

-
铬(Cr)是一种过渡金属元素,亦是一种潜在有毒元素存在于整个环境中[1]。尤其是六价铬(Cr(Ⅵ))被认为是第一类致癌物质[2],Cr(Ⅵ)不仅会影响作物产量和质量[3],还会对人群健康造成有害影响,包括致癌和遗传毒性等[4]。在过去几十年中,土壤Cr污染问题越来越严峻,主要集中于皮革鞣制、金属电镀、不锈钢生产、合金制造、印染、化学品生产等行业地区[5]。据报道,在美国国家优先清单上,Cr污染场地约占11%;日本在污染场地中,Cr(Ⅵ)污染场地占14%[6];而我国Cr污染土壤问题也尤为突出,Cr污染土地面积占污染土地总面积的5.1%[7]。
目前,许多修复Cr污染的方法已被开发并应用于实践,例如固化/稳定[8]、土壤淋洗[9]、电动修复[10]、植物修复[11]和生物修复[12]等。尤其是固化/稳定化修复技术[13],该技术包括固化和稳定化2个过程。其中,固化过程通常是利用水泥基材将Cr污染物通过包埋的形式固定在土壤中。稳定化过程则是向土壤中添加稳定剂(还原剂),将高毒性、迁移性强的Cr(Ⅵ)还原成为低毒性的Cr(Ⅲ),进一步形成沉淀或难溶物质,阻止其迁移,最终稳定在土壤中。由于固化/稳定化修复技术能够以相对较低的成本实现修复目标,且具有周期短、效率高等优点。同时,该技术还能够原位或异位处置各种重金属污染物,在我国已被广泛采用。据统计,仅2017-2018年我国土壤修复工程中固化/稳定化修复技术的选取率就高达48.5%[14]。然而,我国修复场地多数以修复工程竣工验收设为终点,却忽略了土壤修复的长期有效性、缺乏对验收后Cr的环境归趋的关注。此外,Cr在修复后土壤中,具有环境行为多变、机制复杂,易迁移和转化的特点,修复后的环境风险也值得重点关注[15]。
综述了Cr在土壤中的氧化与还原、吸附与解吸、沉淀与溶解等物理化学反应以及被植物、微生物吸收与转化等生物作用的环境行为,并通过调研Cr污染场地长期监测现状,探索污染场地修复后Cr的环境归趋及其影响因素,以期为修复后场地的长期风险管理提供参考依据。
-
由于Cr的核外电子结构为[Ar]3d54s1,Cr存在多种价态与形态,但在土壤介质中主要以Cr(Ⅲ)和Cr(Ⅵ)存在。根据BCR连续提取法[16],可将土壤介质中Cr的赋存形态划分为弱酸提取态、可还原态、可氧化态、残渣态。土壤介质中Cr的赋存形态受到多种因素的影响,如pH、有机质、以及微生物等。刘雪等[17]选用3种不同的Cr污染土壤作为试样,研究了pH和有机质投加量对土壤介质中Cr形态的影响,发现随pH降低和有机质投加量增大,土壤介质中Cr(Ⅵ)含量减少,Cr(Ⅲ)增加。另外,Cr的弱酸可提取态组分占比会减小,而Cr的可氧化态和可还原态组分占比增加,且可氧化态组分增加明显。这表明,酸性条件和有机质有利于Cr(Ⅵ)的还原和弱酸可提取态的Cr向可还原态和可氧化态Cr的转化。另外,郭越宏等[18]探究了腐植酸对李氏禾(Leersia hexandra Swartz)修复红壤中Cr赋存形态的影响。在添加腐植酸后,红壤中Cr的可氧化态组分占比提高,可还原态组分占比降低。
-
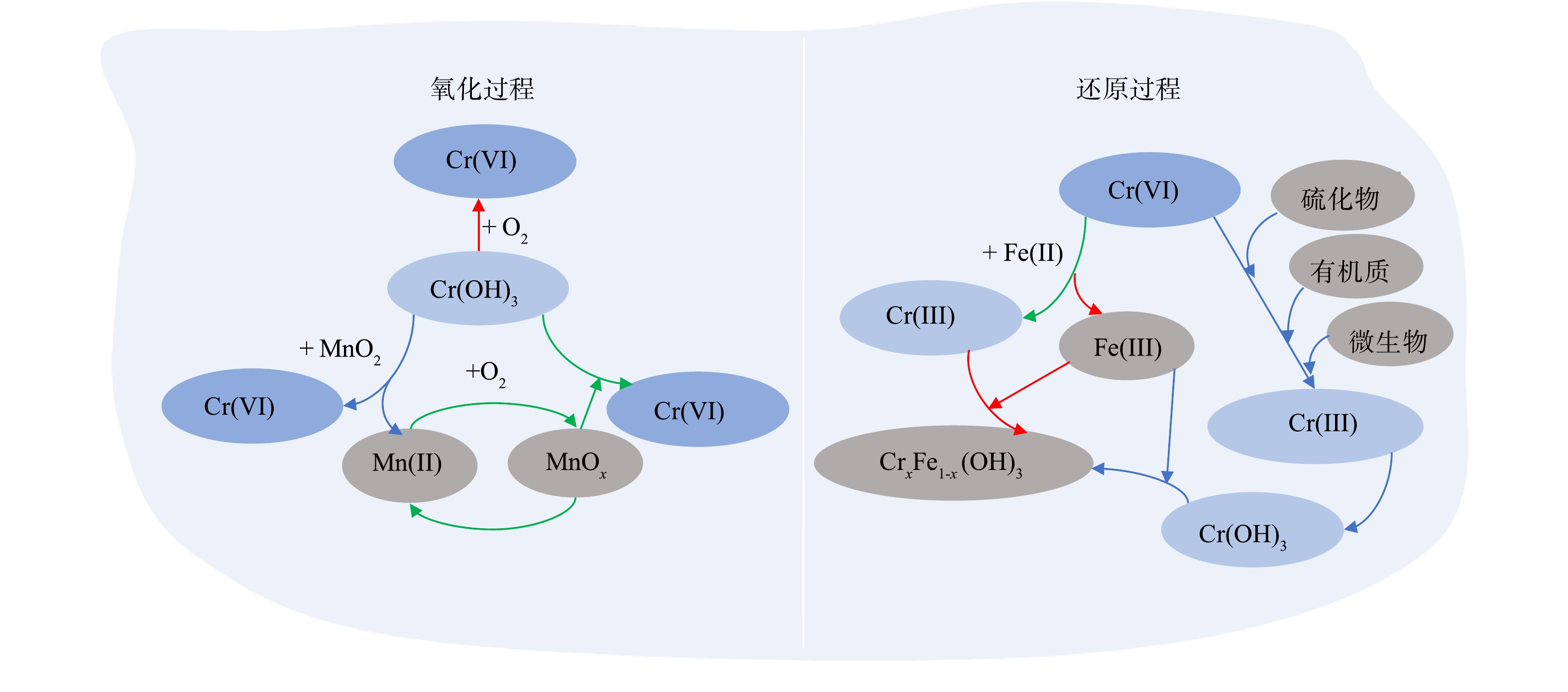
Cr能够与土壤中多种介质发生氧化还原反应(图1)[19]。在酸性环境中,Cr(Ⅵ)是一种强氧化剂,具有非常高的氧化还原电位(E0 (Cr(Ⅵ)/Cr(Ⅲ)) = 1.33 V),而碱性条件下的氧化还原电位为−0.13 V。另外,土壤中溶解氧(E0(O2/OH−) = 0.40 V)和Mn(Ⅳ)(E0(MnO2/Mn2+) = 1.23 V)能够形成一定的氧化环境进而对Cr(Ⅲ)产生氧化作用。PALMER等[20]通过实验证实了溶解氧对Cr(Ⅲ)的氧化是一个缓慢的过程,通常需要数月之久。这说明,溶解氧能够氧化Cr(Ⅲ),但并非占主导地位。而BARTLETT[21]研究发现,正常干燥土壤中Cr(Ⅲ)难被氧化成Cr(Ⅵ),需要有H2O的参与。PAN等[22]发现,锰氧化物与含Cr(Ⅲ)沉积物发生反应,导致地下水中Cr(Ⅵ)浓度升高。LIU等[19]探究了碱性环境中Cr(OH)3的3种氧化机制。结果表明,O2对Cr(OH)3的氧化贡献占比较低,而短时间内(10 d)MnO2贡献占比较高(51%),而在长期(365 d)实验中,MnOx催化氧化贡献占比最高(78%)。
土壤中能够还原Cr(Ⅵ)介质包括Fe(Ⅱ)、硫化物、有机质、微生物等。BARTLETT和JAMES[23]以及THOMAS等[24]研究了不同pH环境下Fe(II)对Cr(Ⅵ)的还原机制。在不同pH范围内呈现2种不同的反应特点。其中,在低pH或中性环境(pH = 1.5 ~ 8.7)下,由于Fe(Ⅱ)具有较低的氧化还原电位(E0 (Fe(Ⅲ)/Fe(Ⅱ) = 0.77 V),对Cr(Ⅵ)表现出高效的还原作用;而在强碱性(pH ≥ 13.5)的环境中,Fe(Ⅱ)能够形成Fe(OH)3−的中间态产物,Fe(OH)3−能与还原生成的Cr(Ⅲ)反应,形成Fe-Cr共沉淀物,从而进一步促进Cr(Ⅵ)的还原。MAHDIEH等[25]研究了4种硫化物(CaSx、FeS、FeS2和Na2S)在不同pH条件下对土壤中Cr(Ⅵ)的还原效率。结果发现,CaSx、Na2S和FeS均能够有效还原Cr(Ⅵ),而FeS2的效果不佳。其中,在碱性土壤中,对Cr(Ⅵ)还原能力的大小顺序为:Na2S > CaSx > FeS > FeS2。在中性土壤中,顺序为:Na2S
>> CaSx ~ FeS > FeS2。在弱酸性土壤中,顺序为:Na2S>> FeS > CaSx ~ FeS2。BARTLETT等[21]在研究土壤中有机质还原Cr(Ⅵ)的过程中,发现包括光照、酸化、冻融、隔绝氧气等环境的变化均能够提高有机质对Cr(Ⅵ)的还原能力。此外,植物和微生物通过根际效应,胞外吸附与吸收、胞内呼吸等作用,还可以将Cr(Ⅵ)富集到生物体内后,经过生化途径将其还原[26-27]。 -
Cr(Ⅲ)很容易被土壤胶体吸附形成极低溶解度的沉淀物,这是由于天然土壤胶体表面带有负电荷,有利于阳离子(如Cr3+)的吸附,其吸附容量是Cr(Ⅵ)的30 ~ 300倍[28]。因此,Cr(Ⅲ)通常以氢氧化物和氧化物的形式存在形成,或吸附在粘土颗粒、土壤有机物、金属羟基氧化物等相对不溶且流动性较差的沉淀物上,进而阻碍其迁移[29]。相反,Cr(Ⅵ)主要以相对易溶的阴离子(如Cr2O72-、HCrO4−和CrO42-)形式存在土壤中,不易被土壤胶体吸附,在土壤中具有强迁移能力[30]。土壤对Cr(Ⅵ)主要的吸附包括非静电引起的专性吸附,以及静电作用形成的非专性吸附。非专性吸附形成的化合物或配合物不稳定,易受到土壤pH值或其它离子组分的影响而发生解吸[31-32]。SHI等[33]研究了Cr(Ⅵ)在14种土壤中吸附行为。对比了土壤性质、pH、Cr(Ⅵ)初始浓度、腐殖酸以及Fe、Al的(氢)氧化物含量等因素对Cr(Ⅵ)吸附的影响,并建立了多位点表面络合模型。结果表明,Fe、Al的(氢)氧化物和腐殖酸是土壤对Cr(Ⅵ)吸附的主要影响因素。ZHANG等[34]通过动态吸附和解吸土壤柱实验研究了土壤中Cr(Ⅵ)的迁移和转化。结果表明,Cr(Ⅵ)初始浓度越高,土壤对Cr(Ⅵ)的吸附能力越差。此外,Cr(Ⅵ)的吸附还明显受pH的影响,在低pH(pH=2)时,土壤对Cr(Ⅵ)的吸附能力会增大。这是因为,pH的变化会导致土壤表面带正电荷的基团与HCrO4−之间的静电吸引力更高,进而增加了Cr(Ⅵ)的吸附。同时,在低pH条件下,Cr(Ⅵ)具有更高的氧化还原电位(1.33 V),易被还原生成Cr3+进而使Cr(Ⅵ)总量减少。此外,解吸实验结果表明,Cr(Ⅵ)不易被土壤固定,更易从土壤中浸出,其吸附过程以弱吸附为主。在环境因素如雨水冲刷下,土壤中的Cr(Ⅵ)也会释放,进而造成污染。
-
土壤中Cr的沉淀形式包括单一沉淀,与其它金属离子形成的共沉淀以及与有机分子形成的络合沉淀等[35-36]。在土壤溶液中,Cr能够与Fe形成Fe-Cr共沉淀。DAI等[37]探究了Cr(Ⅲ)与Fe(Ⅲ)在均质和异质中形成共沉淀的机制。通过X射线分析发现,Fe-Cr共沉淀与Cr(Ⅲ)/Fe(Ⅲ)浓度、(Fex,Cr1-x)(OH)3沉淀物的化学成分、沉淀物的过饱和度以及介质表面电荷等因素有关。进一步用不同的Cr(Ⅲ)/Fe(Ⅲ)比,探究了均质-溶液和异质-石英载体形成(Fex,Cr1-x)(OH)3的沉淀速率。结果表明,在高比值的Cr(Ⅲ)/Fe(Ⅲ)均质溶液中,Cr(Ⅲ)能够吸附在共沉淀物表面,导致溶液中沉淀颗粒生长较慢;而在石英表面上可以形成快速沉淀。这是因为,石英表面的Zeta电位为负有利于Cr(Ⅲ)的沉淀。同时发现,共沉淀颗粒在石英表面上能够快速生长,在1 h内从2 nm增长到4 nm。
影响Cr沉淀溶解包括的因素包括土壤pH、有机酸等[38]。例如,在酸性条件下Cr(OH)3会发生再次溶解、迁移[39-40]。土壤中柠檬酸、富里酸等小分子有机酸可与Cr(Ⅲ)形成可溶性络合物。GUSTAFSSON等[41]利用EXAFS光谱技术探究了Cr(Ⅲ)与天然有机物(黄腐酸)的络合作用。EXAFS结果表明,在低pH(< 5)时,Cr(Ⅲ)与黄腐酸形成单体络合物,仅有Cr-O-C之间的成键作用;而在pH > 5时,会形成多核的Cr(Ⅲ)-黄腐酸络合物(二聚体和四聚体),存在Cr与Cr之间的相互作用。CAO等[42]利用柱实验探究了Cr(Ⅲ)在土壤中的溶出行为。进一步验证了土壤中Cr(Ⅲ)能够与有机酸形成Cr-有机酸的络合物,而提高了Cr(Ⅲ)在土壤中的迁移能力。这是因为,Cr(Ⅲ)-有机酸形成的络合物通常带负电荷或以中性分子的形式存在,与土壤胶体(带负电)形成相互排斥作用,进而增强其在土壤中的迁移能力。
-
如图2所示,土壤中Cr的迁移与转化可以通过微生物或植物作用实现[43-44]。土壤中已被固化/稳定化的Cr可能受植物或微生物的影响而溶出;或在微生物或植物作用下,Cr(Ⅵ)被还原,进而被植物、生物吸收和利用[45]。通常,微生物对还原Cr(Ⅵ)主要发生在胞内、外以及细胞膜上。其中,细胞外还原主要是由胞外还原酶或者胞外多聚物(EPS)与Cr(Ⅵ)作用。WANG等[46]研究发现,Pannonibacter phragmitetus BB分泌的EPS能够有效地还原土壤中Cr(Ⅵ)。这是因为,EPS上含有大量的官能团和氧化还原蛋白,能够结合与还原Cr(Ⅵ)。另外,由于CrO42-与SO42-分子结构具有相似性,Cr(Ⅵ)可以从细胞膜上的硫酸盐转运通道进入到细胞体内,例如拟南芥根系可以通过硫酸盐转运蛋白吸收土壤中的Cr(Ⅵ),进而将其转运至细胞内还原[47]。细胞膜上Cr(Ⅵ)的还原主要由细胞膜上结合的还原酶完成。ZENG等[48]探究了Oceanobacillus oncorhynchi W4中还原Cr(Ⅵ)的关键部位。结果发现,细胞质部分对Cr(Ⅵ)的去除率占比仅为11.1%,而细胞膜部分占比高达82.9%。这表明,其细胞膜是Cr(Ⅵ)还原的主要位置。
许多植物能够富集与稳定Cr(Ⅵ),降低其毒性和迁移性,如Dicoma niccolifera,Sutera fodina以及Leersia hexandra Swartz等植物通过特定的酶作用,促进Cr(Ⅵ)在植物根、茎及叶组织中的积累[49]。JAN等[50]利用番茄植株探究了24-油菜素内酯介导下的植物抗氧化体系对Cr(Ⅵ)的还原作用。结果表明,植物抗氧化过程主要与体内抗氧化剂(如MDHAR、GSH、GSSG和GST等)浓度提高以及部分抗氧化酶(如SOD、APX和GR等)的活性增强有关。VAMERALI等[51]通过调节植物根际的土壤环境(包括降低pH和氧化还原电位)和菌落结构(丛枝菌根和外生菌根),能够使Cr(Ⅵ)富集并被还原为Cr(Ⅲ)降低毒性,减小环境风险。SHI等[11]利用外生菌根真菌Hebeloma vinosophyllum(Cr(Ⅵ)敏感菌株)和Pisolithus sp1(Cr(Ⅵ)耐受菌株)分别探究了Cr对松树的影响。结果表明,Pisolithus sp1能够显著降低土壤中可交换态Cr的含量占比,同时能够改善松树幼苗的生长并增强它们对Cr的耐受性。
-
在Cr污染场地修复中,主要关注毒性高、迁移性强的Cr(Ⅵ)。通常是先将高毒性的Cr(Ⅵ)还原为低毒性的Cr(Ⅲ),再进一步将其固化或稳定化,从而降低Cr在环境中的迁移性和生物可利用性。随着对Cr污染的重视,学者针对Cr污染土壤的治理与修复也新的见解和认识,如表1所示。
目前,Cr(Ⅵ)还原体系包括以铁基(零价铁、Fe(II))、硫基(S2-)、碳基(生物炭,腐殖质)等材料为代表的还原体系、微生物参与的还原体系、以及材料与微生物构成的耦合还原体系3类。其中,铁基材料中的零价铁由于还原能力强、反应活性高和吸附性能优异等特点,被认为是修复Cr(Ⅵ)污染土壤最有前景的材料之一[52]。ZHOU等[53]应用零价铁颗粒和磁选技术来修复Cr(Ⅵ)污染的土壤。结果表明,污染土壤中有超过92%的Cr被吸附到零价铁表面,再通过磁选技术进行零价铁的回收,达到修复Cr(Ⅵ)污染土壤的目的。另外,用于Cr(Ⅵ)还原的硫基材料可分为:可溶性硫化物(如Na2S、多硫化钙(CPS)等)、单质硫、铁硫化物(FeSx)[25]。YUAN等[56]用CPS作为修复药剂,通过机械球磨混匀CPS和Cr(Ⅵ)污染的土壤。结果表明,仅添加土壤质量3%的CPS可显著降低土壤中Cr(Ⅵ)浸出浓度,由115 mg·L−1下降到0.51 mg·L−1。赖冬麟等[57]用FeSO4·7H2O、NaHSO3、CPS和葡萄糖作为还原剂,磷酸盐、CaO、水泥作为固化稳定化剂,对张家口市某机械厂原址电镀污染场地进行土壤修复。该场地修复前土壤中Cr(Ⅵ)的最高含量为37.3 mg·kg−1,经过固化/稳定化修复后,土壤中Cr(Ⅵ)含量下降到低于检出限值,浸出液中Cr(Ⅵ)浓度同样也低于检出限值。用于土壤修复的碳基材料不仅来源广泛、价格低廉,还可以作为改良土壤,改良土壤性质,提高作物产量[59],已被广泛应用。例如,生物炭具有大比表面积、高孔隙率以及丰富的官能团,使得生物炭具有良好的吸附能力和还原性能等[60]。BASHIR等[61]利用甘蔗制生物炭探究了制革厂土壤中Cr污染修复效果。结果表明,投加甘蔗制生物炭能够有效降低土壤中Cr(Ⅵ)的浓度,同时对场地中玉米植株的高度、生物量、叶绿素含量、蛋白质和淀粉含量均有所提高。
利用微生物还原Cr(Ⅵ),具有广阔应用前景[63]。HUANG等[64]发现,环境中无处不在的微生物Aeromonas hydrophila能够有效减少土壤中Cr(Ⅵ)的含量。其还原过程主要包括细胞外和细胞内的还原。BALDIRIS等[65]发现,革兰氏阴性菌株Stenotrophomonas maltophilia不仅能够耐受高浓度Cr(Ⅵ),还能够对Cr(Ⅵ)进行还原。分析发现,该菌株中的铬酸还原酶基因(ChrR)起到了关键作用。
微生物参与的耦合还原修复体系,在Cr污染场地修复方面具有很大的潜力[66-68]。TAN等[66]利用腐植酸负载羧甲基纤维素包裹纳米硫化亚铁协同污泥微生物修复Cr污染土壤。经过90 d修复后,土壤中Cr(Ⅵ)下降了99.16%。ZHANG等[68]利用次生矿物(施氏矿、黄钾铁矾)协同异化铁还原菌(Shewanella Oneidensis MR-1)对Cr(Ⅵ)还原机制进行研究。其还原机制包括:次生矿物对Cr(Ⅵ)的吸附、矿物中Fe(Ⅲ)/Fe(II)氧化还原循环过程中产生电子的间接电子转移,以及矿物中导带电子的间接电子转移。
-
在实际修复中,由于水文地质变化等因素,场地可能溶出Cr(Ⅲ)甚至再生成Cr(Ⅵ),Cr污染发现反弹或迁移[42, 69]。为防范风险,截至2019年,美国环保署(EPA)超级基金项目对18个修复场地开展Cr长期监测,平均监测时间超过20年。如表2所示,在18个长期监测的场地中有11个场地再未发现Cr污染,3个场地(Town Garage/Radio Beacon、BFI Sanitary Landfill、Saco Tannery Waste Pits)存在Cr污染超标。其中,Town Garage/Radio Beacon地下水Cr的轻度超标与采样扰动有关,当采用低流速采样作业时,Cr浓度并未超标。在BFI Sanitary Landfill场地中,通过采取表面屏障、地下水拦截沟、气体收集及处理系统等修复措施,场地持续开展制度控制和长期监测,该场地污染物Cr地下水修复目标限值100 μg·L−1。截至2019年,共完成5轮次周期性的回顾分析。其中,MW-9地下水监测井(浅层基岩地下水)Cr的超标时有发生。自2004年至2019年,在该点位进行29次取样分析发现有18次超标,历史平均监测质量浓度142 μg·L−1,而在2019年春季监测地下水中Cr的质量浓度为267 μg·L−1。对比分析同区域附近点位MW-10(深层基岩地下水)无Cr的超标,推测MW-9中Cr质量浓度的波动是垃圾填埋场中污染物Cr在浅层基岩地下水中迁移的结果。在Saco Tannery Waste Pits场地中,通过对废物堆和排水区覆盖阻隔修复等措施,其地下水中Cr的修复目标限值100 μg·L−1。在1992—2013年,场地基岩水监测井的Cr质量浓度均未超过水修复限值;而在承压水监测井的MW-114B点位Cr质量浓度在2003年出现超标,为160 μg·L−1;同时,沉积物中监测发现靠近排水系统的SED-204和SED-103点位Cr质量浓度也超标,推测该场地Cr污染超标是由于废物堆污染源中Cr泄漏。
-
Cr污染场地固化/稳定化修复后,Cr同样会有氧化还原、吸附、沉淀等各种环境行为,发生如图3所示的迁移与转化。影响Cr污染固化/稳定化修复效果主要原因是,固化体的解体分化与Cr(Ⅲ)的再氧化。前者是污染物迁移的前提,后者是修复后污染风险形成的关键。修复场地环境条件发生变化时,如冻融、酸雨、水位波动、有机酸的形成以及矿物质的风化等,都会导致固化体的解体分化,而诱发Cr的重新溶出[71-74]。HE等[71]使用水泥生石灰固化/稳定化处理Cr(Ⅵ)污染的土壤,经过不同的冻融循环后,探究了固化的土壤抗压强度,毒性浸出等,并揭示了Cr(Ⅵ)污染固化土壤的工程特性的演化规律。结果表明,随着冻融循环次数的增加,固化土的强度会降低,土壤的孔隙会变大,Cr(Ⅵ)浸出浓度会增高。LIAO等[73]研究了在酸雨淋滤条件下改良土壤中Cr的释放特性。研究发现,用微生物(Pannonibacter phragmitetu)改良的土壤中总Cr和Cr(Ⅵ)的浸出量均比用硫酸亚铁改良的土壤浸出量低。其中,酸雨淋溶下碳酸盐结合态,可交换态和有机结合态的Cr是主要释放源。SLEJKO等[75]长期跟踪意大利北部Friuli Venezia Giulia的Cr污染场地。在1997年,发现污染场地地下水Cr(Ⅵ)质量浓度达4500 μg·L−1;在随后的几年中,由于场地修复与Cr污染自然减弱;到2003年时,地下水中Cr(Ⅵ)完全消失;而在2008年观察到地下Cr(Ⅵ)水污染复发,Cr(Ⅵ)质量浓度达到1560 μg·L−1。这是因为,地下水中的Cr(Ⅲ)通过含水层而被土壤中的Mn(Ⅳ)重新氧化形成Cr(Ⅵ)。ZHAO等[76]研究含泥炭土壤地下水位时发现,地下水的波动会形成缺氧-好氧界面,进而会产生羟基自由基(·OH),形成强氧化环境。同样地,JIA[77]等研究地下水时发现,由于地下水水位的波动会捕获土壤孔隙空气中的O2,捕获的O2溶解到孔隙水中与Fe(II)发生反应产生·OH,进而氧化Cr(Ⅲ),引发环境风险。
-
由于固化/稳定化技术特点,无法实现场地土壤中Cr的减量化。而残留在修复后场地中的Cr又会受土壤介质和场地环境因素影响,发生沉淀溶解、吸附解吸,氧化还原、生物吸收等环境行为,而导致Cr在修复后场地中表现为不同的环境归趋[78]。根据文献资料和实际修复案例研究分析可知,修复后场地中Cr的环境归趋途径主要有植物吸收,随雨水下渗向下迁移进入地下水,及随地表径流而迁移形成沉积物等[79]。MOHANTY等[80]研究了禾本科植物巴拉草对Cr的吸收能力。结果表明,巴拉草对Cr的总累计效率为8.2 mg·(kg−1·d−1),其中,在巴拉草根部Cr的累积量为叶稍部位的1 000倍。RANIERI等[81]对比禾本科植物芦苇和落叶乔木臭椿对土壤中Cr的吸收效率。结果表明,在种植360 d后,土壤中Cr由70 mg·kg−1下降至36 mg·kg−1(芦苇)和41 mg·kg−1(臭椿)。其中,芦苇根部Cr的累积量最高达1 910 mg·kg−1(干重),而臭椿根部Cr累积量仅为358 mg·kg−1(干重)。YE等[78]通过批量和柱实验来阐明土壤中Cr(Ⅵ)的吸附和迁移行为。结果表明,在表层土壤中,Cr(Ⅵ)随降雨具有较高的迁移率,同时,在土壤迁移过程中有明显的滞留现象,这进一步说明了Cr(Ⅵ)能与土壤发生多种反应(如吸附与解吸、离子交换、沉淀与溶解)。另外,Cr(Ⅵ)在土壤中迁移符合对流-弥散方程。陈子方等[82]模拟雨水淋溶包气带中Cr(Ⅵ)的释放。结果表明,在粗砂包气带中Cr(Ⅵ)的迁移速率为0.4 cm·d−1,大于在细沙包气带中的迁移速率(0.36 cm·d−1);Cr(Ⅵ)在粗砂和细沙中的淋洗溶出率分别为62.33%和40.36%。童秀菊等[83]通过降雨模拟形成地表径流并探究土壤中Cr(Ⅵ)的迁移情况。结果表明,Cr(Ⅵ)溶解在地表径流溶液中的质量浓度随时间而减小;且在同时形成地下渗流和地表径流时,土壤中Cr(Ⅵ)大部分会向地下迁移。另外,修复后场地地下水形成波动带是Cr在非饱和土壤和饱和含水层之间迁移的主要界面。WEI等[84]通过模拟实验探索了地下水波动期间特定区域中Cr迁移作用。结果发现,Cr(Ⅵ)的浓度在较低的土壤层中减少明显。而Cr(Ⅲ)的浓度会随水位上升和波动幅度的增加而增加。这是由于非饱和土壤底层具有较低的氧化还原电位,能够还原Cr(Ⅵ),生成的Cr(Ⅲ)会随着饱和带向下运移,并在地下水中迁移。
-
在实际修复后污染场地中土壤情况复杂多变,易受外界不确定性因素的影响,故导致Cr的环境行为复杂化。影响重金属Cr在土壤中环境行为诸多,如土壤pH、土壤氧化还原电位、土壤成分(O2、Fe、Mn、有机质、微生物等)、以及土壤类型等。目前,多数研究仅停留在单一因素上,缺乏多因素的耦合联动机制。因此,今后研究中应注重在实际场地中Cr与氧化组分(Mn(Ⅳ)、O2)、还原组分(Fe、S、有机质等)以及迁移和转化组分(微生物、植物)等构成的多元体系下的耦合机制;同时应关注重金属Cr在土壤中的时空、形态分布规律与迁移、转化特征的量化研究。
由于我国修复工程的特点,对修复后场地缺乏长期的关注。当修复后场地的环境因素(冻融、酸雨、地下水等)变化、以及场地的再开发利用过程中的扰动等,会引发重金属Cr在场地内重新活化。因此,修复后场地在管理上需要建立长期跟踪监测机制、改进监管制度、强化风险预警机制、提升阻控水平。尤其是针对容易形成氧化氛围的区域加强监测与跟踪。同时,在物理阻控方面,加强Cr污染土壤的水平防渗,阻隔上层氧气和水分的渗透,防控Cr(Ⅲ)氧化。在化学阻控方面,研发添加固相有机物、铁铝矿物等低成本药剂的复合建材,阻断Cr的迁移与转化。
固化/稳定化修复后场地土壤中铬的环境行为与归趋
Environmental behavior and fate of chromium in the soils of solidification/stabilization post-remediation sites
-
摘要: 土壤重金属铬(Cr)污染形势严峻,对人群健康和生态环境构成了严重威胁。通过固化/稳定化技术降低土壤中金属的毒性或迁移性,是Cr污染场地常用的修复技术之一。由于Cr的环境行为变化多端、机制复杂,故导致固化/稳定化修复后场地存在Cr污染反弹的环境风险。综述了Cr在土壤中的氧化还原、吸附与解吸、沉淀与溶解,以及植物、微生物吸收与转化等多种环境行为;并梳理了已修复的Cr污染场地案例及场地长期跟踪监测数据,探讨了固化/稳定化修复后Cr的环境归趋及其影响因素,以期为修复后场地的风险管理提供参考。Abstract: The chromium (Cr) contamination in soils has posed a serious threat to human health and ecological environment. As the most popular remedial technique addressing Cr contaminated sites, solidification/stabilization can effectively decrease the toxicity and mobility of Cr in soils. Due to the complex and diverse environmental behavior of Cr, however, there are risks of pollution recurrence in post-remediation sites. This review highlighted the environmental behavior of Cr, such as oxidation/reduction, adsorption/desorption, and dissolution/precipitation in soil, as well as the migration and transformation associated with plants and microorganisms. Long-term monitoring data of contaminated sites were also summarized. The environmental fate and key influential factors of Cr in post-remediation sites, where solidification/stabilization was employed, are discussed. This review will provide the critical information for the risk management of post-remediation sites.
-
大气过氧化氢(H2O2)是重要的光化学二次产物,其主要是由HO2与HO2自由基结合生成的[1]。此外,无光情况下已知的能够形成过氧化氢的途径为O3与烯烃的反应以及NO3与有机化合物的反应[2]。过氧化氢唯一的直接排放源为生物质燃烧[3-4]。过氧化氢本身是大气中重要的氧化剂,同时又作为HOx自由基的储库分子,参与HO2和OH自由基的循环[5-6]。作为大气中最重要的氧化剂之一,H2O2在污染大气化学中扮演着重要的作用。一方面,在pH<5的云、雾和雨水中,H2O2被认为是S (Ⅳ)氧化生成硫酸盐最重要的氧化剂,对大气降水酸化和颗粒物的质量增长有重要影响[7-8]。而且,近年来随着研究手段的进步以及对过氧化氢认识的不断深入,有研究发现过氧化氢与气溶胶颗粒物之间存在着复杂的关系。有研究通过外场观测以及模型模拟发现气溶胶表面非均相反应对H2O2可能同时存在源和汇的双重作用[4, 6, 9]。另一方面,有研究将H2O2/HNO3的比值用做评估O3生成对VOCs或NOx敏感性的指示剂,且在多种指示剂评估方法中,认为H2O2/HNO3比值的指示剂方法对O3生成敏感性的评估最合适[10-11]。此外,毒理学研究表明,H2O2能够随细颗粒物进入肺部,进而损伤肺泡,从而对人类健康产生不利影响[12]。因此,对大气中过氧化氢的浓度水平、污染特征、生消过程及影响因素等进行深入研究,对掌握区域光化学氧化剂的污染水平和研究光化学与颗粒物复合污染的形成以及人类健康具有重要意义。
本研究对大气过氧化氢的浓度水平、气象因素和相关环境污染物的大气浓度开展了同步观测,旨在分析影响H2O2生成的因素。着重关注高NOx以及颗粒物污染情况下H2O2的污染特征,通过模式对过氧化氢的源汇进行分析。
1. 实验部分(Experimental section)
1.1 观测地点
本研究的大气过氧化氢观测地点位于北京市海淀区中国科学院生态环境研究中心(40.00°N,116.33°E)综合楼顶楼,采样口距地面20 m。采样点正南方向有一条东西方向公路,采样点周围无工业源,主要为教学园区以及居民区。观测时间为2018年秋季9月22日—10月30日,其中在10月初由于设备故障有部分观测数据缺失。
1.2 方法与设备
本研究使用的大气过氧化氢分析仪器为美国EPA制造的大气过氧化氢分析设备原机,并在使用过程中对设备的数据采集和流量控制部分进行了改进和更换。研究组利用美国NI6002数据采集卡并编写labview程序进行数据采集,并更换了质量流量控制部分的组件。采样气体流速控制在1 L·min−1。仪器对大气过氧化氢的检测原理见文献[13],利用过氧化物与被过氧化物酶催化的对羟基苯乙酸的液相反应,在326 nm波长下激发该反应产生的荧光二聚体,以400—420 nm波长下检测到的荧光强度定量过氧化物浓度。过氧化氢与有机过氧化物的测定通过两个平行通道(Channal A和Chanal B)的设计实现,其中Channal B加入过氧化氢酶选择性破坏过氧化氢。过氧化氢的浓度通过两通道的信号差值计算得到。
观测期间除大气过氧化氢外,配套观测的污染物种类还包括SO2、NOx、CO以及O3等气态污染物。利用Thermo scientific – TEOM1405F实时在线测量大气气溶胶质量浓度;利用URG9000D-AIM分析仪在线测量大气PM2.5上无机水溶性离子及气态氨气、硝酸气和硫酸气浓度;利用TSI-SMPS分析仪实时测量大气气溶胶粒径分布。并在天气晴朗、光化学条件较好的情况下,利用不锈钢采样罐采集全空气样品,使用三级冷阱预浓缩-Dean-switch-GC-FID/FID方法进行了VOCs光化学活性组分(107种)的分析。仪器参数信息见表1。
表 1 同步观测的仪器信息Table 1. The instrument information for observation污染物Pollutants 型号Instruments O3/NOx /CO/SO2 Thermo Fisher Scientific,49i/42i/48i/43i PM2.5 Thermo Fisher Scientific,TEOM-1405F NH3, HNO3, HNO2, H2SO4, Na+,  , K+, Mg2+, Ca2+, F−, Cl−,
, K+, Mg2+, Ca2+, F−, Cl−,  ,
,  ,
, 
Thermo Fisher Scientific,URG-9000D VOCS Entech Instrument Inc.,Entech 7200;Thermo- fisher USA,Trace 1300 Particle size distribution TSI,TSI-3082,TSI-3321 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.3 基于观测的模型(OBM)
基于观测的模型(observation-based model,OBM)是一种以观测数据为基础进行光化学污染过程分析的方法[14]。与传统的空气质量模型相比,其具有不依赖源清单的优势。此外,OBM作为空气质量模型的重要补充,其与化学机理耦合能够模拟光化学产物的生成及衰减过程,获得人们关注的一些光化学产物如O3、H2O2和HNO3等物种生成及消耗途径以及速率的信息。Cardelino和Chameides、以及Wang等[15-17]的研究中对OBM模型在O3生成与前体物关系的应用方面做了详细介绍,而Wang、Qin等[4, 18]的研究中参照O3研究中相对增量反应活性的概念,对H2O2和其前体自由基HO2与NOx、CO和VOC前体物的关系进行了探究。因此,参照相对增量反应活性概念,本研究利用OBM对H2O2生成对前体物变化的敏感性进行了探究,研究中OBM模型采用实测污染物(O3、NO、NO2、SO2、CO、VOCs)和气象因子(T、RH)5 min间隔数据作为限制条件输入。H2O2的净光化学生成速率为H2O2生成速率与损失速率之差。H2O2生成潜势定义为观测地点12 h H2O2净光化学生成速率的总和。特定污染物相对增量反应活性计算见2.2节(式1)。
2. 结果与讨论(Results and discussion)
2.1 浓度水平和变化规律
北京2018年9—10月份观测期间H2O2的平均体积浓度为(0.29±0.36)×10−9。观测期间H2O2的最大体积混合比为1.98×10−9。大气过氧化氢表现出明显的日变化规律(图1),变化规律与同为光化学产物的O3一致,不过H2O2峰值出现时间略晚于O3(图1),过氧化氢在午后15:00左右达到日间峰值。
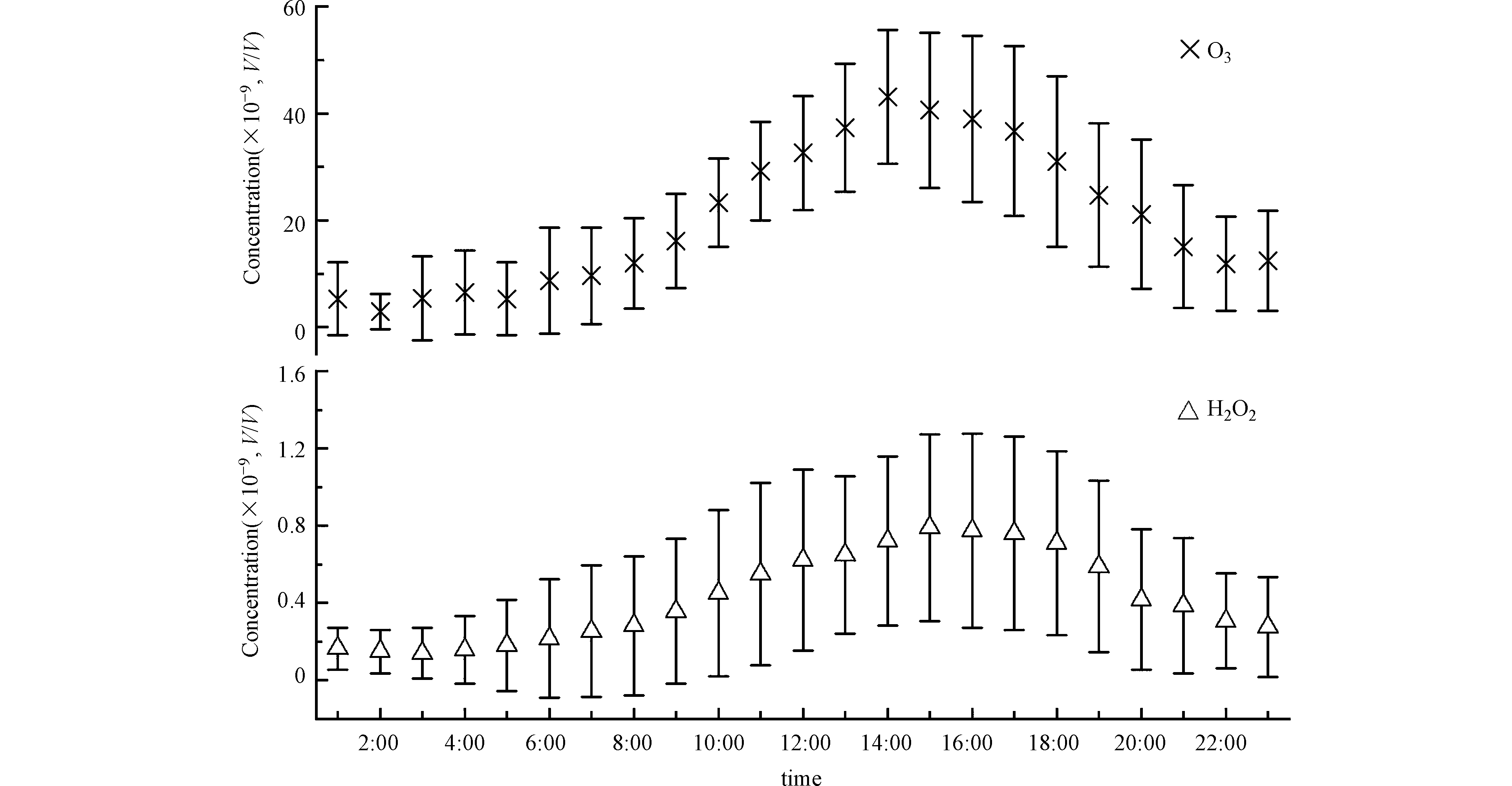 图 1 观测期间H2O2和O3的小时平均日变化Figure 1. Diurnal variation of H2O2 and O3 during the observation
图 1 观测期间H2O2和O3的小时平均日变化Figure 1. Diurnal variation of H2O2 and O3 during the observation北京地区秋季的H2O2观测浓度与国际和国内其他观测所给出的大气过氧化氢的浓度范围基本一致(表2)。文献报告的H2O2的体积浓度范围在0.01×10−9—11.3×10−9[4]之间,由于夏季太阳辐射的增加而导致光化学过程的增强,文献中同一地区的大气过氧化氢的浓度水平常在夏季较高,冬季较低[18]。本次观测期间北京地区以晴朗和多云天气为主导,9月主要为晴朗天气,10月多为多云天气。共捕捉到4次颗粒物污染过程,其中3次发生在10月。观测期间9月H2O2和O3浓度显著高于10月(图2),NOx在10月浓度显著高于9月。10月份较高的NOx浓度通过与HO2自由基结合,可能产生抑制HO2与HO2自由基的结合,而减少H2O2的生成的效应。
表 2 以往观测中大气H2O2的浓度范围Table 2. Concentration range of atmospheric H2O2 in previous observations观测地点Observation site 观测时期Observation time 体积浓度范围Concentration range(×10−9) 文献Reference 北京,中国 2015—2016,春、夏、冬 0.01—2.08 Qin et al.(2018)[18] 北京,中国 2010—2011,夏 D.L—1.59 Liang et al.(2013)[19] 北京,中国 2017—2018,冬 0—0.68 Ye et al.(2018)[20] 珠三角,中国 2006,夏 0.02—4.6 Hua et al.(2008)[7] 北京,中国 2013,秋 0.10—2.4 Zhang et al.(2018)[21] 华北平原,中国 2014,夏 0.01—11.3 Wang et al.(2016)[4] 泰山,中国 2007,春、夏 D.L —1.28 春、D.L —3.55 夏 Ren et al.(2008)[22] 北京,中国 2008,夏 D.L —2.34 He et al.(2010)[2] 萨格勒布,克罗地亚 2004,夏 0.05—6.2 Acker et al.(2008)[23] 首尔,韩国 2002—2004,秋 0.07—0.87 Lee et al. (2008)[24] Toyama Prefecture,日本 2012—2015,夏、冬 0.01—3.5 Watanabe et al.(2018)[25] Whiteface Mountain,北美 1995,夏 0.15—5.15 Balasubramanianand Husain.(1997)[26] 卡罗莱纳州海岸,美国 1986,春 2.4 (max) Barth et al.(1989)[27] 东部,美国 1984,秋 D.L—4.1 Heikes et al.(1987)[28] 加利福尼亚,洛杉矶 2004—2005,春、夏、秋、冬 0.2—3.0 Arellanes et al.(2006)[29] Nikko,日本 1998—2000,夏 0.2—1.6 Takami et al.(2003)[30] | Show TableDownLoad:
CSV
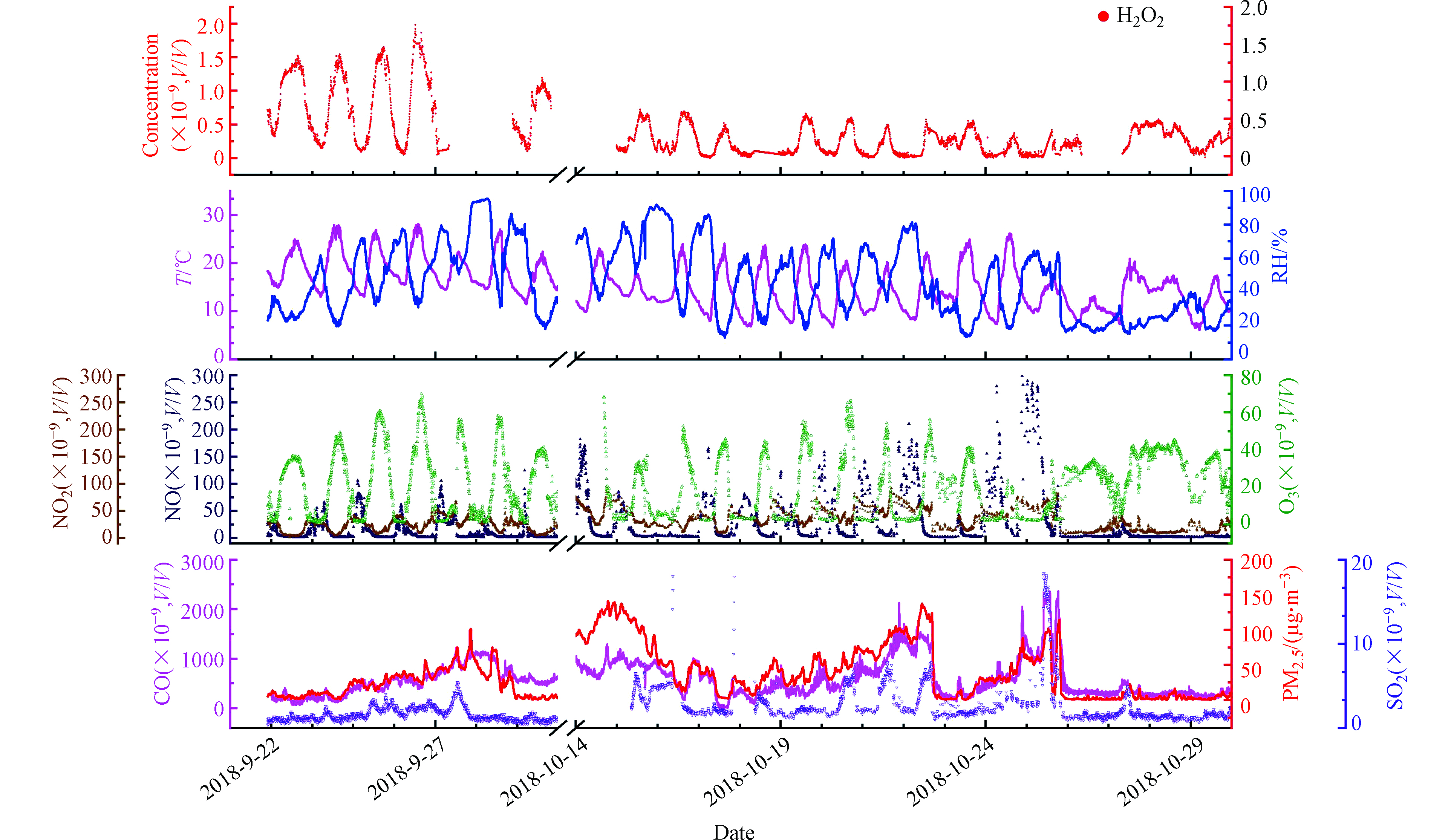 图 2 观测期间H2O2及相关污染物和气象因子的变化Figure 2. Time series of H2O2, trace gaseous species and meteorological data during the observation
图 2 观测期间H2O2及相关污染物和气象因子的变化Figure 2. Time series of H2O2, trace gaseous species and meteorological data during the observationH2O2单日浓度变化曲线并不平滑,经常会有较大的凸起,说明影响大气中H2O2浓度因素的复杂性。一般来说,随着太阳辐射逐渐减弱,光化学产物H2O2以及O3的生成会随着光化学反应活性的降低而逐渐减少。本研究在部分日期的光化学活性较弱或光化学停止的时段观测到明显的H2O2与O3的同步增加的情况(图3)。不同于其它研究观测到的H2O2浓度夜间上升而O3浓度下降,而被推测为臭氧与烯烃的反应或NO3与有机化合物的反应的情况[2],本研究中的O3与H2O2在下午及夜间是同时涨落,更可能是污染气团区域传输的结果。以往对北京城区及近郊地区的大气H2O2的观测表明,北京郊区的夜间O3和H2O2浓度显著高于城区[20],由于北京的特殊地形,山谷风导致平原地区风场日变化为午后偏南风,夜间偏北风的规律,因而图3中夜间H2O2和O3的同步升高现象有可能是北京北部山区气团的水平或垂直方向传输造成的。
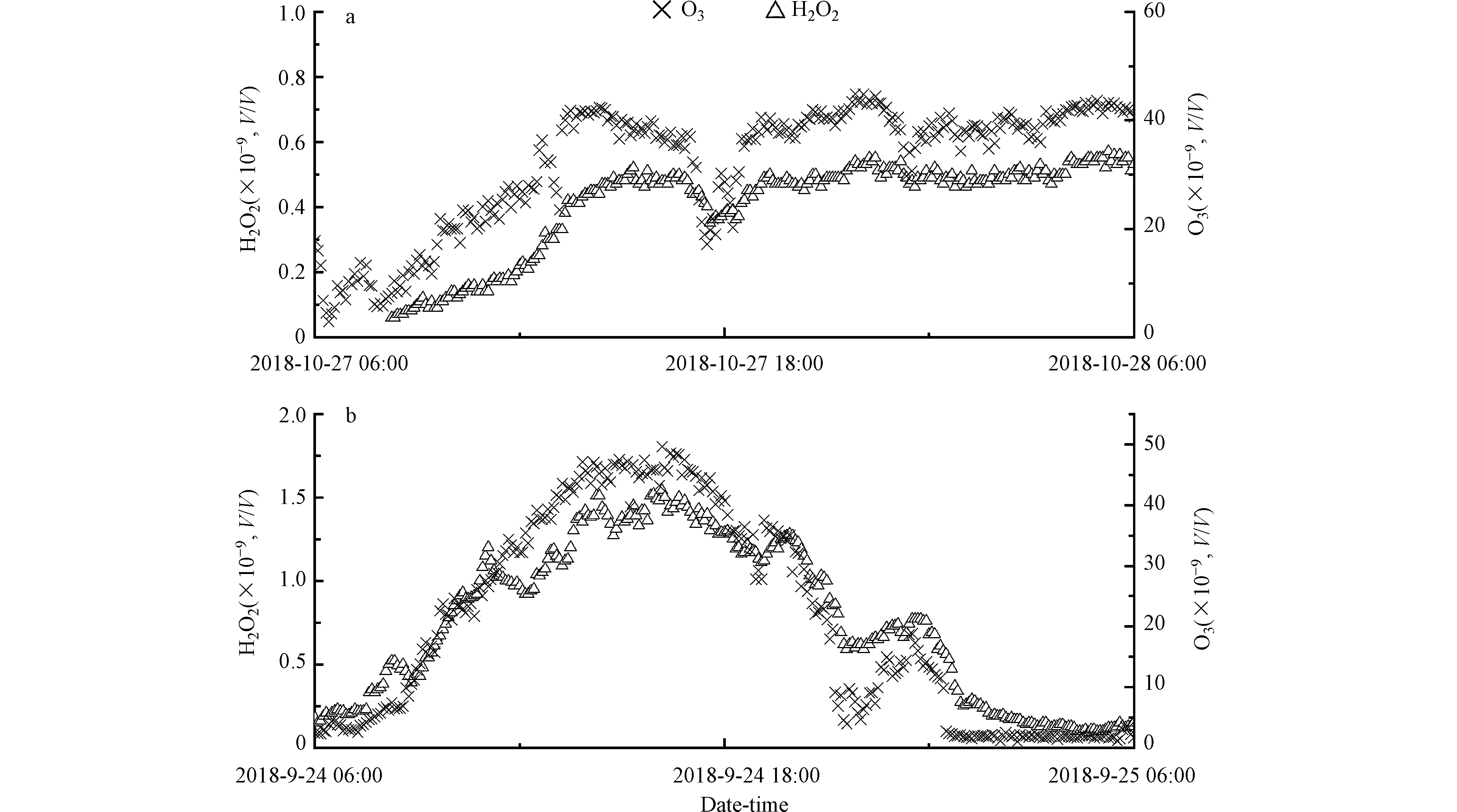 图 3 部分观测日期出现夜间H2O2与O3高值的情况Figure 3. The observed increase of nocturnal H2O2 and O3 during the observation
图 3 部分观测日期出现夜间H2O2与O3高值的情况Figure 3. The observed increase of nocturnal H2O2 and O3 during the observation2.2 H2O2生成对前体物变化的敏感性
大气H2O2主要是HO2与HO2自由基结合生成的,但大气HO2的浓度水平和生成速率则受到其前体物CO、NOx以及VOCs的大气浓度和光照情况影响。有研究发现,霾天TVOC相比非霾天具有显著增加,其中VOCs不同类别烷烃、烯烃、芳烃和炔烃等具有不同的增加比例,增加最快的为烯烃[31]。且也有研究表明,霾天较高水平的VOCs、O3和水汽对H2O2的生成具有较大贡献[18]。考虑到霾天较高VOCs、O3水平和气溶胶非均相作用对H2O2生成产生影响,以及霾天较高相对湿度对H2O2氧化SO2生成硫酸盐对SOA的贡献,了解VOCs各类别及主要单一组分对H2O2生成的影响对提出有效的污染控制措施具有指示意义。目前,对H2O2前体物尤其是VOCs各类别以及主要单一组分对H2O2生成贡献的研究相对较少。相对增量反应活性方法广泛用来研究O3生成对前体物的敏感性。研究中H2O2生成的相对增量反应活性计算参考O3生成的相对增量反应活性计算方法,H2O2相对增量反应活性的计算为,改变H2O2特定前体物源强后,H2O2生成速率变化的百分比与H2O2特定前体物源强变化百分比的比率(式1)[4]。
RIR(X)=ΔPH2O2(X)/PH2O2(X)ΔS(X)/S(X) (1) 式中,X为特定污染物种,S(X)为污染物X的源效应,ΔS(X)为源强变化量,
PH2O2 (X)为7:00—19:00 H2O2生成潜势,ΔPH2O2 (X)为源效应变化ΔS(X)后PH2O2 (X)的变化量。OBM耦合RACM区域大气化学机理用来模拟计算前体物对H2O2生成的相对增量反应活性。RACM中根据排放速率的大小、官能团的相似性以及化合物与OH的反应性对VOCs进行分类。具体分类和机理中涉及的237个反应见文献[32]。按照RACM机理中对VOCs的分类方法将VOCs分为烷烃(ETH、HC3、HC5和HC8)、烯烃(ETE、DIEN、OLI和OLT)和芳烃(XYL和TOL)。各前体物源强削减为10%,不同前体物对H2O2生成的相对增量反应活性结果如图4。结果表明,北京地区秋季H2O2的生成对烯烃最为敏感,在单一前体物中对异戊二烯和反式-2-丁烯最为敏感,此结果与Wang等[4]的研究结果一致,提示在光化学条件较好且具有较高烯烃水平的条件下有利于大气H2O2的生成。反式-2-丁烯主要为机动车排放物种,有效的机动车控制策略可能除了降低大气臭氧和颗粒物污染,也有助于减少H2O2的生成。异戊二烯为植物源排放主要产物,其对大气H2O2的生成有较大的贡献作用。
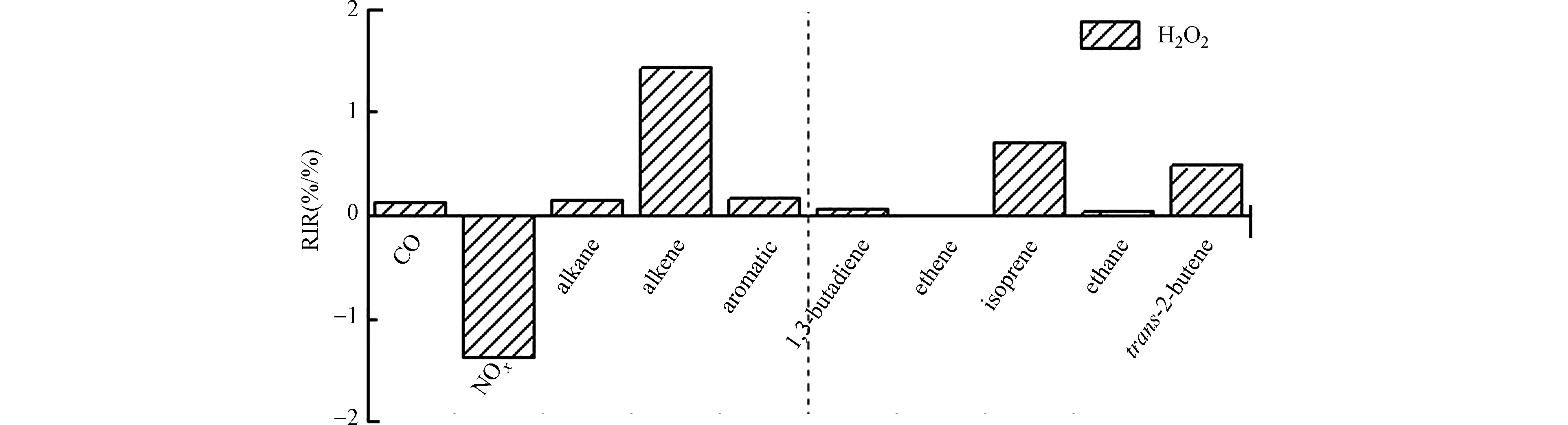 图 4 H2O2生成速率对主要前体物种及个别单一物种的敏感性Figure 4. Sensitivity of hydrogen peroxide production to the major precursor groups and individual species
图 4 H2O2生成速率对主要前体物种及个别单一物种的敏感性Figure 4. Sensitivity of hydrogen peroxide production to the major precursor groups and individual species2.3 H2O2源、汇分析
为了解不同反应途径对大气H2O2生成及损耗的贡献,研究采用了基于观测的OBM模型(observation-based model),耦合气相化学RACM机制(regional atmospheric chemistry mechanism)计算了不同气相反应途径对H2O2生成及损耗的贡献。模式中H2O2的生成途径主要包括HO2与HO2结合的反应、O3和烯烃的反应,损失途径包括H2O2与OH自由基的反应、H2O2的光解反应以及H2O2的干沉降。结果表明,HO2与HO2自由基结合的反应是大气H2O2的主要气相生成途径,贡献了总H2O2气相生成途径的94.23%,O3与烯烃的反应仅占总H2O2生成的5.77%。不同H2O2的去除途径对H2O2消耗的贡献依次为H2O2干沉降、H2O2与OH反应和H2O2光解,依次贡献了总H2O2损失的61.59%、27.40%和11.00%(图5)。
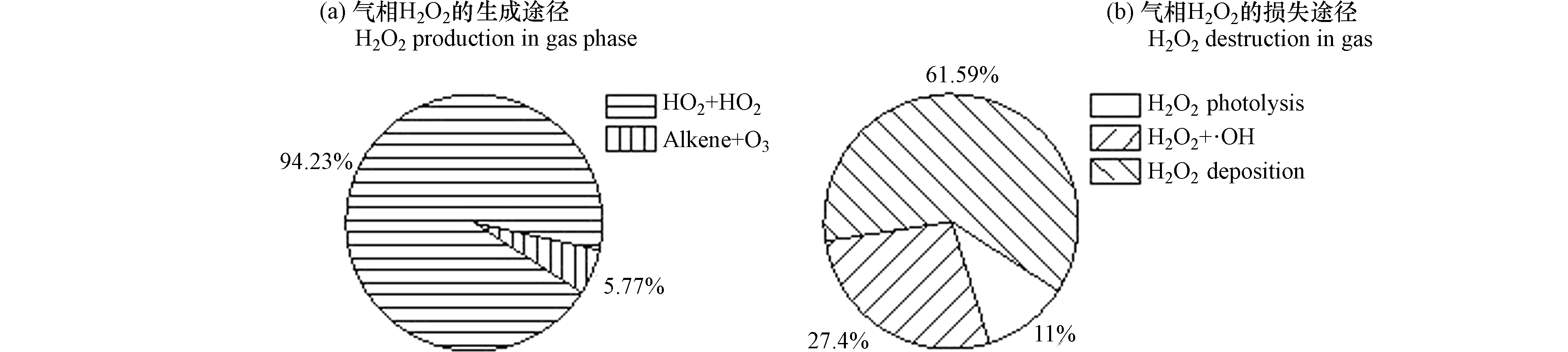 图 5 气相中不同途径对H2O2生成及损失的贡献模拟结果(a) H2O2的气相生成途径 (b) H2O2的气相损失途径Figure 5. Modeled contributions of different pathways to H2O2 production and destruction in gas phase (a) production (b) destruction
图 5 气相中不同途径对H2O2生成及损失的贡献模拟结果(a) H2O2的气相生成途径 (b) H2O2的气相损失途径Figure 5. Modeled contributions of different pathways to H2O2 production and destruction in gas phase (a) production (b) destruction不过,模式中对H2O2生成速率的模拟对比观测结果存在低估的情况。本研究中基于实测环境大气VOCs和NOx浓度限制的RACM机理模拟所给出的HO2自由基日最高浓度为3.61×108 molecule·cm−3,其对应的日最高H2O2生成体积速率约为0.2×10−9·h−1。由模式模拟得到的日间过氧化氢生成体积速率
PH2O2 的累积仅约1.54×10−9,在去除了光解和沉降的去除情况下不足以达到观测到的H2O2峰值体积浓度1.98×10−9。此前在华北地区也有观测发现气溶胶上HO2的非均相转化可能是H2O2的重要源[9]。也有观测表明生物质燃料燃烧可能是局地大气过氧化氢的重要来源[4]。这些额外来源可能贡献了部分的日间过氧化氢生成途径。此外,近年来有研究通过对自由基HO2实测值和模拟值的比较,认为高NOx条件下,模式中对HO2自由基的低估可能高达5倍,且HO2观测值与模拟值的比值随着NOx的增加呈上升的趋势[33]。本研究模拟中H2O2的低估也可能与模式中HO2自由基源的缺失有关。由于本研究未开展气溶胶成分和大气过氧自由基的测量,有关H2O2实测值与模拟值之间的差异,还需未来通过更多观测和模型计算进行深入研究。3. 结论(Conclusion)
本研究对北京秋季9—10月份的大气过氧化氢进行了观测。结果表明,观测期间其体积浓度水平为(0.29±0.36)×10−9;日变化明显,早晚较低,最大值出现在午后15:00—16:00,白天的日变化与O3相似,说明观测期间本地光化学作用主导H2O2生成;夜间H2O2上升,似与污染气团的区域传输有关。H2O2生成速率对光化学前体物的敏感性分析表明,烯烃对H2O2的生成最敏感,光照条件好且烯烃水平高有利于H2O2的生成。大气H2O2生成及损耗的模拟表明,HO2与HO2结合反应是H2O2的主要已知生成途径,H2O2干沉降为主要损失途径。H2O2观测与模拟值的差异可能与机制中缺失颗粒物上的非均相源/汇有关,高NOx条件下模式中HO2自由基源的缺失也可能是导致这种结果的原因。
-

图 2 Cr污染土壤的根际效应和微生物作用
Figure 2. Rhizosphere effect and microbial process of Cr contaminated soil

图 3 修复后场地土壤中Cr的迁移和转化
Figure 3. The migration and transport of Cr in post-remediated soil
表 1 Cr(VI)还原的常用反应体系
Table 1. The common system of Cr(VI) reduction
反应体系 还原类型 还原条件 环境介质 还原剂/还原微生物 还原效果 参考文献 材料体系 铁基材料 材料投加量2.5%,土壤中Cr(Ⅵ)质量分数85.8 mg·kg−1 土壤 零价铁 土壤中Cr(Ⅵ)质量分数降低92% [53] 摩尔比≥ 5∶1,土壤中Cr(Ⅵ)质量分数94 mg·kg−1 土壤 硫酸亚铁 土壤中Cr(Ⅵ)质量分数降低84% [54] pH = 6.0,Cr(Ⅵ)质量浓度4.65 mg·L−1 水溶液 硫化亚铁 1 h,Cr(Ⅵ)质量浓度降低99%以上 [55] 硫基材料 材料投加量5%,Cr(Ⅵ)浸出浓度664.0 mg·L−1 土壤 硫化钠 Cr(Ⅵ)浸出浓度降低99%以上 [56] pH = 7.6,H2S浓度800 μmol·L−1,Cr(Ⅵ)浓度40 μmol·L−1 水溶液 硫化氢 pH = 7.6时,21 min内Cr(Ⅵ) 浓度降低到12.6 μmol·L−1 [58] 碳基材料 pH = 7.5和pH = 5.5,材料投加量5%,土壤中Cr(Ⅵ)质量分数100 mg·kg−1 土壤 生物炭 土壤中Cr(Ⅵ)质量分数分别降低74%和88% [60] pH = 5.8,固液比1g∶100 mL,Cr(Ⅵ)浓度1.1 mmol·L−1 水溶液 泥炭腐殖酸 2 d后Cr(Ⅵ)浓度降低100% [62] 生物体系 还原微生物 pH = 7.2,Cr(Ⅵ)质量浓度20~100 mg·L−1 水溶液 Aeromonas hydrophila ATCC 7966 60 h内Cr(Ⅵ)浓度达到平衡,Cr(Ⅵ)修复速率分别为0.200,0.175,0.125,0.090和0.075 mg·(L−1·h−1) [64] pH = 7.0,Cr(Ⅵ)质量浓度分别在10~70、80~300 、500 mg·L−1 水溶液 Stenotrophomonas maltophilia 各组Cr(Ⅵ)质量浓度分别降低100%,98%~ 99%和92% [65] 耦合体系 材料+微生物 材料投加量1.5%,土壤中Cr(Ⅵ)质量分数100 mg·kg−1,25 ℃,土壤湿度60% 土壤 纳米FeS@HA-Cr-resistant microflora 90 d后土壤中Cr(Ⅵ)质量分数下降99.16% [66] pH = 7.0,初始Cr(Ⅵ)浓度0.1 mmol·L−1,进液Cr(Ⅵ)浓度0.2 mmol·L−1 水溶液 氢氧化铁/三氧化二铁-Shewanella alga strain BrY Cr(Ⅵ)最大还原速率为5.5 μg·h−1 [67]
下载: 导出CSV
表 2 EPA超级基金Cr污染场地修复后的长期监测数据[70]
Table 2. Long-term monitoring data of EPA Superfund Cr contaminated in post-remediation sites[70]
序号 场地名称 涉及污染物 修复措施 监测年份 地下水中Cr是否超标 Cr质量浓度分析 1 Bennington Municipal Sanitary Landfill As、Hg、Cr、有机物等 表面覆盖阻隔、拦截沟、渗滤液收集及处理系统、沉积物清理 1999—2019 否 监测点位Cr质量浓度稍有波动,但未曾超标。 2 BFI Sanitary Landfill As、Cr、有机物等 表面覆盖阻隔、地下水拦截沟、气体收集及处理系统 1994—2019 是 Cr质量浓度基本均低于检测限,仅MW-9中Cr超标时有发生,监测质量浓度总体无明显变化趋势。 3 Elizabeth Mine As、Cr、有机物等 污染区覆盖阻隔,部分污染土固化处理;污染水域填土和覆土 2004— 2019 — 缺乏Cr相关长期监测数据。 4 Parker Sanitary Landfill As、Cr、有机物等 表面屏障、地下水处理系统、气体收集及处理系统、地下水及沉积物定期监测 1999-2019 否 地下水中Cr监测未超标。 5 Pownal Tannery As、Hg、Cr、有机物等 废弃物清挖、固化并进行原位覆盖阻隔,地下水及沉积物定期监测 1999—2019 否 地下水中Cr监测未超标。 6 Tansitor Electronics Inc. As、Cr、有机物等 制度控制,长期监测 1999—2019 — 缺乏Cr相关长期监测数据。 7 Saco Tannery Waste Pits As、Cr、有机物等 废物堆和排水区覆盖阻隔,补偿性湿地系统修复 1993—2019 是 地下水Cr基本均低于检测限,仅MW-114B质量浓度存在超标,监测质量浓度总体无明显变化趋势。 8 Hatheway & Patterson As、Cr、有机物等 拆除建筑物,As污染土原位固化/稳定化,二恶英污染土异位处理 2004-2019 否 地下水中Cr监测未超标。 9 Hocomonoco Pond As、Cr、有机物等 DNAPL收集处理系统,底泥清运,污染土固化。 1999—2019 — 缺乏Cr相关长期监测数据。 10 Nyanza Chemical Waste Dump As、Hg、Cr、有机物等 地下水收集处理系统,底泥清运,污染土固化/稳定化 1990—2019 — 缺乏Cr相关长期监测数据。 11 Silresim Chemical Corporation As、Hg、Cr、有机物等 阻隔装置,土壤真空/气相抽提,地下水处理设施(地下水抽提、气提、碳吸附、热氧化等) 1994—2019 否 地下水中Cr监测未超标。 12 Federal Facility: South Weymouth Naval Air Station As、Cr、有机物等 划为10区分开修复。以As、Cr污染的OU1填埋区为例,清理表土,铺设覆盖层,重建湿地,自然衰减监测 2004—2019 否 地下水中Cr监测未超标。 13 W.R. Grace & Co., Inc As、Cr、有机物等 污染土和底泥固化/稳定化、堆存并表面覆盖,地下水抽提以及自然衰减 1994-2019 否 地下水中Cr监测未超标。 14 Wells G&H, Woburn As、Hg、Cr、有机物等 原位土壤气相抽提,污泥清挖处置,地下水抽提处理,原位化学氧化 1994—2019 否 地下水中Cr测未超标。 15 Federal Facility: Pease Air Force Base As、Hg、Cr、有机物等 固废及底泥清挖异位处置,灌注土壤改良剂,地下水抽提处理及生物修复。土壤和地下水曝气(地下水位以下),土壤气相抽提(地下水位以上) 1994—2019 否 地下水中Cr监测未超标。底泥中位于Paol上游和中游的点位Cr质量浓度存在一次超标,推测与附近污染场地的地表径流相关。 16 Pine Street Canal As、Cr(Ⅵ)、有机物等 污染底泥覆盖阻隔,栖息地恢复以及长期监测 2001-2019 否 2007—2011,地下水中Cr监测未超标;2012—2016,Cr不继续作为关注污染物进行监测。 17 Town Garage/Radio Beacon Cr、有机物等 制度控制,长期监测 1994—2019 是 地下水Cr轻度超标,浓度偏高可能是采样扰动有关。采用低流速地下水采样后,重金属指标均未超标。 18 New Hampshire Plating As、Cr、有机物等 石灰和次氯酸钠处理排水系统,建筑物及废弃物清运,污染土化学稳定化 2004-2019 否 地下水中Cr监测未超标。
下载: 导出CSV
-
[1] YANG Z H, ZHANG X M, JIANG Z, et al. Reductive materials for remediation of hexavalent chromium contaminated soil: A review[J]. Science of the Total Environment, 2021, 773: 145654. doi: 10.1016/j.scitotenv.2021.145654 [2] 王春峰, 姚丹, 陈冠飞, 等. 赤泥重金属和放射性元素的毒性浸出和生物可给性[J]. 环境科学研究, 2017, 30(5): 809-816. [3] ALI S, BAI P, ZENG F R, et al. The ecotoxicological and interactive effects of chromium and aluminum on growth, oxidative damage and antioxidant enzymes on two barley genotypes differing in Al tolerance[J]. Environmental and Experimental Botany, 2011, 70(2/3): 185-191. [4] SU H J, FANG Z Q, TSANG P E, et al. Stabilisation of nanoscale zero-valent iron with biochar for enhanced transport and in-situ remediation of hexavalent chromium in soil[J]. Environmental Pollution, 2016, 214: 94-100. doi: 10.1016/j.envpol.2016.03.072 [5] REALE L, FERRANTI F, MANTILACCI S, et al. Cyto-histological and morpho-physiological responses of common duckweed (Lemna minor L.) to chromium[J]. Chemosphere, 2015, 145: 98-105. [6] SUZUKI T, KAWAI K, MORIBE M, et al. Recovery of Cr as Cr(III) from Cr(VI)-contaminated kaolinite clay by electrokinetics coupled with a permeable reactive barrier[J]. Journal of Hazardous Materials, 2014, 278: 297-303. doi: 10.1016/j.jhazmat.2014.05.086 [7] XIA S P, SONG Z L, JEYAKUMAR P, et al. Characteristics and applications of biochar for remediating Cr(VI)-contaminated soils and wastewater[J]. Environment Geochemistry and Health, 2020, 42: 1543-1567. doi: 10.1007/s10653-019-00445-w [8] 谷庆宝, 马福俊, 张倩, 等. 污染场地固化/稳定化修复的评价方法与标准[J]. 环境科学研究, 2017, 30(5): 755-764. [9] 姚瑶, 张世熔, 王怡君, 等. 3种环保型淋洗剂对重金属污染土壤的淋洗效果[J]. 环境工程学报, 2018, 12(7): 2039-2046. doi: 10.12030/j.cjee.201801198 [10] XU Y F, XU X J, HOU H, et al. Moisture content-affected electrokinetic remediation of Cr(VI)-contaminated clay by a hydrocalumite barrier[J]. Environmental Science and Pollution Research, 2016, 23: 6517-6523. doi: 10.1007/s11356-015-5685-y [11] SHI L, DENG X P, YANG Y, et al. A Cr(VI)-tolerant strain, Pisolithus sp1, with a high accumulation capacity of Cr in mycelium and highly efficient assisting Pinus thunbergii for phytoremediation[J]. Chemosphere, 2019, 224: 862-872. doi: 10.1016/j.chemosphere.2019.03.015 [12] FIELD E K, BLASKOVICH J P, PEYTON B M, et al. Carbon-dependent chromate toxicity mechanism in an environmental Arthrobacter isolate[J]. Journal of Hazardous Materials, 2018, 355: 162-169. doi: 10.1016/j.jhazmat.2018.05.020 [13] LI D N, LI G H, ZHANG D Y. Field-scale studies on the change of soil microbial community structure and functions after stabilization at a chromium-contaminated site[J]. Journal of Hazardous Materials, 2021, 415: 125727. doi: 10.1016/j.jhazmat.2021.125727 [14] SHEN Z T, JIN F, O’CONNOR D, et al. Solidification/stabilization for soil remediation: An old technology with new vitality[J]. Environmental Science & Technology, 2019, 53: 11615-11617. [15] AO M, CHEN X T, DENG T H, et al. Chromium biogeochemical behaviour in soil-plant systems and remediation strategies: A critical review[J]. Journal of Hazardous Materials, 2022, 424: 127233. doi: 10.1016/j.jhazmat.2021.127233 [16] 王亚平, 黄毅, 王苏明, 等. 土壤和沉积物中元素的化学形态及其顺序提取法[J]. 地质通报, 2005(8): 728-734. doi: 10.3969/j.issn.1671-2552.2005.08.009 [17] 刘雪, 王兴润, 张增强. pH和有机质对铬渣污染土壤中Cr赋存形态的影响[J]. 环境工程学报, 2010, 4(6): 1436-1440. [18] 郭越宏, 王建生, 张学洪, 等. 腐植酸对李氏禾-红壤处理系统中的铬赋存形态、微生物群落及酶活性影响[J]. 环境工程, 2021, 39(12): 234-242. [19] LIU W Z, LI J, ZHENG J Y, et al. Different pathways for Cr(III) oxidation: Implications for Cr(VI) reoccurrence in reduced chromite ore processing residue[J]. Environmental Science & Technology, 2020, 54(19): 11971-11979. [20] PALMER C D, WITTBRODT P R, FSIH W. Chromium mineral phases at a highly contaminated hardchrome plating site[J]. Eos Transactions of the American Geophysical Union, 1990, 71(36): 1068. [21] BARTLETT J R. Chromium cycling in soils and water: Links, gaps, and methods[J]. Environmental Health Perspectives, 1991, 92(178): 17-24. [22] PAN C, LIU H, CATALANO J G, et al. Rates of Cr(VI) generation from CrxFe1-x(OH)3 solids upon reaction with manganese oxide[J]. Environmental Science & Technology, 2017, 51(21): 12416-12423. [23] BARTLETT R, JAMES B. Behavior of chromium in soils: III. Oxidation[J]. Journal of Environmental Quality, 1979, 8(1): 31-35. [24] THOMAS H Y, CHEN, TRAINA S J. Inhibited Cr(VI) reduction by aqueous Fe(II) under hyperalkaline conditions[J]. Environmental Science & Technology, 2004, 38(21): 5535-5539. [25] MAHDIEH K, SHAHIN O, NOSRATOLLAH N, et al. Treatment of Cr(VI)-spiked soils using sulfur-based amendments[J]. Archives of Agronomy and Soil Science, 2016, 62(10): 1474-1485. doi: 10.1080/03650340.2016.1152358 [26] BLOOMFIELD C, PRUDEN G. The behaviour of Cr(VI) in soil under aerobic and anaerobic conditions[J]. Environmental Pollution Series A, Ecological and Biological, 1980, 23(2): 103-114. doi: 10.1016/0143-1471(80)90058-6 [27] JOBBY R, JHA P, YADAV A K, et al. Biosorption and biotransformation of hexavalent chromium [Cr(VI)]: A comprehensive review[J]. Chemosphere, 2018, 207: 255-266. doi: 10.1016/j.chemosphere.2018.05.050 [28] 杜晓丽, 刘殿威, 崔申申. 径流入渗土壤胶体释放对重金属截留的影响[J]. 中国环境科学, 2022, 42(3): 1278-1286. doi: 10.3969/j.issn.1000-6923.2022.03.033 [29] 蒋越, 李广辉, 王东辉, 等. 天然有机酸和DTPA组合工艺对Cr(Ⅵ)污染土壤的淋洗修复[J]. 环境工程学报, 2020, 14(7): 1903-1914. doi: 10.12030/j.cjee.201911073 [30] 李东, 贺丽洁, 盛培培. Tessier连续提取法用于土壤铬分析的Cr(Ⅵ)-Cr(Ⅲ)转化及适用性[J]. 环境工程学报, 2021, 15(7): 2368-2378. doi: 10.12030/j.cjee.202012141 [31] MCLEAN J E, BLEDSOE B E. Behavior of metals in soils[M]. Washington, DC: EPA Ground Water Issue, 1992: EPA 540-S-92-018. [32] PUNIA S, WU Lisha, KHODADOUST A P. Adsorption of hexavalent chromium from water using manganese-aluminum coated sand: Kinetics, equilibrium, effect of pH and ionic strength[J]. Journal of Environmental Science and Health, Part A, 2021, 56: 334-345. doi: 10.1080/10934529.2021.1877513 [33] SHI Z Q, PENG S M, LIN X F, et al. Predicting Cr(VI) adsorption on soils: The role of the competition of soil organic matter[J]. Environmental Science:Processes and Impacts, 2020, 22(1): 95-104. doi: 10.1039/C9EM00477G [34] ZHANG X W, TONG J X, WEI W S, et al. Adsorption and desorption for dynamics transport of hexavalent chromium (Cr(VI)) in soil column[J]. Environmental Science and Pollution Research, 2018, 25: 459-468. doi: 10.1007/s11356-017-0263-0 [35] NJOYA O, ZHAO S X, QU Y F, et al. Performance and potential mechanism of Cr(VI) reduction and subsequent Cr(III) precipitation using sodium borohydride driven by oxalate[J]. Journal of Environmental Management, 2020, 275: 111165. doi: 10.1016/j.jenvman.2020.111165 [36] COOK K R. In situ treatment of soil and groundwater contaminated with chromium-technical resource guide[M]. Washington DC: EPA, 2000: EPA 625-R-00-005. [37] DAI C, ZUO X B, CAO B, et al. Homogeneous and heterogeneous (Fex, Cr1-x)(OH)3 precipitation: Implications for Cr sequestration[J]. Environmental Science & Technology, 2016, 50(4): 1741-1749. [38] LEE G, HERING J G. Oxidative dissolution of chromium(III) hydroxide at pH 9, 3, and 2 with product inhibition at pH 2[J]. Environmental Science & Technology, 2005, 39(13): 4921-4928. [39] BARTLETT R J, KIMBLE J M. Behavior of chromium in soils, Ⅰ: Trivalent forms[J]. Journal of Environmental Quality, 1976, 5(4): 379-383. [40] JAMES B R, BARTLETT R J. Behavior of chromium in soils, VII: Adsorption and reduction of hexavalent forms[J]. Journal of Environmental Quality, 1983, 12(2): 177-181. [41] GUSTAFSSON J P, PERSSON I, OROMIEH A G, et al. Chromium(III) complexation to natural organic matter: Mechanisms and modeling[J]. Environmental Science & Technology, 2014, 48(3): 1753-1761. [42] CAO X H, GUO J, MAO J D, et al. Adsorption and mobility of Cr(III)-organic acid complexes in soils[J]. Journal of Hazardous Materials, 2011, 192(3): 1553-1538. [43] KANMANI P, ARAVIND J, PRESTON D. Remediation of chromium contaminants using bacteria[J]. International Journal of Environmental Science and Technology, 2011, 9(1): 183-193. [44] GUO S Y, XIAO C Q, ZHOU N, et al. Speciation, toxicity, microbial remediation and phytoremediation of soil chromium contamination[J]. Environmental Chemistry Letters, 2020, 19(2): 1413-1431. [45] ASATIANI N V, ABULADZE M K, KARTVELISHVILI T M, et al. Effect of chromium(VI) action on Arthrobacter oxydans[J]. Current Microbiology, 2004, 49(5): 321-326. doi: 10.1007/s00284-004-4351-2 [46] WANG Y Y, LIU Y D, ZHENG K X, et al. The role of extracellular polymeric substances (EPS) in the reduction of Cr(VI) by Pannonibacter phragmitetus BB[J]. Journal of Environmental Chemical Engineering, 2021, 9(5): 106163. doi: 10.1016/j.jece.2021.106163 [47] XU Z R, CAI M L, CHEN S H, et al. High-affinity sulfate transporter Sultr1;2 is a major transporter for Cr(VI) uptake in plants[J]. Environmental Science & Technology, 2021, 55(3): 1576-1584. [48] ZENG Q, HU Y T, YANG Y R, et al. Cell envelop is the key site for Cr(Ⅵ) reduction by Oceanobacillus oncorhynchi W4, a newly isolated Cr(Ⅵ) reducing bacterium[J]. Journal of Hazardous Materials, 2019, 368: 149-155. doi: 10.1016/j.jhazmat.2019.01.031 [49] LIU J, DUAN C Q, ZHANG X H, et al. Potential of Leersia hexandra Swartz for phytoextraction of Cr from soil[J]. Journal of Hazardous Materials, 2011, 188(1/3): 85-91. [50] JAN S, NOMAN A, KAYA C, et al. 24-Epibrassinolide alleviates the injurious effects of Cr(VI) toxicity in tomato plants: Insights into growth, physio-biochemical attributes, antioxidant activity and regulation of ascorbate-glutathione and glyoxalase cycles[J]. Journal of Plant Growth Regulation, 2020, 39: 1587-1604. doi: 10.1007/s00344-020-10169-2 [51] VAMERALI T, BANDIERA M, COLETTO L, et al. Phytoremediation trials on metal- and arsenic-contaminated pyrite wastes (Torviscosa, Italy)[J]. Environmental Pollution, 2009, 157(3): 887-94. doi: 10.1016/j.envpol.2008.11.003 [52] QIAN Y T, QIN C D, CHEN M M, et al. Nanotechnology in soil remediation applications vs implications[J]. Ecotoxicology and Environmental Safety, 2020, 201: 110815. doi: 10.1016/j.ecoenv.2020.110815 [53] ZHOU Z C, ALHADIDI Q, DELIZ K Q, et al. Removal of oxyanion forming elements from contaminated soils through combined sorption onto zero-valent iron (ZVI) and magnetic separation: Arsenic and chromium as case studies[J]. Journal of Soil Contamination, 2019, 29(2): 1-12. [54] PALMA L D, GUEYE M T, PETRUCCI E. Hexavalent chromium reduction in contaminated soil: A comparison between ferrous sulphate and nanoscale zero-valent iron[J]. Journal of Hazardous Materials, 2015, 281(8): 70-76. [55] WANG T, QIAN T W, HUO L J, et al. Immobilization of hexavalent chromium in soil and groundwater using synthetic pyrite particles[J]. Environmental Pollution, 2019, 255: 112992. doi: 10.1016/j.envpol.2019.112992 [56] YUAN W Y, XU W T, WU Z B, et al. Mechanochemical treatment of Cr(VI) contaminated soil using a sodium sulfide coupled solidification/stabilization process[J]. Chemosphere, 2018, 212: 540-547. doi: 10.1016/j.chemosphere.2018.08.121 [57] 赖冬麟, 张奇, 陈亭亭, 等. 张家口市某机械厂原址电镀污染场地土壤修复工程实践[J]. 环境工程, 2020, 38(6): 75-80. [58] LAN Y Q, DENG B L, KIM C, et al. Catalysis of elemental sulfur nanoparticles on chromium(VI) reduction by sulfide under anaerobic conditions[J]. Environmental Science & Technology, 2005, 39(7): 2087-2094. [59] SONG Z L, FANG L C, WANG J, et al. Use of biogas solid residue from anaerobic digestion as an effective amendment to remediate Cr(VI)-contaminated soils[J]. Environmental Science and Pollution Research, 2019, 26: 13041-13053. doi: 10.1007/s11356-019-04786-y [60] MANDAL S, SARKAR B, BOLAN N, et al. Enhancement of chromate reduction in soils by surface modified biochar[J]. Journal of Environmental Management, 2017, 186(2): 277-284. [61] BASHIR M S, NAVEED M, AHMAD Z, et al. Combined application of biochar and sulfur regulated growth, physiological, antioxidant responses and Cr removal capacity of maize (Zea mays L. ) in tannery polluted soils[J]. Journal of Environmental Management, 2020, 259: 110051. doi: 10.1016/j.jenvman.2019.110051 [62] ALDMOUR S T, BURKE I T, BRAY A W, et al. Abiotic reduction of Cr(VI) by humic acids derived from peat and lignite: Kinetics and removal mechanism[J]. Environmental Science and Pollution Research, 2019, 26: 4717-4729. doi: 10.1007/s11356-018-3902-1 [63] SU C Q, LI L Q, YANG Z H, et al. Cr(VI) reduction in chromium-contaminated soil by indigenous microorganisms under aerobic condition[J]. 2019, 29: 1304-1311. [64] HUANG X N, MIN D, LIU D F, et al. Formation mechanism of organo-chromium(III) complexes from bioreduction of chromium(VI) by Aeromonas Hydrophila[J]. Environment International, 2019, 129: 86-94. doi: 10.1016/j.envint.2019.05.016 [65] BALDIRIS, ACOSTA N T, MONTES A, et al. Reduction of hexavalent chromium and detection of chromate reductase (ChrR) in Stenotrophomonas Maltophilia[J]. Molecules, 2018, 23(2): 406. doi: 10.3390/molecules23020406 [66] TAN H, WANG C, LI H, et al. Remediation of hexavalent chromium contaminated soil by nano-FeS coated humic acid complex in combination with Cr-resistant microflora[J]. Chemosphere, 2020, 242: 125251. doi: 10.1016/j.chemosphere.2019.125251 [67] BRUCE W L, MIZUBA M M, HANSEL C M, et al. Iron promoted reduction of chromate by dissimilatory iron-reducing bacteria[J]. Environmental Science & Technology, 2001, 35(3): 522-527. [68] ZHANG K, LIAO P, JIN Y W, et al. Conductive property of secondary minerals triggered Cr(VI) bioreduction by dissimilatory iron reducing bacteria[J]. Environmental Pollution, 2021, 286: 117227. doi: 10.1016/j.envpol.2021.117227 [69] JAMES B R, BARTLETT R J. Behavior of chromium in soils, VI: Interactions between oxidation-reduction and organic complexation[J]. Journal of Environmental Quality, 1983, 12(2): 173-176. [70] US Environmental Protection Agency. Search superfund site information [EB/OL]. [2021-11-15] https://cumulis.epa.gov/supercpad/cursites/srchsites.cfm. [71] HE L, WANG Z, GU W B. Evolution of freeze-thaw properties of cement-lime solidified contaminated soil[J]. Environmental Technology & Innovation, 2020, 21: 101189. [72] TU Y M, LIU D Y, WANG T F, et al. Evaluation on later-age performance of concrete subjected to early-age freeze-thaw damage[J]. Construction and Building Materials, 2020, 270: 121491. [73] LIAO Y P, MIN X B, YANG Z H, et al. Assessment of the stability of chromium in remedied soils by Pannonibacter Phragmitetus BB and its risk to groundwater[J]. Journal of Soils and Sediments, 2014, 14: 1098-1106. doi: 10.1007/s11368-014-0860-1 [74] WEN F, HOU H, YAO N, et al. Effects of simulated acid rain, EDTA, or their combination, on migration and chemical fraction distribution of extraneous metals in ferrosol[J]. Chemosphere, 2013, 90(2): 349-357. doi: 10.1016/j.chemosphere.2012.07.027 [75] SLEJKO F F, PETRINI R, LUTMAN A, et al. Chromium isotopes tracking the resurgence of hexavalent chromium contamination in a past-contaminated area in the Friuli Venezia Giulia Region, northern Italy[J]. Isotopes Environ Health Studies, 2019, 55(1): 56-69. doi: 10.1080/10256016.2018.1560278 [76] ZHAO Y P, XIANG W, HUANG C L, et al. Production of hydroxyl radicals following water-level drawdown in peatlands: A new induction mechanism for enhancing laccase activity in carbon cycling[J]. Soil Biology and Biochemistry, 2021, 156: 108241. doi: 10.1016/j.soilbio.2021.108241 [77] JIA M Q, BIAN X, YUAN S H. Production of hydroxyl radicals from Fe(II) oxygenation induced by groundwater table fluctuations in a sand column[J]. Science of the Total Environment, 2017, 584: 41-47. [78] YE T T, LI H B, WANG Z X, et al. Transport and fate of hexavalent chromium in slag-soil system[J]. Environmental Earth Sciences, 2019, 78: 239-250. doi: 10.1007/s12665-019-8245-9 [79] KIMBROUGH D E, COHEN Y WINER A M, et al. A critical assessment of chromium in the environment[J]. Critical Reviews in Environmental Science and Technology, 1999, 29(1): 1-46. doi: 10.1080/10643389991259164 [80] MOHANTY A, PATRA H K. Phytoremediation potential of paragrass-an in situ approach for chromium contaminated soil[J]. International Journal of Phytoremediation, 2012, 14(8): 796-805. doi: 10.1080/15226514.2011.619595 [81] RANIERI E, FRATINO U, PETRELLA A, et al. Ailanthus Altissima and Phragmites Australis for chromium removal from a contaminated soil[J]. Environmental Science and Pollution Research, 2016, 23: 15983-15989. doi: 10.1007/s11356-016-6804-0 [82] 陈子方, 赵勇胜, 孙家强. 铅和铬污染包气带及再释放规律的实验研究[J]. 中国环境科学, 2014, 34(9): 2211-2216. [83] 童菊秀, 刘洋, 孙怀卫. 土壤中Cr(Ⅵ)的地表径流迁移试验研究[J]. 排灌机械工程学报, 2014, 32(3): 271-276. doi: 10.3969/j.issn.1674-8530.13.0133 [84] WEI Y Q, XU X Y, CHEN X, et al. Migration and transformation of chromium in unsaturated soil during groundwater table fluctuations induced by rainfall[J]. Journal of Hazardous Materials, 2021, 45: 126229. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 8163
- HTML全文浏览数: 8163
- PDF下载数: 160
- 施引文献: 0

