




-
近年来,随着城市化进程的加速,坚持人与自然和谐共生、坚持绿色发展已成为普遍共识,大量化工企业被迫搬迁或关停,从而遗留了大量污染地块[1-2],大多数遗留地块具有高污染、高风险的特点,对附近环境和人们的生活带来很大风险,因此污染地块修复已经刻不容缓[3-4]。
在有机污染土壤修复技术中,异位热脱附技术由于修复效果良好,在国内工程应用中比例较高,但对于一些建筑物附近或异味较重的污染地块,异位热脱附技术的应用受到很大限制[5-6]。由于原位热脱附技术具有适用范围广、环境干扰小和可操作性强等优点,该技术近几年受到人们广泛关注[7-10]。燃气热脱附技术(Gas thermal desorption,GTD)在原位热脱附技术中表现优异,GTD以天然气或液化石油气为能源,通过热传导方式加热污染地块,结合抽提装置实现降低地块污染物浓度的目的。GTD具有处理污染物种类多、土壤非均质性影响小、修复工艺简单等突出优点,目前GTD已经成为很多科研机构和环保公司重点关注技术[11-12]。
蒋村等[13]研究了原位热脱附技术对氯苯污染土壤修复影响因素,结果表明,当土壤加热目标温度设定为100 ℃时,90.0%土壤样品中氯苯去除率达99.0%以上,土壤粒径、土壤含水率对土壤氯苯热脱附去除效果也有较大影响。胡正等[14]研究了原位热脱附技术在有机污染地块中的修复效果,热脱附法对有机污染土壤有良好的去除效果,修复后土壤中萘、苯并(a)芘的检出质量分数≤0.8 mg·kg−1,总石油烃检出质量分数≤96.0 mg·kg−1,远小于污染物修复目标值,修复效果较好。张攀等[15]研究表明,在热脱附去除土壤中硝基苯的过程中,加热30 min后脱附效率为86.9%,在目标温度停留20 min后,脱附效率为91.0%,这说明加热至目标温度后的停留时间越长,热脱附效果越好。目前,针对距离加热井不同位置土壤温度峰值问题,冷凝废水中污染物浓度问题以及能耗问题的研究较少,本研究通过土壤温度变化、土壤污染物含量变化、冷凝废水污染物含量变化和能耗等进行分析,研究GTD在有机污染地块中土壤修复效果及影响因素,以为后续类似工程提供实践经验。
-
GTD应用于我国北方某退役煤气厂所遗留污染地块,地块面积为1 400.0 m2,污染深度为−4.0~−7.0 m,待修复地块地下水无污染。场地调查结果显示,主要污染物为TPH(C<16)、苯、萘等挥发性有机污染物,其中TPH、苯、萘最高浓度分别为4 720.0、19.4、529.0 mg·kg−1。3种污染物修复目标分别为826.0、6.4、63.7 mg·kg−1。地质勘察结果表明,污染地块修复区域可分为6个工程地质层,地层岩性分布如表1所示。同时,根据现场地层情况的不同,0~−2.0 m主要以杂填土为主,−2.0 m以下为原状土,分别对其理化性质进行了测试,具体信息见表2。
-
设备主要由加热系统、抽提系统、温度监测系统、冷却系统和尾气处理系统5部分组成。工艺流程图如图1所示。根据本地块污染物与水的共沸点,设定土壤加热的目标温度为150 ℃。首先,在土壤中安置加热管并通过天然气加热升温,高温气体由加热内管进入,然后通过加热外管后直接外排。加热管通过热传导方式加热周围污染土壤,并逐渐升高至目标温度。随着污染土壤温度的升高,目标污染物逐渐挥发,甚至裂解,含有污染物的蒸汽通过抽提井被抽提至地表,再经冷却系统将高温蒸汽进行降温,汽水分离后的气体通过净化处理后排放,液体暂存至储存罐,最后输送至废水处理系统进行无害化处置。在土壤加热过程中,利用压力监测和温度监测系统实时监控修复区域,并通过智能化、自动化控制系统对加热井温度进行实时调控。
-
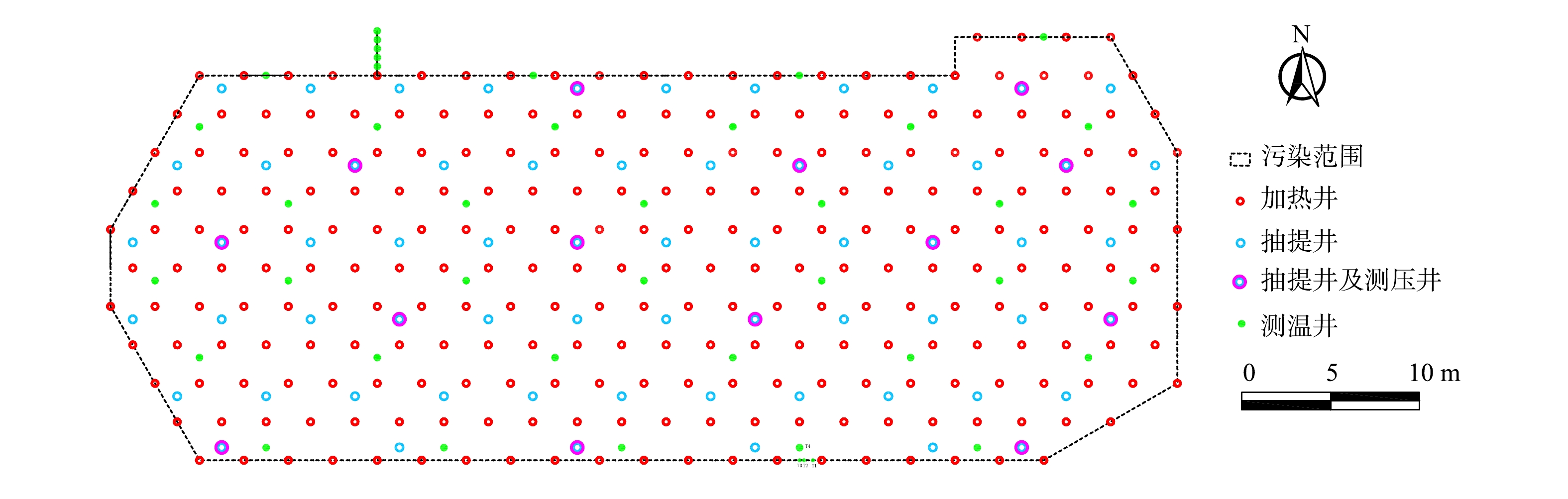
通过地块水文地质勘测,地下水位在-30.0 m以下,无需做止水帷幕和降水处理。根据地块水文地质情况及计算结果,污染地块共布置加热井261口、抽提井63口、测温井43口和压力监测井14口,污染地块井位布置如图2所示。
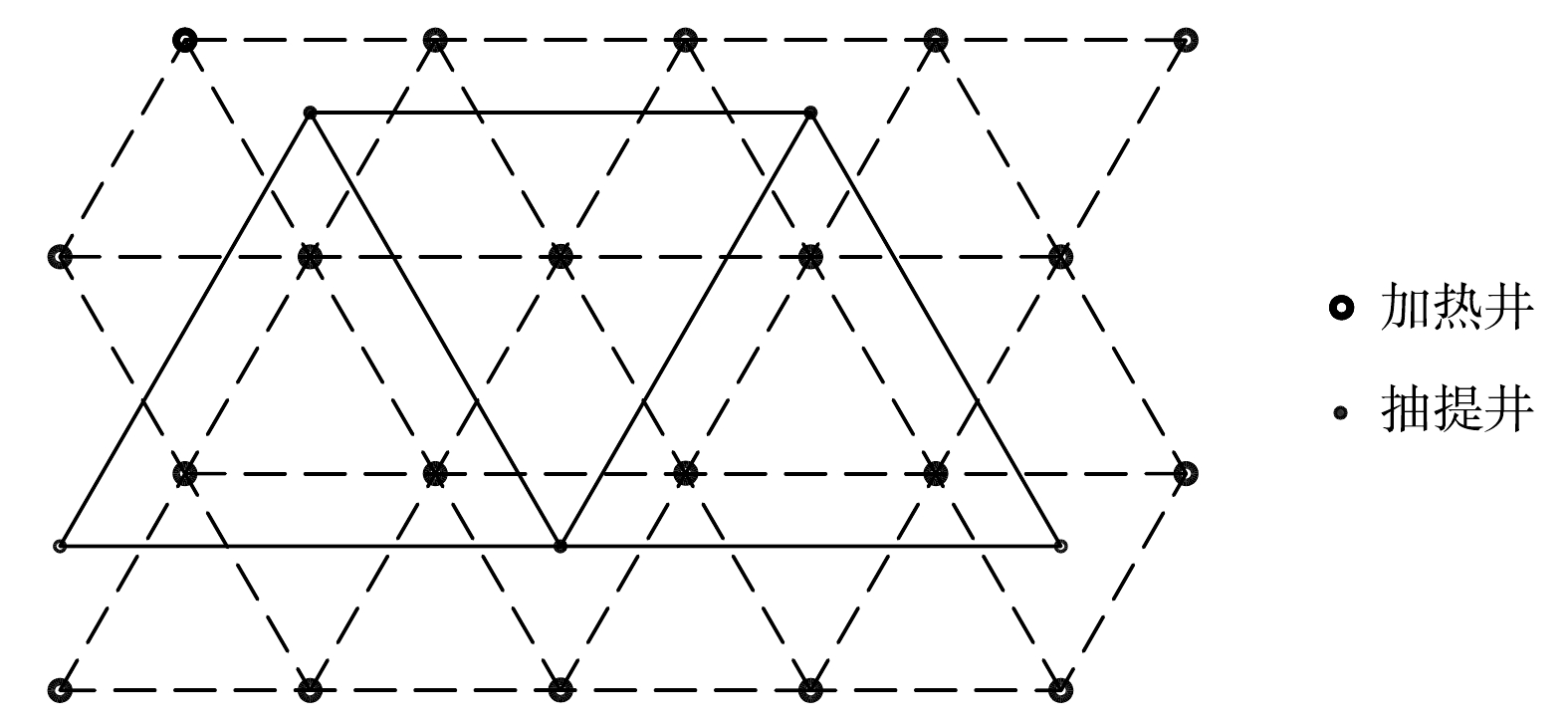
加热井、抽提井布位设计。土壤加热温度与加热井排布方式有关,且加热井排布不适合会导致能源成本大幅度增加。本修复项目采用正三角排布方式对加热井进行布置,通过数值计算和中试实验,设定加热井间距为2.5 m,抽提井间距为5.0 m,抽提井均匀分布于加热井周围,具体排布如图3所示。
测温井布位设计。为了监测污染地块内土壤冷点温度变化,在修复区域内共布置测温井35口,测温井位于正三角排布的3口加热井中心点,如图4所示。测温井内共放置5个热电偶,热电偶深度位置分别为−0.5、−1.5、−3.0、−5.0、−7.0 m。
土壤导热系数较低,为研究单口加热井的周围土壤温度变化情况,在修复区域内选定1口加热井并在其附近设置一系列测温井,测温井与选定加热井的距离分别为0.50、1.00、1.25、1.44 m,具体位置如图5所示。
高温会对周边建筑物地基造成影响,因此需要监测加热井热量传导距离。本研究选择1口边界处加热井,研究其附近土壤温度变化。在加热井同一个方向设置一系列测温井,测温井与加热井的距离分别为0.5、1.0、1.5、2.0、2.5 m。测温井深度为−8.0 m,其中热电偶放置深度为−7.0 m。
-
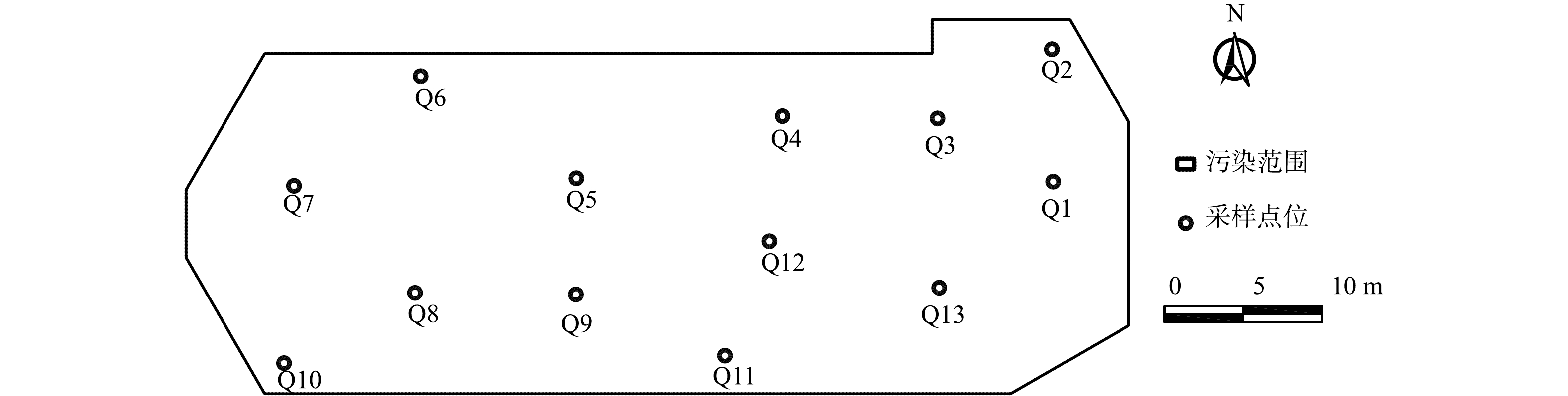
污染地块修复后,在地块内布置13个采样点,采样点均在冷点位置,记为Q1~Q13。上层0~−4.0 m是未污染土壤,在采样点垂直方向上取2个土壤样品,间隔为2.0 m;下层−4.0~−7.0 m是污染土壤,垂直方向上取3个土壤样品,间隔为1.0 m,采样点具体位置如图6所示。
当土壤升至目标温度后,燃气热脱附装置仍维持加热7 d,然后进行土壤样品采集。样品采集后立即放入0~−4 ℃的冷藏箱内保存,并于48 h内送至实验室进行检测。实验室检测指标为苯、萘和TPH,检测设备为气质联用仪GC-MS(EXPEC 5231,杭州谱育科技发展有限公司)和气相色谱仪(GC 2000,杭州谱育科技发展有限公司),检测方法依据HJ 605-2011[16]。
污染地块地下水位较深,但浅层滞水会随土壤温度升高而逐渐蒸发,经抽提井至地表,然后经过冷凝系统形成冷凝废水。每14 d对冷凝废水进行检测1次,监测废水中污染物浓度变化,从而间接反映污染土壤修复效果。实验室检测指标为苯、萘和TPH,检测设备均为气相色谱仪(GC 2000,杭州谱育科技发展有限公司),检测方法分别依据HJ1067-2019[17]、GB/T5750.8-2006[18]和HJ894-2017[19]。
-
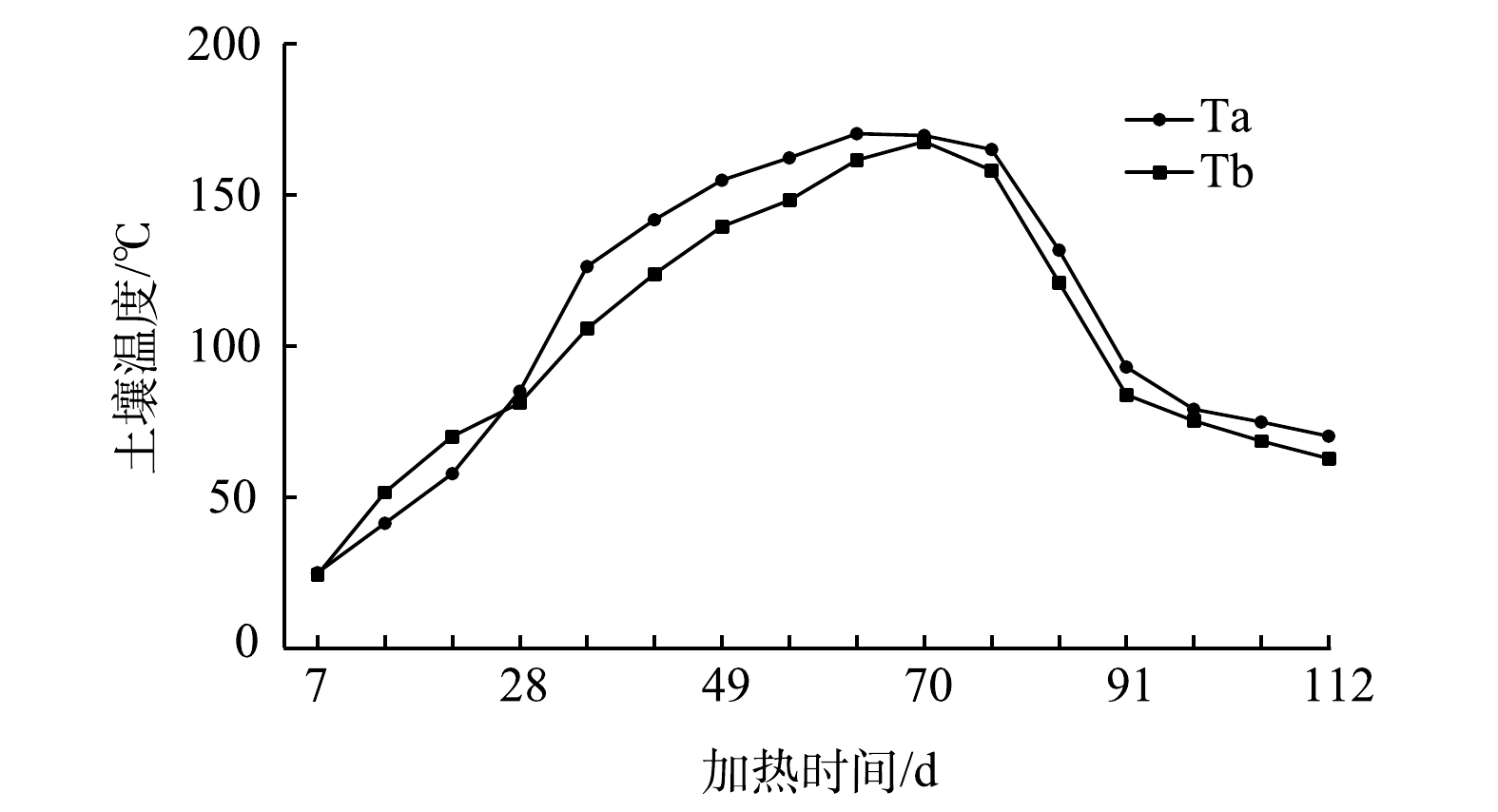
由于本地块内测温井较多,本研究选取2个具有代表性的测温井进行土壤温度变化分析,测温井记为Ta和Tb(具体位置见图4),土壤温度随时间变化如图7所示。由图可知,地块内冷点处土壤升至目标温度150 ℃约需要56 d。土壤升至目标温度后,热脱附装置持续加热7 d后停止加热,直至77 d时Ta和Tb处土壤温度才开始出现下降趋势。这是因为,热量在土壤中的传递需要一定过程,当停止加热时,冷点处土壤温度出现持续上升现象,该现象体现出温度变化的滞后效应。停止加热后,温度监测系统继续对土壤温度进行监测,112 d后土壤温度仍大于60 ℃。
为研究单口加热井附近土壤温度变化情况,在距离加热井0.50、1.00、1.25、1.44 m处分别设置测温井,对附近土壤温度进行监测,测温井记为T1、T2、T3和T4(见图5),温度变化情况如图8所示。测温井距离加热井越近,温度越高,当最远处测温井T4升至目标温度150 ℃时,其他测温井T1、T2、T3温度分别为221、182、173 ℃,均已超过目标温度。停止加热后初始阶段,一定时间内所有测温井温度仍会持续上升,且距离加热井越近的测温点达到温度峰值的时间越早,距离加热井越远的测温点达到温度峰值的时间越晚,温度呈现出明显滞后现象。T1、T2在第63 d分别升至温度峰值221、192 ℃,T3、T4在第70 d分别升至温度峰值188、170 ℃。
有文献表明[4,20],GTD加热井间距一般设置为1.5~4.0 m,通过计算,本项目中设定加热井间距为2.5 m。加热井间距越小,土壤升至目标温度所需时间越少,但所需布置的加热井数量增多,导致前期设备投资增加。同时,加热时间和加热井数量的不同会导致燃气消耗量不同。因此,需要综合考虑工期、成本、技术等各种影响因素,才能设计最佳加热井间距。
为研究加热区域边界外土壤温度变化,距离边界加热井的一侧设置不同距离(0.5、1.0、1.5、2.0、2.5 m)的测温井,土壤温度变化情况如图9所示。由图9可知,测温井距离加热井越近,温度峰值越高;距离加热井越远,温度峰值越低。2.5 m处土壤温度在第70 d时仍为86.8 ℃。为避免土壤热传导对周边建筑物地基造成影响,可根据建筑物地基材料的耐热性能,合理控制加热井与附近建筑物之间的距离。
-
污染地块土壤修复后共设置了13个土壤采样点。选择污染地块内3个具有代表性的取样点进行数据分析,如表3所示。土壤修复前,污染地块0~−4.0 m土壤未受到污染,−4.0~−7.0 m土壤均受到不同程度污染,TPH最高污染浓度为4 762.0 mg·kg−1,处于地下−5.0~−6.0 m;苯的最高污染浓度为18.4 mg·kg−1,处于地下−4.0~−5.0 m;萘的最高污染浓度为388.5 mg·kg−1,处于地下−4.0~−5.0 m。土壤修复后,TPH最高污染浓度为192.8 mg·kg−1,苯为未检出,萘的最高污染浓度为14.4 mg·kg−1,处于地下−6.0~−7.0 m。经过计算TPH去除率均在95%以上,萘去除率超过了92%,苯的去除率约为100%,且污染残留主要集中于抽提井尾端。由此可知,经GTD修复后土壤中大部分污染物已经转化为气相或液相被抽提出来,修复效果良好。
-
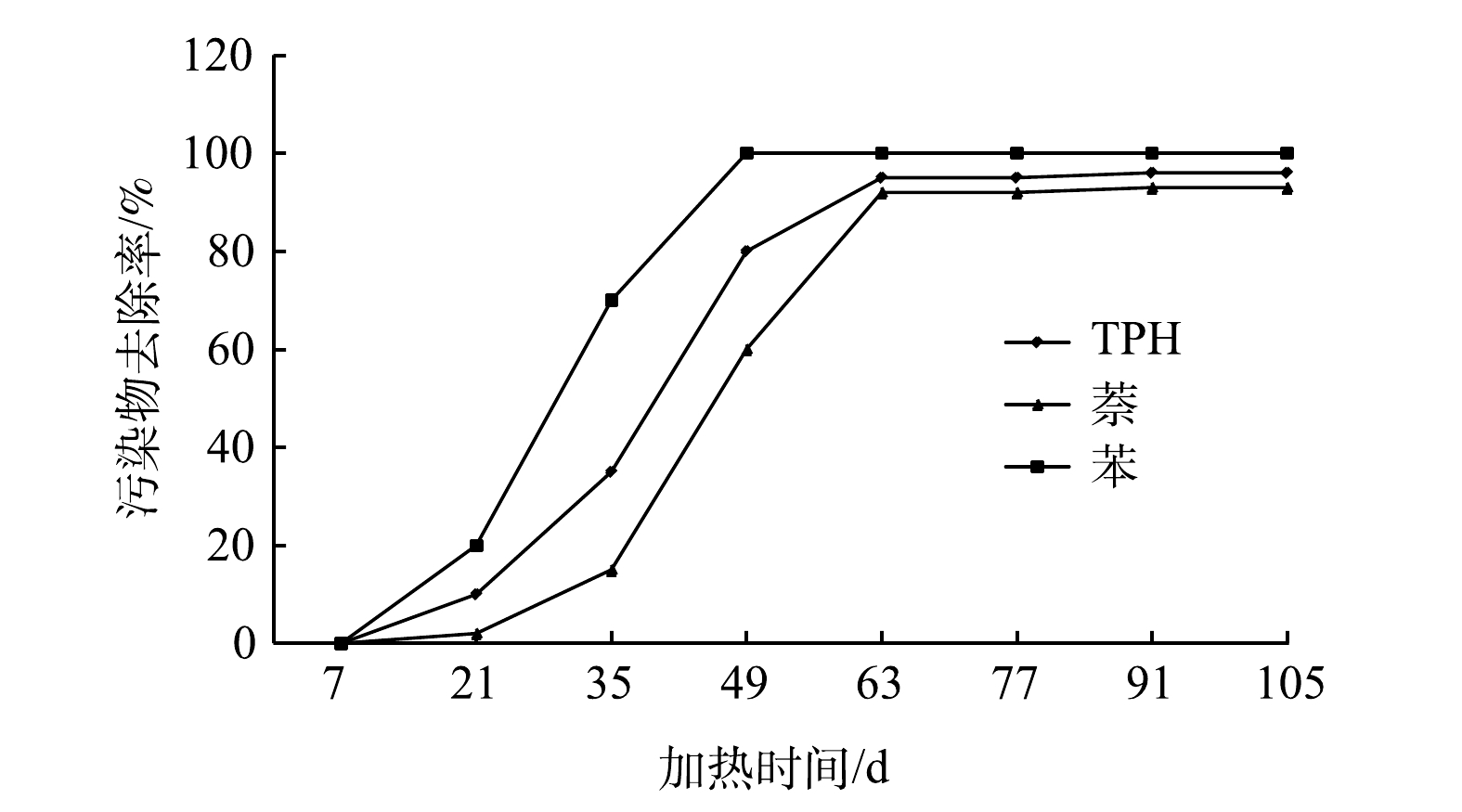
污染地块土壤加热过程中,水分会逐渐蒸发,土壤中污染物会以气体形式随水蒸汽一起通过抽提系统移出地面,经冷凝系统处理后部分气体转化为液体,不凝气通过尾气处理系统达标后排放。修复过程中,每14 d对冷凝的废水取样1次,每次取3个水样送至实验室检测,检测结果如表4所示。第7 d废水样品未检出污染物,这是因为刚开始加热时,土壤温度较低,污染物没有转化为气相被抽出。从第21 d起废水样品中检测出TPH和苯,至第35 d废水中才检测出萘,这是因为,随着加热时间的延长,土壤温度逐渐升高,高沸点污染物开始逐渐向气相转化,并随水蒸汽被抽提井抽出。冷凝废水中不同污染物浓度峰值出现的时间不同,苯浓度在第35 d升至峰值,而TPH和萘的浓度峰值分别出现在第35 d和第63 d,这与污染物沸点有关,沸点越低,废水中污染物浓度峰值出现越早,沸点越高,废水中污染物浓度峰值出现越晚。苯沸点80.1 ℃,萘沸点217.9 ℃,本地块中TPH主要为汽油馏分(C4~C12)和煤油馏分(C10~C16),其对应的沸点范围分别为50~200 ℃和130~250 ℃[21]。冷凝废水中污染物浓度间接反映污染土壤修复效果。
-
本项目自2021年9月20日开始运行,至2021年11月23日加热系统停止运行,从开始加热至停止加热共运行63 d,施工周期内共消耗天然气924 868.6 m3,消耗电量为453 600 kW·h,本项目共修复污染土壤11 200 m3,能耗分析结果如下:每处理1.0 m3污染土壤约消耗天然气82.6 m3,电量40.5 kW·h。
-
影响GTD修复效果的主要因素包括加热温度、停留时间、土壤含水率、土壤渗透性和加热井排布方式等[22-29]。利用温度和污染物含量变化等数据,对GTD修复效果影响因素进行分析与讨论。
1)加热温度。加热温度对污染物去除率影响较大,具体见图10。污染地块目标加热温度与污染物沸点有关,苯、萘和TPH的沸点分别为80.1、217.9 ℃和50~250 ℃。由拉乌尔定律可知,污染物和水混合物的沸点低于单独污染物的沸点,这使得目标加热温度无须超过污染物的沸点[23,30]。苯与水共沸点为69.3 ℃,萘与水的共沸点为98.8 ℃[31]。
当土壤平均温度超过50 ℃时,有机物苯和TPH中低沸点的汽油馏分开始逐渐向气相转化;当土壤平均温度超过100 ℃时,萘以及TPH中的汽油馏分和煤油馏分均开始逐渐向气相转化,随着加热时间增加,土壤温度升高,大量的TPH和萘开始向气相转化,这是由于TPH和萘从土壤中脱附所需的温度更高,开始向气相转化的时间点相对滞后。综上所述,GTD加热温度可直接影响污染物的去除效果。一般而言,目标温度越高,污染物去除效果越好,但污染物去除率与温度并非成线性关系,较高温度往往会伴随着其他副产物的生成和能耗的增加,因此最佳目标温度的确定须要综合考虑项目工期和成本等因素。
2)停留时间。土壤升温前期,加热温度起修复关键作用,当土壤温度升至目标温度后,停留时间则是影响修复效果的主要因素。在本项目中,土壤升至目标温度后继续加热7 d,即在目标温度停留时间为7 d,土壤中不同目标污染物的去除效果均较为理想。萘的最低去除率为92%,TPH的最低去除率为95%,苯的去除率约为100%(具体见图11)。修复后土壤中沸点较高的萘和TPH浓度虽然远低于修复目标值,但仍有少量残留,导致这一现象的原因可能是停留时间较短。综上所述,实际施工过程中,应综合考虑目标温度与停留时间等因素,在施工工期内以最低成本完成污染土壤修复。
3)土壤含水率。在GTD修复土壤过程中,土壤含水率会对升温时间产生影响,含水率越低,土壤升至目标温度所需时间越短。因此加热前降低土壤含水量,可大幅度缩短升温时间。本项目地下水位较深,无须进行降水处置,但须要在场地表层进行混凝土防护,阻止降水对施工区域土壤含水率的影响。此外,土壤含水率还影响污染物去除效果,降低土壤含水率会导致土壤通透性增大,有利于有机污染物的挥发。这是因为,土壤含水率较高液态水会占据大量的空隙,阻碍空气的流通路径。土壤含水率会对能耗产生影响,土壤含水率越高,则土壤升至目标温度所须能量就越多。水的比热容为4.2×103 J·kg−1·℃−1,土壤比热容为0.8×103 J·kg−1·℃−1,水的较热容比土壤大,土壤含水率越高,水分在加热过程中吸收的能量越多,能耗越大。当目标温度高于100 ℃,土壤中水分须经过由液态水转化为气态水的过程,水的汽化热为2.3×106 J·kg−1,远高于液态水和土壤比热容,须消耗更多热能。本地块目标温度为150 ℃,因此,降低含水率可以大幅度降低整体能耗,提高污染土壤修复效率。
4)土壤渗透性。渗透性指由浓度差引起的水分净移动能力,是影响有机污染物在土壤中扩散速度的重要参数。高国龙等[32]研究表明,渗透性较好土壤中的有机物更容易被抽提,去除效率更高[33-34]。周启星等[35]研究表明,当土壤渗透性大于10−4 cm·s−1时,土壤中空气流动较通畅,当土壤渗透性小于10−6 cm·s−1时,土壤中空气流动受到较大影响。经检测,本项目污染地块中地下0~−2.0 m主要为杂填土,−2.0~−8.0 m主要为粉砂土,渗透系数在4.0×10−5 cm·s−1以上,在加热过程中,土壤受热后会出现不同程度裂缝,增大土壤渗透系数,因此,在GTD修复过程中,有机物在土壤孔隙中的扩散速度不会受到较大影响,热脱附效率较高。
综上所述,采用GTD修复有机物污染土壤,须要综合考虑加热温度、停留时间、土壤含水率、土壤渗透性等影响因素。可以根据污染物性质、工期等因素选定合适的目标温度和停留时间。此外,为了避免二次污染,需要对GTD修复过程中产生的废气和废水进行有效收集和无害化处理。
-
1)当燃气热脱附加热井间距为2.5 m,加热时间为56 d,污染土壤升至目标温度150 ℃,停止加热后进行土壤检测,污染物萘、TPH和苯的去除率分别为92%、95%和100%,污染物含量均在修复目标值之下。
2)本项目以天然气和电能为能源,每处理1.0 m3污染土壤约消耗天然气82.6 m3,电量40.5 kW·h。
3)冷凝废水中不同污染物浓度峰值出现的时间不同,苯浓度在第35 d升至峰值,而TPH和萘的浓度峰值分别出现在第49 d和第63 d。沸点越低,污染物去除率峰值出现越早;沸点越高,污染物去除率峰值出现越晚。
燃气热脱附技术土壤修复效果及影响因素
Soil remediation effect and influencing factors of gas thermal desorption technology
-
摘要: 以某退役煤气厂土壤中苯、萘和TPH为目标污染物,基于燃气热脱附技术开展了工程化的修复。当加热井间距为2.5 m,加热56 d,土壤可升至目标温度150 ℃,土壤中苯、萘和TPH的去除率分别为100%、92%和95%,均达到目标修复值。研究发现,沸点越低,污染物去除率峰值出现越早;沸点越高,污染物去除率峰值出现越晚。停止加热后,测温点距离加热井越近,温度升至峰值的时间越早;测温点距离加热井越远,温度升至峰值的时间越晚,温度呈现出明显的滞后现象。通过能耗分析,每处理1.0 m3污染土壤约消耗天然气82.6 m3,电量40.5 kW·h。本研究结果可为利用燃气热脱附技术进行土壤修复工程提供参考。Abstract: Taking benzene, naphthalene and TPH in the soil of a de-commissioned gas plant as the target pollutants, the engineering remediation was carried out based on gas thermal desorption technology in this study. The temperature of the studied soil reached 150 °C after 56 days of heating with the heating wells placed 2.5 m from each other. Accordingly, the removal percentages of benzene, naphthalene and TPH reached 100%, 92% and 95%, respectively, which all met the remediation targets. During the heating process, the time required to reach peak removal percentages for different pollutants was inversely correlated to the boiling points of the pollutants. In terms of the temporal-spatial variation of the temperatures, after the heating was stopped, it has been observed that the time required to reach peak temperature was inversely correlated to the distance from the heating well, and there was an apparent hysteresis effect on temperature profile. A cost-effectiveness study revealed that 82.6 m3 of natural gas and 40.5 kW·h of electricity was needed to treat each cubic meter of polluted soil. This study can provide reference for soil remediation by gas thermal desorption technology.
-
Key words:
- gas thermal desorption /
- soil remediation /
- organics contaminated site /
- gas heating
-
低温等离子体(non-thermal plasma, NTP)具有反应条件温和(常温常压)、适应性广、反应快速等优点,通过产生大量活性物种(O、·OH、O3等)将VOCs降解,受到了广泛关注,然而高的能耗及大量副产物生成限制了该技术工业化应用. 为了克服活性物种寿命短而导致的NTP降解VOCs效率不高、副产物生成的缺点,近年来,研究者开发了多种具有多孔结构的催化剂与NTP协同降解VOCs,通过延长污染物在放电区的停留时间,有效利用副产物O3产生原子氧、过氧自由基等,达到减少副产物的产生、提高能量密度、降低能耗、提高碳平衡等效果[1-7]. 已经研究的与NTP联合的催化剂包括铁电体材料、半导体催化剂、贵金属催化剂以及分子筛[8-11].
金属有机骨架(metal organic frameworks, MOFs)是一种由有机配体和金属离子或金属簇组合成的多孔催化吸附材料,由于其具有可调规整的孔道、大的比表面积和高的孔隙率,应用前景广泛. MOFs材料在获得一定能量时,由于轨道离域而呈现出半导体的特性[12],介质阻挡放电(dielectric barrier discharge, DBD)等离子体中高能电子的能量可达1—20 eV,可以提供足够的能量活化MOFs产生新的活性自由基(电子-空穴对),具有吸“拟光催化过程”和催化效果,从而促进污染物的降解[13];另外,MOFs材料高的比表面积使其具有优异的吸附性能,可有效延长VOCs在反应区的停留时间,提高处理效率,逐渐应用在等离子体和气体吸附中. 例如,Wang等[14]通过水热法制备了MIL-101(Cr)并用于吸附苯表现出良好的吸附性能;Bahri等[15]采用等离子体协同MIL-101、MIL-53材料降解甲苯,发现MOFs材料的加入可以提高甲苯的降解效果、降低副产物O3的生成. 因此,研制适合DBD体系的高性能MOFs催化剂,提高两者的协同作用效果,对于推动该技术工业化应用具有重要的意义.
MOF-74由二价过渡金属(硝酸盐、醋酸盐)和配体2,5-二羟基-对苯二甲酸合成(DOT),具有一维六角形孔洞结构和高密度“开放”金属位点,具有高比表面积、有机配体和金属离子可调性剂以及很好的稳定性,具有优异的电化学性能及光催化特性,在吸附分离、光电催化产氢、化学传感、烟气脱硝等领域受到广泛关注[16-18]. 例如,Chen等[16]采用微波辅助法合成的Ni-MOF-74对CO2吸附性能好:Feng等[8]将MOF-74(Mn-Co-Ni)催化剂与NTP协同应用于甲苯降解,相同条件下甲苯去除率提高了42.9%,并有效控制了副产物O3生成. 目前,将MOF-74材料与DBD协同降解VOCs还鲜有报道[8],催化剂引入DBD后在等离子体内较低温度下O2是否参与吸附活化为O-和O2-等参与催化反应的机理还不明晰.
本研究采用溶剂热法合成制备了Mn基MOF-74(Mn-MOF-74)材料,并通过改变配体为1, 4-苯二甲酸(TPA)制备了Mn-TPA-DMF多孔材料,将两种催化剂引入DBD等离子体降解甲苯气体. 采用XRD、FTIR、SEM、BET和XPS等表征技术对催化剂的结构进行分析,对比了两种Mn基MOFs材料加入DBD后甲苯的降解效果、副产物的形成,推测了DBD催化降解甲苯的反应机理.
1. 实验部分(Experimental section)
1.1 材料
四水合硝酸锰(Mn(NO3)2·4H2O)、2,5-二羟基对苯二甲酸、1,4-苯二甲酸(TPA)、N,N-二甲基甲酰胺(DMF)、乙醇和丙醇等,所有试剂均为分析纯,不需经进一步纯化可以直接使用.
1.2 催化剂的制备
采用溶剂热合成法制备催化剂. 准确称取2.84 mL Mn(NO3)2·4H2O和0.66 g DOT,溶解在15:1:1(V:V:V)的DMF-乙醇-水混合物中,反应混合物超声处理5 min,然后转移到高压釜中,放置在120 ℃热空气烘箱内24 h. 然后取出高压釜在室温下冷却,将获得的棕色晶体用乙醇洗涤后去离子水洗涤多次,然后将样品置于60 ℃烘箱中干燥,制备Mn-MOF-74催化剂.
另外,称取0.92 g Mn(NO3)2·4H2O 和0.90 g TPA,同时溶解在50 mL DMF中,超声处理10 min后,将所得溶液转移到高压釜中,在120 ℃ 的热空气烘箱中保持24 h. 将反应混合物冷却至室温后,得到浅白色结晶化合物,用DMF、甲醇和水洗涤多次,获得的产物在130 ℃下真空干燥,制备Mn-TPA-DMF催化剂.
1.3 催化剂的表征
使用X射线衍射仪(XRD,PANalytical,荷兰),获得制备的催化剂的晶体结构. 催化剂样品的形态通过来扫描电子显微镜(SEM,ZESS,德国的Gemini 300)表征,相应的元素分布通过能量色散X射线光谱仪(EDS)分析系统获得. 使用氮气吸附-脱附实验测定样品的孔隙信息,BET和BJH方程中的吸附数据计算催化剂的比表面积、孔径分布和孔体积. X射线光电子能谱(XPS)通过具有Al-Kα辐射源的Thermo Scientific K α谱仪测量,用C1s为284.8 eV的数据校准测试的元素的结合能数据.
1.4 催化剂的性能评价
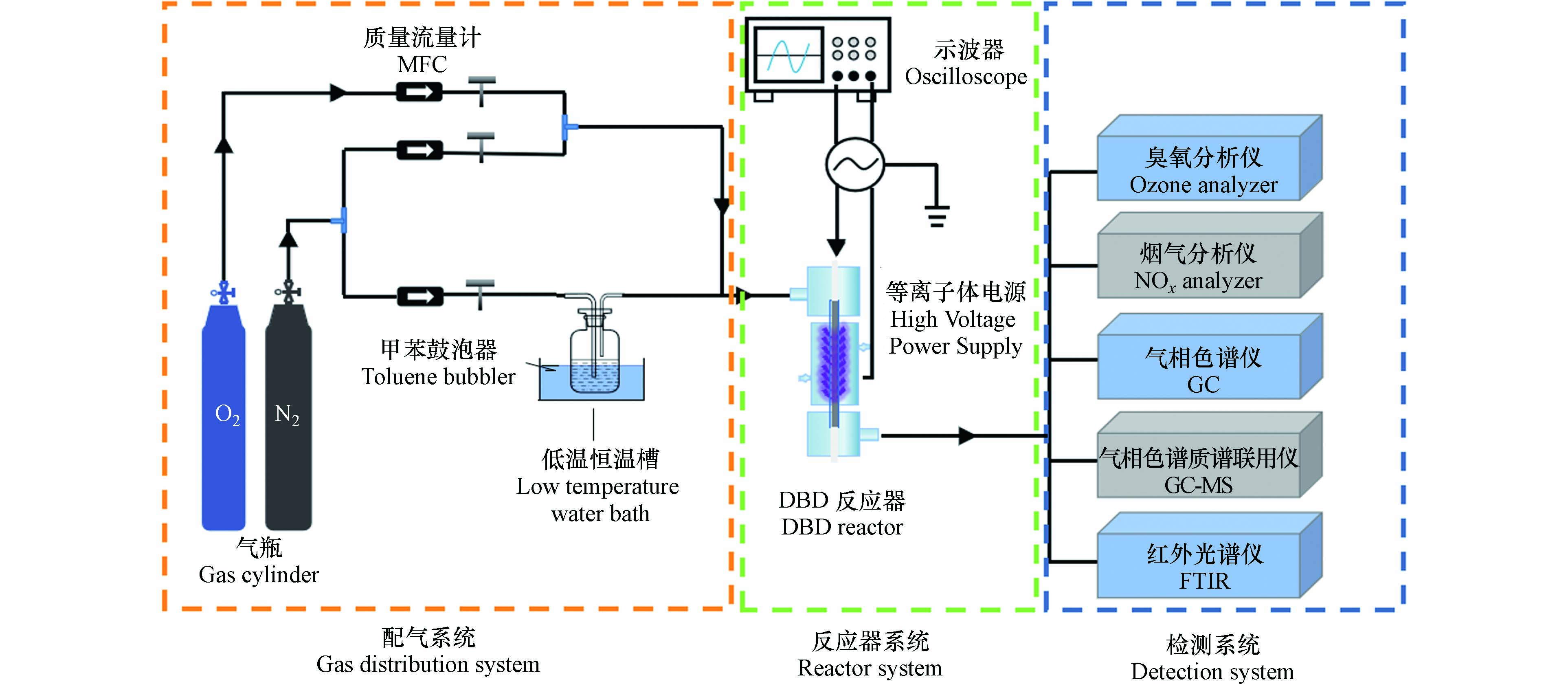
DBD催化降解甲苯气体的工艺流程如图1所示. 该系统由三部分组成:甲苯气体发生器系统、DBD等离子体反应器系统和废气检测系统. 氮气通过装有液体甲苯的气体鼓泡器发生器来生成一定浓度的含甲苯气体. 使用3个质量流量控制器(MFC)分别控制氮气、氧气的流量,得到所需的甲苯初始浓度. 实验时,甲苯初始浓度1368 mg·m−3,气体流量1 L·min−1,氧含量4%(体积分数).
DBD反应器为50 mm放电长度、3 mm放电间隙的同轴结构(图1),采用石英管作为介质层,使用循环水作为接地电极,内管放置1根不锈钢棒作为高压电极. 高压电源为南京苏曼等离子体科技有限公司生产的CTP-2000 K实验电源,输出频率范围为5—20 kHz. DBD催化实验时,将0.3 g催化剂(40—60目)与1.5 g石英砂混合,装入DBD反应器中. 使用高压探头和电流探头测定电压和电流,并通过数字示波器(Tektronix TBS 1000 C)记录放电波形,通过利萨如(V-Q Lissajous)方法计算DBD放电功率P(方程1),借助方程(2)计算能量密度(SED). 本文气体流量(Q)为1 L·min−1,通过调节放电电压来实现SED的变化.
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) 式中,f为放电频率(9.6 kHz),C为电容(0.47 μF),A为利萨如图面积,P为放电功率(W),Q为气体流量
(L⋅min−1) 降解前后的甲苯、CO和CO2的浓度均使用气相色谱仪(GC9790型,浙江福立分析仪器有限公司)在线测定,其中CO和CO2通过Ni转化炉转化为甲烷后由FID检测器测定. O3和NOx(NO2和NO)的浓度分别用O3分析仪(2B Model 106-M,美国)和烟气分析仪(MGA 6-plus,德国)测定. 采用气相色谱-质谱联用仪(GC-MS)对尾气中的有机副产物进行了分析. 尾气用正己烷吸收20 min,再超声1 h. 样品进样至GC-MS中,通过自动进样器进行分析. 甲苯降解率(η)、矿化度(Md)、CO2选择性(
SCO2 stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) stringUtils.convertMath(!{formula.content}) (5) stringUtils.convertMath(!{formula.content}) (6) 式中,Cin和Cout分别为甲苯进口和出口的浓度(mg·m−3),M为甲苯的分子量(g·mol−1),[CO2]和[CO]分别为出口CO2和CO的浓度(mol·m−3),Ey为反应器的能量效率(g·kWh−1).
2. 结果与讨论(Results and discussion)
2.1 催化剂的表征分析
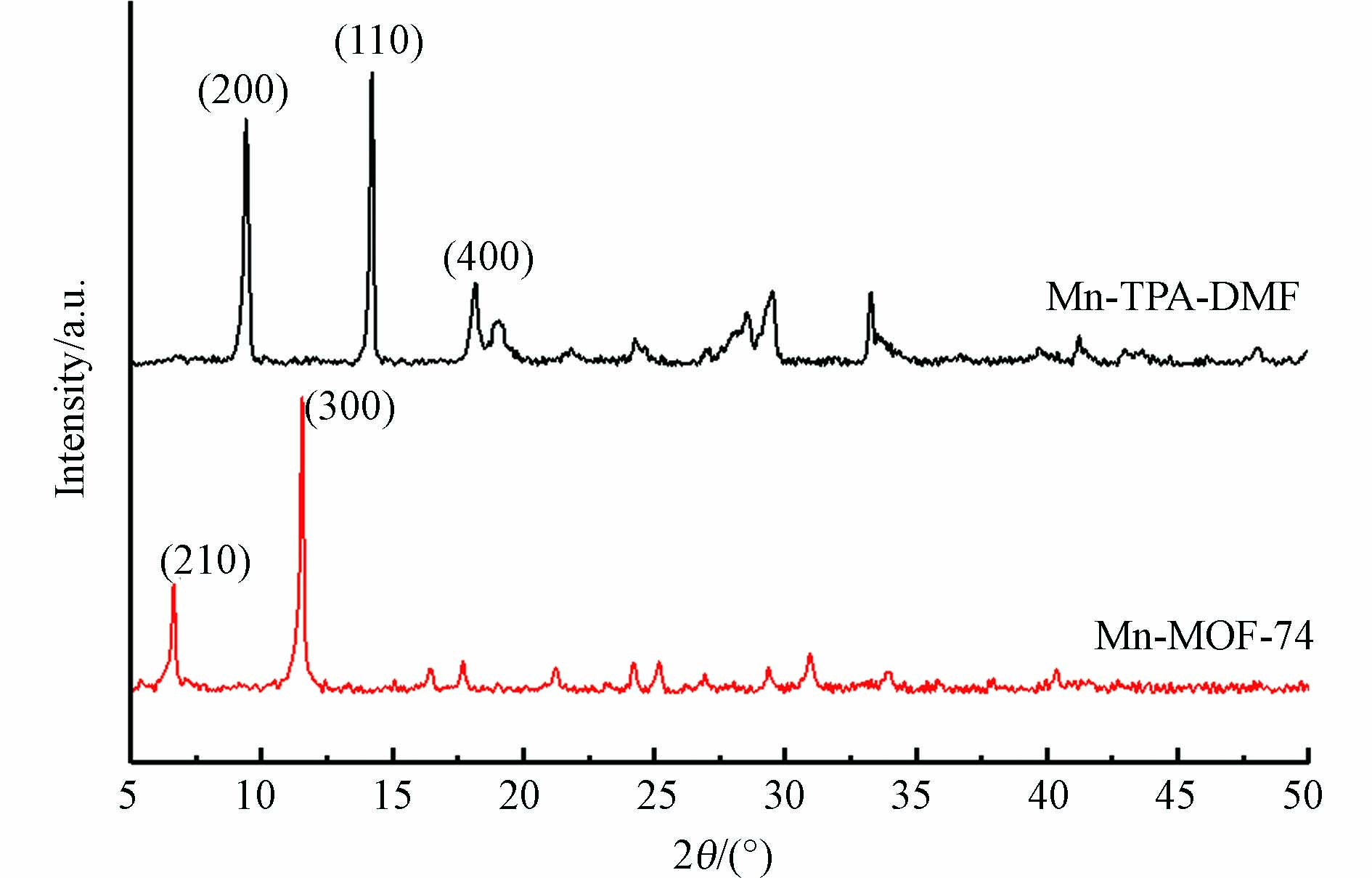
如图2所示,为了确定材料的晶体结构,对制备的样品进行了XRD表征. Mn-MOF-74材料在2θ=6.8°和11.8°附近有两个很强的衍射峰,分别对应于Mn-MOF-74的(210)和(300)晶面[18],其他位置的衍射峰都相对较弱. Mn-TPA-DMF材料在2θ=9.41°、14.22°、18.17°附近的3个衍射峰分别对应材料的(200)、(110)和(400)晶面[19]. 本研究中合成的Mn-MOF-74和Mn-TPA-DMF的XRD谱图的峰位置、强度及顺序与先前文献报道的相一致[20-21],表明材料合成成功.
图3(a)为Mn-MOF-74使用前后的FTIR图. 3422 cm−1附近的特征峰为OH或H2O的伸缩振动吸收峰,表明水或其他溶剂分子存在于Mn-MOF-74通道中[22]. 1567 cm−1和1401 cm−1附近的峰为MOF-74中COO-的不对称和对称伸缩振动峰[23],1240 cm−1附近的峰为C—N键振动吸收峰,说明溶剂DMF吸附在催化剂表面或者孔道内[24],1196 cm−1处的特征峰为C—O单键伸缩振动峰,886 cm−1和806 cm−1处的特征峰为苯环C—H键面内和面外摇摆振动峰,574 cm−1处的特征峰归属于Mn—O伸缩振动峰[25],表明Mn-MOF-74的成功合成.
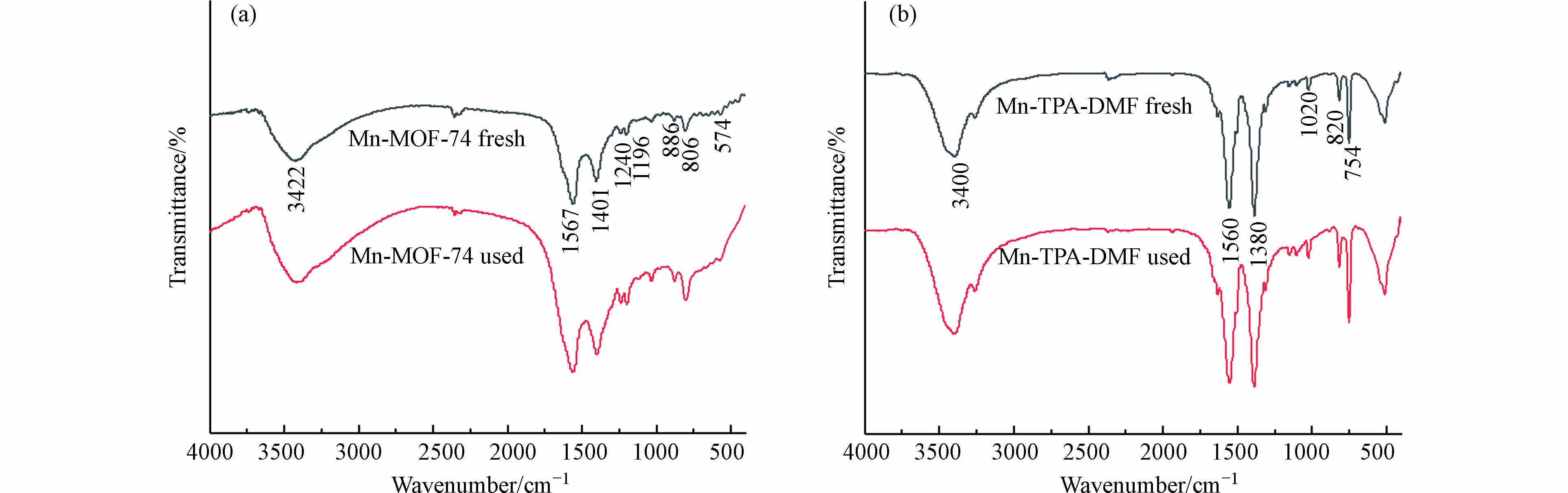 图 3 Mn-MOF-74和Mn-TPA-DMF反应前后的红外图谱Figure 3. FTIR spectra of Mn-MOF-74 and Mn-TPA-DMF before and after catalysis reaction
图 3 Mn-MOF-74和Mn-TPA-DMF反应前后的红外图谱Figure 3. FTIR spectra of Mn-MOF-74 and Mn-TPA-DMF before and after catalysis reaction图3(b)为Mn-TPA-DMF使用前后(fresh 和 used表示)的FTIR图,具有与Mn-MOF-74相似的特征峰,但特征峰向低频(波数)轻微偏移,这可能是由于Mn与2,5-二羟基对苯二甲酸和1,4-苯二甲酸两种不同的有机配体结合引起的. 此外,通过反应前后的红外图谱对比可知,Mn-MOF-74和Mn-TPA-DMF在反应前后没有发生结构上的改变,表明其具有良好的稳定性,适合实际工业应用.
本研究通过SEM分析了催化剂的形貌结构,如图4所示. Mn-MOF-74和Mn-TPA-DMF均呈现出不规则的二维片状结构,表面上有细小的块状颗粒附着. 此外,催化剂的元素映射图表明了Mn、O、C元素的存在,且呈均匀分布.
 图 4 (a—c)Mn-MOF-74和(d—f)Mn-TPA-DMF的SEM和元素映射Figure 4. SEM image and element mapping of (a—c) Mn-MOF-74 and (d—f) Mn-TPA-DMF
图 4 (a—c)Mn-MOF-74和(d—f)Mn-TPA-DMF的SEM和元素映射Figure 4. SEM image and element mapping of (a—c) Mn-MOF-74 and (d—f) Mn-TPA-DMF图5为催化剂的N2吸附-脱附曲线,根据曲线信息计算出的BET比表面积和孔隙体积如表1所示. Mn-MOF-74和Mn-TPA-DMF的N2吸附-脱附曲线均为IV型,并在高的压力下表现出脱附滞后,形成回滞环,说明两种材料都是含有中孔(2—50 nm)结构的材料. 与Mn-TPA-DMF相比(比表面积和孔体积分别为29.123 m2·g−1和0.02421 cm3·g−1),Mn-MOF-74呈现更高的比表面积及孔体积,分别为83.368 m2·g−1和0.17630 cm3·g−1,本文合成的Mn-MOF-74的比表面积高于相似的方法合成的Mg-MOF-74催化剂(18.04 m2·g−1)[26],可以提供更多的表面活性位点.
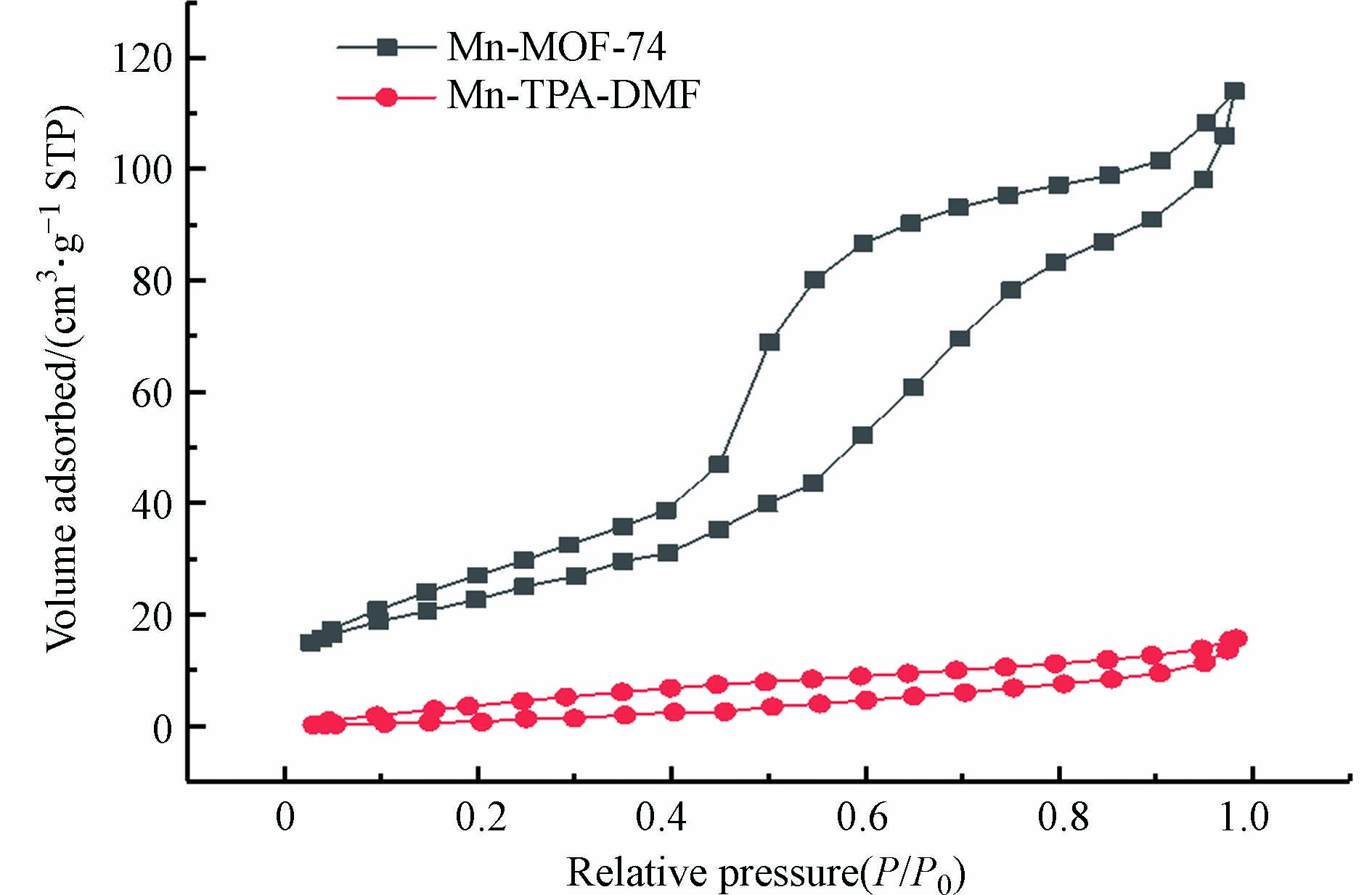 图 5 Mn-MOF-74和Mn-TPA-DMF的氮气吸附-脱附曲线Figure 5. Nitrogen adsorption and desorption curves of Mn-MOF-74 and Mn-TPA-DMF表 1 Mn-MOF-74和Mn-TPA-DMF的比表面积和孔容分析Table 1. BET surface area and pore volume analysis of Mn-MOF-74 and Mn-TPA-DMF
图 5 Mn-MOF-74和Mn-TPA-DMF的氮气吸附-脱附曲线Figure 5. Nitrogen adsorption and desorption curves of Mn-MOF-74 and Mn-TPA-DMF表 1 Mn-MOF-74和Mn-TPA-DMF的比表面积和孔容分析Table 1. BET surface area and pore volume analysis of Mn-MOF-74 and Mn-TPA-DMF催化剂 Catalyst 比表面积/(m2·g−1) Specific surface area 孔容/(cm3·g−1) Pore volume 平均孔径/nm Average diameter Mn-MOF-74 83.368 0.17630 8.460 Mn-TPA-DMF 29.123 0.02421 3.326 | Show Table DownLoad:
CSV
DownLoad:
CSV
图6所示为Mn-MOF-74和Mn-TPA-DMF在甲苯催化反应前后的XPS分析. 图6(a)的全谱分析中显示有Mn、C、N和O元素的存在,表明MOFs材料的成功合成,N元素的存在,说明DMF分子也参与了Mn-MOF-74的配位[27]. 图6(b)为Mn-MOF-74的Mn 2p XPS光谱,位于640.7 eV和652.0 eV的峰分别归因于Mn 2p3/2和Mn 2p1/2的特征峰. 将Mn 2p3/2进行分峰拟合,在结合能为639.3、640.7 、644.7 eV处的峰分别归属于Mn2+、Mn3+和Mn4+[10, 28]. 以上结果表明,Mn以多价态存在,反应过程中可以相互转换,有利于催化反应的进行. 气相中的O2、O3和电子能够借助Mn2+、Mn3+、Mn4+之间的表面转换,被吸附和转移到催化剂表面的甲苯和中间产物上,产生活性粒子(OH、·O等),从而将甲苯进一步矿化为CO2. 如表2所示,通过Mn 2p光谱的定量分析可知,两种Mn基催化剂反应前Mn3+占比都较高,Mn3+的存在能够增加氧空穴,有利于将O2和O3激活为活性氧,进而参与反应;反应后Mn3+占比变化不大,说明Mn3+一直参与甲苯的氧化反应,比较稳定. Mn3+略有下降的原因可能为反应后氧被激活,消耗了部分Mn3+. 在多价态的锰中,高价态的锰可以氧化甲苯,低价态的锰可以通过等离子体催化过程中臭氧的活化而被再氧化. 此外,位于656.0 eV附近的峰归因于Mn 2p特征峰的卫星峰. 反应后Mn的特征峰的强度变低且位置向高结合能的位置轻微偏移,这可能是由于催化剂在放电过程中参与甲苯氧化反应引起的.
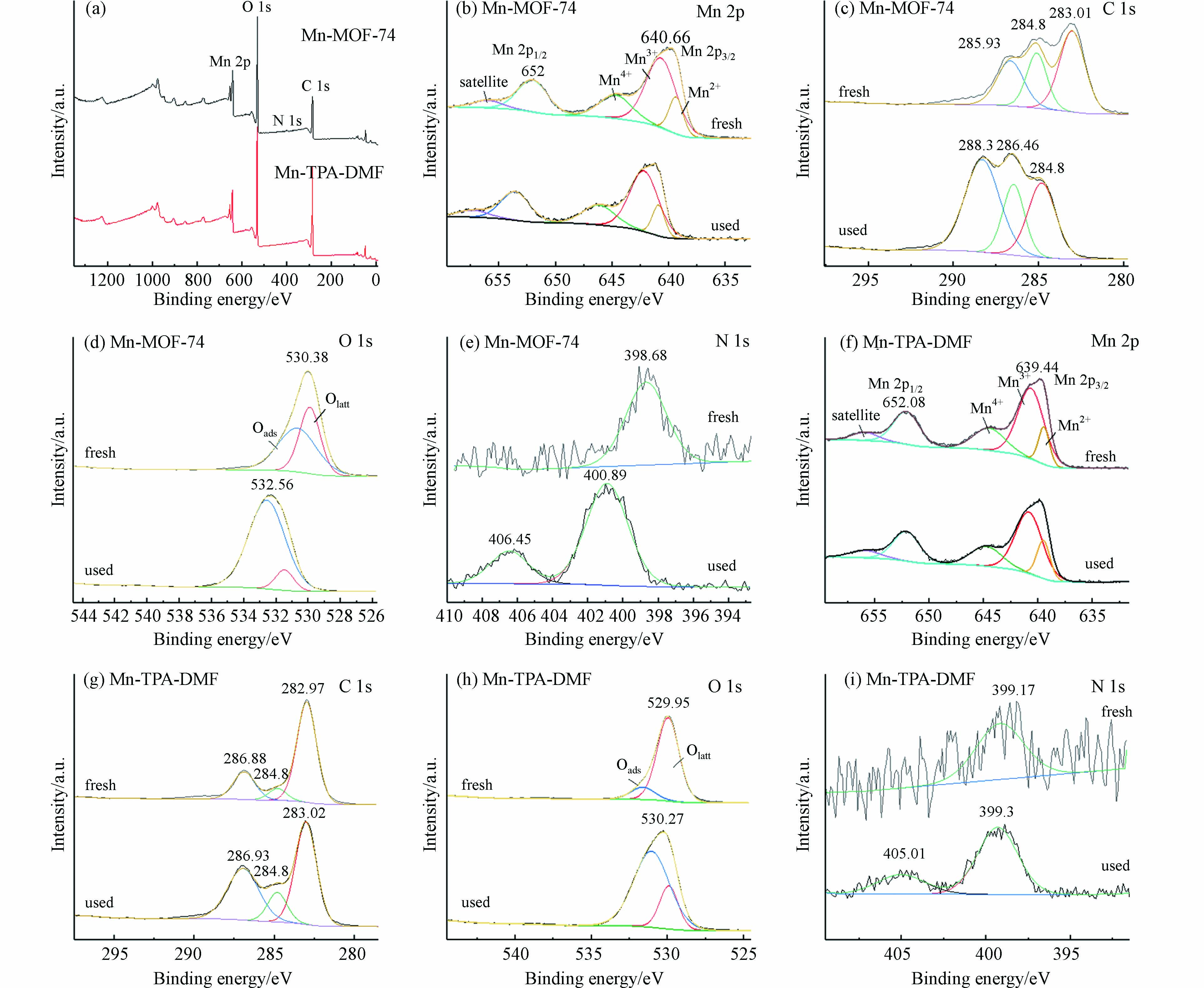 图 6 Mn-MOF-74和Mn-TPA-DMF催化反应前后的XPS分析Figure 6. XPS analysis of Mn-MOF-74 and Mn-TPA-DMF before and after catalytic reaction(a)全谱分析,(b-e)Mn-MOF-74的Mn 2p, C 1s, O 1s, N 1s XPS光谱,(f-i)Mn-TPA-DMF的Mn 2p, C 1s, O 1s, N 1s XPS光谱(a) Full spectrum analysis, and (b-e) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-Muf-74, and(f-i) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-TPA-DMF表 2 Mn-MOF-74和Mn-TPA-DMF各元素价态的组成Table 2. Valence composition of the elements of Mn-MOF-74 and Mn-TPA-DMF
图 6 Mn-MOF-74和Mn-TPA-DMF催化反应前后的XPS分析Figure 6. XPS analysis of Mn-MOF-74 and Mn-TPA-DMF before and after catalytic reaction(a)全谱分析,(b-e)Mn-MOF-74的Mn 2p, C 1s, O 1s, N 1s XPS光谱,(f-i)Mn-TPA-DMF的Mn 2p, C 1s, O 1s, N 1s XPS光谱(a) Full spectrum analysis, and (b-e) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-Muf-74, and(f-i) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-TPA-DMF表 2 Mn-MOF-74和Mn-TPA-DMF各元素价态的组成Table 2. Valence composition of the elements of Mn-MOF-74 and Mn-TPA-DMF催化剂 Catalyst 表面元素比Mn3+/(∑Mnn+) Surface element ratio Mn3+/(∑Mnn+) Oads/Olatt Before After Before After Mn-MOF-74 0.63 0.62 1.23 8.60 Mn-TPA-DMF 0.62 0.60 0.16 6.28 | Show TableDownLoad:
CSV
图6(c)为Mn-MOF-74的C 1s XPS谱,在283.01 eV、285.1 eV和286.65 eV处分峰拟合为3个峰,分别对应于C—C,C—O 和O—C=O[10],反应后特征峰的位置向高结合能的位置轻微偏移,这可能是由于等离子放电或与甲苯及中间产物的结合.
图6(d)为Mn-MOF-74的O 1s XPS光谱,通过分峰拟合可以分为两个特征峰. 位于约529.9 eV处的峰可归因于材料中的晶格氧(O2-)(标记为Olatt),位于约530.7 eV处的峰则归因于表面吸附氧[10, 29]. O2在催化剂上吸附后通常会经过以下变化:吸附氧Oads→O2-(ads)→O-(ads)→Olatt. 一般认为,源自氧空位的Oads比Olatt具有更高的迁移率,并且在VOCs催化氧化过程中比Olatt更有效[30, 31]. 通过表2可知,反应前吸附氧(Oads)的含量比较少,反应后Oads含量明显升高,说明反应过程中催化剂能活化氧物种产生大量Oads,使Oads参与了甲苯的氧化反应. 其中,Mn-MOF-74催化剂显示出较高的Oads/Olatt摩尔比,这表明在Mn-MOF-74催化剂表面上有较高含量的Oads,即表面氧空位密度越高,O2分子越容易在催化剂表面吸附和活化,催化性能越好. 图6(d)表明,发生DBD催化反应后Oads的占比显著增加,表明Mn-MOF-74可以活化氧物种参与甲苯的氧化反应.
图6(e)为Mn-MOF-74的N 1s XPS光谱,在398.7 eV 的峰归属于N(C)3,是由残留DMF引起的. 反应后406.5 eV处出现了1个特征峰,是由等离子体电荷效应引起,说明有少量NOx副产物产生[32].
图6(f-i)为Mn-TPA-DMF的Mn 2p, C 1s, O 1s, N 1s XPS光谱,均呈现和Mn-MOF-74相似的特征峰,从图中也可以分析得出,Mn和表面活性氧物种参与了甲苯的氧化反应.
2.2 DBD催化降解甲苯的效果分析
如图7(a)显示了甲苯在不同DBD催化体系中的降解行为. 由图7可知,甲苯的降解效率随SED的增加也就是放电电压的升高而增加,DBD-催化体系比单独DBD体系具有更高的甲苯去除效率. SED为605.97 J·L−1时,DBD、DBD+Mn-TPA-DMF和DBD+Mn-MOF-74的3个体系中甲苯去除率分别为74.67%、91.23%和94.91%. 能量密度越高,产生的电流脉冲的数量和强度越大,形成的微放电越多,从而通过高能电子和大量活性物质改善了VOCs的降解效果[33-34]. 换句话说,SED升高,增大了高能电子的绝对数量及与气体分子的碰撞几率,导致体系中活性粒子数增加,同时可能诱导催化剂表面发生反应,改变催化剂的特性和微孔放电特性. 此外,两种催化剂相比,Mn-MOF-74比Mn-TPA-DMF表现出更好的催化协同性能. 从表2中可以看出,Mn-TPA-DMF和Mn-MOF-74的金属元素的价态及含量差别不大,但与Mn-TPA-DMF相比,Mn-MOF-74呈现更高的比表面积及孔体积. 结合催化活性测试结果即Mn-MOF-74与等离子体结合时甲苯降解效果更好,说明比表面积和孔体积是影响催化剂活性的主要因素,这与以往的文献报道一致[35].
 图 7 DBD催化体系中不同SED甲苯的去除效率和能量效率Figure 7. (a) Toluene removal efficiency and (b) energy efficiency under different SED in DBD-catalytic systems
图 7 DBD催化体系中不同SED甲苯的去除效率和能量效率Figure 7. (a) Toluene removal efficiency and (b) energy efficiency under different SED in DBD-catalytic systems能量效率可以定义为单位电耗降解甲苯的量,可以说明等离子体催化过程的能耗. 图7(b)可以观察到,能量效率都随着能量密度的升高而降低,说明从经济的角度能量密度越小越好. 与单独DBD相比,相同的SED下DBD催化能量效率有所提高,说明能耗降低了. 当SED为263.68 J·L−1时,单独使用DBD系统的能量效率值为7.43 g·kWh−1,DBD+Mn-MOF-74的能量效率值为11.22 g·kWh−1,说明等离子体与催化剂之间可能存在协同效应. 因此,DBD与催化剂的组合可以提高能量效率,有效降低甲苯降解的能耗,提高DBD系统的经济性.
为了进一步评价DBD-催化体系的性能,进一步研究了CO/CO2浓度、CO2选择性和甲苯矿化度,如图8所示. 如图8(a)所示,在所有情况下,COx浓度都随SED的增加而升高. 此外,DBD催化时生成的CO和CO2的浓度与单独DBD降解时浓度相比显著增加. 当SED为605.87 J·L−1时,DBD+Mn-MOF-74中CO2和CO的浓度分别为1375 mg·m−3和1019.13 mg·m−3,DBD单独体系中为785.71 mg·m−3和712.75 mg·m−3,CO2和CO生成量为单独DBD的175%和143%.
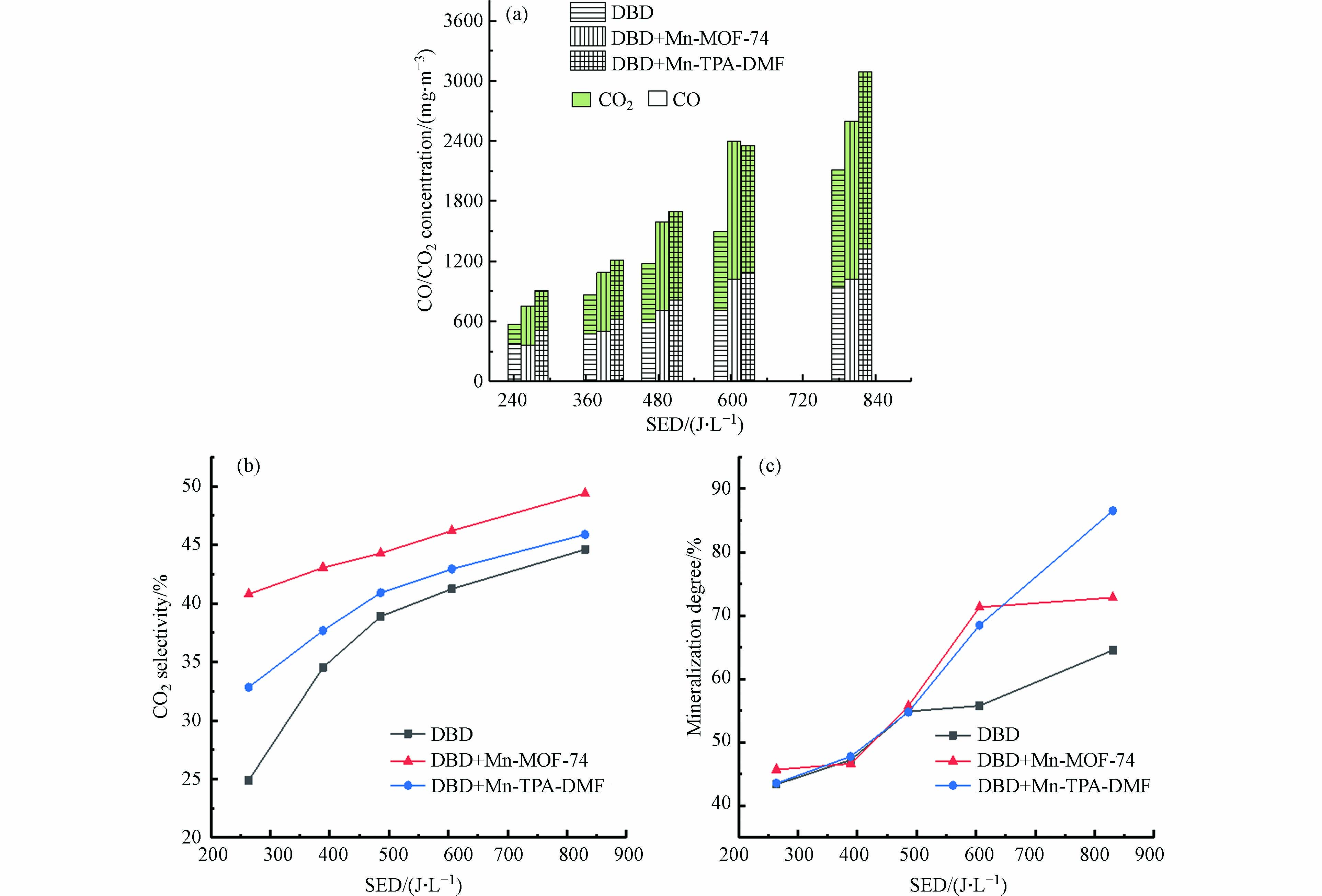 图 8 DBD催化体系(a) CO和CO2浓度(b) CO2选择性和(c)矿化度随SED的变化Figure 8. Changes in (a) CO and CO2 concentration, (b) CO2 selectivity and (c) mineralization degree with SED under DBD catalyst systems
图 8 DBD催化体系(a) CO和CO2浓度(b) CO2选择性和(c)矿化度随SED的变化Figure 8. Changes in (a) CO and CO2 concentration, (b) CO2 selectivity and (c) mineralization degree with SED under DBD catalyst systems图8(b)显示DBD+Mn-TPA-DMF、DBD+Mn-MOF-74和单独DBD体系的CO2选择性随着能量密度的增加而增加. DBD+Mn-MOF-74体系的CO2选择性在能量密度为263.68 J·L−1时为40.79%,比单独DBD体系(24.89%)提高了15.9%.
如图8(c)所示,随着SED的增加,DBD单独体系和DBD-催化剂体系的矿化度都增加. 当能量密度为605.87 J·L−1时,DBD+Mn-TPA-DMF和DBD+Mn-MOF-74体系的矿化率分别为68.48%和71.31%,比单独DBD体系(55.78%)分别提高了12.7%和15.53%. 当SED为830.57 J·L−1时,DBD+Mn-TPA-DMF体系甲苯的矿化度达到最高值86.53%,表明DBD+Mn-TPA-DMF在较高的SED下更有利于甲苯彻底降解为CO2.
NOx和O3是DBD降解VOCs的主要副产物. 图9显示了单独DBD和DBD催化系统中副产物O3和NOx浓度. 在图9(a)中,O3浓度随着SED的增加而降低,这可能是由于O3参与了COx和NOx等其他活性物种的生成[36]. 外加Mn基MOFs催化材料后,DBD降解甲苯气体中副产物O3浓度显著下降,例如,当SED为388.75 J·L−1时,单独DBD系统中O3浓度约为390.64 mg·m−3,而在DBD+Mn-MOF-74和DBD+Mn-TPA-DMF体系中O3浓度分别降至264.64 mg·m−3和303.64 mg·m−3,O3浓度的降低可归因于O3在催化剂表面分解成O2并形成活性原子氧物质(O* )[37]. 就挥发性有机化合物氧化的反应性而言,O* 是比O3更具化学活性的物质,因此,将O3分解为原子氧的能力是等离子体催化过程中挥发性有机化合物降解的重要因素[38]. 此外,Mn2+/Mn3+和Mn4+/Mn3+的氧化还原循环也可以加速表面氧物种的释放,有利于甲苯的氧化[39]. 与Mn-TPA-DMF催化剂相比,Mn-MOF-74催化剂具有较大的比表面积,更有利于O3的吸附和分解,在催化剂表面形成更多的活性氧,提高了催化剂的去除效率和CO2选择性[40].
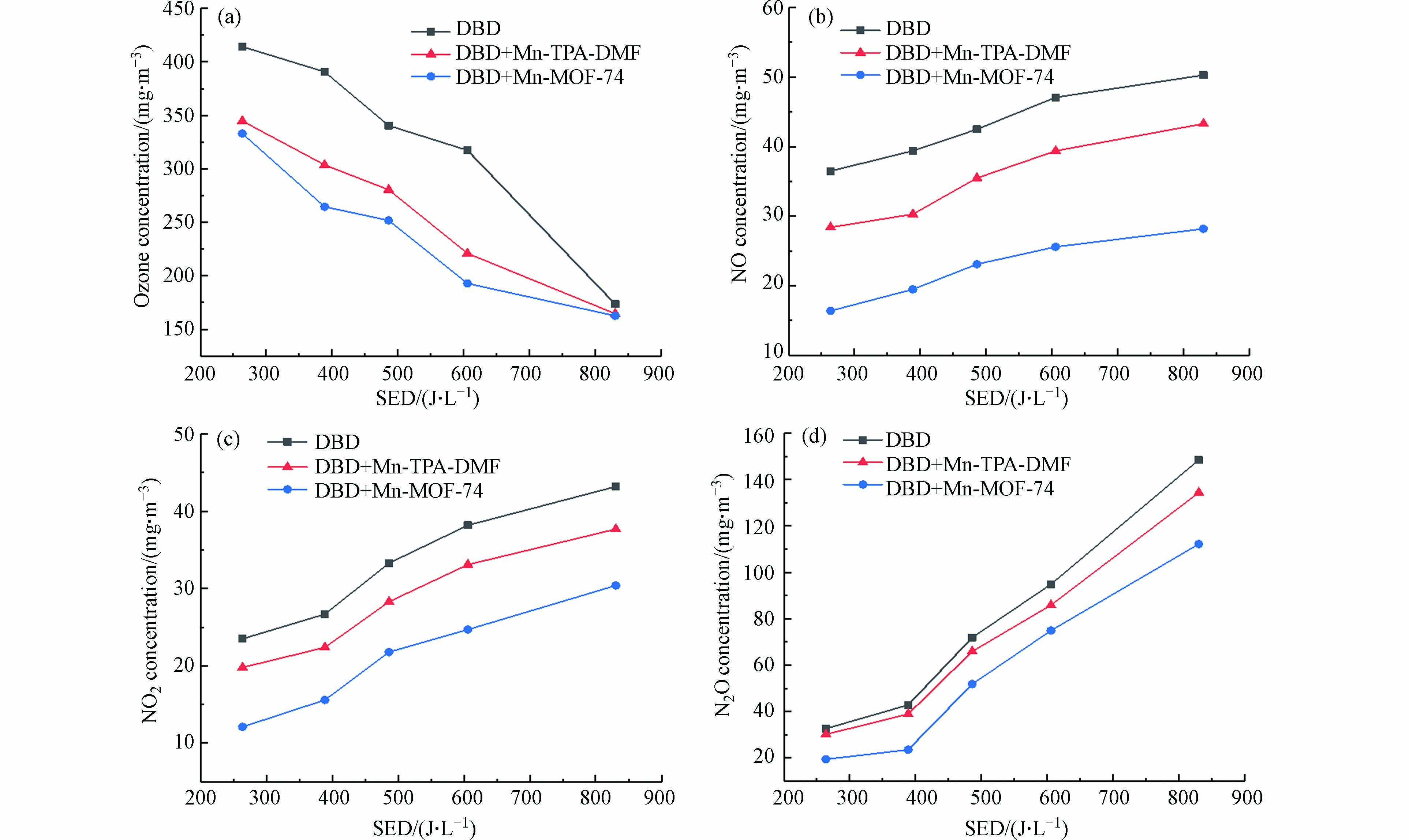 图 9 DBD、DBD+Mn-MOFs催化降解甲苯(a)O3浓度和(b)、(c)、(d) NOx浓度随能量密度的变化Figure 9. DBD and catalyst added (a) O3 concentration and (b) , (c) , (d) NOx concentration as a function of energy density
图 9 DBD、DBD+Mn-MOFs催化降解甲苯(a)O3浓度和(b)、(c)、(d) NOx浓度随能量密度的变化Figure 9. DBD and catalyst added (a) O3 concentration and (b) , (c) , (d) NOx concentration as a function of energy density采用烟气分析仪检测了DBD和DBD-催化体系中3种NOx(NO、N2O和NO2)的浓度,得到NOx生成浓度与SED的关系(图9 b-d). 如图9b-d所示,NO、NO2和N2O的浓度均随SED的增大而增大,与单独DBD系统相比,DBD-催化体系中生成的NOx浓度明显降低,且Mn-MOF-74催化剂的NOx生成量低于Mn-TPA-DMF,表明Mn-MOF-74催化剂更有利于抑制NOx的生成. 例如,当SED为263.68 J·L−1时,单独DBD系统产生的NO2浓度为26.7 mg·m−3,DBD+Mn-TPA-DMF和DBD+Mn-MOF-74体系分别降至22.4 mg·m−3和12.1 mg·m−3. 通过上述结果可知,催化剂可以提高反应的选择性,进而抑制副产物NOx等的生成. 其生成量降低的原因在于Mn基催化剂的引入有利于O3在催化剂的表面生成氧气和表面活性氧,生成的活性氧进而参与甲苯的降解反应,避免了与N2生成NOx. 此外,催化剂的加入还有利于高能电子传输,并降低NOx的解离能进而可以使部分NOx解离为N2[41- 42].
2.3 DBD/Mn-MOFs催化降解甲苯的机理分析
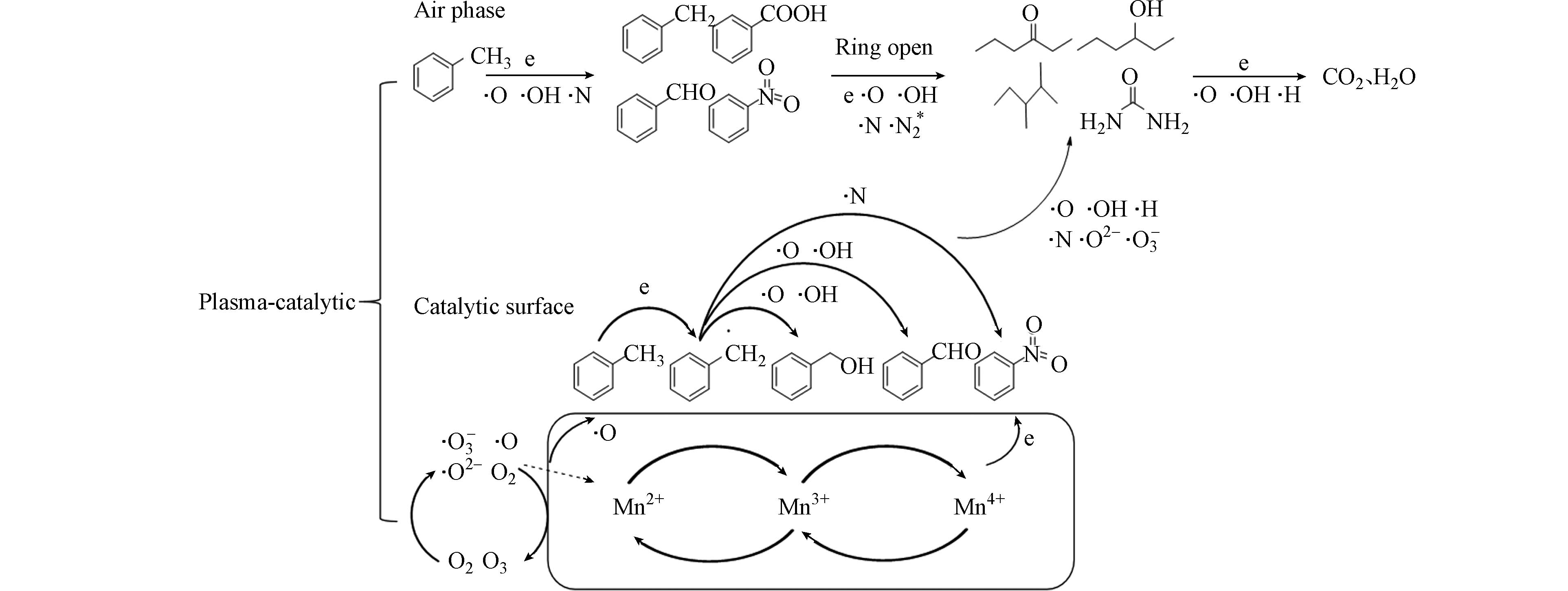
采用气相色谱-质谱联用技术(GC-MS)对DBD及DBD复合催化剂的有机副产物进行了分析. 将反应气体用正己烷吸收20 min,然后将吸收液注入GC-MS仪器进行检测. 结合降解前后材料表征结果及甲苯降解效果,推测DBD/Mn-MOFs催化降解甲苯的机理如下图:由图10可知,等离子体Mn-MOFs催化降解甲苯主要包括3个路径. 等离子体电场内具有丰富的高能电子,高能电子轰击甲苯分子导致甲苯降解是一个主要途径;高能电子与空气中的O2、N2等产生活性粒子(·OH, N, N2*, ·O, N2*, O2- ),活性粒子降解甲苯是第二条途径;再次,催化剂表面的反应取决于甲苯及中间产物的化学吸附、吸附氧的数量及Mn2+、Mn3+、Mn4+之间的转换. 气相中的O2、O3和电子能够借助Mn2+、Mn3+、Mn4+之间的表面转换,被吸附和转移到催化剂表面的甲苯和中间产物上,进一步产生活性粒子(OH、·O等),从而将甲苯进一步矿化为CO2. XPS结果显示催化反应后催化剂的Oads的占比显著增加,也进一步证实Mn基MOFs表面氧空位作为表面缺陷成为催化反应的活性位点,导致O3在催化剂表面分解产生更多的表明氧物种(O和O2),促进甲苯和中间产物的深度氧化[10,31]. 当然,Mn基MOFs催化剂的引入,也会使得等离子体中表面局部场强增大,改变了等离子体放电特性,对降解也会产生影响.
3. 结论(Conclusion)
本文制备了Mn-MOF-74和Mn-TPA-DMF两种Mn基MOFs催化材料,放置在DBD等离子体反应器对甲苯进行催化降解实验. 通过材料表征、降解效果分析等,探索了DBD催化降解机理,结论如下:
1) 与单独DBD相比,DBD与Mn基MOFs催化剂联用显著提高了甲苯的去除率、CO2选择性、矿化度和能量效率,并显著抑制了O3和NOx副产物的生成. DBD+Mn-MOF-74催化降解甲苯效果最好,当SED为605.87 J·L−1时,甲苯降解率达94.91%、CO2选择性达46.2%、矿化度达71.3%,与单独DBD相比,副产物O3和NO生成量下降到50%左右.
2) Mn-MOF-74与Mn-TPA-DMF催化剂相比,表现出更优异的催化活性,这个与Mn-MOF-74具有更高的比表面积、更多的Oads含量、更强的氧化还原性能相关.
3) XPS材料表征表明,Mn基修饰的MOFs材料中Mn作为电荷转移媒介,促进了电荷的循环转移. 通过DBD催化反应前后的催化剂的FTIR图谱可知,Mn-MOF-74和Mn-TPA-DMF在反应前后没有发生结构上的改变,说明催化剂性能稳定.
-
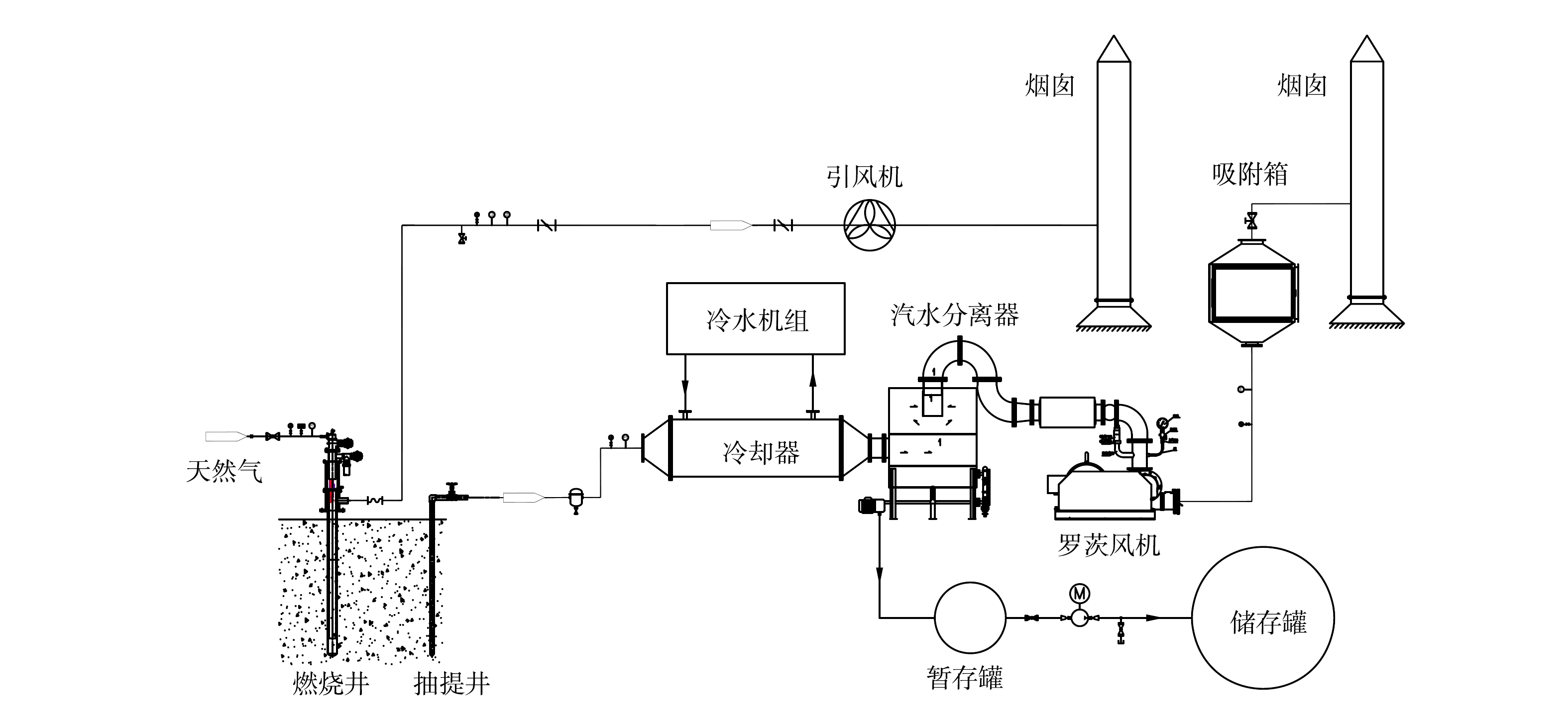
图 1 原位燃气热脱附技术工艺流程图
Figure 1. Process flow diagram of in situ gas thermal desorption technology
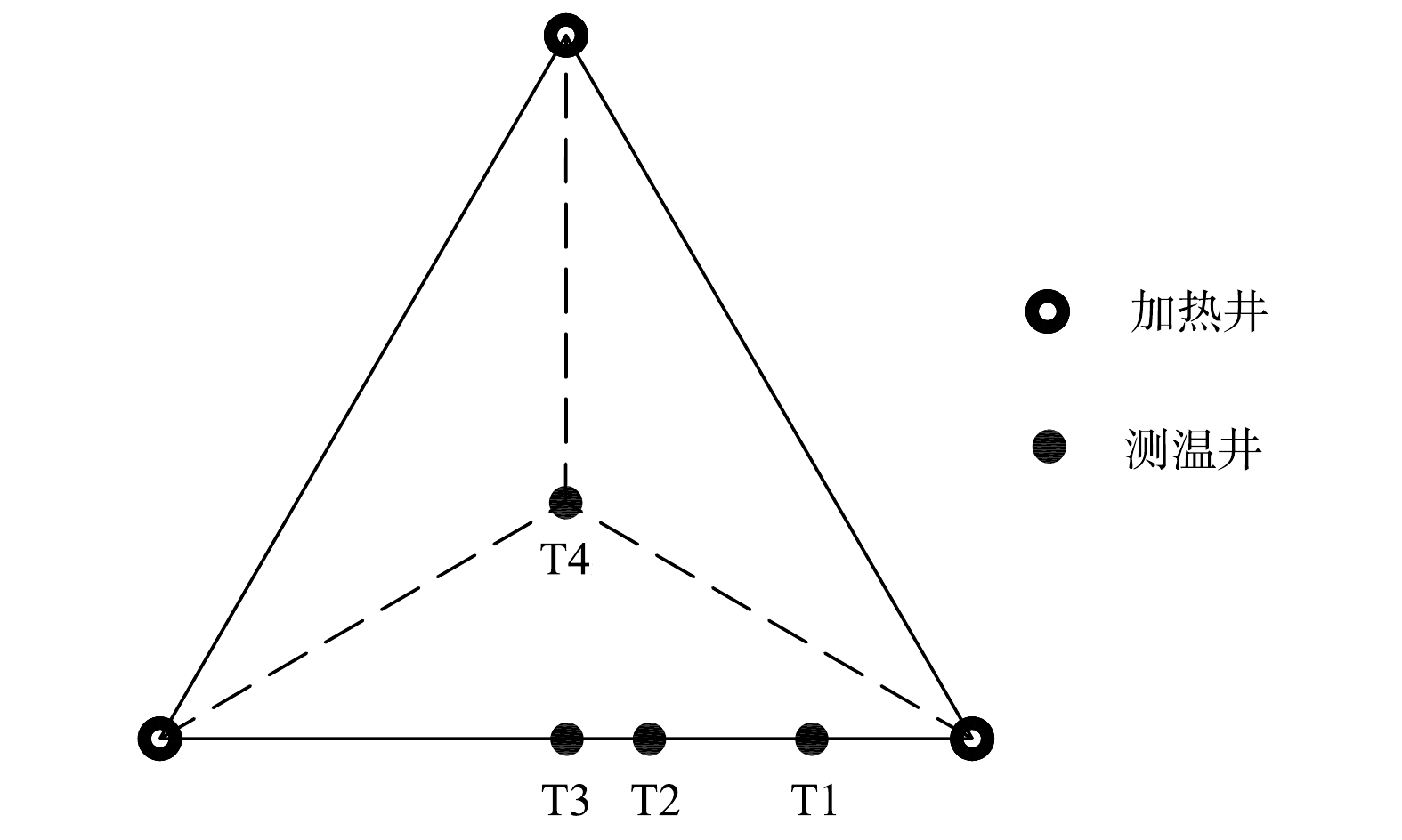
图 5 距离单井热源测温井设计
Figure 5. Design of temperature measuring wells from single well heat source
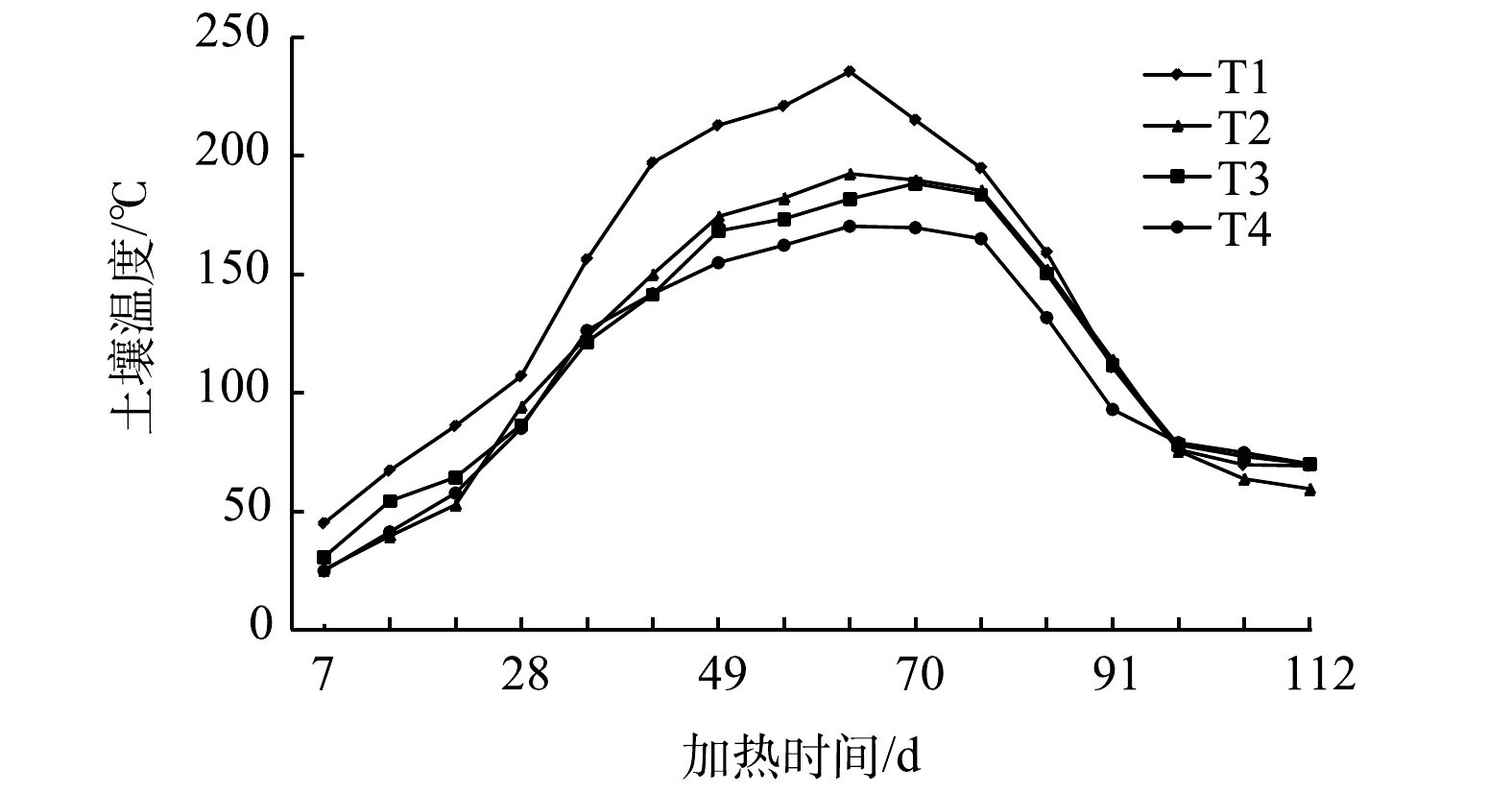
图 8 与加热井不同距离处土壤温度随时间变化情况
Figure 8. Variation of soil temperature with time at different distances from heating well
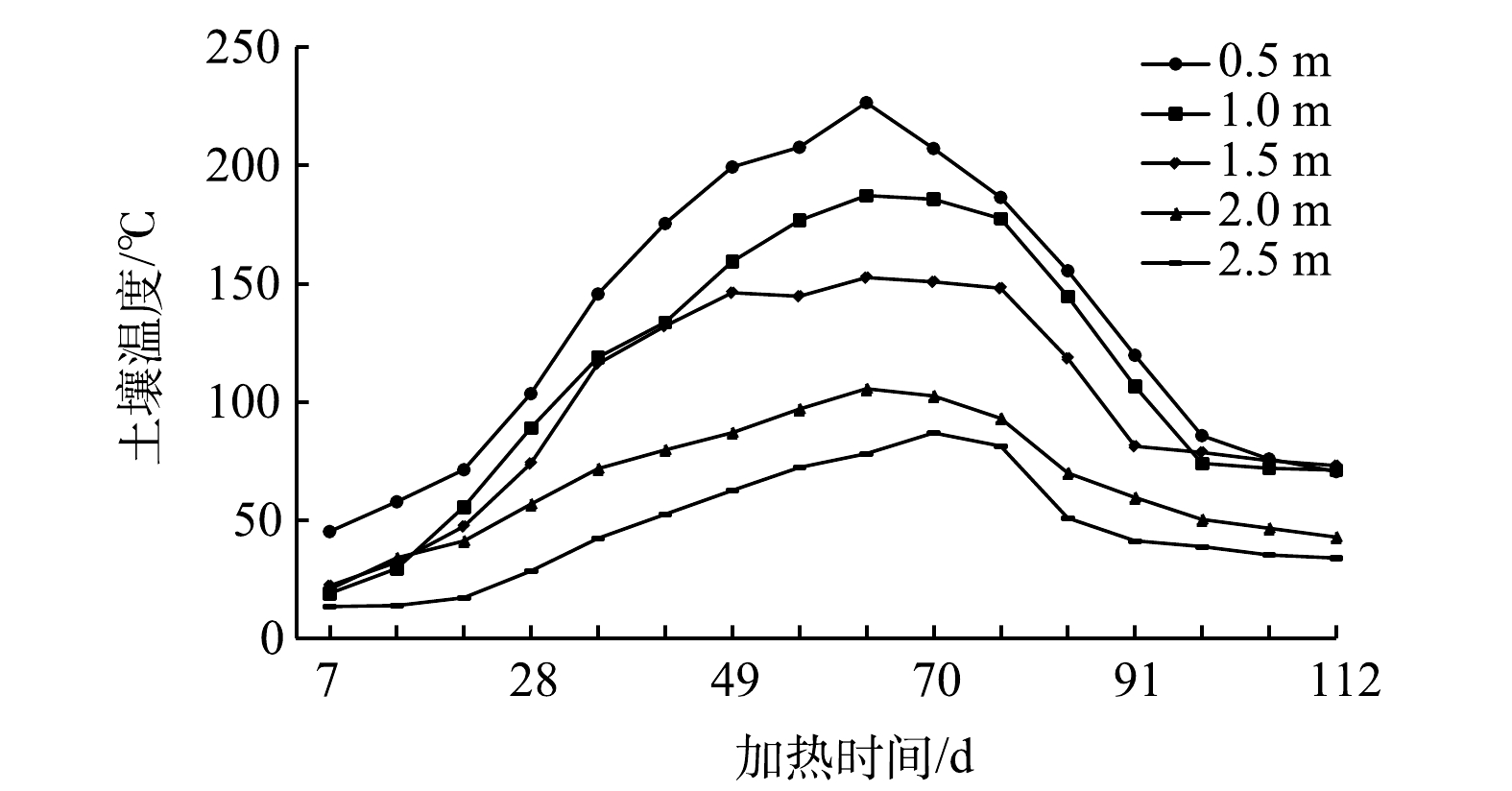
图 9 加热井边界土壤温度随时间变化情况
Figure 9. Variation of soil temperature with time at the boundary of heating well
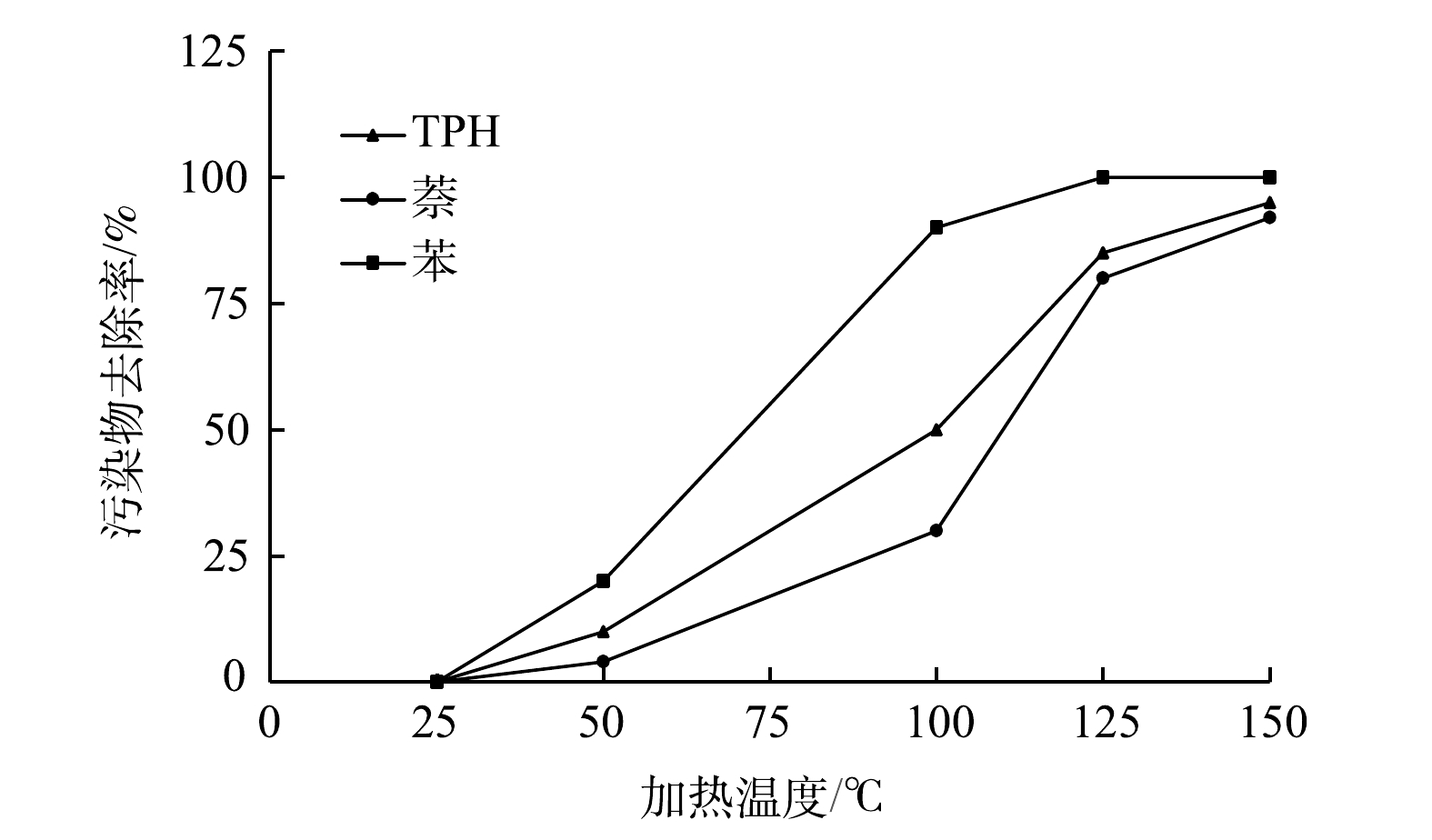
图 10 污染物去除率随加热温度变化情况
Figure 10. Variation of pollutant removal rate with heating temperature
表 1 地块地层岩性分布
Table 1. Stratigraphic lithologic distribution of site
标准层号 分层厚度/m 岩土名称及特征 1 0~1.9 杂填土:呈杂色,主要由建筑垃圾构成,含碎砖、水泥块、砂土、粉土等。无刺激性气味。 2 0.5~1.0 粉土:呈褐黄色,含云母、氧化物及较多石英矿物。常混较多砂质成分,局部夹薄层细沙或粗砾砂。无刺激性气味。 3 1.0~1.9 粗砂砾:呈褐黄,矿物组成主要为石英、云母、长石等,常混有大量粗砂及少量圆砾,局部混有大量细砂。 4 0.5~0.9 粉土:呈褐黄,含云母、氧化物及较多石英矿物等,局部夹薄层粗砂砾,有轻微刺激性气味。 5 4.0~6.6 粗砂砾:呈褐色,矿物组成主要为石英、云母、长石等,级配不良。常混有大量粗砂及少量圆砾,局部混有大量细砂,常夹薄层粉土。有轻微刺激性气味。 6 1.0~2.0 粉土:呈褐黄,含云母、氧化铁等,常混有较多粉质粘土。无刺激性气味。
下载: 导出CSV
表 2 地块各土层理化性质
Table 2. Physical and chemical properties of every soil in site
特征参数土壤 单位 第一层(0~−2.0 m) 第二层(−2.0~−8.0 m) 填土 粉砂土 含水率 — 22.4% 23.6% 湿密度 g·cm−3 1.9 1.9 干密度 g·cm−3 1.6 1.6 饱和度 — 90.9% 93.2% 孔隙比 — 0.6 0.6 渗透系数 cm·s−1 4.3×10−5 5.3×10−5 pH — 8.9 9.3 有机质含量 g·kg−1 10.1 5.9 导热系数 W·(m·K)−1 1.7 1.8
下载: 导出CSV
表 3 土壤中目标污染物浓度变化
Table 3. Concentration variation of target pollutants in soil
目标污染物浓度/(mg·kg−1) 污染物名称 取样编号 0~−2.0 m −2.0~−4.0 m −4.0~−5.0 m −5.0~−6.0 m −6.0~−7.0 m 修复前 修复后 修复前 修复后 修复前 修复后 修复前 修复后 修复前 修复后 TPH Q1 78.6 未检出 24.9 未检出 4 021 19.8 4 286 23.8 3 469 15.6 Q5 50.8 未检出 93.4 未检出 4 439 187.3 4 762 96.3 3 943 192.8 Q9 60.7 未检出 140.7 未检出 2 143 未检出 2 537 12.2 1 814 26.6 苯 Q1 4.5 未检出 未检出 未检出 18.4 未检出 16.2 未检出 12.9 未检出 Q5 未检出 未检出 未检出 未检出 11.3 未检出 14.4 未检出 10.1 未检出 Q9 未检出 未检出 未检出 未检出 9.5 未检出 15.8 未检出 5.8 未检出 萘 Q1 24.6 未检出 19.8 未检出 248.8 未检出 277.3 未检出 183.5 14.4 Q5 未检出 未检出 未检出 未检出 58.9 未检出 87.4 未检出 86.5 未检出 Q9 34.5 未检出 未检出 未检出 388.5 未检出 298.4 未检出 104.3 未检出
下载: 导出CSV
表 4 冷凝废水中目标污染物含量变化
Table 4. Concentration variation of target pollutants in condensate wastewater
污染物名称 单位 编号 目标污染物浓度 第7 d 第21 d 第35 d 第49 d 第63 d 第77 d 第91 d 第105 d TPH mg·L−1 1 未检出 6.08 84.36 134.65 54.26 14.48 4.96 5.83 2 未检出 7.48 99.42 159.49 69.38 8.89 15.58 8.74 3 未检出 5.88 87.17 134.08 60.35 12.72 9.62 0.79 苯 μg·L−1 1 未检出 28.87 66.78 15.78 5.43 未检出 未检出 未检出 2 未检出 25.73 69.31 14.99 4.82 未检出 未检出 未检出 3 未检出 31.49 63.49 14.65 5.04 未检出 未检出 未检出 萘 μg·L−1 1 未检出 未检出 14.59 88.83 134.59 34.53 0.83 0.04 2 未检出 未检出 15.03 79.45 129.86 29.78 0.76 0.04 3 未检出 未检出 14.88 83.92 132.24 29.48 0.77 0.03
下载: 导出CSV
-
[1] 吴嘉茵, 方战强, 薛成杰, 等. 我国有机物污染场地土壤修复技术的专利计量分析[J]. 环境工程学报, 2019, 13(8): 2015-2024. doi: 10.12030/j.cjee.201812195 [2] 王艳伟, 李书鹏, 康绍果, 等. 中国工业污染场地修复发展状况分析[J]. 环境工程, 2017, 35(10): 175-178. [3] CHEN M. Analytical integration procedures for the derivation of risk-based generic assessment criteria for soil[J]. Human and Ecological Risk Assessment, 2010, 16: 1295-1317. doi: 10.1080/10807039.2010.526502 [4] YANG H, HUANG X, THOMPSON J R, et al. China’s soil pollution: Urban brownfields[J]. Science, 2014, 344(6185): 691-692. [5] 陶欢, 廖晓勇, 阎秀兰, 等. 应用多属性决策分析法筛选污染场地土壤修复技术[J]. 环境工程学报, 2017, 11(8): 4850-4860. doi: 10.12030/j.cjee.201707165 [6] 白利平, 罗云, 刘俐, 等. 污染场地修复技术筛选方法及应用[J]. 环境科学, 2015, 36(11): 4218-4224. [7] MYERS K F, KARN R A, ENG D Y, et al. In situ thermal desorption of VOCs in vadose zone soils[J]. Field Analytical Chemistry & Technology, 2015, 2(3): 163-171. [8] KUNKEL A M, SEIBERT J J, ELLIOTT L J, et al. Remediation of elemental mercury using in situ thermal desorption (ISTD)[J]. Environmental Science & Technology, 2006, 40(7): 2384-2389. [9] BONNARD M, DEVIN S, LEYVAL C, et al. The influence of thermal desorption on genotoxicity of multi-polluted soil[J]. Ecotoxicology and Environmental Safety, 2010, 73(5): 955-960. doi: 10.1016/j.ecoenv.2010.02.023 [10] BING Y, XUE N, DING Q, et al. Polychlorinated biphenyls removal from contaminated soils using a transportable indirect thermal dryer unit: implications for emissions[J]. Chemosphere, 2014, 114(22): 84-92. [11] 刘志阳. 一种原位燃气热脱附土壤修复装置: ZL 201820382578.1. X[P]. 2018-12-11. [12] 牛晓阳, 石德升, 赵颖, 等. 污染土壤燃气原位热脱附修复系统: ZL 201811448227.7. X[P]. 2018-11-30. [13] 蒋村, 孟宪荣, 施维林, 等. 氯苯污染土壤低温原位热脱附修复[J]. 环境工程学报, 2019, 13(7): 1720-1726. doi: 10.12030/j.cjee.201810082 [14] 胡正, 沈青. 原位热脱附技术在有机污染地块中的修复效果研究[J]. 环境科技, 2020, 33(6): 30-34 doi: 10.3969/j.issn.1674-4829.2020.06.006 [15] 张攀, 高彦征, 孔火良. 污染土壤中硝基苯热脱附研究[J]. 土壤, 2012, 44(5): 801-806. doi: 10.3969/j.issn.0253-9829.2012.05.015 [16] 中华人民共和国环境保护部. 土壤和沉积物挥发性有机物的测定吹扫捕集/气相色谱-质谱法: HJ 605-2011[S]. 北京: 中国环境科学出版社, 2011. [17] 中华人民共和国生态环境部. 水质苯系物的测定顶空/气相色谱法: HJ 1067-2019[S]. 北京: 中国环境科学出版社, 2019. [18] 中华人民共和国卫生部, 中国国家标准化管理委员会. 生活饮用水标准检验方法有机物指标: GB/T 5750.8-2006[S]. 北京: 中国标准出版社, 2006. [19] 中华人民共和国环境保护部. 水质可萃取性石油烃(C10-C40)的测定气相色谱法法: HJ 894-2017[S]. 北京: 中国环境科学出版社, 2017. [20] 张学良, 廖朋辉, 李群, 等. 复杂有机物污染地块原位热脱附修复技术的研究[J]. 土壤通报, 2018, 49(4): 993-1000. [21] 昊平, 于桂红, 于松峰, 等. 石油馏分组成[J]. 科技信息, 2011, 25: 35-36. doi: 10.3969/j.issn.1001-9960.2011.24.028 [22] ASTON D P, KUEPER B H. Thermal conductive heating in fractured bedrock: Screening calculations to assess the effect of groundwater influx[J]. Advances in Water Resources, 2009, 32(2): 231-238. doi: 10.1016/j.advwatres.2008.10.019 [23] MUNHOLLAND J L, MUMFORD K G, KUEPER B H. Factors affecting gas migration and contaminant redistribution in heterogeneous porous media subject to electrical resistance heating[J]. Journal of Contaminant Hydrology, 2016, 184: 14-24. doi: 10.1016/j.jconhyd.2015.10.011 [24] KROUZEK J, DURDAK V, HENDRYCH J, et al. Pilot scale applications of microwave heating for soil remediation[J]. Chemical Engineering and Processing Process Intensification, 2018, 130: 53-60. doi: 10.1016/j.cep.2018.05.010 [25] ZHAO C, DONG Y, FENG Y, et al. Thermal desorption for remediation of contaminated soil: A review[J]. Chemosphere, 2019, 221: 841-855. doi: 10.1016/j.chemosphere.2019.01.079 [26] HERON G, PARKER K, GALLIGAN J, et al. Thermal treatment of eight CVOC source zones to near nondetect concentrations[J]. Ground Water Monitoring and Remediation, 2009, 29(3): 56-65. doi: 10.1111/j.1745-6592.2009.01247.x [27] 孙磊, 蒋新, 周健民, 等. 五氯酚污染土壤的热修复初探[J]. 土壤学报, 2004, 41(3): 462-465. doi: 10.3321/j.issn:0564-3929.2004.03.021 [28] 赵涛, 马刚平, 周宇, 等. 多环芳烃类污染土壤热脱附修复技术应用研究[J]. 环境工程, 2017, 35(11): 178-181. [29] 王瑛, 李扬, 黄启飞, 等. 温度和停留时间对DDT污染土壤热脱附效果的影响[J]. 环境工程, 2012, 30(1): 116-120. [30] ZHAO C, MUMFORD K G, KUPPER B H. Laboratory study of non-aqueous phase liquid and water co-boiling during thermal treatment[J]. Journal of Contaminant Hydrology, 2014, 164: 49-58. doi: 10.1016/j.jconhyd.2014.05.008 [31] 程能林. 溶剂手册(第四版)[M]. 北京: 化学工业出版社, 2007. [32] 高国龙, 蒋建国, 李梦露. 有机物污染土壤热脱附技术研究与应用[J]. 环境工程, 2012, 30(1): 128-131. [33] FISCHER U, SCHULIN R, KELLER M. Experimental and numerical investigation of soil vapor extraction[J]. Water Resources Research, 1996, 32(12): 3413-3427. doi: 10.1029/95WR02668 [34] ALBERGARIA J T, ALVIM-FERRAZ M C M, DELERUE-MATOS C. Soil vapor extraction in sandy soils: Influence of airflow rate[J]. Chemosphere, 2008, 73(9): 1557-1561. doi: 10.1016/j.chemosphere.2008.07.080 [35] 周启星, 宋玉芳. 污染土壤修复原理和方法[M]. 北京: 科学出版社, 2004. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4656
- HTML全文浏览数: 4656
- PDF下载数: 156
- 施引文献: 0
