

















































-
随着污水厂尾水排放标准的不断提高,化学辅助除磷、介质过滤、臭氧氧化等[1-15]技术被用在深度处理中,以进一步削减污染物,降低出水水质指标。在城镇污水处理厂提标改造新标准要求下,氨氮和总氮的削减成为重点,强化脱氮技术也备受重视。目前普遍采取以下2种措施:一是改造生物反应池强化除氮,再增加深度处理工艺进一步降低氨氮、总氮等指标;二是依靠深度处理技术改造脱氮工艺[14-20]。而针对生物反应池不同改造工艺运行情况开展现场对比实验的研究报道较少,以不同工艺并联运行,进行同时间、同规模现场生产实验的研究几乎未见报道。
本研究在西安市第五污水处理厂进行。在该厂未满负荷运行、先后具备多种工艺的有利条件下,分批次开展了同时间、同规模、不同工艺的现场生产性实验,对几种工艺的运行情况进行了对比研究。西安市第五污水处理厂总设计污水处理规模为40×104 m3·d−1,设计出水水质执行《城镇污水处理厂污染物排放标准》(GB18918-2002)[21]中的一级A标准,采用“预处理+AAO(A系列生物池采用MBBR投加填料)+纤维转盘滤池+次氯酸钠消毒处理”工艺,现状处理水量约30×104 m3·d−1,全厂分为A、B、C、D四个系列生物反应池并联运行,单系列设计规模10×104 m3·d−1。2018年进行了B系列AAO工艺与A系列“AAO+MBBR”工艺的生产性对比实验,2019年A、B系列Ⅳ类提改造后进行了C、D系列AAO工艺与B系列五段式Bardenpho工艺的生产性对比实验。分析比选了不同工艺在强化除氮方面的优缺点,以期为国内城镇污水处理厂准Ⅳ类水质提标改造工程应用提供参考。
-
该厂一期工程(A、B系列)AAO工艺于2010年建成投产,于2013年进行了生物池一级A水质标准提标改造。改造后A系列生物反应池采用“AAO+MBBR工艺”,即在原AAO工艺的基础上,在好氧区中后段增加MBBR悬浮填料区,投加MBBR悬浮填料比例约17%,并增设填料专用推进器、拦网等辅助设施设备;B系列生物池沿用原有AAO工艺,并未进行改造。该厂二期工程(C、D系列)AAO工艺于2018年建成投产。A、B、C、D四个系列AAO工艺生物反应池的水力停留时间均为16.59 h。
2019年,该厂以准Ⅳ类水质标准提标改造为目的,改造为多模式五段式Bardenpho工艺。具体实施内容包括:对A、B系列生物反应池先后进行了工程改造,同时去除A系列MBBR填料;在生物池缺氧区中后段增加隔墙,在好氧区增加2道隔墙;将A、B系列生物反应池均改造为5个区域,即厌氧区、缺氧区、好氧区、第二缺氧区、第二好氧区;C、D系列未进行改造。
-
1)实验进水。即厂区进水原水。进水采样使用自动采样器(sigma900型,哈希公司),每2 h进行自动取样。取样点位于第五污水处理厂进厂市政管网末端10#井。
2)实验出水。即对应实验系列二沉池出水汇流井内或总出水口水样。采样使用取样桶(1 000 mL塑料桶)。取样点位于水下30 cm。每日上午10点取样。
3) MBBR工艺实验用水。该组实验的样品包括A、B系列生物池厌氧区进、出口处混合液,A、B系列生物池缺氧区进、出口处混合液,A、B系列生物池好氧区中段处混合液,以及A、B系列生物池好氧区出口处混合液。采样使用取样桶(1 000 mL塑料桶),取样点位于液面下方1 m处,每日上午10点取样。所有过程样品均使用定量滤纸过滤后,取清液进行检测。
4) Bardenpho工艺实验用水。该组实验的样品包括B系列生物池厌氧区进、出口处混合液,B系列生物池缺氧区进、出口处混合液,B系列生物池好氧区出口处混合液,B系列生物池第二缺氧区出口处混合液,以及B系列生物池第二好氧区出口处混合液。采样使用取样桶(1 000 mL塑料桶),取样点位于液面下方1 m处,每日上午10点取样。所有过程样品均使用定量滤纸过滤后,取清液进行检测。C、D系列未取过程样。
5)分析指标:COD(HJ 828-2017重铬酸钾法)、NH3-N(HJ 535-2009纳氏试剂分光光度法)、
NO−3 -N (HJ/T 346-2007紫外分光光度法)、TN(HJ 636-2012碱性过硫酸钾消解紫外分光光度法)、PO3−4 -P (钼锑抗分光光度法(A))[22]、TP(GB 11893-89钼酸铵分光光度法)、MLSS(CJ/T221-2005重量法)、DO(HJ 506—2009电化学探头法)。 -
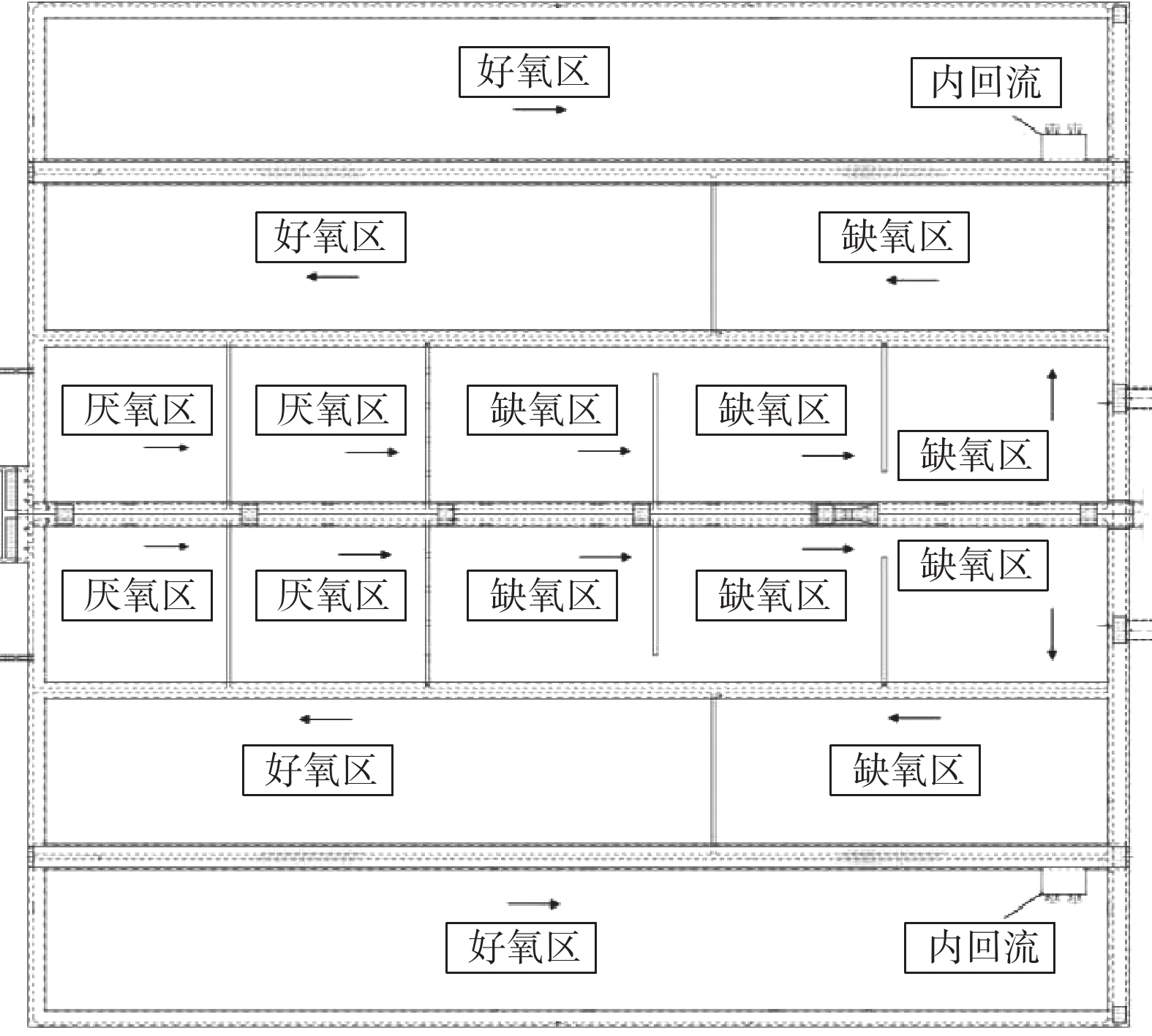
MBBR生产性比选实验在A、B系列同等工况下进行。不投加碳源,在生物池混合液出水口投加除磷药剂。实验的检测日期安排在2018年6月至10月。在这段时间内,随机抽取连续稳定运行11 d的数据。B系列工艺流程图如图1和2所示。实验期间运行水量均为7.5×104 m3·d−1。A、B系列生物池的MLSS控制在5~6 g·L−1;生物池末端DO远程控制在1.0 mg·L−1;污泥回流比为80%;内回流比为200%;除磷药剂的投加量为100 mg·L−1。
A系列生物反应池为“MBBR+AAO工艺”,总水力停留时间16.59 h。其中,厌氧区2.02 h,缺氧区5.53 h,好氧区9.04 h。好氧区MBBR填料区位于好氧段中后段,为循环跑道式填料。填料区的停留时间1.74 h,由4台潜水推流搅拌器进行搅拌及循环推流。B系列生物反应池为AAO工艺,总水力停留时间16.59 h,其中厌氧区2.02 h,缺氧区5.53 h,好氧区9.04 h。
-
2019年,该厂进行准Ⅳ类提标改造,在A、B系列生物反应池中开展Bardenpho工艺的生产性实验,对比B系Bardenpho生物反应池与C、D系列生物反应池的污水处理情况。实验在同等工况下进行,不投加碳源,在生物池混合液出水口投加除磷药剂。实验的检测日期安排在2019年11月至12月。在这段时间内,随机抽取连续稳定运行9 d的数据进行分析。实验期间,B、C、D系列均保持连续稳定运行,工艺流程如图3所示。
本次改造后,B生物池系统总停留时间为16.59 h。其中,厌氧区2.02 h,缺氧区4.10 h,好氧区7.30 h(可调节区好氧运行),第二缺氧区1.92 h,第二好氧区1.25 h。为增强搅拌效果,加强反硝化速率,基于原有搅拌器,在厌氧区、缺氧区、第二缺氧区增加双曲面搅拌器。另外,将生物池混合液回流泵由原好氧区末端移动至第二缺氧区前端,新增回流泵安装隔墙,走道板、回流管道等。
Bardenpho生产性比选实验在B、C、D系列同等工况下运行,不投加碳源,期间一期A系列全部停运。B系列按照Bardenpho模式运行,以区别于C、D系列原有AAO工艺的出水水质。实验期间,B系列与C、D系列均满负荷运行,即B系列运行水量为10×104 m3·d−1,C、D系列运行水量为20×104 m3·d−1。B系列及C、D系列生物池的污泥浓度均控制在5~6 g·L−1;生物池末端的DO远程控制在1.0 mg·L−1;外回流比为70%;内回流比为200%;使用厂家专利除磷药剂,投加量稳定为100 mg·L−1。同时,加强对好氧段末端DO的控制,确保第二缺氧段缺氧状态。
-
实验中进出水水质见表1和2。A系列的出水平均COD较B系列略低,平均值相差不大。计算COD数据的方差,得到A系列方差为6.611,B系列方差为14.049,说明A系列出水的COD指标更稳定。A系列出水平均NH3-N较B系列略高。除10月19日,由于当天控制DO偏低,导致NH3-N升高,其余样品的出水NH3-N均稳定在1.5 mg·L−1以下。计算NH3-N数据的方差,得到A系列方差为0.252,B系列方差为0.772,说明A系列出水的NH3-N指标更稳定。A系列的出水平均TN较B系列低,A系列的TN去除率略高于B系列,去除效果较好。计算TN数据的方差,得到A系列方差为5.796,B系列方差为6.651,说明A系列出水的TN指标更稳定。在除磷药剂投加量相同的情况下,A系列的出水平均
PO3−4 -P值较B系列高0.814 mg·L−1,但A系列的去除率明显低于B系列,除磷效果无明显优势。通过计算方差,发现A系列方差为1.554,B系列方差为0.453,说明B系列出水的PO3−4 P指标更稳定。 -
由于A、B系列工艺的除氮效果存在差异,因此,通过A、B系列
NO−3 -N过程样数据(见图4和5)来分析存在差异的原因。A、B系列的缺氧段均发生了反硝化过程,使得NO−3 -N的质量浓度降低。其中,A系列缺氧段NO−3 -N平均质量浓度降低了2.020 mg·L−1,B系列缺氧段NO−3 -N平均质量浓度降低了1.136 mg·L−1。A系列好氧段中段到末段,部分NO−3 -N的质量浓度有所下降,这可能是好氧区中后段MBBR填料中同步脱氮反硝化反应的体现[23]。并且,A系列缺氧段反硝化效果更佳,可能是由于MBBR工艺悬浮填料中脱落的反硝化细菌等长泥龄微生物部分附着在活性污泥中,经混合液回流至缺氧区,提高了反硝化速率,致使A系列出水硝氮更低。 -
Bardenpho生产实验的进出水水质见表3和4。B系列的出水平均COD较C、D系列略低,去除率相近。计算COD数据的方差,得到B系列方差为8.599,C、D系列方差为22.122,说明Bardenpho工艺中B系列出水的COD指标更稳定。B系列的出水平均NH3-N较C、D系列略高,但B系列NH3-N去除率较低。计算NH3-N数据的方差,得到B系列方差为0.1647,C、D系列方差为0.043 7。就去除效果而言,C、D系列具有微弱优势,且出水NH3-N指标更稳定,但B系列出水NH3-N稳定低于1.5 mg·L−1。B系列的出水平均TN与C、D系列相比略低,且B系列TN去除率较高。计算TN数据的方差,得到B系列方差为1.369,C、D系列方差为0.599。就去除效果而言,B系列具有一定优势,但C、D系列出水的TN指标更稳定。在除磷药剂投加量相同的情况下,B系列出水的平均
PO3−4 -P较C、D系列略高,去除率接近。计算PO3−4 -P数据的方差,得到发现B系列方差为0.006 2,C、D系列方差为0.000 4,说明B系列与C、D系列出水水质指标PO3−4 -P的稳定性接近。 -
由于Bardenpho工艺第二缺氧区停留时间较短,B系列好氧区与第二好氧区的曝气会影响第二缺氧区的缺氧状态,进而影响第二缺氧区的反硝化效果。为确保实验效果,Bardenpho生产性实验中加强了DO控制,以确保第二缺氧段处于缺氧状态。结果亦表明污水处理过程中TN明显降低。在前文关于MBBR实验部分,已对原有AAO工艺中NH3-N、
NO−3 -N的过程样数据进行分析,且Bardenpho工艺比AAO工艺分段多(AAO为3段,Bardenpho为5段),不具备全流程横向比较条件,因此,后面重点分析B系列的NH3-N、NO−3 -N、DO过程样。B系列Bardenpho实验期间NH3-N、NO−3 -N、DO过程样如图6~8所示。分析Bardenpho生产性实验期间B系列生物池过程样的检测结果,发现厌氧区进口至厌氧区出口水样指标NH3-N的平均值下降了2.078 mg·L−1;缺氧区进口至缺氧区出口(剔除11月19日的异常数据)NH3-N的平均值下降了0.327 mg·L−1;缺氧区进口至缺氧区出口
NO−3 -N平均值下降了2.274 mg·L−1,但缺氧区进口水样指标NO−3 -N的平均值仅为3.295 mg·L−1。这说明,系统通过反硝化过程转化了69.01%的NO−3 -N,效果良好,在部分缺氧区出口的样品中已检测不到NO−3 -N。由此可判断,厌氧区和缺氧区的厌氧、缺氧状态良好,处理效果明显。好氧区出口至第二缺氧区出口平均
NO−3 -N下降了2.882 mg·L−1,说明第二缺氧段存在明显的反硝化过程;第一段好氧区出口水样NH3-N指标的平均值为2.106 mg·L−1,而第二缺氧区出口水样比第二好氧区出口水样的NO−3 -N指标有所上升,这可能是由于采集个别样品时第二缺氧区出口水样中DO高于0.5 mg·L−1,此时部分NH3-N在第二好氧区发生了硝化反应。然而,即使考虑该原因的影响,好氧区出口水样比第二好氧区出口水样中平均NO−3 N仍下降了0.836 mg·L−1,故可确定在未投加碳源的情况下,第二缺氧区仍存在一定的反硝化反应。 -
与原有C、D系列AAO工艺相比,B系列Bardenpho工艺的NH3-N去除效果略差,去除率略降低。进一步分析设计参数后发现,B系列Bardenpho工艺好氧区总水力停留时间较C、D系列AAO工艺减少了0.49 h;同时,由于B系列一段、二段好氧区末端的DO明显低于C、D系列AAO工艺好氧区末端的DO,因此,DO可能对出水的NH3-N指标产生较大影响,具体数据如表4所示。
对比原有AAO工艺与Bardenpho工艺,结合实验数据和过程分析,采用硝化、反硝化动力学计算缺氧区容积计算公式(1)进行计算,以核算AAO工艺与Bardenpho工艺的反硝化速率是否符合设计要求。
式中:Vn为缺氧区(池)容积,m3;Q为生物反应池设计流量,m3·d−1;X为生物反应池内混合液悬浮固体平均浓度,g·L−1;△Xv为排除生物反应池系统的微生物量,kg·d−1;Kde为脱氮速率,kg· (kg·d) −1,根据实验资料确定。无实验资料时,一般20 ℃时Kde经验值为0.03~0.06 kg· (kg·d) −1,并按相关公式进行温度修正,若温差不大可忽略。
Bardenpho生产性实验中C、D系列具体相关数据及计算结果见表5和6。
在计算中,C、D系列AAO工艺的反硝化速率以全段水力停留时间计算,B系列五段式Bardenpho工艺的反硝化速率以前3段AAO水力停留时间计算。由于Bardenpho工艺第一缺氧区水力停留时间小于原有AAO工艺缺氧区停留时间,表现为计算中VN不同。另外,Bardenpho工艺的TN取样点在一段好氧区出口,AAO工艺在好氧区出口。根据表6和7,Bardenpho工艺有2 d高于设计下限,AAO工艺有1 d高于设计下限,2种工艺反硝化速率平均值的区别较小,且均低于理论设计负荷0.03~0.06 kg· (kg·d)−1。分析2种工艺的结构,由于Bardenpho工艺有第二缺氧区和第二好氧区进行二次反硝化,且该区域无混合液回流影响,故若进一步增加实际反硝化水力停留时间,可进一步降低出水水样中的TN、NH3-N。
-
1)在同工况、不投加碳源情况下,“AAO+MBBR”工艺相较原有AAO工艺对系统NH3-N的去除效果无明显提升,但对TN、
NO−3 -N有一定的降低效果,且“AAO+MBBR”工艺出水水样中TN更稳定性,对PO3−4 -P的去除效果弱于原有AAO工艺。2)在同工况、不投加碳源情况下,五段式Bardenpho工艺较原有AAO工艺对TN、
NO−3 -N的去除效果有明显提升,出水水样的平均TN降低1.14 mg·L−1,就去除效果而言,B系列去除效果更优。此外,同样条件下五段式Bardenpho工艺对NH3-N的去除效果较差,但指标能稳定低于1.5 mg·L−1,达到准Ⅳ类水质对出水NH3-N、TN的要求。3)在Bardenpho工艺生产实验期间,DO数据不稳定,多次出现第二缺氧段无法稳定处于缺氧状态的情况,在一定程度上影响了出水水质指标。建议使用精确曝气系统代替人工远程调控以加强生物池中DO的控制,以进一步确保NH3-N、TN的出水稳定性。五段式Bardenpho工艺可加强除氮效果,保障出水水质指标NH3-N、TN的稳定达标,在第二缺氧段投加碳源还可加强反硝化效果。
西安市第五污水处理厂生物反应池强化脱氮工艺的比选生产实验
Comparative in-process study on enhanced nitrogen removal processes in biological reaction tank in Xi'an No.5 wastewater treatment plant
-
摘要: 为筛选强化脱氮工艺,利用城镇污水处理厂不同系列的AAO+MBBR工艺、五段式多模式Bardenpho工艺、原有AAO工艺,在生物反应池中进行了比选生产性实验。结果表明:在同工况、同时间、同负荷、不外加碳源情况下,AAO+MBBR工艺与原有AAO工艺相比,出水氨氮相近,总氮降低0.49 mg·L−1,有一定除氮优势;Bardenpho工艺与原有AAO工艺相比,出水氨氮稳定低于1.5 mg·L−1,出水总氮降低1.14 mg·L−1;在不投加碳源情况下,第二缺氧区出现明显的二次反硝化过程。本研究结果可为城镇污水处理厂提标改造工艺的路线选择提供参考。Abstract: To select an enhanced denitrification process in a biological reaction tank, the in-process experiments of biological reaction tank were carried out by using different series of AAO+MBBR processes, the five-stage multi-mode Bardenpho process and the original AAO processes in a urban sewage treatment plant. The experimental results show that compared with the original AAO process the AAO+MBBR process has similar effluent ammonia but low total nitrogen (0.49mg. L-1 lower) under the same working conditions, load and time and without adding any carbon source. Compared with the original AAO process, the Bardenpho process can stabilize the effluent ammonia nitrogen below 1.5 mg•L-1, and the total effluent nitrogen is reduced by 1.14mg•L-1. Without the additional of carbon source, the second anoxic zone shows an obvious secondary denitrification effect. Results in this study can provide reference for the selection of process routes for upgrading and reconstruction of urban sewage treatment plants.
-
随着污水厂尾水排放标准的不断提高,化学辅助除磷、介质过滤、臭氧氧化等[1-15]技术被用在深度处理中,以进一步削减污染物,降低出水水质指标。在城镇污水处理厂提标改造新标准要求下,氨氮和总氮的削减成为重点,强化脱氮技术也备受重视。目前普遍采取以下2种措施:一是改造生物反应池强化除氮,再增加深度处理工艺进一步降低氨氮、总氮等指标;二是依靠深度处理技术改造脱氮工艺[14-20]。而针对生物反应池不同改造工艺运行情况开展现场对比实验的研究报道较少,以不同工艺并联运行,进行同时间、同规模现场生产实验的研究几乎未见报道。
本研究在西安市第五污水处理厂进行。在该厂未满负荷运行、先后具备多种工艺的有利条件下,分批次开展了同时间、同规模、不同工艺的现场生产性实验,对几种工艺的运行情况进行了对比研究。西安市第五污水处理厂总设计污水处理规模为40×104 m3·d−1,设计出水水质执行《城镇污水处理厂污染物排放标准》(GB18918-2002)[21]中的一级A标准,采用“预处理+AAO(A系列生物池采用MBBR投加填料)+纤维转盘滤池+次氯酸钠消毒处理”工艺,现状处理水量约30×104 m3·d−1,全厂分为A、B、C、D四个系列生物反应池并联运行,单系列设计规模10×104 m3·d−1。2018年进行了B系列AAO工艺与A系列“AAO+MBBR”工艺的生产性对比实验,2019年A、B系列Ⅳ类提改造后进行了C、D系列AAO工艺与B系列五段式Bardenpho工艺的生产性对比实验。分析比选了不同工艺在强化除氮方面的优缺点,以期为国内城镇污水处理厂准Ⅳ类水质提标改造工程应用提供参考。
1. 生产性实验方案
1.1 污水处理厂工艺概况
该厂一期工程(A、B系列)AAO工艺于2010年建成投产,于2013年进行了生物池一级A水质标准提标改造。改造后A系列生物反应池采用“AAO+MBBR工艺”,即在原AAO工艺的基础上,在好氧区中后段增加MBBR悬浮填料区,投加MBBR悬浮填料比例约17%,并增设填料专用推进器、拦网等辅助设施设备;B系列生物池沿用原有AAO工艺,并未进行改造。该厂二期工程(C、D系列)AAO工艺于2018年建成投产。A、B、C、D四个系列AAO工艺生物反应池的水力停留时间均为16.59 h。
2019年,该厂以准Ⅳ类水质标准提标改造为目的,改造为多模式五段式Bardenpho工艺。具体实施内容包括:对A、B系列生物反应池先后进行了工程改造,同时去除A系列MBBR填料;在生物池缺氧区中后段增加隔墙,在好氧区增加2道隔墙;将A、B系列生物反应池均改造为5个区域,即厌氧区、缺氧区、好氧区、第二缺氧区、第二好氧区;C、D系列未进行改造。
1.2 水样的采集
1)实验进水。即厂区进水原水。进水采样使用自动采样器(sigma900型,哈希公司),每2 h进行自动取样。取样点位于第五污水处理厂进厂市政管网末端10#井。
2)实验出水。即对应实验系列二沉池出水汇流井内或总出水口水样。采样使用取样桶(1 000 mL塑料桶)。取样点位于水下30 cm。每日上午10点取样。
3) MBBR工艺实验用水。该组实验的样品包括A、B系列生物池厌氧区进、出口处混合液,A、B系列生物池缺氧区进、出口处混合液,A、B系列生物池好氧区中段处混合液,以及A、B系列生物池好氧区出口处混合液。采样使用取样桶(1 000 mL塑料桶),取样点位于液面下方1 m处,每日上午10点取样。所有过程样品均使用定量滤纸过滤后,取清液进行检测。
4) Bardenpho工艺实验用水。该组实验的样品包括B系列生物池厌氧区进、出口处混合液,B系列生物池缺氧区进、出口处混合液,B系列生物池好氧区出口处混合液,B系列生物池第二缺氧区出口处混合液,以及B系列生物池第二好氧区出口处混合液。采样使用取样桶(1 000 mL塑料桶),取样点位于液面下方1 m处,每日上午10点取样。所有过程样品均使用定量滤纸过滤后,取清液进行检测。C、D系列未取过程样。
5)分析指标:COD(HJ 828-2017重铬酸钾法)、NH3-N(HJ 535-2009纳氏试剂分光光度法)、
NO−3 PO3−4 1.3 运行方案及参数
1.3.1 MBBR生产性实验运行方案及参数
MBBR生产性比选实验在A、B系列同等工况下进行。不投加碳源,在生物池混合液出水口投加除磷药剂。实验的检测日期安排在2018年6月至10月。在这段时间内,随机抽取连续稳定运行11 d的数据。B系列工艺流程图如图1和2所示。实验期间运行水量均为7.5×104 m3·d−1。A、B系列生物池的MLSS控制在5~6 g·L−1;生物池末端DO远程控制在1.0 mg·L−1;污泥回流比为80%;内回流比为200%;除磷药剂的投加量为100 mg·L−1。
A系列生物反应池为“MBBR+AAO工艺”,总水力停留时间16.59 h。其中,厌氧区2.02 h,缺氧区5.53 h,好氧区9.04 h。好氧区MBBR填料区位于好氧段中后段,为循环跑道式填料。填料区的停留时间1.74 h,由4台潜水推流搅拌器进行搅拌及循环推流。B系列生物反应池为AAO工艺,总水力停留时间16.59 h,其中厌氧区2.02 h,缺氧区5.53 h,好氧区9.04 h。
1.3.2 Bardenpho生产性实验运行方案及参数
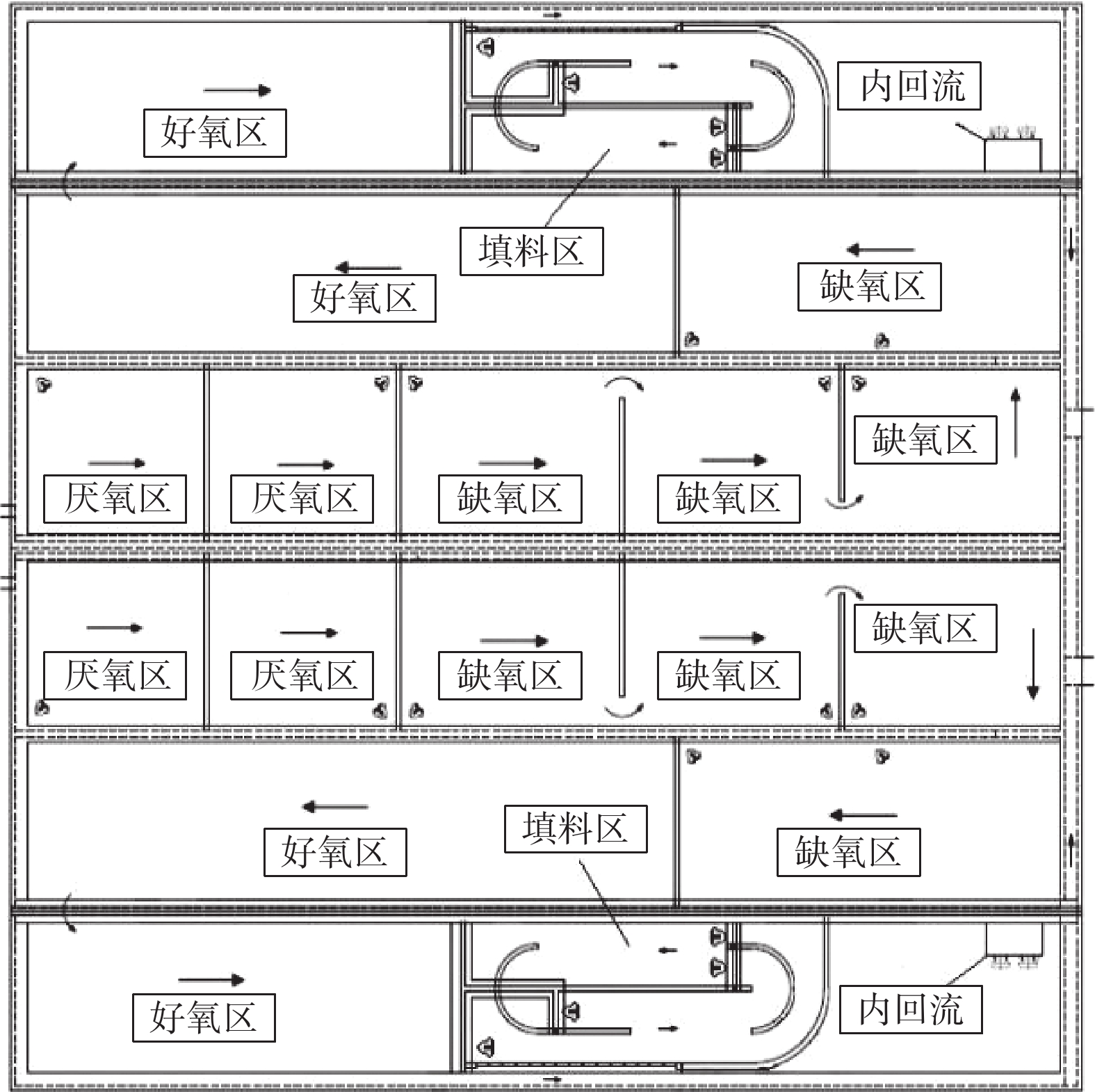
2019年,该厂进行准Ⅳ类提标改造,在A、B系列生物反应池中开展Bardenpho工艺的生产性实验,对比B系Bardenpho生物反应池与C、D系列生物反应池的污水处理情况。实验在同等工况下进行,不投加碳源,在生物池混合液出水口投加除磷药剂。实验的检测日期安排在2019年11月至12月。在这段时间内,随机抽取连续稳定运行9 d的数据进行分析。实验期间,B、C、D系列均保持连续稳定运行,工艺流程如图3所示。
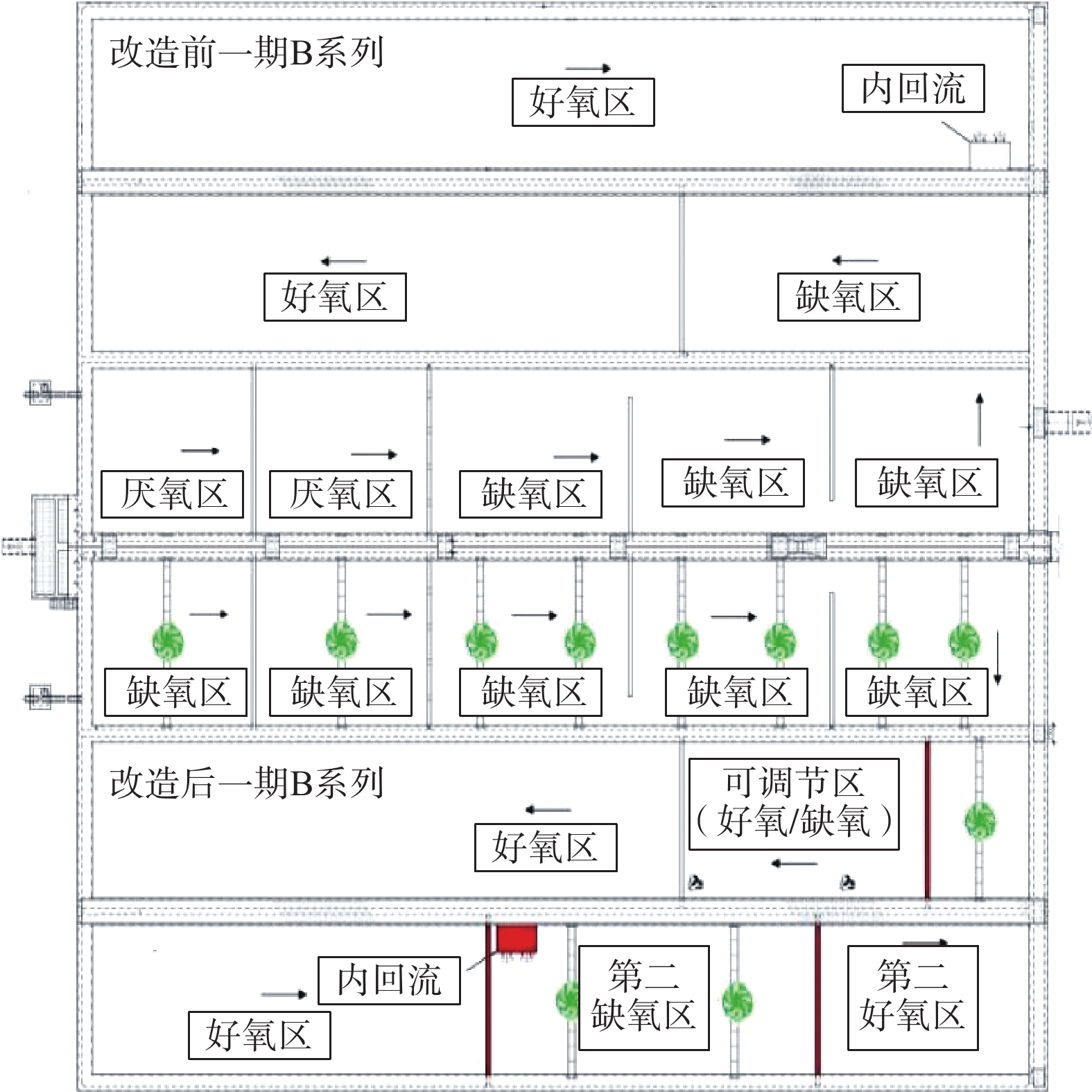 图 3 一期B系列改造前后平面示意图Figure 3. Diagrams of before and after the reconstruction at the plan of B series in phase 1
图 3 一期B系列改造前后平面示意图Figure 3. Diagrams of before and after the reconstruction at the plan of B series in phase 1本次改造后,B生物池系统总停留时间为16.59 h。其中,厌氧区2.02 h,缺氧区4.10 h,好氧区7.30 h(可调节区好氧运行),第二缺氧区1.92 h,第二好氧区1.25 h。为增强搅拌效果,加强反硝化速率,基于原有搅拌器,在厌氧区、缺氧区、第二缺氧区增加双曲面搅拌器。另外,将生物池混合液回流泵由原好氧区末端移动至第二缺氧区前端,新增回流泵安装隔墙,走道板、回流管道等。
Bardenpho生产性比选实验在B、C、D系列同等工况下运行,不投加碳源,期间一期A系列全部停运。B系列按照Bardenpho模式运行,以区别于C、D系列原有AAO工艺的出水水质。实验期间,B系列与C、D系列均满负荷运行,即B系列运行水量为10×104 m3·d−1,C、D系列运行水量为20×104 m3·d−1。B系列及C、D系列生物池的污泥浓度均控制在5~6 g·L−1;生物池末端的DO远程控制在1.0 mg·L−1;外回流比为70%;内回流比为200%;使用厂家专利除磷药剂,投加量稳定为100 mg·L−1。同时,加强对好氧段末端DO的控制,确保第二缺氧段缺氧状态。
2. 结果与讨论
2.1 MBBR工艺生产实验结果
实验中进出水水质见表1和2。A系列的出水平均COD较B系列略低,平均值相差不大。计算COD数据的方差,得到A系列方差为6.611,B系列方差为14.049,说明A系列出水的COD指标更稳定。A系列出水平均NH3-N较B系列略高。除10月19日,由于当天控制DO偏低,导致NH3-N升高,其余样品的出水NH3-N均稳定在1.5 mg·L−1以下。计算NH3-N数据的方差,得到A系列方差为0.252,B系列方差为0.772,说明A系列出水的NH3-N指标更稳定。A系列的出水平均TN较B系列低,A系列的TN去除率略高于B系列,去除效果较好。计算TN数据的方差,得到A系列方差为5.796,B系列方差为6.651,说明A系列出水的TN指标更稳定。在除磷药剂投加量相同的情况下,A系列的出水平均
PO3−4 PO3−4 表 1 MBBR实验进水水质指标Table 1. Influent quality of the MBBR experimentmg·L−1 采样日期 COD NH3-N TN PO3−4-P 2018-06-24 370 36.35 54.8 4.66 2018-06-25 609 39.55 60.8 3.42 2018-06-26 555 42.88 55.6 4.34 2018-06-27 455 31.22 55.5 4.76 2018-07-19 369 32.76 42.5 4.30 2018-07-24 551 40.06 54.1 7.87 2018-08-26 250 33.27 43.9 4.38 2018-09-23 438 37.49 47.9 5.06 2018-10-17 562 35.01 41.9 4.20 2018-10-18 757 27.72 72.7 7.80 2018-10-19 761 29.88 54.5 6.50 平均值 516.09 35.11 53.11 5.21 | Show Table DownLoad:
CSV
表 2 MBBR实验出水水质指标Table 2. Comparison of effluent quality of the MBBR experiment of A and B series
DownLoad:
CSV
表 2 MBBR实验出水水质指标Table 2. Comparison of effluent quality of the MBBR experiment of A and B seriesmg·L−1 取样日期 A系列 B系列 COD NH3-N TN PO3−4-P COD NH3-N TN PO3−4-P 2018-06-24 18 0.847 7.505 1.75 19 0.708 3.450 0.600 2018-06-25 16 1.35 2.758 1.10 17 0.431 4.711 0.430 2018-06-26 21 0.458 5.991 1.25 22 0.347 5.380 0.890 2018-06-27 20 0.347 5.964 3.03 21 0.722 9.172 2.02 2018-07-19 21 0.236 3.394 1.65 19 0.458 5.616 1.71 2018-07-24 18 0.747 8.447 2.48 17 0.347 7.047 0.090 2018-08-26 18 0.458 10.958 4.08 18 0.458 4.825 1.13 2018-09-23 21 0.553 2.973 0.219 27 0.220 8.070 0.108 2018-10-17 15 0.980 6.988 0.044 15 0.520 7.916 0.137 2018-10-18 24 0.270 6.727 0.209 27 0.270 5.788 0.034 2018-10-19 22 1.97 8.06 0.310 22 3.46 13.18 0.024 平均值 19.45 0.747 6.342 1.466 20.36 0.722 6.832 0.652 | Show TableDownLoad:
CSV
2.2 MBBR生产实验中
NO−3 由于A、B系列工艺的除氮效果存在差异,因此,通过A、B系列
NO−3 NO−3 NO−3 NO−3 NO−3  图 4 A系列生产性实验
图 4 A系列生产性实验NO−3 Figure 4.NO−3 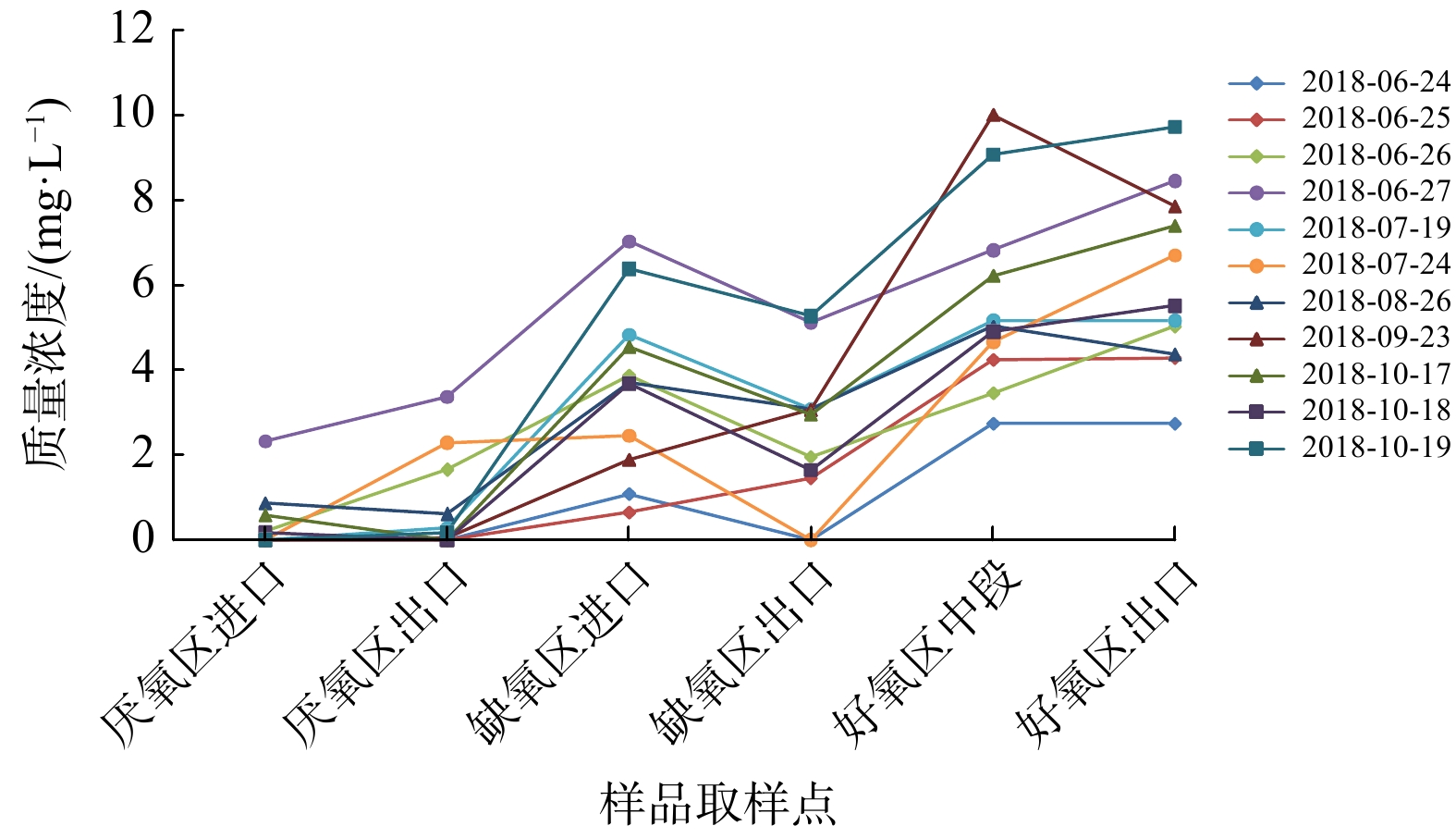 图 5 B系列生产性实验
图 5 B系列生产性实验NO−3 Figure 5.NO−3 2.3 Bardenpho生产实验的结果
Bardenpho生产实验的进出水水质见表3和4。B系列的出水平均COD较C、D系列略低,去除率相近。计算COD数据的方差,得到B系列方差为8.599,C、D系列方差为22.122,说明Bardenpho工艺中B系列出水的COD指标更稳定。B系列的出水平均NH3-N较C、D系列略高,但B系列NH3-N去除率较低。计算NH3-N数据的方差,得到B系列方差为0.1647,C、D系列方差为0.043 7。就去除效果而言,C、D系列具有微弱优势,且出水NH3-N指标更稳定,但B系列出水NH3-N稳定低于1.5 mg·L−1。B系列的出水平均TN与C、D系列相比略低,且B系列TN去除率较高。计算TN数据的方差,得到B系列方差为1.369,C、D系列方差为0.599。就去除效果而言,B系列具有一定优势,但C、D系列出水的TN指标更稳定。在除磷药剂投加量相同的情况下,B系列出水的平均
PO3−4 PO3−4 PO3−4 表 3 Bardenpho实验进水水质Table 3. Influent quality of the Bardenpho experimentalmg·L−1 取样日期 COD NH3-N TN PO3−4-P 2019-11-19 387 40.42 48.0 6.37 2019-11-20 372 42.00 47.1 8.52 2019-11-21 399 57.79 67.9 10.9 2019-11-26 466 43.58 56.3 5.38 2019-11-27 477 42.79 54.2 9.99 2019-11-28 823 33.97 48.4 7.53 2019-12-05 259 40.55 50.9 5.61 2019-12-17 323 40.82 49.2 6.77 2019-12-20 253 45.55 55.3 5.21 平均值 417.7 43.05 53.03 7.36 | Show TableDownLoad:
CSV
2.4 Bardenpho生产实验中脱氮过程分析
由于Bardenpho工艺第二缺氧区停留时间较短,B系列好氧区与第二好氧区的曝气会影响第二缺氧区的缺氧状态,进而影响第二缺氧区的反硝化效果。为确保实验效果,Bardenpho生产性实验中加强了DO控制,以确保第二缺氧段处于缺氧状态。结果亦表明污水处理过程中TN明显降低。在前文关于MBBR实验部分,已对原有AAO工艺中NH3-N、
NO−3 NO−3 NO−3  图 6 B系列Bardenpho生产性实验过程样NH3-N的质量浓度Figure 6. NH3-N Process samples of B seriesBardenpho production experiments
图 6 B系列Bardenpho生产性实验过程样NH3-N的质量浓度Figure 6. NH3-N Process samples of B seriesBardenpho production experiments 图 7 B系列Bardenpho生产性实验过程样
图 7 B系列Bardenpho生产性实验过程样NO−3 Figure 7.NO−3 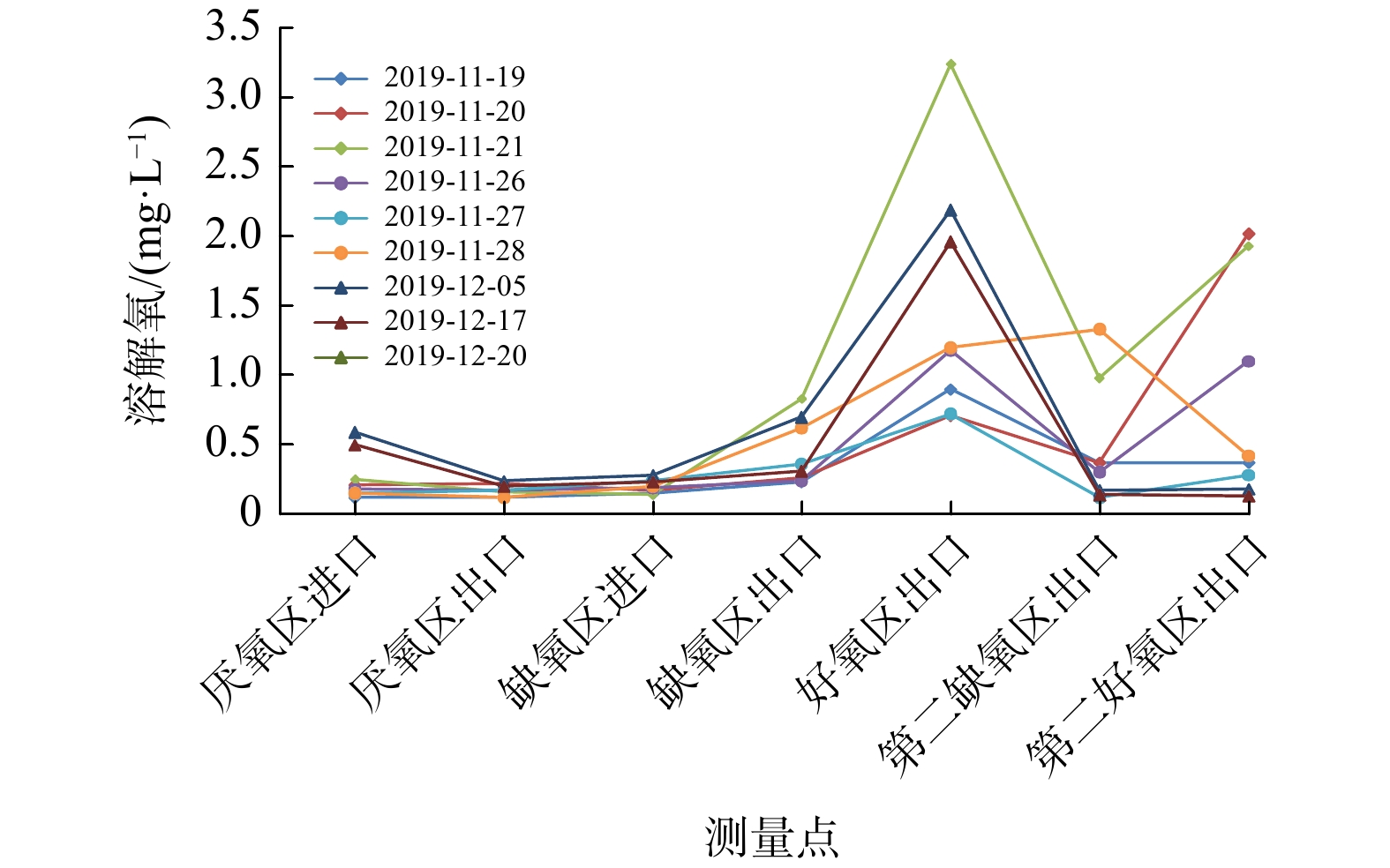 图 8 B系列Bardenpho生产性实验过程样取样点DO测量值Figure 8. measurement value of dissolved oxygenat sampling points of series BBardenpho production experiments
图 8 B系列Bardenpho生产性实验过程样取样点DO测量值Figure 8. measurement value of dissolved oxygenat sampling points of series BBardenpho production experiments分析Bardenpho生产性实验期间B系列生物池过程样的检测结果,发现厌氧区进口至厌氧区出口水样指标NH3-N的平均值下降了2.078 mg·L−1;缺氧区进口至缺氧区出口(剔除11月19日的异常数据)NH3-N的平均值下降了0.327 mg·L−1;缺氧区进口至缺氧区出口
NO−3 NO−3 NO−3 NO−3 好氧区出口至第二缺氧区出口平均
NO−3 NO−3 NO−3 2.5 Bardenpho生产实验运行参数分析
与原有C、D系列AAO工艺相比,B系列Bardenpho工艺的NH3-N去除效果略差,去除率略降低。进一步分析设计参数后发现,B系列Bardenpho工艺好氧区总水力停留时间较C、D系列AAO工艺减少了0.49 h;同时,由于B系列一段、二段好氧区末端的DO明显低于C、D系列AAO工艺好氧区末端的DO,因此,DO可能对出水的NH3-N指标产生较大影响,具体数据如表4所示。
对比原有AAO工艺与Bardenpho工艺,结合实验数据和过程分析,采用硝化、反硝化动力学计算缺氧区容积计算公式(1)进行计算,以核算AAO工艺与Bardenpho工艺的反硝化速率是否符合设计要求。
Vn=0.001QΔN−0.12ΔXvKdeX (1) 式中:Vn为缺氧区(池)容积,m3;Q为生物反应池设计流量,m3·d−1;X为生物反应池内混合液悬浮固体平均浓度,g·L−1;△Xv为排除生物反应池系统的微生物量,kg·d−1;Kde为脱氮速率,kg· (kg·d) −1,根据实验资料确定。无实验资料时,一般20 ℃时Kde经验值为0.03~0.06 kg· (kg·d) −1,并按相关公式进行温度修正,若温差不大可忽略。
Bardenpho生产性实验中C、D系列具体相关数据及计算结果见表5和6。
表 5 Bardenpho生产性实验中各好氧区末端的DOTable 5. Measured values of dissolved oxygen at the end of aerobic zone of the Bardenpho production experimentmg·L−1 采样日期 B系列一段好氧区 B系列二段好氧区 C好氧区 D好氧区 2019-11-19 0.900 0.370 3.615 2.315 2019-11-20 0.710 2.020 3.595 2.985 2019-11-21 3.240 1.930 4.275 2.895 2019-11-26 1.180 1.100 3.725 1.560 2019-11-27 0.720 0.280 3.935 4.360 2019-11-28 1.200 0.420 4.390 2.720 2019-12-05 2.190 0.180 3.255 1.420 2019-12-17 — — 4.190 1.940 2019-12-20 1.960 0.130 3.770 2.275 | Show TableDownLoad:
CSV
表 6 Bardenpho生产性实验C、D系列相关参数Table 6. Experimental parameters of C and D series Bardenpho in-process experiments采样日期 进水温度/℃ VN/m3 进水TN/(mg·L−1) 出水TN/(mg·L−1) △Xv/(kg·d−1) VSS比例 X/(g·L−1) Kde/(kg·(kg·d)−1) 2019-11-19 19.9 23 041 48 7.00 2 413 0.5 5 458 0.021 2019-11-20 19.9 23 041 47.1 6.85 2 445 0.5 5 885 0.019 2019-11-21 19.5 23 041 67.9 7.45 2 493 0.5 6 090 0.032 2019-11-26 18.9 23 041 56.3 6.27 1 972 0.5 6 445 0.026 2019-11-27 18.8 23 041 54.2 7.69 2 774 0.5 6 465 0.020 2019-11-28 18.8 23 041 48.4 6.61 3 593 0.5 6 380 0.014 2019-12-05 18.4 23 041 50.9 5.28 3 748 0.5 6 190 0.016 2019-12-17 18.1 23 041 49.2 5.19 1 963 0.5 6 140 0.023 2019-12-20 18.0 23 041 55.3 7.21 1 990 0.5 5 920 0.027 | Show TableDownLoad:
CSV
在计算中,C、D系列AAO工艺的反硝化速率以全段水力停留时间计算,B系列五段式Bardenpho工艺的反硝化速率以前3段AAO水力停留时间计算。由于Bardenpho工艺第一缺氧区水力停留时间小于原有AAO工艺缺氧区停留时间,表现为计算中VN不同。另外,Bardenpho工艺的TN取样点在一段好氧区出口,AAO工艺在好氧区出口。根据表6和7,Bardenpho工艺有2 d高于设计下限,AAO工艺有1 d高于设计下限,2种工艺反硝化速率平均值的区别较小,且均低于理论设计负荷0.03~0.06 kg· (kg·d)−1。分析2种工艺的结构,由于Bardenpho工艺有第二缺氧区和第二好氧区进行二次反硝化,且该区域无混合液回流影响,故若进一步增加实际反硝化水力停留时间,可进一步降低出水水样中的TN、NH3-N。
表 4 Bardenpho实验出水水质情况对比表Table 4. Comparison of effluent quality of Bardenpho experimentmg·L−1 取样日期 B系列 C、D系列 COD NH3-N TN PO3−4-P COD NH3-N TN PO3−4-P 2019-11-19 14 0.137 3.56 0.084 16 0.111 7.00 0.132 2019-11-20 21 0.768 4.55 0.044 33 0.400 6.85 0.132 2019-11-21 16 0.347 4.47 0.264 17 0.268 7.45 0.100 2019-11-26 22 1.321 5.75 0.070 19 0.821 6.27 0.132 2019-11-27 21 0.584 5.49 0.120 20 0.295 7.69 0.153 2019-11-28 25 1.421 7.34 0.264 15 0.216 6.61 0.111 2019-12-05 19 0.611 3.90 0.224 21 0.716 5.28 0.163 2019-12-17 19 0.216 6.93 0.064 20 0.295 5.19 0.163 2019-12-20 15 1.350 7.30 0.244 22 0.189 7.21 0.163 平均值 19.11 0.751 5.477 0.153 20.33 0.368 6.617 0.139 最大值 25 1.421 7.340 0.264 33 0.821 7.690 0.163 最小值 15 0.216 3.900 0.044 15 0.189 5.190 0.100 | Show TableDownLoad:
CSV
表 7 Bardenpho实验B系列相关参数Table 7. Experimental parameters of B series Bardenpho in-process experiments采样日期 进水温度/℃ VN/m3 进水TN/(mg·L−1) 出水TN/(mg·L−1) △Xv/(kg·d−1) VSS比例 X/(g·L−1) Kde/(kg·(kg·d)−1) 2019-11-19 19.9 17 083 48 5.530 2 268 0.5 5 995 0.028 2019-11-20 19.9 17 083 47.1 4.870 4 460 0.5 6 160 0.015 2019-11-21 19.5 17 083 67.9 7.840 4 463 0.5 5 695 0.034 2019-11-26 18.9 17 083 56.3 11.010 2 163 0.5 5 995 0.032 2019-11-27 18.8 17 083 54.2 15.160 2 879 0.5 6 135 0.021 2019-11-28 18.8 17 083 48.4 8.820 3 363 0.5 6 355 0.018 2019-12-05 18.4 17 083 50.9 2.560 3 906 0.5 6 490 0.022 2019-12-17 18.1 17 083 49.2 14.750 4 260 0.5 6 740 0.008 2019-12-20 18.0 17 083 55.3 7.690 3 657 0.5 6 875 0.022 | Show TableDownLoad:
CSV
3. 结论与建议
1)在同工况、不投加碳源情况下,“AAO+MBBR”工艺相较原有AAO工艺对系统NH3-N的去除效果无明显提升,但对TN、
NO−3 PO3−4 2)在同工况、不投加碳源情况下,五段式Bardenpho工艺较原有AAO工艺对TN、
NO−3 3)在Bardenpho工艺生产实验期间,DO数据不稳定,多次出现第二缺氧段无法稳定处于缺氧状态的情况,在一定程度上影响了出水水质指标。建议使用精确曝气系统代替人工远程调控以加强生物池中DO的控制,以进一步确保NH3-N、TN的出水稳定性。五段式Bardenpho工艺可加强除氮效果,保障出水水质指标NH3-N、TN的稳定达标,在第二缺氧段投加碳源还可加强反硝化效果。
-
图 3 一期B系列改造前后平面示意图
Figure 3. Diagrams of before and after the reconstruction at the plan of B series in phase 1
图 4 A系列生产性实验
NO−3 -N过程样Figure 4.
NO−3 N Process samples of A series MBBR in-process experiments
图 5 B系列生产性实验
NO−3 N过程样Figure 5.
NO−3 N Process samples of B seriesMBBR in-process experiments
图 6 B系列Bardenpho生产性实验过程样NH3-N的质量浓度
Figure 6. NH3-N Process samples of B seriesBardenpho production experiments
图 7 B系列Bardenpho生产性实验过程样
NO−3 -N的质量浓度Figure 7.
NO−3 N Process samples of B series Bardenpho production experiments
图 8 B系列Bardenpho生产性实验过程样取样点DO测量值
Figure 8. measurement value of dissolved oxygenat sampling points of series BBardenpho production experiments
表 1 MBBR实验进水水质指标
Table 1. Influent quality of the MBBR experiment
mg·L−1 采样日期 COD NH3-N TN PO3−4-P 2018-06-24 370 36.35 54.8 4.66 2018-06-25 609 39.55 60.8 3.42 2018-06-26 555 42.88 55.6 4.34 2018-06-27 455 31.22 55.5 4.76 2018-07-19 369 32.76 42.5 4.30 2018-07-24 551 40.06 54.1 7.87 2018-08-26 250 33.27 43.9 4.38 2018-09-23 438 37.49 47.9 5.06 2018-10-17 562 35.01 41.9 4.20 2018-10-18 757 27.72 72.7 7.80 2018-10-19 761 29.88 54.5 6.50 平均值 516.09 35.11 53.11 5.21
下载: 导出CSV
表 2 MBBR实验出水水质指标
Table 2. Comparison of effluent quality of the MBBR experiment of A and B series
mg·L−1 取样日期 A系列 B系列 COD NH3-N TN PO3−4-P COD NH3-N TN PO3−4-P 2018-06-24 18 0.847 7.505 1.75 19 0.708 3.450 0.600 2018-06-25 16 1.35 2.758 1.10 17 0.431 4.711 0.430 2018-06-26 21 0.458 5.991 1.25 22 0.347 5.380 0.890 2018-06-27 20 0.347 5.964 3.03 21 0.722 9.172 2.02 2018-07-19 21 0.236 3.394 1.65 19 0.458 5.616 1.71 2018-07-24 18 0.747 8.447 2.48 17 0.347 7.047 0.090 2018-08-26 18 0.458 10.958 4.08 18 0.458 4.825 1.13 2018-09-23 21 0.553 2.973 0.219 27 0.220 8.070 0.108 2018-10-17 15 0.980 6.988 0.044 15 0.520 7.916 0.137 2018-10-18 24 0.270 6.727 0.209 27 0.270 5.788 0.034 2018-10-19 22 1.97 8.06 0.310 22 3.46 13.18 0.024 平均值 19.45 0.747 6.342 1.466 20.36 0.722 6.832 0.652
下载: 导出CSV
表 3 Bardenpho实验进水水质
Table 3. Influent quality of the Bardenpho experimental
mg·L−1 取样日期 COD NH3-N TN PO3−4-P 2019-11-19 387 40.42 48.0 6.37 2019-11-20 372 42.00 47.1 8.52 2019-11-21 399 57.79 67.9 10.9 2019-11-26 466 43.58 56.3 5.38 2019-11-27 477 42.79 54.2 9.99 2019-11-28 823 33.97 48.4 7.53 2019-12-05 259 40.55 50.9 5.61 2019-12-17 323 40.82 49.2 6.77 2019-12-20 253 45.55 55.3 5.21 平均值 417.7 43.05 53.03 7.36
下载: 导出CSV
表 5 Bardenpho生产性实验中各好氧区末端的DO
Table 5. Measured values of dissolved oxygen at the end of aerobic zone of the Bardenpho production experiment
mg·L−1 采样日期 B系列一段好氧区 B系列二段好氧区 C好氧区 D好氧区 2019-11-19 0.900 0.370 3.615 2.315 2019-11-20 0.710 2.020 3.595 2.985 2019-11-21 3.240 1.930 4.275 2.895 2019-11-26 1.180 1.100 3.725 1.560 2019-11-27 0.720 0.280 3.935 4.360 2019-11-28 1.200 0.420 4.390 2.720 2019-12-05 2.190 0.180 3.255 1.420 2019-12-17 — — 4.190 1.940 2019-12-20 1.960 0.130 3.770 2.275
下载: 导出CSV
表 6 Bardenpho生产性实验C、D系列相关参数
Table 6. Experimental parameters of C and D series Bardenpho in-process experiments
采样日期 进水温度/℃ VN/m3 进水TN/(mg·L−1) 出水TN/(mg·L−1) △Xv/(kg·d−1) VSS比例 X/(g·L−1) Kde/(kg·(kg·d)−1) 2019-11-19 19.9 23 041 48 7.00 2 413 0.5 5 458 0.021 2019-11-20 19.9 23 041 47.1 6.85 2 445 0.5 5 885 0.019 2019-11-21 19.5 23 041 67.9 7.45 2 493 0.5 6 090 0.032 2019-11-26 18.9 23 041 56.3 6.27 1 972 0.5 6 445 0.026 2019-11-27 18.8 23 041 54.2 7.69 2 774 0.5 6 465 0.020 2019-11-28 18.8 23 041 48.4 6.61 3 593 0.5 6 380 0.014 2019-12-05 18.4 23 041 50.9 5.28 3 748 0.5 6 190 0.016 2019-12-17 18.1 23 041 49.2 5.19 1 963 0.5 6 140 0.023 2019-12-20 18.0 23 041 55.3 7.21 1 990 0.5 5 920 0.027
下载: 导出CSV
表 4 Bardenpho实验出水水质情况对比表
Table 4. Comparison of effluent quality of Bardenpho experiment
mg·L−1 取样日期 B系列 C、D系列 COD NH3-N TN PO3−4-P COD NH3-N TN PO3−4-P 2019-11-19 14 0.137 3.56 0.084 16 0.111 7.00 0.132 2019-11-20 21 0.768 4.55 0.044 33 0.400 6.85 0.132 2019-11-21 16 0.347 4.47 0.264 17 0.268 7.45 0.100 2019-11-26 22 1.321 5.75 0.070 19 0.821 6.27 0.132 2019-11-27 21 0.584 5.49 0.120 20 0.295 7.69 0.153 2019-11-28 25 1.421 7.34 0.264 15 0.216 6.61 0.111 2019-12-05 19 0.611 3.90 0.224 21 0.716 5.28 0.163 2019-12-17 19 0.216 6.93 0.064 20 0.295 5.19 0.163 2019-12-20 15 1.350 7.30 0.244 22 0.189 7.21 0.163 平均值 19.11 0.751 5.477 0.153 20.33 0.368 6.617 0.139 最大值 25 1.421 7.340 0.264 33 0.821 7.690 0.163 最小值 15 0.216 3.900 0.044 15 0.189 5.190 0.100
下载: 导出CSV
表 7 Bardenpho实验B系列相关参数
Table 7. Experimental parameters of B series Bardenpho in-process experiments
采样日期 进水温度/℃ VN/m3 进水TN/(mg·L−1) 出水TN/(mg·L−1) △Xv/(kg·d−1) VSS比例 X/(g·L−1) Kde/(kg·(kg·d)−1) 2019-11-19 19.9 17 083 48 5.530 2 268 0.5 5 995 0.028 2019-11-20 19.9 17 083 47.1 4.870 4 460 0.5 6 160 0.015 2019-11-21 19.5 17 083 67.9 7.840 4 463 0.5 5 695 0.034 2019-11-26 18.9 17 083 56.3 11.010 2 163 0.5 5 995 0.032 2019-11-27 18.8 17 083 54.2 15.160 2 879 0.5 6 135 0.021 2019-11-28 18.8 17 083 48.4 8.820 3 363 0.5 6 355 0.018 2019-12-05 18.4 17 083 50.9 2.560 3 906 0.5 6 490 0.022 2019-12-17 18.1 17 083 49.2 14.750 4 260 0.5 6 740 0.008 2019-12-20 18.0 17 083 55.3 7.690 3 657 0.5 6 875 0.022
下载: 导出CSV
-
[1] 孙悦. 城镇污水处理厂尾水排放水环境影响及对策[J]. 资源节约与环保, 2019(5): 70. doi: 10.3969/j.issn.1673-2251.2019.05.065 [2] 解宇峰, 李文静, 李维新, 等. 江苏省城市污水处理厂尾水时空排放特征研究[J]. 环境工程, 2014, 32(8): 33-37. [3] 汪锋, 钱庄, 张周, 等. 污水处理厂尾水对排放河道水质的影响[J]. 安徽农业科学, 2016, 44(14): 65-68. doi: 10.3969/j.issn.0517-6611.2016.14.024 [4] 许赟溢. 污水处理厂尾水排江环境影响研究[J]. 环境与发展, 2014, 26(3): 37-39. doi: 10.3969/j.issn.1007-0370.2014.03.014 [5] 杨国钰. 以拟建杨台子污水处理厂为例谈——污水处理厂尾水排放对受纳区域水环境的影响及对策[J]. 治淮, 2009(6): 14-15. doi: 10.3969/j.issn.1001-9243.2009.06.007 [6] 吴雪, 何佳, 徐晓梅, 等. 滇池流域污水厂尾水污染负荷特征分析[J]. 中国给水排水, 2018, 34(17): 69-73. [7] 吴钦. 城镇污水处理厂尾水排放对水环境影响及对策[J]. 环境与发展, 2019, 31(6): 29. [8] 环境保护部. 水污染防治行动计划: 中英文对照[M]. 北京: 人民出版社, 2015. [9] 崔朋, 章诗璐, 万年红, 等. 高效气浮工艺深度除磷试验研究[J]. 住宅产业, 2019(11): 143-148. [10] 沈怡雯. 高效沉淀池在污水处理厂UNITANK工艺强化除磷中的应用[J]. 净水技术, 2019, 38(S1): 139-142. [11] 洪铁. V型滤池基本构造及实际运用[J]. 科技风, 2019(23): 201. [12] DINGP, CHUL, WANGJ. Advanced treatment of petrochemical wastewater by combined ozonation and biological aerated filter[J]. Environmental Science & Pollution Research, 2018, 25(4): 9673-9682. [13] LI XW, SHIH C, LI K X, et al. Combined process of biofiltration and ozone oxidation as an advanced treatment process for wastewater reuse[J]. Frontiers of Environmental Science & Engineering, 2015, 9(6): 1076-1083. [14] 戴仲怡, 王雪, 彭建国, 等. 曝气缺氧/多级AO工艺用于大型污水厂提标改造[J]. 中国给水排水, 2019, 35(18): 50-54. [15] 赖辉辉, 乐华斌, 胡雁新. 高浓度氨氮及总磷进水的污水厂准地表Ⅳ类提标改造工程[J]. 广东化工, 2018, 45(17): 146-147. doi: 10.3969/j.issn.1007-1865.2018.17.070 [16] 刘浩, 杨俊杰, 于宁. Bardenpho五段法/MBBR用于青岛李村河污水厂三期扩建[J]. 中国给水排水, 2016, 32(24): 62-66. [17] 杨宇星, 吴迪, 宋美芹, 等. 新型MBBR用于类地表Ⅳ类水排放标准升级改造工程[J]. 中国给水排水, 2017, 33(14): 93-98. [18] 孙欣, 崔洪升. Bardenpho+深床滤池工艺用于半地下污水处理厂工程[J]. 中国给水排水, 2017, 33(16): 82-85. [19] 胡香, 张辉, 许光远, 等. 反硝化深床滤池深度脱氮效果研究[J]. 中国给水排水, 2017, 33(21): 13-17, 24. [20] CHUDOBA P, PUJOL R. Technical solutions for upgrading high rate and mediumloaded activated sludge plants for nutrient removal.[J]. Water Science &Technology, 2000, 41(9): 131-138. [21] 国家环境保护总局, 国家质量监督检验检疫总局. 城镇污水处理厂污染物排放标准: GB18918-2002[S]. 北京: 中国环境科学出版社, 2002. [22] 国家环境保护总局《水和废水监测分析方法》编委会. 水和废水监测分析方法[M]. 4版. 北京: 中国环境科学出版社, 2002. [23] YANG X P, WANG S M, ZHANG D W, et al. Isolation and nitrogen removal characteristics of an aerobic heterotrophic nitrifying-denitrifying bacterium, Bacillus subtilis A1[J]. Bioresource Technology, 2011, 102(2): 854-862. doi: 10.1016/j.biortech.2010.09.007 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5188
- HTML全文浏览数: 5188
- PDF下载数: 93
- 施引文献: 0


























