


 下载:
下载:



-
抗生素被广泛的应用于医疗、畜牧业、农作物养殖等方面。抗生素的大量使用会导致其在环境中积累,并在城市污水处理厂中被检测到[1-2]。由于抗生素具有生物毒性,故其对生物降解具有很高的抗性[3]。这导致含有抗生素的医疗废水、生活废水等排放至污水处理厂却无法得到高效的去除,危害着生态以及人类健康[4-6]。
光催化技术可用于抗生素的强化去除,但也存在降解不彻底、反应不易控制、能耗较高等缺点[7]。因此,在难降解废水处理中,常将光催化作为前处理,提高废水可生化性,并与生物法耦合,提高污染物的生化降解效率。LI等[8]将TiO2负载在改进的多孔载体上,以提高三氯苯酚的降解,三氯苯酚通过吸附、光降解、光催化和生物降解4个途径得到去除。ZHOU等[9]通过将Er3+:YAlO3/TiO2负载到多孔载体上,将光催化与生物降解耦合,提高了含酚废水的降解效率。DING等[10]选择紫外光催化对含有抗生素的废水进行预处理,发现光催化有助于分解抗生素中的主要官能团以消除其抗菌活性,有利于后续生物处理。FU等[11]成功将TiO2附着在载体表面并通过光催化协同去除水体中的异味物质2-甲基异冰片和土臭素,发现该系统能保持较好的去除效率和稳定性。然而,目前该领域仍然存在一些待解决的问题。例如,光催化剂在载体表面的高效附着、光催化对生物的毒性作用、光催化与生物降解耦合后对污染物的去除途径和机理等。
好氧颗粒污泥(aerobic granular sludge, AGS)是由微生物、胞外聚合物(extracellular polymeric substances, EPS)、无机盐等聚集而成的生物聚集体。其内部环境相对稳定、菌落丰富,在处理难降解废水时具有一定的优势。HUANG等[12]利用AGS与光催化相结合处理染料废水,染料通过AGS吸附、脱附后在自净洗脱液中光解。鉴于AGS较强的吸附能力,且其紧凑的颗粒结构能为内部微生物提供很高的保护,将纳米二氧化钛(TiO2 P25)负载到AGS表面制备生物纳米材料,通过表面光催化与内部生物降解耦合,有望强化抗生素的降解。本研究综合考虑吸附动力学和负载TiO2对AGS内生物活性等方面的影响,确定了最佳TiO2负载量;考察了该生物纳米材料对磺胺类抗生素磺胺嘧啶(sulfadiazine, SDZ)的降解性能,并通过检测磺胺嘧啶及中间产物,确定了降解途径。
全文HTML
-
实验的接种污泥取自污水处理厂二沉池,在序批式反应器(sequencing batch reactor, SBR)中以模拟废水培养AGS。SBR主体为内径8 cm、高100 cm的有机玻璃,工作体积为4 L。反应周期为4 h,包括进水12 min、曝气210 min、沉淀加出水18 min,体积交换率为50%。曝气阶段空气通过曝气泵从底部微孔曝气头通入,并用气体转子流量计维持气体流速在0.25~0.30 m3·h−1。
SBR中初始混合液污泥质量浓度(mixed liquor suspended solid, MLSS)约为5 g·L−1。模拟废水的具体组分如下:641 mg·L−1 CH3COONa、469 mg·L−1 C6H12O6、191.1 mg·L−1 NH4Cl、23 mg·L−1 KH2PO4、60 mg·L−1 CaCl2、300 mg·L−1 NaHCO3、24 mg·L−1 MgSO4·7H2O、21 mg·L−1 FeSO4·7H2O、20 mg·L−1 EDTA、1.6 mg·L−1 FeCl3·6H2O以及0.5 mL·L−1微量元素。其中微量元素包括240 mg·L−1 MnCl3·4H2O、300 mg·L−1 Na2MoO4·2H2O、60 mg·L−1 CuSO4·5H2O、300 mg·L−1 H3BO3、60 mg·L−1 KI、240 mg·L−1 ZnCl2和116 mg·L−1 CoCl2·6H2O。模拟废水中碳氮比为100:5,NaHCO3将进水pH控制在7.1~7.9。反应器在室温下(约为25 ℃)运行长达160 d,以使污泥达到性能稳定。培养后的AGS粒径在1~5 mm,MLSS约为10 g·L−1,混合液挥发性悬浮固体质量浓度(mixed liquor volatile suspended solids, MLVSS)与MLSS的比值在0.8右,污泥体积指数(sludge volume index, SVI)为37 mL·g−1,COD以及
NH+4 -N的去除率均达97%以上。 -
选择TiO2 P25为光催化剂,在4个锥形瓶中分别加入MLSS约为1.42 g·L−1的AGS。初始TiO2质量浓度分别为10、20、50、100 mg·L−1。以150 r·min−1在30 ℃进行AGS上TiO2的吸附动力学实验。吸附动力学实验完成后,将污泥用去离子水清洗后恢复至原体积,在30 ℃下以150 r·min−1振荡进行24 h的脱附实验并检测水相中的TiO2。
通过吸附动力学分析吸附机理,采用伪一级动力学方程(式(1))、伪二级动力学方程(式(2))分别对不同质量浓度TiO2的吸附实验数据进行拟合[13]。
式中:t为时间,h;k1为伪一级动力学吸附速率常数,g·(mg·h)−1;k2为伪二级动力学吸附速率常数,g·(mg·h)−1;qe和qt分别为平衡状态和t时的吸附容量,mg·g−1。
-
为了探究光照条件对附着TiO2后的生物纳米材料性能的影响,并考察其对SDZ的降解性能和途径,进行了一系列批实验。实验所用废水初始COD值为500 mg·L−1,
NH+4 -N质量浓度为35 mg·L−1,并按照SBR内污泥培养条件加入其他元素组分,MLSS为5 g·L−1。反应器上方布置紫外灯,功率为10 W,波长254 nm。在100 mL的烧杯中,研究不同光照条件对生物纳米材料微生物活性的影响。设置不同光照模式:(I)紫外光照射0 h+可见光照射4 h;(II)紫外光照射1 h+可见光照射3 h;(III)紫外光照射2 h+可见光照射2 h。设置不同紫外光光照时间:分别设置0、1、2 h 的紫外光光照时间进行实验。通过调节紫外光光源距离调整光照强度,分别为0、0.3、0.45 mW·cm−2。
在工作体积为1 L的有机玻璃反应器内探究SDZ的降解途径以及降解产物。选用没有被TiO2负载的原始AGS和以20 mg·L−1 TiO2负载的AGS (T-AGS),进行8组10 h的批实验,分别为:AGS在紫外光下运行为A组;AGS在可见光下运行为B组;T-AGS在紫外光下运行为C组;T-AGS在可见光下运行为D组;不添加AGS和T-AGS在紫外光下运行为E组;不添加AGS和T-AGS在可见光下运行为F组;AGS加NaN3可见光下运行为G组;T-AGS加NaN3可见光下运行为H组。
-
COD、
NH+4 -N、MLSS和MLVSS均采用标准方法[14]测定。扫描电镜根据参考方法[15]制备样品。EPS采用加热提取法[16]并采用Lowry法[17]和苯酚-硫酸法[18]对样品进行测定。TiO2的浓度采用紫外分光光度法进行测定[19]。死活细胞分析利用激光共聚焦显微镜进行观察[20]。污泥比耗氧速率(specific oxygen uptake rate, SOUR)的测定参考LIU等[21]的方法。SDZ及中间产物使用高效液相色谱(HPLC, Shimadzu, LC-20AT)测定,流动相使用体积比为25∶75的乙腈和超纯水溶液,并用乙酸调节pH至4.0,流速1 mL·min−1,检测波长269 nm。
1.1. AGS的培养

1.2. 吸附实验及脱附实验
1.3. 反应装置的运行

1.4. 分析方法

-
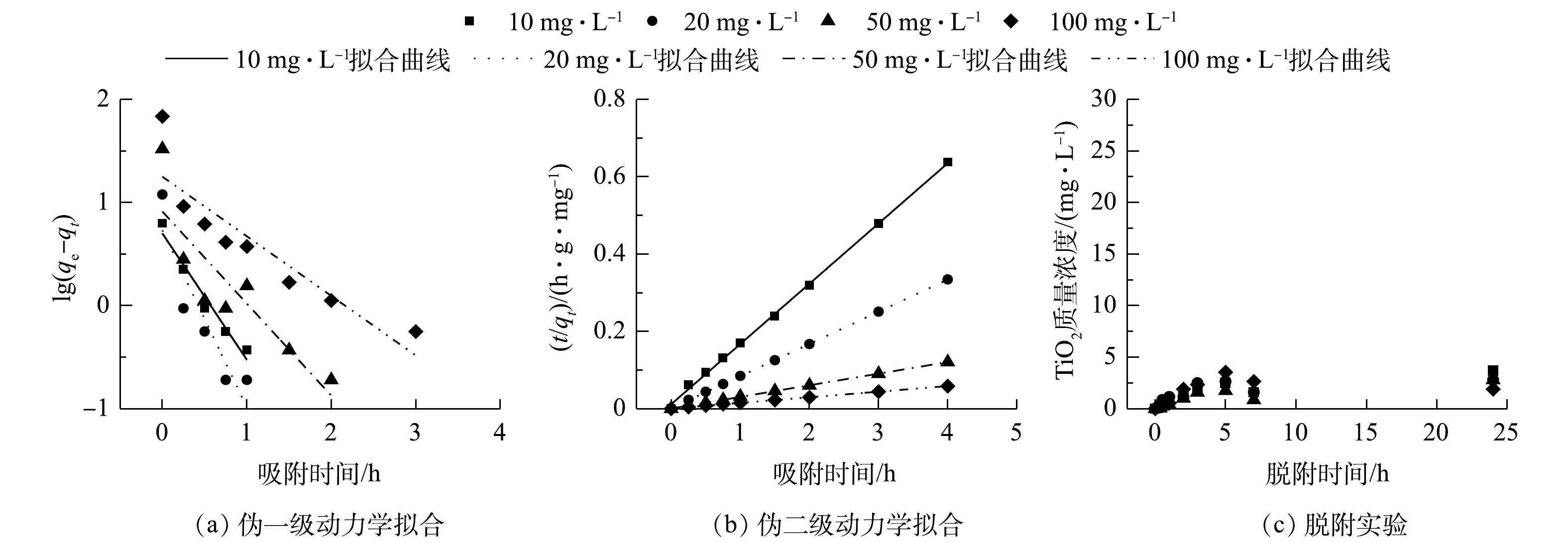
对AGS吸附负载TiO2的过程进行了动力学拟合。伪一级动力学拟合结果分别以t和lg(qe−qt)为横、纵坐标作图,计算30 ℃下不同TiO2初始浓度下的k1和qe。结果如图1(a)和表1所示。伪一级动力学方程的拟合情况较差,可决系数低;伪二级动力学拟合中不同浓度TiO2吸附实验对应的R2均大于0.99,远高于伪一级模型的拟合结果。考虑到模型拟合和实验观察到的吸附能力之间的良好一致性以及较高的可决系数,说明TiO2在AGS上的吸附反应较符合伪二级动力学模型。该动力学模型包括表面吸附、颗粒内扩散和液膜扩散等吸附过程,能够全面地反映AGS吸附TiO2的动力学过程机制。在最初的0.5 h内,TiO2的吸附速率很快,随着反应的进行,吸附位点也越来越少,随后达到吸附平衡。投加量为20 mg·L−1的TiO2拥有最快的伪二级动力学吸附速率。
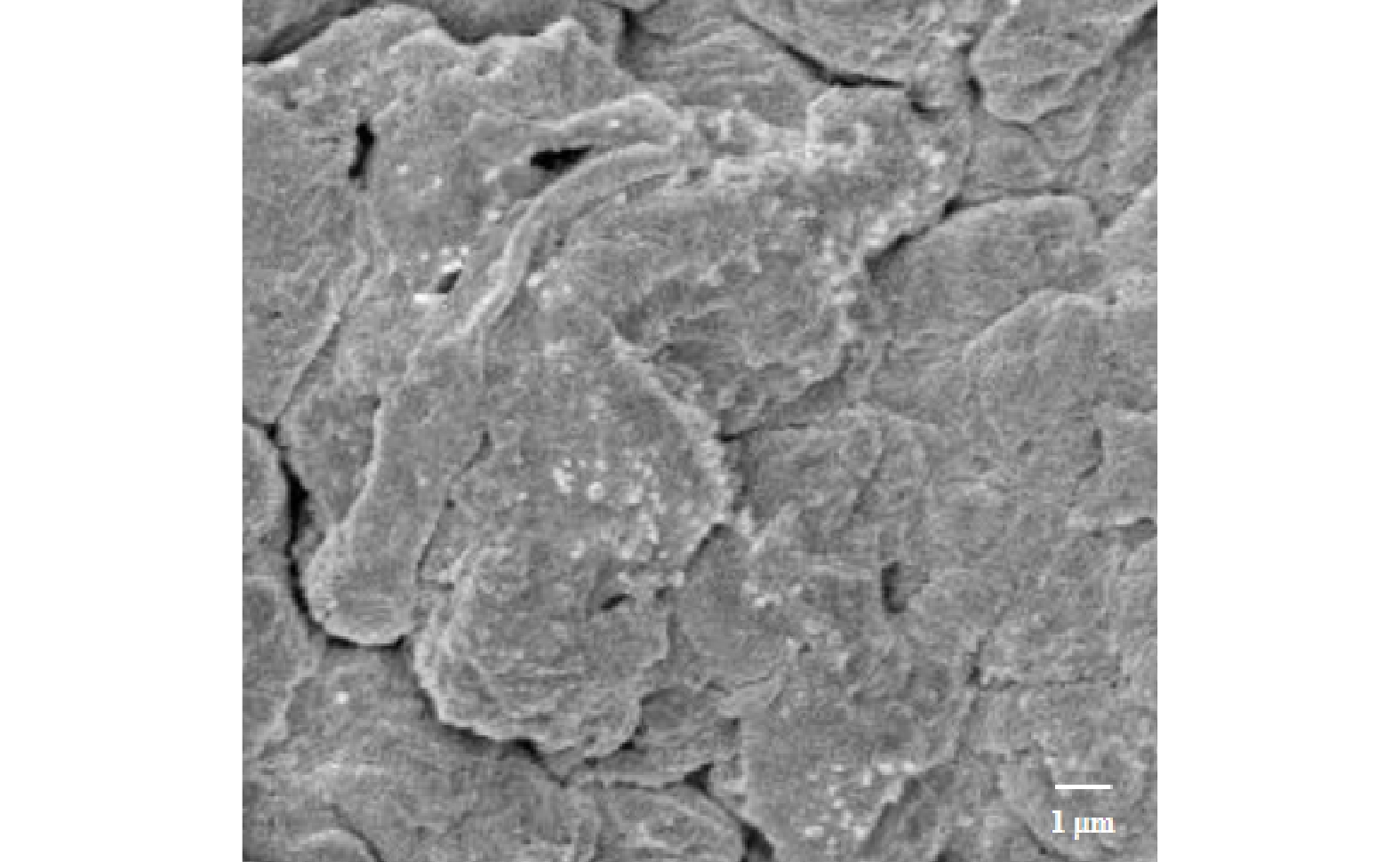
为了研究负载后的TiO2是否会随着反应的运行而从AGS上脱附,从而影响后续实验,故开展了24 h的脱附实验。由图1(c)可知,负载10、20、50、100 mg·L−1 TiO2的AGS在24 h振荡后几乎没有发生TiO2脱附,脱附反应可以忽略不计。这说明本实验制备的生物纳米材料可以在反应器中长期运行。对负载20 mg·L−1 TiO2后的AGS进行SEM分析。由图2可以看出,TiO2在AGS的表面比较清晰,大部分呈球形颗粒。有些模糊的部分可能是因为被颗粒表面EPS所包裹所致。
-
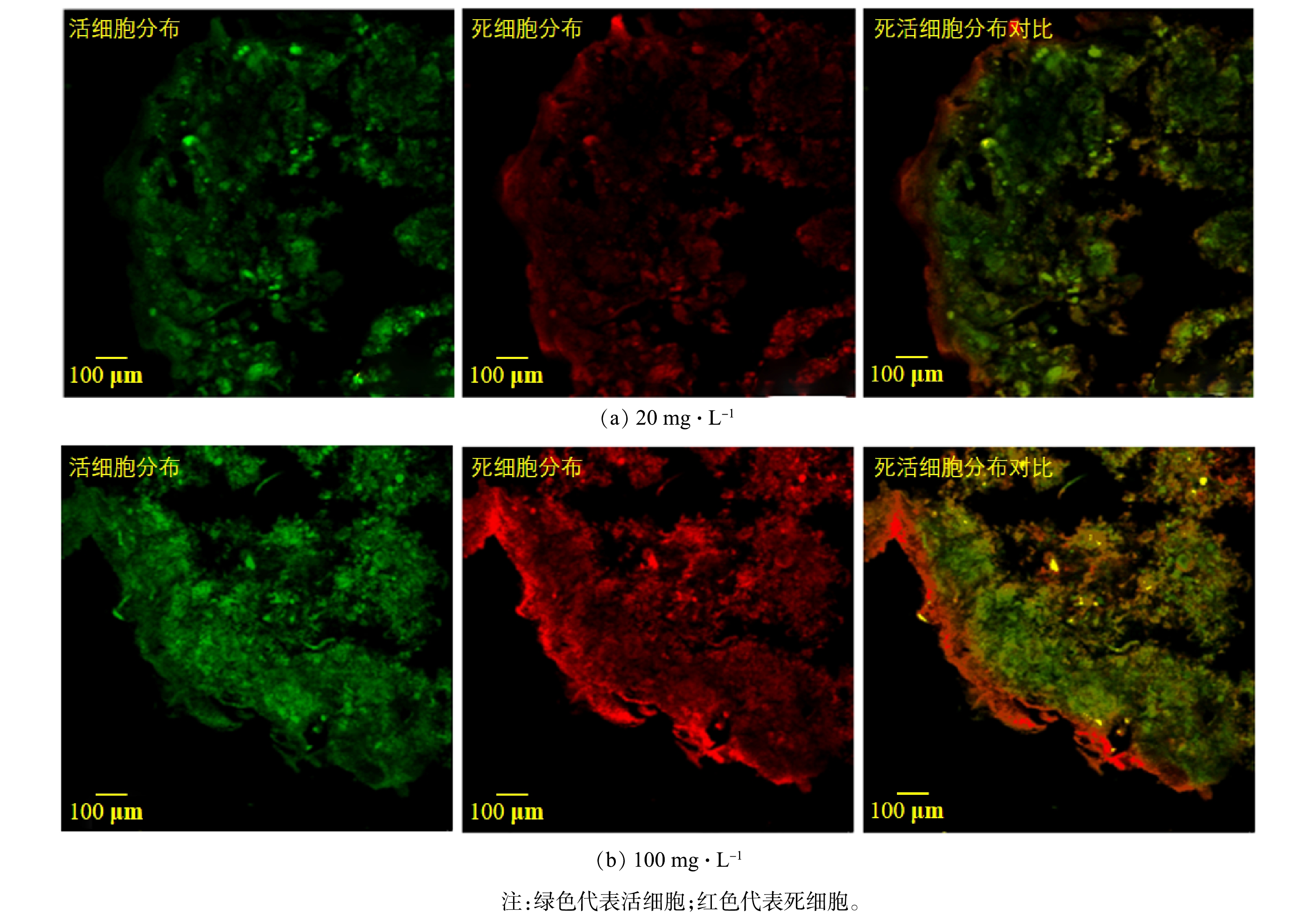
为了观察负载的TiO2对AGS内细胞生物的影响,对AGS吸附TiO2 12 h后切片观察死活细胞分布。图3(a)和图3(b)分别是在20 mg·L−1和100 mg·L−1 TiO2条件下制备的生物纳米材料,其中绿色代表活细胞,红色代表死细胞。结果表明,AGS最外侧的细菌受到严重影响,这可能是由于纳米颗粒引起微生物的氧化损伤,从而导致细胞膜失去完整性[22]。前述吸附动力学研究结果表明,TiO2在AGS上的吸附包括颗粒内部扩散,所以AGS内部的微生物也会受到影响[20]。与20 mg·L−1相比,在负载100 mg·L−1 TiO2的AGS内部微生物受到的伤害成都明显增加,颗粒内外大量细胞已经死亡。
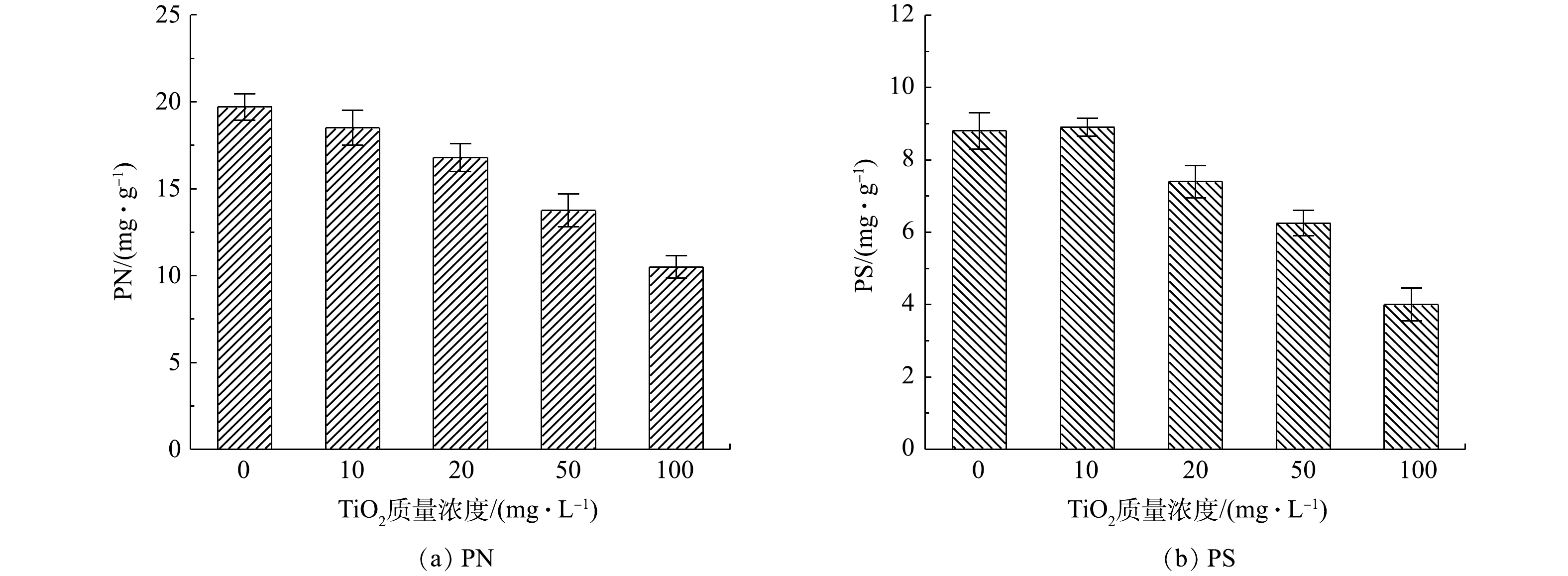
EPS是AGS的重要组成部分,对AGS的结构稳定起着重要的作用[23]。如图4所示,原始AGS中蛋白质(protein, PN)和多糖(polysaccharide, PS)含量分别为19.7 mg·g−1和8.8 mg·g−1。随着TiO2质量浓度的增加,污泥内PN和PS逐渐减少。当初始TiO2投加量为50 mg·L−1时,PN和PS含量已经显著下降到13.8 mg·g−1和6.3 mg·g−1。EPS能够保护细胞,减少外界有毒有害物质的影响,在较低的EPS含量下,其保护作用逐渐减弱。这可能是图3中100 mg·L−1 TiO2投加量下制备的生物纳米材料内部死细胞明显增加的原因所在。同时EPS的减少对AGS的结构稳定性造成影响,同样会增加微生物与TiO2的接触,对微生物活性产生抑制[24]。综合AGS对TiO2的吸附动力学结果和TiO2对AGS细胞和EPS的影响,本实验选择20 mg·L−1 TiO2条件下制备的生物纳米材料进行后续研究。
-
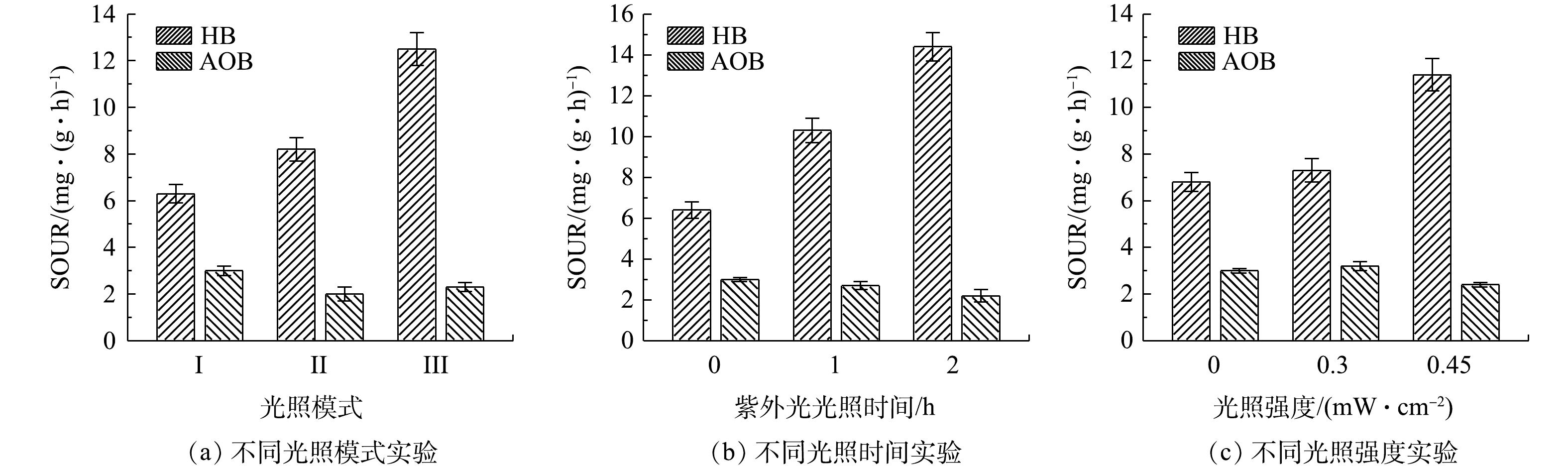
图5所示为不同光照条件对负载20 mg·L−1 TiO2的AGS (T-AGS)活性的影响。分别对异养菌(heterotrophic bacteria, HB)及氨氧化菌(ammonia oxidizing bacteria, AOB)的SOUR进行了测定。由图5(a)可知,从不同光照模式下SOUR的变化情况来看,模式I(紫外光照射0 h+可见光照射4 h)中HB的SOUR最低、AOB的活性最高;在模式II(紫外光照射1 h+可见光照射3 h)和模式III(紫外光照射2 h+可见光照射2 h)中,引入紫外光照射提高了HB活性,但对AOB活性有一定的抑制作用。图5(b)中仅有紫外光照射时的实验结果也验证了紫外光对HB活性的促进和对AOB活性的抑制作用。另外,与图5(a)中模式III(紫外光照射2 h+可见光照射2 h)相比,图5(b)中仅有紫外光照射2 h后AOB的SOUR略有提高。这可能是由于经紫外光照射后的AOB在可见光光照下发生了光复活性,例如酶促DNA修复使紫外光下造成的阻碍基因正常复制的二聚体裂开,损伤的细胞自我恢复活性使SOUR回升[25]。进一步研究了不同紫外光光强对T-AGS中SOUR的影响。如图5(c)所示,在0.3 mW·cm−2光强下,T-AGS中HB和AOB的活性均有小幅上升;而将紫外光光强提升至0.45 mW·cm−2时,虽然HB活性有所提高,但AOB活性受到抑制。因此,后续实验中选择0.3 mW·cm−2的光照强度。
-
在探究SDZ降解途径以及降解产物的批实验中,首先对10 h后反应器中剩余的耗氧有机物(以COD计)和
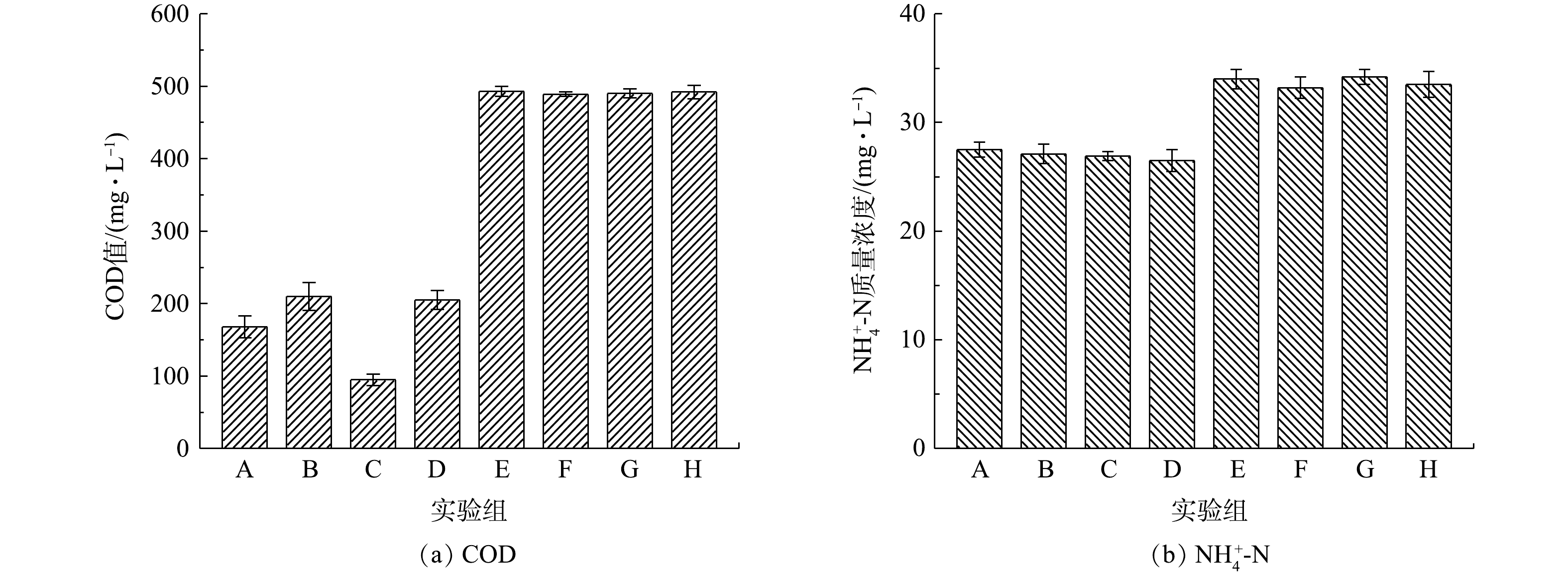
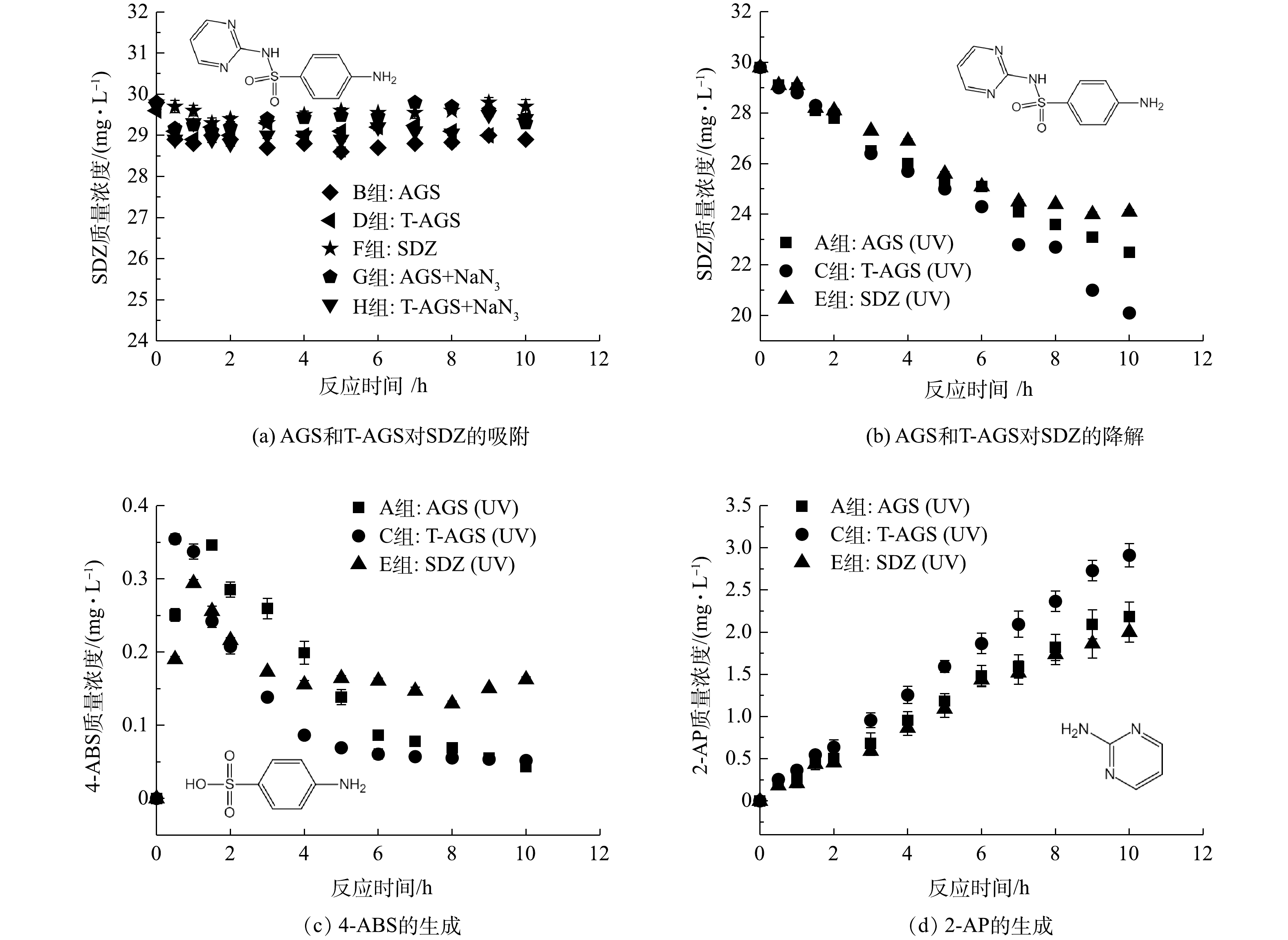
NH+4 -N的质量浓度进行测定,以探究制备的T-AGS是否会影响常规污染物的去除,结果如图6所示。A组和B组分别为普通AGS在紫外光和可见光下运行,其对COD的去除率分别为66%和58%,说明紫外光照射时普通AGS内HB活性有所增加。B组和D组(T-AGS在可见光下运行)中COD值相近,表明负载TiO2对T-AGS的耗氧有机污染物去除性能没有明显影响。与A组相比,C组(T-AGS在紫外光下运行)中耗氧有机污染物(以COD计)去除率显著提升,达到81%,说明紫外光催化显著提高了耗氧有机污染物的降解。不添加AGS和T-AGS的E组(紫外光下运行)和F组(可见光下运行),以及添加NaN3进行微生物灭活组G和组H中耗氧有机污染物几乎不通过光照和吸附去除。图6(b)是NH+4 -N的去除情况,A、B、C及D组对NH+4 -N的去除率均为23%左右,负载TiO2和紫外光光照对NH+4 -N去除效果的影响较小。而无生物活性的E、F、G及H组中几乎没有NH+4 -N的降解。上述结果表明,与原始AGS相比,T-AGS对于耗氧有机污染物(以COD计)以及NH+4 -N的去除没有抑制作用;相反,由于紫外光刺激了HB的活性,COD的去除率有所提高。图7显示了各实验组中SDZ及其中间产物的质量浓度随时间的变化情况。B组在可见光下运行的普通AGS和D组T-AGS几乎无法降解SDZ(图7(a)),这是由于SDZ具有生物毒性,其无法诱导生物体内的相应酶或辅因子对SDZ进行生物降解[26]。F组是不添加AGS和T-AGS在可见光下运行的反应,表明SDZ几乎不被可见光光解。G组和 H组由于加入NaN3进行生物失活,AGS与T-AGS在前0.5 h内通过物理吸附去除了1 mg左右的SDZ,随后达到吸附平衡,SDZ不再减少。由图7(b)可见,在紫外光照射下,A、C和E组内SDZ质量浓度随时间逐渐降低,在10 h内SDZ的平均降解速率分别为0.73、0.97、0.57 mg·(L·h)−1。不添加生物质的E组内SDZ可以在紫外光照射下得到少量降解,而在A组中由于紫外光刺激提高了普通AGS中HB的生物活性,使得SDZ去除率稍有提升。T-AGS在紫外光光照下具有最好的SDZ降解效果,这是由于负载在颗粒表面的TiO2在紫外光下生成了羟基自由基,因而加速了SDZ的降解。
图7(c)和图7(d)是对SDZ降解过程中可能的中间产物(氨基苯磺酸(4-ABS)和2-氨基嘧啶(2-AP))进行了检测。在SDZ降解过程中,由于苯环和嘧啶环之间相连的磺酰胺基团中的S与N之间的化学键断裂而分离,分别形成4-ABS和2-AP。4-ABS在SDZ降解初期生成量较大,而后逐渐降低。这可能是由于4-ABS不稳定,被微生物或紫外光进一步降解,且其降解速率大于SDZ降解生成4-ABS的速率,导致其无法在体系内积累。添加生物质的A组及C组中4-ABS的积累明显少于E组。这说明生物降解对于4-ABS去除的重要性,也证明了生物与光催化相结合更有助于SDZ及中间产物的去除。相比之下,2-AP更不易被进一步降解,其质量浓度不断积累,表明2-AP无法被微生物利用且紫外照射下较稳定。2-AP具有较强的毒性,通过羟基化反应使其进一步转化[27],或可通过高锰酸钾氧化为2-硝基嘧啶,以降低其生物毒性[28]。
2.1. TiO2的吸附与脱附
2.2. 负载TiO2对生物的影响
2.3. 光照条件对AGS的影响
2.4. SDZ降解途径及产物






-
1)利用AGS吸附TiO2制备了稳定性较好的新型生物纳米材料,AGS对TiO2的吸附过程符合伪二级动力学。
2)综合考虑吸附动力学和负载TiO2对AGS内生物活性等方面的影响,确定了最佳TiO2负载量为20 mg·L−1。
3) 0.3 mW·cm−2强度的紫外光会促进T-AGS中HB的活性,提高对耗氧有机污染物(以COD计)的去除率,且不会对AOB及
NH+4 -N的去除造成较大的影响。4)紫外光照射促进了T-AGS对SDZ的降解及其中间产物的去除,中间产物中4-ABS易被进一步降解,而2-AP较为稳定。

