


 下载:
下载:







-
挥发性有机污染物(volatile organic compounds,VOCs)是指在常温下饱和蒸汽压大于70 Pa、常压下沸点在50~260 ℃以内的有机化合物[1]。VOCs作为PM2.5和O3的重要前驱体,会对大气环境和人类健康造成危害[2-3]。近年来,工业源VOCs排放量有所上升[4],已成为我国VOCs排放的主要来源[5]。企业工厂VOCs排放除了有组织污染排放源外,还存在较多的无组织排放源。无组织排放的VOCs具有物质种类多、排放无规则、排放总量大等特点,其连续在线监测难度大,很难对其进行监测和管理[6-8]。若对无组织排放VOCs的监测不及时,易造成恶臭扰民、厂界排污纠纷等问题。因此,对VOCs排放集中的区域进行实时无组织排放VOCs监测,及时了解环境中不同位置VOCs变化情况,防止其对环境以及人体健康造成危害,具有重要意义。
目前,化工厂VOCs监测方式可分为点、线、面3大类[9]。点监测又可以分为离线点监测和在线点监测。离线点监测为利用苏玛罐[10-11]、气袋[12-13]和吸附管[14-15]等采样工具采集某个位置样品,然后将样品送回实验室进行分析。这种监测方法常规简便,且所用到的实验室仪器如气相色谱/质谱联用仪[16-17]等精度较高、性能优异。然而,该监测方法所采集的样品为某一时间段的VOCs,其特征不具有时效性;且该方法无法监控区域VOCs浓度的长时期时空变化[18]。在线点监测为GC-MS[19-20]、传感器[21]、便携式GC[22]等在线分析仪器对某个位置进行监测分析。这种监测方法通常能够长期自动采样监测,数据具有时效性。但是该方法监测范围单一,难以通过一个位置的VOCs组成与浓度情况实时反映该区域所有位置的情况。线监测为开放光路长光程傅里叶变换红外光谱仪[23]等对直线上最远1 km的距离进行监测。线监测方法速度快,范围比点监测要广,数据有时效性,但能够识别的物质较少,且不能对大面积范围进行采样监测。面监测则为红外气体相机[24]等对最小直径1 km范围内的空间进行监测。红外气体相机监测覆盖范围较大,分析速度快,能够连续在线监测,但无法进行组分定性分析。
综上所述,现有VOCs点与线监测方法中存在监测覆盖面小,而面监测存在无法对未知污染物进行组分分辨的难题。因此,需要结合点监测所用到的高灵敏度、高分辨率仪器与面监测所覆盖的较大监测范围,进行点面联合在线连续监测。使用单台仪器对大范围多个点位VOCs监测的系统有Sentinel哨兵系统[25]等。Sentinel 哨兵系统能够在1 h以内监测50个不同点位VOCs,采样直径可达500 m。但是,该系统检测限为1~100 μmol·mol−1,无法进行低浓度VOCs监测,且只能针对特定物质进行定量分析,无法对未知混合VOCs进行定性分析。
为实现单台分析仪器对化工厂多个不同点位的VOCs连续在线监测,本课题组使用多管路长距离的采样装置进行连续采样,将采样装置与高性能飞行时间质谱仪搭建为一套远距离多通道VOCs连续在线监测系统(以下简称监测系统)。监测系统通过对PAMS与有机硫标气的采样分析,得到其性能指标,以期为化工厂VOCs在线监测提供参考。
全文HTML
-
远距离多通道VOCs连续采样在线监测系统主要由远距离多通道VOCs连续采样装置(以下简称采样装置)与真空紫外灯单光子电离源飞行时间质谱仪(以下简称质谱仪)组成,原理结构示意图如图1所示。监测系统从采样到分析需要通过抽气、储气、质谱仪检测3步完成。1)抽气阶段:气体样品从外界依次经过采样管路、储气罐与抽气泵,保障外界气体样品被采集到;2)储气阶段:在储气罐与外界气压差的作用下,气体样品在储气罐中储存起来,直到储气罐内气压值达到设置的储气气压值,保障储气罐内气体样品恒压进样;3)质谱仪检测阶段:储气罐内气体进入质谱仪进行检测。储气罐气压值达到设置的检测气压时,质谱仪完成检测,保障质谱仪检测到的气体量一致。监测系统连续运行时,2个储气罐交替使用,从而提高监测效率。
-
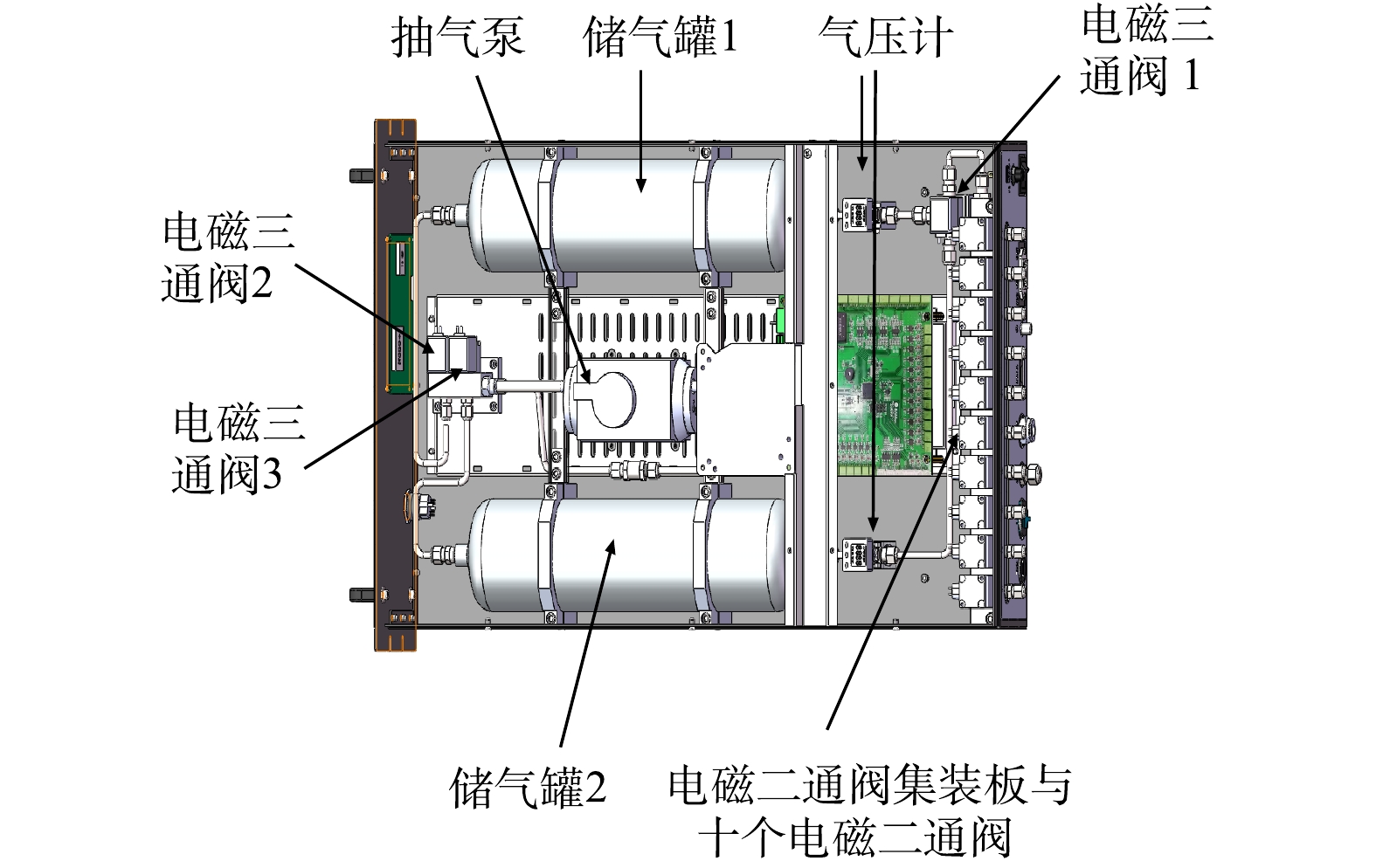
本研究设计的采样装置需在1 h内对10个不同点位样品按时间顺序分别采样。每个点位采样时间小于6 min。采样装置设计如图2所示,主要包含采样管、储气罐、气压计、电磁阀等部件。
基于VOCs远距离采样的需求,要求采样管路内壁材料本身无VOCs物质挥发、对VOCs物质吸附性较低。使用聚四氟乙烯材质采样管路(SMC有限公司,日本),聚四氟乙烯采样管对有机物吸附性较小,能耐腐蚀和老化,价格比钝化的金属管要便宜。系统设计中要求在较短时间内完成10个不同点位的采样,需要直径较大采样管和功率较大采样泵,同时要综合考虑选用泵、采样管的体积和价格。选用采样管外径为6.35 mm,管壁厚1 mm,管长500 m;抽气泵(气海机电制造有限公司,中国)最大流量为28.0 L·min−1。抽气泵对500 m采样管抽气时,采样管前端在整个采样管中流速最小,为1.9 L·min−1。500 m采样管体积为7.4 L,以最小流速作为整段采样管流速计算样品经过采样管的时间为234 s,能在较短时间内采集到样品。
由于采样装置有10个采样通道和2个储气通道,采样装置通过电磁阀开关控制采样顺序。10个采样通道与2个储气通道相连需要多个电磁阀与管路。通过电磁二通阀集装板让不同点位采集到的样品汇集到同一条管路;从同一条管路进入储气罐,减少采样装置内的多余管路与阀。
在对远距离空气样品进行采样时,进样气压较低导致质谱仪灵敏度低,不同长度采样管采样下进样气压不一致,质谱仪难以定量分析。采样过程中需要监测储气罐内气压变化,从而得知进样气压与进样量,本研究使用气压计来记录储气罐的气压变化情况。用2 L储气罐(世伟洛克公司,美国)能够储存气体并在恒定气压值下进行样测。质谱仪采样时流量为0.8 L·min−1,信号强度稳定时间约为10~15 s,信号强度稳定后需要再采样10~15 s进行检测,即整个采样检测时间最多需要30 s。质谱仪开始采样时,储气罐内气压为101 kPa,在采样30 s后,储气罐中气压为81 kPa。检测系统可在45 min内完成对10个不同点位的在线分析。
-
本研究选用真空紫外灯单光子电离源飞行时间质谱仪(广州禾信仪器股份有限公司,中国)作为VOCs分析器,其性能与原理已有多篇相关报道[26-27]。该质谱仪能够对多种VOCs进行定性定量分析,响应时间为秒级。
-
实验仪器:本课题组自主研制的远距离多通道VOCs连续采样在线监测系统;4010L型气体动态稀释仪(Sabio公司,美国);BMSN-2氮气气体纯化器(安捷伦科技有限公司,美国)。
监测样品:浓度为1 μmol·mol−1的57种组分PAMS标准气体(大连大特气体有限公司,中国);浓度为1 μmol·mol−1的9种组分有机硫标准气体(大连大特气体有限公司,中国);纯度为99.999%氮气(广州粤佳气体有限公司,中国)。
监测条件:氮气前装有氮气纯化器过滤杂质,1 μmol·mol−1浓度的标准气体通过气体动态稀释仪进行稀释,稀释仪流量为2.0 L·min−1。每次样品检测之间监测系统用氮气进行吹洗30 min。采样装置抽气时间260 s,储气气压值为低于标准大气压20 kPa,检测气压值为低于标准大气压33 kPa。
-
对监测系统进行性能表征以获得稳定性、检测限、线性、残留影响、信号强度衰减率、定量误差等性能指标。监测系统有500 m长采样管,每次从采样端对监测系统进行校准会耗费大量的标气与氮气,且在外场进行监测时,对不同位置的采样端口进标气校准费时费力,可通过对质谱仪端校准代替监测系统采样端校准,监测系统采样端校准方式需要对系统的样品残留和信号衰减情况来进行计算。
500 m采样管中样品残留会在采样过程中被洗脱出来,进而影响分析结果;500 m管中有样品残留,且进样气压值低于标准大气压,监测系统采样端进样时检测到的信号强度会低于质谱仪端进样分析时信号强度。需对监测系统的残留影响与信号强度衰减率进行测试,得到信号强度转换的指标。
残留影响是残留在采样过程中洗脱出来的信号强度占原样品信号强度的百分比。监测系统的残留影响由式(1)计算得到。
式中:Si为第i次VOCs残留影响;Ii为监测系统第i次吹洗氮气时的信号强度;M为监测系统检测VOCs时的信号强度。
信号强度衰减率是VOCs经过监测系统时对于SPI-MS直接检测VOCs时的减少程度与SPI-MS直接检测VOCs时的比值。信号强度衰减率计算公式如式(2)所示。
式中:D为信号强度衰减率;y为SPI-MS直接检测VOCs时信号强度;M为监测系统检测VOCs时的信号强度。
1.1. 系统原理
1.2. 采样装置
1.3. 质谱仪
1.4. 仪器样品与条件
1.5. 监测系统性能表征
-
用监测系统对0.06 μmol·mol−1 PAMS与有机硫标准气体分别进行重复采样,并分析7次,分别计算各质荷比物质的相对标准偏差,得到PAMS标准气体和有机硫标准气体稳定性结果(见表1)。由表1中可知,所有物质RSD≤7%。
从低浓度到高浓度依次检测标气,其中PAMS标准气体梯度浓度为0.02、0.06、0.10、0.17、0.24、0.31及0.38 μmol·mol−1。有机硫标准气体梯度浓度为0.01、0.06、0.17、0.24、0.31、0.45及1 μmol·mol−1。对PAMS标准气体中的15种质荷比物质和有机硫标准气体中的5种质荷比物质进行分析得到回归方程和判定系数,其结果列于表1。由表1可知,所有物质判定系数均大于0.99。
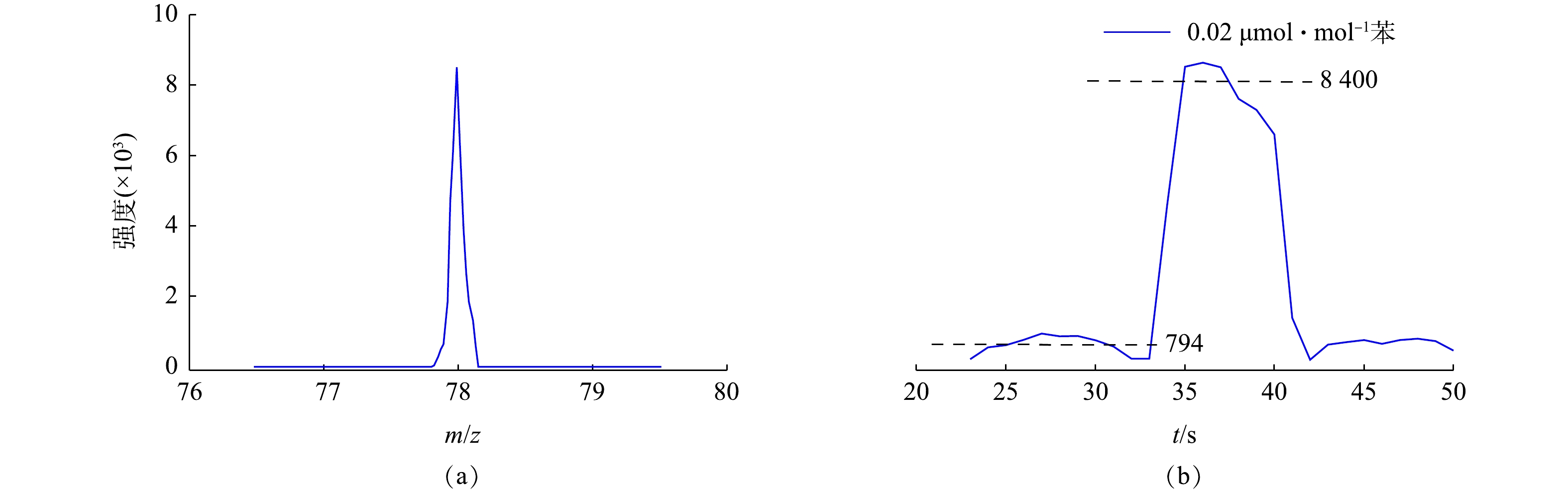
监测系统分别对0.02 μmol·mol−1 PAMS和氮气进行采样分析,得到PAMS标准气体中15种质荷比物质信号强度与本底噪声。同样条件下,分别对0.02 μmol·mol−1有机硫和氮气进行采样分析,得到有机硫标准气体中5种质荷比物质信号强度与本底噪声。PAMS标准气体中苯质谱图如图3(a)所示,苯离子信号强度变化图如3(b)所示。根据检测限的信噪比为3来计算各物质检测限[28],检测限计算结果列于表1,结果表明各物质在浓度为0.013 μmol·mol−1以上均能检测到,最优检测限为0.004 μmol·mol−1。
-
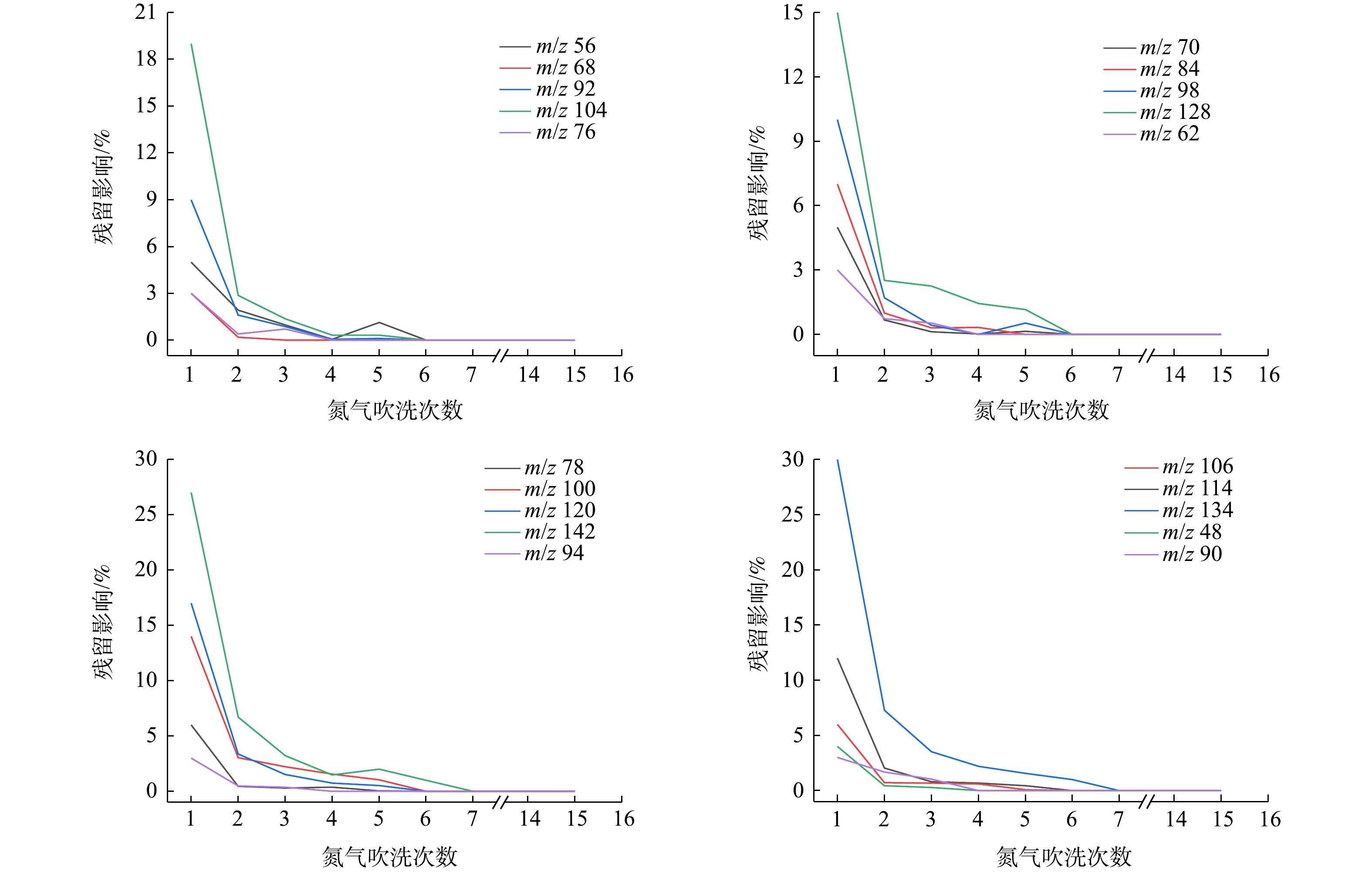
监测系统对PAMS(浓度0.1 μmol·mol−1)或有机硫标准气体(浓度为0.1 μmol·mol−1)进行采样分析,并用纯净氮气连续清洗系统15次,通过公式(1)计算可得各物质的残留影响(见图4)。分析图4数据发现,20种物质在氮气吹洗第7次后残留影响可降至1%以下。
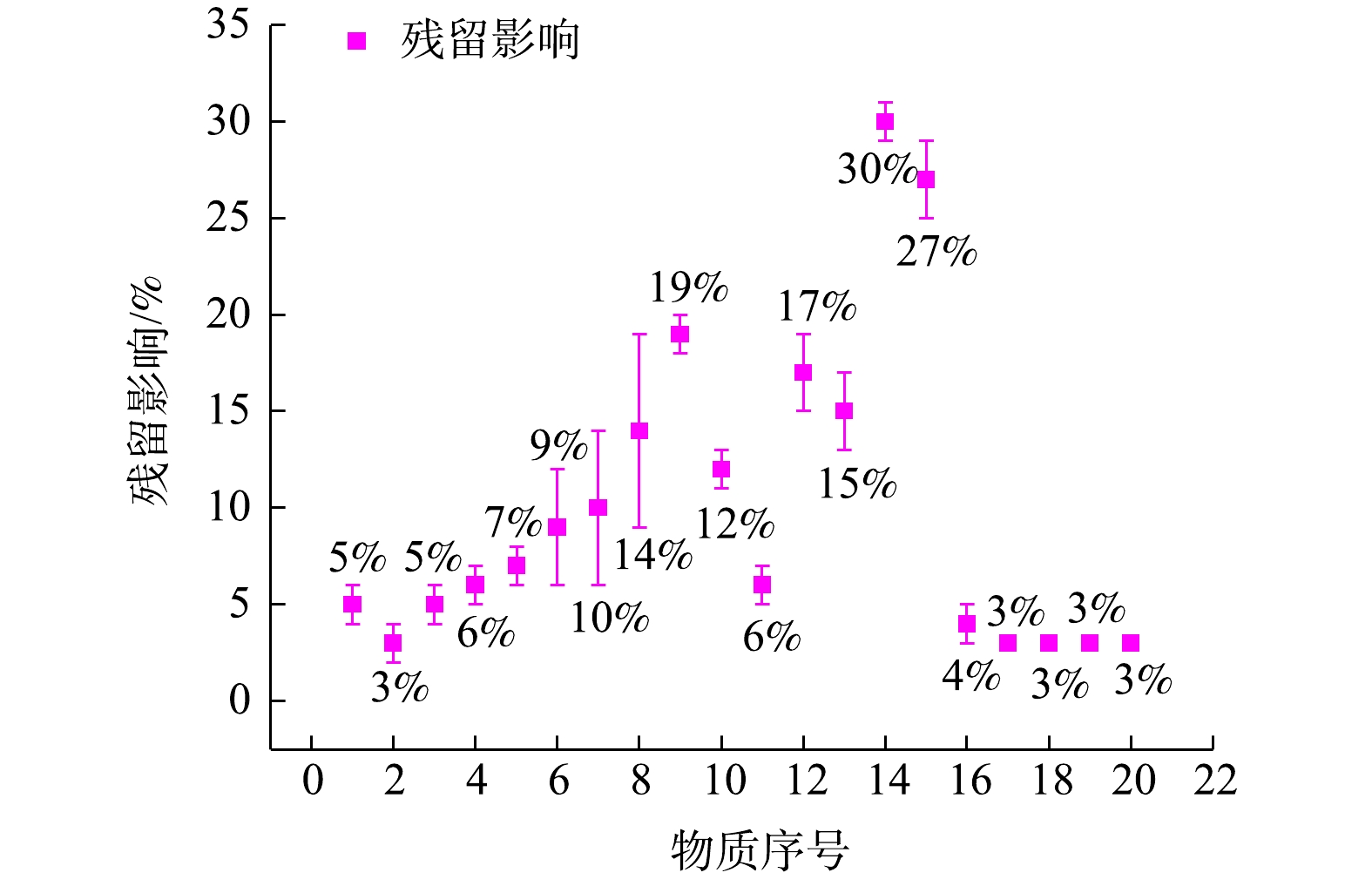
监测系统对PAMS(浓度0.1 μmol·mol−1)或有机硫标准气体(浓度为0.1 μmol·mol−1)与氮气交替分别进行采样分析,一共7次。通过公式(1),计算得到20种物质的残留影响。PAMS与有机硫标准气体中20种物质残留影响及波动范围如图5所示,图5中各物质序号与表1一致。由图5中数据可知,监测系统中所有物质的残留影响在1%~33%,整个监测系统的残留影响波动范围小于10%。
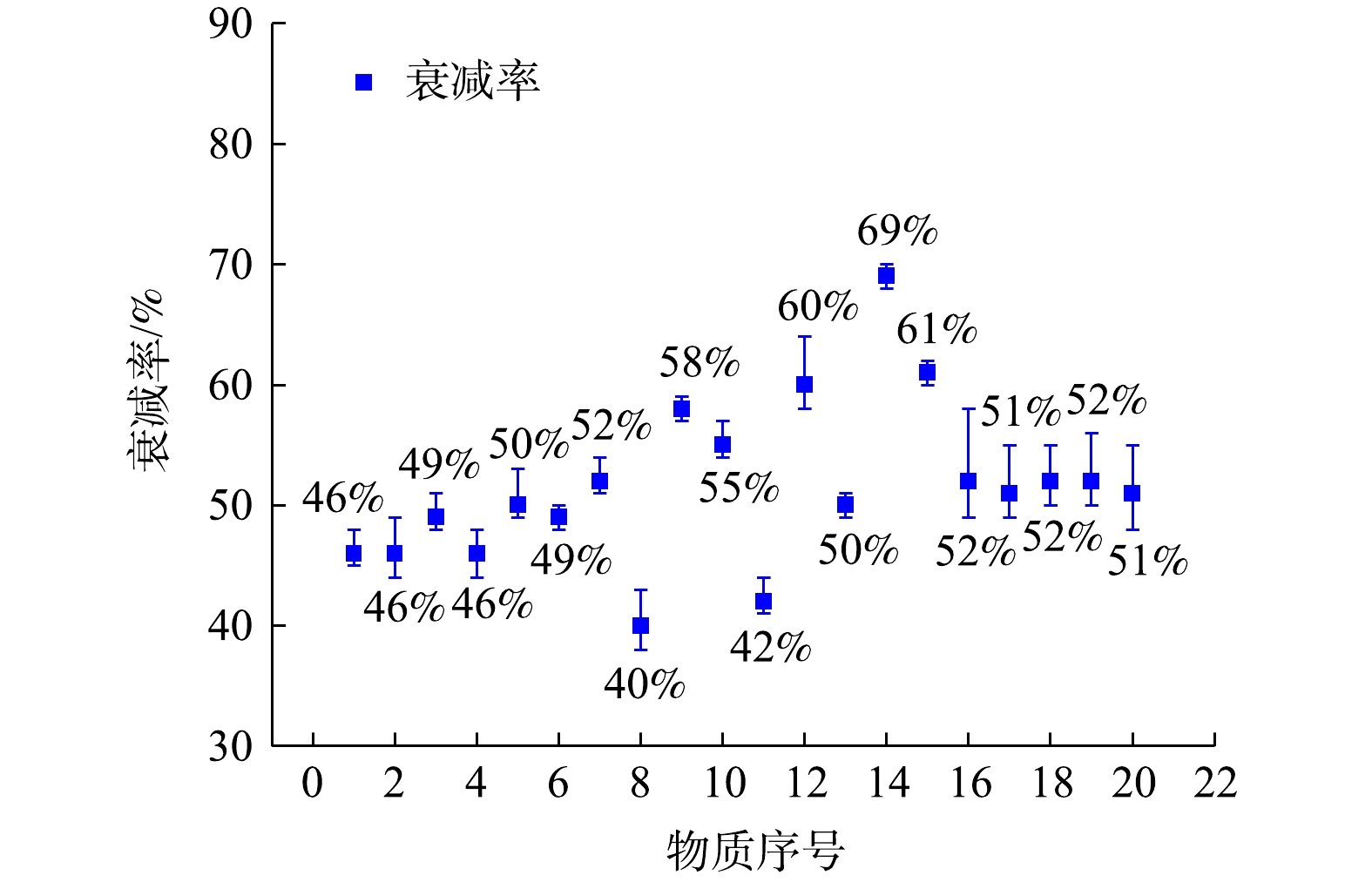
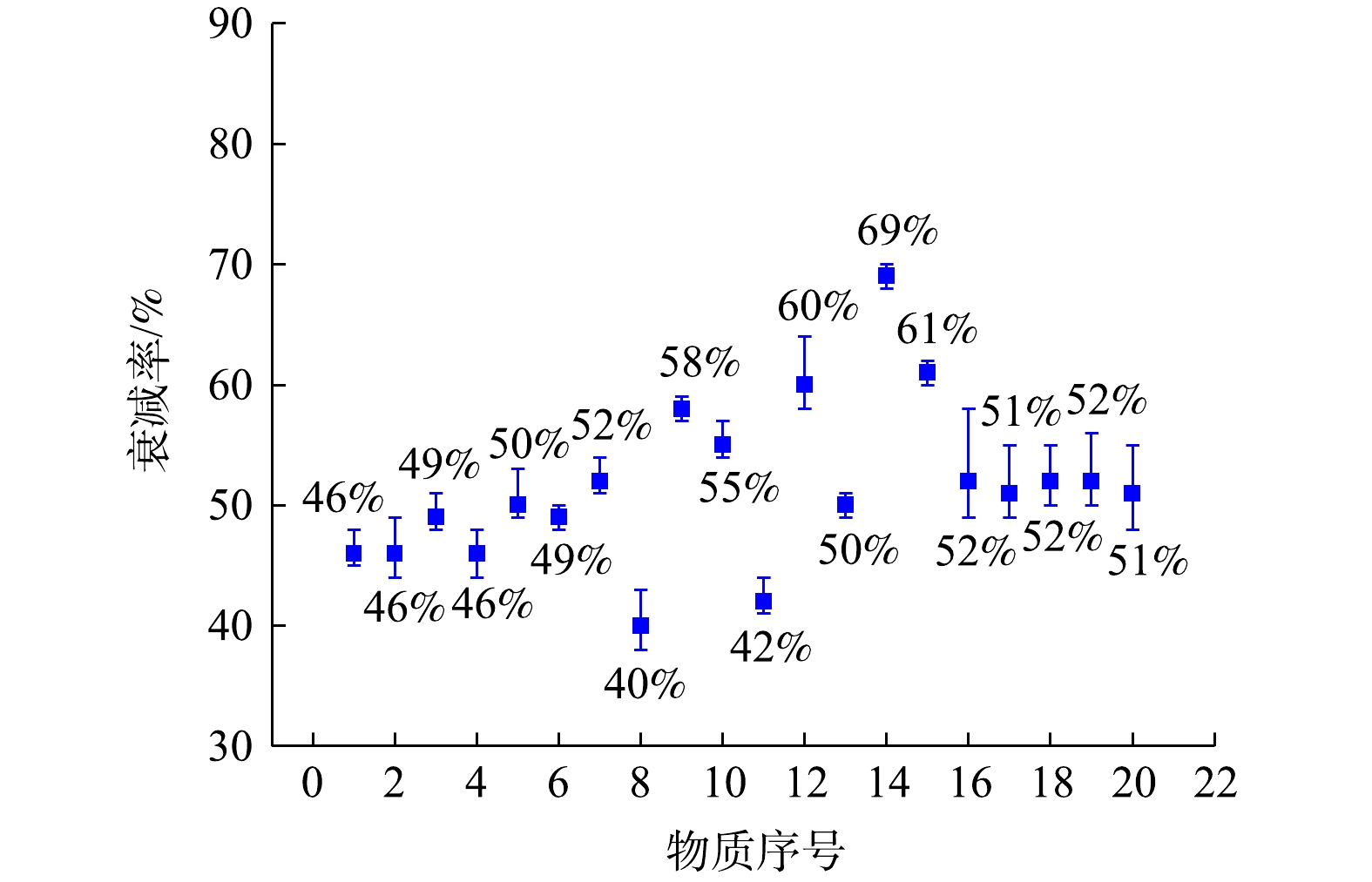
用质谱仪对PAMS(浓度0.1 μmol·mol−1)与有机硫标准气体(浓度为0.1 μmol·mol−1)进行了分析,经7次后得到无衰减的VOCs信号强度。再用监测系统对相同条件的PAMS与有机硫标准气体分别进行采样分析,7次后得到衰减后的VOCs信号强度。最后,通过式(2)计算得到20种物质各自的信号强度衰减率。PAMS与有机硫标准气体中20种物质信号强度衰减率以及波动范围的分析结果如图6所示,图中各物质序号与表1一致。由图6数据分析可知,所有物质信号强度衰减率在28%~70%,各物质信号强度衰减率的波动范围小于12%。
定量误差分析方式:监测系统监测到的信号强度(Mn)转换为SPI-MS直接检测时的信号强度(y),再使用表1中各SPI-MS线性公式计算得到各物质浓度。定量公式如(3)所示。
式中:y为SPI-MS直接检测时的信号强度;Mn为监测系统第n次检测的信号强度强度;Si为第i次VOCs残留影响;
6∑i=1Mn−i⋅Si 为VOCs残留信号强度;D为VOCs信号强度衰减率。利用该监测系统分别对0.17 μmol·mol−1 PAMS与有机硫标准气体进行了连续监测。分析了PAMS与有机硫标准气体的定量误差(见表2),结果表明,各物质定量误差为−13%~25%。
2.1. 监测系统的稳定性、线性分析、检测限
2.2. 监测系统的残留影响、信号强度衰减率的变化、定量误差分析
-
1)本研究研制的远距离多通道VOCs连续采样在线监测系统采样距离可达500 m,并可在1 h内完成10个不同点位的采样,对于范围较大的化工园区可以使用单台质谱仪在较短时间内完成多个点位的连续在线监测。
2)该系统可实现对C4~C10烃类、有机硫化物和苯系物定性定量分析;所测全部物质RSD≤7%;监测系统最优检测限为0.004 μmol·mol−1;在浓度为0.01~1 μmol·mol−1时各物质判定系数大于0.99;定量误差为−13%~25%。定量误差为−13%~25%,可以满足VOCs成分复杂多变的化工园区检测要求。
