



-
近年来,随着原油轻质化和清洁能源发展趋势盛行,制氢成为石油化工、储能等支柱产业关注的核心焦点;煤焦气化制氢作为当前最为成熟和经济的生产工艺,目前已在全国大规模推广应用。在煤焦制氢工艺中,水煤浆气化产生的粗煤气混合物经脱盐水洗涤后,产生大量富含Ca2+/Mg2+及酚类等有机物的高硬度难降解气化废水,其高效达标处理成为抑制煤焦制氢产业持续发展的瓶颈[1]。
煤焦气化制氢产生的高硬度难降解有机废水处理通常采用以生化为核心的组合工艺,但因生化好氧单元产生的CO2与来水中的Ca2+/Mg2+反应产生大量无机灰分,且常规沉降无法实现无机灰分与活性污泥的持续有效分离,导致生化池中灰分在污泥细胞外集聚并形成隔离保护层,从而对生化系统产生不同程度的抑制作用[2]。生化池生成的无机灰分可能影响其微生物群落结构[3]、优势菌群丰度[4]、微生物代谢途径以及丝状菌生长趋势[5]等;无机灰分也能引起污泥胞外聚合物(EPS)分泌增加[6-7],多糖中相应增加的亲水羧基使得细胞与EPS间的斥力增加[8-9],从而导致活性污泥形态松散、密实度降低直至污泥沉降性降低[10];无机灰分还可能导致污泥微生物细胞在高渗透压下失活死亡,从而显著影响生化系统综合降解效率。
针对来水钙硬在生化系统中生成无机灰分、并从多方面影响生化效能的问题,目前普遍采取化学沉淀[11]、絮凝[12]、电化学[13]等方法从源头大幅降低来水钙硬,但这些方法普遍存在除钙硬不彻底、处理成本高、产生二次污染等问题,无法根本性避免来水硬度在后续生化池中的持续性集聚[14]。通过生化系统排泥来避免生化池中无机灰分集聚是相对合理的选择,但定向排出无机灰分的关键在于先探明活性污泥与灰分的结合形式,并采取经济有效的方法实现泥灰分离。有研究表明,钙镁等无机矿化物与活性污泥的结合作用力主要是静电作用力[15-16],该作用力促使灰分表面层在活性污泥表面或孔道中形成了液桥[17],但无法形成稳固的络合键[18],这为积极探索泥灰原位分离方法实现生化池调节提供了理论支撑。城市污泥中衍生磷与灰分的分离为磷资源化回收提供新思路的同时,也验证了活性污泥与无机灰分之间的作用力为弱结合力[19]。常规的离心分离过程因其流场均匀性,不仅无法实现定向分离,还可能加剧无机灰分在污泥絮体中的渗透[20]。超声过程虽然能利用油相表面属性差异将污泥与带油的无机组分分开,但也无法实现污泥与无机质的直接分离[21]。而旋流器中因流场剪切和边界效应引起的分散相自转运动[22],可适当脱附活性污泥表面或孔道中夹带的颗粒物或胞外聚合物[23],这为实现活性污泥与无机灰分的分离提供了借鉴[24]。
针对高硬度有机废水生化处理过程生成的无机灰分影响其处理效率的问题,目前尚缺乏对生化池中活性污泥与无机灰分的结合形式及分离途径的系统性实验研究。本研究以高硬度难降解有机废水为对象,首先设置并列SBR并测试了不同硬度废水的生化处理效率和所生成的无机灰分对污泥性状的影响,然后采取离线实验测试了生化池活性污泥与无机灰分的结合形式及潜在分离途径,最后考察了旋流处理对泥灰混合物中无机灰分的分离效率,并通过实际工程侧线实验验证无机灰分旋流分离对生化系统综合效能的改善效果,以期为实际高硬度废水生化处理提标改造提供新思路。
-
为考察来水Ca2+浓度对生化池中污泥性状及其生化降解效率的影响,搭建了6组SBR,各自配制进水的Ca2+浓度分别为0、200、400、800、1 600和2 400 mg·L−1,对应的SBR分别记为R1、R2、R3、R4、R5和R6。此外,进水还通过乙酸钠和碳酸氢铵配制,确保其耗氧有机物(以COD计)和氨氮质量浓度分别为600 mg·L−1和150 mg·L−1;活性污泥则接种自华东理工大学污水处理厂,初始污泥质量浓度均为3 500 mg·L−1,污泥中有机质占比(MLSS/MLVSS)为0.67,污泥的体积指数(SVI)为103 mL·g−1。现场污泥旋流脱灰强化生化侧线实验期间,生化池进水平均COD、氨氮和TN的质量浓度分别为486、151和195 mg·L−1,Ca2+和Mg2+平均质量浓度分别为1 715 mg·L−1和121 mg·L−1;旋流分离实验前池内固体悬浮物质量浓度(SS)则为11.5 g·L−1,其中有机组分和无机组分质量浓度分别为2.4 g·L−1和9.1 g·L−1。
-
Ca2+浓度对污泥性状及生化效率影响实验共设置了6组SBR,并开展150 d的同步对比实验。单个反应器的有效容积为6 L,反应器单一周期操作时长为:进水20 min、曝气4 h、沉淀70 min、排水30 min;设置70 min沉淀时间主要是为了满足后续反应器中生成无机灰分后的污泥充分沉降需求,也与现场工程条件一致。后续泥灰混合液单一离心分离实验和测试取样以及循环旋流分离实验均依托这6组SBR。
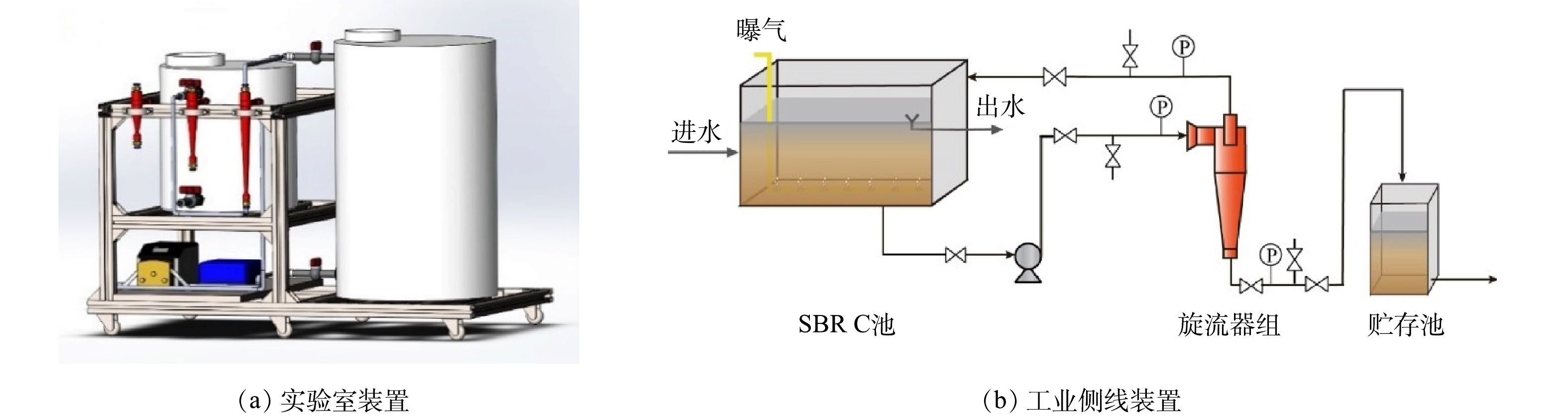
6组SBR池经过150 d的连续运行后,进一步对反应器中的混合液开展泥灰旋流分离实验,并根据6组SBR连续运行过程中的降解效率和泥灰混合液组成特性确定具体适用于旋流分离的SBR。为确保旋流分离实验过程中充分取样,分离前先将SBR池中所有泥水混合液稀释至200 L。泥灰离线旋流分离实验室装置如图1(a)所示。
污泥旋流脱灰强化生化侧线实验则依托中石化某120 m3·h−1煤制氢气化废水SBR开展,现场工业侧线装置如图1(b)所示。SBR由3间独立的生化池组成,实验仅对其中的C列池体增设提升泵、内回流管线和旋流器,并在其曝气阶段对泥水混合液实施旋流分离处理。同步并行实验确保3列生化池的Ca2+浓度和钙硬完全一致,确保了平行实验的可对比性。此外,其他两列生化池定期排泥不同,C列生化池将旋流分离底流作为剩余污泥持续外排。
-
为验证旋流过程对生化池泥灰混合液分离的可行性,共设计了锥角分别为6°、8°和10°共3种旋流器(图2),各自的溢流口直径、底流口直径、柱段直径、柱段长度、溢流管插入深度分别为7、3、25、35和8 mm,进口宽和高分别为5 mm×10 mm、7 mm×7 mm和6 mm×8 mm。泥灰混合液旋流分离的实验过程中,旋流器的处理量和分流比分别设置为200 L·h−1和5%;污泥旋流脱灰强化生化侧线实验中的旋流器则根据小试实验优选结果等比例放大4倍得出(锥角不变)。
-
采用原子力显微镜(AFM, Bruker, Dimension Icon, 配套Scan Asyst-Air探针)对污泥表面形貌粗糙度进行测试,显微镜设置为QNM模式且扫描频率为1 Hz,并用Rq表征均方根粗糙度(即在取样长度内样品形貌轮廓偏离平均线的均方根值);SBR降解过程中生成的无机组分则采用X射线衍射仪(XRD, Bruker, D8 Focus)进行定性分析;泥灰混合液中溶解性钙离子浓度采用电感耦合等离子体发射光谱仪(ICP-OES)进行定量测试。生化处理过程中COD、氨氮、TN等水质指标,以及污泥沉降性、比耗氧速度、体积指数等指标等,则采用通用标准或参考示例方法进行测试[25-26]。
-
来水Ca2+质量浓度分别为0~2 400 mg·L−1时,经过150 d的连续运行,6组SBR中污泥有机质占比和污泥体积指数变化趋势如图3所示。在初始条件、进水条件(Ca2+浓度除外)和操作条件完全相同的前提下,如图3(a)所示,除了R6反应器以外,其他5组生化池内的污泥有机质占比与污泥沉降性变化趋势基本一致,即生化池内污泥有机质占比从初始值0.67逐渐降低,而污泥沉降指数在初期略微升高后持续降低;当进水Ca2+质量浓度达到2 400 mg·L−1时,生化池内的有机质占比降低至0.39,并呈现持续降低的趋势。XRD测试结果表明,与对照组R1反应器不同,其他5组反应器中污泥样品衍射图谱均出现了明显的碳酸钙特征峰,表明来水中的Ca2+在生化池好氧段与CO2反应持续产生了钙盐,并使得污泥中的无机灰分绝对量大幅增加,而有机质占比持续降低。生化池中生成的无机灰分附着或粘附在活性污泥上,增加了池内悬浮物的密度(图3(b)),这使得生化池中污泥的体积指数不断下降,并在进水Ca2+质量浓度为2 400 mg·L−1时最低降至11.7 mL·g−1以内。
来水中Ca2+在生化池生成的无机灰分在影响污泥有机质占比和沉降性能的同时,也对SBR系统的降解效率产生了显著影响。图4(a)和图4(b)分别为出水COD和氨氮浓度的变化曲线,不考虑前15 d各反应器驯化阶段存在的降解效率波动,相较对照组R1反应器,随着进水Ca2+浓度的增加,其他反应器对COD和氨氮的去除率随之略微降低,并在进水Ca2+质量浓度达到2 400 mg·L−1时,R6反应器在实验末期几乎对COD没有去除效果,而氨氮去除率仅为20.7%、较R1反应器低约70%。这主要与来水盐分抑制微生物代谢、以及生成的无机灰分混入污泥后抑制微生物细胞与水体中溶解氧和营养物的传质效率等因素相关。
-
为探明生化池中生成的无机灰分与活性污泥的结合形式,在第100 天取R4反应器中适量的泥灰混合液,以300 r·min−1转速离心5 min后分成等量的上清液和底物。如图5所示,在上清液和底物中添加足量盐酸溶液溶解后,针对固体灰分的质量浓度为3 230 mg·L−1、Ca2+质量浓度为3 208 mg·L−1的泥水混合液,上清液与底物中Ca2+浓度的差异明显大于上清液与底物中固体灰分的质量浓度差异,这表明离心过程引起的碳酸钙灰分与活性污泥沉降趋势存在差异,混合液中更多的碳酸钙灰分沉降至离心管底部。由此可证实,R4的泥灰混合液中钙灰与活性污泥之间为非稳态结合,而且离心等物理法即可实现泥灰分离。
进一步通过原子力显微镜观测不同SBR中活性污泥与无机灰分的结合差异。如图6所示,分别选取R1和R4中的泥灰混合液样品,并对2个反应器中的样品各扫描2个不同区域。其中R1反应器中的样品1和2的表面粗糙度分别为20.5 nm和25.9 nm;而R4反应器中样品1和2的表面粗糙度分别为38.2 nm和35.6 nm。R4中样品粗糙度有所增加,这主要是由于来水Ca2+生成的无机灰分附着或包裹在污泥表面所致。
-
考虑到述泥灰混合液离心后污泥与无机灰分的分离效果,并为了满足实际工程的连续运行及高效分离需求,设计了锥角分别为6°、10°和20°的3种旋流器开展泥灰混合液分离实验。结合图3和图4的结果可以发现,钙硬过高时生化池降解效率过低,旋流分选后存在污泥严重失活风险;而足够的进水Ca2+浓度可引起无机灰分累计,便于充分验证旋流过程对泥灰混合液的分离效果,因此,选取R4反应器中的泥灰混合液作为分离对象,R4反应器中MLSS和MLVSS质量浓度分别为18.8 g·L−1和11.4 g·L−1。
经过稀释后,针对固体灰分的质量浓度为462 mg·L−1、其中有机质质量浓度为79 mg·L−1的泥灰混合样品,经过3种旋流器单次分离或10次循环分离后的底流与溢流样品悬浮物组分如表1所示。单次分离实验结果表明,3种结构旋流器对泥灰混合液中固相的分离效率分别为5.2%、11.5%和14.7%;与离心分离类似,旋流分离除了实现固相分离以外,其底流和溢流中的钙灰比例也发生了变化。3种工况下溢流中悬浮物的有机质占比较进料均有略微提高,表明稀释的泥灰混合液经旋流处理后一定程度实现了活性污泥与无机灰分的定向分选效果。进一步对单次旋流分离的溢流进行循环旋流分离,并不断持续10次,最佳旋流器(锥角为10°)可使最终溢流中的有机质占比从0.17提升至0.37。由此证实了旋流分选过程对泥灰混合液中活性污泥和灰分的富集作用,而且循环分选过程能不断强化对活性污泥和灰分的富集。
-
依托实际SBR工程装置C列在线开展侧线实验,在其4 h的曝气阶段通过测流内循环形式、采用柱段直径为100 mm、锥角为10°的优选旋流器对SBR池B列中的240 m3泥水混合液实施灰分分离,以确保单一生化周期内可100%处理生化池中的泥水混合液。旋流分选分流比为5%,现场SBR池的A列和B列则作为实验对照样。
SBR生化池旋流脱灰改造降解效率对比结果如图7所示。经过3个月的连续运行,在A列和B列池内固体悬浮物浓度和有机质占比不变的条件下,C列池经过旋流分离定向排泥使池内有机质占比显著提升至0.45,且通过加大旋流底流排泥量使池内MLSS降至4.7 mg·L−1。这主要是因为旋流底流污泥中的无机灰分比例高于生化池,即定向外排污泥以无机灰分为主;此外,随着生化池内无机灰分比例的降低,避免了活性污泥上无机灰分附着引起的污泥过快沉降,污泥体积指数也相应提升至67.3 mL·g−1。随着C列池内泥灰总量和比例的降低,生化池对污水的降解效率也逐渐改善;相较A列和B列池体(考虑到A列和B列池体出水水质接近,因此,将这2列出水混合后取平均值,再与C列进行对比),C列池体内的泥灰混合液经过侧流分选。C列池的COD和氨氮平均去除率分别提高了3.3%和9.4%,使出水平均耗氧有机物(以COD计)和氨氮质量浓度分别降低了17.1 mg·L−1和14.3 mg·L−1,说明提标效果显著。
-
1)来水Ca2+对生化系统污泥中的有机质占比和污泥沉降性影响显著,SBR池经过150 d连续降解,进水Ca2+浓度越高,生化池中有机质占比越低,污泥体积指数越小,则生化系统耗氧有机物(以COD计)和氨氮去除率越低,且变化趋势随实验持续而递进。
2)来水Ca2+在生化系统生成以钙盐为主的无机灰分,并导致污泥表界面粗糙度显著增加,证实了无机灰分与活性污泥之间为非稳态结合,且通过离心等物理法即可实现分离。
3)针对来水Ca2+质量浓度为800 mg·L−1且经过了150 d连续降解的SBR池泥灰混合液实施旋流分离,在最佳结构条件下,单次和10次循环分离后旋流溢流样品中的污泥有机质占比从0.17分别提高至0.24和0.37。
4)依托120 m3·h−1煤制氢废水处理SBR开展侧线旋流脱灰实验,经过90 d的连续运行,对比3列池体表明,改造生化池中污泥有机质占比提高至0.447、污泥体积指数提升至67.3 mL·g−1,且出水平均耗氧有机物(以COD计)和氨氮质量浓度分别降低至17.1 mg·L−1和14.3 mg·L−1。
高硬度废水处理生化池中泥-灰旋流分离实验
Hydrocyclone separation of activated sludge and inorganic ash in the biological tank treating high hardness wastewater
-
摘要: 针对高硬度废水生化处理过程中生成的无机灰分导致污泥活性降低和沉降性差等问题,采用旋流分离方法实现污泥原位脱灰以改善废水综合处理效能。通过离线和在线实验,探究了污泥与无机灰分的结合形式、泥灰混合物旋流分离效率以及旋流处理对高硬度废水生化处理效率的影响。结果表明,6组不同Ca2+浓度的来水经过150 d生化实验,生化池污泥有机质占比从进水Ca2+浓度为0 mg·L−1时的0.75降至Ca2+浓度为2 400 mg·L−1时的0.39,生化系统COD和氨氮去除率也相应降低11%和60%;原子力显微镜测试结果表明,来水含钙条件下生化池污泥表面因无机灰分附着导致其粗糙度从无钙来水下的20.5 nm增至38.2 nm,且活性污泥与无机灰分间的非稳态结合可通过离心等物理法实现分离;来水Ca2+浓度为800 mg·L−1时,泥灰混合液经过最佳结构旋流器10次循环分离,其有机质占比从0.17升高至0.37;依托120 m3·h−1煤制氢废水处理SBR进行旋流脱灰侧线实验,经过3个月连续运行,改造生化池污泥有机质占比较对比样从0.21提升至0.45,且出水平均COD和氨氮分别降低17.1 mg·L−1和14.3 mg·L−1。活性污泥在线旋流分离调理可为高硬度废水生化处理提标改造提供参考。Abstract: The generated inorganic ash in biological reactor treating high hardness wastewater decreased the activity and settleability of activated sludge. Hydrocyclone application was expected to in-situ separate the inorganic ash from the activated sludge and increase its biological treatment efficiency. The attachment pattern and separation efficiency between inorganic ash and activated sludge were investigated by offline and online experiments, as well as the effect of inorganic ash removal on the biological treatment efficiency of simulated high hardness wastewater. The results showed that the organics ratio decreased from 0.75 in SBR reactor without calcium ion to 0.39 with influent calcium ion concentration of 2 400 mg·L−1 after 150 d continuous biological treatment of 6 groups of influent with different Ca2+ concentrations, COD and NH4-N removal efficiencies also decreased by 11% and 60%, respectively. The atomic force microscope (AFM) images illustrated that the generated inorganic ash increased the surface roughness of activated sludge from 20.5 nm for Ca2+-free influent to 38.2 nm for Ca2+-containing influent. The surface-attached inorganic ash can be removed from the activated sludge by centrifuge. The mixture of inorganic ash and activated sludge in SBR with influent calcium ion concentration of 800 mg·L−1 was circularly separated for 10 times by hydrocyclone with the optimum structure, and the organics ratio in SBR increased from 0.17 to 0.37. The side-flow hydrocyclone separation experiments were conducted for 120 m3·h−1 SBR in a coal-to-hydrogen gasification wastewater treatment plant, three months running caused the increase of the organics ratio from 0.21 in control SBR to 0.45 in modified SBR, the effluent COD and NH4-N decreased by 17.1 mg·L−1 and 14.3 mg·L−1, respectively. The new approach of online hydrocyclone separation conditioning provides a reference for the upgrade of the biological treatment efficiency of high hardness wastewater.
-
Key words:
- high hardness wastewater /
- biological process /
- inorganic ash /
- hydrocyclone separation /
- MLVSS/MLSS
-
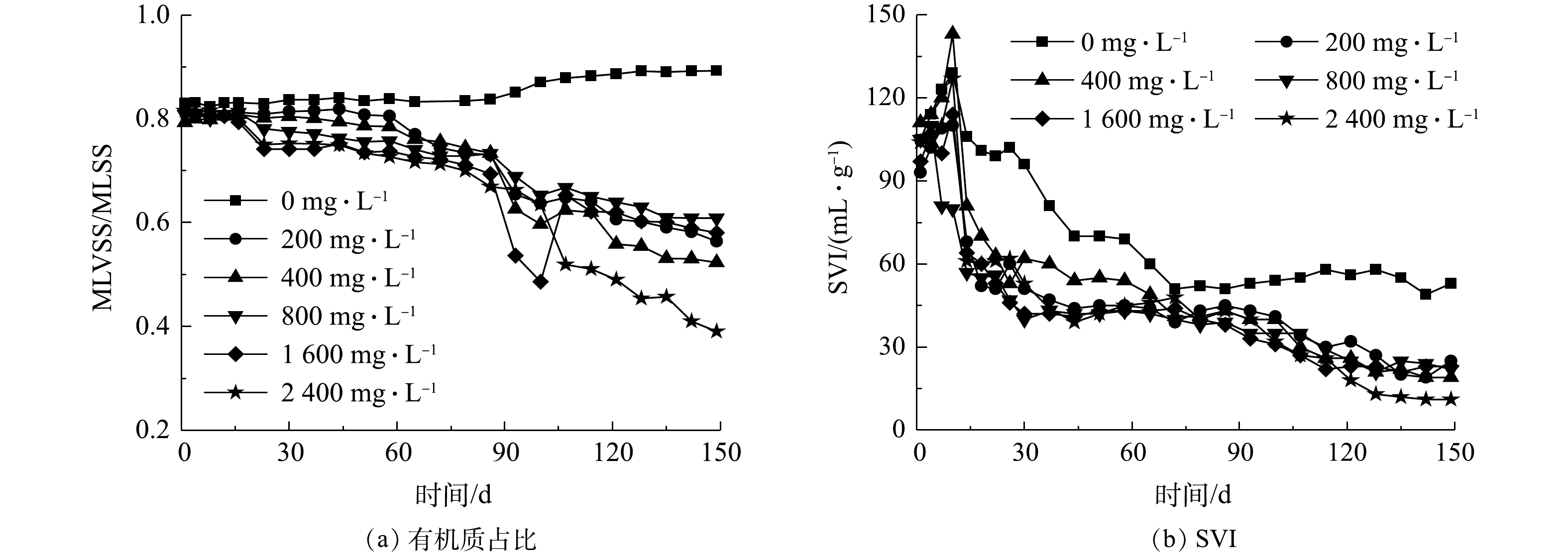
图 3 来水Ca2+浓度对生化池参数的影响
Figure 3. Effect of influent Ca2+ concentration on MLVSS/MLSS and SVI
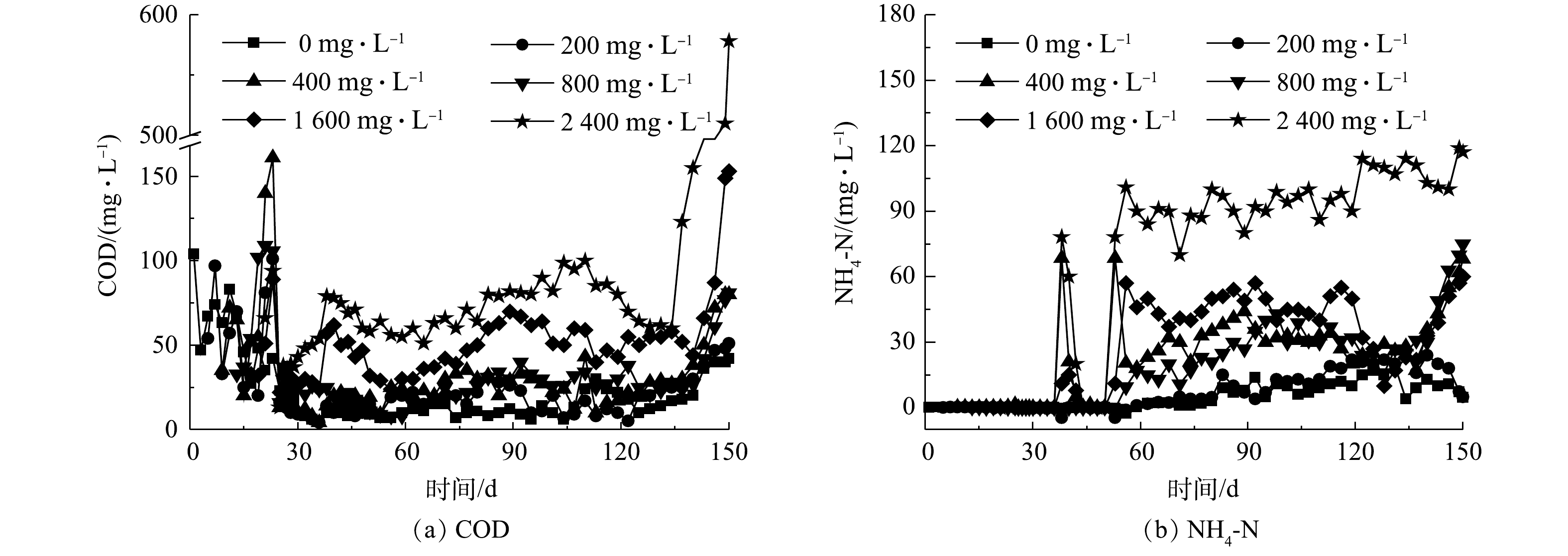
图 4 不同Ca2+浓度条件下SBR降解效率
Figure 4. COD and NH4-N removal efficiencies in SBRs at different influent Ca2+ concentrations
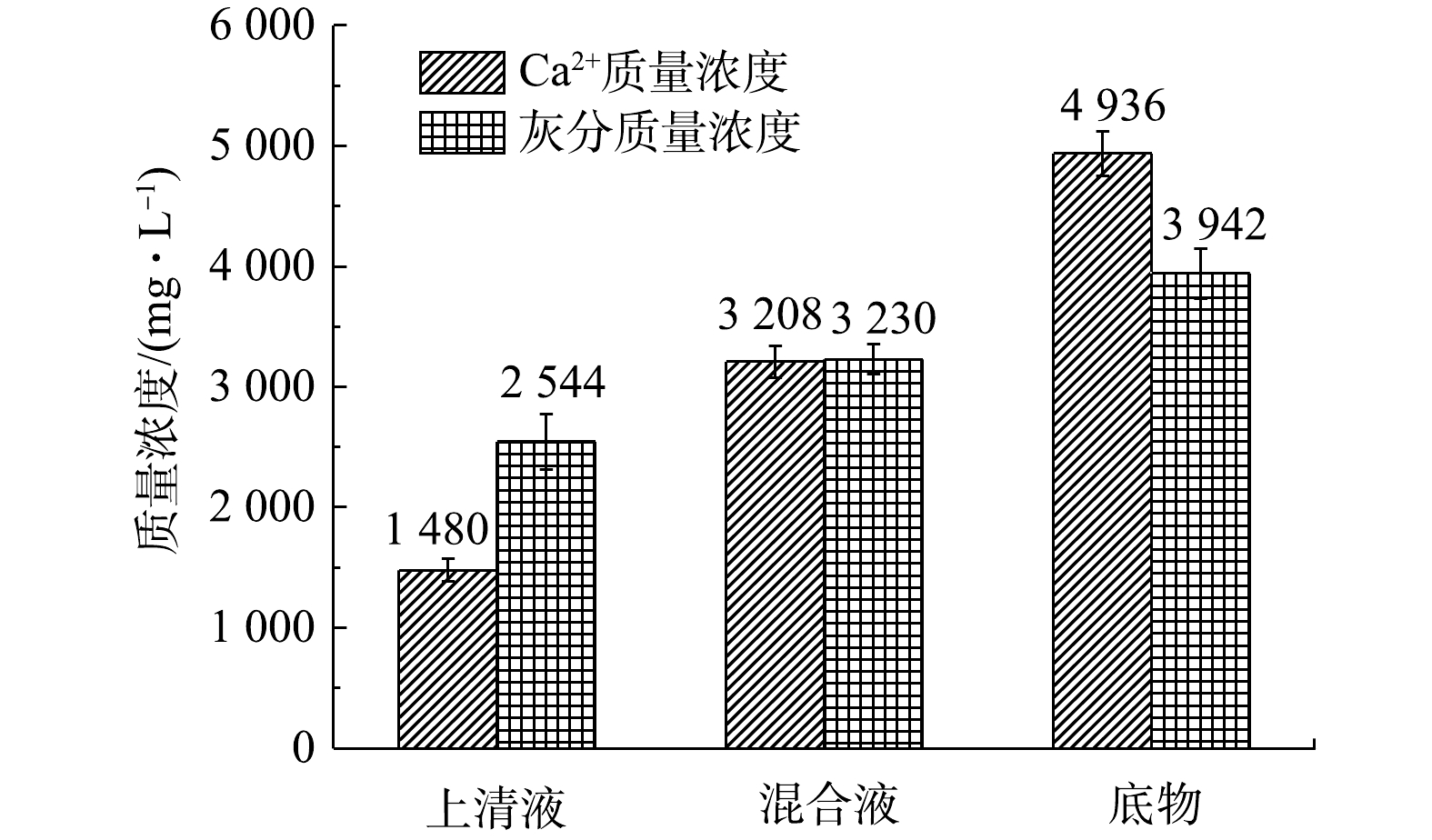
图 5 活性污泥离心分离前后Ca2+质量浓度及灰分质量浓度
Figure 5. Ca2+ and inorganic ash concentrations in activated sludge before and after centrifugation
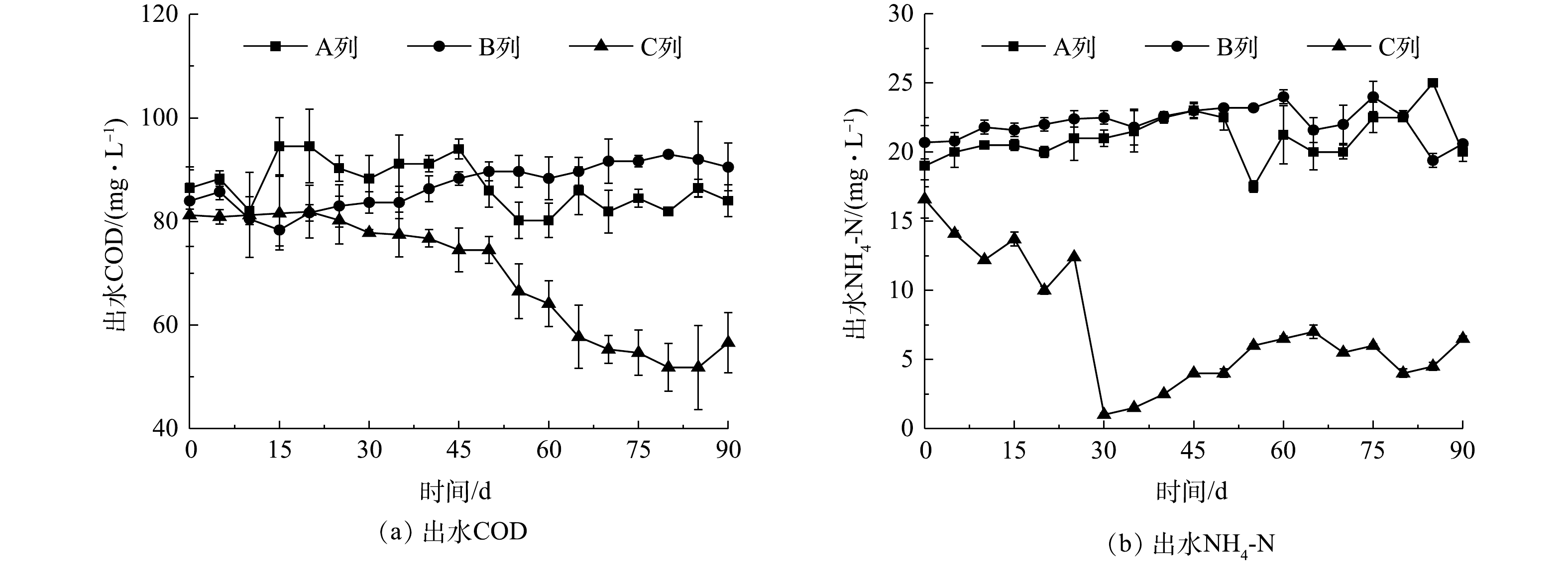
图 7 SBR生化池旋流脱灰改造降解效率对比
Figure 7. Effect of hydrocyclone classification on biological treatment efficiency in SBR
表 1 泥灰混合液旋流分离效率
Table 1. Hydrocyclone separation efficiency of the mixture of activated sludge and inorganic ash
旋流器结构 旋流溢流泥灰单次分离 旋流溢流泥灰10次循环分离 旋流底流泥灰单次分离 固体灰分质量浓度/(mg·L−1) 有机质占比 固体灰分质量浓度/(mg·L−1) 有机质占比 固体灰分质量浓度/(mg·L−1) 有机质占比 6°旋流器 438 0.21 205 0.27 559 0.13 10°旋流器 409 0.24 171 0.37 581 0.10 20°旋流器 394 0.26 85 0.31 672 0.05  下载: 导出CSV
下载: 导出CSV
-
[1] 潘咸峰. 煤制氢污水处理方法: CN105565582A[P]. 2014-10-08. [2] LAY C W, LIU Y, FANE G A. Impacts of salinity on the performance of high retention membrane bioreactors for water reclamation: A review[J]. Water Research, 2010, 44(1): 21-40. doi: 10.1016/j.watres.2009.09.026 [3] ADAV S S, LEE D J, LAI J Y. Microbial community of acetate utilizing denitrifiers in aerobic granules[J]. Applied Microbiology & Biotechnology, 2010, 85(3): 753. [4] SCHMIDT J E, AHRING B K. Granular sludge formation in upflow anaerobic sludge blanket (UASB) reactors[J]. Biotechnology & Bioengineering, 2015, 49(3): 229-246. [5] BARAT R, MONTOYA T, BORRAS L, et al. Calcium effect on enhanced biological phosphorus removal[J]. Water Science & Technology, 2006, 53(12): 29-37. [6] WAN C L, YANG X, LEE D J, et al. Aerobic granulation of aggregating consortium X9 isolated from aerobic granules and role of cyclic di-GMP[J]. Bioresource Technology, 2014, 152: 557-561. doi: 10.1016/j.biortech.2013.11.052 [7] WAN C L, YANG X, LEE D J, et al. Partial nitrification of wastewaters with high NaCl concentrations by aerobic granules in continuous-flow reactor[J]. Bioresource Technology, 2014, 152: 1-6. doi: 10.1016/j.biortech.2013.10.112 [8] LIAO B Q, ALLEN D G, DROPPO I G, et al. Surface properties of sludge and their role in bioflocculation and settleability[J]. Water Research, 2001, 35(2): 339-350. doi: 10.1016/S0043-1354(00)00277-3 [9] WILEN B M, JIN B, LANT P. The influence of key chemical constituents in activated sludge on surface and flocculating properties[J]. Water Research, 2003, 37(9): 2127-2139. doi: 10.1016/S0043-1354(02)00629-2 [10] 于德爽, 李津, 陆婕. MBR工艺处理含盐污水的试验研究[J]. 中国给水排水, 2008, 24(3): 5-8. doi: 10.3321/j.issn:1000-4602.2008.03.002 [11] KU Y, JUNG I L. Photocatalytic reduction of Cr(VI) in aqueous solutions by UV irradiation with the presence of titanium dioxide[J]. Water Research, 2001, 35(1): 135-142. doi: 10.1016/S0043-1354(00)00098-1 [12] 李凌春. 聚丙烯酰胺(PAM)的共聚合成及表征应用[D]. 重庆: 重庆大学, 2008. [13] 顾平, 武耘羽, 刘阳, 等. 废水中除铜研究的最新进展[J]. 工业水处理, 2017, 37(5): 1-5. [14] 党敏. 超滤/纳滤双膜工艺处理南四湖水中试研究[D]. 哈尔滨: 哈尔滨工业大学, 2015. [15] SHENG G P, XU J, LI W H, et al. Quantification of the interactions between Ca2+, Hg2+ and extracellular polymeric substances (EPS) of sludge[J]. Chemosphere, 2013, 93(7): 1436-1441. doi: 10.1016/j.chemosphere.2013.07.076 [16] HONG L, FANG H. Characterization of electrostatic binding sites of extracellular polymers by linear programming analysis of titration data[J]. Biotechnology & Bioengineering, 2010, 80(7): 806-811. [17] SOBECK D C, HIGGINS M J. Examination of three theories for mechanisms of cation-induced bioflocculation[J]. Water Research, 2002, 36(3): 527-538. doi: 10.1016/S0043-1354(01)00254-8 [18] GUIBAUD G, COMTE S, BORDAS F, et al. Comparison of the complexation potential of extracellular polymeric substances (EPS), extracted from activated sludges and produced by pure bacteria strains, for cadmium, lead and nickel[J]. Chemosphere, 2005, 59(5): 629-638. doi: 10.1016/j.chemosphere.2004.10.028 [19] WANG Q, LI J, POON C. Novel recycling of phosphorus-recovered incinerated sewage sludge ash residues by co-pyrolysis with lignin for reductive/sorptive removal of hexavalent chromium from aqueous solutions[J]. Chemosphere, 2021, 285: 131434. doi: 10.1016/j.chemosphere.2021.131434 [20] GAO N, DUAN Y, LI Z, et al. Hydrothermal treatment combined with in-situ mechanical compression for floated oily sludge dewatering[J]. Journal of Hazardous Materials, 2021, 402: 124173. doi: 10.1016/j.jhazmat.2020.124173 [21] JIA H, LIU B, ZHANG X, et al. Effects of ultrasonic treatment on the pyrolysis characteristics and kinetics of waste activated sludge[J]. Environmental Research, 2020, 183: 109250. doi: 10.1016/j.envres.2020.109250 [22] HUANG Y, LI J, ZHANG Y, et al. High-speed particle rotation for coating oil removal by hydrocyclone[J]. Separation and Purification Technology, 2017, 177: 263-271. doi: 10.1016/j.seppur.2016.12.001 [23] ZUBROWSKA-SUDOL M, WALCZAK J. Effects of mechanical disintegration of activated sludge on the activity of nitrifying and denitrifying bacteria and phosphorus accumulating organisms[J]. Water Research, 2014, 61: 200-209. doi: 10.1016/j.watres.2014.05.029 [24] LIU Y, WANG H, XU X, et al. Achieving enhanced denitrification via hydrocyclone treatment on mixed liquor recirculation in the anoxic/aerobic process[J]. Chemosphere, 2017, 189: 206-212. doi: 10.1016/j.chemosphere.2017.09.056 [25] APHA. Standard Methods for the Examination of Water and Wastewater, 21th edition[S]. American Public Health Association, 2005, Washington, DC. [26] LI H, JIN Y, RASOOL B, et al. Effects of ultrasonic disintegration on sludge microbial activity and dewaterability[J]. Journal of Hazardous Materials, 2019, 161: 1421-1426. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4334
- HTML全文浏览数: 4334
- PDF下载数: 65
- 施引文献: 0
