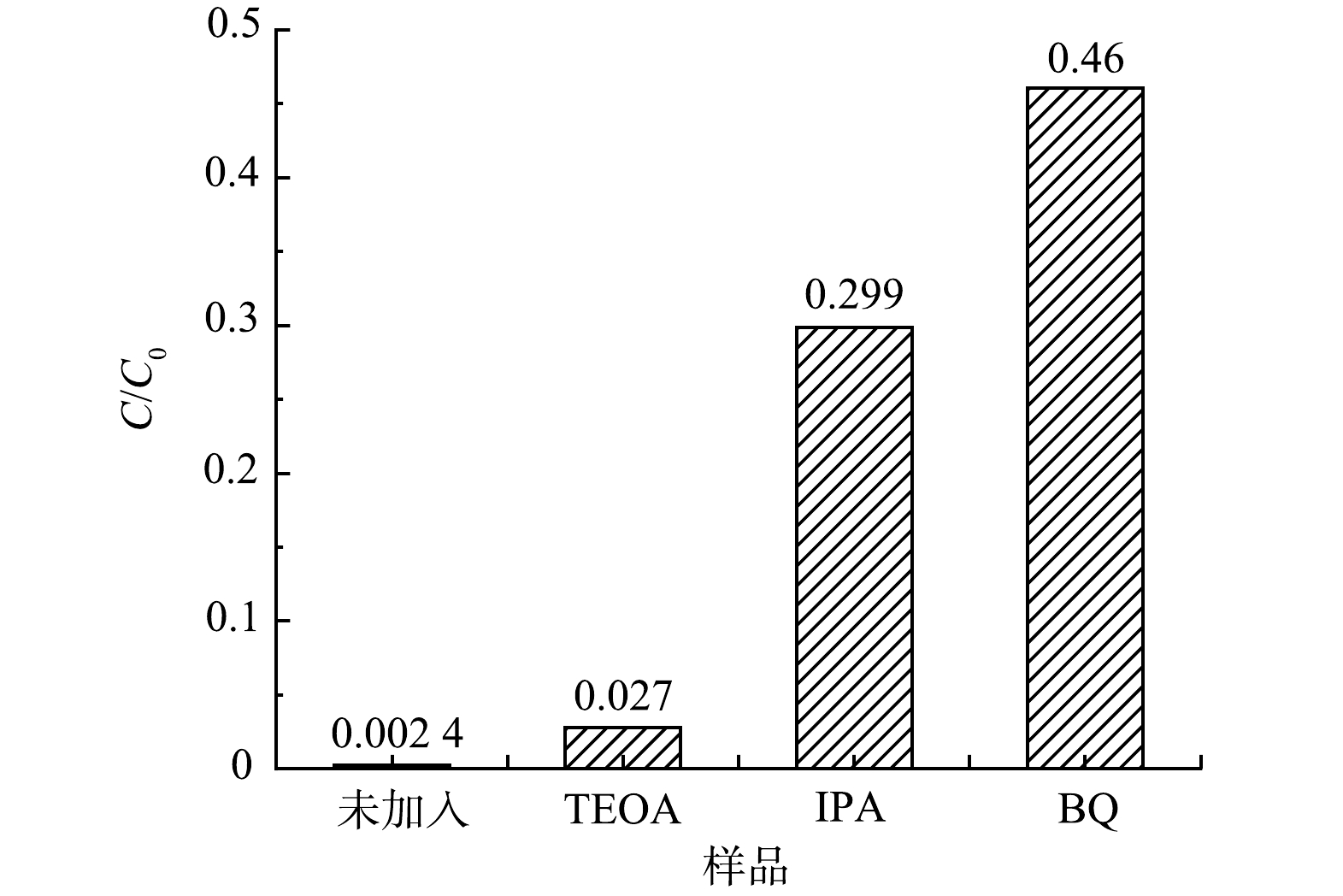
为了研究55%-PCN光催化降解RhB过程中的主要物种,分别用三乙醇胺(TEOA)、苯醌(BQ)和异丙醇(IPA)作为光催化降解体系中空穴(h+)、超氧自由基(·O2−)、羟基自由基(·OH)的捕获剂[36]来进行实验,结果如图11所示。当加入TEOA后,降解率未发生明显变化,即h+被捕获后催化活性变化不大,表明h+不是RhB降解的主要活性物种;在降解体系中加入IPA后,降解率下降明显,说明·OH对RhB的降解具有一定的促进作用;当加入BQ后,光降解效率明显下降,光催化活性被抑制的最明显,表明·O2−是RhB降解时最主要的活性物种。基于以上分析,在RhB降解时,最主要的活性物种是·O2−,其次是·OH,h+的活性可忽略不计。
本研究对降解后RhB溶液进行高分辨质谱测试,以探究55%-PCN光催化降解RhB的具体过程。在图12(a)中,m/z=444表示RhB的阳离子峰;m/z=416、388、359、331表示RhB脱去1、2、3、4个乙基的分子离子峰;m/z=399表示RhB脱去1个羧基的离子峰;m/z=302可能是脱去4个乙基和1个羧基后被1个·OH羟基化所得产物的离子峰;m/z=319可能是脱去4个乙基和1个羧基后被2个·OH羟基化获得产物的离子峰[37];推测其余离子峰也是各个大分子不断被·OH多次氧化后的产物。由图12(b)可以看出,m/z>150的各处离子峰强度都有一定程度上的降低,而m/z<150的各个离子峰强度明显升高。这说明55%-PCN光催化降解RhB的过程是一个随着光照时间的升高,RhB开始脱乙基和羧基,同时·OH不断进攻苯环使其断裂、氧化,最终经过多次氧化反应生成各个小分子的过程。
基于以上结果,推测55%-PCN光催化降解RhB的机理如图13所示。可以看出55%-PCN的价带主要由N原子的N2p轨道构成[32,38]。在光照射下,N原子上的电子充当电子供体被激发到导带生成光生e−,价带上留下大量h+(式(3))。导带上e−的还原电势较E(O2/·O2−)=−0.046 eV更负,与水中溶解的O2反应生成·O2−(式(4)),超氧自由基进一步反应生成过氧氢根(HOO·)和过氧化氢(H2O2)(式(5)和式(6)),接下来继续又被还原成具有强氧化性的·OH[32](式(7))。55%-PCN的孔结构给RhB的降解反应提供了更多边缘反应位点,RhB在脱乙基和脱羧基过程中不断与·OH接触,发生羟基化氧化反应。另外,氰基是常见的吸电子基团,常被用作电子受体[39]。当g-C3N4的结构中存在氰基时,氰基与相邻的N原子之间会形成局部分子内供体-受体(D-A)体系。D-A体系可以有效转移分子内的电荷,使光生e−和h+分离开来,从而降低载流子重组率[40]。故55%-PCN中的氰基的引入进一步给光催化降解RhB提供更多的活性位点,最终使RhB被氧化为NH4+、CO2、H2O等小分子。
-
1) 气体模板NH4Cl的引入可以在不破坏g-C3N4基本结构的基础上制备PCN,表面介孔数量增多,大大增加其比表面积和孔体积,给后续反应提供更多的活性位点。同时有利于提高吸光性能,经过测试其可见光吸收范围相比g-C3N4变宽。
2) 当前驱体中NH4Cl添加量为55%时所制备样品降解性能最佳。55%-PCN在50 min内对RhB的降解率为98%,伪一级动力学速率常数为6.07×10−2 min−1,是g-C3N4的5倍;并且其对RhB的TOC去除率比g-C3N4高,说明其可以更好地将RhB大分子吸附并降解。
3) 自由基捕获实验表明,在RhB降解时,·O2−和·OH参与了反应,在反应中,·O2−为最主要的活性物种。吸电子基团氰基与相邻的N原子之间会形成局部分子内D-A体系,使光生电子和空穴更好地分离,增加载流子寿命。55%-PCN的孔结构以及氰基的引入,均给RhB的降解反应提供了更多反应活性位点,在脱乙基和羧基过程中不断与·OH发生氧化反应,最终产物为NH4+、CO2、H2O等小分子。
基于气体模板法制备的多孔g-C3N4对罗丹明B的降解效果及机理
Degradation effect and mechanism of rhodamine B by porous g-C3N4 prepared by gas template method
-
摘要: 为了处理废水中的染料大分子有机污染物,以NH4Cl为气体模板,通过高温煅烧法制备多孔氮化碳(PCN),使用XRD、SEM、TEM、FT-IR、UV-vis、XPS和BET等分析方法对催化剂进行了表征,并以水中罗丹明B(RhB)的降解率为评价标准,考察了样品的光催化性能和活性物种。结果表明:PCN在不改变氮化碳(g-C3N4)基本结构的基础上显著提高其光吸收能力和光催化活性;与g-C3N4相比,当前驱体中NH4Cl添加量为55%时,样品的比表面积由13.878 m2·g−1增至28.548 m2·g−1,TOC去除率由85.7%增至95.8%,降解速率和光电流密度分别是g-C3N4的2倍和2.5倍;在光催化降解RhB的过程中,·O2−是起主要作用的活性物种。多孔结构有利于提高比表面积以吸附更多的有机物大分子,并且为光催化反应提供更多的活性位点。煅烧过程中氰基的生成减少了光生电子和空穴的复合,载流子的寿命变长,二者的共同作用使PCN的光催化性能大幅提升。本研究成果可为开发新型光催化剂和建立RhB的降解方法提供参考。Abstract: In order to treat the dye macromolecular organic pollutants in wastewater, porous g-C3N4 (PCN) was prepared by the high temperature calcination method using NH4Cl as gas template. The photocatalysts were characterized by XRD, SEM, TEM, FT-IR, UV-vis, XPS, BET and other analytical methods. Photocatalytic performance and active species were investigated based on the degradation rate of rhodamine B (RhB) in water. The results showed that PCN could greatly enhance light absorption ability and photocatalytic activity of g-C3N4 without changing its basic structure. Compared with g-C3N4, when the addition amount of NH4Cl in precursor was 55%, the specific surface area of PCN sample increased from 13.878 m2·g−1 to 28.548 m2·g−1, TOC removal rate increased from 85.7% to 95.8%, degradation rate and photocurrent density were 2 times, 2.5 times of g-C3N4, respectively. In the process of photocatalytic degradation of RhB, ·O2− was the main active species. Porous structure was beneficial to increase specific surface area, adsorb more organic macromolecules and provide more active sites for photocatalytic reactions. The generation of cyano groups during calcination process reduced recombination of photogenerated electrons and holes, and extended the lifetime of carrier. Their combined effect could greatly improve the photocatalytic performance of PCN. The results of this study can provide a reference for the development of new photocatalysts and the establishment of RhB degradation methods.
-
Key words:
- g-C3N4 /
- gas template /
- porous /
- photocatalytic degradation /
- rhodamine B(RhB)
-
目前全球染料年产量超过700×104 t,染料品种已经超过10×104余种,常用染料有2 000种以上,而且每年人工合成的新型染料也层出不穷,各地的江河湖泊都受到不同程度的污染[1-2]。而且这些染料大多为酚类化合物、苯类化合物[3],其结构复杂、难以生物降解、对生态环境危害极大[4-5]。因此,处理有机废水中的染料大分子是当前必须解决的热点问题。目前常用于有机废水治理的方法主要有物理吸附法、化学法、生物处理法、膜分离技术等[6-7]。但是这些方法往往对系统条件要求苛刻,成本和能耗高,需要二次维护[8-9]。与上述处理方法相比,光催化技术具有操作简单、成本低廉、循环性好等优点[10-11]。
g-C3N4作为一种通过π-π共轭形成的可见光响应型催化剂,通过范德华力作用堆砌形成二维层状结构,与石墨烯的层状结构类似,其具有优异的光稳定性和热稳定性[12]、良好的生物相容性、合适的能带结构以及优异的光电转化性能,常用于光催化分解水制氢[13-15]、二氧化碳还原[16-17]、有机污染物降解[18-19]等。然而,g-C3N4也有不可避免的缺陷,如比表面积小导致对有机污染物的吸附性能差[20];光生电子-空穴对复合率高导致催化活性差等,严重限制了其对有机污染物的降解性能[21-22]。目前,有研究者通过制备三维多孔氮化碳改善了上述问题。LIU等[23]以三聚氰酸-三聚氰胺超分子和离子液体分别做为前体和模板,合成了三维多孔超薄g-C3N4纳米片。其中,3D多孔结构增大了g-C3N4的比表面积,暴露了更多的活性位点,超薄结构的纳米片降低了载流子传输距离,抑制了载流子的复合率。WANG等[24]在软模板P-123存在下对超分子前驱体进行水热处理,制备出由空心气泡组成的三维g-C3N4催化剂。硬模板法[25]在控制造孔孔径大小和孔径分布上具有明显优势,但脱除模板的过程中要用到强酸或强碱进行处理,容易使氮化碳的—N—、═NH和—NH2官能团发生质子化作用,最终破坏其缩聚结构,且模板脱除过程产生的废酸、废碱过多[26-27]。软模板法可选择大多数表面活性剂以及低沸点分子充当模板,但是实验过程中需要调控的因素很多,生成的孔不如硬模板法整齐,并且表面活性剂可能不随高温完全分解从而残留于样品表面[28]。与上述方法相比,NH4Cl作为气体模板辅助造孔,无需在后续过程中去除模板,实验操作步骤更加简便,且不容易改变g-C3N4的基本结构。此外,以NH4Cl为模板辅助制备多孔g-C3N4不仅有利于对污染物的吸附[29],还可在煅烧前驱体的过程中促进CN−的生成,进而抑制光生电子空穴对的复合,提高对有机污染物的降解性能。
本研究采用气体模板NH4Cl辅助制备出多孔g-C3N4,通过XRD、FT-IR、UV-vis和XPS等表征研究样品的化学结构和晶相结构,通过SEM和TEM表征样品的表面形貌和微观结构,通过光催化降解水中RhB评价样品的光催化性能和循环性能,利用荧光光谱仪、瞬态荧光光谱仪和电化学工作站研究光生电子与空穴的复合和迁移状况,最后通过自由基捕获实验和高分辨质谱仪测试分析其光催化降解机理,旨在为开发新型光催化剂和建立RhB的降解方法提供参考。
1. 材料与方法
1.1 实验原料
实验所用的药品均为分析级试剂,使用前无需纯化。三聚氰胺和RhB购自天津科密欧科技有限公司,NH4Cl购自上海麦克林生化科技有限公司,对苯醌购自上海麦克林生化科技有限公司,三乙醇胺购自天津市大茂化学试剂公司,无水乙醇和异丙醇购自天津富宇精细化工有限公司,去离子水由超纯水仪处理所得。
1.2 实验仪器
马弗炉(KSL-1400X-A1,合肥科晶材料技术有限公司),X射线衍射仪(D2 PHASER,德国Bruker公司),X射线光电子衍射仪(ESCALAB 250XI,美国Thermo Fisher Scientific Inc公司),场发射扫描电镜(SU5000,日本Hitachi公司),透射电子显微镜(FEI Tecnai G2 F20 S-TWIN,美国FEI公司),紫外-可见分光光度计(UV-2600,日本岛津公司),傅里叶红外光谱仪(INVENIO,德国Bruker公司),BET比表面积测试仪(Autosorb-IQ-II,陕西盖卓电子科技有限公司),荧光光谱仪(FS5,英国爱丁堡公司),电化学工作站(CHI600E,上海辰华仪器有限公司),氙灯光源(CEL-HXF300-T3,北京中教金源有限公司),总有机碳分析仪(vario TOC cube,Elemental仪器公司),高分辨质谱仪(Thermo Scientific Q Exactive,美国Thermo Scientific公司)。
1.3 实验方法
1) g-C3N4的制备。以三聚氰胺为前驱体,通过高温煅烧法制备g-C3N4。取1.5 g三聚氰胺,用锡箔纸包裹后,置于有盖坩埚中,在马弗炉以550 ℃煅烧3 h。自然冷却所得黄色即为g-C3N4,研磨后进行光催化性能测试。
2) 多孔g-C3N4的制备。采用高温煅烧三聚氰胺和NH4Cl混合物的方法制备多孔g-C3N4。将1.5 g三聚氰胺分别与相应质量(0.675、0.75、0.825、0.9、0.975 g)的NH4Cl溶于50 mL去离子水中,磁力搅拌4 h,放入70 ℃烘箱中,蒸干水分。随后用锡箔纸包裹后,放入有盖的坩埚内,在马弗炉以550 ℃煅烧3 h,自然冷却所得黄色固体即为x%-PCN(x%表示前驱体中NH4Cl的质量分数),研磨后进行光催化性能测试。
1.4 表征和性能测试
1) 各项表征和测试。XRD测试:以Cu Kα为辐射源,扫描范围为2θ=5°~80°,扫描速度为2(°)·min−1。FT-IR测试:样品与KBr以1:100质量比混合研磨,压成半透明薄片测样,波数为400~4 000 cm−1。SEM测试:取少量样品贴于黑色导电胶上,真空喷金后测样。TEM测试:将样品溶于无水乙醇,超声混匀后滴在铜网上,装样测试。BET测试:样品以150 ℃脱气6 h。UV-vis测试:采用紫外-可见分光光度计,扫描波长为200~800 nm。PL测试:以波长为385 nm的激发光测量样品波长为390~640 nm的发射光谱。光电流-阻抗测试:采用三电极体系,0.1 g催化剂溶于10 mL溶剂(V水∶V无水乙醇=9∶1),超声后取上清液涂膜于FTO玻璃上,以此作为工作电极,参比电极为饱和甘汞电极,铂片电极为对电极,电解液为0.5 mol·L−1的Na2SO4溶液。高分辨质谱:取1.5 mL降解后RhB液体,甲醇为溶剂,EIS为离子源。
2) 催化剂降解性能测试。取0.1 g催化剂,加入100 mL质量浓度为30 mg·L−1的RhB溶液中,在黒暗环境下磁力搅拌20 min,以达到吸附-脱附平衡。随后用300 W氙灯模拟太阳光照射,每隔10 min取5 mL溶液,离心后取上清液,用UV-vis分光光度计测试吸光度。将使用过后的催化剂用水和乙醇洗至中性,烘干后按照上述步骤重复4次,以考察催化剂的稳定性。
2. 结果与讨论
2.1 催化剂表征
1) XRD分析。由图1可以看出,g-C3N4在13.5°和27.5°处有2个特征衍射峰,13.5°处的峰是面内三嗪环之间相互连接的特征峰,对应g-C3N4的(100)晶面,27.5°处的峰是环状芳香物层与层之间的堆积特征峰,对应g-C3N4的(002)晶面。与g-C3N4相比,PCN的2个特征峰均未发生明显改变,说明以气体模板法辅助制备PCN没有改变g-C3N4的晶相结构。此外,55%-PCN与g-C3N4相比,27.5°处的峰向左偏移至27.16°,根据布拉格公式(2dsinθ=nλ)[30]计算g-C3N4和55%-PCN的晶面层间距,分别为0.324 nm和0.329 nm,说明层间距变大。这可能是由于,三聚氰胺在高温下团聚脱氨的过程中,NH4Cl同时受热分解为NH3和HCl气体,导致生成的g-C3N4层与层之间的堆积作用变弱[31]。以上分析表明,气体模板的加入可以使g-C3N4片层与片层之间分离开来,从而进一步影响样品的性能。
2) 形貌分析。由图2(a)和图2(b)可以观察到g-C3N4整齐的片层堆叠,由图2(e)也可以观察到其表面光滑完整的大块结构。由图2(c)可以看出,55%-PCN的形貌与g-C3N4相比发生很大的改变,大片层破裂为小碎片,并且由图2(d)也可以看到样品表面产生很多孔径为50~100 nm的介孔。由图2(f)也可以看出孔的生成。
3) FT-IR分析。如图3所示,在g-C3N4的FT-IR光谱中,3 000~3 500 cm−1的宽峰为前驱体中未聚合的氨基(—NH2或═NH)的伸缩振动峰,1 200~1 640 cm−1对应于三嗪环间C—N和C═N键的特征峰,位于810 cm−1附近的吸收峰对应三嗪环的伸缩振动。与g-C3N4相比,PCN在1 200~1 600 cm−1和810 cm−1处的特征峰没有发生明显的变化,表明以NH4Cl为模板并未破坏g-C3N4的主体结构,在3 000~3 500 cm−1处的吸收峰变宽,可能是NH4Cl分解时的氨基与g-C3N4边缘位置未聚合的氨基或羟基结合,导致吸收范围变宽。值得注意的是,PCN在2 173 cm−1处出现1个明显的吸收峰,此为—C≡N的特征吸收峰[32]。并且随着前驱体中NH4Cl添加量的增多,峰强度越强,说明改性后的多孔结构有利于氰基的形成,使g-C3N4的面内形成更多的孔道结构。
4) UV-vis表征。如图4(a)所示,g-C3N4在波长小于470 nm处的蓝紫光和紫外光区吸收较强,可见光区的吸收较弱。55%-PCN在紫外光区和可见光区的吸收明显增强。此外,禁带宽度可利用UV-vis光谱数据、按照Kubelka-Munk函数由式(1)计算获得。
 图 4 g-C3N4和PCN的UV-vis吸收光谱和带隙图Figure 4. UV-vis absorption spectra and band gap diagrams of g-C3N4 and PCN
图 4 g-C3N4和PCN的UV-vis吸收光谱和带隙图Figure 4. UV-vis absorption spectra and band gap diagrams of g-C3N4 and PCNαhν=A(hν−Eg)1/2 (1) 式中:α为吸光度;h代表普朗克常数;ν为频率;Eg为禁带宽度;A为常数。以(αhν)2为纵坐标,hν为横坐标作图,对所得曲线取切线,切线与横坐标的交点即为Eg,即所对应的样品的禁带宽度[33]。由图4(b)可以看出,55%-PCN的禁带宽度为2.73 eV,与g-C3N4禁带宽度(2.78 eV)相似。这说明改性后的PCN具有较强光吸收性能且带隙宽度变化不大。
5) XPS分析。由图5(a)可以看出,g-C3N4和55%-PCN的C1s、N1s、O1s峰均出现在285、400、520 eV左右,表明二者的元素组成一致。在图5(b)中,284.8、286.6、288.3 eV的3个峰分别是g-C3N4的C—C键、C—O键和N═C—N键的特征峰。这表明,以NH4Cl为模板,只在g-C3N4中产生了较多的孔道结构,没有影响到C的键合状态。在图5(c)中,g-C3N4和55%-PCN在398.7、399.5、401.1 eV处的3个峰,分别对应三嗪环内C═N—C键、环与环之间的H—N—(C)3键和末端氨基上的C—N—H键,表明N的键合状态也未受到气体模板的影响。由图5(d)可以看出,g-C3N4和55%-PCN的价带电位分别为+1.14 eV和+0.88 eV。采用文献中的方法[34]计算出g-C3N4和55%-PCN的导带电位,分别为−1.64 eV和−1.85 eV。这表明多孔结构不仅有利于提高g-C3N4的光吸收性能,还使导带上电子的还原电势更负。
6) BET分析。如图6(a)所示,二者的曲线趋势为Ⅳ型等温曲线,表明g-C3N4和55%-PCN均为中孔材料。另外,测试结果显示,55%-PCN的比表面积(28.548 m2·g−1)与g-C3N4(13.878 m2·g−1)相比有所提高,55%-PCN的孔体积(0.143 cm3·g−1)与g-C3N4(0.046 cm3·g−1)相比也有所提高。这表明PCN的表面性能得到了改善。由图6(b)可以看出:g-C3N4的孔径集中分布于5 nm左右;55%-PCN的孔径分布比较宽泛,增加了10 nm左右和10~80 nm孔道结构。这表明以NH4Cl为模板可增加g-C3N4的孔道结构,有利于提高催化剂对有机污染物的吸附性能。
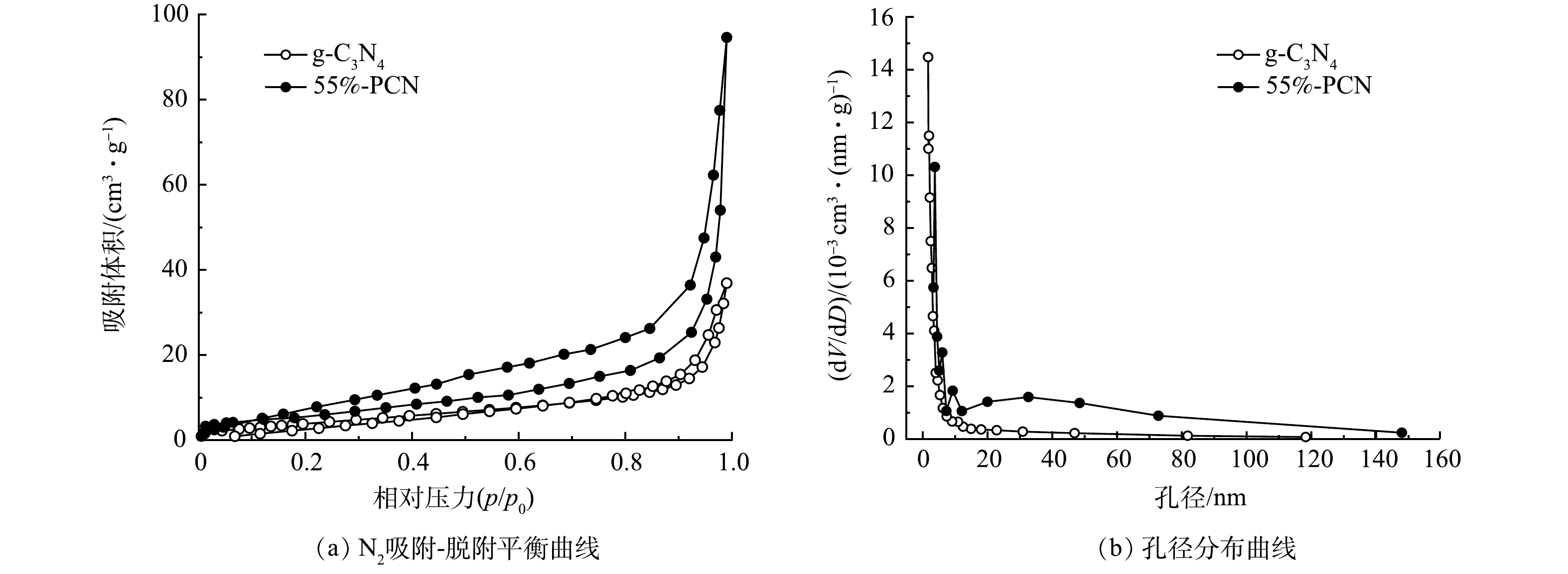 图 6 g-C3N4和55%-PCN的氮气吸附-脱附平衡曲线和孔径分布曲线Figure 6. N2 adsorption-desorption curves and pore size distribution curves of g-C3N4 and 55%-PCN
图 6 g-C3N4和55%-PCN的氮气吸附-脱附平衡曲线和孔径分布曲线Figure 6. N2 adsorption-desorption curves and pore size distribution curves of g-C3N4 and 55%-PCN2.2 性能分析
1) 光致发光(PL)光谱和瞬态荧光光谱分析。PL峰强度越强表明光生电子空穴对的复合率越高。由图7(a)可以看出,55%-PCN的发射峰强度较g-C3N4明显降低,表明光生电子空穴对的复合率受到一定程度的限制,有利于光催化性能的提升。图7(b)是g-C3N4和55%-PCN的瞬态荧光光谱图。当荧光强度衰减到最大值的1/e时所用的时间为荧光寿命,即光生电子的平均寿命,其寿命越长,越有利于光催化性能的提升。可以看出,55%-PCN的平均寿命为1.42 ns,接近g-C3N4的7倍(0.21 ns),表明55%-PCN中载流子具有较长的寿命来参与光催化反应。氰基是一种常见的吸电子基团,可以更好地使55%-PCN表面的光生电子和空穴分离[32],提升载流子的寿命,最终影响其光催化性能。
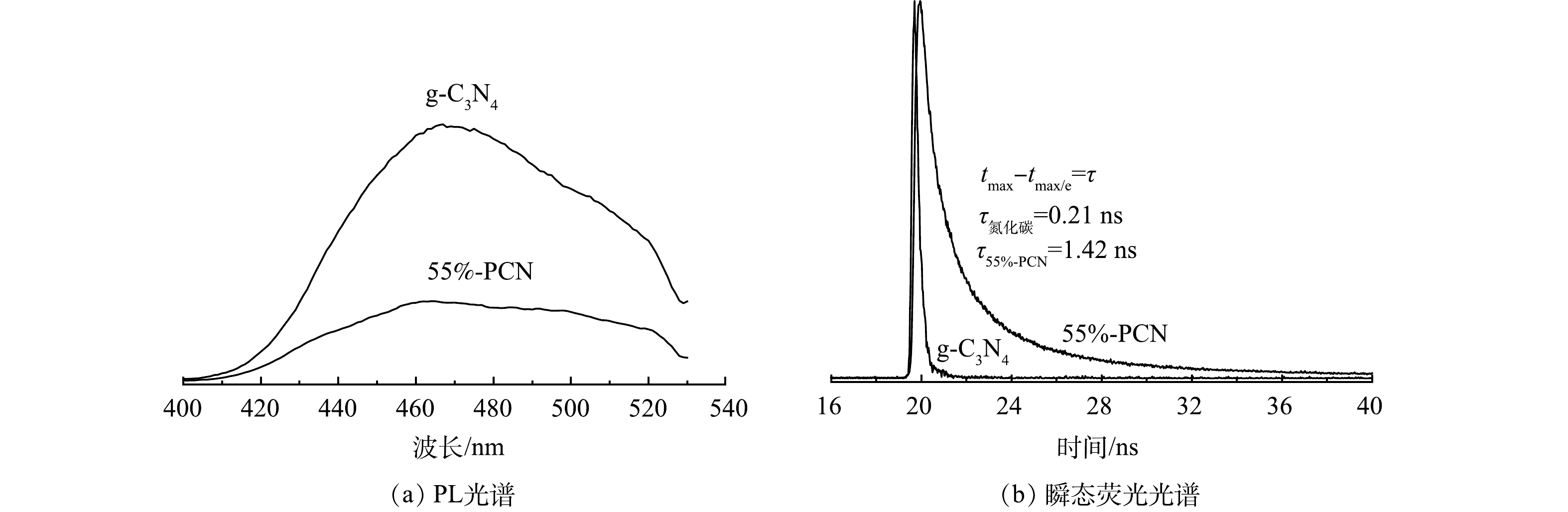 图 7 g-C3N4和55%-PCN的PL光谱和瞬态荧光光谱Figure 7. Photoluminescence (PL) spectra and fluorescence lifetime spectra in g-C3N4 and 55%-PCN
图 7 g-C3N4和55%-PCN的PL光谱和瞬态荧光光谱Figure 7. Photoluminescence (PL) spectra and fluorescence lifetime spectra in g-C3N4 and 55%-PCN2) 光电性能。虽然通过PCN的制备可以抑制光生电子空穴对的复合,但光生电子-空穴对参与到光催化反应的数量尚不清楚。采用瞬态光电流强度和电化学阻抗谱研究光生电子的迁移效率和迁移阻力,结果如图8所示。在图8(a)中,光电流强度越大,表示催化剂中的激发电子向导电玻璃表面的迁移效率越高,越有利于光催化性能的提升,PCN-55%的光电流强度最大,是g-C3N4的2.5倍,表明其电子迁移效率最高。电化学阻抗谱的圆弧半径越小,电阻越小,电荷转移效率更高效。由图8(b)同样可以看出,55%-PCN的电子迁移阻力最小,可提高光生电子向催化剂表面的迁移效率。综合以上分析,55%-PCN样品中的电子迁移阻力较小,有利于激发电子的迁移。
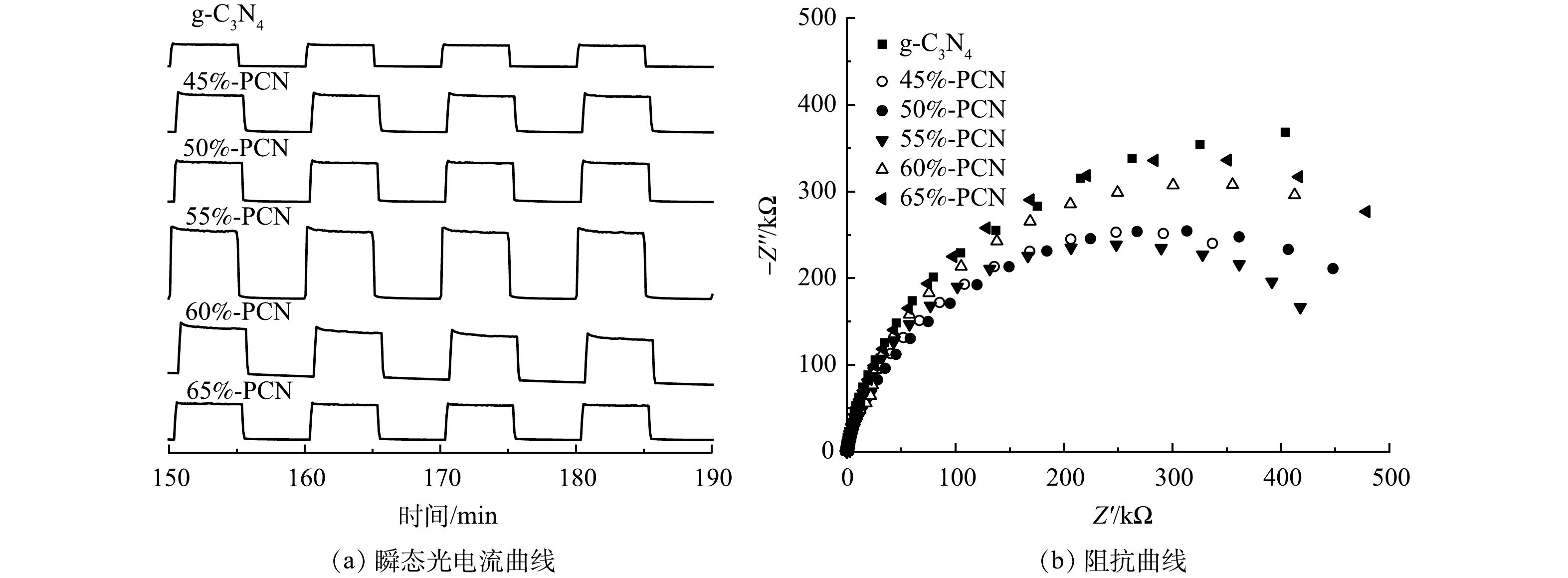 图 8 g-C3N4和PCN的光电流曲线和阻抗曲线Figure 8. Photocurrent curves and impedance curves of g-C3N4, PCN
图 8 g-C3N4和PCN的光电流曲线和阻抗曲线Figure 8. Photocurrent curves and impedance curves of g-C3N4, PCN3) 光催化降解罗丹明B性能。在图9(a)中,−20~0 min表示催化剂在黑暗环境下对RhB的吸附过程,0~50 min表示光照下光催化降解RhB的过程。可以看出,不加催化剂时,RhB在光照50 min后的自去除率只有7.4%,g-C3N4的暗吸附率为2.8%,而45%-PCN、50%-PCN、55%-PCN、60%-PCN和65%-PCN的暗吸附率分别为5.2%、7.7%、8.9%、10.3%、11.5%。这表明,PCN中的孔道随气体模板NH4Cl的增加而增多,同时对RhB的吸附性能也越强。55%-PCN在50 min内即可将RhB完全降解,而在相同时间内g-C3N4的降解率只有50%左右。
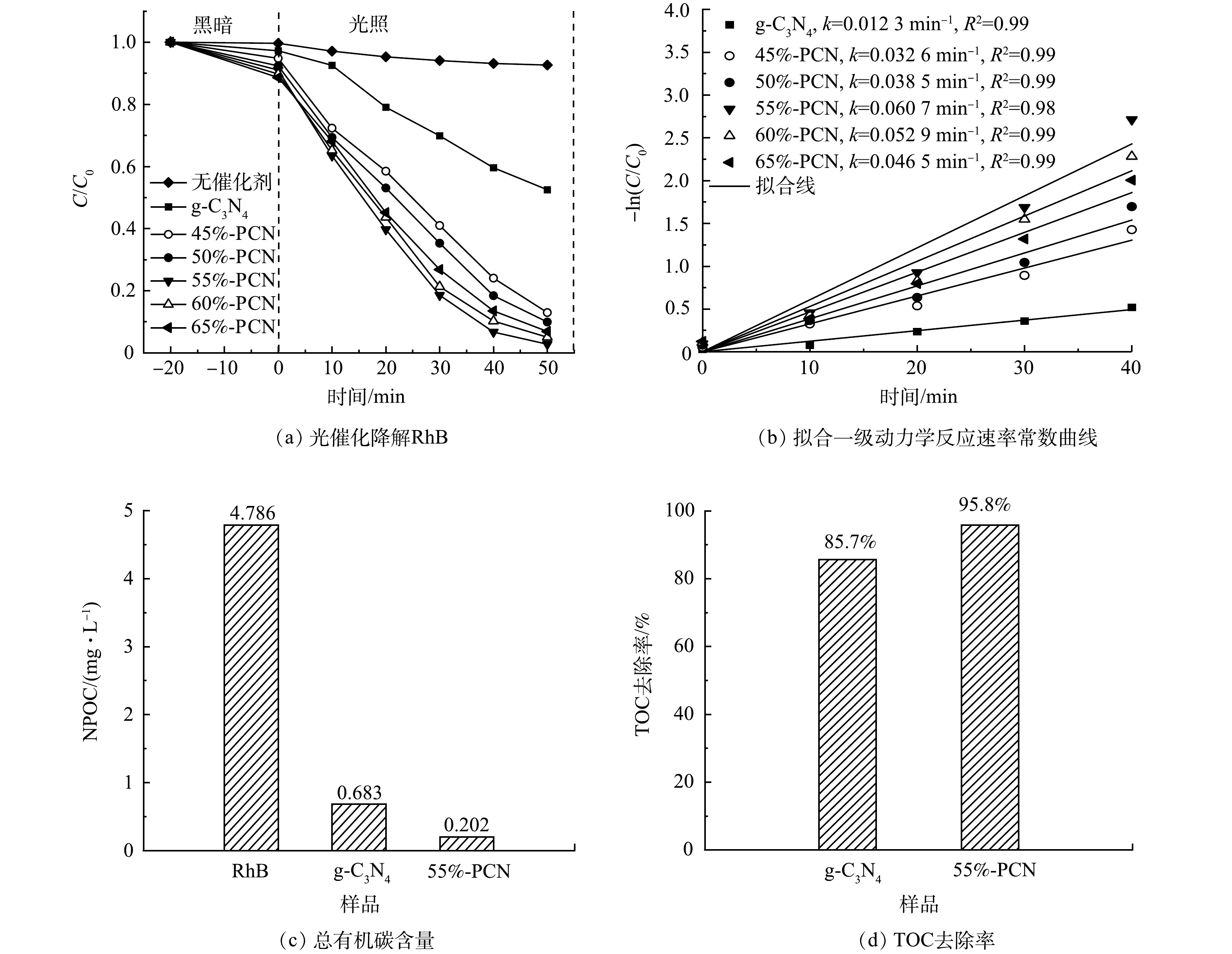 图 9 g-C3N4和PCN对RhB降解效果的对比Figure 9. Comparison of the degradation effects on RhB of g-C3N4 and PCN
图 9 g-C3N4和PCN对RhB降解效果的对比Figure 9. Comparison of the degradation effects on RhB of g-C3N4 and PCN为了更直观地比较样品在光照下降解RhB溶液的光催化性能,可根据拟合伪一级动力模型[35]计算反应的表观速率常数。光催化降解RhB的伪一级动力学速率常数和降解率关系如式(2)所示。
lnCC0=−kt (2) 式中:k为表观速率常数,min−1;C0和C分别为RhB的初始质量浓度和光照时间t时的质量浓度,mg·L−1。由于0~40 min的降解数据更符合伪一级动力学方程,故选择此范围数据,根据文献中的方法[32]作图。以ln(C0/C)为纵坐标,t为横坐标做图,斜率为k,计算所得的k值标记于图9(b)中。g-C3N4的k值为1.23×10−2 min−1,55%-PCN的k值最大(6.07×10−2 min−1),是g-C3N4的5倍。
根据有机碳含量计算g-C3N4和55%-PCN降解RhB的TOC去除率,g-C3N4为85.7%,55%-PCN为95.8%(图9(c)和图9(d)),所以RhB有机物大分子基本被全部分解为无机物小分子。55%-PCN与g-C3N4相比,去除率升高,说明其可以更好地将RhB大分子吸附并降解。
2.3 催化剂稳定性分析
以上研究表明,55%-PCN具有对RhB优异的吸附性能和降解性能,但催化剂的稳定性是限制其能否工业化应用的又一关键指标。由图10(a)可以看出,55%-PCN在相同循环时间内重复使用4次的降解率分别为97.4%、95.4%、93.6%、92.7%,对水中RhB的降解率未发生明显变化。由图10(b)可以看出,55%-PCN使用前后的XRD图谱出峰位置不变,说明反应前后样品的结构和化学组成也没有发生改变。以上均表明55%-PCN具有稳定的光催化活性。
 图 10 55%-PCN光催化降解RhB的稳定性Figure 10. Stability of 55%-PCN photocatalytic degradation on RhB
图 10 55%-PCN光催化降解RhB的稳定性Figure 10. Stability of 55%-PCN photocatalytic degradation on RhB2.4 光催化机理分析
为了研究55%-PCN光催化降解RhB过程中的主要物种,分别用三乙醇胺(TEOA)、苯醌(BQ)和异丙醇(IPA)作为光催化降解体系中空穴(h+)、超氧自由基(·O2−)、羟基自由基(·OH)的捕获剂[36]来进行实验,结果如图11所示。当加入TEOA后,降解率未发生明显变化,即h+被捕获后催化活性变化不大,表明h+不是RhB降解的主要活性物种;在降解体系中加入IPA后,降解率下降明显,说明·OH对RhB的降解具有一定的促进作用;当加入BQ后,光降解效率明显下降,光催化活性被抑制的最明显,表明·O2−是RhB降解时最主要的活性物种。基于以上分析,在RhB降解时,最主要的活性物种是·O2−,其次是·OH,h+的活性可忽略不计。
本研究对降解后RhB溶液进行高分辨质谱测试,以探究55%-PCN光催化降解RhB的具体过程。在图12(a)中,m/z=444表示RhB的阳离子峰;m/z=416、388、359、331表示RhB脱去1、2、3、4个乙基的分子离子峰;m/z=399表示RhB脱去1个羧基的离子峰;m/z=302可能是脱去4个乙基和1个羧基后被1个·OH羟基化所得产物的离子峰;m/z=319可能是脱去4个乙基和1个羧基后被2个·OH羟基化获得产物的离子峰[37];推测其余离子峰也是各个大分子不断被·OH多次氧化后的产物。由图12(b)可以看出,m/z>150的各处离子峰强度都有一定程度上的降低,而m/z<150的各个离子峰强度明显升高。这说明55%-PCN光催化降解RhB的过程是一个随着光照时间的升高,RhB开始脱乙基和羧基,同时·OH不断进攻苯环使其断裂、氧化,最终经过多次氧化反应生成各个小分子的过程。
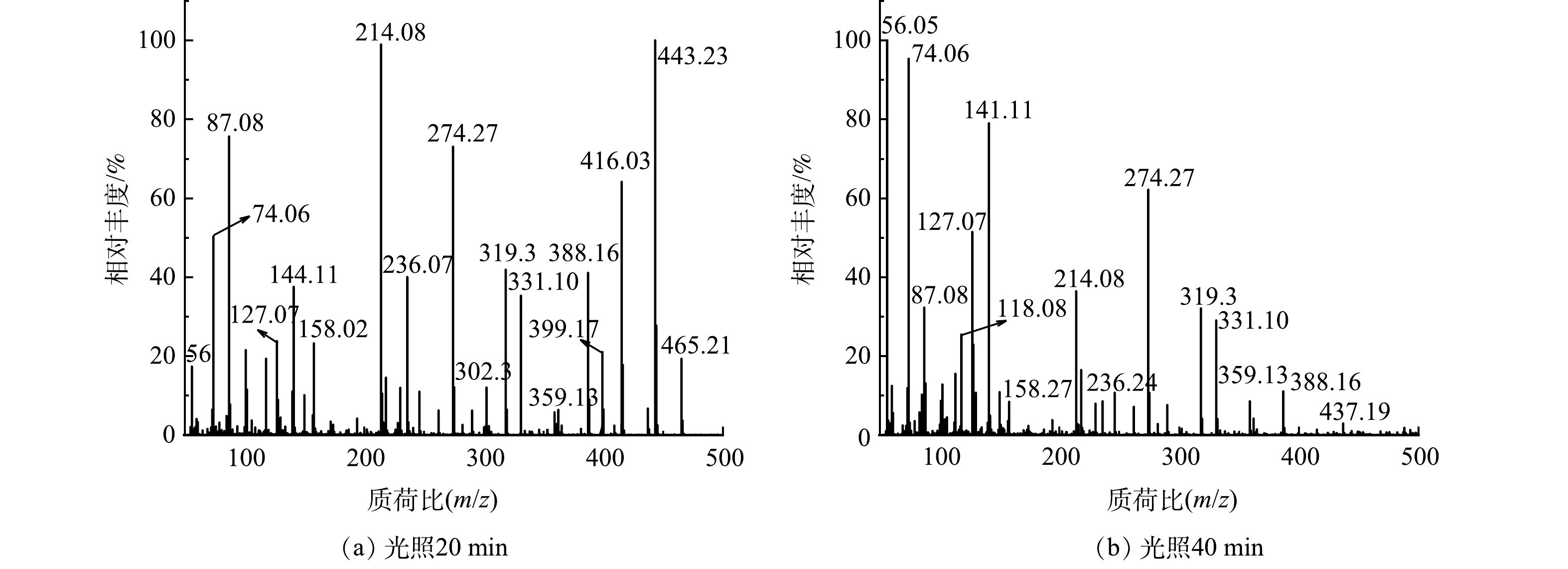 图 12 55%-PCN光催化降解RhB的质谱图Figure 12. Mass spectra of photocatalytic degradation on RhB by 55%-PCN
图 12 55%-PCN光催化降解RhB的质谱图Figure 12. Mass spectra of photocatalytic degradation on RhB by 55%-PCN基于以上结果,推测55%-PCN光催化降解RhB的机理如图13所示。可以看出55%-PCN的价带主要由N原子的N2p轨道构成[32,38]。在光照射下,N原子上的电子充当电子供体被激发到导带生成光生e−,价带上留下大量h+(式(3))。导带上e−的还原电势较E(O2/·O2−)=−0.046 eV更负,与水中溶解的O2反应生成·O2−(式(4)),超氧自由基进一步反应生成过氧氢根(HOO·)和过氧化氢(H2O2)(式(5)和式(6)),接下来继续又被还原成具有强氧化性的·OH[32](式(7))。55%-PCN的孔结构给RhB的降解反应提供了更多边缘反应位点,RhB在脱乙基和脱羧基过程中不断与·OH接触,发生羟基化氧化反应。另外,氰基是常见的吸电子基团,常被用作电子受体[39]。当g-C3N4的结构中存在氰基时,氰基与相邻的N原子之间会形成局部分子内供体-受体(D-A)体系。D-A体系可以有效转移分子内的电荷,使光生e−和h+分离开来,从而降低载流子重组率[40]。故55%-PCN中的氰基的引入进一步给光催化降解RhB提供更多的活性位点,最终使RhB被氧化为NH4+、CO2、H2O等小分子。
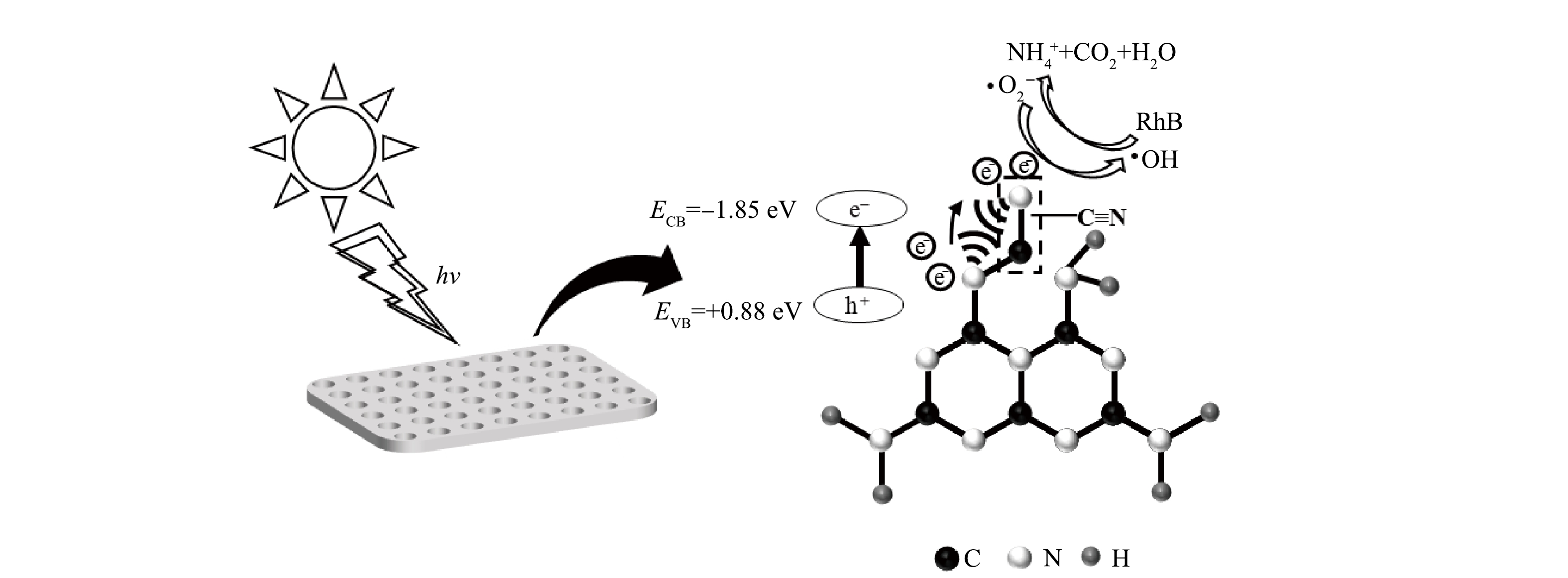 图 13 55%-PCN光催化降解RhB机理Figure 13. Mechanism of photocatalytic degradation on RhB by 55%-PCN
图 13 55%-PCN光催化降解RhB机理Figure 13. Mechanism of photocatalytic degradation on RhB by 55%-PCNPCN+hν→h++e− (3) e−+O2→·O−2 (4) ·O−2+H+→HOO⋅ (5) e−+HOO⋅→H2O2 (6) e−+H2O2→⋅OH (7) 3. 结论
1) 气体模板NH4Cl的引入可以在不破坏g-C3N4基本结构的基础上制备PCN,表面介孔数量增多,大大增加其比表面积和孔体积,给后续反应提供更多的活性位点。同时有利于提高吸光性能,经过测试其可见光吸收范围相比g-C3N4变宽。
2) 当前驱体中NH4Cl添加量为55%时所制备样品降解性能最佳。55%-PCN在50 min内对RhB的降解率为98%,伪一级动力学速率常数为6.07×10−2 min−1,是g-C3N4的5倍;并且其对RhB的TOC去除率比g-C3N4高,说明其可以更好地将RhB大分子吸附并降解。
3) 自由基捕获实验表明,在RhB降解时,·O2−和·OH参与了反应,在反应中,·O2−为最主要的活性物种。吸电子基团氰基与相邻的N原子之间会形成局部分子内D-A体系,使光生电子和空穴更好地分离,增加载流子寿命。55%-PCN的孔结构以及氰基的引入,均给RhB的降解反应提供了更多反应活性位点,在脱乙基和羧基过程中不断与·OH发生氧化反应,最终产物为NH4+、CO2、H2O等小分子。
-
图 4 g-C3N4和PCN的UV-vis吸收光谱和带隙图
Figure 4. UV-vis absorption spectra and band gap diagrams of g-C3N4 and PCN
图 6 g-C3N4和55%-PCN的氮气吸附-脱附平衡曲线和孔径分布曲线
Figure 6. N2 adsorption-desorption curves and pore size distribution curves of g-C3N4 and 55%-PCN
图 7 g-C3N4和55%-PCN的PL光谱和瞬态荧光光谱
Figure 7. Photoluminescence (PL) spectra and fluorescence lifetime spectra in g-C3N4 and 55%-PCN
图 8 g-C3N4和PCN的光电流曲线和阻抗曲线
Figure 8. Photocurrent curves and impedance curves of g-C3N4, PCN
图 9 g-C3N4和PCN对RhB降解效果的对比
Figure 9. Comparison of the degradation effects on RhB of g-C3N4 and PCN
图 10 55%-PCN光催化降解RhB的稳定性
Figure 10. Stability of 55%-PCN photocatalytic degradation on RhB
图 12 55%-PCN光催化降解RhB的质谱图
Figure 12. Mass spectra of photocatalytic degradation on RhB by 55%-PCN
-
[1] WANG T, SUN D L, ZHUANG Q, et al. China's drinking water sanitation from 2007 to 2018: A systematic review[J]. Science of the Total Environment, 2020, 757: 143923-143933. [2] MENON P, SINGH T A, PANI N, et al. Electro-Fenton assisted sonication for removal of ammoniacal nitrogen and organic matter from dye intermediate industrial wastewater[J]. Chemosphere, 2021, 269: 128739-128750. doi: 10.1016/j.chemosphere.2020.128739 [3] SHI Y F, LI S N, WANG L Y, et al. Compositional characteristics of dissolved organic matter in pharmaceutical wastewater effluent during ozonation[J]. Science of the Total Environment, 2021, 778: 146278-146287. doi: 10.1016/j.scitotenv.2021.146278 [4] LI J X, XU Y Q, DING Z Z, et al. Photocatalytic selective oxidation of benzene to phenol in water over layered double hydroxide: A thermodynamic and kinetic perspective[J]. Chemical Engineering Journal, 2020, 388: 124248. doi: 10.1016/j.cej.2020.124248 [5] YASEEN D A, SCHOLZ M. Treatment of synthetic textile waste water containing dye mixtures with microcosms[J]. Environmental Science and Pollution Research, 2018, 25(2): 1980-1997. doi: 10.1007/s11356-017-0633-7 [6] 姚悦, 李桂菊, 马万瑶. 电絮凝法深度处理制革废水的实验研究[J]. 天津科技大学学报, 2019, 34(6): 66-70. doi: 10.13364/j.issn.1672-6510.20180002 [7] AKARSU C, DEVEECI E U, GONEN C, et al. Treatment of slaughterhouse wastewater by electrocoagulation and electroflotation as a combined process: Process optimization through response surface methodology[J]. Environmental Science and Pollution Research, 2021, 28: 34473-34488. doi: 10.1007/s11356-021-12855-4 [8] JIANG T J, LUO C W, XIE C, et al. Synthesis of oxygen-doped graphitic carbon nitride and its application for the degradation of organic pollutants via dark Fenton-like reactions[J]. RSC Advances, 2020, 10: 32906-32918. doi: 10.1039/D0RA05202G [9] WANG W L, ZHAO J M, SUN Y Y, et al. Facile synthesis of g-C3N4 with various morphologies for application in electrochemical detection[J]. RSC Advances, 2019, 9: 7737-7746. doi: 10.1039/C8RA10166C [10] DONG C, QU Z P, JIANG X, et al. Tuning oxygen vacancy concentration of MnO2 through metal doping for improved toluene oxidation[J]. Journal of Hazardous Materials, 2020, 391: 122181. doi: 10.1016/j.jhazmat.2020.122181 [11] HUANG H R, ZHANG Z J, GUO S K, et al. Interfacial charge-transfer transitions enhanced photocatalytic activity of TCNAQ/g-C3N4 organic hybrid material[J]. Materials Letters, 2019, 255: 126546. doi: 10.1016/j.matlet.2019.126546 [12] YU Y, YAN W, WANG X F, et al. Surface engineering for extremely enhanced charge separation and photocatalytic hydrogen evolution on g-C3N4[J]. Advanced Materials, 2018, 30(9): 1705060. doi: 10.1002/adma.201705060 [13] YANG C, ZHANG S S, HUANG Y, et al. Sharply increasing the visible photoreactivity of g-C3N4 by breaking the intralayered hydrogen bonds[J]. Applied Surface Science, 2020, 505: 144654. doi: 10.1016/j.apsusc.2019.144654 [14] LI C M, WU H H, DU Y H, et al. Mesoporous 3D/2D NiCoP/g-C3N4 heterostructure with dual Co-N and Ni-N bonding states for boosting photocatalytic H2 production activity and stability[J]. ACS Sustainable Chemistry & Engineering, 2020, 8: 12934-12943. [15] LIAO J Z, CUI W, LI J Y, et al. Nitrogen defect structure and NO+ intermediate promoted photocatalytic NO removal on H2 treated g-C3N4[J]. Chemical Engineering Science, 2020, 379: 122282. doi: 10.1016/j.cej.2019.122282 [16] SUN Z X, WANG H Q, WU Z B, et al. g-C3N4 based composite photocatalysts for photocatalytic CO2 reduction[J]. Catalysis Today, 2018, 300: 160-172. doi: 10.1016/j.cattod.2017.05.033 [17] LIU M J, WAGEH S, Al-GHAMDI A A, et al. Quenching induced hierarchical 3D porous g-C3N4 with enhanced photocatalytic CO2 reduction activity[J]. Chemical Communications, 2019, 55: 14023-14026. doi: 10.1039/C9CC07647F [18] LI C M, YU S Y, ZHANG X X, et al. Insight into photocatalytic activity, universality and mechanism of copper/chlorine surface dual-doped graphitic carbon nitride for degrading various organic pollutants in water[J]. Journal of Colloid and Interface Science, 2019, 538: 462-473. doi: 10.1016/j.jcis.2018.12.009 [19] LI Y H, GU M L, SHI T, et al. Carbon vacancy in g-C3N4 nanotube: Electronic structure, photocatalysis mechanism and highly enhanced activity[J]. Applied Catalysis B, 2020, 262: 118281. doi: 10.1016/j.apcatb.2019.118281 [20] LIU S H, LIN W X. A simple method to prepare g-C3N4-TiO2/waste zeolites as visible-light responsive photocatalytic coatings for degradation of indoor formaldehyde hazard[J]. Journal of Hazardous Materials, 2019, 368: 468-476. doi: 10.1016/j.jhazmat.2019.01.082 [21] GUO F S, HU B, YANG C, et al. On-Surface polymerization of in-Plane highly ordered carbon nitride nanosheets toward photocatalytic mineralization of mercaptan gas[J]. Advanced Materials, 2021, 33(42): 2101466. doi: 10.1002/adma.202101466 [22] XIAO Y T, TIAN G H, LI W. Molecule self-assembly synthesis of porous few-layer carbon nitride for highly efficient photoredox catalysis[J]. Journal of the American Chemical Society, 2019, 141(6): 2508-2515. doi: 10.1021/jacs.8b12428 [23] LIU Y P, ZHAO S, WANG Y Y, et al. Controllable fabrication of 3D porous carbon nitride with ultrathin nanosheets templated by ionic liquid for highly efficient water splitting[J]. International Journal of Hydrogen Energy, 2021, 46(49): 25004-25014. doi: 10.1016/j.ijhydene.2021.05.018 [24] WANG X L, LIU Q, YANG Q, et al. Three-dimensional g-C3N4 aggregates of hollow bubbles with high photocatalytic degradation of tetracycline[J]. Carbon, 2018, 136: 103-112. doi: 10.1016/j.carbon.2018.04.059 [25] CHENG J S, HU Z, LI Q, et al. Fabrication of high photoreactive carbon nitride nanosheets by polymerization of amidinourea for hydrogen production[J]. Applied Catalysis B, 2019, 245: 197-206. doi: 10.1016/j.apcatb.2018.12.044 [26] DUAN Y Y, LI X F, LV K, et al. Flower-like g-C3N4 assembly from holy nanosheets with nitrogen vacancies for efficient NO abatement[J]. Applied Surface Science, 2019, 492: 166-176. doi: 10.1016/j.apsusc.2019.06.125 [27] WANG J H, ZAHNG C, SHEN Y F, etal. Environment-friendly preparation of porous graphite-phase polymeric carbon nitride using calcium carbonate as templates, and enhanced photoelectro chemical activity[J]. Journal of Materials Chemistry A, 2015, 3(9): 5126-5131. doi: 10.1039/C4TA06778A [28] 巩正奇, 闫楚璇, 宣之易, 等. 制备类石墨相氮化碳多孔光催化剂的模板法发展[J]. 工程科学学报, 2021, 43(3): 345-354. [29] GUO Q Y, ZAHNG Y H, ZHANG H S. 3D foam strutted graphene carbon nitride with highly stable optoelectronic properties[J]. Advanced Functional Materials, 2017, 27(42): 1703711. doi: 10.1002/adfm.201703711 [30] 杨锋. CH3NH3PbI3钙钛矿材料的制备及性能研究[D]. 绵阳: 西南科技大学, 2017. [31] ZHOU B, WAQAS M, YANG B, et al. Convenient one-step fabrication and morphology evolution of thin-shelled honeycomb-like structured g-C3N4 to significantly enhance photocatalytic hydrogen evolution[J]. Applied Surface Science, 2020, 506: 145004. doi: 10.1016/j.apsusc.2019.145004 [32] LI F, YUE X Y, ZHOU H P, et al. Construction of efficient active sites through cyano-modified graphitic carbon nitride for photocatalytic CO2 reduction[J]. Chinese Journal of Catalysis, 2021, 42(9): 1608-1616. doi: 10.1016/S1872-2067(20)63776-7 [33] 宁湘, 武月桃, 王续峰, 等. 石墨相氮化碳/二氧化锡复合材料的制备及光催化性能[J]. 无机化学学报, 2019, 35(12): 2243-2252. doi: 10.11862/CJIC.2019.272 [34] WEI W J, WANG Y B, HUANG Y F, et al. Constructing isotype CN/s-CN heterojunction with enhanced photocatalytic performance[J]. Diamond and Related Materials, 2020, 101: 107616. doi: 10.1016/j.diamond.2019.107616 [35] 陈甜. g-C3N4/活性炭复合材料中g-C3N4光催化活性和活性炭再生性能改善的研究[D]. 太原: 太原理工大学, 2020. [36] 王新哲. 多孔氮化碳制备及可见光催化增强的机制研究[D]. 吉林: 东北电力大学, 2021. [37] 刘华俊, 彭天右, 彭正合, 等. Dy/WO3光催化降解罗丹明B的反应机理[J]. 武汉大学学报, 2007, 53(2): 127-132. [38] HE Q C, ZHOU F, ZHAN S, et al. Enhancement of photocatalytic and photoelectrocatalytic activity of Ag modified Mpg-C3N4 composites[J]. Applied Surface Science, 2017, 391: 423-431. doi: 10.1016/j.apsusc.2016.07.005 [39] YAO C, YANG Y Z, LI L, et al. Elucidating the key role of the cyano (−C≡N) group to construct environmentally friendly fused-ring electron acceptors[J]. The Journal of Physical Chemistry C, 2020, 124(42): 23059-23068. doi: 10.1021/acs.jpcc.0c08022 [40] OU H H, CHEN X R, LIN L H, et al. Biomimetic donor-acceptor motifs in conjugated polymers for promoting exciton splitting and charge separation[J]. Angewandte Chemie International Edition, 2018, 57(28): 8729-8733. doi: 10.1002/anie.201803863 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4815
- HTML全文浏览数: 4815
- PDF下载数: 73
- 施引文献: 0
