

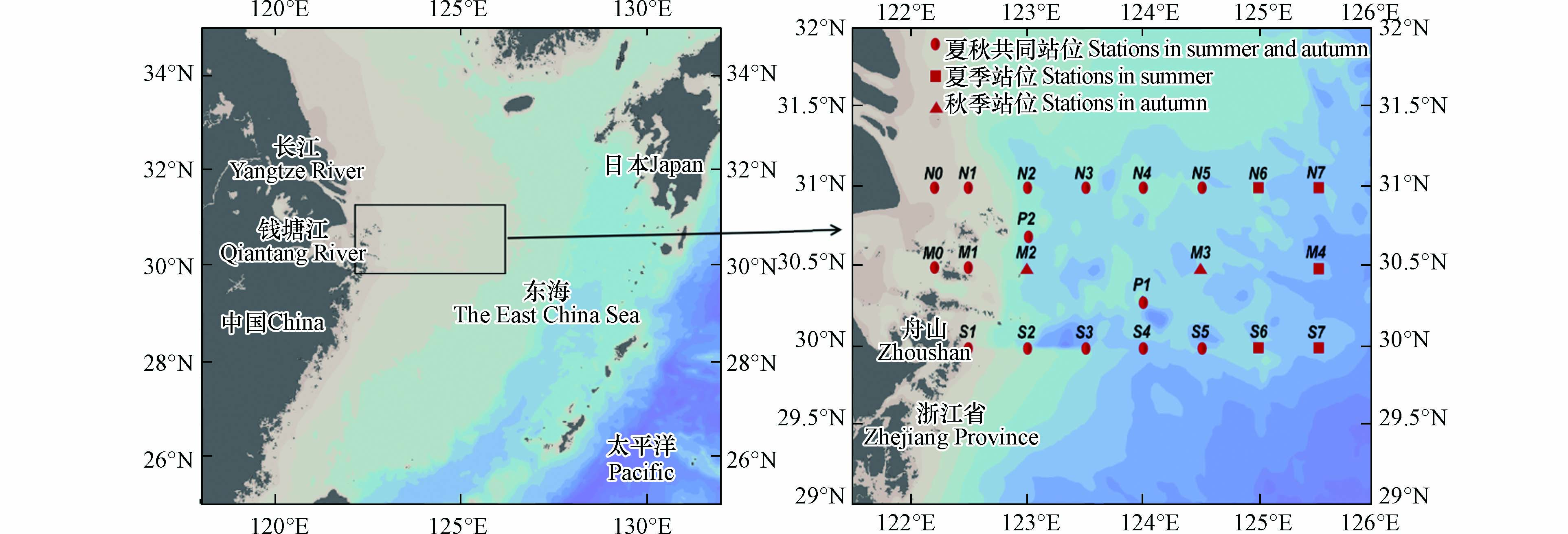
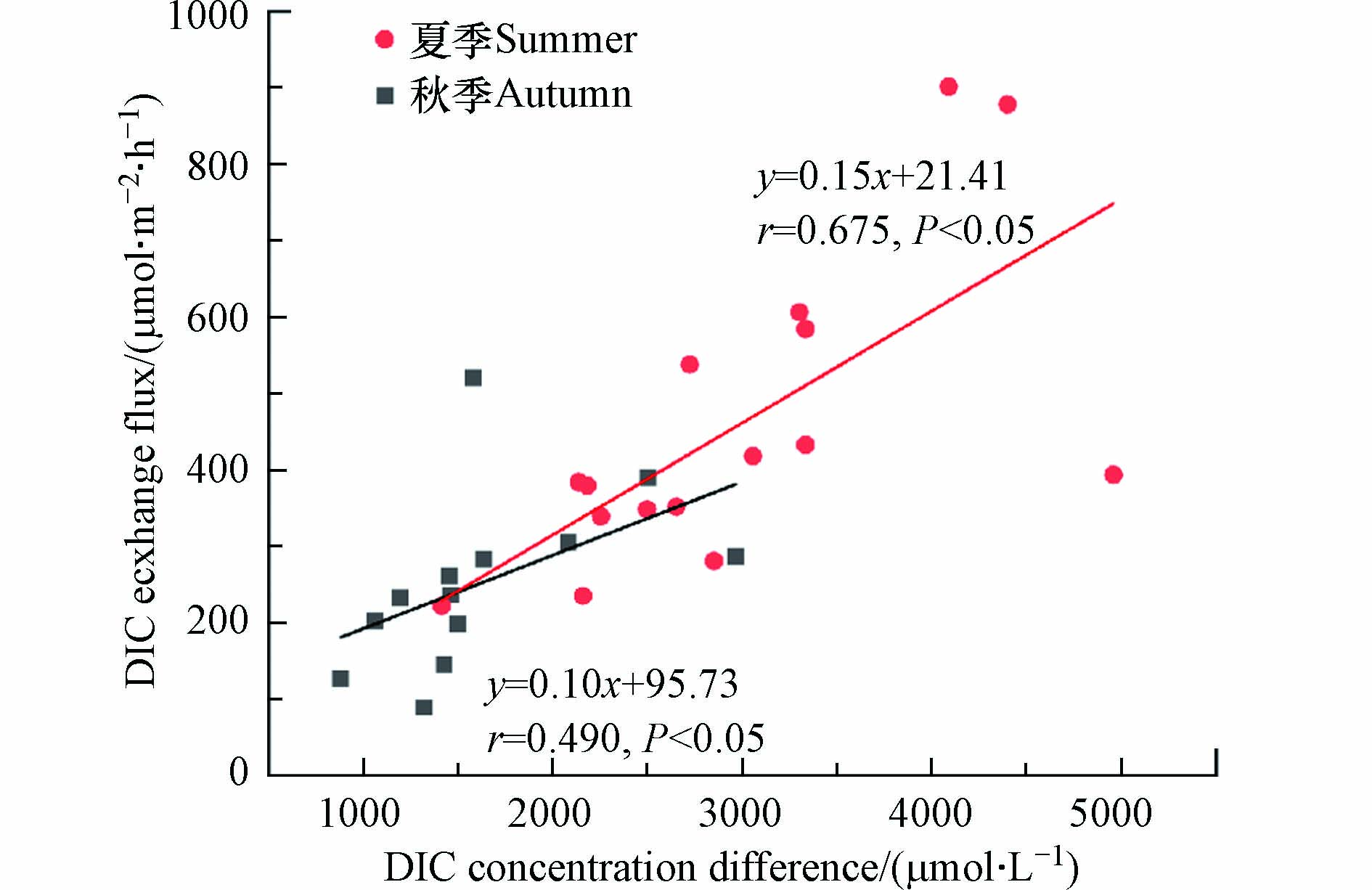
 下载:
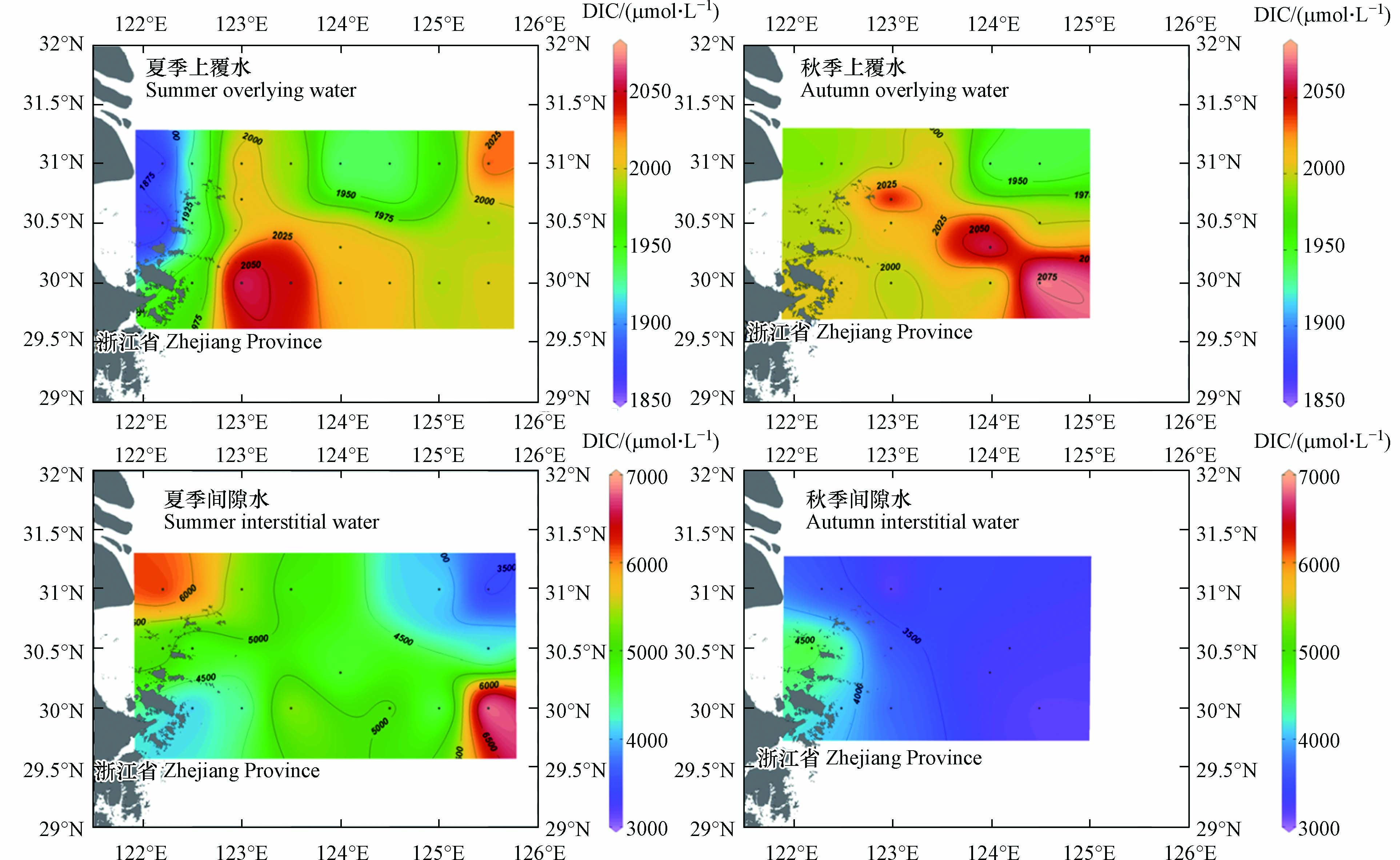
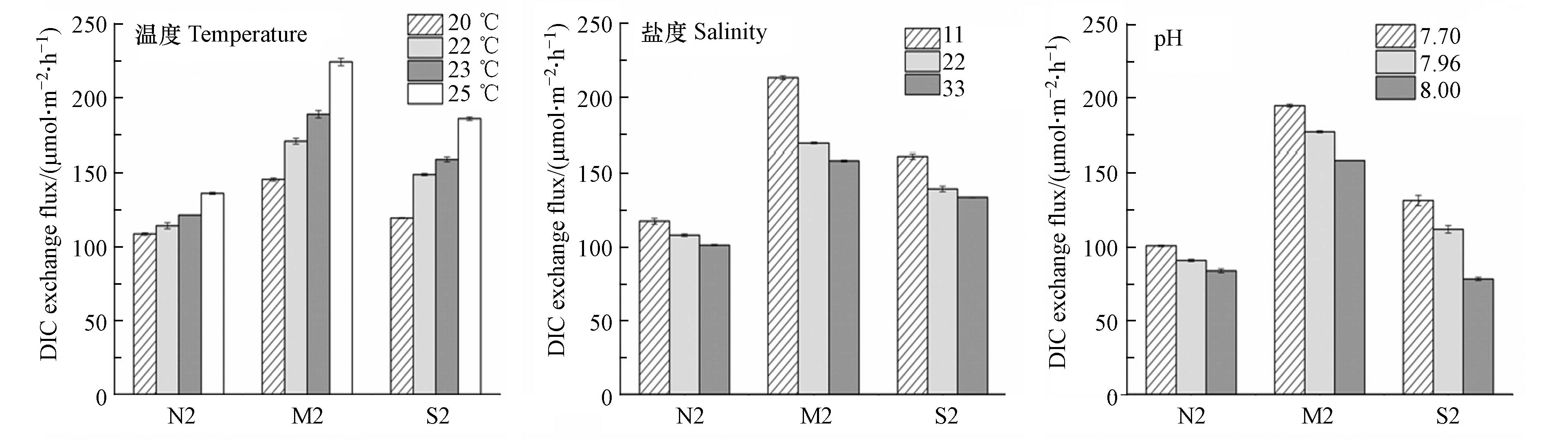
下载:
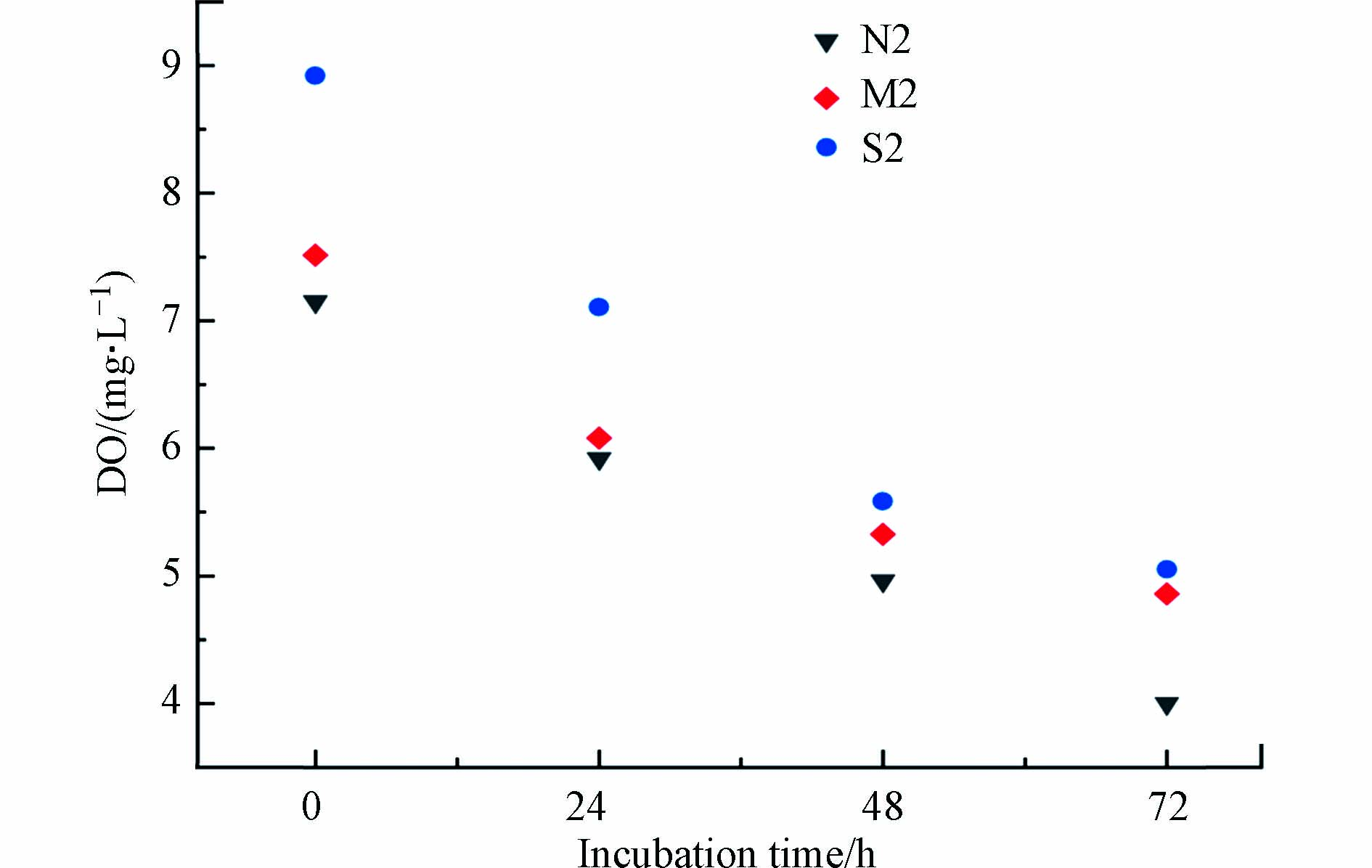
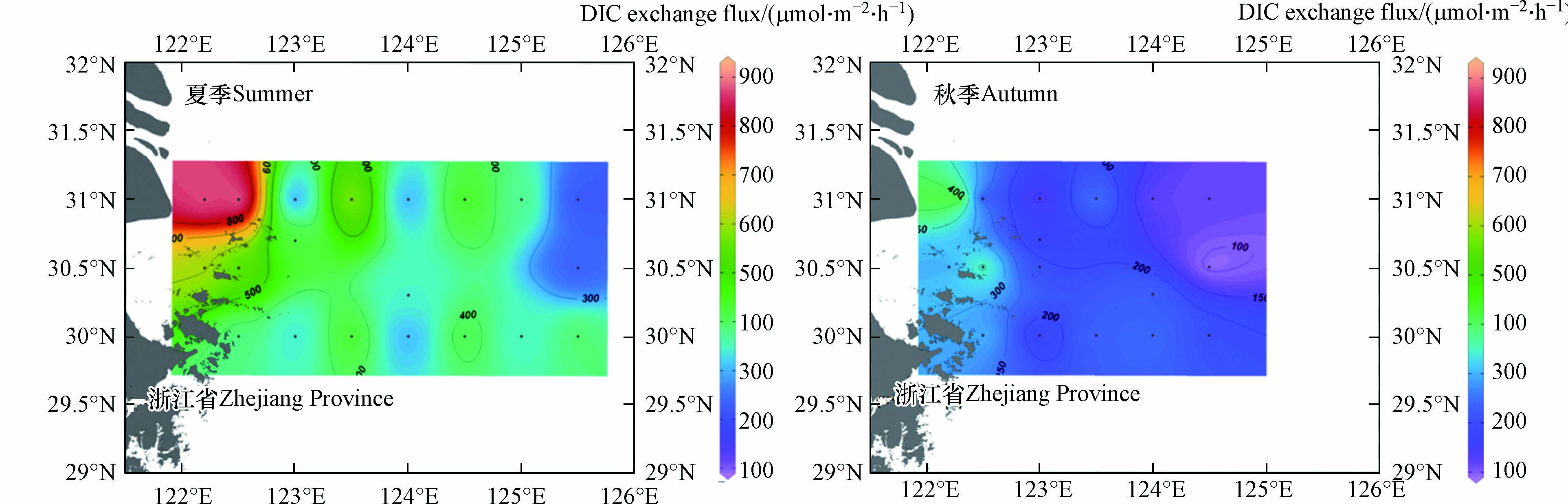




-
原位化学氧化是通过原位注射或添加氧化剂与污染物反应达到修复污染土壤和地下水的目的[1]。众多氧化剂中,过硫酸盐(PS)因产生的自由基氧化能力强、对环境友好等优点被广泛应用于有机污染土壤和地下水的修复[2-3]。通常情况下,PS可通过加热[4]、紫外照射[5]或过渡金属离子[6]等方式活化而产生硫酸根自由基(
SO⋅−4 )和羟基自由基(·OH)[7],以实现有机污染物的高效去除。但针对有机污染土壤和地下水原位化学氧化修复,以上活化方式均存在一定缺陷。例如,加热费用比较昂贵、能耗高;紫外照射活化不适合土壤修复等[8]。相比传统PS活化方式,土壤和地下水基质活化PS降解有机污染物更符合当前绿色修复理念[9-10]。过硫酸盐在25 ℃时的半衰期约为600 d,因而可以传输到其他氧化物质不易到达的污染源区和迁移距离较远的污染羽,但传统活化方式对PS的影响范围有限,因此,土壤和地下水基质对PS的活化对土壤和地下水修复更具优势[11]。土壤中含有大量的Fe/Mn矿物、天然有机质和钒矿物。有研究发现Fe/Mn矿物和天然有机质均可有效活化PS降解有机污染物[12-14],例如,Fe/Mn矿物存在时PS产生
SO⋅−4 和·OH的速率相比无Fe/Mn矿物存在时提高了2~20倍。钒(V)是土壤中普遍存在的微量元素,平均含量约为90 mg·kg−1[15-16];同时V也是一种变价金属,主要以+3、+4、+5价的形式存在于土壤环境中[17]。最近,FANG等[18]考察了不同钒矿物存在时H2O2对污染物的降解,发现在水溶液和泥浆中钒矿物均能有效催化H2O2的分解,达到高效降解邻苯二甲酸二乙酯的目的。综合以上结果可推断,采用PS原位化学氧化修复土壤或地下水时,钒矿物可能会加速PS的分解,有效促进污染物的降解,缩短修复时间。但迄今为止,有关钒矿物对PS降解有机污染物的影响研究较少,其机理也不清楚。2,4-二硝基甲苯(2,4-DNT)作为重要的化工原料,广泛用于医药、染料、农药等行业,在生产、储运和使用过程中如发生渗漏、溢出等事故,则会严重污染土壤和地下水,从而对环境和人体健康造成较大危害。鉴于上述原因,该物质已被列为美国EPA、欧盟以及我国所制定的优先控制有毒有机污染物[19]。基于此,本研究以2,4-DNT为目标污染物,考察3种钒氧化物对PS降解2,4-DNT的影响,并借助自由基淬灭实验和电子自旋共振技术(ESR)阐述2,4-DNT降解过程中的反应机制;随后研究了钒氧化物浓度、PS浓度和初始pH对PS降解2,4-DNT的影响,以期为土壤和地下水基质在PS原位化学氧化中的应用提供参考。
全文HTML


-
三氧化二钒(V2O3)、二氧化钒(VO2)和五氧化二钒(V2O5)购置于北京百灵威科技有限公司;2,4-DNT(98%)购置于日本东京化学工业公司;氢氧化钠(NaOH)、硫酸(H2SO4)、甲醇(MeOH)、乙醇(EtOH)、叔丁醇(TBA)和PS购置于国药集团化学试剂有限公司;5,5-二甲基-1-吡咯啉-N-氧化物(DMPO)购自安普试剂有限公司(上海中国)。实验室pH计购自梅特勒-托利多仪器有限公司(中国上海);低温恒温振荡器 (SHA-2)购自常州菲普实验仪器厂;高效液相色谱仪(LC-20AT)购自日本岛津公司;电子自旋共振波谱仪(ESR,EMXplus-10/12)购自布鲁克股份有限公司(美国)。
-
移取100 mL浓度为14.0 μmol·L−1的2,4-DNT溶液至150 mL锥形瓶后,采用0.1 mol·L−1的H2SO4和NaOH溶液调节pH;随后投加0.060 g PS和0.015 g 钒氧化物于锥形瓶中,在25 ℃下水平振荡。在不同反应时间点(0、1、2、4、7、10 h)分别取2 mL样品,其中1 mL样品甲醇淬灭后测定2,4-DNT浓度,1 mL样品进行ESR-DMPO分析。在自由基淬灭实验中,反应开始前分别加入乙醇或叔丁醇,以保证每种试剂浓度为1.0 mmol·L−1和5.0 mmol·L−1,其他实验条件不变。
在以上反应体系中,分别考察V2O3浓度(0.5、1.0、2.0、5.0和10.0 mmol·L−1)、PS浓度(0.5、1.0、2.0、5.0和10.0 mmol·L−1)、2,4-DNT浓度(5.6、14.0、28.0、56.0和112.0 μmol·L−1)以及初始pH(3.0、5.0、7.0、9.0和11.0)对2,4-DNT降解效果的影响。每组实验均重复进行3次。
-
2,4-DNT浓度分析:采用日本岛津LC-20A高效液相色谱仪测定2,4-DNT浓度,配有SPD-10Avp紫外检测器。色谱柱为Syncronis (240 mm × 4.6 mm,5 μm) C18反相色谱柱,流动相为甲醇∶水= 75%∶25%,检测波长254 nm,流动相流速为1 mL·min−1,柱温35 ℃。
SO⋅−4 和·OH鉴定:采用DMPO作为SO⋅−4 和·OH的捕获剂;反应1 h后,取45 µL反应溶液,在5 s内将5 µL 200 mmol·L−1浓度的DMPO加入到反应溶液中并振荡2 min;随后将注入样品的毛细管置于ESR检测器中进行自由基分析。仪器操作参数:微波频率9.86 GHz、微波功率20 mW、调制频率100 kHz、扫描时间59.39 s[20]。 -
2,4-DNT降解率的计算如式(1)所示,准一级动力学方程[21]如式(2)所示。
式中:X为降解率;C0和Ct分别为反应时间为0和t时刻溶液中2,4-DNT的浓度,μmol·L−1;kobs为准一级动力学常数,h−1。
1.1. 材料与仪器
1.2. 实验方法
1.3. 分析方法


1.4. 相关模型及计算方法
-
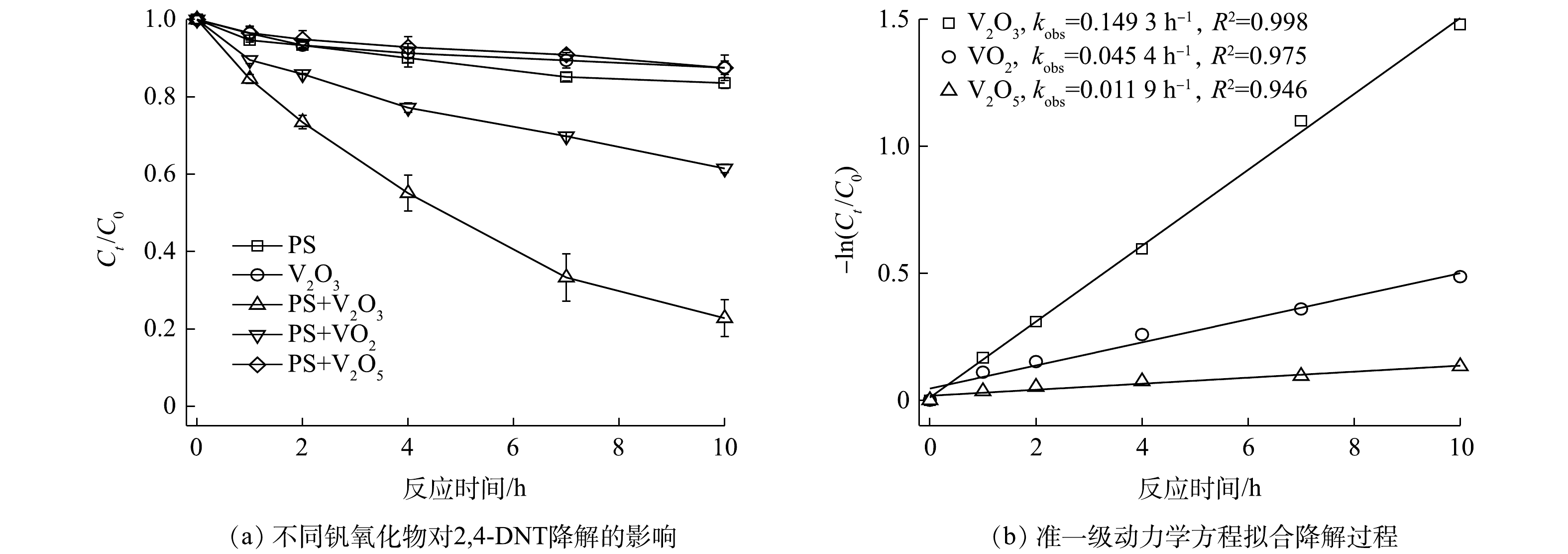
不同钒氧化物对PS降解2,4-DNT的影响情况如图1所示。由图1(a)可见,不同氧化体系中2,4-DNT的降解率差异较为明显,其中PS/V2O3体系表现出较强的氧化能力,而PS/V2O5体系对2,4-DNT的降解效果最差。当PS浓度、钒氧化物浓度、2,4-DNT浓度和初始pH分别为5.0 mmol·L−1、10.0 mmol·L−1、14.0 μmol·L−1和5.0时,反应10 h后,PS/V2O3、PS/VO2和PS/V2O5体系中2,4-DNT的降解率分别为77.2%、38.5%和12.48%。此外,为明确PS/V2O3、PS/VO2和PS/V2O5体系对2,4-DNT的降解过程,采用准一级动力学方程对2,4-DNT的降解数据进行了拟合(图1(b))。结果表明,准一级动力学方程可很好地描述不同体系中2,4-DNT的降解过程(R2>0.94),计算得到的准一级动力学常数kobs分别为0.149 3、0.045 4和0.011 9 h−1,动力学拟合结果进一步表明PS/V2O3体系对2,4-DNT的降解效果最佳。PS/V2O3体系更好的降解性能主要是因为,相比VO2和V2O5,V2O3具有更强的还原能力,会持续活化PS产生较多的
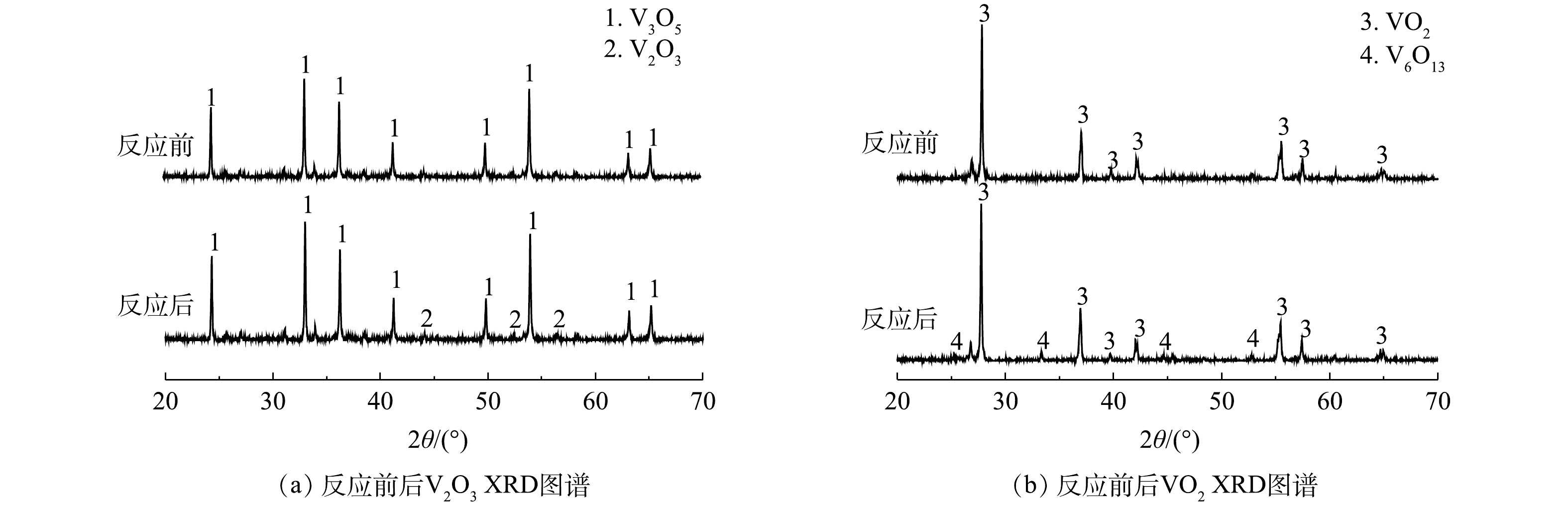
SO⋅−4 和·OH。为了阐明钒氧化物活化PS的机理,借助X射线衍射仪(XRD)对体系中钒氧化物反应前后的晶体结构进行了表征。如图2所示,反应前V2O3和VO2中仅含有V(Ⅲ)和V(Ⅳ),随着PS的加入,反应体系中分别检测出V3O5和V6O13晶体结构,其中V3O5代表V(Ⅲ)和V(Ⅳ)混合物,V6O13代表V(Ⅳ)和V(Ⅴ)混合物,表明钒氧化物主要是通过V(Ⅲ)、V(Ⅳ)和V(Ⅴ)之间的电子转移进行PS的活化。如式(3)~式(9)所示,V2O3表面的V(Ⅲ)失电子氧化为V(Ⅳ),V(Ⅳ)进一步失电子氧化为V(Ⅴ),通过以上连续的电子转移为
S2O2−8 提供电子使其生成SO⋅−4 ,SO⋅−4 进而与H2O或OH−反应生成·OH,以实现对2,4-DNT的有效降解。 -
类似于Fenton反应,PS也会通过V2O3的活化产生
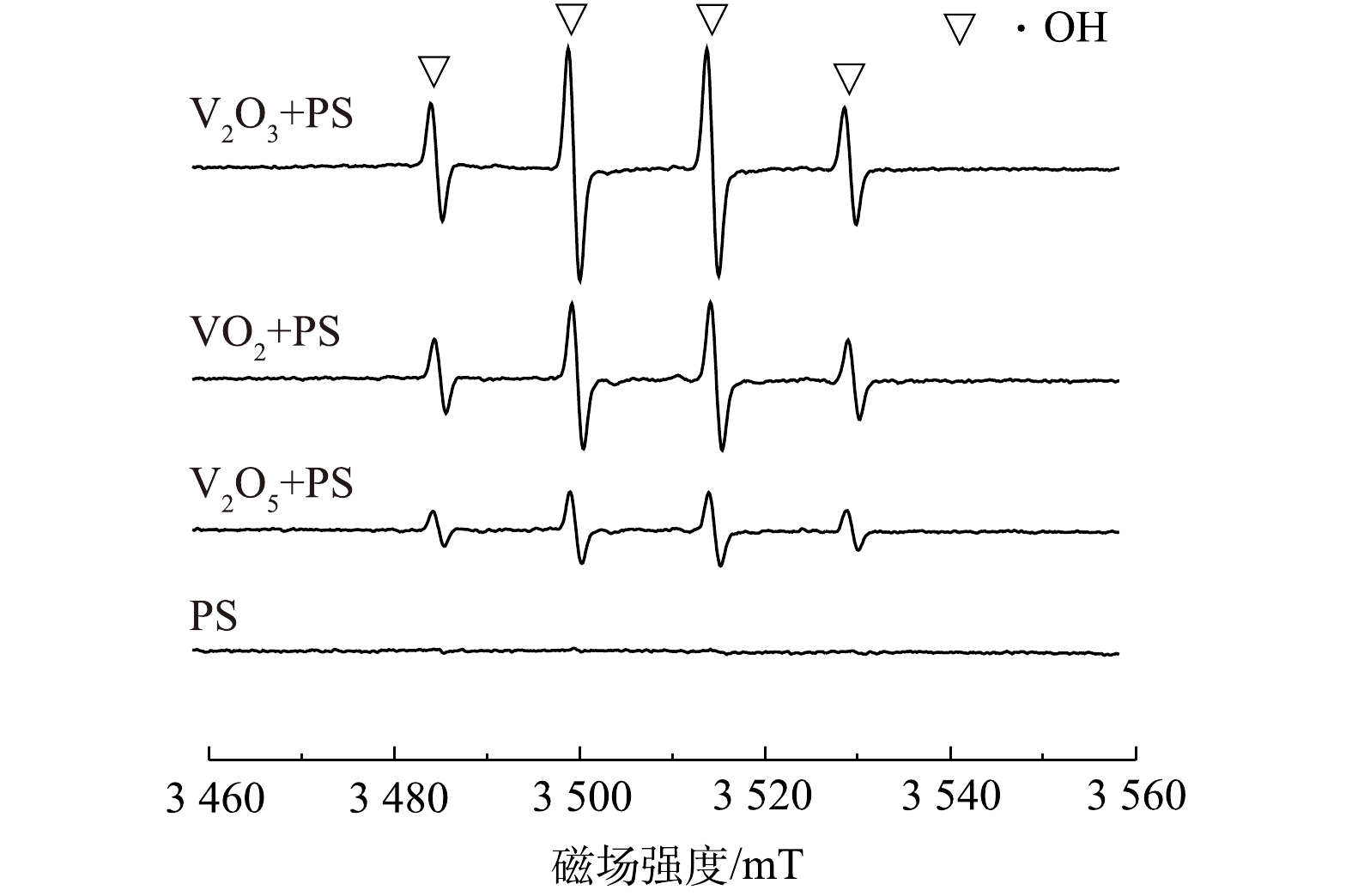
SO⋅−4 ,SO⋅−4 进而与H2O或OH−反应生成·OH。本研究以DMPO为捕获剂,采用ESR技术对PS、PS/VO2、PS/V2O3和PS/V2O5体系中的自由基进行鉴定。由于DMPO-SO⋅−4 信号在高背景下不容易被检测到,故采用DMPO-·OH来鉴定自由基的产生[22]。由图3可以看出,单独PS体系中并未检测到明显的DMPO-·OH特征峰,这表明在25 ℃时PS基本不分解。但当加入VO2、V2O3和V2O5时,反应体系中均出现不同强度的DMPO-·OH特征峰信号,且在PS/V2O3体系中检测到的DMPO-·OH信号强度最高。这表明VO2、V2O3和V2O5对PS均有一定的活化能力,但与VO2和V2O5相比较,V2O3的活化能力最强,这也是在PS/V2O3体系中2,4-DNT降解效果最佳的原因。为进一步明确
SO⋅−4 和·OH对2,4-DNT降解的贡献,以PS/V2O3降解2,4-DNT为例,采用EtOH和TBA进行自由基淬灭实验。通常情况下,EtOH与SO⋅−4 和·OH的反应速率常数分别为(1.6~7.7)×107 L·(mol·s)−1和(1.2~2.8)×109 L·(mol·s)−1;TBA与SO⋅−4 和·OH的反应速率常数分别为(4.0~9.4)×105 L·(mol·s)−1和(3.8~7.6)×108 L·(mol·s)−1 [23]。可见,EtOH可淬灭SO⋅−4 和·OH,而TBA只能淬灭·OH[24-25]。如图4所示,当EtOH和TBA浓度为1.0 mmol·L−1时,2,4-DNT的降解受到显著抑制,在反应10 h时,在加入2种淬灭剂的体系中,2,4-DNT的降解率由80.46%分别降低至15.99%和19.14%;当EtOH和TBA浓度为5.0 mmol·L−1时,2,4-DNT降解几乎完全抑制,这表明在PS/V2O3氧化2,4-DNT过程中,·OH是降解2,4-DNT的主要活性物质。 -
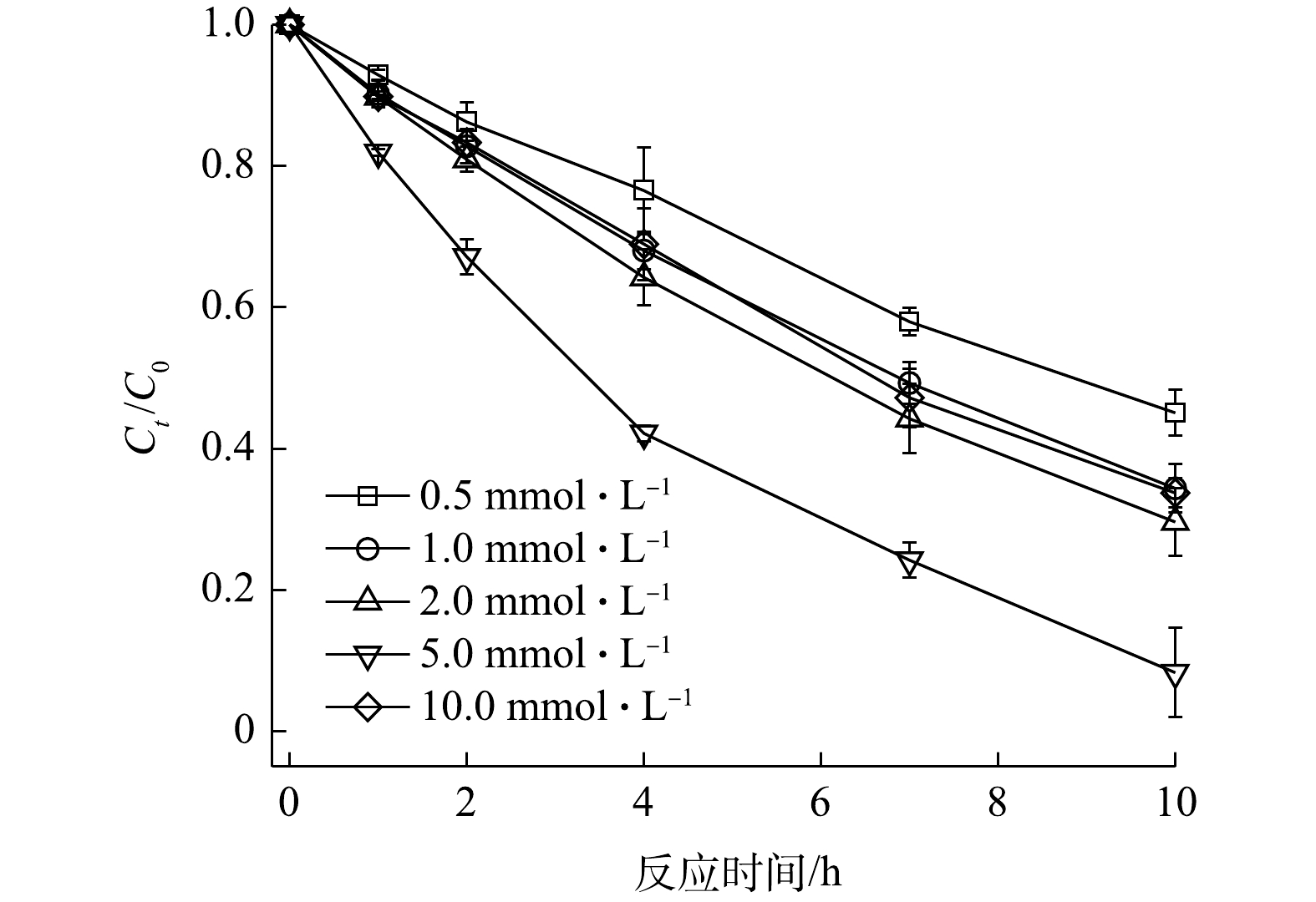
1)V2O3浓度的影响。V2O3浓度对2,4-DNT降解的影响如图5所示。当PS浓度、2,4-DNT浓度和初始pH分别为5.0 mmol·L−1、14.0 μmol·L−1和5.0时,反应10 h后,在V2O3浓度为0.5、1.0、2.0、5.0和10.0 mmol·L−1体系中2,4-DNT的降解率分别为54.94%、65.63%、80.38%、91.70%和66.27%。由图5可以看出,当V2O3浓度为0.5~5.0 mmol·L−1时,2,4-DNT的降解率随V2O3浓度的升高而增加;但当V2O3浓度为10 mmol·L−1时,2,4-DNT降解率明显降低,呈现出抑制作用。这是因为,高浓度的V2O3会使其表面活性点位数量的增加,可以促进
SO⋅−4 和·OH的生成;但过量的V2O3会消耗体系中的SO⋅−4 和·OH,进而抑制2,4-DNT的降解[26]。这与Fe2+活化PS降解污染物的规律一致,即:适当增加Fe2+的浓度可提高PS的活化效率,以增强污染物的降解;但超过一定阈值后,过量的Fe2+会对2,4-DNT的降解产生抑制[27]。2)初始PS和2,4-DNT浓度的影响。当V2O3浓度和2,4-DNT浓度分别为2.0 mmol·L−1和14.0 μmol·L−1,初始pH为5.0时,不同浓度PS对2,4-DNT的降解效果如图6(a)所示。随着PS浓度的升高,PS/V2O3体系中2,4-DNT的降解率显著提升。当反应10 h时,2,4-DNT的降解率由PS浓度为0.5 mmol·L−1时的15.13%提升至PS浓度为10.0 mmol·L−1时的98.74%。采用准一级动力学方程对不同PS浓度时2,4-DNT的降解数据进行拟合,计算得到PS浓度为0.5、1.0、2.0、5.0和10.0 mmol·L−1时2,4-DNT降解的kobs分别为0.015 7、0.024 8、0.038 5、0.174 3和0.425 4 h−1,相关系数R2大部分在0.95以上。反应体系中PS浓度增加会导致溶液中
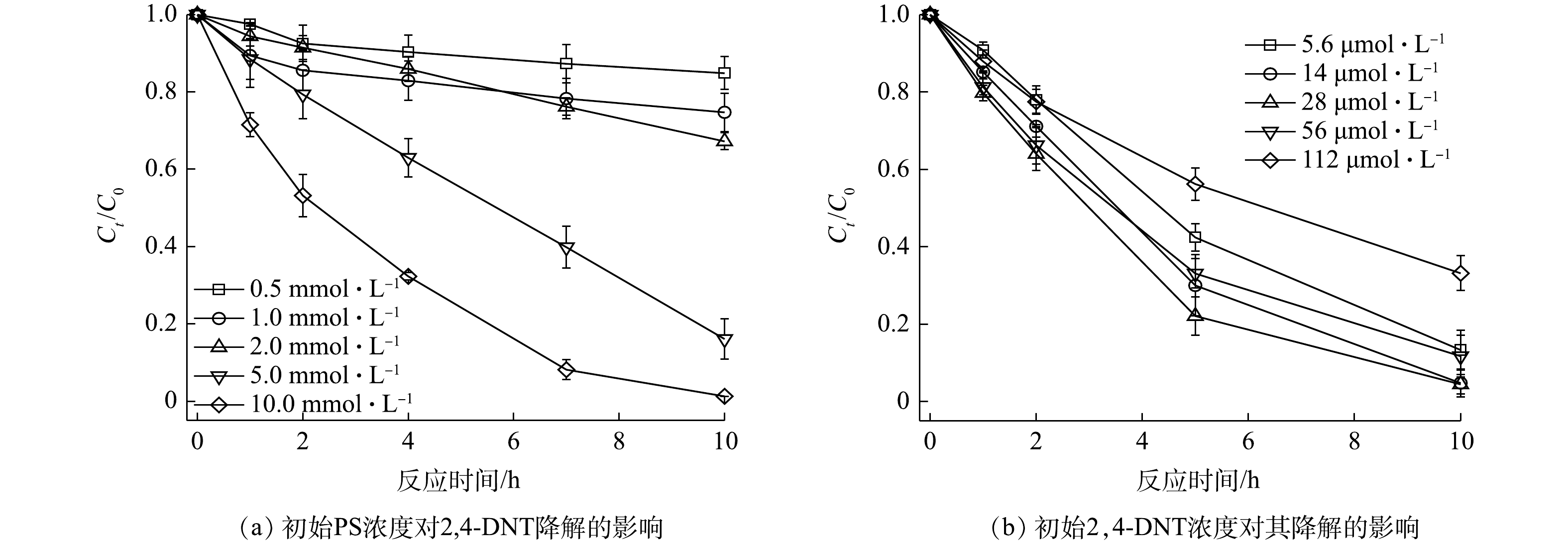
SO⋅−4 或·OH含量升高,从而有效提升2,4-DNT的降解效率。之前的研究也报道了类似的现象,即PS浓度越高,产生的活性自由基含量也相应增加[28-29]。在V2O3和PS的浓度分别为2.0 mmol·L−1和5.0 mmol·L−1、初始pH为5.0的条件下,不同2,4-DNT浓度对其降解的影响如图6(b)所示。研究结果表明,当2,4-DNT浓度分别为5.6、14.0、28.0和56.0 μmol·L−1时,2,4-DNT表现出较高的降解率,分别为86.62%、95.33%、95.54%和88.29%。但当2,4-DNT浓度增加至112.0 μmol·L−1时,2,4-DNT的降解率略有下降,反应10 h时的降解率仅为66.85%。这主要是因为,一定量的PS和V2O3产生的自由基含量有限,难以实现高浓度2,4-DNT的完全降解。
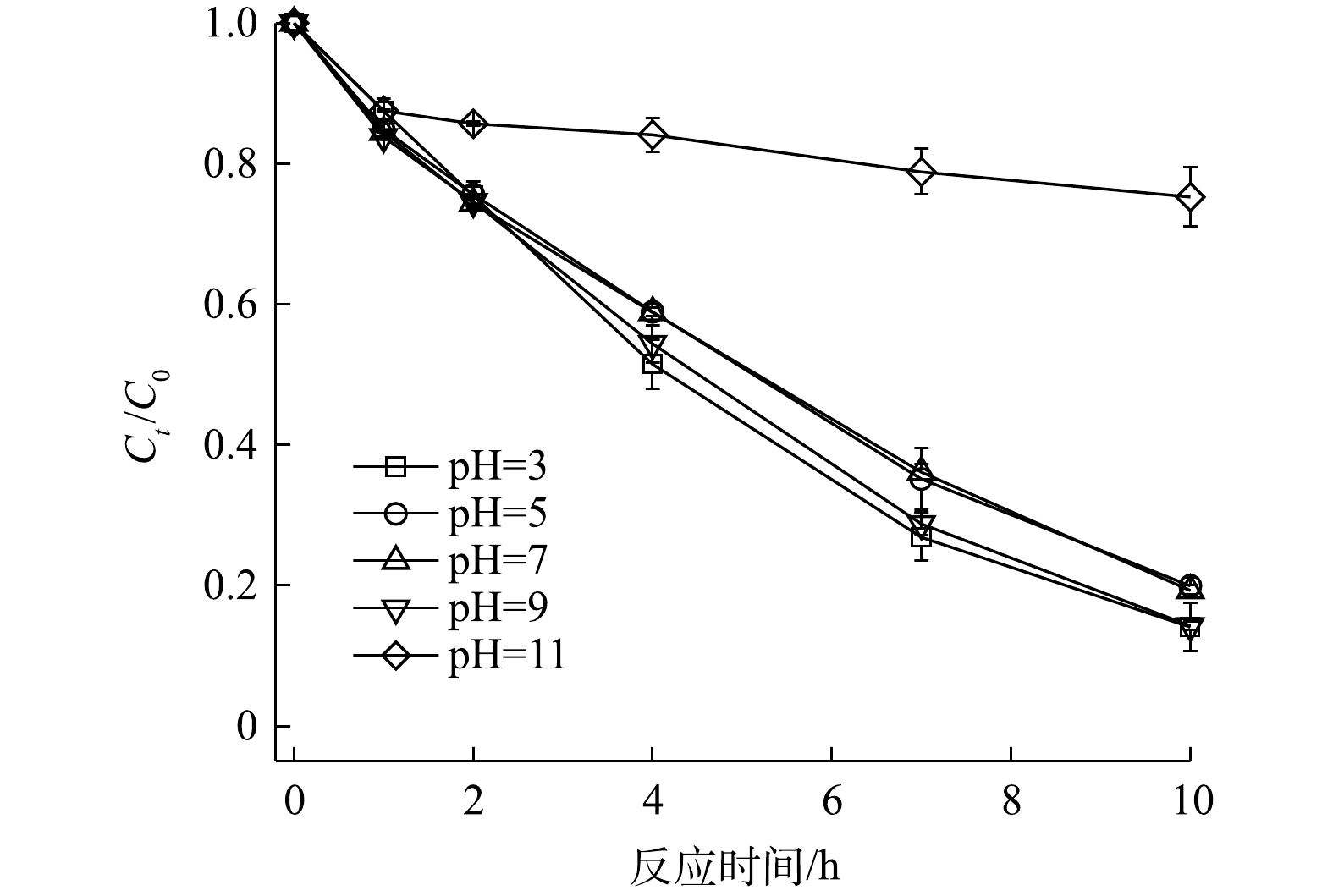
3)初始pH的影响。反应体系的pH在PS原位化学氧化过程中扮演着重要角色。图7为不同pH条件下PS/V2O3体系对2,4-DNT的降解曲线。当PS、V2O3和2,4-DNT浓度分别为5.0 mmol·L−1、2.0 mmol·L−1和14.0 μmol·L−1时,在初始pH为3.0~9.0时,2,4-DNT降解率的变化并不明显,反应10 h时2,4-DNT的降解率分别为85.91%、80.07%、80.72%和85.72%;但当初始pH为11.0时,PS/V2O3体系的降解效果受到明显抑制,反应10 h时2,4-DNT的降解率仅为24.73%。这表明酸性和弱碱性环境有利于V2O3活化PS降解2,4-DNT。其主要原因可能是:在酸性或弱碱性条件下
SO⋅−4 和·OH反应活性较高,而在强碱性条件下,pH的升高会导致V2O3活化PS的能力降低,同时SO⋅−4 转化·OH效率较低,因此,导致强碱性条件下2,4-DNT的降解效果明显受到抑制[30-31]。
2.1. 不同钒氧化物对PS降解2,4-DNT的影响




2.2. PS/钒氧化物体系中自由基鉴定







2.3. PS/V2O3体系降解2,4-DNT的影响因素





-
1)不同钒氧化物体系对2,4-DNT的降解效果呈显著区别。其中,PS/V2O3体系表现出较强的氧化能力,当反应10 h时,2,4-DNT的降解率为77.2%。采用准一级动力学模型可很好的描述PS/V2O3体系对2,4-DNT的降解过程。
2)在加入VO2、V2O3和V2O5后,PS/钒氧化物体系中均出现不同强度的DMPO-·OH特征信号峰,且在PS/V2O3体系中的DMPO-·OH信号强度最高。这说明·OH是降解2,4-DNT的主要活性物质。
3) 2,4-DNT的降解率随V2O3浓度的升高先升高后降低,当V2O3浓度为5.0 mmol·L−1时降解效果最佳(91.70%);随着PS浓度的升高,PS/V2O3体系中2,4-DNT的降解率相应显著提升;当初始pH分别为3.0、5.0、7.0和9.0时,反应10 h后V2O3活化PS对2,4-DNT的降解率分别为85.91%、80.07%、80.72%和85.72%。
