



-
近年来,我国城市化进程和产业转型日益加快,许多企业(尤其是化工、农药、冶金等污染企业)为落实国家政策陆续迁至郊区、工业园区或关闭停产,致使城区内遗留了大量废弃的工业污染场地[1-3]。各类土壤污染物中,有机污染物的种类繁多,其具有毒性强、易致癌、易迁移等特点[4-5],对人群健康和生态环境的潜在危害大,应优先控制[6]。因此,针对有机污染场地的治理和修复工作已刻不容缓。
热脱附技术是一种近年来被广泛采用的有机污染场地修复技术,该技术通过加热升温使土壤中的有机污染物挥发、分离并对其集中处理[7]。目前,采用热脱附技术能将土壤加热至500 ℃以上(超过大多数有机污染物的沸点)[8]。该方法具有适用范围广、修复时间短、修复效果好等优点[7],但因为加热土壤需消耗大量能源,所以该技术的应用成本较高[9]。在土壤热脱附修复工程中,只有精确掌握污染场地土壤的热物性以及污染物迁移和相变对场地温度分布的影响规律,才能有效指导加热井的合理布置以及加热功率的即时调整,进而找到降低能耗和成本的途径。目前,关于土壤修复的研究主要关注技术联用、反应机理和脱除效率等[10-12],鲜有土壤热物性对热脱附过程传热和能耗的研究。同时,虽已有大量关于土壤热物性的研究,但其背景多为农业、林业和地源热泵等领域,关于有机污染场地土壤热物性的数据较少。
为了解有机污染场地土壤热物性的规律,本研究以苏州市某修复场地示范区域内的表层土壤为研究对象,用探针式导热仪探究了有机污染土壤在热脱附前后的热导率差异以及表观密度状态(松散或压实)和温度(10~90 ℃)对热脱附后土壤热导率的影响规律。
-
土壤采样点位于苏州市某复合有机污染修复场地编号为G01、G06和G09的示范区域,其各自相距约30~70 m,采样深度为0~3 m。前期场地调查已获悉,该场地的主要污染物为挥发性和半挥发性有机物,包括苯、乙苯、二甲苯、氯苯、二氯苯、二氯乙烯、三氯乙烯和石油烃C6~C20等。各区域0~3 m表层土壤的主要物性参数如表1所示。
在采集过程中,用KD2 Pro探针式导热仪、Takeme-10EC探针式水分仪、环刀和天平等仪器和工具分别测定土壤在原位状态下的热导率、含水率和密度。采集完毕后,抽取部分样品,用TM-85型土壤密度计检测其粒径分布情况,用气相色谱-质谱法检测其各类有机污染物的质量浓度。
-
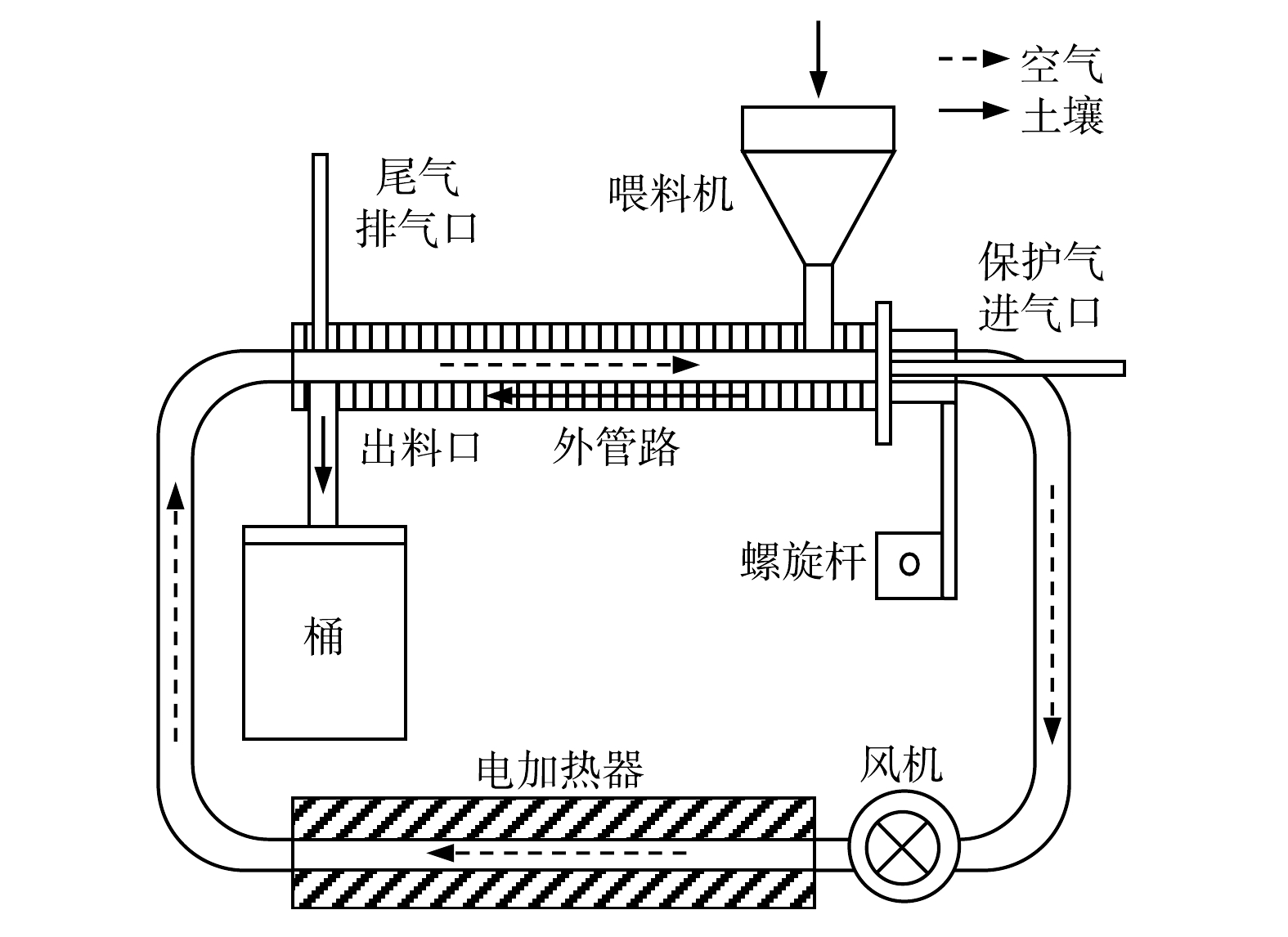
为了防止土壤中大块石砾造成热脱附管路堵塞,首先对运回实验室的土壤样品进行预处理,即将其风干、研磨后过2 mm筛。预处理后,用图1所示的实验室小型热脱附装置对土壤进行热脱附处理。外管路中土壤的运动方向与内管路中热空气的流动方向相反,因此二者可以充分换热,从而达到热脱附的目的。
在土壤已检出的主要污染物中,石油烃C20的沸点最高,为334.8 ℃[13]。因此,本实验将热脱附温度设定为350 ℃,可将主要污染物全部脱除,同时也会使土壤中的自由水全部蒸发。热脱附持续1 h后,抽取部分样品,检测其各类有机污染物的质量浓度以判断污染物是否被全部脱除,同时检测其粒径分布。此外,用D8 DISCOVER型X射线衍射仪和重铬酸钾容量法分别检测土壤中矿物质和有机质的质量分数(亦即土壤的化学组成),以探究其对土壤热导率造成的影响。
-
为探究热导率的变化规律,本研究用3个区域经热脱附后的土壤制备了2类试样。
1)经过热脱附后,土壤中的有机污染物和水分发生了迁移和相变。为探究热脱附前后土壤热导率的差异,制备了密度、含水率等参数与1.1节中在示范场地采样时原位测试的土壤基本相同的试样,在(25±1) ℃(与原位测试时的温度相同)的条件下测试其热导率。
2)在热脱附过程中,当温度低于水的沸点(标准大气压下为100 ℃)时,由于水的比热较大,故大部分能量用于加热土壤水分,致使土壤单位温升所需的能耗较高,因此需要对水的沸点以下土壤的热导率随温度的变化规律进行研究。为此,制备了一批不同表观密度状态(松散和压实)的试样,在10~90 ℃(以10 ℃为间隔)的条件下测试其热导率。试样的质量含水率均为1.9%±0.2%,系热脱附后土壤的自然含水率(绝干土壤易吸收空气中的水分,自然含水率不为0);松散试样的干密度均为(1.10±0.03) g·cm−3,系热脱附后土壤的自然堆积状态,用于与压实试样进行对比;压实试样的干密度均为(1.40±0.03) g·cm−3,参照该修复场地G01区域土壤的实际干密度(如表1所示),以期测试结果能为修复工程提供参考。
-
如图2所示,为了控制测试过程中土壤试样的温度,本实验将土壤试样盛放于特制的不锈钢圆筒容器内,并将容器置于水浴锅中。圆筒容器的顶盖通过法兰结构与圆筒连接,TR-1型探针和热电偶通过顶盖上开设的孔位竖直地插入土壤试样内部,用防水胶带固定探针和热电偶的位置以防止其松动的同时隔绝水分。连接好装置后,设定水浴锅的温度,当热电偶的读数稳定在设定温度附近达到1 h后,即认为试样内部温度分布均匀,可开始进行热导率测试。每次测试完毕后,纪录试样的热导率λ和测试过程中试样的平均温度T。为保证测试结果的可重复性,每个温度点做多次测试,每次测试均更换探针的位点。实际测试过程中,土壤的平均温度与设定值相差在1 ℃以内。
-
从表2中可知:1)经过350 ℃热脱附后,3个区域土壤中的各污染物均被脱除至低于仪器检出下限,即各污染物脱除率接近100%;2)该场地同一深度的污染情况具有显著的不均匀性,与另外2个区域相比,G01区域的污染物种类更多且浓度更高。
从表3中可知,3个区域的表层土壤均为黏壤土,经过350 ℃热脱附后,粒径均有一定程度增大且各区域之间无显著差异。其中,粒径小于0.005 mm的黏粒占比从超过30%减少至不足15%;0.005~0.075 mm的粉粒占比无明显变化;0.075~0.250 mm的细砂粒占比则从不足10%增加至超过20%,且出现了占比约1%的0.250~0.500 mm的中砂粒。陈王若尘[14]的研究也有类似发现,即温度低于300 ℃时经热脱附后土壤的粒径会减小,而高于300 ℃时粒径则会增大。土壤在受热升温时会出现大颗粒破碎[15]和小颗粒团聚[16]这两种现象,当破碎作用占优时粒径减小,而团聚作用占优时则粒径增大[14]。
在密度、含水率和测试温度都基本相同的情况下,热脱附后与热脱附前(原位测试时)土壤热导率的对比如图3所示。与热脱附前相比,G01和G06区域土壤的热导率在热脱附后增大7.6%和3.8%,而G09区域则基本保持不变,其测量平均值基本在1.4~1.5 W·(m·℃)−1的范围内。对热脱附前后的热导率进行方差分析,得到热脱附前后热导率的均方值之比F = 2.989,低于相应显著性水平(α = 0.05)下的临界值Fcrit = 7.709,这说明热脱附前后土壤的热导率无显著差异。
在不同表观密度状态和温度的条件下,经热脱附后质量含水率为1.9%±0.2%的土壤的热导率随温度的变化曲线如图4所示。从图中可知,在10~90 ℃以内,3个区域松散状态和压实状态土壤的热导率均随温度的升高而增大。这与HIRAIWA等[17]以及陈正发等[18]的研究结果相似。经计算,在2种表观密度状态下,热导率与温度的Pearson积矩相关系数均大于0.970(显著性水平α = 0.05),呈正相关关系。土壤属于非金属材料,其导热主要依靠晶格振动为载体,温度越高则晶格振动越剧烈,越有利于热量传递。此外,压实土壤比松散土壤的热导率大,即土壤的热导率随干密度的增大而增大。干密度反映土壤固体颗粒的密实程度,干密度增大意味着土壤固体颗粒占比增大而空气占比(孔隙率)减小。由于土壤固体颗粒的热导率比空气的热导率大2个数量级,因此,干密度越大的土壤热导率越大。分别就温度和干密度对热导率的影响进行了方差分析,得到温度与热导率的F值为29.763,干密度与热导率的F值为191.348。后者大于前者,这表明干密度对热导率的影响比温度对热导率的影响更显著。
用不同的公式对热导率λ和温度T拟合后发现,在10~90 ℃,最简单的线性式即具有足够好的拟合程度,因此,本研究给出线性拟合公式供热修复工程参考。λ-T拟合公式和拟合优度R2如表4所示。
值得注意的是,密度、含水率和温度均一致时,经热脱附后3个区域土壤的热导率却有显著差异(α = 0.05),其中,F = 3.604 > Fcrit = 2.152。最大差异出现在温度为80 ℃且表观密度状态为压实时,此时G01区域土壤的热导率为0.219 W·(m·℃)−1,而G09的则为0.274 W·(m·℃)−1,相差达到0.055 W·(m·℃)−1。热脱附后不同区域土壤的热导率比较为:G06 > G09 > G01。
3个区域土壤的采样深度、预处理方法和热脱附条件均相同,污染物浓度均低于检出下限且粒径分布无显著差异(α = 0.05),因此,造成上述热导率差异的主要原因并非污染物或粒径分布差异,而更可能是土壤化学组成的差异。土壤有机质以腐殖质为主,其热导率的常用值为1.26 W·(m·℃)−1[19]。土壤矿物质中,石英的热导率为7.69 W·(m·℃)−1[20],其他各类矿物质的热导率在1.53~5.51 W·(m·℃)−1 [20],矿物质热导率的常用值为4.43 W·(m·℃)−1[19]。因为矿物质热导率的约为有机质的3.5倍,所以矿物质含量高而有机质含量低的土壤的热导率更大。JOHANSEN模型[21]是计算土壤热导率的经典模型之一,其公式表明土壤的热导率随石英含量的升高而增大。此外,ABU-HAMDEH等[22]通过实验发现,黏壤土的热导率随有机质含量的升高而减小,并指出研究有机质对土壤热导率影响的文献十分匮乏,无法与已有结果进行比较。如表5所示,土壤经热脱附后,矿物质质量分数G06 > G09 > G01,而有机质质量分数G06 < G09 < G01,这是导致不同采样区域土壤热导率对比呈现G06 > G09 > G01这一趋势的主要原因。
-
1)经350 ℃热脱附处理后,土壤中的主要污染物被全部脱除;受高温热处理时的团聚作用的影响,热脱附后土壤的粒径略有增大,其热导率与热脱附前相比则无显著变化,平均为1.4~1.5 W·(m·℃)−1;土壤热导率随温度升高或干密度增大均呈增大趋势,且干密度对热导率的影响比温度更显著。
2)温度的土壤热导率拟合计算公式具有较高精度,可供该场地或类似黏壤土质地的原位热修复工程作为热物性参考数据。
3)土壤中矿物质质量分数的变化是导致同一场地不同区域土壤热导率呈现较大差异的主要原因。3个示范区域两两相隔仅30~70 m,但热导率的最大差值达0.055 W·(m·℃)−1,这说明对污染场地土壤热物性空间分布的精准把握有助于指导原位热修复工程的开展。
致谢 感谢浙江大学能源清洁利用国家重点实验室李晓东教授、吴昂键讲师和王博硕士在热脱附装置使用上的指导与帮助。
有机污染黏壤土热脱附后热导率的变化特性
Changes in thermal conductivity of organic contaminated clay loam after thermal desorption
-
摘要: 为探究有机污染土壤热脱附后热导率的变化特性,采集了苏州市某原位热脱附修复场地编号为G01、G06和G09的示范区域深度为0~3 m的土壤(系黏壤土),并利用实验室的小型热脱附装置在350 ℃的条件下对污染土壤试样进行了1 h热脱附;对其热脱附前后的粒径分布以及热脱附后的化学组成(矿物质和有机质的质量分数)进行了表征,并用探针式导热仪测试了其热导率。结果表明,在高温热脱附处理过程中,土壤颗粒的团聚作用比破碎作用更强,导致热脱附后土壤粒径增大;当密度、含水率和温度等条件保持一致时,热脱附后土壤的热导率较场地原位测试时无显著变化,平均值在1.4~1.5 W·(m·℃)–1;随温度升高或干密度增大,土壤热导率均增大,且干密度对热导率的影响比温度更加显著。此外,3个采样区域的土壤热导率呈现一定的差异,其中,G06区域的热导率最大而G01区域最小,最多相差0.055 W·(m·℃)–1,这主要是由不同区域土壤中矿物质(其热导率是有机质的3倍以上)质量分数的变化所致。本研究结果可为实际热修复场地的地层温升预测提供参考。Abstract: In order to explore the changes in thermal conductivity of organic contaminated soil after thermal desorption, soils (clay loam) with a depth of 0~3 m at the demonstration areas numbered G01, G06 and G09 in an in-situ thermal desorption remediation site in Suzhou City were collected, and then were deal with thermal desorption at 350 ℃ for 1 h by the lab-scale thermal desorption apparatus. Particle size distributions before and after thermal desorption and chemical compositions (mass percentage of minerals and organic matters) after thermal desorption of the soil samples were characterized, and the thermal conductivity was measured by a probe-type thermal conductivity meter. The results showed that the increase of soil particle size after thermal desorption was due to the fact that the agglomeration effect of soil particles was stronger than the fragmentation effect during the high-temperature treatment. When density, moisture content and temperature were kept constant, the thermal conductivity of the soil samples after thermal desorption had no significant changes compared with the in-situ measured results, with the average results being 1.4~1.5 W·(m·℃)−1 Soil thermal conductivity increased with either increasing the temperature or increasing the dry density, and the effect of dry density was more pronounced. In addition, there was a variation of the thermal conductivity of soil samples among the three areas, where G06 area was the highest and G01 area was the lowest with the maximum discrepancy being 0.055 W·(m·℃)−1. Such variation was mainly due to the changes in mass percentage of minerals, which had a thermal conductivity that was over 3 times greater than that of organic matters. These results could provide a reference for predicting the temperature rise in practical thermal remediation sites.
-
Key words:
- organic contamination /
- clay loam /
- thermal desorption /
- thermal conductivity
-
磷是所有生物生存所必需的营养元素,也是现代工农业生产中不可缺少的资源[1]。作为一种珍贵的不可再生资源,全球磷资源的日益短缺和枯竭令人担忧。据估计,2040年后,全球磷需求量将超过全球磷供应量[2],农业磷需求将在2050年达到峰值[3]。以目前的磷矿储量和消耗速度计算,世界磷矿资源将在100~200年内消耗殆尽[4],届时将发生无磷可用的“磷危机”现象[5]。我国磷资源短缺问题同样严重,截至2020年我国的磷矿储量不到世界储量的5%,每年的磷矿消耗量却达世界水平的40%~50%,因此磷资源回收成为我国可持续发展的战略性需求[2]。污水处理厂剩余污泥中富集了大量的磷[6],这使得污泥成为潜力巨大的“二次磷资源”。从剩余污泥中回收磷既可以解决磷资源紧缺与水体富营养化问题,又可以实现废物的资源化利用。然而金属、致病菌等有毒害污染物质的存在,使富磷污泥不能直接作为磷肥使用。目前,污泥中的磷先释放再回收成为磷回收的主要手段,其中磷的有效释放是磷回收的关键。
污泥水热碳化技术是指高含水率污泥在一定温度、反应时间和自产生压力下,经过水解、脱水、脱羧和缩聚等一系列复杂反应,最终产生富碳固体—水热炭,以及液体产品—水热液[7]。污泥水热碳化反应对磷的迁移及形态转化影响较大。随着水热温度的升高污泥胞外聚合物及细胞结构破坏,胞内聚磷酸盐、磷脂等被释放至水热液中,并进一步分解为无机磷(P2O74−和PO43−)[8],而暴露在液相中的P2O74−和PO43−会与污泥中溶出的钙、镁、铝、铁等金属离子结合生成相应的金属磷酸盐,经沉淀或吸附于固体物质表面形成吸附态磷,并经溶解和重组/重结晶形成更加稳定的磷化物,从而使磷不断富集在固相水热炭中[9]。为从污泥水热炭中回收磷,研究人员开展了大量研究。如PÉREZ等[10]采用有机酸浸取污泥水热炭中的磷,发现有机酸可以将磷的浸取率提高至75%以上,其中草酸在5 min时几乎可以将水热炭中全部的磷释放至液相。SHETTIGONDAHALLI等[11]研究了在污泥水热碳化过程中和水热碳化后加酸(H2SO4)对磷浸出的影响,结果表明在水热碳化之后加入硫酸具有更高的磷浸出效果。XUE等[7]研究了HCl对城市污泥水热碳化过程中磷转移和转化的影响,结果发现在水热碳化过程中加入0.5%~2.5%HCl,可使富集在水热炭中的磷含量由污泥总磷含量的99.5%降低至91.8%,非磷灰石无机磷由34%增长到94%,这是由于盐酸提供了低pH环境,促进了钙相关磷沉淀物溶解并释放磷至液相所至。
乙二胺四乙酸(EDTA)是一种金属络合剂,在污泥水热碳化过程中可与释放的金属离子络合形成可溶性金属络合物,从而可避免金属离子与磷酸根形成沉淀沉积在固相中,便于磷释放至液相。例如,MA等[12]采用EDTA与高压脉冲放电相结合的方法预处理污泥,在破坏污泥结构的同时防止金属磷酸盐沉淀的形成,显著提高了污泥厌氧发酵过程中磷的释放。ZOU等[13]的研究表明,在厌氧消化过程中添加EDTA可以损伤细胞膜并与金属离子络合,有效提高了磷的释放效率(达82%),并且对Fe-P中磷的释放效果优于对Al-P中磷的释放效果。陈树俊[14]、史可等[15]在污泥低温热水解过程中添加EDTA,磷的释放率分别达到65%和77.9%,远高于原污泥中磷的释放效果(分别为5%和39.7%)。此外,EDTA对EPS具有一定的溶解破坏作用,能改变微生物的细胞结构,促进胞外物质与污泥细胞的分离。刘博文等[16]采用热碱-EDTA耦合法强化破解污泥,破解后SCOD、TN、TP、多糖和蛋白质溶出量均升高,污泥残渣中VS含量小于热碱破解法。肖倩等[17]利用EDTA对消化污泥胞外紧密型EPS(TB-EPS)进行提取,发现EDTA对TB-EPS具有一定的溶解破坏作用,并造成细胞膜损伤。因此,采用EDTA与污泥水热碳化相结合对污泥溶胞释磷以及避免磷与金属离子形成沉淀,促进磷从固相向液相转移将具有非常积极的作用,但目前这方面的研究有限。
本研究采用EDTA强化污泥水热碳化释磷,研究不同条件下磷的释放特性,并通过SMT法提取水热炭中不同形态的磷,分析磷的形态分布规律,探讨磷的迁移转化行为和内在机制,这对从污泥水热碳化过程中回收磷资源具有重要意义。
1. 材料与方法
1.1 试验原料
试验所用污泥取自天津市某污水厂二沉池的剩余污泥,取回后剔除杂质经2 h自由沉淀后去除上层清液,冷藏于4 ℃冰箱待用。污泥性质如表1所示。
表 1 污泥的基本性质Table 1. Basic properties of sludgepH SS/(mg·L−1) VSS 含水率 SCOD/(mg·L−1) TP/(mg·g−1) 6.24±0.91 12 000±2 000 72.50%±2.60% 95.20%±3.60% 102.00±78.00 19.97±5.87 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 试验方法
将污泥样品搅拌均匀,取70 mL转移到反应釜中,投加EDTA 0~35 mmol·L−1,设置反应温度150~250 ℃,反应0~6 h。反应结束后自然冷却至室温,采用0.45 μm滤膜对样品进行抽滤,分别得到固相和液相产品,将固相产品放入电热恒温干燥箱中105 ℃干燥至恒重,经充分研磨后,得水热炭;液相进行相关指标测定。
1.3 分析方法
采用Flash EA
1112 型全自动元素分析仪(美国)测定污泥和水热炭中C、H、N、S元素,O元素含量采用差减法计算;根据《煤的工业分析》(GB/T212—2008)测定污泥的灰分,根据Dulong公式计算水热炭的高位热值;采用Optima8300 电感耦合等离子体光谱仪(美国)测定金属(Fe、Al、Mg、Ca)浓度。利用SMT法提取污泥及水热炭中五种形态的磷,分别为总磷(TP)、有机磷(OP)、无机磷(IP)、磷灰石无机磷(AP,通常为与Ca和Mg结合的各种磷)和非磷灰石无机磷(NAIP,通常为与Fe、Mn和Al氧化物及其氢氧化物结合的磷)[18-19]。磷含量采用钼锑抗分光光度法测定。
2. 结果与讨论
2.1 温度对EDTA强化污泥水热碳化释磷的影响
在EDTA投加量为5 mmol·L−1、水热碳化时间为30 min时,研究了不同反应温度对污泥释磷的影响,如图1所示。由图1(a)可知,液相中TP和PO43−的浓度随温度的升高而增加,210 ℃后趋于平缓。这是由于温度是有机大分子降解和高活性化学键断裂和重组的主要能量来源,在150~190 ℃时主要发生水解和脱水反应,此阶段主要以有机物的释放为主;210~250 ℃脱羧、缩聚和芳香化等反应加剧[20]。高温高压环境破坏污泥絮体结构,水解有机物,使得细胞内及胞外聚合物中的磷大量释放至液相,由于同时释放的金属离子与EDTA络合,使得磷以离子态形式大量存在于液相中。但在较低温度时,由于不能提供足够的能量,使得150~190 ℃时磷的释放速率小于210 ℃;在210 ℃时,高温促进了反应的剧烈进行,提高了磷的释放效率并基本达到最大值,此时水热液中TP和PO43−为178.8和154.3 mg·L−1,磷的浸出率为64.4%(图1(b))。随着反应温度的继续升高,磷从固相转移到液相的释放量不再发生明显变化。由于温度过高会增加能耗,加大成本,因此本研究选择210 ℃作为最佳水热碳化温度。
2.2 时间对EDTA强化污泥水热碳化释磷的影响
在水热碳化温度为210 ℃,EDTA投加量为5 mmol·L−1的条件下,探究了不同水热时间对TP和PO43−的影响,见图2。图2(a)显示,液相中磷的浓度随反应时间的增加呈先增加后少量降低的趋势。这是由于反应过程中发生的污泥破解释放磷和金属,以及释放的金属离子与EDTA发生络合反应置换磷等过程均需要一定的反应时间。因此,随着反应时间的增加,磷浓度呈线性增长,并在4 h时达到峰值,此时液相中TP和PO43−的浓度分别达到300.6和295.59 mg·L−1,对应磷的浸出率可达79.9%(图2(b))。此时大部分污泥细胞已经破裂,胞内物质被释放出来,同时液相中EDTA与溶出的金属离子络合,阻止了金属离子与磷再结合形成金属磷酸盐沉淀又向固相中沉积的过程。随着反应时间的延长,释放的金属离子增多,而EDTA有限,导致多余的金属与液相中的磷结合,从而使得部分磷重新形成金属磷酸盐沉淀或结晶[21],液相中磷释放量少量下降。可见在一定范围内延长反应时间对磷的释放具有促进作用,但过长时间不利于磷释放,因此本研究选择4 h作为磷释放的最佳反应时间。
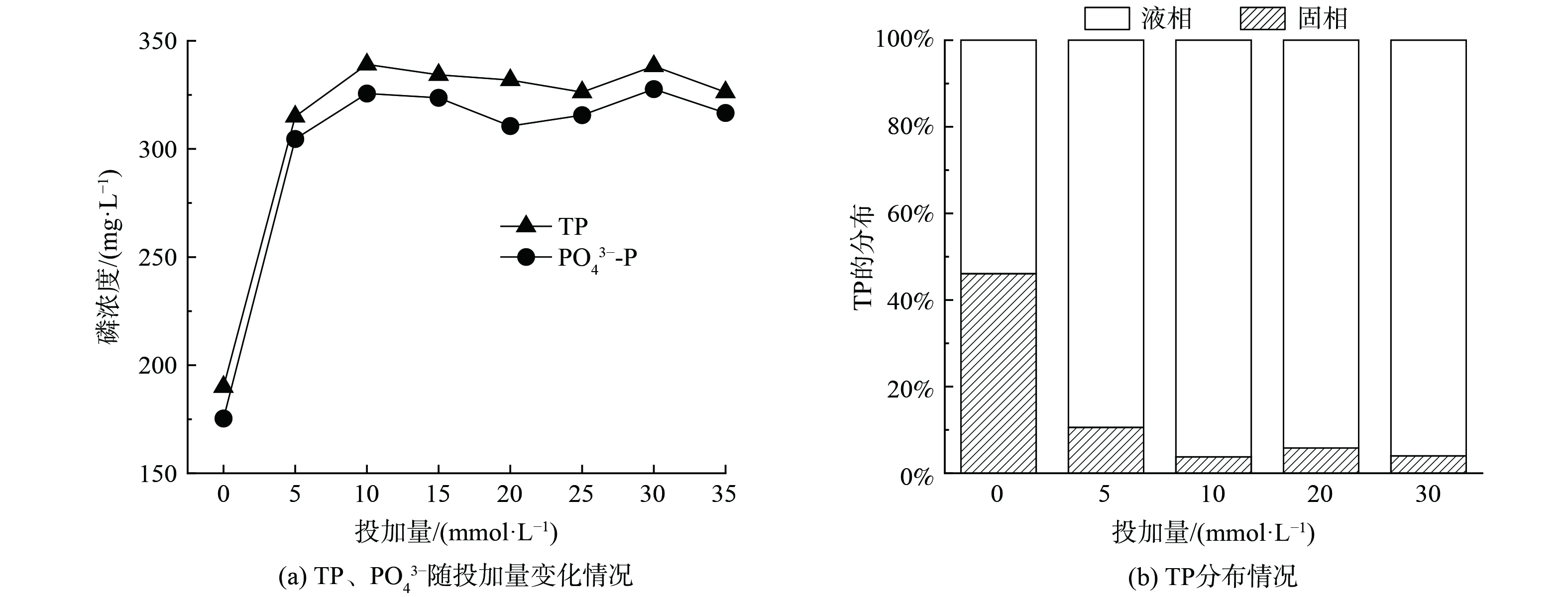
2.3 EDTA投加量对污泥水热碳化释磷的影响
在温度210 ℃,时间4 h条件下,考察不同EDTA投加量对污泥中TP和PO43−释放的影响,情况如图3所示。由图3(a)可知,磷的浓度随EDTA投加量的增加而升高,在投加量为10 mmol·L−1时,TP和PO43−的浓度达到最大值,分别为339.1和325.6 mg·L−1,约是未投加EDTA时的2倍,此时磷的释放率达到96.2%(图3(b));此后继续增加EDTA用量,液相中的磷浓度基本趋于稳定,但仍有约4%的磷存在于固相中,并以AP的形式存在。这可能是由于过量EDTA使溶液呈酸性,导致小部分与钙、镁结合的磷无法释放出来。
图3结果说明EDTA可以有效促进磷从固相向液相大量转移,显著高污泥释磷效果。这是由于:一方面EDTA螯合大量金属离子,防止磷与金属离子沉淀;另一方面,EDTA可以分解EPS并溶解细胞膜,使细胞部分暴露在污泥中并释放胞内有机物,增强污泥中磷的释放[12]。因此,在污泥水热碳化阶段加入EDTA不仅可以掩蔽金属离子对磷释放的影响,还可以溶解EPS,增强释磷效果。
2.4 污泥浓度对EDTA强化污泥水热碳化释磷的影响
在温度为210 ℃,时间为4 h,EDTA投加量为10 mmol·L−1的条件下,考察了不同污泥浓度(3.7~33.6 g·L−1)对TP和PO43−释放的影响,结果见图4。由图4(a)可知,随着污泥浓度的降低,TP和PO43−的浓度减少,其中由33.6 mg·L−1降低到8.4 mg·L−1时,TP和PO43−的下降速率较快,8.4 mg·L−1降低到3.7 mg·L−1时,TP和PO43−的下降速率变缓。这主要是由于污泥浓度降低导致反应物中磷的总含量降低,从而使得污泥浓度减少时上清液中磷的释放量降低。而从图4(b)可以看出,污泥浓度对TP在固相和液相之间的转移影响不大,不同污泥浓度的反应液在试验条件下磷的释放率均在92%~95%范围,这是由于在EDTA辅助污泥水热碳化的作用下可以将污泥中大部分的磷释放出来。
 图 4 污泥浓度对污泥释磷的影响Figure 4. Effect of sludge concentration on phosphorus release from sludge
图 4 污泥浓度对污泥释磷的影响Figure 4. Effect of sludge concentration on phosphorus release from sludge2.5 EDTA对固相中磷形态的影响
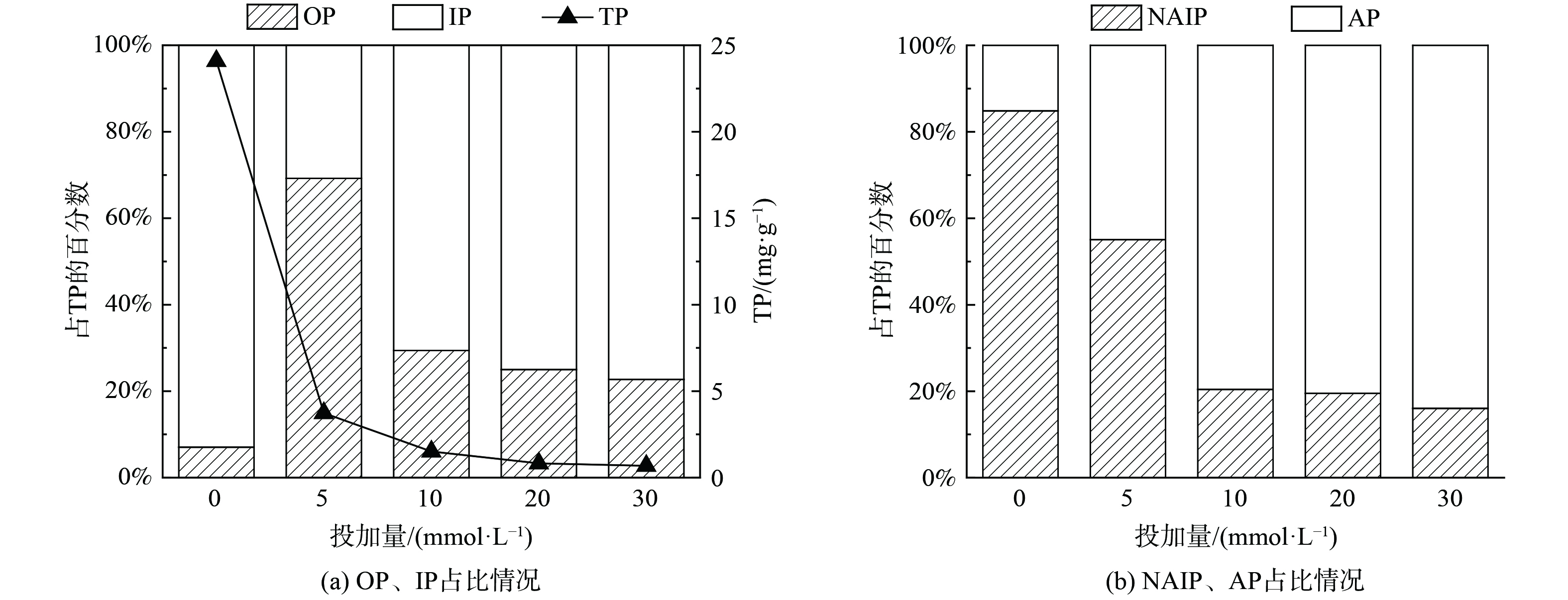
图5显示了不同EDTA投加量下水热炭中磷的形态分布。由图5(a)可知,水热炭中TP的质量分数随投加量的增加而降低,这与前文结论一致(图3)。该结果说明在水热碳化过程中投加EDTA促进了固相中的磷向液相转移。水热炭中的磷可分为有机磷(OP)和无机磷(IP),其中OP主要包括正磷酸单酯、磷酸二酯等,而IP主要包括正磷酸盐和焦磷酸盐等[22-23]。
图5(a)显示,当直接将污泥水热碳化后(EDTA=0 mmol·L−1),水热炭中IP占比较高,约为92.8%,OP占比较少为7.2%。这是由于原泥中含有大量金属成分,如Ca、Mg、Al、Fe等,其与磷结合形成无机金属磷酸盐等形态存在于污泥中,同时污泥中含有细胞物质,部分磷以卵磷脂、磷壁酸、聚磷酸等有机磷形式存在,且经水热碳化后,从细胞中释放的金属离子会与从有机磷中释放的磷结合形成金属磷酸盐,使得固相中大部分磷以IP形态存在。当EDTA投加量为5 mmol·L−1时,水热炭中的OP占比升高,IP降低,这是由于EDTA能与溶胞释放的金属结合,部分金属被EDTA络合进入液相,同时释放的磷也进入液相,从而导致水热炭中IP的占比降低。随着EDTA浓度的升高,水热炭中的IP升高,OP降低,这是由于水热碳化使污泥中的有机聚磷酸盐在高温下降解,导致短链聚磷酸盐的数量增加,并为金属络合提供了额外的P-O键,从而使IP占比增多;同时,OP和焦磷酸盐水解,转化为正磷酸盐[24],而水解形成的正磷酸盐与金属结合形成的含磷沉淀物在EDTA的作用下向液相中释放出磷,从而导致OP占比降低。由图5(b)可知,水热炭中AP占TP的比例不断升高,NAIP相反。说明EDTA促进了NAIP向AP转化。这可能是由于EDTA与金属离子形成稳定络合物的酸度范围不同,与Ca2+和Mg2+在碱性范围络合,而污泥经EDTA强化水热碳化处理后,溶液pH呈酸性,EDTA与Ca2+和Mg2+络合减少,导致AP在TP中的占比升高。
2.6 EDTA对水热碳化过程中金属浸出的影响
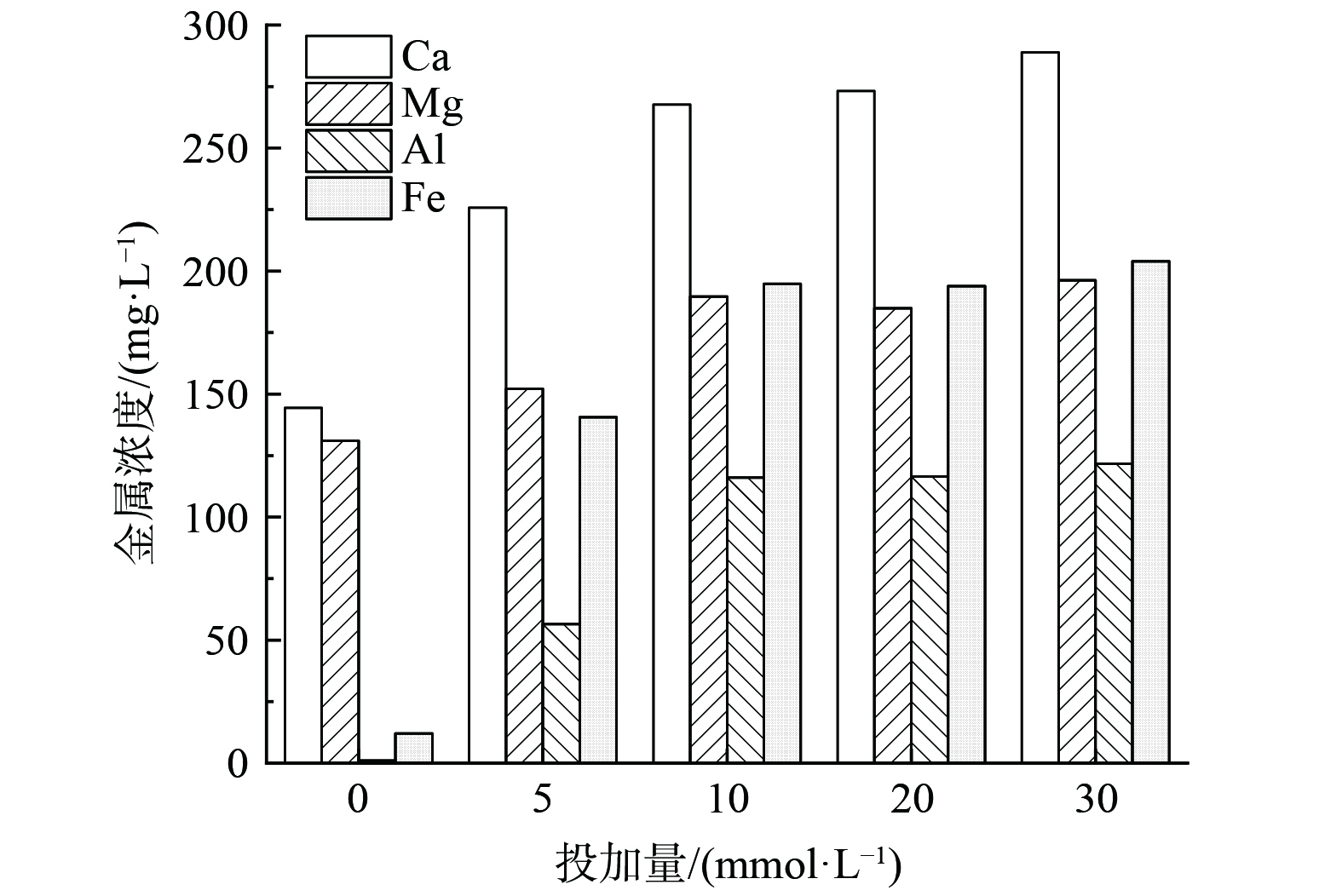
由图6可知,随着EDTA投加量的增加,Ca2+、Mg2+、Al3+和Fe3+的浓度呈现先增加后趋于平缓的趋势。当对污泥单独进行水热碳化处理时,液相中Ca2+、Mg2+、Al3+和Fe3+含量分别从原始污泥的84.5、19.5、0.9和0 mg·L−1增加至144.4、131.0、1.0和12.1 mg·L−1,说明单独水热碳化处理对Ca2+和Mg2+的释放有一定影响,而Al3+和Fe3+只有少量释出。当EDTA投加量为10 mmol·L−1时,Ca2+、Mg2+、Al3+和Fe3+分别增加至267.7、189.6、116.1和194.8 mg·L−1。该结果表明,EDTA的投加对Al3+和Fe3+的释出量影响较大,Ca2+和Mg2+的浸出量受水热碳化和EDTA的双重影响。Ca2+、Mg2+、Al3+和Fe3+在污泥中的赋存形态不同,导致其浸出量之间的差异。Mg2+主要存在于细胞内,Fe3+、Ca2+和Al3+主要储存在细胞内和EPS中[25]。由于污泥中Ca2+含量高,处于EPS外围吸附能力弱的部分,Ca2+在静置条件下被释放进入液相,导致原始污泥上清液中Ca2+含量较高。经水热碳化处理后,细胞膜破裂,EPS结构被破坏,存在于细胞内的Ca2+、Mg2+等金属离子被释放,但由于Ca2+与EPS之间的架桥作用,且部分从细胞内释放的Fe3+、Al3+被EPS捕获,因而只经过水热碳化处理后Fe3+、Ca2+和Al3+的释放量小于Mg2+。当加入EDTA后,EDTA会进一步造成细胞膜破裂和EPS裂解,并与细胞内和胞外聚合物释放的金属离子络合形成可溶性物质进入液相,由于EDTA与Fe3+、Ca2+和Al3+离子的络合能力大于Mg2+,所以与单独水热碳化处理相比,液相中Fe3+、Ca2+和Al3+的释放量大于Mg2+。随着EDTA的增加,大量的金属离子不断释出与其发生络合反应,进而释放出大量的磷酸根进入液相中,此结果也解释了EDTA的添加可以有效地促进污泥中磷的释放,为后续磷的高效回收创造了有利条件。
2.7 水热炭理化性质
对原污泥和经EDTA强化污泥水热碳化后水热炭的元素组成和热值进行了分析,探讨EDTA对水热炭性质的影响,见表2。污泥经过EDTA强化水热碳化处理后,水热炭中的C、H、O、N均有不同程度的下降,这是由于在水热碳化过程中大量有机物发生水解、脱水、脱羧、再聚合等化学反应,分解成小分子物质[26],各元素溶解进入液相,尽管污泥中大量的金属及磷溶出,但仍有部分矿物质(如SiO2)及其它无机组分富集在固相产物中,导致水热炭中各元素含量整体下降,而灰分含量增加[27]。水热炭中的H/C和(O+N)/C降低,说明水热炭的芳香性增强而极性减弱,疏水性更好,有利于提高污泥的脱水性能[28-29]。O/C的值降低,说明随着脱水、脱羧、脱羟基等反应的发生,水热炭表面含氧官能团减少。同时H/C和O/C的降低,使得水热炭有靠近褐煤特性的趋势,而O含量的降低有利于其充分燃烧,提高了其类煤性能。经水热处理之后,O、H含量降低使得水热炭的高位热值升高,也表明发生了脱水和脱羧反应。总体而言,经过EDTA强化污泥水热碳化处理后,水热炭的热值增加、燃烧性能增强。
表 2 污泥及水热炭的元素组成和热值Table 2. Elements composition and calorific value of sludge and hydrochar样品 元素分析(wt) 元素比 HHV/(MJ·kg−1) C H N S O 灰分 H/C O/C (O+N)/C 污泥 30.30% 4.47% 5.48% 0 34.81% 24.94% 0.15 1.15 1.33 10.44 水热炭 29.28% 3.92% 4.61% 0 23.43% 38.76% 0.13 0.80 0.96 11.36 | Show TableDownLoad:
CSV
2.8 水热炭表征分析
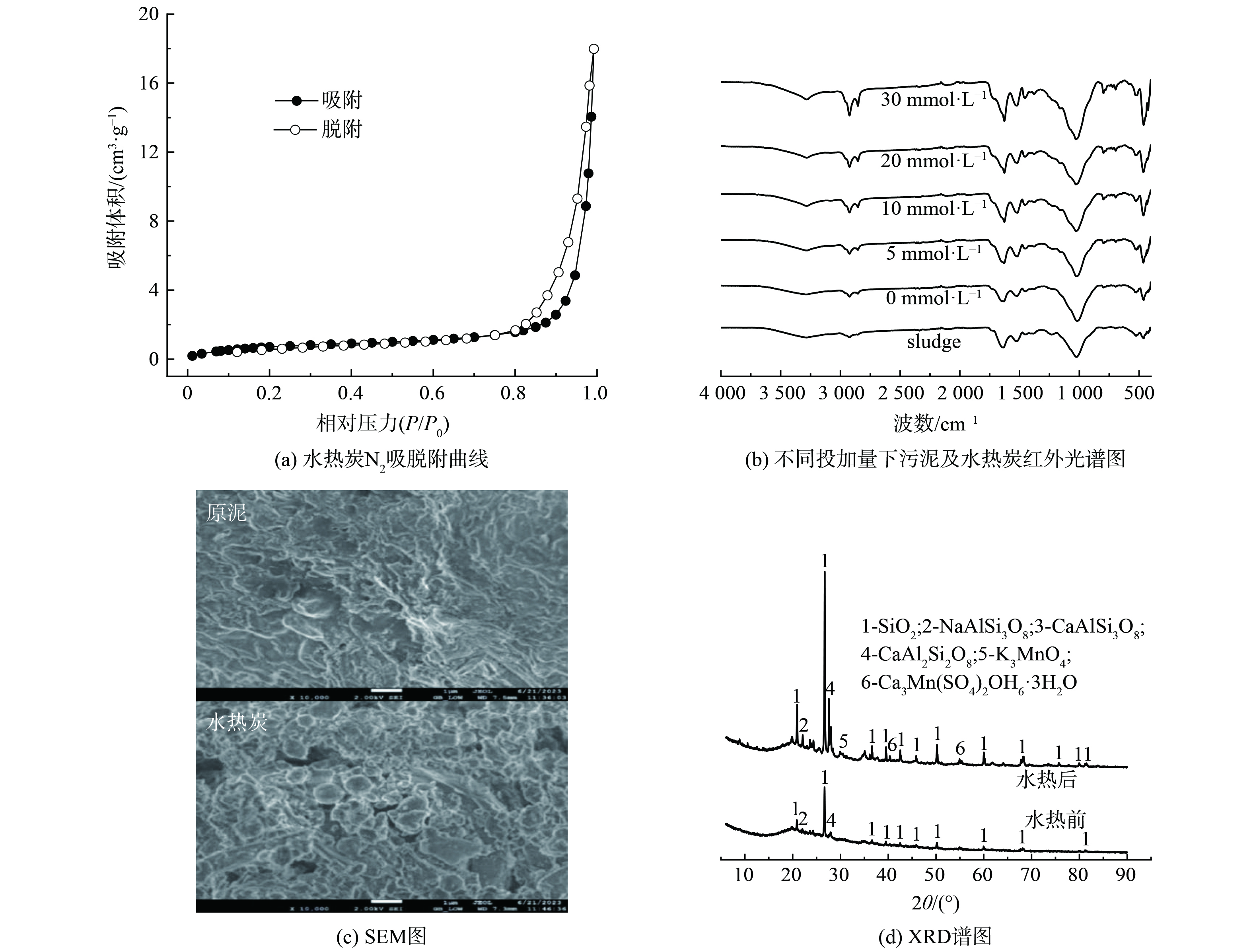
图7(a)为EDTA强化污泥水热碳化处理后得到的水热炭的氮气吸-脱附等温曲线。从图7(a)中可知,吸附曲线与脱附曲线不一致,吸脱附过程中存在滞后环,根据国际纯粹与应用化学联合会(IUPAC)的分类标准,该等温曲线符合Ⅳ型曲线特征,表明水热炭属于介孔材料,孔径在2~50 nm之间。
使用傅里叶转换红外光谱(FTIR)对水热炭主要有机官能团进行分析测定,结果如图7(b)所示。从图中可以看到,不同谱图之间的官能团种类没有明显差异,说明水热碳化过程保留了污泥原有的官能团结构,但官能团峰值强度有所变化。3 200~3 500 cm−1处的吸收峰为O—H伸缩振动峰[30],说明水热炭中存在醇类、羧酸类等物质[31]。随着EDTA投加量的增加,3 282 cm−1处的缔合O—H伸缩振动峰逐渐突出,可能是由于发生了脱水反应,羧基逐渐降解,使得O—H伸缩振动增强[30]。2 924和2 854 cm−1吸收峰分别为C—H拉伸和伸缩振动峰,吸收峰随EDTA用量增加逐渐增强,表明有脂肪物质生成[30]。1 023 cm−1处的吸收峰由SiO2引起的[31],经EDTA强化水热碳化处理后比原污泥增强。说明水热炭中SiO2的相对含量增加。
图7(c)为污泥与经EDTA强化水热碳化处理后得到的水热炭的表面形貌图(SEM)。图7(c)结果显示,处理前后固体表面微观结构发生了比较明显的变化。处理前,原污泥表面光滑平整,呈致密的片状结构;处理后,水热炭表面变得疏松、凹凸不平且有孔洞,说明污泥絮体结构和细胞结构在EDTA强化水热碳化过程中被破坏。水热碳化过程是在高温高压环境里进行的,处于次临界的水相经氧化作用进入污泥内部并大量溶解其中的有机物,在温度降低和释压后,这些水相则与污泥迅速分离,最终使得水热炭呈现出松散多孔的蜂窝状结构[32],这也更有利于磷的释出。
污泥经水热炭后,灰分含量仍占有很大比例,这些灰分主要为一些无机盐成分。使用X-射线衍射(XRD)的方法,对水热炭中存在的无机晶相进行成像分析,对比水热碳化前后污泥中主要组分的变化,见图7(d)。从图7(d)中可以看出,对于污泥和水热炭,出峰主要集中在19°~85°之间。污泥中主要成分包括石英(SiO2)、钠长石(NaAlSi3O8)、方解石(CaAlSi3O8)、钙长石(CaAl2Si2O8),由于这些物质在水热炭化过程中热稳定性强,大部分晶体结构仍然以灰分的形式被保留在水热炭中。与原污泥相比,经水热碳化反应后,增加了钾锰氧化物(K3MnO4)和钙锰钒。
3. 结论
1)经过EDTA强化污泥水热碳化处理后,污泥絮体结构被破坏,胞内有机物质被释放,大分子物质水解,大量磷从固相转移至液相,使水热液中TP和PO43−含量显著增加。在210 ℃,时间4 h,EDTA投加量10 mmol·L−1的条件下,TP和PO43−的浓度分别可达339.1和325.6 mg·L−1,释放率为96.2%。
2)在水热碳化过程中,高温促使污泥破解释放金属离子和磷至液相,EDTA的加入促进了EPS和细胞膜的溶解,并阻止了金属离子与磷再形成难溶沉淀物,促进了磷的释放;增加EDTA投加量会促使大量的OP转化为IP,NAIP转化为AP。
3)经过EDTA强化污泥水热碳化后,水热炭保留了污泥中原有的官能团,燃烧性能和热值均有所提高,芳香性增强而极性减弱,同时出现了孔结构。
-
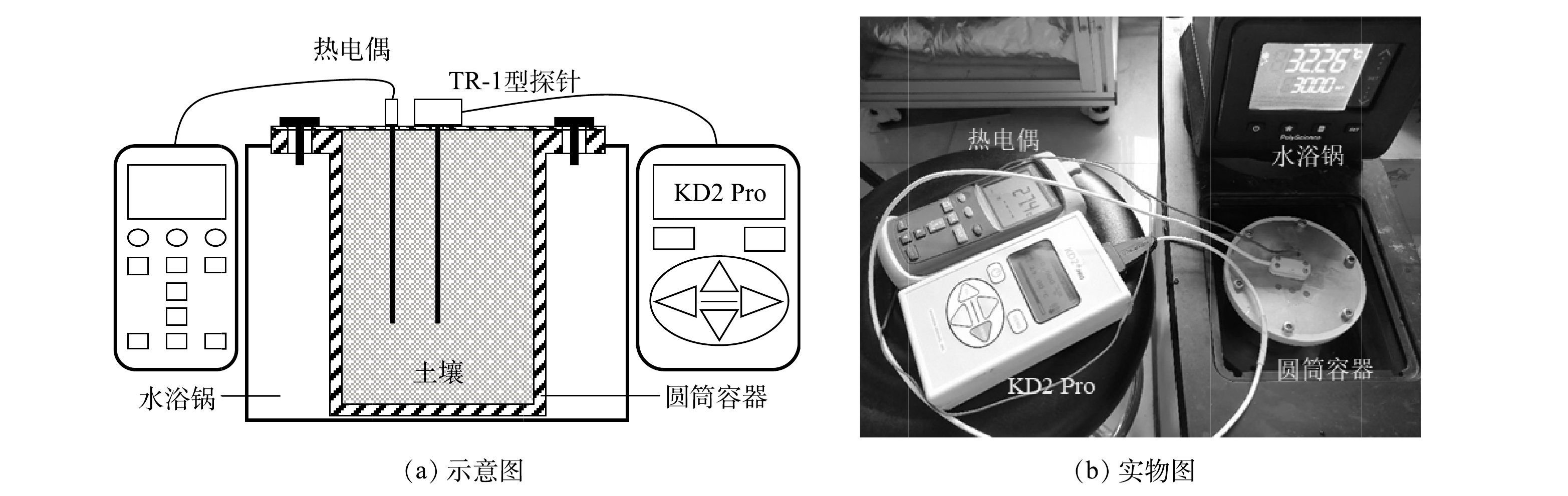
图 2 土壤热导率的探针法测试装置
Figure 2. Probe-type thermal conductivity measurement instrument for soil samples
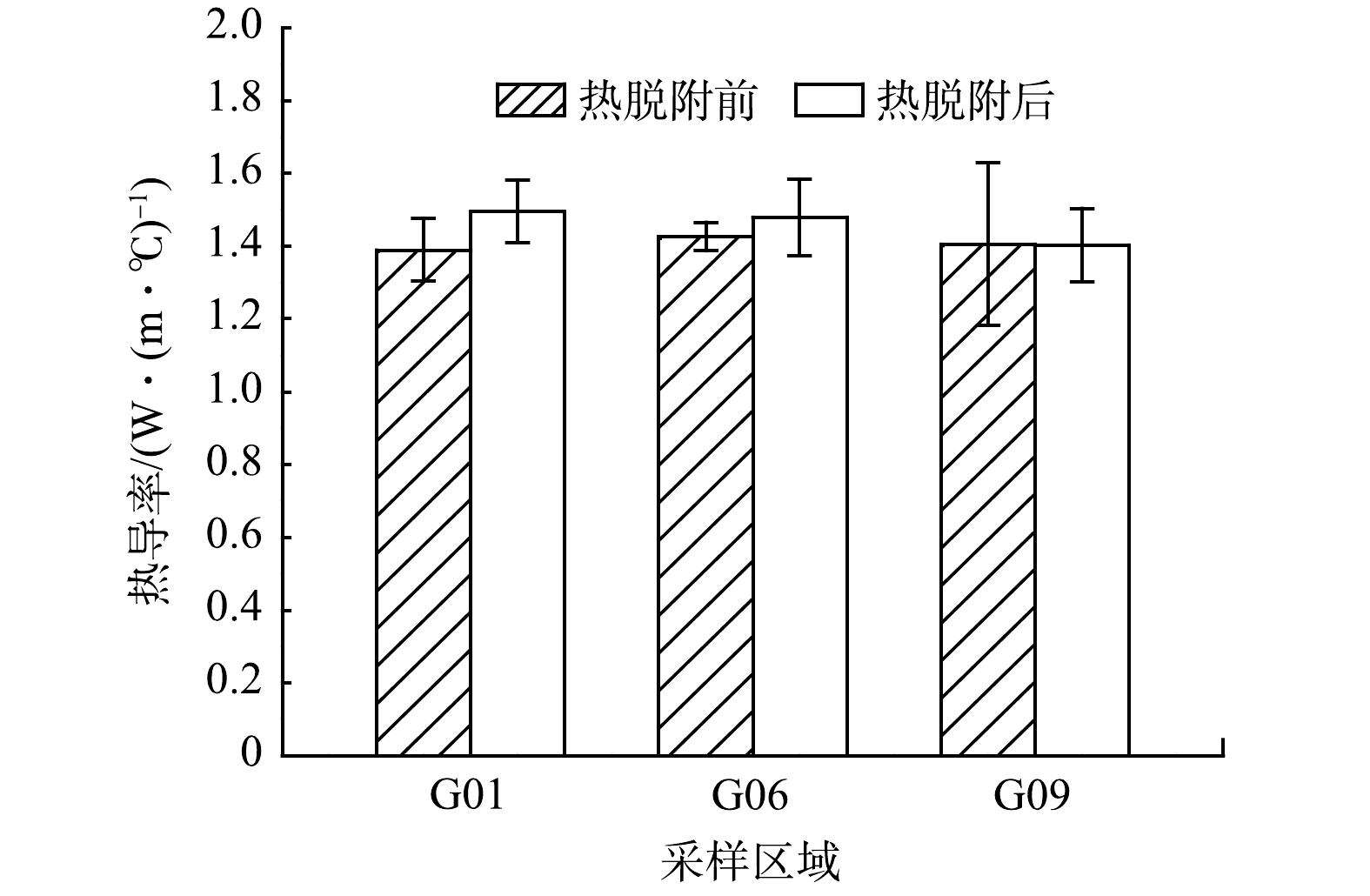
图 3 相同参数条件下热脱附前后土壤试样的热导率对比
Figure 3. Thermal conductivity of soil samples with same physical properties before and after thermal desorption
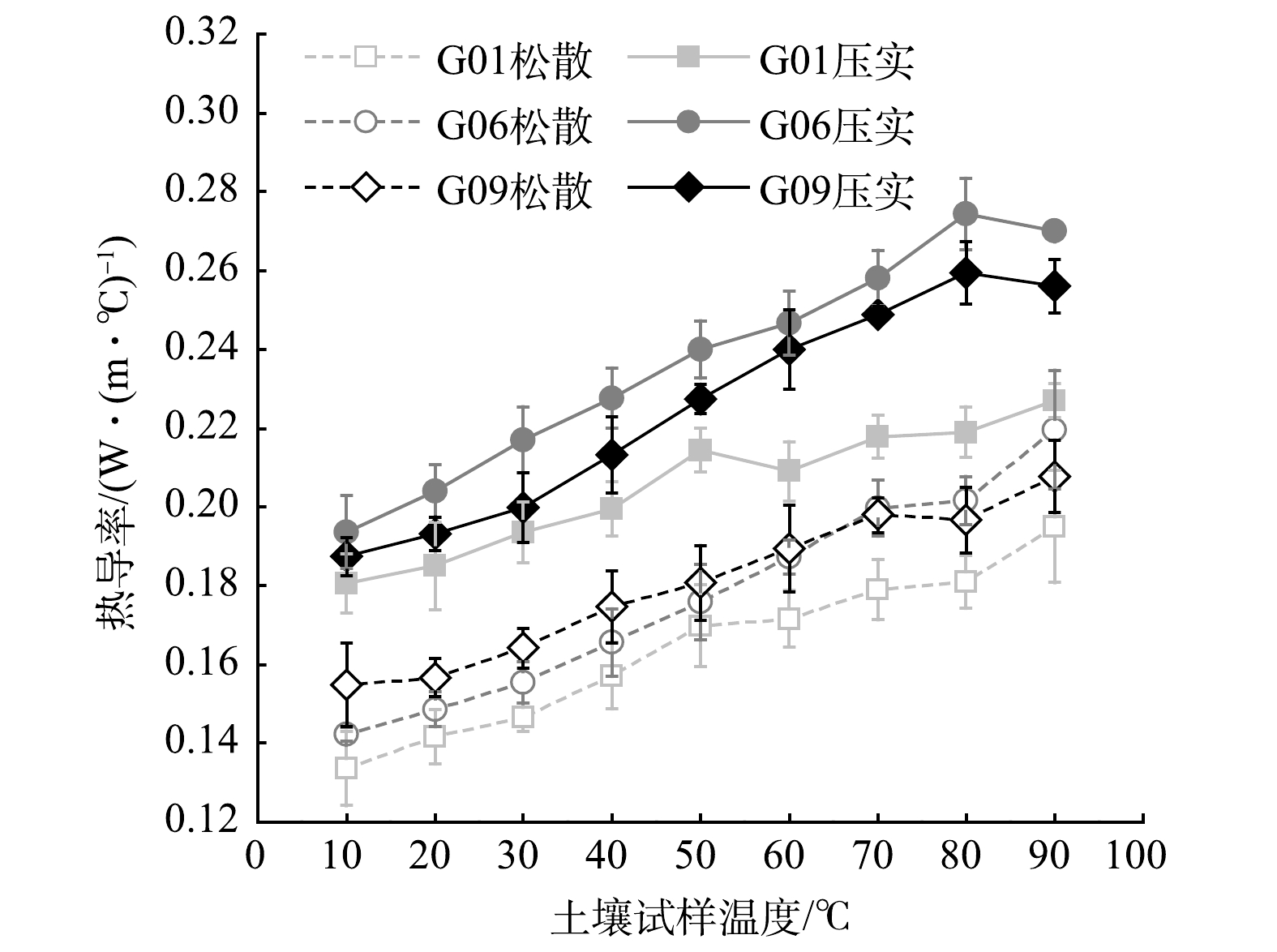
图 4 热脱附后土壤试样的热导率-温度曲线
Figure 4. Thermal conductivity-temperature curve of soil samples after thermal desorption
表 1 表层土壤的主要物性参数
Table 1. Main physical properties of soil samples in superficial layer
采样区域 ρw/(g·cm−3) ρd/(g·cm−3) ρg/(g·cm−3) e θ/% Sr/% pH G01 1.85 1.40 2.75 0.97 32.6 92.0 7.40 G06 1.83 1.36 2.75 1.03 35.0 94.0 7.86 G09 1.91 1.52 2.75 0.81 25.8 87.0 7.15 注:ρw为湿密度,ρd为干密度,ρg为土粒比重,e为孔隙比,θ为质量含水率,Sr为饱和度。
下载: 导出CSV
表 2 热脱附前后土壤试样的污染物浓度
Table 2. Concentrations of contaminants in soil samples before and after thermal desorption
污染物名称 检出下限/(mg·kg−1) 热脱附前质量分数/(mg·kg−1) 热脱附后质量分数/(mg·kg−1) G01 G06 G09 G01 G06 G09 苯 0.05 3.56 ND ND ND ND ND 乙苯 0.05 1.48 0.39 ND ND ND ND 间/对-二甲苯 0.05 5.29 0.35 ND ND ND ND 邻-二甲苯 0.05 8.50 0.07 ND ND ND ND 1,2,4-三甲苯 0.05 7.41 0.13 ND ND ND ND 正丁基苯 0.05 3.27 0.07 ND ND ND ND 正丙苯 0.05 ND 0.06 ND ND ND ND 氯苯 0.05 0.44 ND 0.60 ND ND ND 三氯甲烷 0.05 1.31 ND ND ND ND ND 三氯乙烯 0.05 6.03 ND ND ND ND ND 石油烃C6~C20 6 7 15 19 ND ND ND 注:ND表示该污染物浓度低于仪器检出下限。
下载: 导出CSV
表 3 热脱附前后土壤试样的粒径分布
Table 3. Particle size distribution of soil samples before and after thermal desorption
热脱附状态 采样区域 颗粒质量分数/% 黏粒<0.005 mm 粉粒0.005~0.075 mm 细砂粒0.075~0.250 mm 中砂粒0.250~0.500 mm 粗砂粒0.500~2.000 mm 热脱附前 G01 30.6 62.9 6.5 0 0 G06 34.0 64.8 1.2 0 0 G09 44.2 55.3 0.5 0 0 热脱附后 G01 14.4 60.8 24.1 0.7 0 G06 12.9 60.8 25.1 1.2 0 G09 14.5 63.1 21.5 0.9 0
下载: 导出CSV
表 4 热脱附后土壤试样的热导率-温度拟合公式
Table 4. Thermal conductivity-temperature fitting formula of the soil samples after thermal desorption
采样区域 表观密度状态 λ-T拟合公式 R2 G01 松散 λ = 7×10−4T + 0.127 0.979 压实 λ = 6×10−4T + 0.176 0.946 G06 松散 λ = 1×10-3T + 0.129 0.988 压实 λ = 1×10−3T + 0.185 0.980 G09 松散 λ = 7×10−4T + 0.146 0.980 压实 λ = 1×10−3T + 0.175 0.970
下载: 导出CSV
表 5 热脱附后土壤试样的化学组成
Table 5. Chemical compositions of the soil samples after thermal desorption
采样区域 矿物质质量分数/% 有机质质量分数/% 其他物质质量分数/% 石英 钾长石 斜长石 方解石 菱铁矿 辉石 云母 黏土 总计 G01 62.55 3.13 10.38 1.43 0 0 0 12.00 89.49 8.26 2.25 G06 57.70 4.15 10.52 1.94 0 1.11 1.12 15.88 92.42 5.13 2.55 G09 59.58 3.18 8.81 1.00 0.91 0.82 1.00 15.53 90.83 6.75 2.42 注:其他物质指土壤中的空气、自由水等。
下载: 导出CSV
-
[1] YANG H, HUANG X J, THOMPSON J R, et al. China's soil pollution: Urban brownfields[J]. Science, 2014, 344: 691-692. [2] 骆永明. 中国污染场地修复的研究进展、问题与展望[J]. 环境监测管理与技术, 2011, 23(3): 1-6. doi: 10.3969/j.issn.1006-2009.2011.03.002 [3] 宋昕, 林娜, 殷鹏华. 中国污染场地修复现状及产业前景分析[J]. 土壤, 2015, 47(1): 1-7. [4] SAKSHI, SINGH S K, HARITASH A K. Polycyclic aromatic hydrocarbons: Soil pollution and remediation[J]. International Journal of Environmental Science and Technology, 2019, 16: 6489-6512. doi: 10.1007/s13762-019-02414-3 [5] 焦文涛, 韩自玉, 吕正勇, 等. 土壤电阻加热技术原位修复有机污染土壤的关键问题与展望[J]. 环境工程学报, 2019, 13(9): 2027-2036. doi: 10.12030/j.cjee.201905138 [6] 邓忆凯, 韩彪, 黄世友, 等. 挥发性有机物污染土壤修复技术研究[J]. 科技创新与应用, 2020(28): 163-164. [7] O'BRIEN P L, DESUTTER T M, CASEY F X M, et al. Thermal remediation alters soil properties: A review[J]. Journal of Environmental Management, 2018, 206: 826-835. doi: 10.1016/j.jenvman.2017.11.052 [8] 缪周伟, 吕树光, 邱兆富, 等. 原位热处理技术修复重质非水相液体污染场地研究进展[J]. 环境污染与防治, 2012, 34(8): 63-68. doi: 10.3969/j.issn.1001-3865.2012.08.014 [9] 李书鹏, 焦文涛, 李鸿炫, 等. 燃气热脱附技术修复有机污染场地研究与应用进展[J]. 环境工程学报, 2019, 13(9): 2037-2048. doi: 10.12030/j.cjee.201905108 [10] 夏天翔, 姜林, 魏萌, 等. 焦化厂土壤中PAHs的热脱附行为及其对土壤性质的影响[J]. 化工学报, 2014, 65(4): 1470-1480. doi: 10.3969/j.issn.0438-1157.2014.04.043 [11] 隋红, 姜斌, 黄国强, 等. 生物通风修复含石油污染物土壤过程[J]. 化工学报, 2004, 55(9): 1488-1492. doi: 10.3321/j.issn:0438-1157.2004.09.027 [12] 杨悦锁, 陈煜, 李盼盼, 等. 土壤、地下水中重金属和多环芳烃复合污染及修复研究进展[J]. 化工学报, 2017, 68(6): 2219-2232. [13] MACKAY D, WAN Y S, MA K C, et al. Handbook of physical-chemical properties and environmental fate for organic chemicals[M]. Boca Raton: CRC Press Inc., 2006. [14] 陈王若尘. 添加剂对焦化污染土壤性质及热脱附行为影响研究[D]. 杭州: 浙江大学, 2020. [15] ARARUNA J T, PORTES V L O, SOARES A P L, et al. Oil spills debris clean up by thermal desorption[J]. Journal of Hazardous Materials, 2004, 110(1/2/3): 161-171. doi: 10.1016/j.jhazmat.2004.02.054 [16] 赵海波, 郑楚光. 多分散结构团聚体烧结的数值模拟[J]. 中国电机工程学报, 2012, 32(8): 58-63. [17] HIRAIWA Y, KASUBUCHI T. Temperature dependence of thermal conductivity of soil over a wide range of temperature (5-75℃)[J]. European Journal of Soil Science, 2010, 51(2): 211-218. [18] 陈正发, 朱合华, 闫治国, 等. 高温环境下土的导热系数研究[J]. 地下空间与工程学报, 2016, 12(6): 1532-1538. [19] 黄昌勇, 徐建明. 土壤学[J]. 3版. 北京:中国农业出版社, 2010. [20] 肖衡林, 吴雪洁, 周锦华. 岩土材料导热系数计算研究[J]. 路基工程, 2007(3): 54-56. doi: 10.3969/j.issn.1003-8825.2007.03.026 [21] JOHANSEN O. Thermal conductivity of soils[D]. Trondheim: University of Trondheim,1975. [22] ABU-HAMDEH N H, REEDER R C. Soil thermal conductivity: Effects of density, moisture, salt concentration, and organic matter[J]. Soil Science Society of America Journal, 2000, 64(4): 1285-1290. doi: 10.2136/sssaj2000.6441285x -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4170
- HTML全文浏览数: 4170
- PDF下载数: 66
- 施引文献: 0
