


 下载:
下载:



 百度学术
百度学术


-
臭氧是一种高效氧化剂,可选择性攻击废水中不饱和有机物。因此,在难降解废水处理及废水深度处理中得到了广泛应用[1-2]。但单独臭氧氧化工艺存在氧化能力有限、气-液传质速率慢、水中溶解度较低、具有选择性等缺陷,成为制约臭氧化技术应用的关键问题。
多相催化臭氧化工艺可通过将臭氧转化为强氧化性自由基或通过催化剂对污染物的吸附来提高污染物去除率[3-5],从而克服单独臭氧氧化工艺氧化能力有限的问题。催化材料中的活性炭材料具有高比表面积(500~1 500 m2·g−1)、耐腐蚀性、吸附容量大、廉价等优点,是一种应用广泛的催化剂[6],可有效促进臭氧分解生成羟基自由基(·OH),从而提高难降解有机设计污染物的去除率[7-8]。
但目前催化臭氧还存在臭氧溶解度低和气液传质速率慢等局限性,产生小的臭氧气泡是一种解决上述问题可行的方法。微气泡是直径小于50 μm的微小气泡,在溶液中具有气-液接触面积大,气-液接触时间(即液相停留时间)长、气含率高、气-液传质速率高等优势。此外,微气泡表面存在电荷聚集效应,小于50 μm的微气泡在纯水中的Zeta(ζ)电位平均值约为−35 mV[9],在微气泡收缩过程中表面电荷密度和ζ电位急剧升高,并在破裂时产生·OH,具有类催化效应。而臭氧微气泡的类催化效应更为显著[10]。TAKAHASHI等[11]的研究表明,微气泡破裂可产生自由基。KHUNTIA等[12]采用对氯苯甲酸(pCBA)作为探针对臭氧微气泡系统中·OH浓度进行检测计算,结果发现,酸性条件下臭氧微气泡产生·OH的效率高于碱性条件下。
本研究采用微气泡催化臭氧化(以活性炭为催化剂)处理酸性大红3R,并与单独微气泡臭氧化和传统气泡臭氧化进行比较,考察了微气泡催化臭氧分解特性以及处理酸性大红3R脱色和TOC的去除效能,分析了微气泡催化臭氧化强化·OH氧化过程,探究了酸性大红3R臭氧化降解途径,以期为微气泡催化臭氧化技术在印染废水处理中的应用提供参考。
全文HTML
-
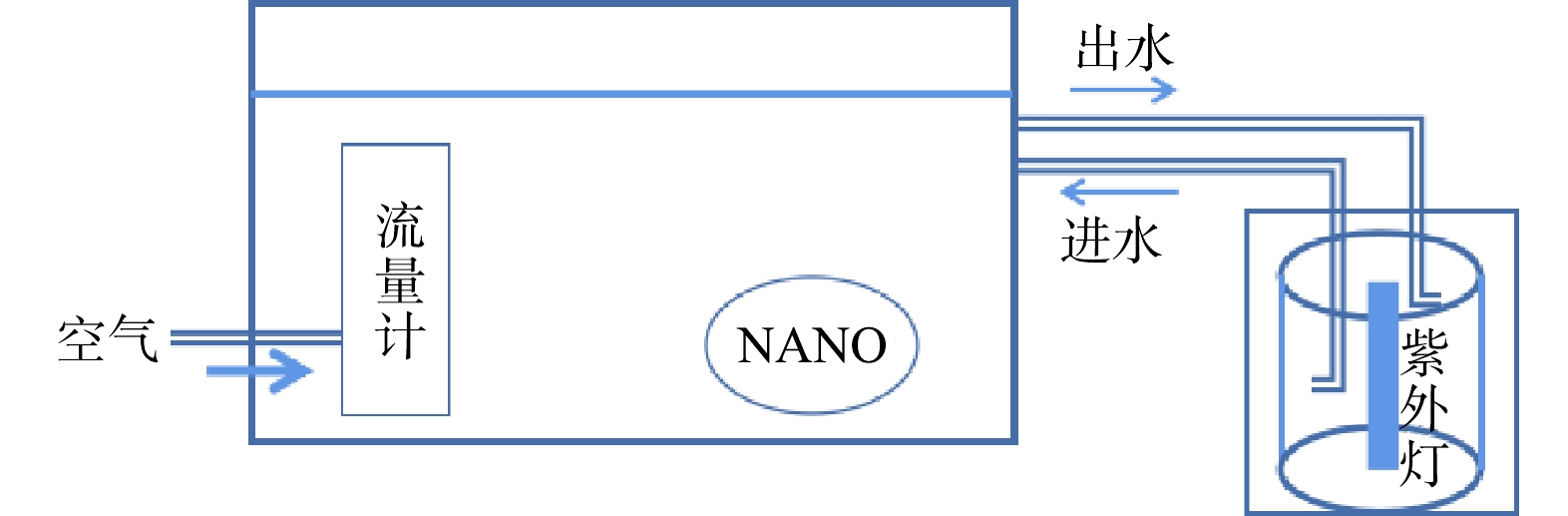
实验装置如图1所示。臭氧发生器(石家庄冠宇环保,5 g·h−1)以纯氧为气源产生臭氧气体,通过流量计控制臭氧气体流量。臭氧气体和循环液体进入微气泡发生器(北京晟峰恒泰科技有限公司),通过气液混合-水力剪切-气液分离作用产生臭氧微气泡,进入反应器底部。实验中,控制臭氧气体流量保持为0.3 L·min−1,在此流量下,臭氧微气泡平均直径为51.4 µm[13],臭氧投加量为12.5 mg·min−1。反应器采用直径200 mm,高500 mm的密封有机玻璃容器,反应器设计处理水量为10 L。在反应器中部填充经处理后的商品柱状煤质颗粒活性炭。同时,在反应器底部采用微孔曝气盘产生臭氧传统气泡。尾气中的臭氧采用碘化钾溶液进行吸收。
-
将酸性大红3R(C20H11N2O10S3·3Na,分子质量为604.48)(青岛安吉达化学试剂公司)溶于去离子水中,配置浓度为100 mg·L−1酸性大红3R模拟废水;催化剂选用长度为4~8 mm商品柱状煤质颗粒活性炭,使用前通过去离子水多次洗涤并在100 ℃烘箱干燥24 h。活性炭表面主要物理化学性质[14-17]如表1所示。
-
1)臭氧分解速率系数(Kd)的确定。在气流量0.3 L·min−1、臭氧投加量12.5 mg·min−1、活性炭投加量2 g·L−1酸性大红3R初始浓度为100 mg·L−1的条件下,进行微气泡催化臭氧曝气、微气泡臭氧曝气以及传统气泡臭氧曝气,溶解臭氧浓度达到饱和后停止曝气,并测定臭氧分解过程中溶解臭氧浓度c随时间t的变化,其符合一级反应动力学方程,如式(1)所示。由式(2)得到ln(cs/c)与t的线性关系,其斜率即为臭氧分解系数Kd[18]。
将边界条件t=0、c=cs代入式(1)并积分得式(2)。
式中:cs为饱和溶解臭氧浓度,mg·L−1;c为t时刻水中溶解臭氧浓度,mg·L−1;Kd为臭氧分解系数,min−1。
2)臭氧化处理酸性大红3R。分别采用微气泡催化臭氧化、微气泡臭氧化及传统气泡臭氧化处理酸性大红3R。酸性大红3R初始浓度100 mg·L−1,臭氧气流量0.3 L·min−1、臭氧投加量12.5 mg·min−1,活性炭投加量2 g·L−1,测定处理过程中的溶解臭氧浓度、尾气臭氧散逸量、脱色率、TOC去除率、溶液pH、H2O2浓度随时间变化。采用GC-MS分析酸性大红3R降解过程中间产物。微气泡催化臭氧化中,分别加入相同浓度(15 mmol·L−1)的NaCl、Na2SO4和Na2CO3作为·OH捕获剂,考察·OH捕获剂对TOC去除效果的影响。
酸性大红3R脱色和TOC去除符合一级反应动力学方程,采用式(3)分别计算脱色和TOC去除反应速率常数。通过测定液相臭氧浓度和平均尾气臭氧逸散量,并采用式(4)计算臭氧利用效率[19]。
式中:c0为酸性大红3R或TOC初始浓度,mg·L−1;c为t时刻酸性大红3R或TOC的浓度,mg·L−1;k为反应速率常数,min−1。
式中:QG为臭氧投加量,mg·min−1;QAe为臭氧逸散量,mg·min−1;t为反应时间,min;CAb为溶解臭氧浓度,mg·L−1;VL为溶液体积,L;η为臭氧利用率。
-
采用能量色散光谱测量仪器(EDS)进行 (S-4800-I,Hitachi,Japan)测定活性炭表面1~2 μm深处元素分布[20]。采用Boehm滴定法测定活性炭样品中SOG的含量[21]。采用比表面积分析仪(D/MAX-2500,Ri-gaku,Japan)测定活性炭样品比表面积。根据Noh和Schwarz法测定活性炭样品pHPZC[22]。采用碘量法测定气相臭氧浓度[23];靛蓝比色法测量溶解臭氧浓度[24];pH采用酸度计(上海雷磁,pHs-3c)进行测定。采用TOC分析仪器(TOC-VCPN,Shimadzu Corporation,日本)测量TOC浓度。采用过氧化物酶-DPD法测定H2O2的浓度[25]。采用GC-MS (DSQⅡ,Thermo,America)分析处理中有机物种类[26]。
1.1. 实验装置
1.2. 材料
1.3. 实验过程
1.4. 分析方法
-
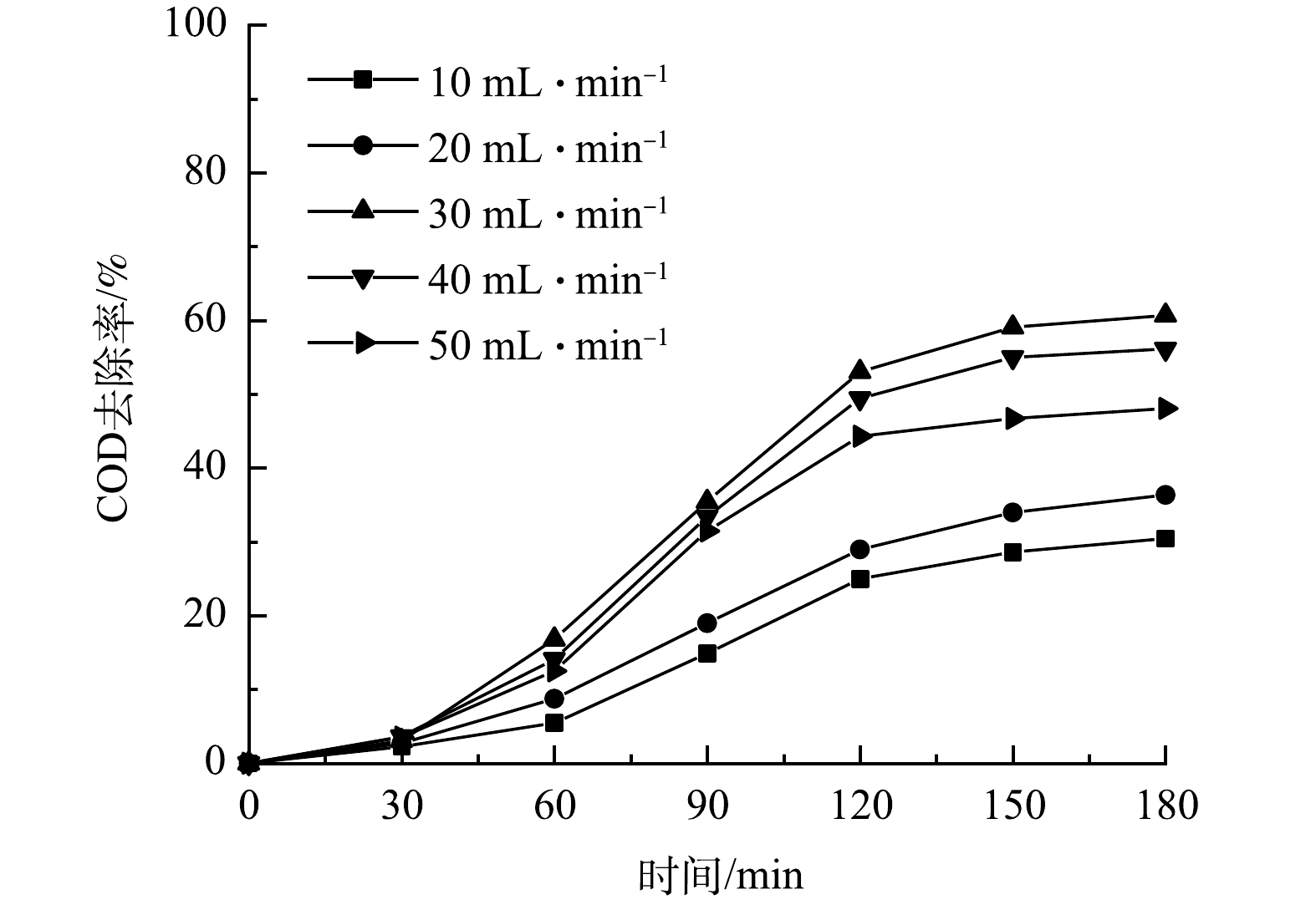
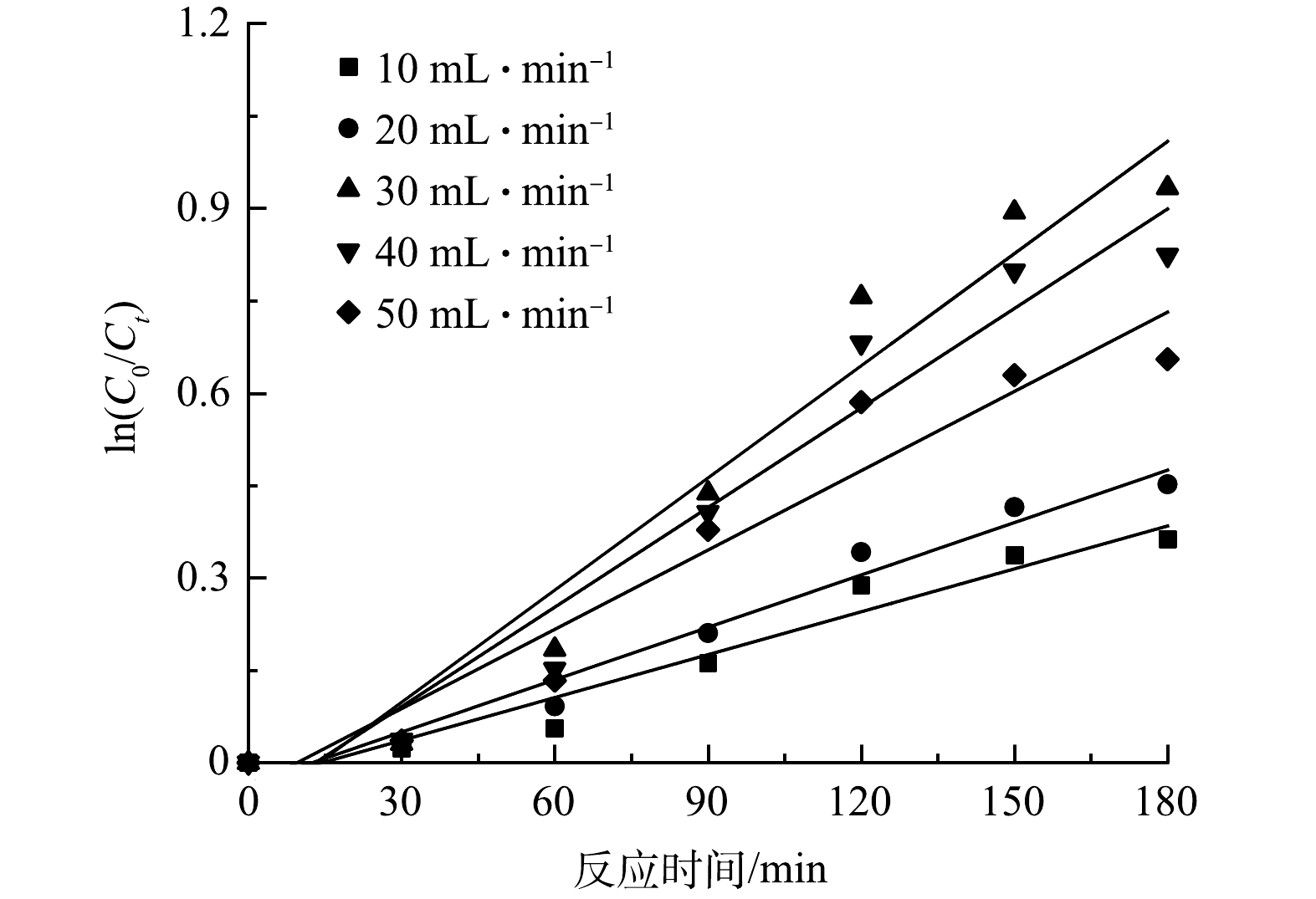
在微气泡催化臭氧曝气与微气泡臭氧曝气各25 min、传统气泡臭氧曝气60 min后,此时溶解臭氧浓度达到饱和,然后停止曝气,溶解臭氧浓度随时间变化如图2所示。由图2可看到,由于臭氧分解作用,溶解臭氧浓度均逐渐降低。根据此过程中溶解臭氧浓度随时间的变化,计算得到微气泡催化臭氧曝气、微气泡臭氧曝气以及传统气泡臭氧曝气中的臭氧分解系数分别为0.093、0.049和0.013 min−1。可见,采用微气泡曝气可显著提高臭氧分解效率。这可能是由于臭氧微气泡收缩破裂过程可促进·OH产生,从而提高了臭氧分解链式反应速率。此外,在微气泡催化臭氧曝气过程中,由于活性炭催化与微气泡收缩破裂的协同效应,可产生更多·OH,从而进一步提高了臭氧分解效率,臭氧分解速率分别为微气泡和传统气泡臭氧曝气的1.9倍和7.2倍。
-
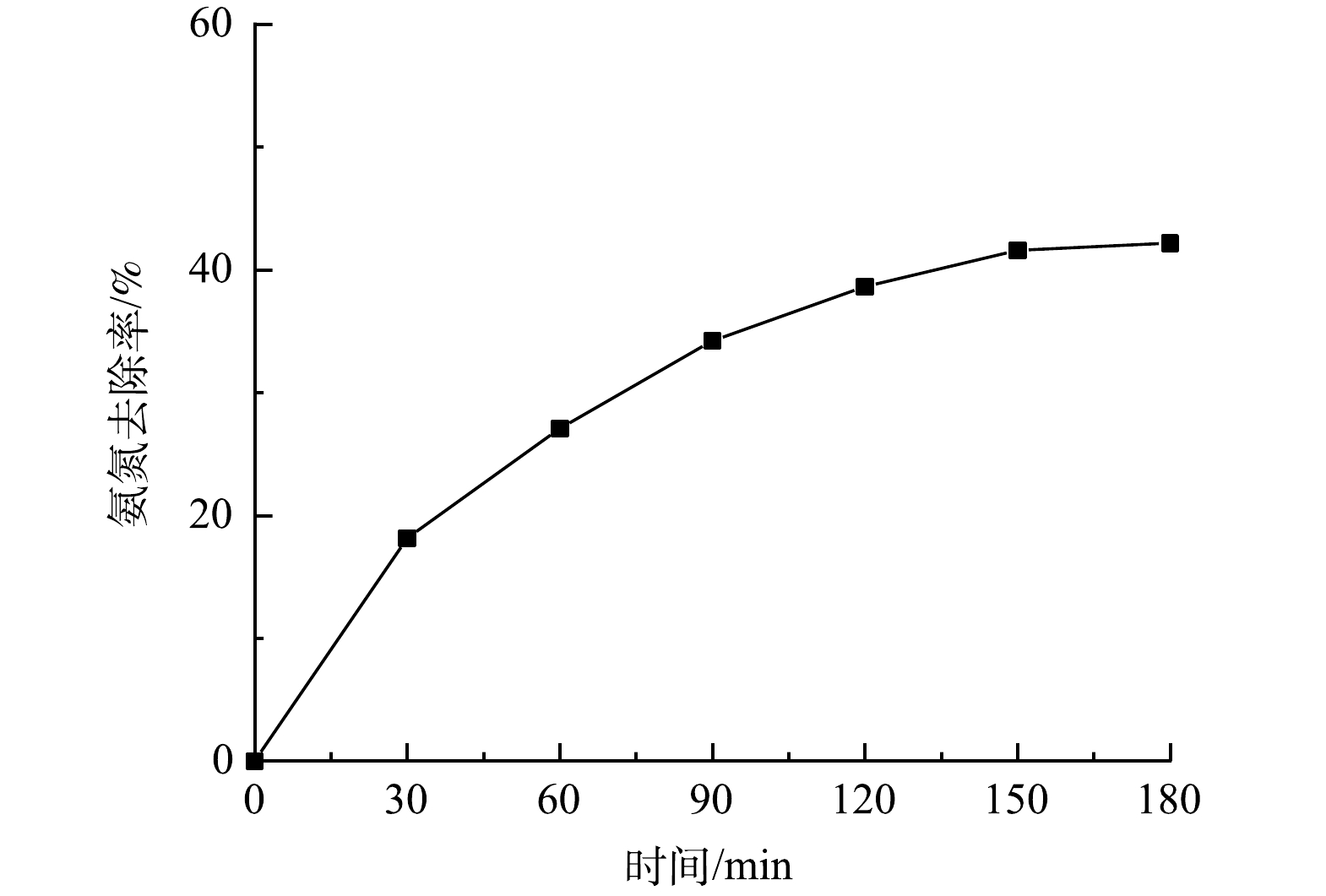
微气泡催化臭氧化、微气泡臭氧化以及传统气泡臭氧化处理酸性大红3R过程中染料分子脱色率的变化如图3所示。计算出的相应脱色反应动力学常数分别为0.342、0.173和0.056 min−1。偶氮基(—N=N—)是酸性大红3R的发色基团,容易被·OH或O3氧化断裂[27],故微气泡催化臭氧化可有效提高酸性大红3R脱色速率。此外,仅有活性炭吸附下的酸性大红3R的脱色率在120 min后仅为14.1%。因此,在微气泡催化臭氧化反应过程中,氧化作用对酸性大红3R脱色起到主要作用。
-
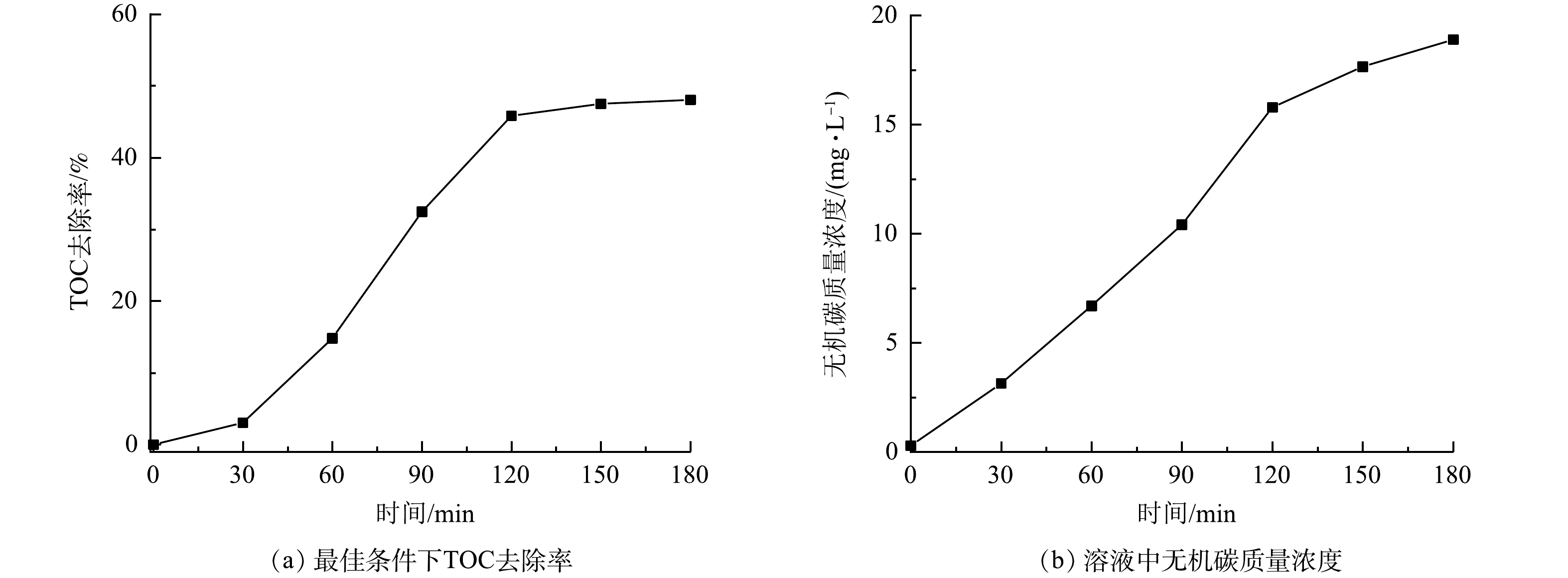
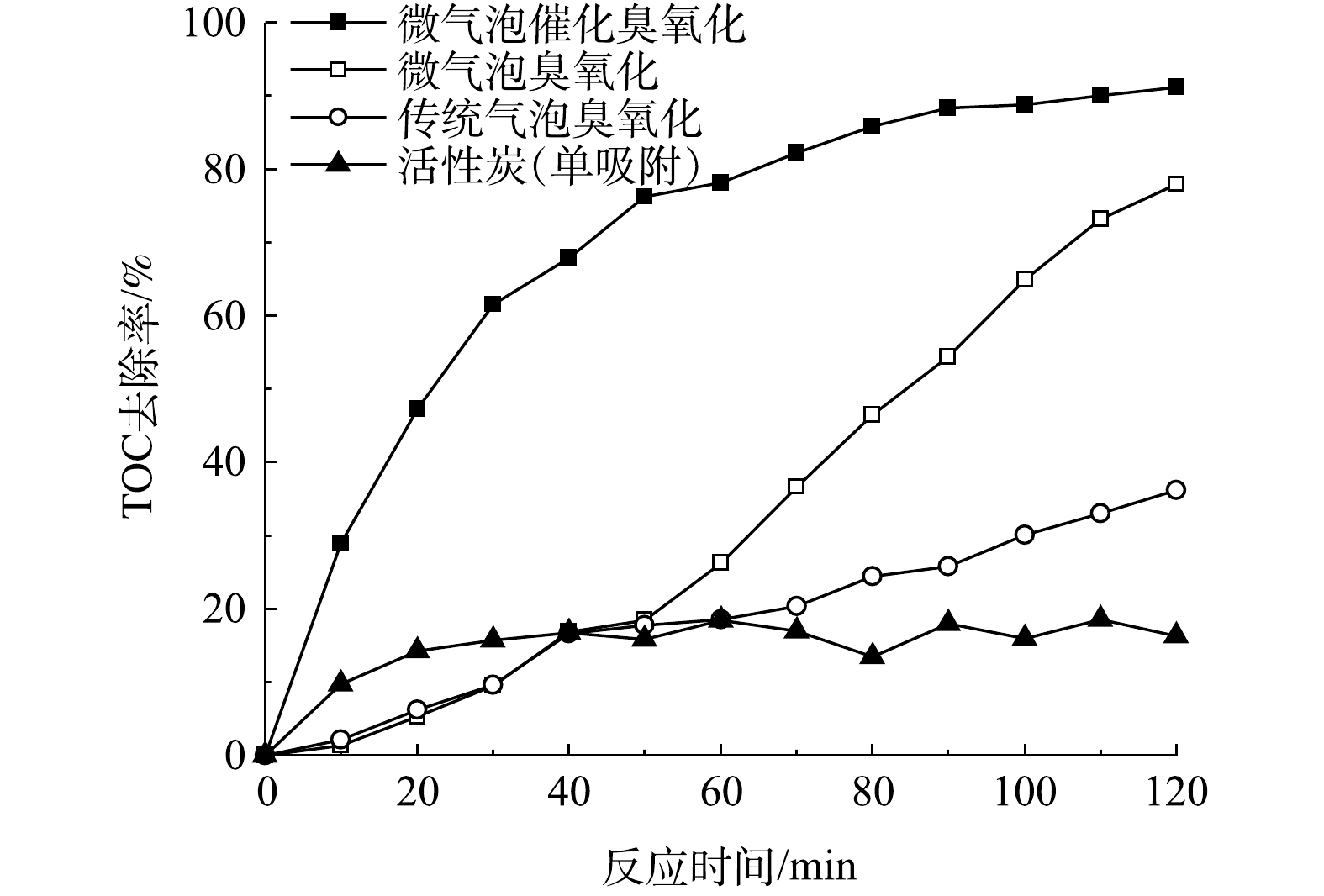
在微气泡催化臭氧化、微气泡臭氧化以及传统气泡臭氧化处理酸性大红3R过程中,TOC去除率随时间的变化如图4所示。可以看到,在处理时间为120 min时,其TOC去除率分别为91.2%、77.9%和36.2%。微气泡臭氧化处理前60 min的TOC去除率仅为26.2%,其原因可能是臭氧主要用于直接氧化脱色,·OH氧化反应较弱,在脱色完成后,TOC的去除率显著提高,在120 min时TOC去除率可达到77.9%。微气泡催化臭氧化处理中TOC去除率随处理时间的延长显著提高,处理60 min时TOC去除率已高达78.2%,120 min时达到91.2%(活性炭吸附40~120 min的平均TOC去除率为15.3%)。由此可见,活性炭催化与微气泡臭氧化存在协同效应,通过强化产生·OH,提高TOC去除速率。
分别计算了微气泡催化臭氧化、微气泡臭氧化、传统气泡臭氧化以及活性炭吸附中TOC去除反应速率常数,为0.018 7、0.013 6、0.003 6和0.000 9 min−1。TOC去除臭氧化反应包括以下4个过程:臭氧直接氧化 (Ⅰ) ;臭氧自分解产生·OH氧化 (Ⅱ) ;臭氧微气泡收缩破裂产生·OH氧化 (Ⅲ) ;活性炭催化臭氧分解产生·OH氧化 (Ⅳ) 。其中,传统气泡臭氧化包括过程 (Ⅰ) 和 (Ⅱ) ,微气泡臭氧化包括过程 (Ⅰ) 、 (Ⅱ) 和 (Ⅲ) ,微气泡催化臭氧化包括上述所有过程以及活性炭吸附。通过比较反应速率常数,估算出上述过程对微气泡催化臭氧化TOC去除的贡献率:过程 (Ⅰ) 和 (Ⅱ) 为19.25%,过程 (Ⅲ) 为53.48%,过程 (Ⅳ) 为22.46%,活性炭吸附为4.81%。由此可见,大约75%的TOC去除与·OH氧化有关,且臭氧微气泡收缩破裂产生·OH最为关键。如果仅考虑TOC快速去除阶段,则微气泡臭氧化60~120 min中TOC去除反应速率常数为0.020 6 min−1,略低于微气泡催化臭氧化0~60 min的0.024 2 min−1。
微气泡催化臭氧化处理中,活性炭经过5次循环使用后,TOC去除反应动力学常数分别为0.023 8、0.022 6、0.023 1、0.021 8和0.021 3 min−1,平均值为0.022 5 min−1,标准偏差为0.001 0 min−1,这表明活性炭催化活性在5次循环使用中基本保持稳定。经过5次循环使用后,对活性炭表面的主要元素及其质量分数进行了测定,结果分别为C 23.2%、O 44.9%、Si 13.4%、Al 5.9%、Fe 4.0%、Sb 1.3%和Mg 1.2%。与原始活性炭相比,经过5次循环使用后的活性炭表面主要元素及其质量分数基本不变,这可能是活性炭催化活性保持稳定的重要原因。
-
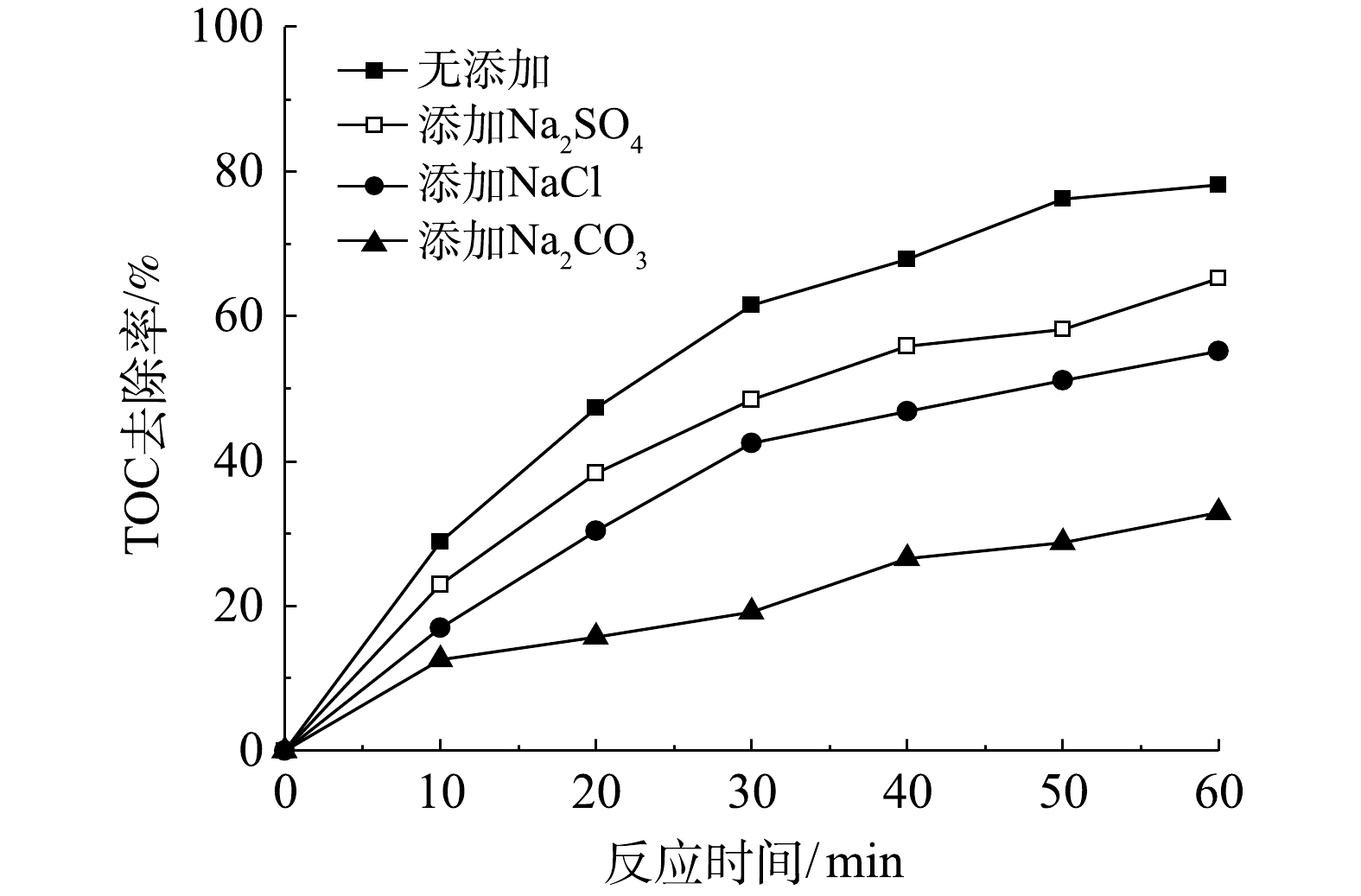
实际工业废水通常盐度较高,存在Cl−、
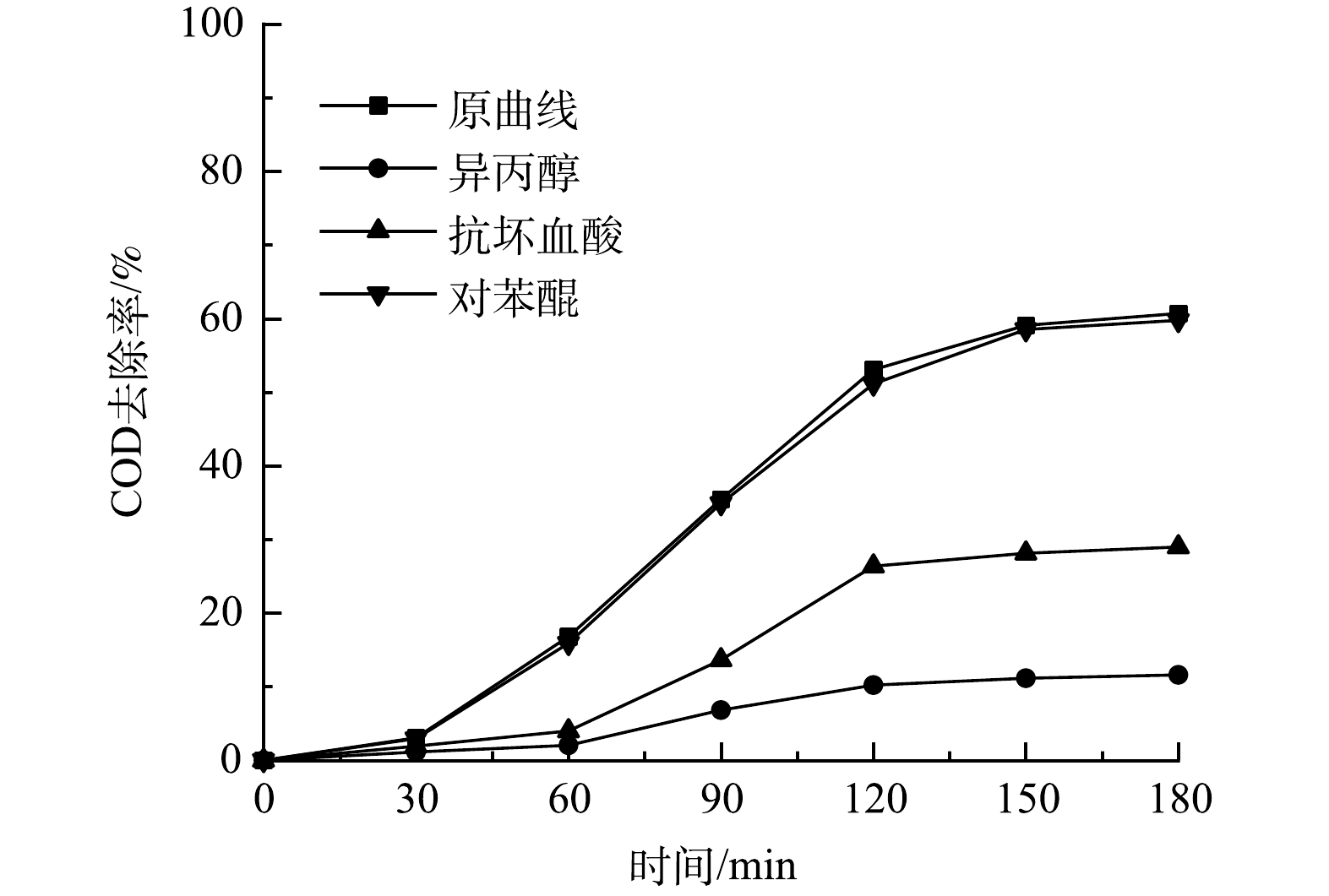
SO2−4 、CO2−3 等无机阴离子,将会对·OH氧化反应产生不利影响[28]。在微气泡催化臭氧化处理中,投加相同浓度(15 mmol·L−1) NaCl、Na2SO4和Na2CO3,TOC去除率变化如图5所示。由图5可以看到,投加NaCl、Na2SO4和Na2CO3后,TOC去除率均有所下降,在0~60 min 内TOC去除反应速率常数分别由0.024 2 min−1下降至0.015 1 min−1 (Na2SO4)、0.012 1 min−1 (NaCl)和0.005 5 min−1 (Na2CO3)。可以看出,Na2CO3的抑制作用最强,这是由于CO2−3 与·OH反应生成CO−3 ·的反应速率常数较高(k=3.9×108 L·(mol·s)−1),可快速消耗·OH。此外,无机阴离子会降低微气泡表面的ξ电位,使得微气泡收缩破裂产生·OH效率下降[9]。此结果亦表明,·OH氧化反应在TOC去除中具有重要作用。 -
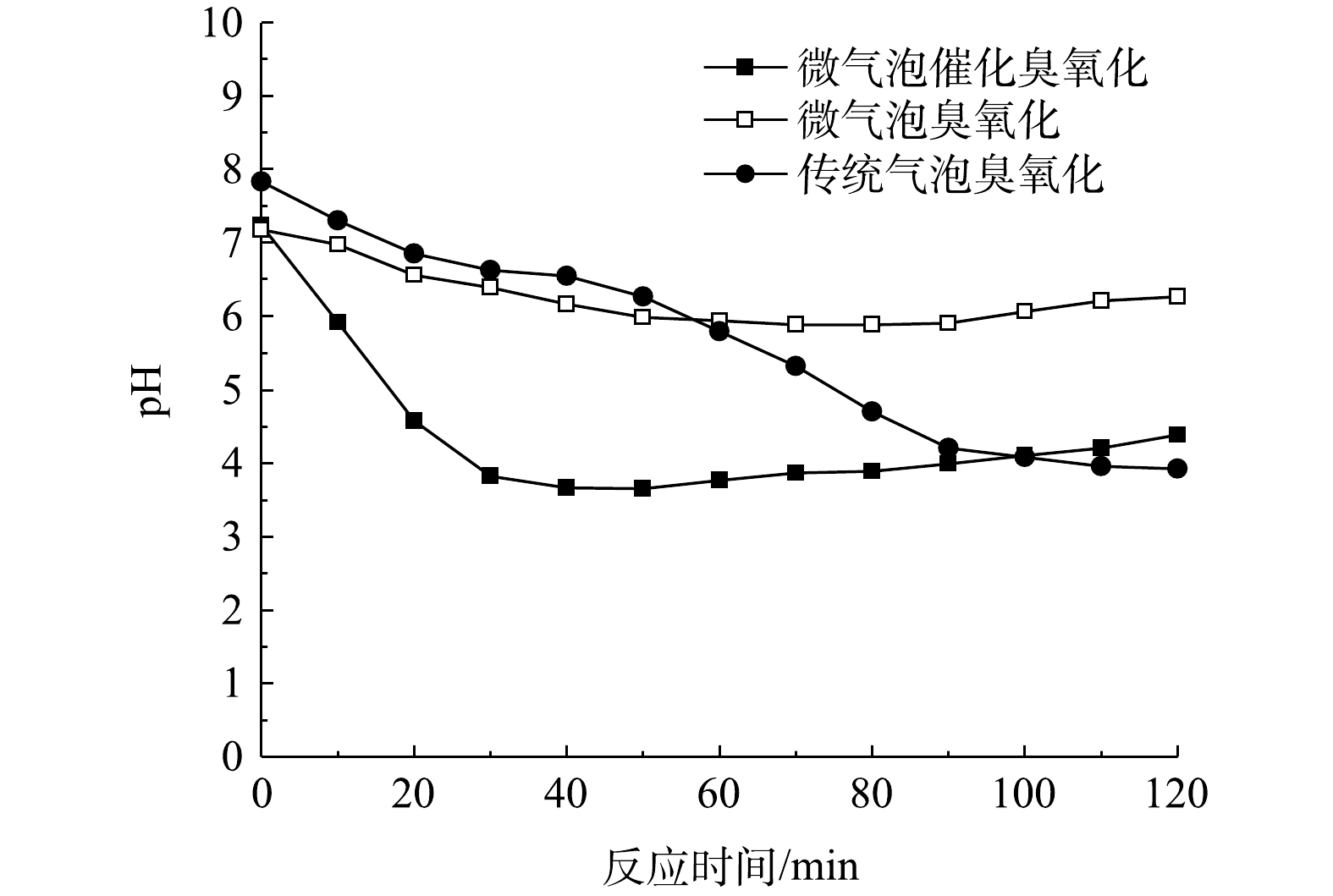
微气泡催化臭氧化、微气泡臭氧化以及传统气泡臭氧化处理中pH随时间的变化如图6所示。可以看出,在传统气泡臭氧化处理过程中pH由7.84下降至3.92;在微气泡臭氧化处理80 min时,pH由7.18降至5.88,120 min时回升至6.27;在微气泡催化臭氧化40 min时,pH由7.24降至3.65,120 min时回升至4.38。在微气泡催化臭氧化与微气泡臭氧化处理中,pH均呈现先下降后升高的趋势。这可能是由于反应初期酸性大红3R分子首先降解为小分子有机酸,且其生成速率高于降解速率,造成小分子有机酸积累,因此pH不断下降;而到反应后期,由于小分子有机酸不断被·OH氧化矿化,pH变化出现拐点,开始不断升高。同时,微气泡催化臭氧化过程中,pH变化拐点早于微气泡臭氧化过程,这表明活性炭催化与臭氧微气泡协同促进·OH氧化反应,该过程的变化与TOC变化趋势相一致。
-
·OH氧化反应对TOC去除具有关键作用,其产生过程所涉及的液相主体反应如式(5)~式(14)所示,活性炭表面反应见式15)~式(19)。
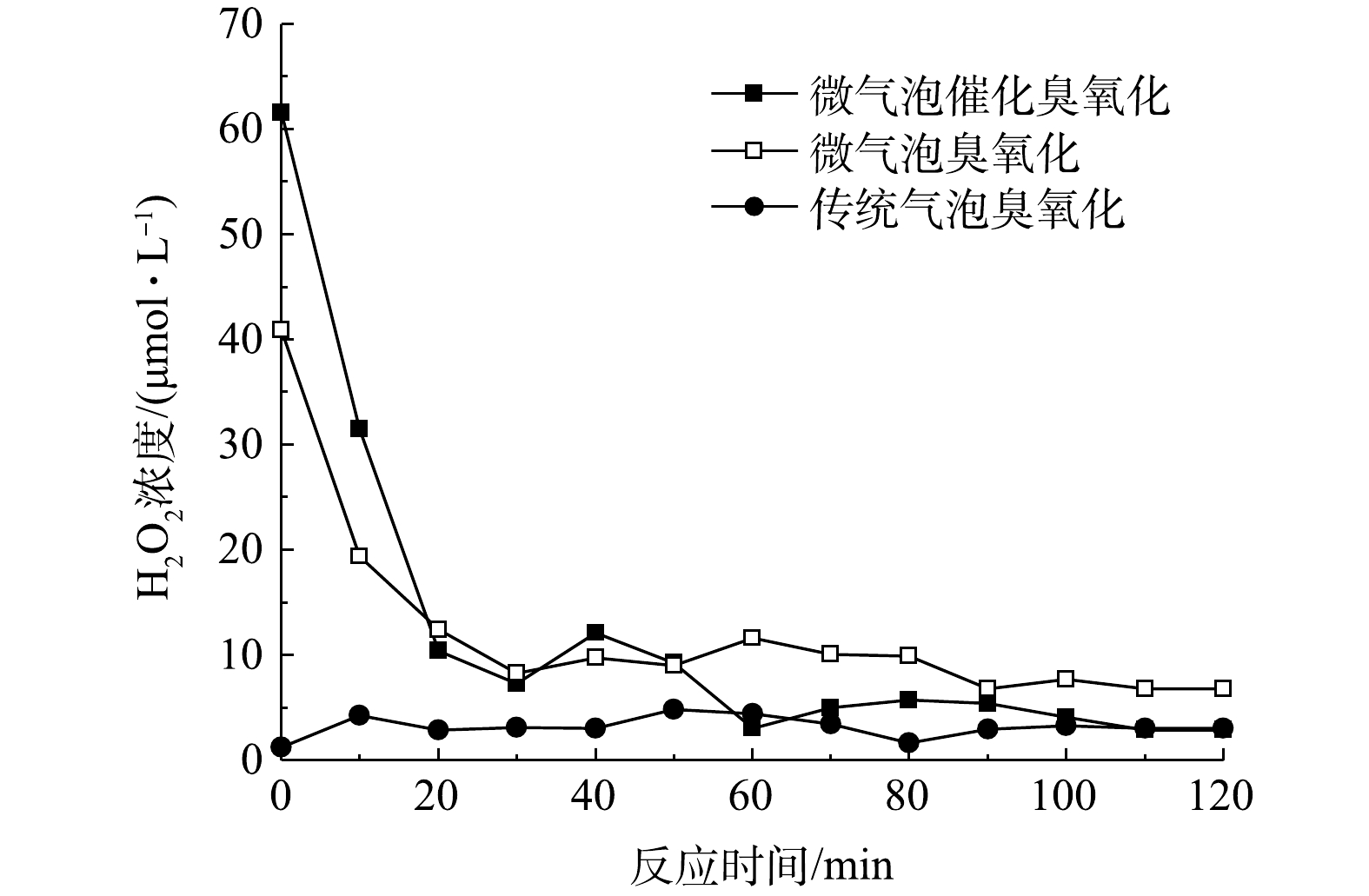
可以看出,H2O2作为臭氧分解产生·OH的前体物,对于·OH的产生有着重要的影响。微气泡催化臭氧化、微气泡臭氧化以及传统气泡臭氧化处理中H2O2浓度随时间变化如图7所示。在传统气泡臭氧化中H2O2浓度极低,为1.2~4.8 μmol·L−1,平均值为3.2 μmol·L−1。传统气泡臭氧化中H2O2由式(5)生成,通过式(6)~式(11)引起臭氧自分解生成·OH。同时,由式(5)可知,碱性条件有利于引发臭氧分解,在传统气泡臭氧化过程中的pH持续降低,不利于臭氧分解反应,导致TOC去除率较低。同时,由图7可以看出,微气泡臭氧化与微气泡催化臭氧化H2O2初始浓度均较高,分别为40.9 μmol·L−1和61.6 μmol·L−1;此后,H2O2浓度均呈现明显下降趋势并在40 min后趋于稳定,在反应40~120 min后H2O2平均浓度分别为6.1 μmol·L−1和8.9 μmol·L−1。臭氧微气泡收缩破裂快速产生·OH(式(12)),进而·OH与O3反应生成H2O2(式(13)和式(14)),这可能是微气泡臭氧化初期H2O2浓度较高的主要原因。微气泡催化臭氧化中存在活性炭与O3的反应(式(17)和式(19)),从而进一步提高初期H2O2浓度。在反应后期,微气泡臭氧化与微气泡催化臭氧化均出现H2O2浓度的持续下降,这主要是由于,H2O2在臭氧分解链式反应中被消耗生成·OH,以及H2O2与活性炭反应生成·OH。
-
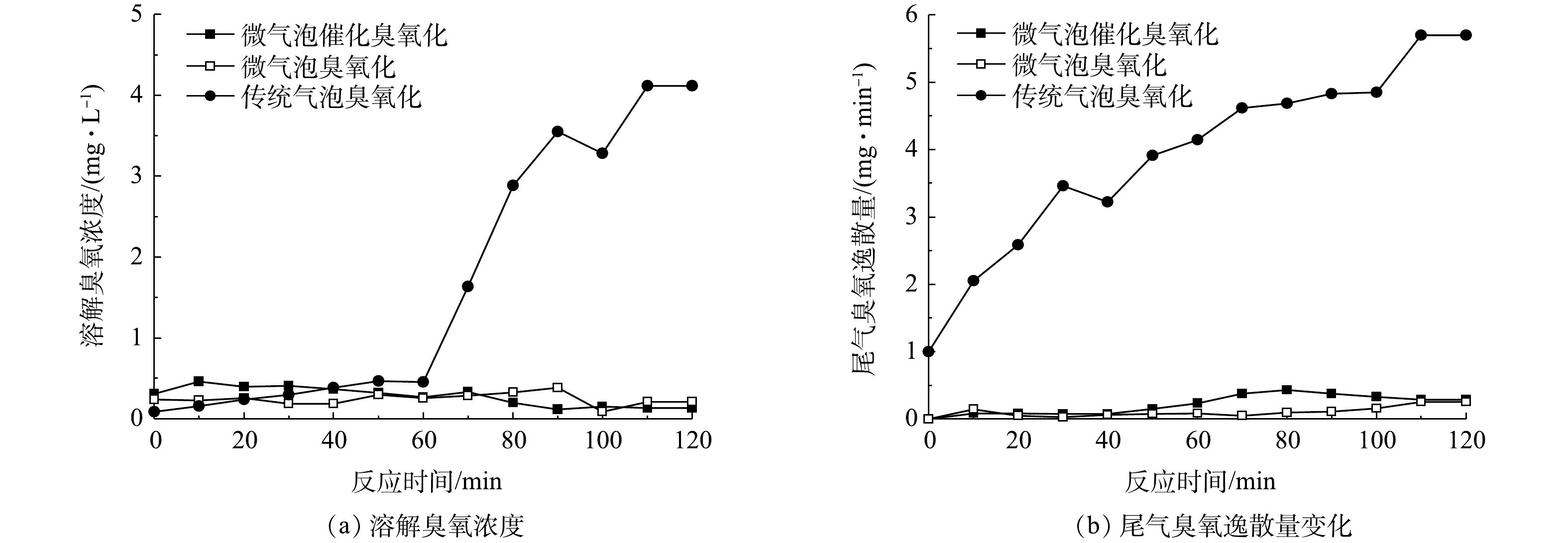
微气泡催化臭氧化、微气泡臭氧化以及传统气泡臭氧化处理中溶解臭氧浓度和臭氧逸散量随时间的变化如图8(a)和图8(b)所示。可以看出,在传统气泡臭氧化处理70 min后,溶解臭氧浓度由0.45 mg·L−1迅速升高至4.11 mg·L−1。其原因可能是,pH不断降低,从而抑制溶解臭氧分解,同时臭氧反应消耗量下降,造成了溶解臭氧累积。微气泡催化臭氧化与微气泡臭氧化均具有高臭氧传质效率和高臭氧化反应效率,处理过程中溶解臭氧浓度和尾气臭氧逸散速率始终较低,其平均溶解臭氧浓度分别为0.29 mg·L−1和0.25 mg·L−1,平均尾气臭氧逸散量分别为0.21 mg·min−1和0.09 mg·min−1,平均臭氧利用率分别为98.3%和99.3%,臭氧利用率约为传统气泡臭氧化的1.7倍。
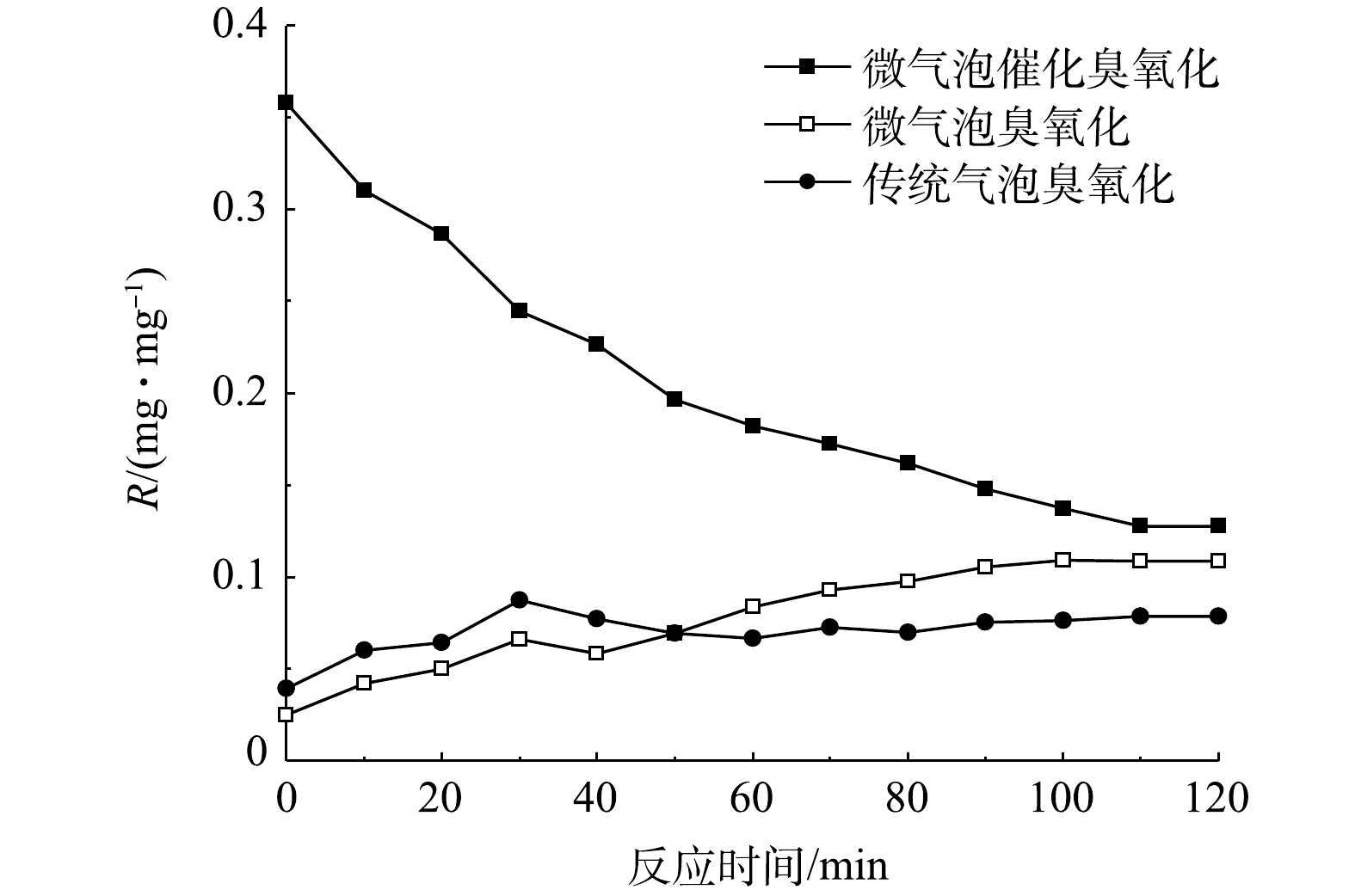
在微气泡催化臭氧化、微气泡臭氧化以及传统气泡臭氧化处理进程中,累积TOC去除量与累积臭氧消耗量的比值(R)[11]随时间变化如图9所示。由图9可以看出,微气泡臭氧化和传统气泡臭氧化处理初期R值均较低,此后逐渐增加,在120 min时R值分别升至0.079 mg·mg−1和0.109 mg·mg−1。其原因是,反应初期微气泡臭氧化和传统气泡臭氧化过程中臭氧主要用于氧化脱色,脱色反应结束后TOC的去除开始增加,R值得以升高。微气泡臭氧化R值高于传统气泡臭氧化,这表明微气泡臭氧化由于促进·OH氧化更有利于TOC去除。微气泡催化臭氧化反应初期即可实现TOC的快速去除,R值高达0.358 mg·mg−1(10 min),至反应结束(120 min)时R值降至0.128 mg·mg−1 (不含活性炭吸附),这亦证实微气泡催化臭氧化可明显改善TOC的氧化去除率。由此可见,尽管臭氧微气泡产生过程会增加能耗,但微气泡催化臭氧化在增强氧化能力、提高臭氧反应效率和臭氧利用率方面具有显著优势,与传统催化臭氧化相比,仍有提升效率、污染物减排整体效益以及降低成本的巨大潜力。同时,相比于芬顿等高级氧化技术,微气泡催化臭氧化在处理过程的清洁性、环境友好性、操作维护简易性等方面亦具有明显的应用优势,在难降解工业废水深度处理中具有良好的应用前景。
-
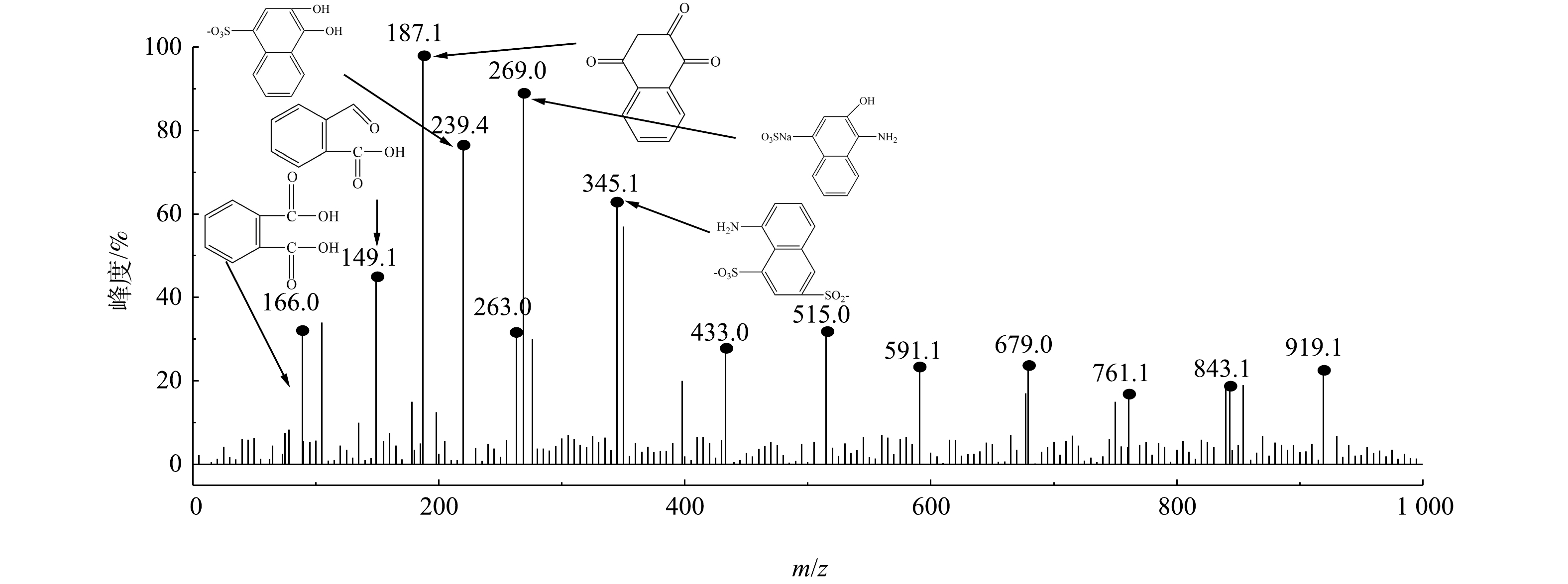
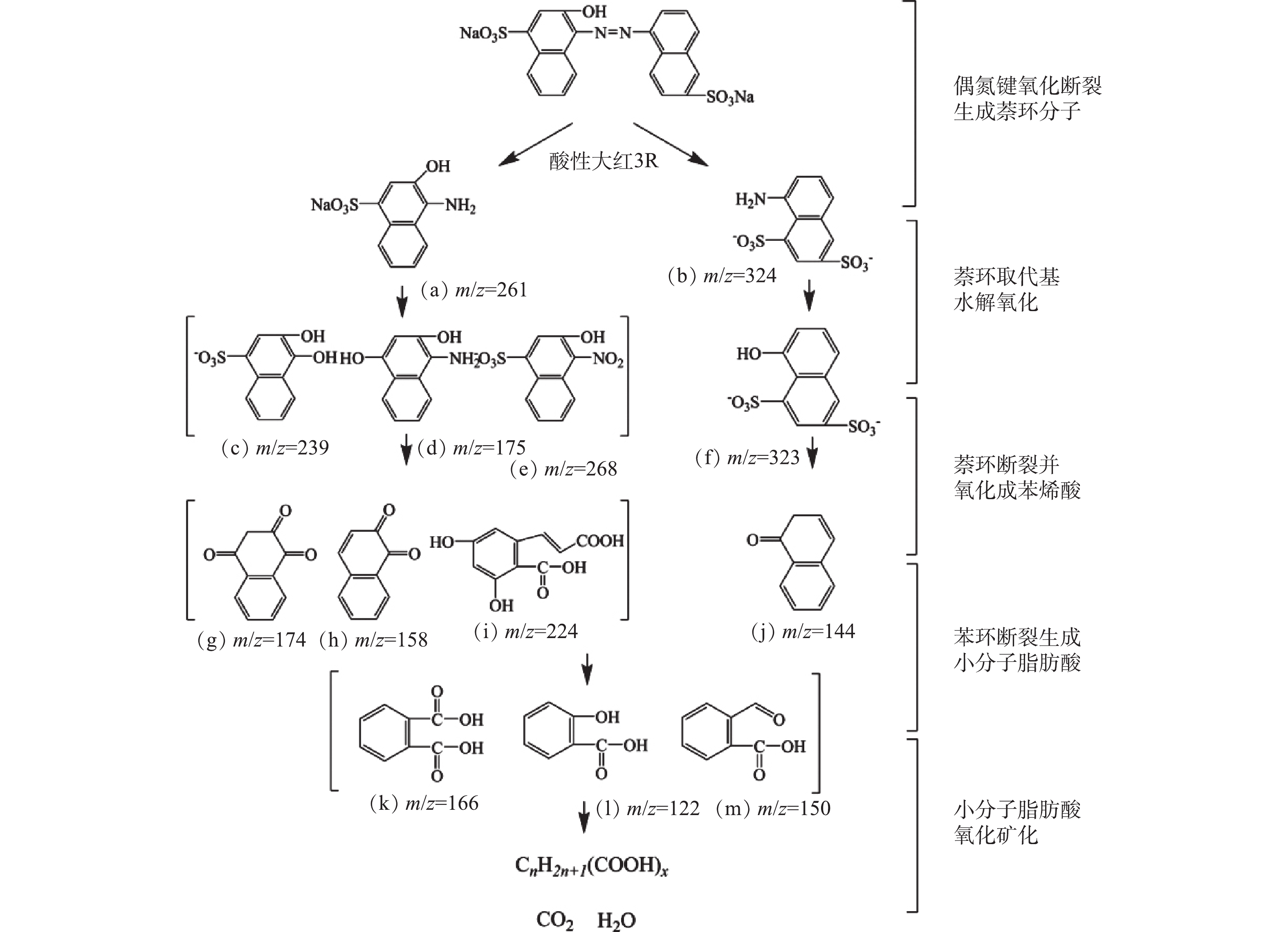
采用GC-MS对微气泡催化臭氧化处理酸性大红3R在90 min时刻的中间产物进行了分析,质谱检测结果和酸性大红3R可能的降解途径分别如图10和图11所示。由图10可知,在降解过程中可能存在的中间产物分别为3-羟基-4-氨基萘磺酸盐(a)、5-氨基-1,4-萘二磺酸盐(b)、3,4-二羟基萘磺酸盐(c)、1,3-二羟基-4-氨基萘(d)、3-羟基-4-硝基萘磺酸盐(e)、5-羟基-1,3-萘二磺酸(f)、1,2,4-萘三酮(g)、1,2-萘二酮(h)、3,5-二羟基-6-羧基肉桂酸 (i) 、1-萘酮(j)、邻苯二甲酸(k)、水杨酸(l)、2-甲酰基苯甲酸(m)。由图11可知,微气泡催化臭氧化降解酸性大红3R途径为:偶氮键最先断裂生成萘和苯系化合物,进而萘和苯系化合物发生氧化、开环、断裂,生程小分子有机酸,最终小分子有机酸被彻底矿化。
2.1. 臭氧分解速率变化
2.2. 脱色率的变化
2.3. TOC去除率的变化
2.4. 无机阴离子的影响
2.5. pH的变化
2.6. H2O2和·OH的变化
2.7. 臭氧利用率
2.8. 酸性大红3R可能的降解途径
-
1)与微气泡臭氧化和传统气泡臭氧化相比,微气泡催化臭氧化可加速溶解臭氧分解并强化·OH氧化能力,可显著提高对酸性大红3R的催化性能,其臭氧分解系数为0.093 min−1,脱色和TOC去除反应速率常数分别为0.342 min−1和0.024 min−1,TOC去除与臭氧消耗比值R为0.128 mg∙mg−1,平均臭氧利用率为98.3%。
2)在臭氧微气泡破裂和活性炭催化协同作用下,微气泡催化臭氧化产生·OH的能力显著增强,对TOC去除贡献率约为75%;·OH捕获剂的存在使得TOC去除率下降,其中 Na2CO3抑制作用最为显著。
3)微气泡催化臭氧化降解酸性大红3R途径为偶氮键最先断裂生成萘和苯类化合物,进而萘和苯类化合物氧化、开环、断裂,产生小分子有机酸,最终小分子有机酸彻底矿化。
