


-
随着我国生活污水的处理量和处理效率的不断提升,污水处理厂产生的污泥量剧增。截至2020年,全国干污泥产生量高达1.16×107 t[1]。污泥既是“污染物”,也是“资源”,如处理不当,将造成二次污染和资源浪费。污泥中有机物组分复杂,主要含有蛋白质、多糖、脂肪以及纤维素等,其中,蛋白质组分占污泥有机物的40%~60%[2],污泥中的溶解性有机物(dissolved organic matter, DOM)是微生物利用的直接碳源,DOM的组成特性与污泥脱水以及厌氧消化产酸(产酸细菌的直接底物)、产甲烷密切相关[3]。污泥中的复杂有机物(特别是不溶性有机物)主要存在于微生物细胞及其胞外聚合物中,其生物可利用性低,因其表面官能团束缚水分子,导致污泥厌氧消化和脱水等过程处理效率低。
污泥预处理是促进污泥胞内有机物释放和胞外聚合物破解的主要方式[4]。如高温热水解预处理[5]、微波预处理[6]等对污泥的溶胞破壁作用明显,预处理后可显著提升溶解性有机物浓度,进而提升污泥厌氧消化产甲烷量。但是,污泥预处理过程所释放的溶解性有机物也可能发生进一步转化反应,如美拉德反应[5,7],从而导致大量难降解大分子有机物的生成(超过100 kDa)。这些难降解溶解性有机物难以被微生物降解利用,对后续污泥厌氧消化[5]及其脱水滤液的处理会带来不利影响。因此,进一步明确污泥在预处理过程中除溶解性有机物释放外的进一步转化反应,探求一种基于污泥中有机物转化定向调控的预处理方式对解决污泥处理处置难题至关重要。近年来,硫酸根自由基由于其具有强氧化性,且过硫酸盐(peroxodisulfate,PDS)适用范围广和易于储存等优点,使得基于过硫酸盐的高级氧化技术受到关注[8-9]。有研究表明,Fe活化过硫酸盐能有效提升污泥的脱水性能[10],并且对污泥发酵产酸[11]有促进作用。此外,过硫酸盐在Fe(Ⅱ)和碳质材料的不同活化作用下具有不同的氧化途径[10,12]。其中,Fe(Ⅱ)通过均相方式活化过硫酸盐产生硫酸根自由基和羟基自由基等自由基,而碳质材料通过非均相活化包括自由基和非自由基两种氧化途径[13]。此外,硫酸根自由基对有机物的氧化作用具有一定靶向性,可对具有不饱和键和芳香族结构的有机物进行选择性矿化[14-15],破坏荧光类官能团(例如芳香族蛋白质、色氨酸类蛋白质物质等)[10],对羧基、芳香类有机物[16](酚羟基)、含氮基团和烯烃[16]等都有较强的作用,而这类有机物组分及其官能团结构与污泥的可生物降解性、脱水性能、厌氧消化直接相关[17]。有研究表明,在厌氧消化过程中,蛋白质物质优先被降解,其次是含羧基和芳香性的物质[3]。其中,色氨酸和络氨酸类蛋白质是脂肪酸生成的重要底物[3,18]。
利用活化过硫酸盐的多种途径和对有机物氧化作用的选择性,将实现污泥中溶解性有机物释放后的进一步氧化反应,避免热处理中大分子、难降解有机物的形成。同时,也有助于深入认识污泥中释放的溶解性有机物的官能团特性。然而,以铁、炭及其组合作用下活化过硫酸盐对污泥预处理过程有机物的释放特征及选择性作用仍缺乏认识,尚不清楚铁和炭对过硫酸盐在不同的活化途径下,污泥溶解性有机物释放特性和组分降解的差异。为此,本研究利用氯化亚铁Fe(Ⅱ)和椰壳活性炭(Activated Carbon,AC)活化过硫酸盐预处理剩余污泥,研究Fe(Ⅱ)和活性炭活化过硫酸盐不同方式下,对污泥溶解性有机物释放特征的影响,以及对污泥溶解性有机物组分的选择性作用。
-
剩余污泥取自北京市清河污水处理厂二沉池污泥,取回后在4 ℃下自然沉降24 h,除去上清液后过18目筛去除杂质,置于4 ℃保存备用。浓缩后污泥经6 000 r∙min−1离心10 min,上清液经0.45 µm滤膜过滤后分析测定相关指标,剩余污泥基本特性如表1所示。
氯化亚铁和过硫酸钾均为优级纯,椰壳活性炭的孔径特征如表2所示。活性炭经研磨取60~100目,用超纯水洗涤至上清液无色(pH为中性),放入烘箱中在105 ℃干燥2 h,取出放干燥器中备用。
-
1) PDS投加量对污泥预处理的影响。研究不同PDS投加量对污泥预处理过程中溶解性有机物释放的影响,分为5个实验组:原污泥对照组、单独PDS实验对照组、单独Fe(Ⅱ)活化PDS组、单独AC活化PDS组、Fe(Ⅱ)+AC活化PDS组。其中,Fe(Ⅱ)∶PDS=1∶1(摩尔比),AC∶PDS=1∶1(质量比)。根据樊雅欣等[19]的研究,预处理时间统一为1 h。
2) Fe(Ⅱ)投加量和投加方式的影响。当PDS投加量为2 mmol∙g−1(以VSS计)、PDS∶AC=1∶1(质量比)时,研究不同的Fe(Ⅱ)投加量(0.6、0.9、1.2、1.5、1.8、2.1 mmol∙g−1)和分次投加Fe(Ⅱ)(将0.6、1.2、1.8 mmol∙g−1的Fe(Ⅱ)分别在0、40 、80 min分3次加入)对污泥预处理效果的影响。共设置7组实验(表3):原污泥为实验对照组,记为A实验组;不同Fe(Ⅱ)投加量(0.6、0.9、1.2、1.5、1.8、2.1 mmol∙g−1)记为B、C、D、E、F、G实验组。
所有实验均在250 mL的锥形瓶中进行。首先往锥形瓶里面放100 mL污泥,然后依次加入适量过硫酸钾、椰壳活性炭、氯化亚铁摇匀后置于恒温振荡箱中,在25 ℃、200 r∙min−1振荡,在设定时间取样,每次取10 mL,样品用0.45 μm的滤膜过滤,取其滤液。每组做2个平行实验,取平均值。
-
污泥pH和电导率通过pH计(FE20K,Mettler Toledo)和电导率仪(Multi 3420)进行测定;TS、VS、TSS、VSS通过重量法[20]测定。COD通过哈希管消解及DR2 800分光光度计(HACH,USA)测定。溶解性蛋白质和多糖分别采用修正的Lowry法[21]和DuBious[22]分光光度法测定。总有机碳采用总有机碳分析仪(TOC-L,Japan)测定。采用三维荧光光谱仪(3D-EEMs,HITACHI,Japan)对溶解性有机物DOM进行三维荧光光谱分析。生物炭的孔径结构采用比表面积及孔径分析仪进行测定。
-
污泥中的有机物主要存在于微生物细胞及其胞外聚合物(EPS)中,预处理能够使胞内和污泥絮体中的有机物溶解释放[19]。如图1(a)所示,PDS的强氧化性明显强化了污泥中溶解性有机物的释放,而经过单独活性炭和单独Fe(Ⅱ)活化均能进一步提升PDS的氧化能力,促进污泥中微生物细胞和胞外聚合物的破解。其中,单独Fe(Ⅱ)活化PDS预处理污泥对TOC释放提升作用最为明显,在PDS的投加量为2 mmol∙g−1时,单独Fe(Ⅱ)活化PDS使污泥TOC释放量达到231.65 mg∙L−1,是单独PDS预处理污泥后TOC质量浓度(129.05 mg∙L−1)的1.8倍。单独AC对PDS的活化同样发挥了促进污泥中TOC释放的作用。但与单独Fe(Ⅱ)活化PDS不同,单独AC活化PDS对污泥中溶解性有机物的释放作用随着PDS投加量的增加而呈线性增加。在PDS投加量低于1.2 mmol∙g−1时,采用单独活性炭活化对污泥中溶解性有机物的释放作用不如单独Fe(Ⅱ)活化作用明显;但当PDS投加量达到1.2 mmol∙g−1以上,单独活性炭活化与单独Fe(Ⅱ)活化基本达到相同的促进污泥中溶解性有机物释放的作用。在PDS的投加量为2 mmol∙g−1时,单独活性炭活化和单独Fe(Ⅱ)活化作用下污泥TOC的质量浓度分别为224.6和245.8 mg∙L−1,基本一致。当采用Fe(Ⅱ)和活性炭共同活化PDS时,随着PDS投加量的增加,TOC质量浓度反而明显下降。在PDS投加量小于0.4 mmol∙g−1时,Fe(Ⅱ)和活性炭共同活化PDS具有更强的释放溶解性有机物能力;但当PDS投加量超过0.8 mmol∙g−1时,TOC释放的作用明显削弱。
Fe(Ⅱ)和活性炭共同活化PDS对促进污泥溶解性有机物释放作用的削弱,可能是由于共同活化以及较高的PDS投加量下,PDS活化反应加速,短时间内产生大量的硫酸根自由基。一方面,如公式(4)[23]所示,快速作用于污泥中释放的溶解性有机物,使释放的部分溶解性有机物进一步氧化分解,从而降低了TOC质量浓度;另一方面,短时间内过量的硫酸根自由基会与Fe(Ⅱ)或硫酸根自由基发生反应,从而导致自由基的清除(公式(2)、公式(3))[12,24-25],抑制硫酸根自由基与污泥胞外聚合物和微生物细胞发生作用。根据硫酸根自由基与溶解性有机物、Fe(Ⅱ)以及硫酸根自由基反应速率常数可知,硫酸根自由基与Fe(Ⅱ)反应速率最快,速率常数达4.6×109 mol∙(L·s)−1,比与有机物反应速率常数(2.35×107 mol∙(L·s)−1,mol∙L−1是基于C的摩尔浓度)高2个数量级。但由于污泥中的溶解性有机物浓度高于Fe(Ⅱ)和硫酸根自由基,其更易与硫酸根自由基发生反应。因此,短时间内过量硫酸根自由基与释放的溶解性有机物的氧化分解反应,可能是导致Fe(Ⅱ)和活性炭共同活化PDS促进污泥溶解性有机物释放作用削弱的主要原因。
活性炭活化的TOC质量浓度随PDS投加量增加呈线性增加,而单独PDS、Fe(Ⅱ)和Fe(Ⅱ)+AC活化PDS对污泥溶解性有机物释放的影响呈指数增长关系(表4),PDS的过量投加将不利于溶解性有机物的释放。如图1(b)所示,除活性炭活化PDS外,TOC质量浓度增量随着PDS投加量提高呈明显的下降趋势。在低PDS投加量下,促进TOC释放的作用最为明显。如图1(b)所示,Fe(Ⅱ)活化在PDS投加量为2.7 mmol∙g−1,TOC质量浓度增量降低到10 mg∙L−1,而Fe(Ⅱ)+AC活化在PDS投加量为1.6 mmol∙g−1时,TOC质量浓度增量便降低到10 mg∙L−1。这说明,Fe(Ⅱ)+AC活化PDS促进TOC释放作用衰减最为明显。单独活性炭活化PDS对溶解性有机物释放影响明显不同,其TOC质量浓度增量随PDS投加量增加几乎不变。这说明单独活性炭在不同PDS投机量下均能较为稳定的促进污泥中溶解性有机物的释放。这可能是由于单独活性炭活化过硫酸盐是通过非自由基途径而实现。已有研究表明,该非自由基氧化体系能在复杂的水基质中长期保持较高的氧化效率[12]。
在PDS投加量为1.6 mmol∙g−1时,通过对比单独Fe(Ⅱ)活化、单独活性炭活化以及Fe(Ⅱ)+活性炭活化PDS预处理污泥的TOC释放动力学过程(图2),可进一步证明Fe(Ⅱ)活化过硫酸盐是较快速的反应,而活性炭活化PDS的非自由基途径则反应较为缓慢。具体地,单独Fe(Ⅱ)活化PDS在反应进行40 min基本达到TOC的最大质量浓度;Fe(Ⅱ)+AC活化PDS则在20 min便达到最大TOC质量浓度;而单独活性炭活化PDS进程缓慢,在90 min达到最高TOC质量浓度。Fe(Ⅱ)对PDS的活化作用主要是液相的Fe(Ⅱ)与PDS直接发生作用,属于均相体系;与非均相体系相比,均相体系具有较低的相间传质阻力[24],因而能够实现较高的反应速率。然而,活性炭对PDS的活化机理相对复杂,并且PDS活化反应的电子传递过程是在活性炭的固体表面上进行,存在液固两相的界面过程,属于非均相体系,反应进程则较为缓慢。有研究表明,在Fe(Ⅱ)活化过硫酸盐反应中有2个阶段,包括快速阶段和慢速阶段;初期反应物充足,自由基生成快,初期反应速度快,而随着反应进行,由于Fe(Ⅱ)的消耗,自由基生成速度变缓[25-26]。尽管活性炭活化PDS的反应进程相对较慢,但将活性炭加入到Fe(Ⅱ)活化PDS的体系中,进一步加快了Fe(Ⅱ)活化速率,短时间内产生的大量自由基与溶解有机物发生反应,使溶解性有机物分解矿化。
-
为证明短时间内Fe(Ⅱ)+AC活化PDS过程大量自由基生成会降低TOC释放的质量浓度,进一步开展了Fe(Ⅱ)投加量和分批次投加方式对TOC释放的质量浓度影响。如图3(a)所示,Fe(Ⅱ)+AC活化PDS(PDS投加量为2 mmol∙g−1、AC∶PDS=1∶1(质量比))预处理污泥,随着Fe(Ⅱ)投加量从0.6 mmol∙g−1升高到2.1 mmol∙g−1,TOC质量浓度由263.8 mg∙L−1降低到193.8 mg∙L−1。这一定程度上说明了Fe(Ⅱ)过量投加反而导致释放溶解性有机物的矿化分解。而在活化反应过程中,将总量相等的Fe(Ⅱ)分别在0、40、80 min分3次加入,TOC质量浓度得到了提高(图3(b))。在Fe(Ⅱ)投加量为1.8 mmol∙g−1时,分批次的投加方式使TOC释放质量浓度由204.05 mg∙L−1提升到244.25 mg∙L−1。VICENTE等[27]采取多次铁投加的方式,敌草隆被氧化和矿化的速率提高。此外,在达到相同的TOC质量浓度,活性炭的投加能够降低Fe(Ⅱ)的投加量。如图3(a)所示,在Fe(Ⅱ)+炭活化PDS下,当Fe(Ⅱ)的投加量在0.6 mmol∙g−1时,释放的TOC质量浓度为263.8 mg∙L−1,而单独Fe(Ⅱ)活化PDS,在Fe(Ⅱ)投加量达2 mmol∙g−1,TOC的质量浓度为245.8 mg∙L−1。
在PDS投加量为2 mmol∙g−1、AC:PDS=1∶1(质量比)时,随着Fe(Ⅱ)投加量的增加,污泥上清液中释放的溶解性蛋白质、多糖质量浓度变化如图4所示。其中,溶解性多糖质量浓度随着Fe(Ⅱ)投加量增加而增加,与释放TOC质量浓度的变化趋势不同;而释放的溶解性蛋白质质量浓度随着Fe(Ⅱ)投加量增加出现先升高后下降的趋势,与TOC质量浓度变化趋势较为接近。这说明,Fe(Ⅱ)+AC活化PDS预处理对溶解性有机物不同组分产生不同作用。Fe(Ⅱ)活化PDS产生的硫酸根自由基可促进污泥胞外和胞内有机化合物的分子结构断裂,使蛋白质和多糖等有机物释放,但大量的硫酸根自由基快速与释放的溶解性有机物发生进一步反应。GUO等[28]用电解Fe0的方式活化过一硫酸盐(PMS)预处理污泥发现,硫酸根自由基可能发生对蛋白质类物质的矿化作用,而对多糖作用较弱。WEI等[10]用Fe(Ⅱ)活化过硫酸盐同样也发现硫酸根自由基优先与蛋白质反应,特别是亲水性蛋白质被高度破坏。同时,多糖质量浓度随Fe(Ⅱ)含量的逐渐增加也证明了TOC质量浓度的降低并非主要源于硫酸根自由基与Fe(Ⅱ)和自身反应发生的清除作用。蛋白质质量浓度的变化则证明:随Fe(Ⅱ)投加量的增加,短时间内产生的大量硫酸根自由基使部分蛋白质组分矿化分解。在更高的Fe(Ⅱ)投加量下,由于污泥絮体和微生物细胞破解作用的加强,释放的溶解性蛋白质仍然要高于被自由基矿化分解的蛋白质组分,因此表现出释放溶解性蛋白质浓度重新增加(图3(a))。
结合三维荧光光谱(3D-EEM)对溶解性有机物组成进行了表征(图5)。如(图5(a))所示,未处理污泥中荧光类有机物主要包括可溶性微生物副产物(峰A,Ex=280,Em=310)和色氨酸类蛋白质(峰B,Ex=230,Em=350)[29-30]。经过预处理后,这些荧光类蛋白质组分被明显矿化分解。这与之前的研究一致。例如,BIAN等 [31]用超声激活过硫酸盐处理污泥,发现硫酸根自由基具有选择性地与芳香族类、色氨酸类蛋白质反应,破解该类有机物分子结构。已有研究表明[10],污泥胞外聚合物中的芳香族聚合物(例如:木质素酚、愈创木基结构、多氯联苯、谷氨酸脱氢酶和膜支架蛋白、木质素、疏水性氨基酸、不饱和脂肪链等)会优先与硫酸根自由基反应,破坏分子结构。此外,随着Fe(Ⅱ)投加量的增加,出现了一种新的荧光类有机物为类黄腐酸类物质(图(f)中峰C,Ex=210,Em=400),该类有机物通常来源于死亡微生物分解产生的副产物[31],表明微生物细胞被氧化破裂。Fe(Ⅱ)投加量的增加导致更多的微生物细胞破裂,释放了该类有机物。因此,荧光类有机物的特定官能团是硫酸根自由基的靶向有机物,Fe(Ⅱ)投加量的增多对该类有机物的氧化作用明显,可导致TOC释放浓度的降低。
为了进一步证明实验结果的准确性,以苯酚、牛血清白蛋白和葡萄糖为反应物,通过PDS和活性炭活化PDS(AC∶PDS=1∶1)对其进行氧化处理(预处理120 min)。结果表明,未活化的PDS对有机物的降解作用较弱,而经过活性炭的活化之后,对苯酚和牛血清白蛋白降解作用明显,但葡萄糖仍未被降解(图6)。这与污泥被活化PDS预处理后DOM中蛋白质、多糖的降解规律相一致。同时,结合牛血清白蛋白三维荧光光谱(图7)可发现,被活化的过硫酸盐可以对荧光类蛋白质进行矿化分解。这说明活化PDS对污泥中荧光类蛋白质具有氧化降解作用。
用Fe(Ⅱ)-炭活化过硫酸盐方法预处理污泥,通过Fe(Ⅱ)-炭投加方式的调整,可以实现溶解性有机物的不同释放速率,并且该高级氧化预处理对DOM组分中蛋白类物质具有选择性。这对于精确的调控污泥预处理后DOM的释放及组成特性具有重要意义,DOM组分直接影响污泥脱水以及污泥厌氧消化过程。例如,LIU等[6]发现,预处理后释放的DOM中低分子量(104~105 Da)的蛋白质类物质直接影响污泥的脱水性能。XIAO等[32]亦发现,低分子量蛋白质(< 20 kDa)影响污泥脱水性能的关键参数。DOM作为微生物的直接碳源和能源,在厌氧消化中起着至关重要的作用。在厌氧消化中,蛋白质物质优先被降解[3]。其中,色氨酸和络氨酸类蛋白质是脂肪酸的重要底物[3,18]。
-
1) Fe(Ⅱ)、活性炭和两者联合活化PDS预处理剩余污泥能够有效地促进污泥中有机物溶出;但在不同活化方式下,溶解性有机物的释放速率不同,担负活性炭活化PDS过程溶解性有机物的释放较慢但稳定。
2) Fe(Ⅱ)和活性炭共同活化PDS时,其促进污泥溶解性有机物释放作用较低。其主要原因是:活性炭进一步加强了Fe(Ⅱ)在均相体系下,短时间内形成的过量
SO−4⋅ 与释放的溶解性有机物发生氧化分解反应。3)活化过硫酸盐产生的自由基反应可对污泥中溶解性有机物组分选择性地氧化分解,且主要与具有荧光基团的蛋白质类物质和芳香类物质反应,而不氧化分解糖类有机物。
铁(II)-炭活化过硫酸盐对剩余污泥中溶解性有机物释放的影响
Influence on DOM release from sewage sludge by Fe(II)-AC activating persulfate
-
摘要: 通过预处理调控污泥溶解性有机物(DOM)的释放及其组分特性,对提升污泥后续厌氧消化、脱水等处理效率至关重要。为此,以Fe(Ⅱ)和活性炭活化过硫酸盐(PDS)预处理污泥,研究了不同活化PDS氧化路径下污泥DOM的释放特性及组分变化特征。结果表明,Fe(II)活化在PDS投加量为2.7 mmol∙g−1时,TOC释放增量降低到10 mg∙L−1;Fe(Ⅱ)+AC活化在PDS投加量为1.6 mmol∙g−1时,TOC的释放增量降低至10 mg∙L−1;而在活性炭活化下,TOC的释放量随PDS投加量的增加呈线性增加。在活性炭活化下,污泥DOM的释放较缓慢但稳定,而在Fe(Ⅱ)活化下DOM释放速率快;同时,被活化的过硫酸盐很容易对DOM中具有荧光基团的蛋白质类物质和芳香类物质氧化分解,但糖类有机物不被氧化。通过Fe(Ⅱ)和活性炭的组合和对投加量的控制,可以选择性地调控污泥中溶解性有机物的释放速率和有机组成。本研究结果可为强化污泥厌氧消化和污泥深度脱水而选择性地调控污泥预处理过程提供参考。Abstract: Control of the release and components of dissolved organic matter (DOM) in sludge by pretreatment is important to improve the efficiency of subsequent anaerobic digestion and sludge dewatering. Therefore, Fe(Ⅱ) and activated carbon (AC) activating persulfate (PDS) were used to pretreat sludge, thus to investigate the release and changes of organic components of DOM from sludge under different PDS oxidation pathways. Results showed that when the dosage of PDS in Fe(Ⅱ) activation was 2.7 mmol∙g−1, the increment of TOC release decreased to 10 mg∙L−1. While the activated carbon activation increased the TOC release linearly with the increase of the PDS dosage. The release of DOM from sludge under AC activation was slow but stable, and the release rate of DOM under Fe(Ⅱ) activation was very fast. In the meantime, protein and aromatic substances within fluorescent groups in DOM were easily oxidized and decomposed by the activated persulfate, but carbohydrates could not be oxidized. Therefore, through the combination of Fe(Ⅱ) and AC and its dosage, the release rate and organic composition of DOM from sludge could be selectively adjusted. The results of this study can provide a reference for selectively regulating the sludge pretreatment process for strengthening anaerobic digestion and deep dewatering of sludge.
-
Key words:
- persulfate /
- sludge pretreatment /
- dissolved organic matter /
- advanced oxidation /
- Fe(Ⅱ)-AC
-
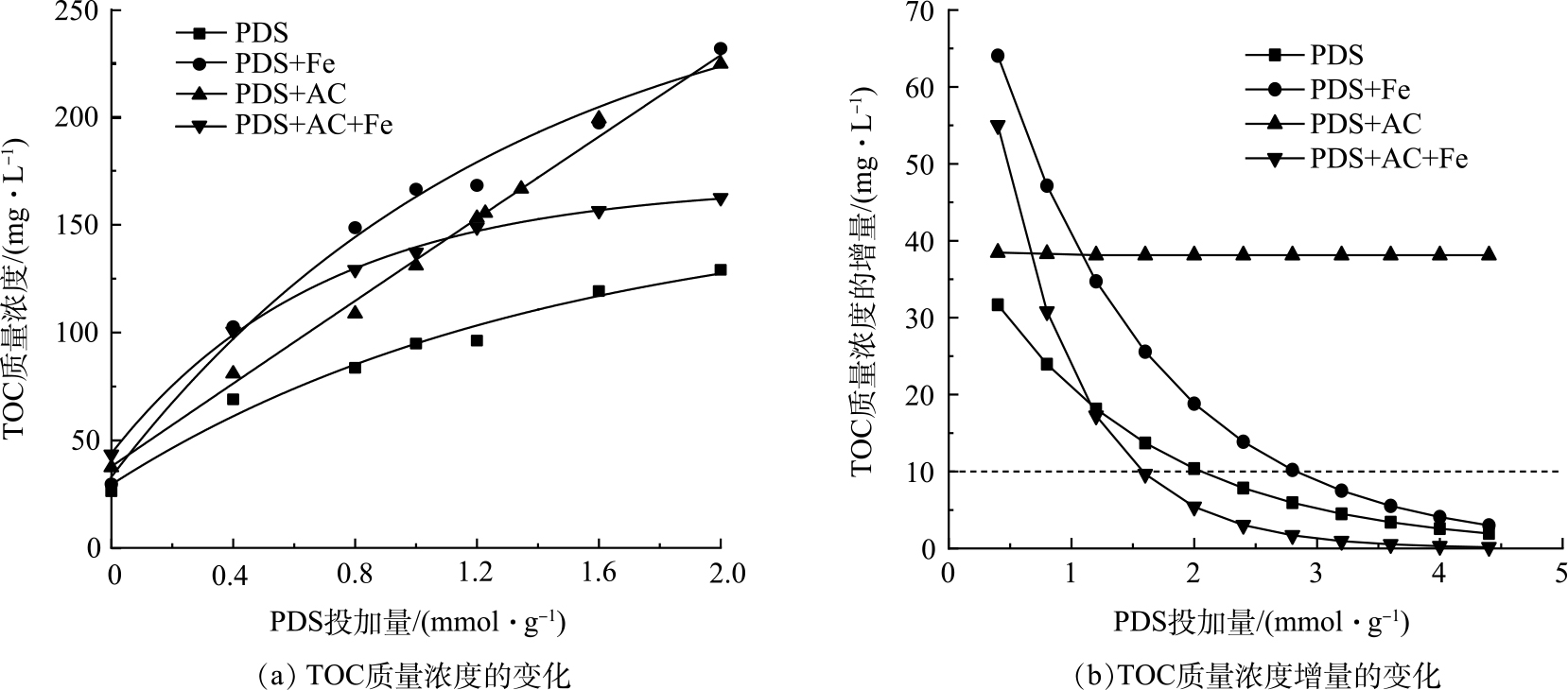
图 1 Fe(II)-炭活化PDS预处理污泥的TOC质量浓度和TOC质量浓度增量与PDS投加量关系
Figure 1. Relationship between TOC mass concentration and TOC mass concentration increase of Fe(II)-carbon activated PDS pretreated sludge and PDS dosage
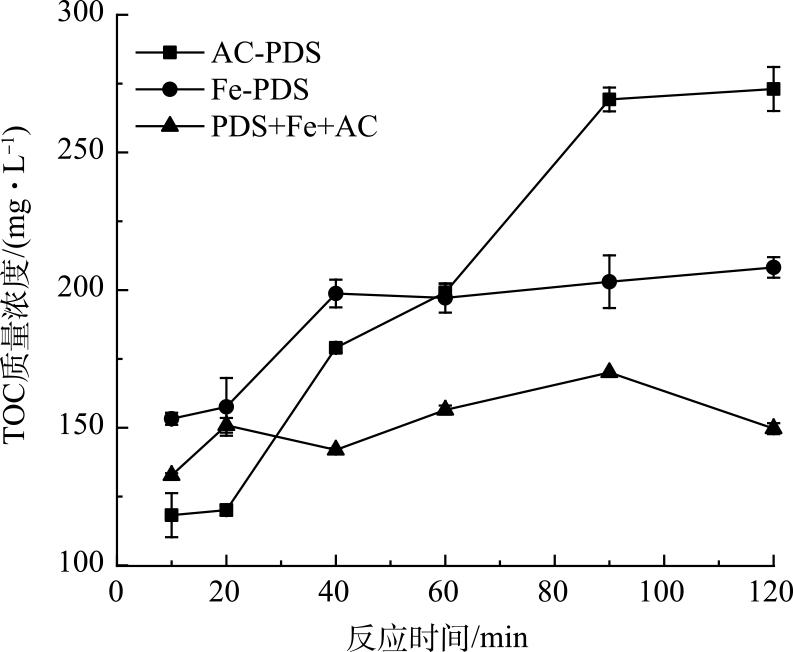
图 2 Fe(Ⅱ)-炭活化PDS预处理污泥TOC释放过程的变化图
Figure 2. TOC release process of Fe(Ⅱ)-carbon activated PDS pretreatment sludge

图 3 Fe(Ⅱ)投加量和批次投加方式对活化过硫酸盐预处理污泥TOC释放的质量浓度的影响
Figure 3. Influence of Fe(Ⅱ) dosage and batch dosage method on TOC release concentration of activated persulfate pretreatment sludge
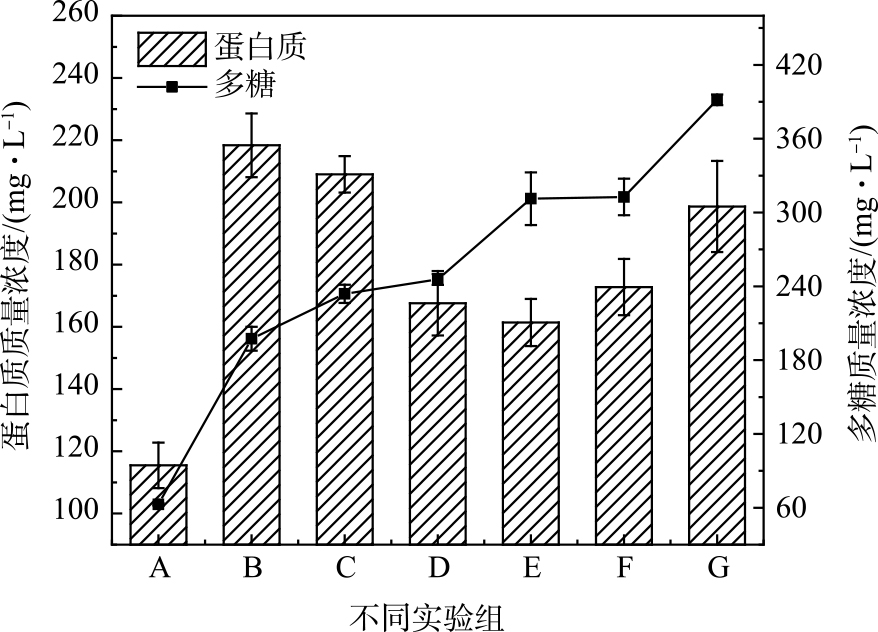
图 4 Fe(Ⅱ)投加量对PDS预处理污泥释放溶解性多糖和蛋白质浓度的影响
Figure 4. Influence of Fe(Ⅱ) dosage on concentration of soluble polysaccharides and protein released by PDS pretreatment sludge
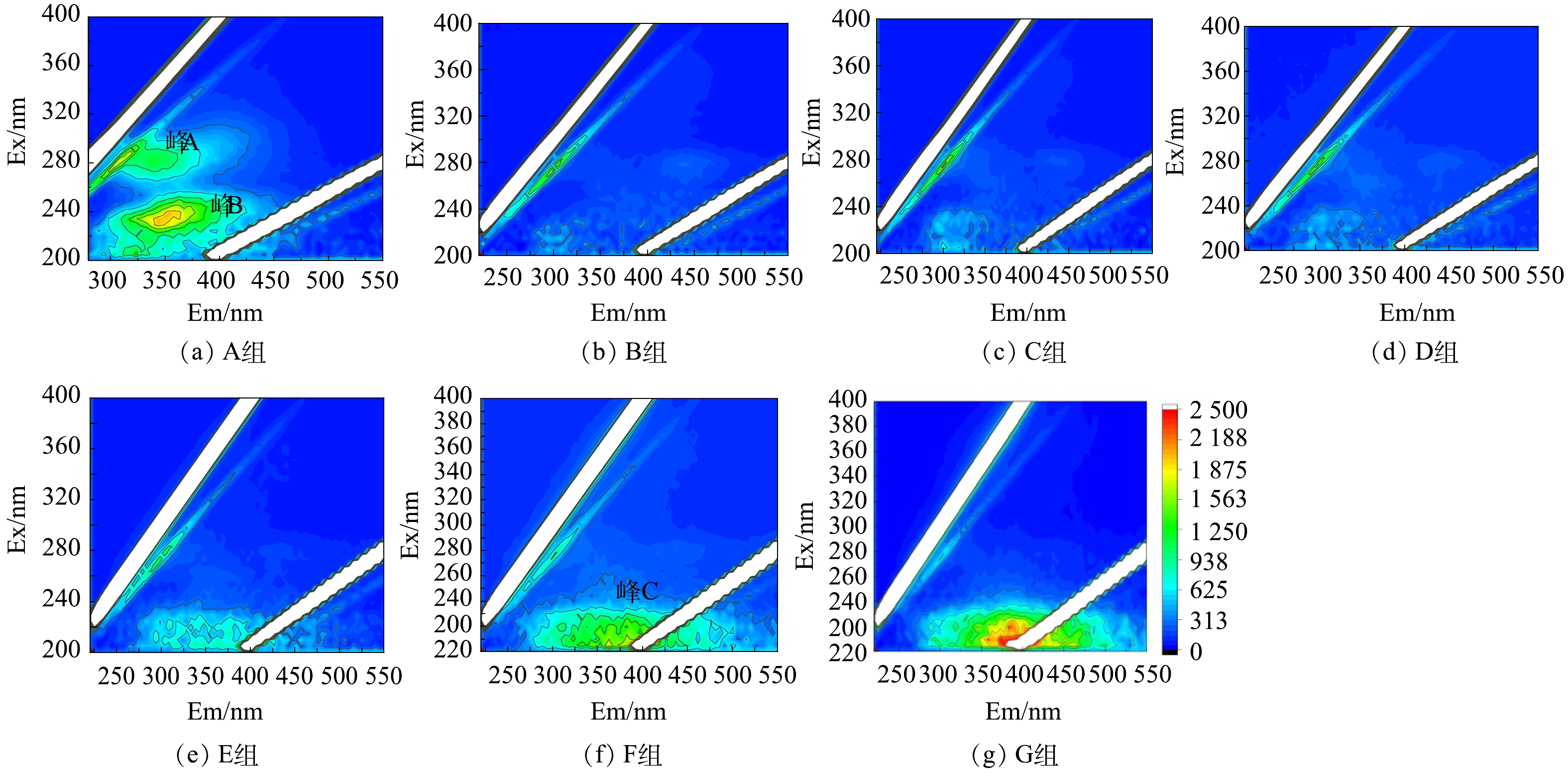
图 5 Fe(II)投加量对PDS预处理污泥中溶解性有机物的三维荧光光谱
Figure 5. Three-dimensional fluorescence spectrum of Fe(II) dosage bovine serum albumin in the PDS pretreated sludge
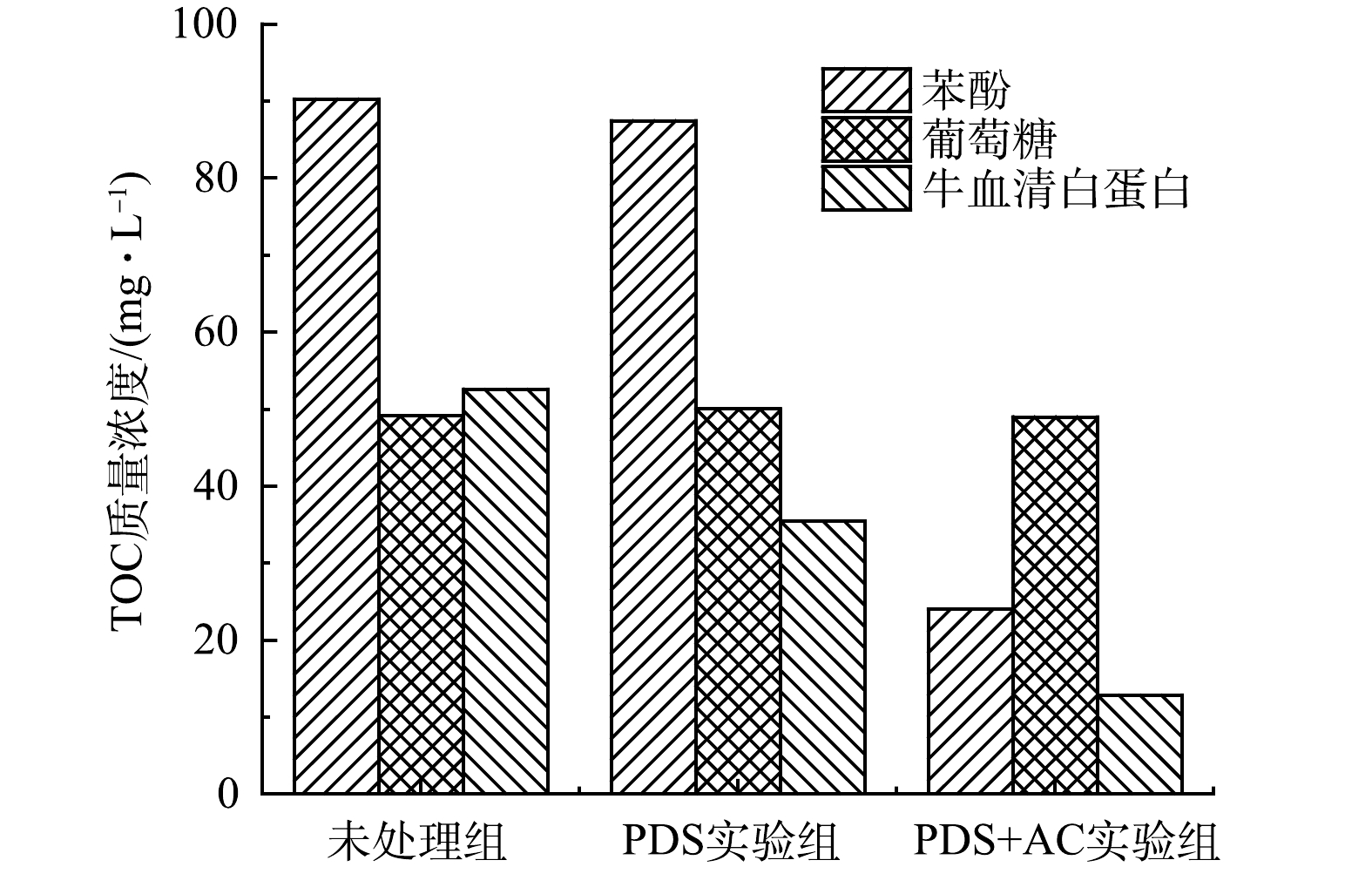
图 6 活化PDS对苯酚、牛血清白蛋白和葡萄糖的降解作用
Figure 6. Degradation effect of activated PDS on phenol, bovine serum albumin and glucose

图 7 牛血清蛋白降解的三维荧光谱
Figure 7. three-dimensional fluorescence spectra of degradation of bovine serum albumin
表 1 剩余污泥的基本特性
Table 1. Basic characteristics of excess sludge
pH 电导率/(μS∙cm−1) TCOD/( mg∙L−1) SCOD/( mg∙L−1) TOC/( mg∙L−1) TSS/(g∙L−1) VSS/(g∙L−1) TS/% VS/% 含水率/% 6.93 1,002 544 49 29 30.22±0.1 20.3±0.1 3.14±0.03 2.09±0.02 96.86±0.03  下载: 导出CSV
下载: 导出CSV
表 2 活性炭的孔径特征
Table 2. Pore size characteristics of activated carbon
BET比表面积/(m2∙g−1) 微孔内比表面积/(m2∙g−1) 外比表面积/(m2∙g−1) 总孔容/(cm3∙g−1) 微孔孔容/(cm3∙g−1) 平均孔径/nm 1 438.82 790.49 647.33 0.73 0.32 2.70
下载: 导出CSV
表 3 实验设计
Table 3. Design of experiments
实验组 污泥 PDS投加量/(mmol∙g−1) AC投加量/(g∙L−1) Fe(Ⅱ)投加量/( mmol∙g−1) A 原泥 0 0 0 B 原泥 2 10.97 0.6 C 原泥 2 10.97 0.9 D 原泥 2 10.97 1.2 E 原泥 2 10.97 1.5 F 原泥 2 10.97 1.8 G 原泥 2 10.97 2.1
下载: 导出CSV
表 4 Fe(Ⅱ)-炭活化PDS预处理污泥TOC质量浓度(CTOC)与PDS投加量(x)关系
Table 4. Relationship between TOC mass concentration (CTOC) of Fe(Ⅱ)-carbon activated PDS pretreatment sludge and PDS dosage (x)
实验组 拟合方程 R2 PDS CTOC = 33.3+126.29×(1-exp(-(x -0.04)/1.43)) 0.981 Fe(II)活化PDS CTOC = 39.4+126.29×(1-exp(-(x -0.04)/1.31)) 0.988 AC活化PDS CTOC = 38.25+95.33x 0.994 Fe(II)+AC活化PDS CTOC = 40.3+128.67×(1-exp(-(x +0.02)/0.69)) 0.999
下载: 导出CSV
-
[1] 中华人民共和国住房和城乡建设部. 2020年城乡建设统计年鉴[EB/OL]. [2021-10-12]. http://www.mohurd.gov.cn/xytj/tjzljsxytjgb/jstjnj/w02021101222731370199062500.zip,2020. [2] 杨倩茜. 超声破解预处理对剩余污泥厌氧消化的适应性研究[D]. 武汉: 华中科技大学, 2011. [3] XIAO K K, ABBT-BRAUN G, HORN H. Changes in the characteristics of dissolved organic matter during sludge treatment: A critical review[J]. Water Research, 2020, 187: 116441. doi: 10.1016/j.watres.2020.116441 [4] 董嘉琦. Fe(Ⅱ)/PS预处理剩余污泥及其对污泥厌氧消化的影响研究[D]. 南昌: 南昌大学, 2020. [5] NGO P L, UDUGAMA I A, GERNAEY K V, et al. Mechanisms, status, and challenges of thermal hydrolysis and advanced thermal hydrolysis processes in sewage sludge treatment[J]. Chemosphere, 2021, 281: 130890. doi: 10.1016/j.chemosphere.2021.130890 [6] LIU J B, WEI Y S, LI K, et al. Microwave-acid pretreatment: A potential process for sludge dewaterability[J]. Water Research, 2016, 90: 225-234. doi: 10.1016/j.watres.2015.12.012 [7] 肖雄. 高含固污泥热水解工艺优化及其对厌氧消化的影响研究[D]. 北京: 北京化工大学, 2020. [8] 万甜, 闫幸幸, 任杰辉, 等. Fe(II) 活化过硫酸盐改善污泥脱水性能[J]. 环境工程学报, 2020, 14(1): 189-196. doi: 10.12030/j.cjee.201902067 [9] ZHOU A J, WEI Y L, FAN Y X, et al. Sulfate reduction-mediated syntrophic microbiomes accelerated waste-activated sludge fermentation on the basis of SO•−4 oxidation and eliminated superfluous sulfate[J]. ACS Sustainable Chemistry & Engineering, 2020, 8(25): 9325-9334.[10] WEI L L, XIA X H, ZHU F Y, et al. Dewatering efficiency of sewage sludge during Fe2+-activated persulfate oxidation: Effect of hydrophobic/hydrophilic properties of sludge EPS[J]. Water Research, 2020, 181: 115903. doi: 10.1016/j.watres.2020.115903 [11] LUO J Y, WU L J, FENG Q, et al. Synergistic effects of iron and persulfate on the efficient production of volatile fatty acids from waste activated sludge: Understanding the roles of bioavailable substrates, microbial community & activities, and environmental factors[J]. Biochemical Engineering Journal, 2019, 141: 71-79. doi: 10.1016/j.bej.2018.10.010 [12] CHENG X, GUO H G, ZHANG Y L, et al. Non-photochemical production of singlet oxygen via activation of persulfate by carbon nanotubes[J]. Water Research, 2017, 113: 80-88. doi: 10.1016/j.watres.2017.02.016 [13] USHANI U, LU X Q, WANG J H, et al. Sulfate radicals-based advanced oxidation technology in various environmental remediation: A state-of-the-art review[J]. Chemical Engineering Journal, 2020, 402: 126232. doi: 10.1016/j.cej.2020.126232 [14] DUAN X G, AO Z M, ZHOU L, et al. Occurrence of radical and nonradical pathways from carbocatalysts for aqueous and nonaqueous catalytic oxidation[J]. Applied Catalysis B:Environmental, 2016, 188: 98-105. doi: 10.1016/j.apcatb.2016.01.059 [15] HU P D, LONG M C. Cobalt-catalyzed sulfate radical-based advanced oxidation: A review on heterogeneous catalysts and applications[J]. Applied Catalysis B:Environmental, 2016, 181: 103-117. doi: 10.1016/j.apcatb.2015.07.024 [16] VARANASI L, COSCARELLI E, KHAKSARI M, et al. Transformations of dissolved organic matter induced by UV photolysis, hydroxyl radicals, chlorine radicals, and sulfate radicals in aqueous-phase UV-based advanced oxidation processes[J]. Water Research, 2018, 135: 22-30. doi: 10.1016/j.watres.2018.02.015 [17] LI K, WANG J X, LIU J B, et al. Advanced treatment of municipal wastewater by nanofiltration: Operational optimization and membrane fouling analysis[J]. Journal of Environmental Sciences, 2016, 43: 106-117. doi: 10.1016/j.jes.2015.09.007 [18] LI X W, DAI X H, TAKAHASHI J, et al. New insight into chemical changes of dissolved organic matter during anaerobic digestion of dewatered sewage sludge using EEM-PARAFAC and two-dimensional FTIR correlation spectroscopy[J]. Bioresource Technology, 2014, 159: 412-420. doi: 10.1016/j.biortech.2014.02.085 [19] 樊雅欣, 刘红燕, 潘凌峰, 等. 活化方式对过硫酸盐强化剩余污泥发酵的影响[J]. 中国环境科学, 2019, 39(6): 2460-2466. doi: 10.3969/j.issn.1000-6923.2019.06.028 [20] 国家环境保护总局. 水和废水监测分析方法[M]. 4版. 北京: 中国环境科学出版社, 2002. [21] FROLUND B, GRIEBE T, NIELSEN P H. Enzymatic activity in the activated-sludge floc matrix[J]. Applied Microbiology and Biotechnology, 1995, 43(4): 755-761. doi: 10.1007/BF00164784 [22] DUBOIS M, GILLES K A, HAMILTON J K, et al. Colorimetric method for determination of sugars and related substances[J]. Analytical Chemistry, 1956, 28(3): 350-356. doi: 10.1021/ac60111a017 [23] XIE P C, MA J, LIU W, et al. Removal of 2-MIB and geosmin using UV/persulfate: Contributions of hydroxyl and sulfate radicals[J]. Water Research, 2015, 69: 223-233. doi: 10.1016/j.watres.2014.11.029 [24] XIAO S, CHENG M, ZHONG H, et al. Iron-mediated activation of persulfate and peroxymonosulfate in both homogeneous and heterogeneous ways: A review[J]. Chemical Engineering Journal, 2020, 384: 123265. doi: 10.1016/j.cej.2019.123265 [25] WANG S L, WU J F, LU X Q, et al. Removal of acetaminophen in the Fe2+/persulfate system: Kinetic model and degradation pathways[J]. Chemical Engineering Journal, 2019, 358: 1091-1100. doi: 10.1016/j.cej.2018.09.145 [26] BU L J, SHI Z, ZHOU S Q. Modeling of Fe(II)-activated persulfate oxidation using atrazine as a target contaminant[J]. Separation and Purification Technology, 2016, 169: 59-65. doi: 10.1016/j.seppur.2016.05.037 [27] VICENTE F, SANTOS A, ROMERO A, et al. Kinetic study of diuron oxidation and mineralization by persulphate: Effects of temperature, oxidant concentration and iron dosage method[J]. Chemical Engineering Journal, 2011, 170: 127-135. doi: 10.1016/j.cej.2011.03.042 [28] GUO S D, HUANG Y X, ZHOU L, et al. Improvement of sludge dewaterability and disintegration efficiency using electrolytic zero-valent iron activated peroxymonosulfate[J]. Water Science & Technology, 2021, 84(2): 458-468. [29] CHEN W, WESTERHOFF P, LEENHEER J A, et al. Fluorescence excitation-emission matrix regional integration to quantify spectra for dissolved organic matter[J]. Environment Science& Technology, 2003, 37(24): 5701-5710. [30] 陈诗雨, 李燕, 李爱民. 溶解性有机物研究中三维荧光光谱分析的应用[J]. 环境科学与技术, 2015, 38(5): 64-68. [31] CHANG B, BIAN C, GE D D, et al. Enhancement of waste activated sludge dewaterability by ultrasound-activated persulfate oxidation: Operation condition, sludge properties, and mechanisms[J]. Chemosphere, 2021, 262: 128385. doi: 10.1016/j.chemosphere.2020.128385 [32] XIAO K K, CHEN Y, JIANG X, et al. Comparison of different treatment methods for protein solubilisation from waste activated sludge[J]. Water Research, 2017, 122: 492-502. doi: 10.1016/j.watres.2017.06.024 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5023
- HTML全文浏览数: 5023
- PDF下载数: 50
- 施引文献: 0

