

















 下载:
下载:
















 百度学术
百度学术

















-
目前在大型工业园区的突发排放废水高效经济应急处置仍然是一个重大的难题,尤其对于突发难降解有机物废水的处理,目前主要是投加大量氧化药剂进行去除,这不仅造成药剂的浪费和出水残余氧化性过高,而且过量的氧化剂还可能会抑制对目标物的去除。同时,由于应急废水组成往往高度复杂,含有大量无机离子和有机物,会对氧化处理效率产生不利影响,甚至会产生有毒有害的副产物。ZHANG等[1]的研究表明,卤素离子(X−),如氯离子可以与
SO−4⋅ 快速反应,生成活性较弱的卤离子自由基(Cl·、Cl−·、Cl−2⋅ ),从而降低有机物的降解效率。但CHEN等[2]的研究表明,Cl−会促进过硫酸盐氧化去除双氯芬酸钠。MA等[3]发现,水体中的HCO−3 会与SO−4⋅ 和·OH反应生成活性较低的碳酸盐自由基(HCO−3⋅ ),导致目标污染物的降解率降低。因此,研究不同影响因素对突发废水的应急处理具有现实意义。近年来,基于硫酸根自由基(
SO−4⋅ )的高级氧化技术受到越来越多的关注和重视,SO−4⋅ 具有很强的氧化性,能氧化水体中的大部分有机物。过硫酸盐在室温下呈固态,易于储存和输送,价格也相对较低,而且能对有机物进行快速处理,这些特征使其在废水的应急处理过程中具有广阔的应用前景[4]。有研究[5]表明,室温下过硫酸盐氧化污染物的作用并不明显,需要在过渡金属、光、加热等条件下活化。与其他活化方式相比,Fe2+活化过硫酸盐所需的激活能量较低,而且Fe2+无毒无害、不会对环境造成污染和在自然界存量丰富,因此,Fe2+活化过硫酸盐氧化去除有机污染物被认为是较为经济有效的方法。苯胺(AN)是重要的有机化工原料及产品,被广泛应用于国防、塑料、油漆、印染等行业,同时也是严重污染环境和危害人体健康的有害物质,是三致物质[6]。由于AN对生物具有毒性效应,已经被列入中国环境优先污染物黑名单中[6]。本研究选择AN作为目标污染物,利用Fe2+活化过硫酸钠(PS)氧化体系去除AN,研究了不同的PS投加量、Fe2+投加量、溶液pH、水体中常见阴离子(Cl−、
HCO−3 、NO−3 )以及有机物对AN去除效果的影响;同时,利用自由基淬灭剂和电子自旋共振技术(electron paramagnetic resonance, EPR)对反应体系中自由基的种类进行了鉴定,并提出一种简便可行的对反应体系中自由基进行定量的方法,以期为不同方式活化过硫酸盐降解有机物的降解机理提供参考。
全文HTML









-
过硫酸钠(≥99%)、氢氧化钠(≥99%)和硫酸(95%~98%)均购于天津光复精细化工研究所。硝酸钠(≥99%)、氯化钠(≥99.8%)、碳酸氢钠(NaHCO3)和七水硫酸亚铁(FeSO4·7H2O,98%)购于天津市大茂化学试剂厂。苯胺、苯酚(98%)和硝基苯购于上海TCI试剂公司。5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)为色谱纯购自于上海阿拉丁生化科技股份有限公司。甲醇(MeOH,色谱纯)和叔丁醇(TBA,色谱纯)购自Fisher公司。所用超纯水电导率为18.2 Ω·cm−1。
-
所有实验均在50 mL棕色玻璃瓶中进行,搅拌速度600 r·min−1,反应温度25 ℃。在棕色玻璃瓶中加入0.1 mmol·L−1的AN溶液和一定浓度的PS溶液。通过使用10 mmol·L−1的H2SO4或NaOH溶液将其调节至所需初始pH。然后加入一定浓度的FeSO4·7H2O溶液,反应开始进行,反应过程中不对溶液pH进行调节。在一定的时间间隔取1 mL样品,用甲醇终止反应,然后用0.45 µm滤膜过滤后放入液相小瓶中分析。为了进一步研究Fe2+活化PS过程的反应机理,使用甲醇(MeOH)和叔丁醇(TBA)作为自由基淬灭剂。由于MeOH与
SO−4⋅ (1.6×107~7.7×107 L·(mol·s)−1)和·OH (1.2×109~2.8×109 L·(mol·s)−1)反应速率较快,而TBA与·OH反应较快(3.8×108~7.6×108 L·(mol·s)−1),但与SO−4⋅ (4.0×105~9.1×105 L·(mol·s)−1)反应速率较慢。所以,MeOH可以同时淬灭SO−4⋅ 和·OH,而TBA选择性淬灭·OH,利用这2种自由基淬灭剂对自由基选择性的不同,可识别溶液中参与反应的自由基种类。DMPO可用作标记化合物,捕集氧化反应过程中生成的自由基(SO−4⋅ 和·OH),形成稳定的自旋加合物(DMPO-SO4和DMPO-OH),并通过EPR检测确定是否含有自由基。所有实验至少进行2次,取平均值且计算AN去除率。 -
AN浓度的测定均采用高效液相色谱(LC-2010型,日本岛津)直接进样的方式测定,反相C18柱(250 mm×4.6 mm),流动相为乙腈/缓冲盐(v∶v)=65∶35,柱温为25 ℃,流速为1.0 mL·min−1,紫外检测波长为280 nm。溶液pH采用pHS-3C型pH计(上海雷磁)测定。利用Phoenix 8000总有机碳分析仪(美国泰克玛)测定有机碳(TOC)。
-
化学反应动力学是定量描述化学随时间变化的基础理论,表达了反应速率及其影响参数之间的函数关系。一般有以下影响因素:反应物浓度、温度、绝对压力以及催化剂的浓度和种类等[7]。一级动力学方程被广泛应用于催化剂化学反应中,其一级反应动力学速率方程如式(1)所示。
式中:r为反应动力学速率;C为AN浓度,mmol·L−1;t为反应时间,min;k为反应速率常数,min−1;基于初始变量t=0和C=C0,分离变量、积分,则得一级反应动力学方程如式(2)和式(3)所示。
式中:C0为AN初始浓度,mmol·L−1;Ct为AN在t时刻时的浓度,mmol·L−1;k为一级反应动力学常数,min−1。以ln(Ct/C0)为纵坐标,以时间t为横坐标作图,若各点之间呈线性关系,且其拟合的可决系数较高,R2接近于1,则说明其反应机理符合一级动力学模型。
-
根据已有的文献,在Fe2+/PS体系中的主要活性物质是·OH和
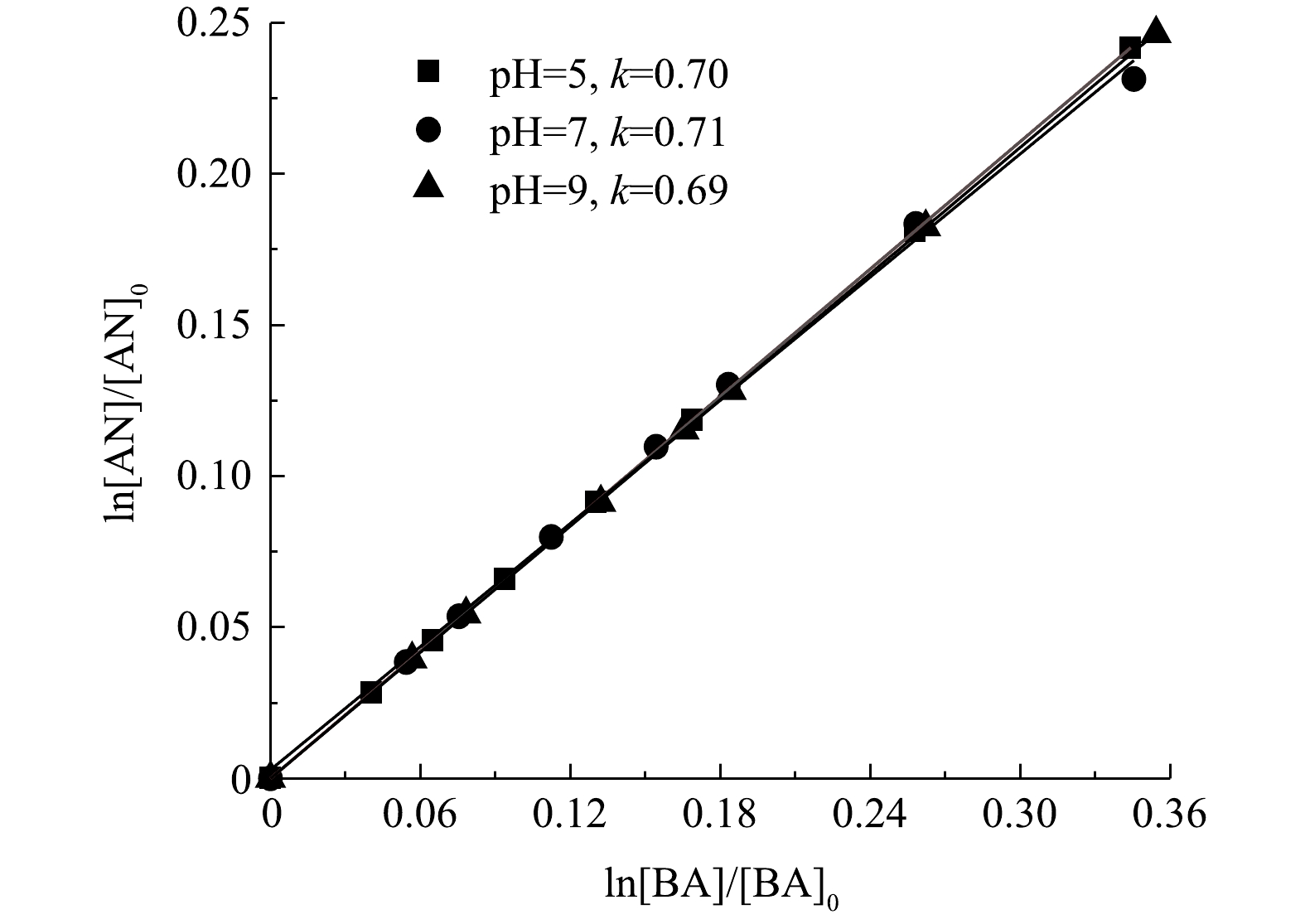
SO−4⋅ [8-9]。为了更准确地对这2种自由基进行定量分析,需得到AN与·OH和SO−4⋅ 的二级反应速率常数。已有文献报道,AN与SO−4⋅ 的二级反应速率常数为(7.7±0.5)×109 L·(mol·s)−1[10]。但是,对AN与·OH的二级反应速率常数目前尚未见报道,需要通过实验来确定。本研究通过Fe2+/H2O2氧化AN,利用相对速率法[9]测定AN与·OH的二级反应速率常数,苯甲酸(BA)作参比化合物。BA与的·OH二级反应速率常数为5.9×109 L·(mol·s)−1[9]。图1为不同pH下AN与·OH的反应速率常数测定结果。由图1可知,当反应溶液pH为5、7、9时,AN和·OH的二级反应速率常数与BA和·OH的二级反应速率常数比值分别为0.70、0.71和0.69。由此可见,AN与·OH的二级反应速率常数基本不受pH影响,经计算为(3.78±0.35)×109 L·(mol·s)−1。根据AN浓度相对于
SO−4⋅ 和·OH稳态浓度的变化率,可得到动力学表达式(4)。假设PS的投加量过量,可通过利用添加TBA前后的一级反应动力学常数的比值得到SO−4⋅ 对AN降解所占比率为a(式(5))。综合式(4)和式(5)可得式(6),进一步对式(6)化简可得到式(7);由式(7)化简可求得SO−4⋅ 的浓度(式(8))。同理可以求得·OH的浓度(式(9))。式中:CAN表示t时刻AN的浓度,mol·L−1;
CSO−4⋅ 表示t时刻SO−4⋅ 的浓度,mol·L−1;C⋅OH 表示t时刻·OH的浓度,mol·L−1;t为反应时间,s;kAN+SO−4⋅ 和kAN+·OH表示AN与SO−4⋅ 和·OH的二级反应速率常数,L·(mol·s)−1;a表示SO−4⋅ 对AN降解所占比率;kapp是AN降解的一级反应动力学常数,s−1。根据以上建立的自由基稳态浓度计算方法,可以估算在AN降解过程中·OH和
SO−4⋅ 的生成量,并同样可利用以上方法求得在不同过硫酸盐活化体系中·OH和SO−4⋅ 的生成量。
1.1. 材料和试剂
1.2. 实验装置和实验方法




1.3. 分析方法
1.4. 氧化动力学分析
1.5. 稳态自由基浓度的估算














-
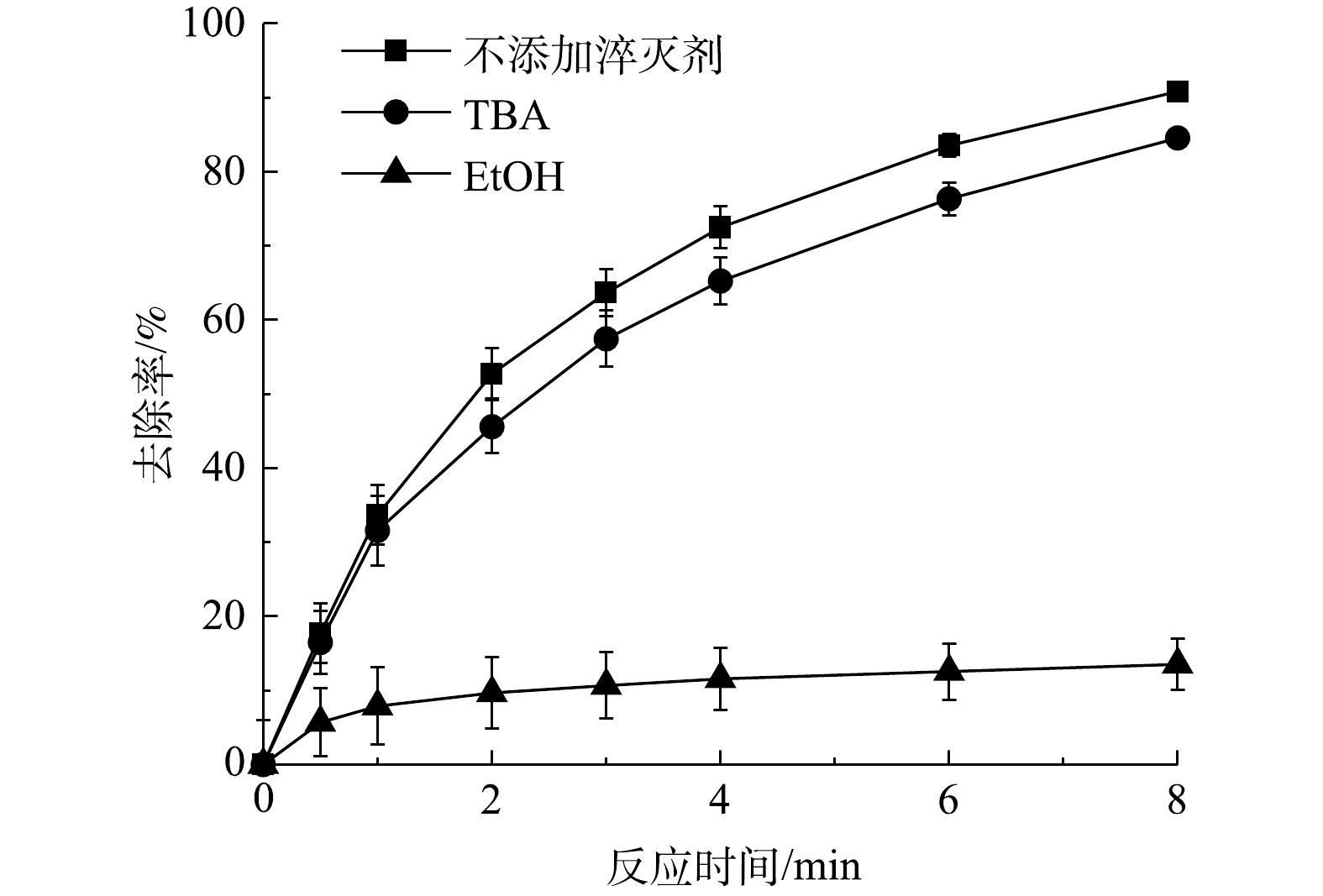
图2为添加淬灭剂后AN的去除率变化情况。由图2可知,在反应体系中加入10 mmol·L−1 MeOH后,AN的去除率降低了77.32%;当加入10 mmol·L−1 TBA后,AN的去除率仅降低了6.29%。由此可见,MeOH和TBA的加入均可抑制AN的降解。此结果表明,在反应中·OH和
SO−4⋅ 同时存在,且SO−4⋅ 起主导作用。SO−4⋅ 是由Fe2+与PS反应产生。而·OH的形成原因主要有2点:一方面是由于PS在酸性条件下水解生成H2O2,然后Fe2+与H2O2发生氧化反应生成·OH;另一方面是由于生成的SO−4⋅ 通过链引发反应生成·OH。为了进一步验证反应体系中同时存在·OH和
SO−4⋅ ,利用EPR对其进行了检测和鉴定,结果如图3所示。反应中加入0.1 mmol·L−1 DMPO后表现出典型的DMPO-SO4和DMPO-OH特征谱峰,根据其精细结构常参数,可以确定2种自由基同时存在。CHEN等[8]的研究结果同样表明,在FeS活化PS降解2,4-二氯苯氧乙酸时,反应过程中同时生成了·OH和SO−4⋅ ,但·OH在降解过程中起主要作用。可见,活化PS对不同有机物物降解时,均会生成·OH和SO−4⋅ ,但对于不同有机物起主要作用的自由基种类并不完全相同。 -
氧化剂的用量是影响污染物降解的重要因素,也是评价处理技术经济效益的重要指标。在AN为0.1 mmol·L−1、Fe2+的投加量为2 mmol·L−1时,分别控制PS的浓度为2、5、8、10 mmol·L−1,AN的去除率变化结果如图4所示。由图4可知,随着反应的进行,在反应4 min后,当PS浓度由2 mmol·L−1增加到8 mmol·L−1时,AN的去除率由49.14%增加到97.73%。这表明随着PS的投加量的增加,自由基的生成量也逐渐增加。但是,进一步增加PS的投加量反而导致AN的去除率下降。当PS浓度由8 mmol·L−1增加到10 mmol·L−1后,在反应4 min后,AN的去除率由97.73%降低至82.21%。由图4也可看出,尽管Fe2+可以通过混凝去除部分有机物,但相比于Fe2+/PS氧化体系对AN的去除率低。可见,适当的PS投加量有助于自由基的产生,从而能促进AN的降解。对于大多数有机污染物,增大氧化剂浓度可加速有机物的降解。但由于在PS体系中,过量的
SO−4⋅ 之间以及SO−4⋅ 与过量的PS之间通过式(10)和式(11)发生反应[8],导致SO−4⋅ 有效利用率降低,进而影响有机物的降解效果,且过高的PS投加量会增加费用。因此,本研究中选择PS的最佳投加量应为8 mmol·L−1。图5为在不同PS浓度下,添加TBA后AN去除率的变化结果。对图4和图5不同浓度PS下降解AN的反应进行一级降解动力学拟合,反应速率常数如表1所示。由表1可知,Fe2+/PS体系对AN的降解符合一级降解动力学,且当PS浓度为2、5、8、10 mmol·L−1时,AN的反应速率系数分别为1.7、149.5、291.7、924.2和353.7×10−3 min−1,可以看出PS投加量为8 mmol·L−1时反应速率最快。由表1可以计算得出,
SO−4⋅ 对AN的降解贡献占比约为80%,说明在Fe2+/PS体系降解AN的过程中,SO−4⋅ 起主要作用。利用表1中所得的数值和1.5中稳态自由基的估算方法,可以得到不同PS浓度下自由基的生成量(图6)。由图6可以得出,当PS浓度为8 mmol·L−1时,SO−4⋅ 和·OH的浓度最高,分别为16.45×10−13 mmol·L−1和7.14×10−13 mmol·L−1。一般来说,·OH容易通过去氢或加成的方式参与反应[11]。相反,SO−4⋅ 氧化目标物主要是通过电子转移的方式,并倾向于攻击目标物的富电子部分[12]。因此,氨基作为供电子基团更容易受到SO−4⋅ 的攻击。此外,SO−4⋅ 氧化还原电位高达2.5~3.1 V,比·OH (1.8~2.7 V)氧化能力强[13]。在实际废水的应急处理中,不能过量地投加氧化剂,要根据实际情况进行确定,否则不仅会造成药剂浪费和出水氧化性太高,也会导致有机物去除率降低。同时,还需要注意的是,在过硫酸盐氧化有机物过程中可能会生成过量的SO2−4 ,有研究者为了降低SO2−4 带来的二次污染做了相关的研究。CASSIDY等[14]利用水体中微生物硫酸盐还原菌(SRB),还原PS生成的SO2−4 得到H2S,可有效降低SO2−4 的二次污染风险。 -
常温下过硫酸盐与有机物反应速率较慢,对有机物的氧化效果并不显著,一般需要催化剂对其进行活化[5]。Fe2+催化PS发生的是类似于Fenton氧化的反应(式(12)),能够快速生成强氧化性的
SO−4⋅ ,从而氧化去除难降解有机物[9]。Fe2+是影响过硫酸盐降解的重要影响因素,图7为不同Fe2+浓度时AN的去除率。由图7可知,在AN和PS初始浓度分别为0.1 mmol·L−1和8 mmol·L−1时,当Fe2+投加量为1~6 mmol·L−1时,AN的去除率从47.14%先增高到100%然后又降低至86.61%,其中Fe2+投加量为4 mmol·L−1时AN的去除率最高为100%。同时,由图7可以看出,当不投加Fe2+时,PS氧化性很弱,不能对AN有效去除。可见,本实验中Fe2+最佳投加量为4 mmol·L−1,而且过量的Fe2+投加量会导致AN的去除率降低。WANG等[15]开展的同样研究表明,当用Fe2+活化PS降解甲氧苄氨嘧啶时,过量的Fe2+会抑制甲氧苄氨嘧啶的降解。这可能是由于过量的Fe2+会与SO−4⋅ 和·OH发生反应(13)和(14),导致对SO−4⋅ 和·OH的消耗降低了AN的去除效率。同时,由于PS会将Fe2+迅速氧化为Fe3+,从而降低Fe2+的浓度,故需要增加Fe2+的投加浓度;而如果投加高浓度的Fe2+,会导致Fe2+与目标有机物对SO−4⋅ 产生竞争。因此,为了使Fe2+在溶液中保持一定浓度,需加入一定量的络合剂,如乙二胺四乙酸(EDTA)、乙二胺二琥珀酸三钠(EDDS)和柠檬酸(CA)等,通过络合剂对Fe2+的屏蔽效应,调控Fe2+参与反应的速率。但是,该方法仍存在部分络合剂对Fe2+循环转化效率低,以及具有生物毒性,难以降解,带来二次污染的问题,故需要进一步开发高效、环境友好的络合剂[5]。 -
由于pH既会改变自由基的分布也会影响目标污染物的赋存状态,故溶液pH对活化过硫酸盐氧化有机物具有重要影响[16]。为了避免加入的缓冲液(PBS)中其他离子对降解过程产生干扰,所以在本实验利用NaOH和H2SO4溶液调节pH。在AN浓度为0.1 mmol·L−1,Fe2+和PS的投加量分别为4 mmol·L−1和8 mmol·L−1条件下,不同初始pH对AN去除影响的情况如图8所示。其中,pH=2.5为未对溶液pH进行调节时的初始值。由图8可以看出,当pH为2.5、3、5、7、9、11时,反应8 min后AN的去除率分别为100%、95.06%、92.99%、78.63%、58.74%和30.86%。由此可知,酸性条件更有利于AN的去除,这与其他研究所得结论一致[17-19]。但是,本研究发现pH在2.5~5时,AN的去除率相差不大,而在中性和碱性条件下AN的去除率明显降低。LI等[17]利用PS降解二苯胺(DPA)过程中发现,酸性条件下有利于DPA的去除,但不同pH下DPA的降解率差异并不显著,当溶液pH为2.2、3.8、7.4、8.0和9.8时,DPA的降解率为10%~21%。RASTOGI等[19]利用Fe2+活化PS氧化降解多氯联苯(PCBs)过程中发现,当溶液为弱酸性条件(pH=5.0)、中性条件(pH=7.0)和碱性条件(pH=9.0)时,PCBs的降解率分别为42%、40%和20%,而在强酸性条件(pH=3.0)时PCBs的降解率为80%。可见,尽管在酸性条件下Fe2+活化PS氧化有机物的效率更高,但对于不同的有机物在不同pH时降解效果的差异不尽相同。这可能是由于以下3点原因造成的:当溶液的pH大于4.0时,Fe2+和Fe3+由于会生成沉淀而减少[20],且形成的Fe2+和Fe3+的羟基氧化物(式(15)~式(18))活化PS生成
SO−4⋅ 的效率很低[21];由式(19)可知,当在强碱性条件下,SO−4⋅ 会与HO−反应生成·OH,但是·OH的氧化活性会受到阴离子(SO2−4 )的抑制而降低[9];AN的解离常数(pKa)为4.6,这表明:当pH小于4.6时AN主要以阳离子状态存在(AN阳离子);而当pH大于4.6时,主要以分子态存在[22]。由于电斥力作用,SO−4⋅ 与分子态有机物反应速率较慢[23],所以在pH<4.6时,更能够促进对AN的降解。值得注意的是,本研究发现,当不添加Fe2+时,不同pH下PS仍可以氧化去除AN,不同pH下AN的去除率变化见图9。由图9可知,随溶液pH的升高,AN去除率随之逐渐升高,在pH为11时去除率最高,去除率为66.6%。这表明碱性条件可以活化PS,但碱性条件活化PS去除AN的效率较Fe2+活化PS低。朱杰等[24]利用碱活化PS对水中氯苯进行去除,当pH=10.28时,反应5 h后氯苯的去除率为53.75%。QI等[25]同样发现,碱活化过一硫酸盐(PMS)能够有效地去除水中有机污染物,如酸性橙7、苯酚、对氯苯酚、双酚A和磺胺甲恶唑等,并通过添加淬灭剂(TBA、MeOH、叠氮化物和对位苯醌)和ESR实验得出反应中起主要作用的是超氧游离基(
O⋅−2 )和单线态氧(1O2)。LI等[26]在利用过硫酸盐降解苯酚时发现,碱性条件(pH=9.0)下较中性条件(pH=7.4)和酸性条件(pH=2.7)下的苯酚去除率高,并通过EPR分析得出,起主要作用的是单线态氧(1O2)。葛勇建等[27]的研究表明,在NaOH活化PMS氧化水中环丙沙星时,超氧游离基(O⋅−2 )和单线态氧(1O2)是主要的活性氧物种,而并非是SO−4⋅ 与·OH。由此可见,碱性条件能够促进PS降解有机物,可能是由于反应过程中生成了O⋅−2 和1O2。在实际废水处理应用中,当水体的pH较高时,可不投加Fe2+而直接投加过硫酸盐处理有机废水。例如在造纸、化工、纺织、食品和石化等许多工业部门,均会产生高浓度的碱性废水,可直接添加过硫酸盐降解废水中的有机污染物,过硫酸盐对此类废水的处理具有明显的优势。如果实际废水中还需要同时去除重金属,可以先将废水调节为碱性条件,然后再投加过硫酸盐。这样一方面可以利用碱性条件对重金属沉淀去除,另一方面可以利用碱性条件活化过硫酸盐氧化去除有机物。笔者之前在处理天津市某垃圾渗滤液废水时(pH约为7.8,COD约为780 mg·L−1,重金属浓度约为7.42 mg·L−1),通过将渗滤液废水pH调节至11后投加约1.6 g·L−1过硫酸盐,发现重金属和COD值的去除率分别可达85%和62%。可见,利用碱性条件活化过硫酸盐对有机污染物进行降解,在环境污染治理领域具有潜在的应用价值。
本研究还进一步考察了反应过程中pH的变化情况,结果见图10。由图10可知,当初始pH分别为2.5、3、5、7、9和11时,反应后最终pH为2.3、2.5、3.1、4.8、5.8和7.2,可见溶液的pH均发生了下降。这可能是由于:PS分解产生
HSO−4 ,HSO−4 容易水解生成H+导致pH降低(式(20)和式(21));PS发生水解(式(22)和式(23)),产生H+,进而导致pH下降。 -
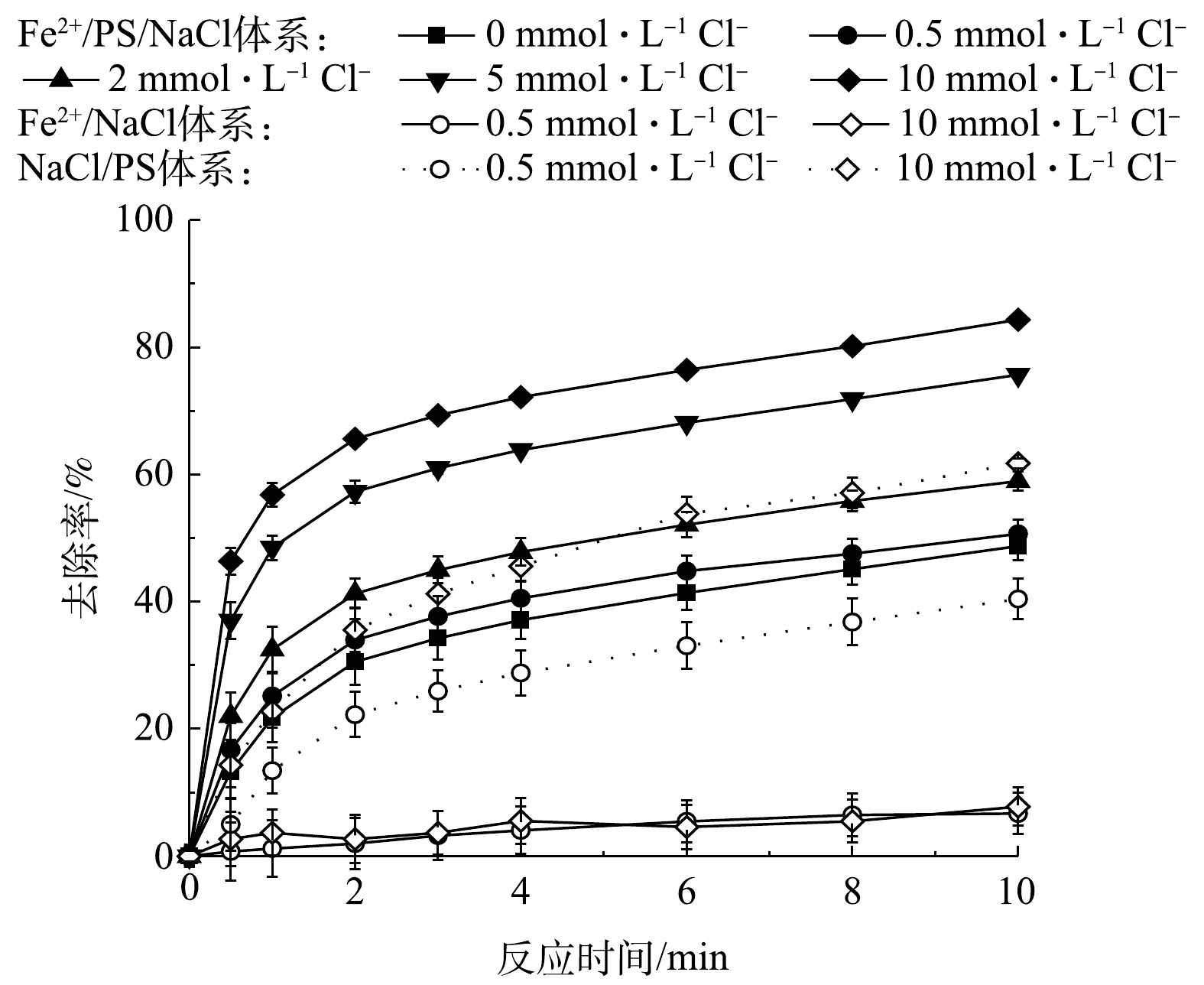
Cl−由于会与
SO−4⋅ 和·OH发生反应,生成氯自由基,所以,在基于SO−⋅4 氧化去除有机物的过程中Cl−起重要作用[1]。图11为在不同Cl−浓度下,Fe2+活化PS对AN的去除率变化。由图11可知,在AN初始浓度为0.1 mmol·L−1、PS浓度为8 mmol·L−1、Fe2+投加量为1 mmol·L−1的条件下,分别添加0.5、2、5和10 mmol·L−1的Cl−,随着Cl−投加量的增加,反应10 min后AN的去除率由51.68%逐渐升高至84.36%。有研究[28-29]表明,Cl−会消耗SO−4⋅ 形成Cl·、Cl−2⋅ 和其他氯自由基,由于这些物质的氧化能力较SO−4⋅ 和·OH弱,故会降低Fe2+/PS体系对目标物的去除效率(式(24)~式(28))。但本研究中的结果表明,Cl−会促进AN的去除,当Cl−浓度为5 mmol·L−1和10 mmol·L−1时,AN的去除率分别为75.69%和84.36%。且当只有PS和Cl−存在时,Cl−仍能够促进PS对AN的去除。LI等[26]的研究表明,在酸性条件下,Cl−的存在能显著促进PMS对苯酚的降解;进一步研究表明是由于Cl−与PS反应过程中生成了游离氯(HClO、Cl2和ClO−),最终产物以4-氯苯酚和2,4-二氯苯酚为主;该研究中还发现降解过程中尽管Cl−可以有效促进PS对苯酚的降解,但对TOC降解的效果并不明显。同时,结合图11中只有Fe2+和Cl−存在时AN去除率的结果可推测,Cl−的加入能够促进PS对有机物的降解是由于反应过程中生成了含氯的氧化物质,但并不能像SO−4⋅ 和·OH可将有机物完全矿化,且NaCl/PS体系可能会生成有害的氯代副产物。综上所述,在处理实际高浓度含Cl−废水中的有机物时,可以直接投加PS,利用溶液中的Cl−对PS活化去除有机物,但需要注意氯代副产物的影响。 -
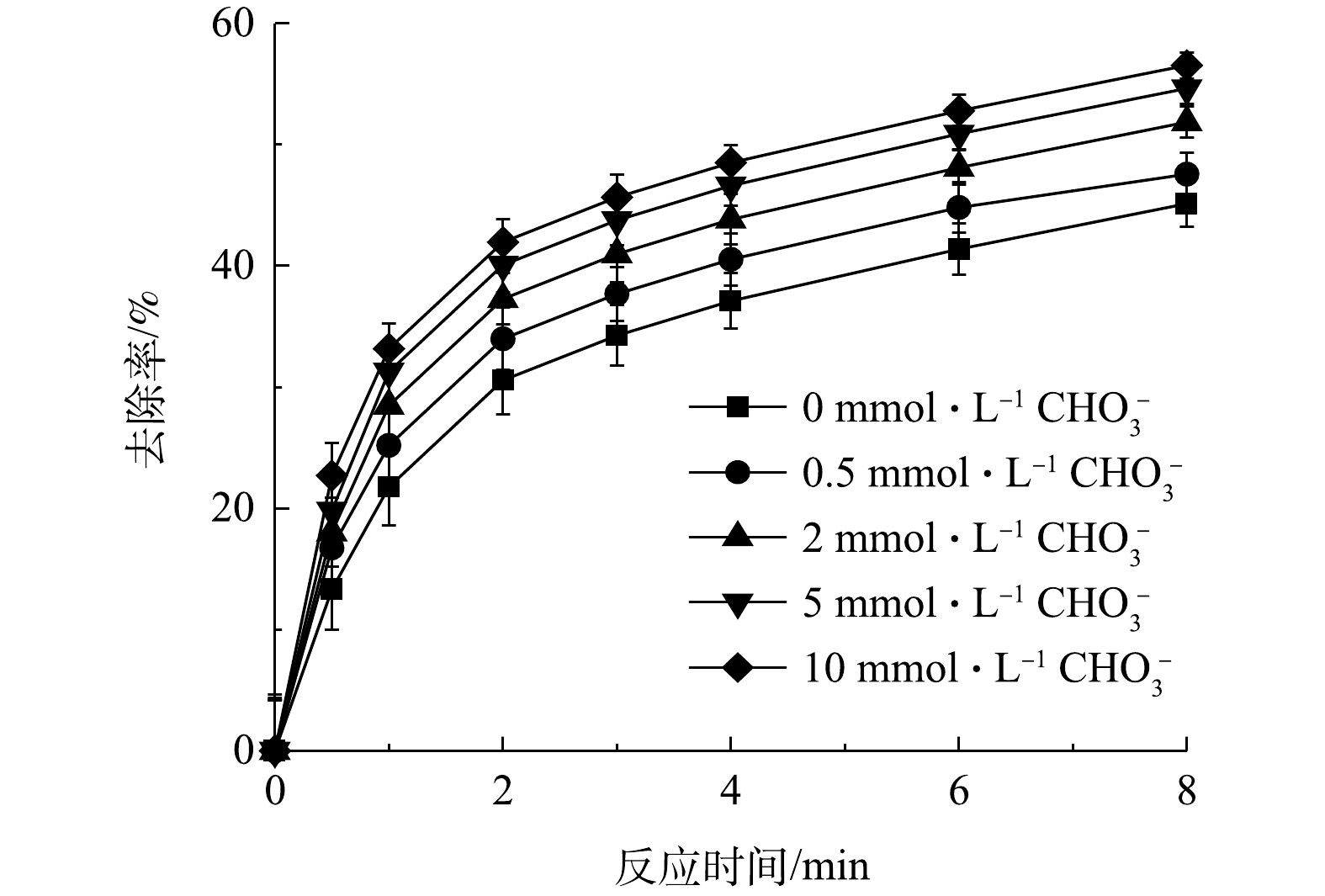
在AN初始浓度为0.1 mmol·L−1、PS浓度为8 mmol·L−1,Fe2+投加量为1 mmol·L−1的条件下,分别添加0.5、2、5和10 mmol·L−1的
NO−3 。由图12可以看出,随着NO−3 浓度的升高,AN的去除率没有明显降低。MA等[3]通过热活化PS降解苯、甲苯乙苯和二甲苯时发现,不同NO−3 浓度基本不会影响这些有机物的去除效果。PENG等[30]使用CuS活化PS降解农药阿特拉津时发现,NO−3 对阿特拉津的去除同样没有影响。理论上NO−3 可以与·OH和SO−4⋅ 发生反应,生成氧化性较·OH和SO−4⋅ 更弱的NO⋅2 和NO⋅3 (式(29)和式(30))[31-32],但该反应的速率很慢(k=5.5×104 L·(mol·s)−1)[28],所以NO−3 的抑制作用可以忽略。CO−3 和HCO−3 是导致自然水体碱度和缓冲能力的主要因素。一般认为,HCO−3 会与SO−4⋅ 和·OH反应生成活性较低的碳酸盐自由基(HCO3⋅ )(式(31)和式(32)),由于HCO3⋅ 与有机物的反应速率较SO−4⋅ 和·OH低[33],所以加入HCO−3 可导致有机物的去除率明显降低。在AN初始浓度为0.1 mmol·L−1、PS浓度为8 mmol·L−1、Fe2+投加量为3 mmol·L−1的条件下,分别添加0.5、2、5和10 mmol·L−1的HCO−3 ,此时AN的去除率变化如图13所示。由图13可知,随着HCO−3 离子浓度的升高,AN的去除率相应降低。当加入10 mmol·L−1HCO−3 时,反应8 min后AN的去除率约下降了11.4%,可见AN的去除率没有显著降低。这可能是由于:一方面,HCO3⋅ 的氧化性不如SO−4⋅ 和·OH (低2~3个数量级[8]),但其能够以较快的反应速率与富电子化合物(与AN的反应速率为6×108 L·(mol·s)−1)发生反应[34],这在一定程度上抵消了因HCO−3 对SO−4⋅ 和·OH的消耗而造成的AN的去除率降低。SONG等[35]研究Fe2+活化PS降解阻燃剂磷酸三苯酯(TPhP)时发现,当加入HCO−3 后虽然会抑制TPhP的降解,但随着HCO−3 浓度的升高,TPhP的降解率会随之升高。这可能是由于TPhP是富电子化合物,导致生成的HCO3⋅ 能够降解TPhP,因而提高了TPhP的去除率。另一方面,HCO3⋅ 可以催化PS分解生成SO−4⋅ [30]从而促进AN的降解。GUAN等[36]利用磁性孔铜铁氧体(CuFe2O4)活化PMS降解阿特拉津时发现,在HCO−3 浓度较低时其对阿特拉津去除有促进作用,但当浓度较高时反而会产生抑制作用。GUAN等[36]研究结果表明,这是由于在HCO−3 浓度较低时,一方面HCO−3 能够活化PMS生成SO−4⋅ ;另一方面HCO−3 可以与CuFe2O4发生络合作用促进阿特拉津的去除;在高浓度HCO−3 时会对反应中生成的SO−4⋅ 淬灭,导致阿特拉津的去除率降低。 -
实际废水中含有大量的其他有机物,这些有机物往往会对目标有机物的降解有竞争作用,造成目标有机物的降解效率降低。为了研究其他有机污染物对PS降解AN的影响,分别投加不同浓度的苯酚和硝基苯且考察AN的去除率。在AN初始浓度为0.1 mmol·L−1、PS浓度为8 mmol·L−1,Fe2+投加量为1 mmol·L−1的条件下,分别添加0.5、2、5和10 mmol·L−1的苯酚和硝基苯,AN的去除率变化如图14所示。由图14(a)可以看出,随着硝基苯浓度的增大,AN的去除率逐渐降低。这可能是由于硝基苯的加入对溶液中的活性自由基产生了竞争反应,导致AN的去除率逐渐降低。但由图14(b)中可以看出,随着苯酚浓度的增大,AN的去除率也有所增大,这可能是由于苯酚对PS进行了活化。苯酚活化PS可能是通过以下途径:苯酚还原PS产生了
SO−4⋅ (式(33))[37];PS在氧化苯酚过程中,苯酚通过亲核攻击分解PS产生HO−2 (式(34))。生成的HO−2 通过式(35)促进PS生成SO−4⋅ ,从而促进AN的去除[37]。由此可见,针对能够活化过硫酸盐的含有酚类的有机物废水,可直接添加过硫酸盐对其进行去除。 -
在实际难降解有机废水处理中,常用的氧化技术有活化PS、KMnO4、O3和Fenton氧化。以天津滨海新区某大型工业园区污水处理厂二沉池出水(TOC为4.45 mg·L−1,pH为7.12)作为背景水体。本研究中AN的初始浓度为0.1 mmol·L−1,PS、KMnO4和H2O2的投加浓度分别为2、5、8和10 mmol·L−1,Fe2+浓度为2 mmol·L−1,反应时间为10 min,此条件下不同氧化剂对AN的去除率如图15所示。由图15可知,在实际废水中AN的去除率明显低于纯水中。由上文所得结果可知,水体中不同有机物和阴离子对AN的去除影响不同,均可导致AN去除率降低。在实际废水处理中,需要投加较纯水过量的PS和适当延长反应时间。由图15也可以看出,当PS、H2O2和KMnO4投加量为10 mmol·L−1时,AN的去除率均为最高。此时,PS、H2O2和KMnO4对AN的去除率分别为80.56%、62.90%和31.11%,可见在实际废水中PS对有机物的氧化去除效果最好。由图15也可以得出,PS、H2O2和KMnO4对TOC的去除率分别为78.52%、54.95%和22.62%,均低于对AN的去除率,但Fe2+/PS对AN的矿化程度明显较芬顿氧化体系高。CAO等[38]发现,Fe2+/PS能够完全氧化杀虫剂林丹。RASTOGI等[19]发现,Fe2+/PS甚至能够彻底矿化多氯联苯(PCBs)。同时,PS比O3和H2O2具有更强的稳定性,不会因为挥发导致氧化剂利用效率降低,有利于环境修复过程中的传质过程。相比于H2O2,PS在水体和土壤中停留时间更长,在一定程度上有利于难降解有机物的去除,这些特点使其在有机污染物的处理应用中具有广阔的前景。
2.1. Fe2+/PS体系中自由基的识别







2.2. 不同PS投加量对AN去除率的影响













2.3. 不同Fe2+投加量对AN去除率的影响




2.4. 不同pH对AN去除率的影响










2.5. Cl−对AN去除率的影响






2.6.
NO−3 和HCO−3 对AN去除效率的影响







































2.7. 其他有机物对AN去除效率的影响




2.8. 不同氧化技术对AN实际废水的处理
-
1)
SO−4⋅ 在AN去除过程中起主要作用,对AN的去除贡献占比约为80%。2)随着PS和Fe2+初始浓度的升高,AN的去除率逐渐升高,但投加过量反而会抑制AN的去除,氧化去除AN的过程符合一级降解动力学。
3)酸性环境有利于Fe2+活化PS氧化去除AN,在不加入Fe2+时,碱性条件同样可活化PS,但对有机物的去除率较Fe2+活化PS有所降低。
4)
HCO−3 和硝基苯的加入会抑制AN的去除,而NO−3 对AN的去除基本没有影响,苯酚的加入会促进AN的去除。5) Cl−不仅可以在Fe2+/PS体系中促进对AN的氧化去除,同时在仅有PS存在时也可促进对AN的去除,但可能并不像
SO−4⋅ 和·OH可将有机物完全矿化,且会生成有害的氯代副产物。




