







 下载:
下载:




















-
工业废水种类繁多,成分复杂,常含有毒有害物质,须经处理达到相关标准,才能够排放。加强工业废水的有效处理和达标排放是实施生态环境保护的重要内容[1]。其中,电力、炼油、油气开采、焦炭、皮革、冶金、造纸、农药等行业产生的难处理高含盐废水,除了常规的水质指标(如悬浮物质、COD、BOD、pH、重金属离子等)之外,还应将可溶性盐(含量常大于1%)和有毒有害有机物质作为重点去除对象[2-4]。
高含盐废水中有机物的去除方法主要有物理吸附[5-6]、膜分离[7]、生物降解[8]、高级氧化技术(advanced oxidation processes,AOPs)等[9-10]。AOPs技术通过产生具有强氧化能力的活性氧化物(reactive oxidant species,ROS),可快速氧化降解大多数有机物(如有机染料[11]、表面活性剂[12]、烃类[13]、酚类[14]、药物活性成分[15]、农药[16]等),是深度处理含盐废水的主要手段[17-19]。其中,应用较多的AOPs有Fenton氧化技术[20-21]、臭氧催化氧化技术[22-23]、“臭氧+双氧水”氧化技术[24-25]、活化过硫酸盐技术[26-27]等,起主要作用的ROS为羟基自由基(·OH)或硫酸根自由基(
SO−4⋅ )。后文将基于·OH的AOPs称为HR-AOPs(hydroxyl radical based AOPs),将基于SO−4⋅ 的AOPs称为SR-AOPs(sulfate radical based AOPs),基于非自由基的AOPs称为NR-AOPs(non-radical based AOPs)。氯离子(Cl−)是大多数高含盐工业废水中的主要阴离子[28]。有研究结果表明,高浓度的Cl−对HR-AOPs和SR-AOPs有不同程度的抑制作用[29-32]。由于Cl−与·OH和
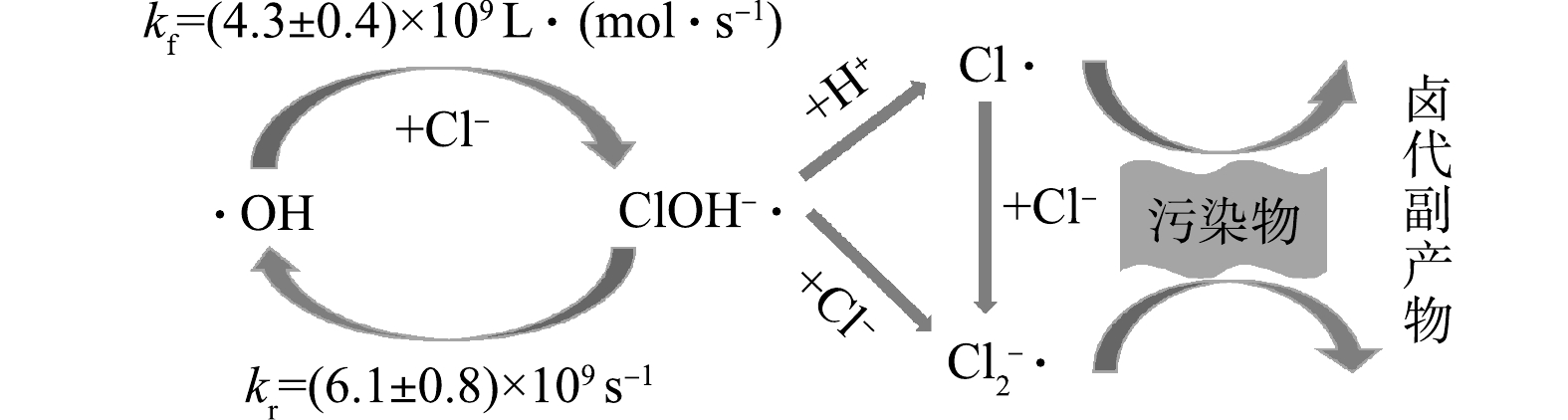
SO−4⋅ 反应均有较高的反应速率,反应如式(1)~(5)所示[32-36],基本机理为Cl−与·OH和SO−4⋅ 经过一系列反应生成Cl·,而Cl·与Cl−具有高反应速率(反应(5)),因此在高浓度Cl−环境中,极易生成氧化活性相对较弱的Cl−2⋅ ,从而可能降低整个氧化反应对有机物的去除效率[29, 32, 37],增大氧化剂用量成本。同时,反应过程中产生的一系列活性氯(Cl·、Cl−2⋅ 等)等次生自由基也具有一定的氧化能力,与有机物反应后,可能导致多种有毒甚至致癌的氯代有机副产物的生成[29, 37-39]。已有研究[20, 22-27]大多未考虑Cl−对COD去除率和氧化剂效率的影响,氯代有机副产物的生成亦被忽略。同时,结合反应式(1)~(5)的正、逆反应速率常数可发现,主要反应物(如Cl−、
SO2−4 、H+、OH−)的浓度可能会影响各反应中的物质平衡浓度,进而可调节Cl−对AOPs效率的影响。本文梳理了HR-AOPs、SR-AOPs及NR-AOPs在氧化去除有机物时受Cl−影响的机理,从Cl−与不同自由基(·OH、
SO−4⋅ )的系列反应及正逆反应速率常数、Cl−浓度与pH的复合影响2方面出发,探讨了Cl−对3类AOPs工艺去除废水中有机物的不同影响及作用机理,提出了调控Cl−对有机物去除效率的抑制和控制氯代有机副产物生成的可能途径,以期为提高AOPs工艺去除高氯盐废水中有机物的效率提供参考。
全文HTML








-
以·OH为主要ROS的HR-AOPs包括Fenton氧化[40]、类Fenton氧化[41]、O3催化氧化[42]、能量方式(紫外光、微波、超声等)[43-45]活化H2O2氧化等技术。·OH的氧化还原电位(E0)与pH紧密相关:中性条件下E0为1.8 V,氧化能力较弱;酸性条件下E0为2.7 V[46],对大多数有机污染物具有非选择性氧化能力;且·OH能够引入含氧官能团,这使得污染物更易被生物降解[47]。在高氯废水中,Cl−能与·OH迅速发生反应(见式(2)~(5)),生成多种含氯自由基(Cl·、
Cl−2⋅ 、ClOH−·)。其中,Cl·和Cl−2⋅ 的氧化还原电位分别为2.59 V[48]和2.30 V[49],具有较强的氧化能力,能够一定程度减小Cl−清除·OH导致体系有机物去除速率降低的影响。Cl·亲电性强,容易加成到有机物分子的不饱和键上,Cl−2⋅ 则会通过单电子转移和抽氢反应与不饱和有机物及中间产物发生氯化作用,其选择性比·OH对有机物的去除更强[32],但会导致氯代有机物副产物的生成。由于物质的各种状态会影响反应平衡过程,因此,Cl−在体系中的浓度是影响上述反应的重要因素。Cl−与·OH发生的主要反应(式(2))是可逆反应。基于该反应,可认为Cl−浓度对正逆反应中·OH和ClOH−·的相对比例有重要影响,故Cl−清除·OH和产生一系列氯代自由基与Cl−浓度密切相关。当Cl−清除·OH和ClOH−·自分解的反应速率相等(见反应式(6))时,将式(2)正逆反应速率常数代入式(6),计算得到的Cl−浓度约为1.42 mol·L−1(约50 000 mg·L−1)。理论上,当[Cl−]≥1.42 mol·L−1时,正反应更具优势,ClOH−·有净累积,·OH减少故污染物降解反应受到抑制。而Cl−浓度较低时,式(2)的逆反应占主导,ClOH−·会更快地自分解重新生成·OH,此时体系中ClOH−·累计较少,理论上,污染物降解速率受Cl−的影响较小。因此,HR-AOPs处理含氯废水时,对低浓度Cl−的抑制作用具有一定耐受能力。
以上基于反应式(2)的计算表明,HR-AOPs处理含氯盐有机废水时受Cl−浓度差异的影响较大,许多研究结果也证实了这一点。ZHANG等[50]采用UV/H2O2法降解4-硝基酚,当水溶液中Cl−浓度从0增加到15 mmol·L−1时对4-硝基酚降解速率有所抑制,但效果并不显著,且明显弱于
NO−3 等其他阴离子。王广生等[51]考察了Cl−对UV/NO−3 光化学降解磺胺甲噁唑(sulfamethoxazole,SMX)的影响时也发现,0~7 mmol·L−1的Cl−对SMX的降解并不产生明显抑制。许入义等[52]研究光电催化氧化体系降解苯胺类污染物的同步耦合反应机制时,通过加入1 000~8 000 mg·L−1的NaCl探究Cl−的影响,当NaCl质量浓度为1 000~6 000 mg·L−1时,单光与光电体系的污染物去除速率同幅度持续上升,而当NaCl浓度进一步增加至8 000 mg·L−1时反应速率下降,这是由于产生了活性Cl·,其氧化活性不如·OH所导致。文献[50-52]表明,HR-AOPs对低浓度Cl−的抑制作用具有一定的耐受性,而不同研究体系中耐受Cl−影响的范围并不相同,且均低于理论计算值(1.42 mol·L−1,约50 000 mg·L−1)。这是由于Cl−与·OH在实际反应过程中,除式(2)中的逆反应消耗ClOH−·外,式(3)~(4)的反应也会消耗ClOH−·;当式(2)的反应产生ClOH−·后,无论ClOH−·有无净累计,式(3)~(5)的反应都会消耗ClOH−·,产生Cl·和
Cl−2⋅ 。YANG等[37]采用动力学模拟软件Kintecus拟合计算UV/H2O2体系产生的自由基种类及稳态浓度时发现,纯水和540 mmol·L−1 NaCl体系中·OH的稳态浓度在pH为7时分别为9.83×10−13和9.82×10−13 mol·L−1,在pH为3时分别为1.08×10−12和4.42×10−13 mol·L−1。该结果说明,在中性条件下,即使Cl−在较高浓度时,其对HR-AOPs中·OH稳态浓度的影响也较小,但酸性环境会明显降低·OH的稳态浓度,进而抑制对有机物的去除速率。在NaCl浓度为540 mmol·L−1的UV/H2O2体系中,产生Cl−2⋅ 的稳态浓度在pH 7和pH 3时分别为3.88×10−14和4.89×10−11 mol·L−1,是同体系中·OH稳态浓度的1/30和110倍,这表明酸性条件有利于Cl−2⋅ 生成,且中性条件时也有较明显的Cl−2⋅ 产生,而Cl−2⋅ 与有机物反应会生成氯代有机物副产物。因此,HR-AOPs处理含氯废水时,尽管理论上反应速率受低浓度Cl−的抑制作用影响较小,但仍会有较高浓度Cl−2⋅ 的生成,具有生成氯代有机物副产物的风险;且由于pH的影响(见反应式(3)),在低pH下,即使Cl−在较低浓度时也会对有机物去除速率产生明显抑制,且生成氯代有机物副产物的风险增大。 -
Cl−浓度对HR-AOPs体系的影响与pH密切相关。根据氧化还原电位(E0)的大小,在中性条件下,Cl·(E0=2.59 V)[48]和
Cl−2⋅ (E0=2.30 V)[49]的氧化能力均强于·OH(E0=1.8 V),而酸性条件下·OH(E0=2.7 V)氧化有机物的能力更强。因此,单从ROS的氧化还原能力来看,酸性条件下Cl−的反应效率会下降,中性(甚至碱性)条件反而会促进反应。不同类型HR-AOPs中,Fenton氧化反应最佳pH为3~4,而臭氧催化氧化、类Fenton氧化、能量方式活化H2O2、光催化氧化等AOPs的最佳pH范围则为中性或偏碱性。Cl−对Fenton氧化技术的抑制作用较后几种更明显。许多研究结果也证实了这一点:邬莎娜等[53]采用Fenton法处理N,N-二甲基甲酰胺模拟废水时发现,Cl−对体系中COD的去除有很强的抑制效果,出水COD从90 mg·L−1上升至250 mg·L−1;吴广宇等[54]采用Fe0非均相UV/Fenton技术处理水中阿特拉津(Atrazine,ATZ)时,却发现Cl−对ATZ的去除具有明显促进作用,在pH为3时,200 mg·L−1的Cl−能将ATZ的去除率从3 min达到的90.8%提高至为1 min后达到的91.2%。Cl−与·OH发生式(2)的反应会首先产生ClOH−·。因为ClOH−·在酸性条件下能够与H+快速反应生成Cl·(式(3)),所以抑制了污染物的降解;而在中性或碱性条件下,通过式(2)的逆反应可重新生成·OH。当pH为1时,将式(3)的速率常数代入式(7)后可发现,反应速率常数k′f3(2.6×10−9 s−1)与式(2)的逆反应速率常数kr2(6.1×10−9 s−1)为同一数量级,部分ClOH−·能够通过式(3)转化为Cl·。然而,随着pH升高,ClOH−·通过式(3)的反应转化为Cl·的比例会明显减少,Cl−对HR-AOPs去除有机物的抑制作用减弱。BOUTITI等[55]利用UV/H2O2降解1-己基-3-甲基咪唑时发现,在天然环境的pH条件下,当Cl−浓度大于0.1 mol·L−1时,污染物的降解速率不受影响,但在酸性环境下会抑制污染物的降解。PIGNATELLO等[56]也发现了类似情况,当pH为2.8且Cl−浓度超过0.01 mol·L−1时,利用Fenton反应降解2,4-二氯苯氧乙酸的反应会受到明显抑制,此亦是由于清除了·OH,活性物质减少而致。KIWI等[57]发现,在酸性条件下通过Fenton反应产生·OH来降解偶氮染料,而当Cl−存在时,其降解率也明显下降。由此说明,pH对HR-AOPs处理高盐废水有较大影响,且在酸性条件下受Cl−的抑制作用更加明显。YANG等[37]的模拟实验也证明了这一现象,即当pH为3时,UV/H2O2处理无卤素的纯水体系中仅存在·OH一种自由基,其稳态浓度为1.08×10−12 mol·L−1,而在投加540 mmol·L−1的NaCl后,体系中·OH、Cl·和
Cl−2⋅ 的稳态浓度分别为4.42×10−13、1.40×10−15、4.89×10−11 mol·L−1;中性条件下,纯水与含氯体系中·OH稳态浓度几乎相同,含氯体系中产生的Cl−2⋅ 也仅为同体系·OH的1/30,而在酸性条件下纯水体系中的·OH是含氯体系中·OH的2.44倍,含氯体系中产生的Cl−2⋅ 则是同体系·OH的110倍,故酸性条件下Cl−对UV/H2O2去除有机物的抑制作用将更加明显。Cl−和pH对HR-AOPs的影响机理如图1所示。在实际采用HR-AOPs进行水处理时,Cl−的影响机制与体系pH具有复合作用,而HR-AOPs对低浓度Cl−的抑制作用有一定耐受能力,在酸性条件下受Cl−抑制作用大于中碱性条件。
1.1. Cl−浓度对HR-AOPs的影响











1.2. Cl−与pH对HR-AOPs的复合影响





-
因
SO−4⋅ 具有氧化还原电位高(2.5~3.1 V)、pH适用范围广[58]、存留时间(30~40 μs)比·OH更长[59]等优点,故SR-AOPs倍受关注。SO−4⋅ 与有机物的反应主要通过夺取电子来实现,对有机物的氧化更具选择性,对某些有机物的氧化效率比·OH更高。SR-AOPs主要是通过活化过一硫酸氢钾(KHSO5,Potassium peroxymonosulfate,PMS)或过硫酸钠(Na2S2O8,Sodium peroxydisulfate,PDS)产生SO−4⋅ 进而氧化降解有机物。由于Na2S2O8比KHSO5更稳定,且在水中溶解度更高,因此,在实际污染修复中用得更多。活化方式可分为2大类:能量活化(如热、紫外光、超声等)[60-62]和催化剂活化(过渡金属等)[63]。 -
在采用SR-AOPs处理含氯有机废水时,Cl−会与有机污染物竞争消耗
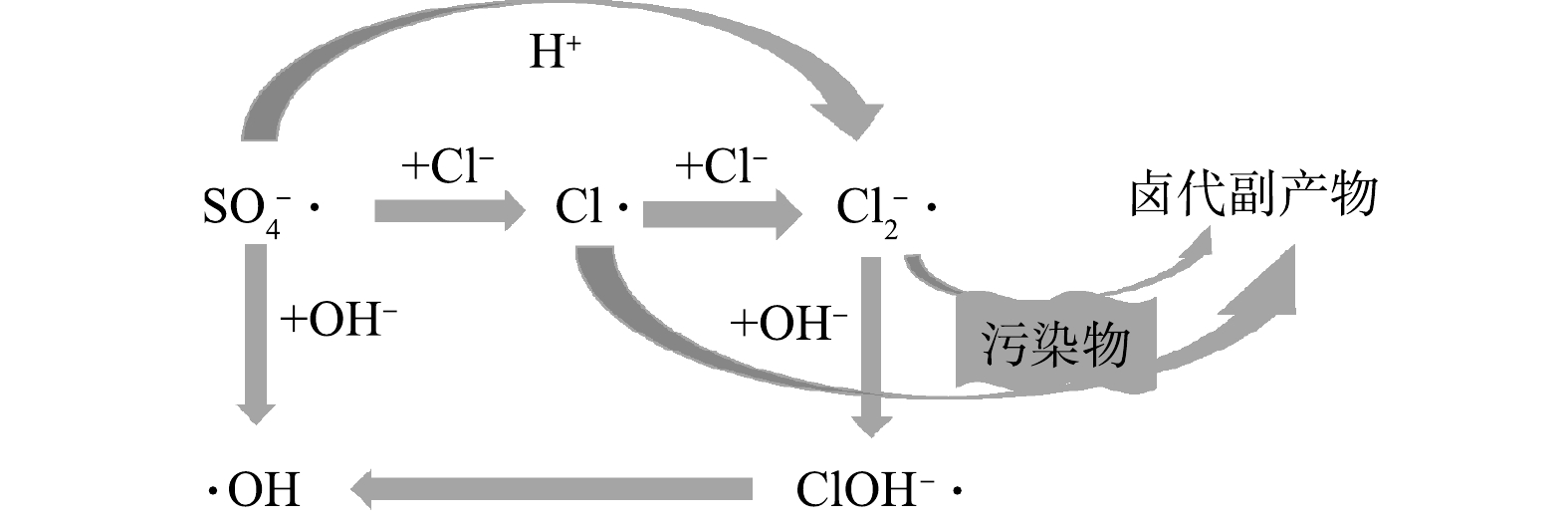
SO−4⋅ ,直接反应生成Cl·(式(1)),Cl·再进一步与其他Cl−反应生成Cl−2⋅ (式(5))[64]。不同于Cl−在HR-AOPs中与·OH经过一系列反应才生成Cl·,Cl−与SO−4⋅ 反应直接生成Cl·。从主要反应来看,SR-AOPs比HR-AOPs更易受Cl−的影响,同样也受Cl−浓度和pH的影响,但具体表现有较大差别。Cl−通过发生式(1)的反应清除
SO−4⋅ 而生成Cl·,由于式(1)的正反应速率kf1大于其逆反应速率kr1,因此,从理论上来说,利用SR-AOPs处理含氯盐废水时,在低Cl−浓度下有机物的去除会受到较大抑制,即Cl−产生抑制作用的浓度阈值更低。以热活化PDS的相关研究为例,Cl−的存在对酮洛芬[65]、阿特拉津[66]、磺胺-氯吡嗪[67]、三氯乙烯[68]和四氯化碳[69]等的降解均产生了明显抑制。CHAN等[70]采用活化过氧单硫酸盐(PMS)法降解阿特拉津并探讨了Cl−的影响,发现Cl−在反应中具有明显抑制作用,其原因可能是由于Cl−清除了SO−4⋅ ,并且产生了氧化活性较弱的Cl·。当Cl−浓度持续增大时,积累的Cl·将与Cl−进一步发生反应(式(5))生成Cl−2⋅ 。当溶液中的自由基种类增加,对污染物降解的影响则更加复杂。而当Cl−浓度增加到一定程度时,则对污染物降解的影响呈先抑制后促进的双重作用。WANG等[71]在钴活化过氧单硫酸盐体系(Co/PMS)中发现,当Cl−(浓度为0~10 mmol·L−1)存在时,
SO−4⋅ 对偶氮染料的降解作用被明显抑制;而当Cl−浓度较高(>100 mmol·L−1)时,偶氮染料的降解作用被增强;这可能是由于Cl−浓度较低时会优先清除SO−4⋅ ,而产生活性更小的Cl·阻碍染料的脱色;当Cl−浓度进一步增大,Cl−通过双电子转移直接与PMS氧化反应产生活性卤素Cl2或HClO,活性卤素能进一步与染料反应可能产生卤代有机化合物。周骏等[72]在UV/PMS体系降解硝基氯芬的动力学及机理研究中也提出了Cl−的双重作用,Cl−为0.05~10 mmol·L−1时对污染物的降解起抑制作用,而当Cl−从30 mmol·L−1增加到100 mmol·L−1,体系反应速率迅速上升,甚至能达到无氯体系的8.5倍。在热/PDS降解二氯苯氧氯酚[73]和头孢氨苄[74]等的研究中,也都发现Cl−存在类似先抑制后促进的双重作用。 -
SR-AOPs受pH的影响主要表现在体系中的自由基成分及含量在不同pH条件下差异较大。有研究表明,pH<9时
SO−4⋅ 为主要自由基成分;当9<pH<11时,体系中通常出现SO−4⋅ 与·OH共存的情况;当pH>11时,则主要是·OH[75]。这主要是由于,在偏碱性条件下,SO−4⋅ 能够与OH−发生反应(8)产生·OH[76-77]。在pH与Cl−的复合影响下,较低pH条件时SR-AOPs体系中主要的自由基为SO−4⋅ ,其能迅速与Cl−发生反应(见式(1)和式(5)),生成Cl−2⋅ ;当pH较高时,SO−4⋅ 生成·OH而导致Cl−2⋅ 浓度降低,同时Cl−2⋅ 与OH−发生式(9)的反应,使得Cl−2⋅ 浓度进一步降低,ClOH·−浓度增加[78],并且ClOH−·会继续发生自分解产生更多·OH(式(2)的逆反应)[79]。FANG等[80]研究SO−4⋅ 降解多氯联苯时发现,Cl−的存在会影响SO−4⋅ 氧化过程中不同条件下自由基的种类分布,并抑制多氯联苯的降解。WU等[81]研究氯化物对SR-AOPs降解贝扎菲酯和卡马西平影响时也发现类似规律。YUAN等[82]在探究UV/PDS降解染料废水时同样发现,染料在碱性介质中更容易被破坏,这可能是由于·OH为该pH条件下的优势自由基,对污染物的去除选择性不强。因此,Cl−对SR-AOPs的影响主要为
SO−4⋅ 与Cl−反应后形成多种卤素自由基,从而降低体系氧化能力[83-84];同时,在中性或碱性pH条件下,SO−4⋅ 会生成·OH,形成SO−4⋅ 与·OH共存并与Cl−作用的复杂体系[85]。Cl−和pH对SR-AOPs的影响机理如图2所示。综上所述,在含氯盐有机废水处理中SR-AOPs比HR-AOPs更易受Cl−的影响,即使较低浓度即会对有机物去除速率产生明显抑制,且易产生有毒氯代有机物副产物,从而使可吸附有机卤素(absorbable organic halogen,AOX),大量积累,毒性增强[86]。因此,在采用SR-AOPs处理含氯盐废水的研究和应用中还应重视氯代副产物的产生和控制。



2.1. Cl−浓度对SR-AOPs的影响








2.2. Cl−与pH对SR-AOPs的复合影响














-
近年来,非自由基氧化途径(NR-AOPs)逐渐成为AOPs的新兴研究方向。非自由基氧化对污染物的选择性更好,且对氧化剂的利用率也更高[87]。目前,对NR-AOPs的研究大多采用过氧一硫酸盐(PMS)为氧化剂。这是由于PMS分子中(—O3S—O—O—H)过氧键呈非对称结构,且只带一个负电荷,比呈对称结构的过氧二硫酸盐(PDS)更易被活化[88]。PMS非自由基氧化的主要途径包括PMS自分解或与有机物作用产生1O2 [89-90]、PMS直接氧化[91-93]降解有机物。由于这2种途径均未大量产生·OH或
SO−4⋅ ,不存在Cl−竞争消耗自由基而导致污染物降解过程受抑制,因此,PMS的非自由基氧化途径比HR-AOPs和SR-AOPs受Cl−影响更小。 -
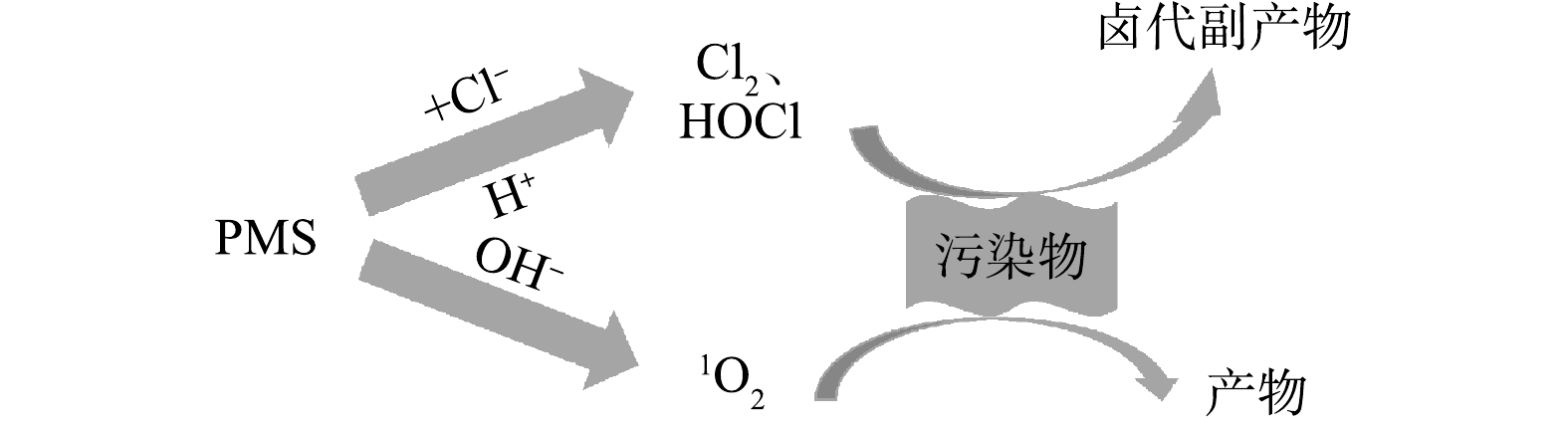
以PMS为氧化剂的NR-AOPs工艺在处理含盐废水时,PMS与Cl−会发生式(10)~(11)的反应而生成HClO和Cl2[94]。HClO和Cl2都是常用漂白剂,具有较强的氧化性(E0(HClO/Cl−)=1.50 V,E0(HClO/Cl2)=1.63 V)[95]。因此,理论上Cl−对PMS非自由基氧化途径降解有机污染物是有一定促进作用的;且随着Cl−浓度增加,HClO、Cl2也更多积累,有机物降解效率也随之增加。LEI等[93]在研究未活化PMS脱色阳离子染料时发现,通过增加Cl−剂量,可进一步缩短RhB的脱色时间,同时还能有效促进TOC降低,这也间接证明无氯代副产物生成。丁曦等[96]采用非活化PMS降解柳氮磺胺吡啶(Sulfasalazine,SSZ)时,发现Cl−的存在同样能明显提高SSZ的降解效率。古振川等[97]在采用Cl−/PMS降解甲氧苄啶(Trimethoprim,TMP)时发现,当Cl−投加量从1.0 mmol·L−1增至7.0 mmol·L−1时,TMP的降解率也随之呈指数型增大。
-
在酸性条件下,PMS的NR-AOPs体系中会发生式(11)~(13)的反应,存在的活性氯物质以HClO为主。ZENG等[98]在探究Cl−/PMS对2,4-二氯苯酚的协同降解作用时,发现降解产物为2,4,6-三氯苯酚,据此提出的机理为Cl−/PMS体系在酸性条件下通过Cl−与
HSO−5 而非SO2−5 反应生成HClO,进而与2,4-二氯苯酚反应生成2,4,6-三氯苯酚;同时,其对照实验表明,Cl−不能活化H2O2和PDS去除2,4-二氯苯酚。FANG等[99]在采用PMS处理2,4,6-三溴酚(2,4,6-tribromophenol,TBP)含盐废水时发现,在pH小于7时会产生有毒卤化衍生物,因此,该技术并不是净化TBP废水的最佳选择。LI等[100]在研究PMS氧化降解苯酚时发现,在酸性条件(pH为2.7)下,Cl−能够促进苯酚降解,并检测到多种活性氯成分,说明在酸性条件下Cl−与PMS反应产生了活性氯物质(HClO和Cl2)(式(10)~(11))进而促进苯酚降解。该降解过程中还检测到4-CP、2,4-DCP等氯代中间产物生成;在中性条件下还检测出活性氯成分,而在碱性条件(pH为9.0)下未检测到活性氯成分;而在中性和碱性条件下,无论有无Cl−均比酸性条件下苯酚降解明显更快,且有无Cl−对苯酚的降解速率影响非常小。通过淬灭实验和电子顺磁共振证实,中性及碱性条件下活性物质为1O2,且在中性及碱性条件时均未发现中间氯代产物生成,苯酚降解更为彻底。因此,PMS非自由基氧化处理含盐废水时,在酸性条件下通过产生多种活性氯物质(HClO和Cl2)以促进污染物降解,而在碱性条件下则主要通过PMS自分解或与有机物作用产生1O2,从而促进污染物降解(见式(14))[89]。综上所述,基于PMS的NR-AOPs是一种新型有效处理含氯盐废水的方法,Cl−和pH对该过程的影响机理如图3所示。与HR-AOPs和SR-AOPs不同,Cl−在偏酸性条件下与PMS反应生成活性氯(主要为HClO和Cl2),能明显促进氧化降解有机污染物,但会生成卤代副产物;在偏碱性条件下,PMS通过自分解或与某些有机物作用产生1O2,从而能有效氧化降解有机物,受Cl−影响小,且无卤代副产物产生。因此,采用PMS非自由基氧化技术处理含氯盐有机废水时,需严格控制pH,可通过调节pH至碱性以减少氯代副产物的生成。PMS非自由基对高氯盐有机废水处理去除有机物有一定应用前景。

3.1. Cl−浓度对PMS非自由基氧化的影响
3.2. Cl−与pH对PMS非自由基氧化的复合影响


-
Cl−对自由基(
SO−4⋅ 、·OH)型AOPs去除有机物有较大影响,故需进一步研究Cl−竞争反应及氯代副产物产生的机理,增加对AOX、生物毒性、TOC等指标的考量,在更深层面探究Cl−对AOPs去除有机物的影响。此外,还需要探索更多的非自由基(1O2、O−2⋅ )途径氧化降解污染物,研究活性物质种类、来源、机理、反应条件优化、受干扰离子影响机理等因素对反应体系的影响。研究非均相催化材料在AOPs中的应用,可通过在催化材料界面产生SO−4⋅ 、·OH与目标污染物反应,减弱自由基与Cl−的反应,以提高AOPs处理含盐有机废水对Cl−影响的抗性。



