



 下载:
下载:


































 百度学术
百度学术



-
尿液是生活废水中营养成分的主要来源,虽然其体积仅占生活废水总体积的1%,却贡献了其中80%的氮和50%的磷[1]。尿液废水直接排放不但会导致水源污染、水体富营养化,还具有携带病原体、传播传染病的风险[2]。传统的尿处理方式是排入生活废水中通过污水处理厂进行集中处理,这种混合处理方式具有基础设施投资大、运营成本高的缺点,故不适合在居民分散、经济欠发达地区应用[3-4]。因此,对尿液单独分离并进行原位处理的源分离、分散式尿处理技术日益引起业界的关注[5-8]。
电化学氧化技术具有占地面积小、建造成本低、运营需求少等优点,特别适合在分散式旱厕污水、畜禽养殖废水等小型、分散式水处理设施中应用[9]。多项研究证明:电化学氧化方法同时具备脱氮、矿化、消毒功能,被认为是最具有应用前景的尿液处理技术之一[10-12]。HOFFMANN等[13]开发了基于电化学氧化技术的“Eco-san”原位处理厕所系统,尿液废水经过4 h的处理后COD去除率达到88.3%,其出水水质满足冲洗厕所或排放的要求。
有研究[14-15]表明,在电化学氧化尿液过程中,氯离子(Cl−)阳极氧化产生的活性氯(Cl·、
Cl−⋅2 、Cl2、HClO/ClO−等)在氨氮去除、有机物降解以及灭菌过程中发挥了重要作用。然而,氯与有机物之间会发生反应生成消毒副产物(DBPs)。DIBRA等[11]发现,使用硼掺杂金刚石电极(BDD)电化学氧化模拟尿液时,TOC去除效率远高于钌、铱氧化物涂层电极,但采用BDD电极存在将Cl−氧化成无机消毒副产物(ICBPs)的问题,其主要成分为高氯酸盐(ClO−4 )。JASPER等[16]采用铱氧化物涂层电极电化学氧化尿液废水,发现DBPs的主要成分为有机消毒副产物(OCBPs)以及氯酸盐(ClO−3 )。现有研究的重点主要集中在脱氮、脱磷、灭菌以及降低能耗等问题[10-12],虽然有些研究注意到产生DBPs的问题,然而电极反应与尿液中复杂有机成分的降解反应互相耦合,特别是电极性能的差异使上述过程更加复杂[10-11],导致目前对DBPs产生机理的认识尚不充分。由于多种DBPs具有三致效应[17],因此,如何减少和去除DBPs的问题历来受到高度重视[18-20]。然而,作为一种潜在的来源,电化学氧化后的尿液中残留DBPs的问题尚未得到充分关注和认识,给该技术的广泛应用带来了潜在的风险。基于上述原因,本研究从电化学析氯以及氯“催化”降解有机物与去除氨氮的机理入手,重点探究了电化学氧化尿液过程中OCBPs、ICBPs生成与转化的基本规律,并以此为基础进行电化学工艺优化。针对电化学处理后的尿液中残留的DBPs成分主要为有机物的情况,采用适合有机物去除的活性炭吸附方法进行后处理技术研究。本研究结果可为电化学氧化技术在尿处理以及畜禽养殖废水预处理等类似领域的应用提供参考。
全文HTML



-
1)模拟尿液。由于尿液的成分和浓度受个体差异、饮食、代谢等情况的影响较大,为保证不同批次实验结果的可比性,采用根据文献[21]报道的成分和浓度自行配制的模拟尿液进行实验,详见表1。
2)实验试剂。所用试剂均为分析纯,除尿酸购自Sigma-Aldrich公司外,其余均购自国药试剂有限公司。配液用水为超纯水。颗粒活性炭购自国药试剂有限公司,其苯吸附率≥30.0%、碘值为1 000 mg·g−1、灰度为9.8%、抗磨强度≥80.0%、装填密度≥350.0 g·L−1、比表面积≥500 m2·g−1、pH为7.0~9.0。
-
1)电化学氧化。采用单室反应器,通过恒温水浴控制反应温度为(25±2) ℃,工作模式为恒电流、批处理,每批处理量为360 mL。电流密度为25、50、100 mA·cm−2,电解时间分别为22、16、8 h。阳极采用自制的钌铱金属氧化物涂层钛电极(Ti/RuOx-IrOx),有效面积为40 mm×90 mm,阴极采用与阳极同等规格的不锈钢板,阳、阴极间距为3 mm。电解过程中消耗的电量采用施加在单位体积模拟尿液上的比电量来评估,比电量按式(1)计算。
式中:Q为比电量,Ah·L−1;I为电流,A;t为反应时间,h;V为体积,L。
2)微生物灭活。对经过电化学氧化处理、未处理的模拟尿液采用标准方法进行培养并检查微生物的繁殖情况,通过比较微生物繁殖情况来考察灭活效果。样品培养依据GB/T 5750.12-2006《生活饮用水标准检验方法 微生物指标》[22]进行,微生物繁殖情况采用菌落计数法进行评估。
3)活性炭吸附。在电化学处理后的模拟尿液中投加活性炭,以探究OCBPs的吸附去除情况(电化学处理的工艺条件采用通过工艺优化确定的最佳条件)。取若干份电化学氧化处理后的模拟尿液,每份100 mL,分别加入到250 mL棕色具塞锥形瓶中,投加定量活性炭后放置在恒温振荡器上25 ℃、150 r·min−1摇匀,定时采样分析。吸附量和去除率依次按式(2)和式(3)进行计算。
式中:qei为活性炭对成分i的平衡吸附量,mg·g−1;ηe为去除率,%;C0i为成分i的初始质量浓度,mg·L−1;Cei为成分i的平衡质量浓度,mg·L−1;V为溶液体积,L;m为活性炭质量,g。
-
总有机碳(TOC)、氨氮、氯离子采用标准方法测定[23]。游离氯、总氯采用N,N-二乙基-对苯二胺(DPD)/KI分光光度法测定,DPD试剂采用WTW试剂套装(00599,WTW,德国)。
由于尿液中基本不含溴,因此,有机、无机DBPs均不考虑含溴物质,重点研究代表性的有机、无机氯代消毒副产物(OCBPs、ICBPs)。其中,OCBPs包括三氯甲烷(TCM)、一氯乙酸(MCAA)、二氯乙酸(DCAA)及三氯乙酸(TCAA),均采用气相色谱法[24]进行检测,气相色谱仪(Trace 1300,赛默飞,美国)配套的色谱柱型号为TR-FFAP,检测器为电子捕获检测器(ECD)。具体方法为:水样经过酸化、盐析后用甲基叔丁基醚萃取,取1 mL萃取液经过无水硫酸钠脱水后直接进行TCM检测。另取2 mL萃取液经酸化甲醇衍生化后,经饱和碳酸氢钠溶液洗涤、无水硫酸钠脱水后进行上述3种氯乙酸(CAAs)的检测。ICBPs包括
ClO−3 和ClO−4 ,采用离子色谱仪(ICS-5000,戴安,美国)测定,配套的色谱柱型号为IonPac AS23,淋洗液为15 mmol·L−1和90 mmol·L−1 NaOH溶液。
1.1. 试剂与材料
1.2. 实验、评估方法
1.3. 检测分析方法


-
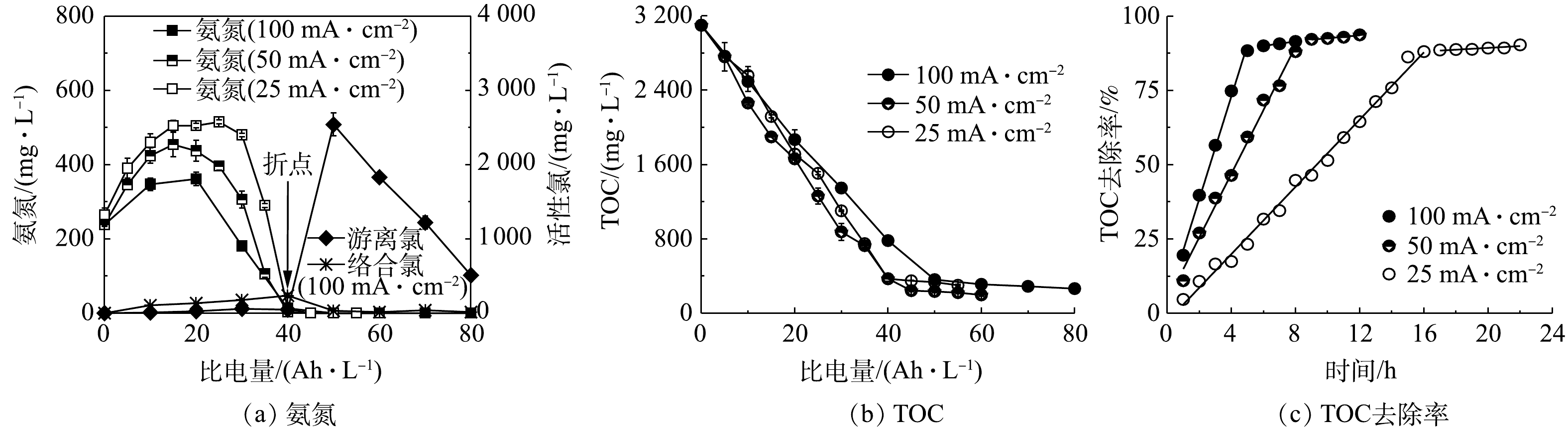
图1为氨氮、TOC质量浓度随时间的变化情况。如图1(a)所示,氨氮浓度呈先升高后降低的变化趋势。这是因为电化学处理前模拟尿液中的氨氮来源于NH4Cl,开始电解后,溶液中的Cl−被电化学氧化为游离氯,尿素与游离氯发生氯代、水解反应,部分转化为
NH+4 [25],其与溶液中原有的NH+4 累加导致了氨氮浓度的升高。当电流密度分别为25、50、100 mA·cm−2时,氨氮的峰值质量浓度分别为515.0、454.0、361.5 mg·L−1,表现为随电流密度升高而降低的趋势。这是因为氨氮浓度同时受NH+4 生成和转化的影响。游离氯优先与尿素反应生成NH+4 ,其次与NH+4 反应“催化”其转化、去除。当电流密度较低时,产生的游离氯较少,用于“催化”NH+4 去除的部分也较少,因此,NH+4 累加效应更明显;当电流密度升高时,游离氯产量增加,其“催化”NH+4 转化的比例增加,导致NH+4 累加量减少。氨氮的浓度在尿素完全分解时达到峰值,继续电解,游离氯全部参与NH+4 去除反应,导致氨氮浓度快速降低,在比电量达到40 Ah·L−1时降低到检测限以下。在反应初期,游离氯与尿素、氨氮反应形成含氯络合物(即络合氯),导致游离氯的质量浓度升高不明显,而络合氯的质量浓度快速升高。络合氯不稳定,通过水解、歧化等反应分解为CO2、N2、
NO−3 等产物,同时游离氯被还原为Cl−。在上述过程中,Cl−通过先被氧化、后被还原,在价态不变的情况下促使有机物、氨氮去除,起到了类似于催化剂的作用。当氨氮被完全降解后,其与游离氯反应生成络合氯的反应结束,游离氯开始析出,导致在氨氮被完全去除的同时出现游离氯浓度由基本不变到快速升高的转折点,该过程与在高氮废水中加入氯气进行脱氮处理的原理相同(简称为“折点加氯法”[26]),上述转折点即为“氯化折点”。由图1(a)可知,达到折点时所消耗的比电量与电流密度无关。如图1(b)所示,在电流密度为25、50、100 mA·cm−2时,溶液中TOC的质量浓度呈连续降低的变化趋势,在折点时,TOC的去除率分别为88.0%、88.0%、74.8%。继续电解,在比电量达到50 Ah·L−1时,TOC的去除率趋于稳定,分别为89.2%、92.5%、88.4%。经过进一步分析发现:TOC去除率与电解时间呈线性关系(图1(c)),说明当采用恒流模式进行电解时,矿化速率保持不变,即TOC降解近似零级反应。其原因为:电化学氧化尿液时,虽然通过直接电极反应和通过游离氯为媒介的间接氧化对有机物降解均有贡献,但间接氧化起主要作用[13]。在氯“催化”反应中[27],阳极析氯是限速步骤。由于Ti/RuOx-IrOx具有良好的析氯活性和选择性,在电流密度25~100 mA·cm−2时,在阳极产生的游离氯浓度随电流密度线性增加。在电流恒定而且有机物过量时,游离氯的生成速率不变,使得TOC降解也近似匀速,表现为零级反应。MINEO等采用铂铱电极电化学氧化尿液废水进行脱氮研究时也发现了类似的变化规律[28]。
-
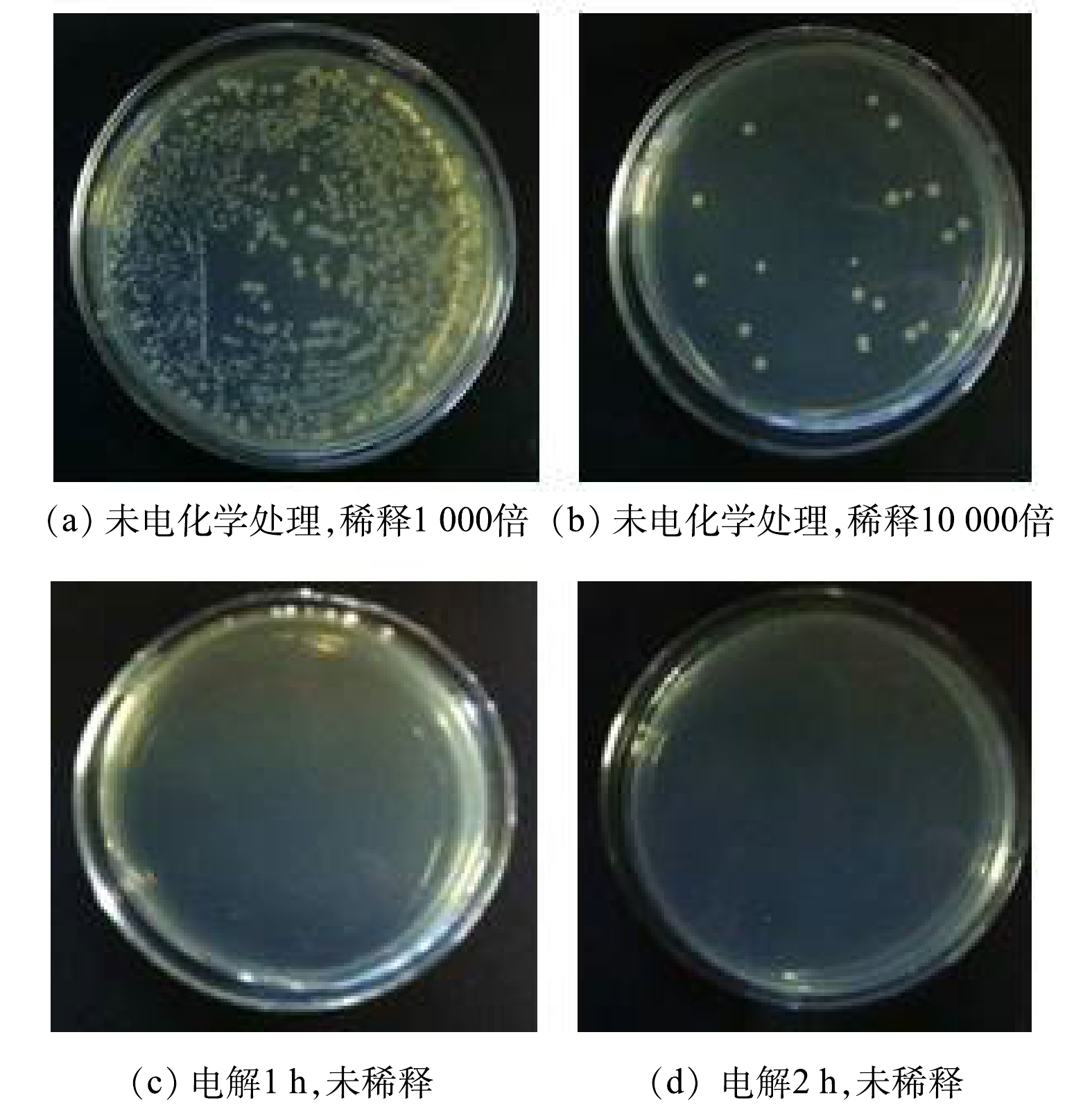
如图2(a)、图2(b)所示,未电化学处理的模拟尿液,按标准方法培养后菌落总数达到2×105 CFU·mL−1,说明由于尿液中含有丰富的营养物质而且具有良好的生物相容性,排入环境后其中的微生物会大量繁殖。如果尿液被大肠杆菌、血吸虫卵等病原体污染就会造成传播腹泻、血吸虫等疾病的风险[29]。在电流密度为100 mA·cm−2时,通过1 h的电化学处理后,模拟尿液中总氯(包括游离氯和络合氯)的质量浓度达到115.0 mg·L−1,采用同样的方法检测,未发现微生物滋生,这是因为游离氯、络合氯均具有高效、广谱的杀菌效果。MINEO等[30]的研究结果表明,当溶液中总氯的质量浓度保持在20 mg·L−1以上时,能够使微生物彻底灭活。在折点时,尿液中总氯的质量浓度达到284.5 mg·L−1,能够达到可靠灭菌的效果。
-
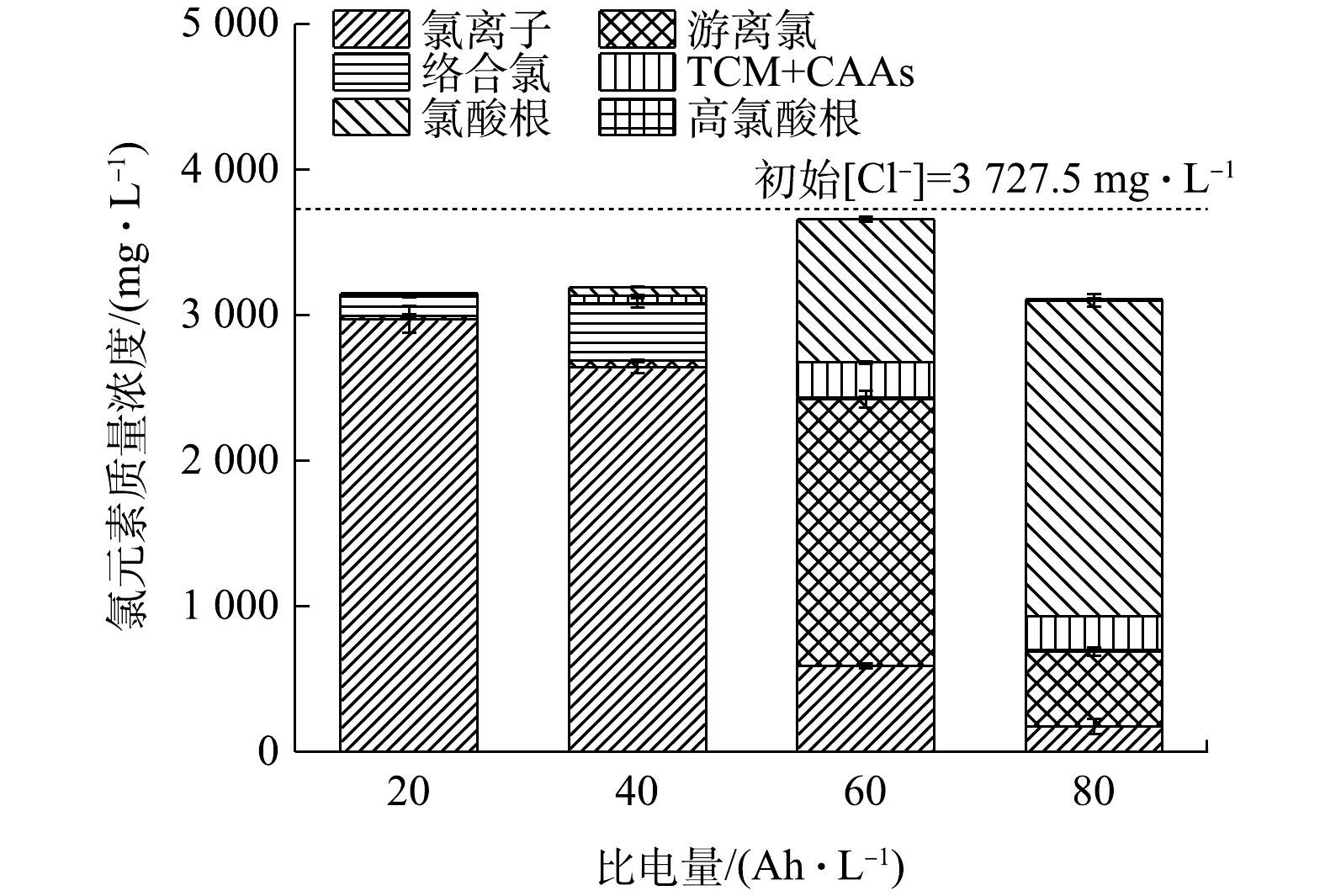
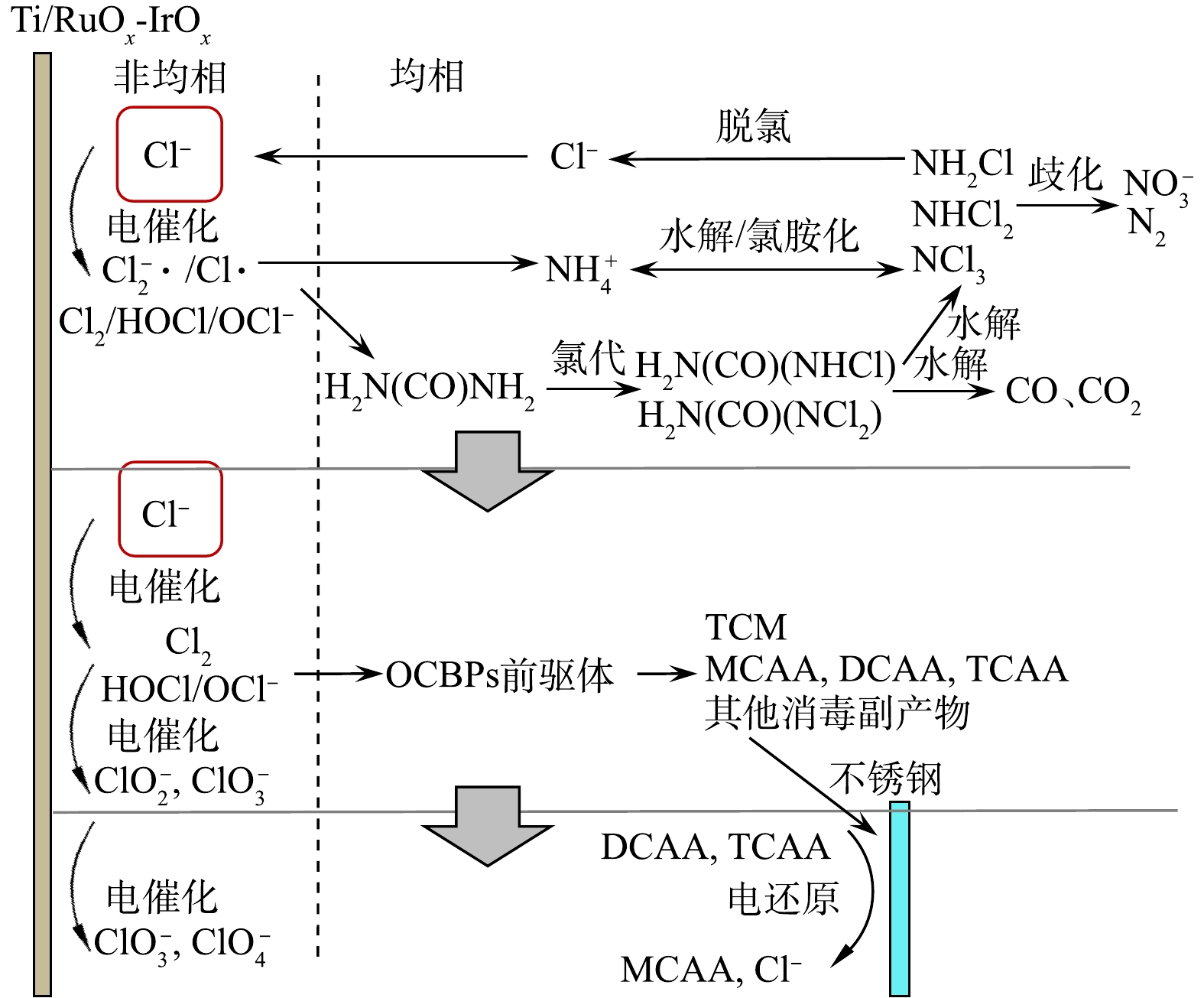
图3为电化学氧化处理模拟尿液过程中阳极析氯和氯元素的转化情况,图4为根据析氯和氯元素转化规律得出的氯“催化”尿素、氨氮降解以及OCBPs、ICBPs生成、转化的历程。根据体系中发生的主要反应,可将反应历程大致分为3个阶段。
第1阶段(比电量为0~40 Ah·L−1),被电化学氧化的Cl−生成了游离氯,后者分别与尿素、
NH+4 发生氯代和氯胺化反应,通过氯代(氯胺化)、水解、歧化系列反应,实现了尿素的矿化和NH+4 的去除[27]。在本阶段,溶液中氯元素的主要存在形式仍然是Cl−。这是因为:虽然Cl−在阳极被电化学氧化生成了游离氯,但其与尿素、NH+4 反应,又被还原为Cl−。溶液中络合氯的浓度有所升高,在比电量为40 Ah·L−1时,其含有的氯元素质量占溶液中全部氯元素质量的11.0%。这是因为络合氯是尿素、氨氮降解过程的中间产物,在分解为小分子产物前有一定的积累。OCBPs总浓度保持在较低水平,这是因为大部分OCBPs是活性前驱体与游离氯反应的产物[31]。本阶段游离氯更容易与浓度高、易分解的尿素、NH+4 等小分子反应,而这些物质并不是OCBPs的活性前驱体,因此,产生的OCBPs较少。在比电量为40 Ah·L−1时,在溶液中检测到微量的ClO−3 (其含有的氯元素质量占溶液中全部氯元素质量的1.5%)、未检测到ClO−4 。其原因为:ClO−3 和ClO−4 是游离氯被电化学氧化的产物。在折点之前,溶液中游离氯的浓度较低,导致其与电极接触进而被氧化为ClO−3 、ClO−4 的部分较少[14]。第2阶段(比电量为40~60 Ah·L−1),随着尿素、氨氮完全降解,游离氯不再被还原为Cl−,导致析氯成为本阶段的主要反应。在比电量为50 Ah·L−1时,游离氯的质量浓度达到峰值(图1(a)),其对溶液中全部氯元素的质量占比为68.1%。随着游离氯浓度升高,其与尿酸、肌酸等成分的接触增加,这些大分子有机酸具有较高的OCBPs生成势[16,32],与游离氯反应产生了大量的TCM以及CAAs类物质。在比电量为70 Ah·L−1时,TCM、MCAA、DCAA、TCAA的质量浓度之和达到峰值,此时上述4种物质含有的氯元素的质量之和占溶液中全部氯元素质量的6.0%。溶液中
ClO−3 的质量浓度开始增加,在比电量为60 Ah·L−1时,其含有的氯元素质量达到氯元素总质量的26.4%。出现上述现象的原因为:Ti/RuOx-IrOx电极能够将游离氯电化学氧化为ClO−3 ,随着溶液中游离氯浓度的升高,有更多的游离氯被氧化为ClO−3 。第3阶段(比电量>60 Ah·L−1),溶液中游离氯的质量浓度达到峰值后快速降低,而
ClO−3 的质量浓度随之升高。上述现象说明溶液中发生的主要反应为游离氯被氧化,生成了ClO−3 。当比电量为80 Ah·L−1时,ClO−3 中含有的氯元素质量占溶液中全部氯元素质量的58.2%,仅检测到微量的ClO−4 (其含有的氯元素质量占溶液中全部氯元素质量的0.3%)。这是由于Ti/RuOx-IrOx为活性电极,氧化能力较弱[12],难以将ClO−3 进一步氧化为ClO−4 。TCM、MCAA、DCAA、TCAA含有的氯元素质量之和有所减少,其主要原因是TCM的挥发以及DCAA、TCAA的电化学脱氯作用[32]。 -
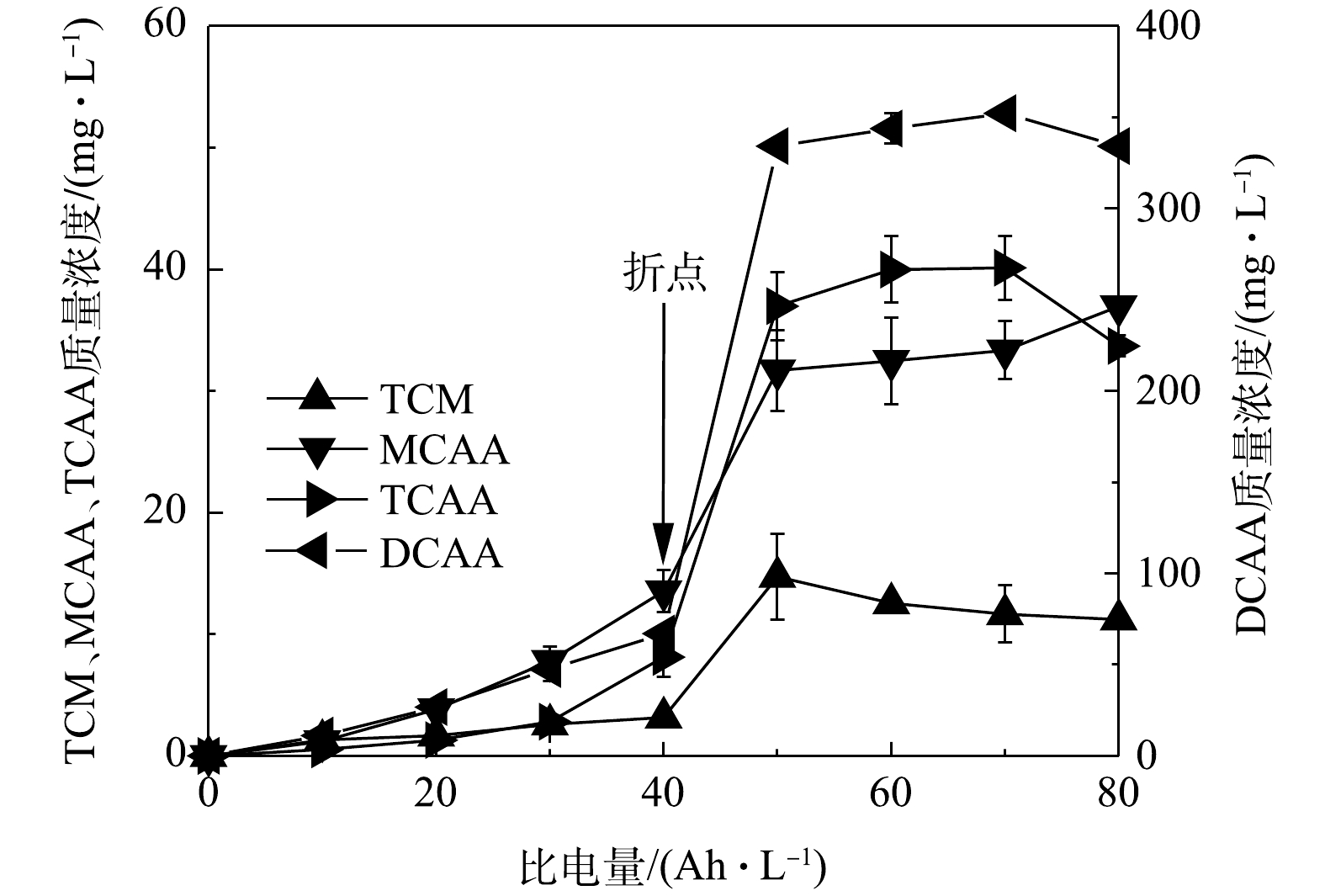
1) OCBPs的质量浓度随电解时间的变化情况。如图5所示,在电流密度为100 mA·cm−2时,TCM、MCAA、DCAA及TCAA的质量浓度均在折点时出现突变,呈显著升高的趋势,说明OCBPs的生成与游离氯浓度密切相关[29]。在折点之前,游离氯被尿素、
NH+4 氯代反应消耗,其在溶液中的浓度保持在较低水平,从而抑制了OCBPs的产生;在折点之后,游离氯浓度快速升高并与尿酸、肌酸等大分子充分反应,生成大量TCM、CAAs等OCBPs成分[33-34]。其中,TCM的质量浓度在比电量为50 Ah·L−1时达到峰值,由折点时的3.17 mg·L−1升高到14.73 mg·L−1;此后,TCM的质量浓度持续降低。吹脱实验(不电解)结果表明,导致TCM质量浓度降低的主要原因是该物质的挥发作用。DCAA是3种CAAs中的主要成分,其质量浓度在折点时为67.08 mg·L−1,在比电量为70 Ah·L−1时达到最大值,为352.19 mg·L−1。MCAA与TCAA的质量浓度接近,在比电量为70 Ah·L−1时分别为36.95 mg·L−1和40.13 mg·L−1。此后继续电解,DCAA、TCAA浓度有缓慢下降趋势,而MCAA的浓度略有增加。这是因为:DCAA、TCAA可以在阴极发生脱氯反应,转化为MCAA,但MCAA不能被电化学分解[32]。对比类似的研究,在折点时,电化学处理后的模拟尿液中TCM和CAAs的质量浓度为电化学处理厕所废水中相应成分的5~10倍[16]。出现这种情况的原因是:厕所废水中的有机物被冲洗水稀释,导致TCM与CAAs的前驱体浓度降低,从而减少了TCM和CAAs的生成量。如果采用相同的倍数对电化学氧化处理的模拟尿液进行稀释,其TCM和CAAs的质量浓度与电化学氧化厕所废水中相应成分的质量浓度相当。
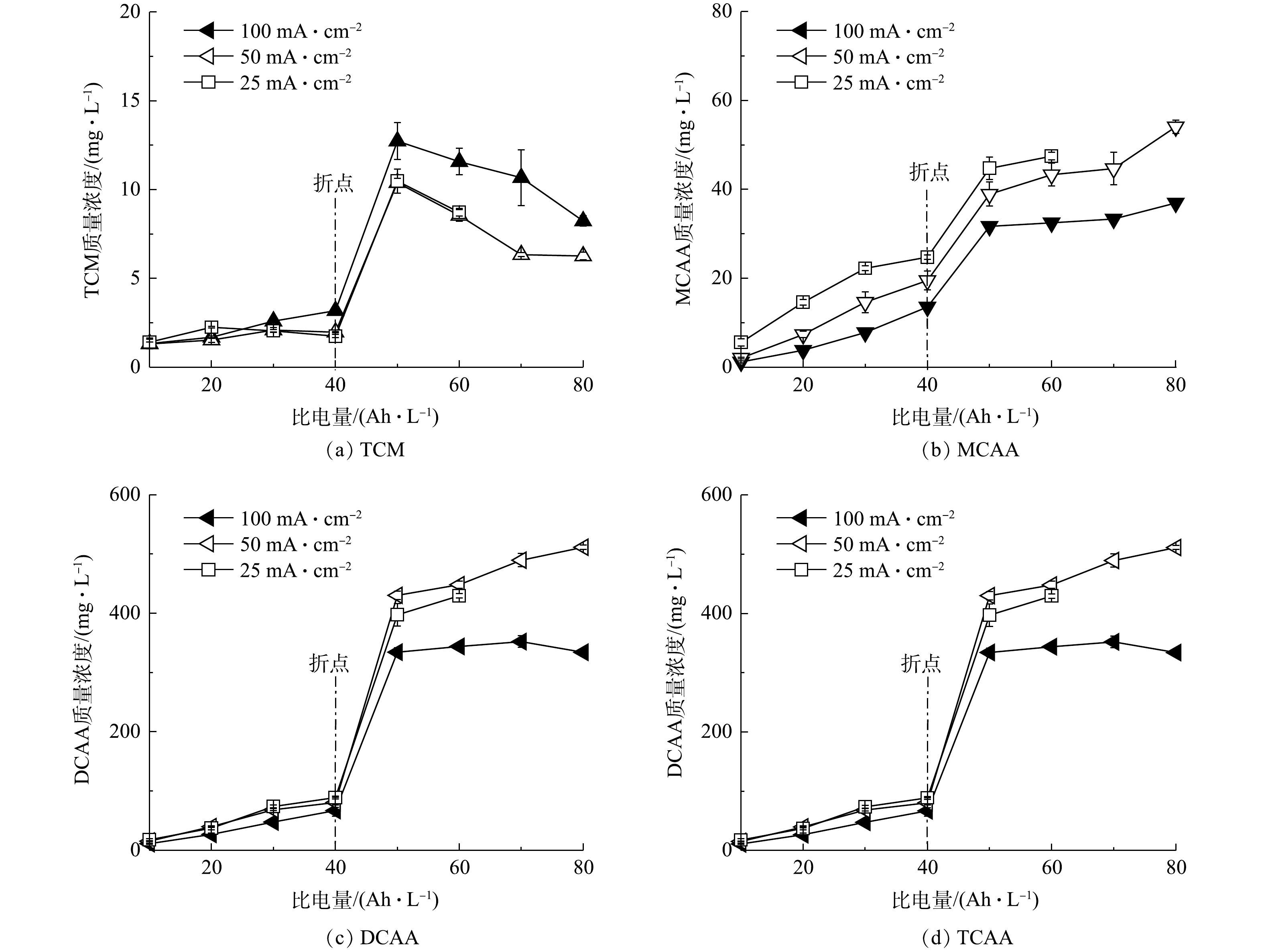
2)电流密度对OCBPs浓度的影响。如图6所示,当电流密度分别为25、50、100 mA·cm−2时,在折点之前,TCM的浓度受电流密度的影响不明显,均保持在较低水平(图6(a))。其原因是:TCM的生成速率主要受游离氯浓度的影响[31],在折点之前,无论电流密度高低,溶液中游离氯的浓度均比较低,因此,TCM的生成速率也比较低。在折点之后,溶液中TCM的浓度随电流密度提高而增加。这是因为,影响TCM浓度的因素包括2方面:一是游离氯与前驱体反应生成TCM,导致其浓度升高;二是TCM在N2、CO2等气体产物的吹脱作用下出现挥发,导致其浓度降低。当比电量相同时,电流密度较低时吹脱时间较长,导致TCM挥发、减少的作用更加突出。
对于非挥发性的CAAs类物质,MCAA的质量浓度在电解过程中呈连续上升的趋势,尤其是在折点之前,游离氯浓度较低的情况下,其质量浓度仍然连续升高。这说明MCAA不仅是前驱体与游离氯反应的产物,也是氯代尿素、氯胺降解的副产物,其生成机理与DCAA、TCAA的生成机理有差异[33]。MCAA的质量浓度在整个电解过程中随电流密度的提高而减小(图6(b)),其原因是电流密度较大时尿素与
NH+4 降解更快,导致MCAA的积累量较少。DCAA与TCAA质量浓度的变化规律类似(图6(c)、图6(d))。在折点之前,受电流密度影响不大,其质量浓度均保持在较低水平,说明DCAA和TCAA的生成速率主要受游离氯浓度的影响[31]。在折点之后,DCAA、TCAA的质量浓度均随电流密度提高而减少。这是因为DCAA和TCAA能够在阴极发生电化学还原脱氯反应,而且电流密度越高,反应速度越快[32]。在折点时,DCAA、TCAA的浓度受游离氯浓度升高、电化学脱氯的共同影响,随电流密度变化的规律有所不同,当电流密度为25、50、100 mA·cm−2时,DCAA的质量浓度分别为88.96、80.72、67.08 mg·L−1,TCAA的质量浓度分别为5.28、7.30、8.13 mg·L−1,说明采用较高的电流密度有利于减少折点时溶液中DCAA和TCAA的总量。
-
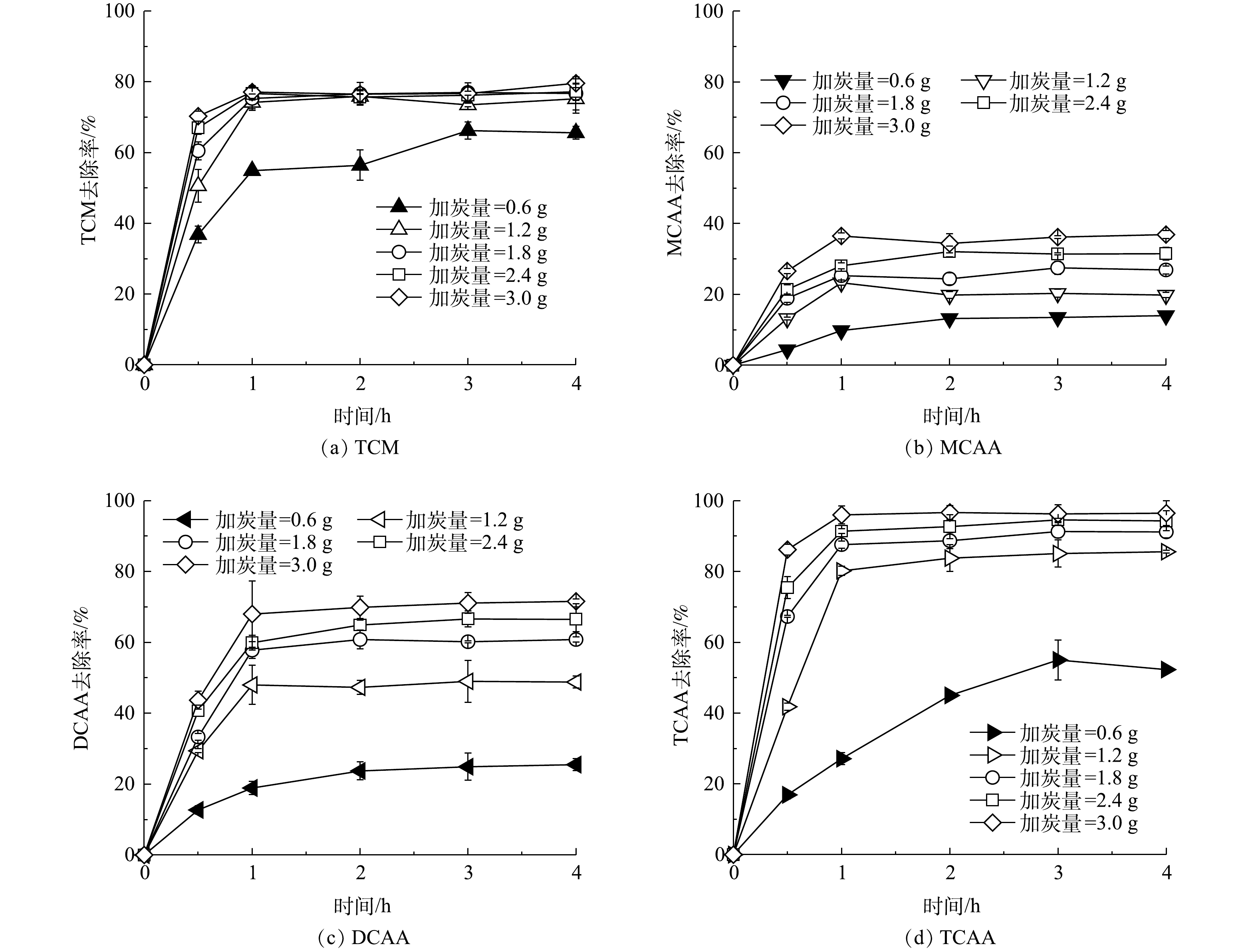
如图7所示,当投加活性炭的质量为0.6~3.0 g时,TCM、MCAA、DCAA及TCAA均在投加活性炭3 h后达到吸附平衡。在投加活性炭前,上述4种成分在溶液中的质量浓度分别为3.48、13.55、67.08、8.13 mg·L−1,当加炭量为1.2 g时,该4种成分的去除率分别为75.2%、19.9%、48.8%、85.6%;当活性炭投加量增加到3.0 g时,其去除率分别增加到79.6%、36.9%、71.6%、96.5%。上述结果表明,活性炭对主要污染物DCAA、TCAA具有较好的吸附效果。优先吸附TCAA,其次为DCAA。上述结果与叶必雄等的研究结果一致[35]。出现这种现象的原因是,活性炭为非极性吸附剂,易吸附弱极性成分。在CAAs分子中,氯原子的强吸电子效应使乙酸根的负电荷得到分散,导致其极性减弱,由于引入的氯原子越多,该物质的极性越弱,因此,TCAA比DCAA更容易被活性炭吸附[36]。
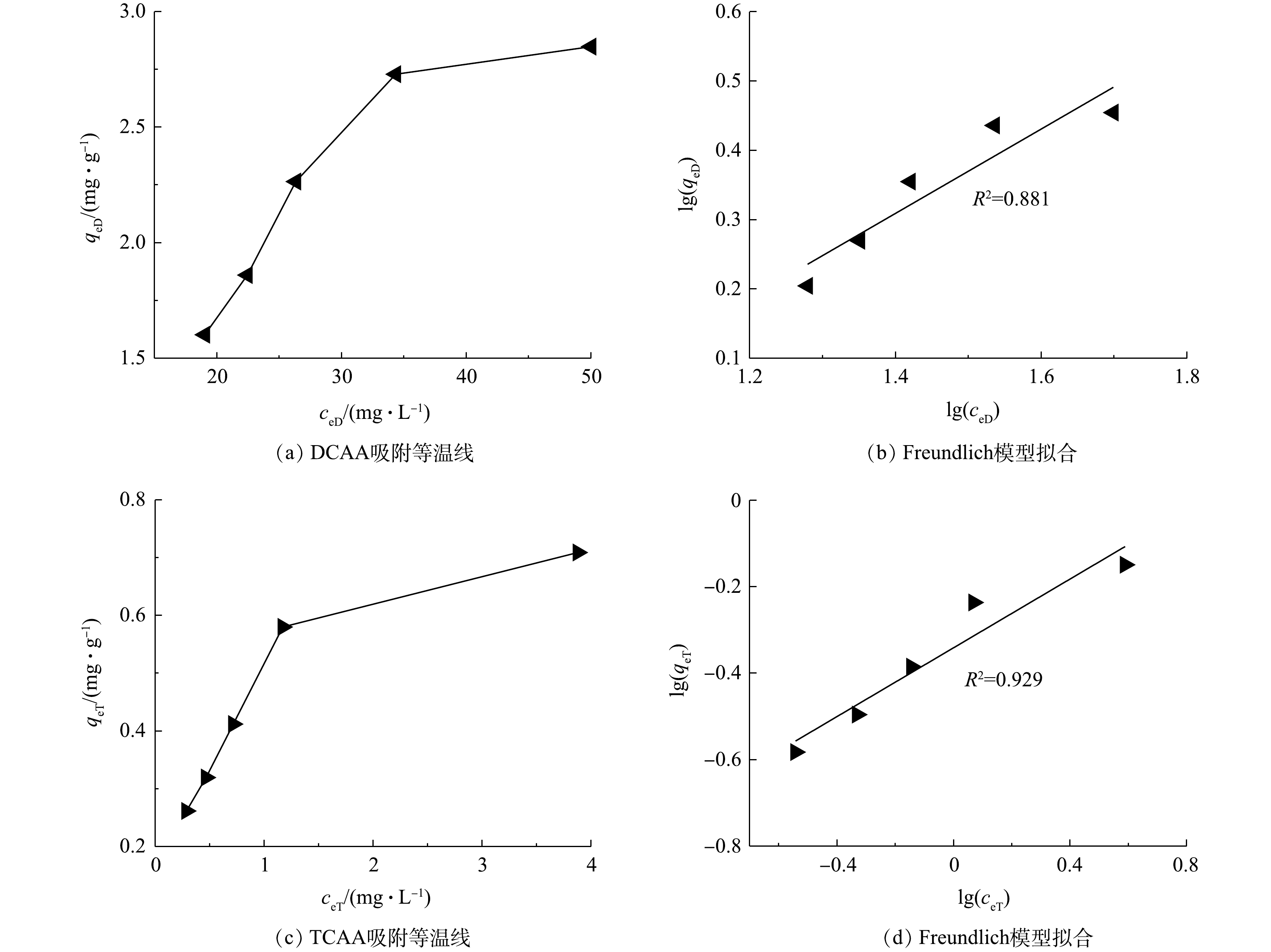
图8为DCAA、TCAA的吸附等温线。如图8所示,当活性炭的投加量为0.6~3.0 g时,DCAA的平衡浓度ceD为19.03~50.00 mg·L−1,其平衡吸附量qeD为1.60~2.85 mg·g−1(图8(a));TCAA的平衡浓度ceT为0.29~3.88 mg·L−1,其平衡吸附量qeT为0.26~0.71 mg·g−1(图8(c))。上述结果表明,在电化学处理的模拟尿液中,DCAA的平衡吸附量高于TCAA,这与DCAA、TCAA单组分吸附实验的结果不同[37]。出现这种现象的原因是,在电化学处理后的模拟尿液中存在多种有机成分,这些成分同时被吸附时会对活性位点产生竞争效应,导致各种组分的平衡吸附量均低于单组分吸附过程。沈忠等[38]的研究结果表明,对于多组分吸附的情况,每种组分的平衡吸附量近似与其平衡浓度成正比,亦即平衡浓度越高,该组分的平衡吸附量越大。在电化学处理的模拟尿液中,DCAA的平衡浓度为TCAA的数十倍,导致DCAA的平衡吸附量也高于TCAA。
采用Langmuir、Freundlich等模型对DCAA、TCAA的吸附特性进行分析,均未取得很好的拟合结果(图8(b)、图8(d))。其原因同样为多种吸附质互相干扰,导致各组分的吸附特性偏离了该物质的单组分等温吸附规律。
2.1. 氨氮去除与有机物降解











2.2. 微生物灭活
2.3. 析氯和氯元素转化规律



















2.4. OCBPs生成的控制


2.5. 活性炭吸附OCBPs的效果
-
1)采用Ti/RuOx-IrOx电极电化学氧化模拟尿液,在折点之前,阳极产生的游离氯优先与尿素、氨氮等小分子反应,阻止了游离氯与OCBPs的前驱体接触(生成OCBPs)以及其被进一步电化学氧化(生成ICBPs),从而抑制了OCBPs、ICBPs的形成;在折点之后,小分子被完全降解,对游离氯的竞争反应结束,导致其浓度快速升高,OCBPs、ICBPs的浓度也随之升高。
2)通过优化工艺能够有效减少电化学氧化处理的尿液中残留的DBPs。优化后的工艺条件为:电流密度为100 mA·cm−2,处理时间为4 h(在折点时结束电解)。此时尿液中的氨氮完全去除、TOC去除率为74.8%、微生物完全灭活,残留的主要DBPs成分为OCBPs,其浓度约为峰值浓度的20%,基本不含ICBPs。
3)活性炭对电化学氧化处理后尿液中残留的OCBPs具有较好的吸附效果。在加炭量为30 g·L−1时,OCBPs中主要成分DCAA、TCAA的去除率分别为71.6%、96.5%,这为减少电化学处理的尿液中残留的DBPs、控制排放风险提供了可行的方案。
