









-
环境DNA[1](environmental DNA, eDNA)通常指从环境样品(如水样、泥样等)直接获取的DNA。环境样品中一般包含一定区域内存在的生物组织、排泄物以及分泌黏液等,通常被认为是环境DNA的主要来源[2]。从环境样品中提取DNA之后,通过聚合酶链式反应(polymerase chain reaction, PCR)扩增目标DNA片段,进一步检测分析环境DNA所携带的信息,以达到获取某一区域环境生物信息的目的[3-4]。在过去的十几年,环境DNA技术已经被逐步应用于生态环境监测(如判断某些物种的存在与否)的研究中。RONDON等[5]从土壤样品中提取环境DNA研究土壤微生物的多样性,并首次提出了环境DNA这一术语。严格来讲,环境DNA技术是于2008年开始被研究者应用于监测大型生物的[6]。从2010年开始,随着实时荧光定量PCR技术(qPCR)和DNA条形码技术的引入,环境DNA技术的应用从定性分析物种存在与否逐步扩展到定量分析物种丰度[1-2, 7-9]。
近年来,有研究[7]表明,存在于某一区域的生物可以通过环境样品(如水样、泥样等)并采用环境DNA技术检测到,这一新兴检测技术有可能显著提升生态监测效率。与传统生态监测方法相比,环境DNA技术有如下优点:该技术不需要捕获、捕杀目标生物,因此,是更为环境行为友好的监测手段;该技术通过PCR技术扩增目标DNA片段,对DNA的检出限低于1 pg,与传统方法相比,对低丰度生物有更灵敏的检出限;该技术对于物种的鉴别是基于基因序列而非传统方法的人眼判断,与传统方法相比,对于物种鉴别的准确性更强。由于采样方式与判别标准不同于传统生态监测技术,环境DNA技术有望大幅提升生态监测的可行性与准确性。然而作为新兴生态监测手段,环境DNA技术仍然存在一些缺陷,如样品处理与检测手段有待进一步优化,环境DNA在环境中的迁移变化行为尚不明确等。着力解决这些问题可促进该技术的推广与应用,这些问题也是相关研究领域的重点研究课题。而对环境DNA降解动力学及环境影响因素的研究,可提高环境DNA数据对于周边环境的指示意义,如在不同的地域环境下,可根据周边环境特征,通过获取环境DNA数据,更准确地推断周边环境生物的丰度。
作为新兴技术,目前环境DNA的研究还局限于对环境水样的分析检测。有研究报道,温度[10-11]、pH[12]等环境变量是水体环境DNA变化的影响因素。底栖生物是重要的环境质量指示生物,多数底栖生物生活在表层沉积物中,因此,表层沉积物中的环境DNA同样值得关注。目前,对于表层沉积物中的环境DNA关注较少[9],环境DNA在表层沉积物中变化的主要影响因素仍然未知,需要进一步研究探索。本研究通过小试实验引入并培养底栖生物,以底栖生物为目标生物,通过采集表层沉积物提取环境DNA的方法,探究了模拟自然环境下水体表层沉积物中环境DNA含量变化与周边环境变量的关系,揭示了表层沉积物中的环境DNA对于周边环境变化的指示意义,为环境DNA变化的动力学研究提供参考。
-
本研究以日本大螯蜚(Grandidierella japonica)作为目标生物,引入不同的生物丰度,共进行了4组小试规模的实验研究,每组实验包括3个平行样本和1个空白样本,结果见表1。实验设置与部分实验数据参考本课题组已有的研究[9]。本研究所使用的日本大螯蜚来自实验室水族箱养殖。
每个小试装置为1个250 mL烧杯(DURAN,德国),内装有70 g石英砂(HARIO WS-10BR,日本)作为沉积物和180 mL人工海水(配方见US EPA人工合成海水配方[13])。石英砂和海水在使用之前均已在灭菌锅中进行高温灭菌。实验启动时,4组小试装置中的平行样分别引入10、20、30和50只日本大螯蜚并依次命名为A组、B组、C组和D组,所有小试装置放入25 ℃恒温箱中运行并通过空气泵持续曝气。在小试装置中养殖日本大螯蜚4 d后全部取出,之后启动实验。在实验启动后的第0、6、12、18、24、72、144、264、384小时进行取样,每次取样从每个实验组烧杯中取出1.5 g表面石英砂和0.3 mL水样。石英砂用于环境DNA提取与qPCR分析,水样用于细菌数分析。在实验启动后的第0、72、144、264、384小时,各检测1次烧杯内水中的pH、DO(溶解氧)、盐度、电导率等指标。4个空白样只含有高温灭菌过后的石英砂和人造海水,在实验开始和结束后进行取样分析检测。
-
在实验过程中采集的石英砂沉积物样品采用CTAB(cetyltrimethylammonium bromide)方法提取环境DNA,具体操作流程参见文献中的方法[8-9],最终将每个环境DNA样品分别溶于200 μL TE缓冲液。采用DNeasy Blood & Tissue Kit套件(QIAGEN, 德国)提取日本大螯蜚的动物组织DNA,采用Qubit 3.0 Fluorometer仪器(Thermo Fisher Scientific, 美国)测定从动物组织提取的DNA浓度。
-
在进行qPCR标准物制备时,本研究共使用3对PCR引物,通过美国国立生物技术信息中心(NCBI)数据库Primer-BLAST工具设计[8, 14],引物序列及PCR退火温度见表2。qPCR标准物为使用引物413F和413R通过传统PCR反应扩增纯化后的产物,标准物片段长度为413 bp。纯化后的标准物浓度通过Qubit 3.0 Fluorometer仪器测定,进而通过式(1)计算得出标准物中的DNA片段拷贝数[15],之后分成小份,于−20 ℃保存备用。
式中:c为标准物中DNA拷贝数,copies·µL−1;a为纯化后的标准物浓度,g·µL−1;b为DNA片段长度,bp;NA为阿伏伽德罗常数。
在进行qPCR检测沉积物样品中环境DNA片段含量时,每个提取出的环境DNA样品与平行样均进行2次平行qPCR检测,每个96孔反应板上包括2组拷贝数为1、10、100、1 000、1 000的qPCR标准物和3个空白对照样品。每个qPCR反应体系为20 μL,包括10 μL LightCycler® 480 SYBR Green I Master (ROCHE, 瑞士),正反引物126F与126R各0.3 μmol L−1,5 μL经超纯水10倍稀释的环境DNA提取液。qPCR反应在LightCycler® 480 Instrument II仪器上进行,首先在95 ℃下变性3 min;之后进入扩增程序并循环50次:95 ℃下解旋30 s,在60 ℃下退火30 s,在72 ℃下延伸1 min;循环结束后,进入熔解曲线程序,根据熔解曲线筛选出有效的qPCR反应并计算原环境DNA样品的拷贝数。
-
水样的盐度、电导率和温度通过Mother Tool CD-4307 SD便携式手持检测仪测定,溶解氧通过Fuso DO-5509溶氧仪测定,pH通过LAQUA Twin pH计测定,水样中活细菌总数通过BD Accuri™ C6流式细胞仪检测。
-
为探究环境变量对环境DNA含量变化的影响,本研究采用广义线性模型(general linear model,GLM)进行回归分析。假设环境DNA降解规律服从指数降解模型,以qPCR检测出的表层沉积物环境DNA片段含量在t时刻的降解速率(由指数降解模型求得[9])作为因变量,以环境变量中的生物丰度、总活菌量、pH、盐度、溶解氧作为自变量,缺失数据由前后2个时间点均值代替。由于水质电导率与盐度为相关非独立关系变量,因此,回归分析中未将电导率纳入自变量中。本研究所涉及的数据分析均在R语言(版本3.6.0)[16]及其集成开发环境R Studio[17]上进行。
-
本研究在实验中设置的空白样经qPCR检测,反应均为阴性,因此,认为本研究中不同实验组之间未受到DNA污染。实验期间,由qPCR检测出的环境DNA片段变化如图1所示,服从指数降解特征,对于实验期间环境DNA指数降解动力学研究可参见已有报道[9],4组实验中环境DNA的指数降解系数分别为0.044、0.021、0.029和0.005 5 h−1。4组实验组表层沉积物中的环境DNA含量在日本大螯蜚移除之后(0 h)迅速下降,在实验进行72 h后,基本降低至5 000 copies·g−1以下,并保持在这一较低含量水平。除此以外,在生物丰度较低的实验组(A,B)表层沉积物中,环境DNA片段含量低于生物丰度较高的实验组(C,D)。
本研究中4组实验组表层沉积物中的环境DNA含量在日本大螯蜚移除之后迅速下降,在实验进行72 h后,降低至较低水平。已有研究[18-20]表明,水体中环境DNA在目标生物移除后在1~14 d之后,降低至无法检出的水平,其变化速度与本研究基本处于同一水平。然而对于深层底泥中的环境DNA,有相关研究[21-25]显示可持续2 000、4 000、6 000、12 600 a后仍被检出。本研究与以往研究的主要区别为,本研究中环境DNA载体介质为表层沉积物而非深层底泥,这说明表层沉积物中的环境DNA与水体中的环境DNA可能具有相似的变化特征。可能的原因是表层沉积物与水体直接接触,其所携带的环境DNA所处的自然环境与水环境相似,因此,二者含有的环境DNA具有类似的变化特征。
-
实验期间,各实验组水样中总活菌量变化如图2所示。各实验组水样中的活菌含量在日本大螯蜚移除之后(0 h)迅速下降,在实验进行72 h后基本降低至较低含量水平。除此以外,在生物丰度较低的实验组(A,B,C)水样中,总活菌含量低于生物丰度较高的实验组(D),这可能由于生物丰度较高的实验组内含有较多的有机质,导致活菌含量较高[9]。
实验期间,各实验组水样中pH、盐度、溶解氧、电导率变化如图3所示。各实验组水样中的pH在日本大螯蜚移除之后(0 h)缓慢上升,在实验进行264 h后pH保持在8左右;除此以外,在生物丰度较低的实验组(A)水样中,pH低于生物丰度较高的实验组(B,C,D)。各实验组水样中的盐度在日本大螯蜚移除之后(0 h)缓慢上升,在实验进行264 h后,盐度保持在2.6%~2.7%左右;除此以外,生物丰度较低的实验组(A)水样中盐度低于生物丰度较高的实验组(B,C,D)。各实验组水样中的电导率在日本大螯蜚移除之后(0 h)缓慢上升,在实验进行264 h后,电导率保持在4.5 S·m−1左右;除此以外,在生物丰度较低的实验组(A)水样中,电导率低于生物丰度较高的实验组(B,C,D)。各实验组在实验期间一直处于曝气状态,因此,水样中的溶解氧在整个实验期间保持相对稳定,未表现出明显变化趋势。
在4组实验所检测的水质参数中,除溶解氧以外均表现出较明显的变化趋势。总活菌量表现出随时间下降的趋势,可能的原因是生物从反应器中移除后,反应器内部的有机物质随时间逐步被降解,导致微生物数量逐渐降低。pH、盐度、电导率随时间推移呈现初步上升趋势,可能的原因是反应器中的部分水分随着时间的推移蒸发,导致反应器内溶解性物质浓度上升。研究发现表层沉积物中环境DNA含量与实验组生物丰度呈正相关关系。有研究[18, 26-32]报道,物种丰度与环境DNA含量呈现一定程度的线性或指数正相关关系,可决系数为0.03~0.93。尽管环境DNA与生物丰度之间精确的数量关系目前尚未明确,但目前的研究发现至少证明了环境DNA存在定量估计生物丰度的可能性。
-
本研究中表层沉积物中环境DNA片段降解速率与环境变量(包括生物丰度、总活菌量、pH、盐度、溶解氧)回归关系如表3所示。水质参数中盐度与环境DNA片段降解速率呈显著负相关关系(回归系数=−3 673,P=0.005),pH与环境DNA片段降解速率呈显著正相关关系(回归系数=830.2,P=0.04)。生物丰度、总活菌量、溶解氧等水质参数与环境DNA片段降解速率呈正相关关系但不显著,回归系数分别为3.34、0.000 006、300。
通过广义线性回归分析发现,环境DNA降解速率与水质盐度呈显著负相关关系,在盐度较高的环境下降解速率降低;与pH呈显著正相关关系,在pH较高的情况下降解速率上升。并与总活菌量、溶解氧等水质参数呈不显著正相关关系。由于实验模拟水质为常见海水水质,因此,环境变量的研究结果对于河口海湾底泥的环境DNA监测具有一定的参考价值。
-
环境DNA技术是近几年新兴的生态环境监测手段,目前的相关研究主要集中于水样与鱼类监测,而对于沉积物与底栖生物的研究相对缺乏。底栖生物经常被认为是环境质量指标生物,但其具有难以辨认捕捉的缺点,通过提取分析表层沉积物样品中的环境DNA,可以克服底栖生物难以辨认捕捉的缺点,从而提升监测效率。本研究发现表层沉积物中的环境DNA与水体中的环境DNA具有相似的变化特征,因此,同样可以用来表征某一区域较短时间内的环境质量变化。WEI等[33]对日本东京湾表层沉积物中的环境DNA进行了为期1年的持续取样研究,也发现表层底泥环境DNA的变化对于1个月以内的自然环境有较强的指示意义。
目前,有关环境DNA的研究主要来自国外报道,来自国内的研究报道还较少。我国幅员辽阔,生态环境多样,随着社会经济的发展,我国环境监测的指标要求将逐步从理化性指标扩展到生物性指标,在这一趋势下,环境DNA技术有望对提高环境监测效率和效果起到较大的推进提升作用。
-
1)通过小试实验模拟海水底泥环境发现,水体表层沉积物中的环境DNA在源生物体移除后72 h内,能降低至较低含量水平,其变化特征与水体中的环境DNA较为相似。
2)通过广义线性回归分析发现,环境DNA降解速率与水质盐度存在显著负相关关系,与pH存在显著正相关关系。这表明,表层沉积物中的环境DNA对于周边环境变化具有一定的指示意义。
致谢:感谢日本东京大学工学院都市工学系中岛典之教授、飞野智宏讲师提供的技术支持与指导!
环境DNA在监测表层沉积物中的运用及其与环境变量的关系
Application of environmental DNA in monitorining surface sediment and its relationship to environment variables
-
摘要: 环境DNA技术是近几年出现的新兴环境生态监测技术,为研究环境变量对表层沉积物中环境DNA变化的影响,通过小试实验模拟海水环境并以日本大螯蜚作为目标生物,引入4组不同的生物丰度,运用环境DNA技术研究了表层沉积物中环境DNA含量变化与周边环境变量的关系。在小试装置中养殖日本大螯蜚4 d后全部取出,之后启动实验。在实验启动后的第0、6、12、18、24、72、144、264、384小时进行取样,提取出的环境DNA片段含量通过实时荧光进行定量PCR检测。结果表明,表层沉积物中的环境DNA在源生物移除后72 h内降低至较低含量水平,与水体中的环境DNA有较为相似的变化特征。通过广义线性回归分析,发现环境DNA降解速率与水质盐度呈显著负相关(P=0.000 5),与pH呈显著正相关(P=0.04),说明表层沉积物中的环境DNA对于周边环境变化具有一定指示意义。上述结果为进一步推动环境DNA技术的应用及其对环境变量影响作用的深入研究提供参考。Abstract: Environmental DNA (eDNA) is an emerging tool for environmental and ecological monitoring in recent years. To clarify the effects of environment variables on the variation of eDNA in surface sediment, through the lab-scale experiments which could simulate marine environment and choose benthic organism Grandidierella japonica as target species with 4 groups of different bioabundance, the relationship between the variation of eDNA in surface sediment and ambient environment variables was investigated by using environmental DNA technology. After Grandidierella japonica were cultured for 4 days, and they were taken out from the experimental devices, then the following experiments start-up. The surface sediment samples and water samples were collected at 0, 6, 12, 18, 24, 72, 144, 264, 384 h from the start-up of the eperiments, the eDNA was extracted from these surface sediment samples and target eDNA copy numbers were determined by quantitative PCR with species-specific primers. The results showed that after removal of Grandidierella japonica, environmental DNA in surface sediment decreased to low level within 72 hours, which was similar to the decreasing characteristics of environmental DNA in water. The general linear modelling regression showed that the eDNA decay rate was significantly and negatively (P=0.000 5) related to the water salinity and significantly and positively (P=0.04) related to the pH value, indicating that environment DNA in surface sediment could reflect the changes of surrounding environment at a certain degree. This study provide a reference for promoting the application of eDNA and profoundly studying its effects on environment variables.
-
可挥发性有机化合物(volatile organic compounds, VOCs)是大气污染物中一大类[1-3]。浓度较高的VOCs气体会刺激人的眼睛、鼻子或咽喉等,导致干咳头晕、恶心疲劳等症状。长期生活在受VOCs污染的环境中,人体的神经系统会被损害,并诱发癌症,故VOCs的治理刻不容缓[4-6]。
传统VOCs处理技术主要有燃烧法、催化氧化法、吸收吸附法等。其中,燃烧法的操作较为简单,但因危险性较高,故对安全防护的要求较高;催化氧化法不需要额外试剂,且产生污染物较少,但同时存在催化剂稳定性和寿命等限制[7-8];吸收法可将VOCs回收再利用,但需根据待处理VOCs种类使用特定吸收剂,普适性较差;吸附法常用活性炭作为吸附剂,净化率高,但活性炭使用寿命很短,需频繁更换。
低温等离子体(non-thermal plasma, NTP)技术是一种新型VOCs处理技术,相较于传统VOCs处理技术,具有适用性广、响应快速等特点,因而受到广泛关注[9-11]。在众多产生NTP的放电形式中,介质阻挡放电(dielectric barrier discharge, DBD)因其结构简单、可通过改变放电参数调控等离子体能量密度,且能处理较大流量气体等优势而被广泛研究。王保伟等[12]通过研究放电间距对单介质阻挡放电(single dielectric barrier discharge, SDBD) 等离子体降解甲苯的影响,发现随放电间距的增大,甲苯转化率和CO2选择性呈先增后降趋势。ZHAO等[13]使用双介质阻挡放电(double dielectric barrier discharge, DDBD)等离子体降解多种芳烃、烷烃、酮和酯类VOCs,发现电离能是影响所有VOCs降解效率的重要参数,电离能越大,降解效率越低。相较于SDBD放电腔,DDBD放电腔可很好地保护放电电极不受工作气体污染。
为进一步优化NTP技术,提升VOCs转化率,并降低NTP降解VOCs过程中产生的臭氧与有机副产物产量,催化剂协同技术被越来越多应用于NTP降解VOCs的体系中[10-11, 14-17]。在众多研究中,对催化剂性能的表征大多使用单种VOCs进行。然而,实际情况下待处理的VOCs组分复杂,催化剂在多组分VOCs的处理中的表现还鲜有报道。
本研究拟使用双介质阻挡放电(DDBD)反应器产生低温等离子体,以甲苯、丙酮及乙酸乙酯的混合气体作为待降解模拟VOCs混合废气[18-19],并制备常用于协同NTP降解VOCs的Mn2O3/γ-Al2O3催化剂,以研究NTP降解复杂成分VOCs的特性,以及催化剂对NTP降解混合VOCs的影响,以期为NTP降解VOCs的实际应用提供参考。
1. 实验部分
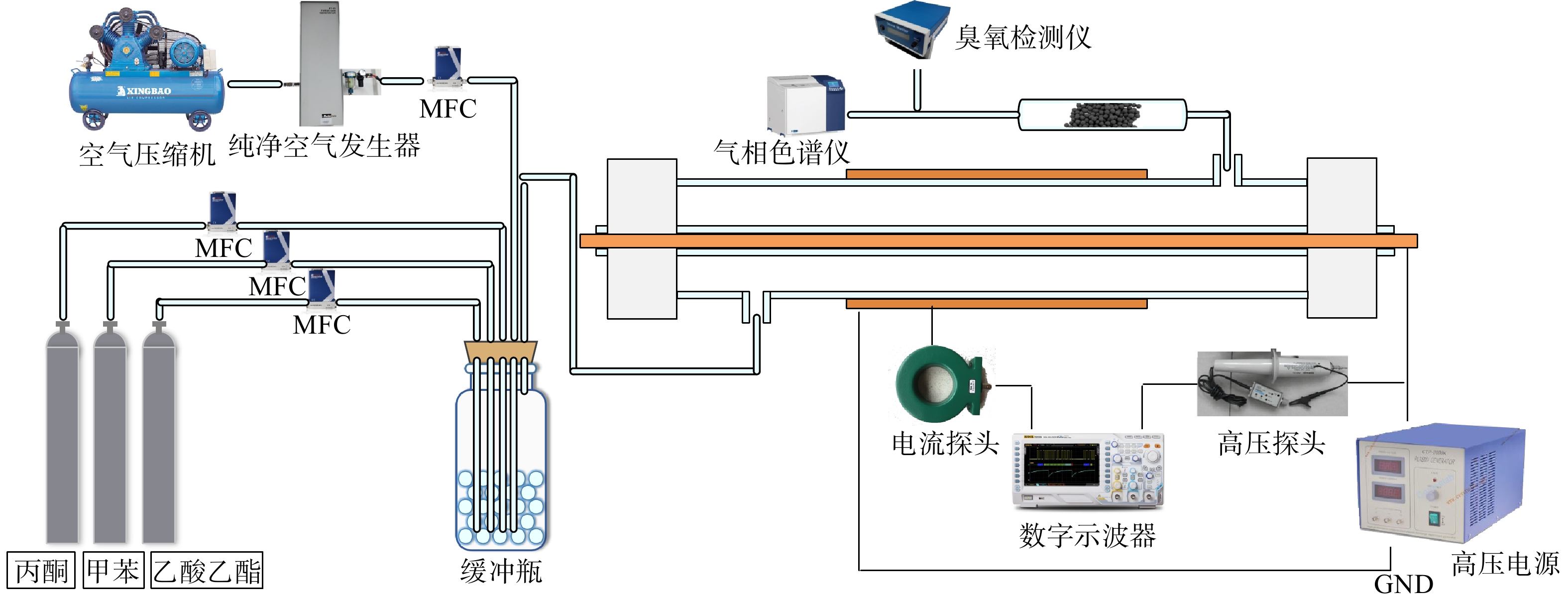
1.1 实验装置
实验装置及流程图如图1所示。模拟混合VOCs废气由高浓度丙酮、甲苯及乙酸乙酯标气经稀释得到。稀释标气所用气体为经过纯净空气发生器干燥后的压缩空气。使用4个质量流量控制器(mass flow controller, MFC)分别控制丙酮、甲苯、乙酸乙酯及压缩空气的流量,以得到实验所需的各VOCs组分的初始浓度。模拟混合VOCs经缓冲瓶混合后通入DDBD反应器降解,模拟VOCs的流速固定在1 L ∙ min−1。在VOCs单独降解实验中,甲苯、丙酮及乙酸乙酯的初始体积分数均为(33±2)×10−6。在混合VOCs降解实验中,甲苯、丙酮及乙酸乙酯的初始体积分数也均为(33±2)×10−6。
DDBD反应器的2层介质分别为1根外径为20 mm,内径为17 mm的石英管(外管),以及1根外径为8 mm、内径为6 mm的石英管(内管)。内管中放置1根直径6 mm的铜棒作为高压电极,外管缠绕宽度为10 cm的铝箔作为接地电极。模拟混合VOCs经DDBD反应器进气口进入反应器内进行低温等离子体降解。经初步降解后的废气由反应器出气口进入催化剂反应管进行进一步反应。该催化剂反应管为内径5 mm、长度30 cm的石英管。
降解前后的VOCs、CO和CO2体积分数均使用气相色谱仪(GC2060ⅢA,上海锐敏仪器有限公司)在线测定。其中,VOCs的体积分数使用配置有HT-5型毛细管柱(柱长30 m,内径0.32 mm)的火焰离子化检测器(flame ionization detector, FID)检测;CO和CO2的体积分数使用配有甲烷化转化炉的FID检测器检测。气相色谱仪的检测条件设定为:炉温60 ℃,检测器温度140 ℃,进样器温度120 ℃,甲烷转换炉320 ℃。反应过程中生成的臭氧体积分数使用臭氧检测仪(GT-2000-k3, Korno)测定。
1.2 催化剂的制备
将一定量的Mn(NO3)2(AR,国药集团化学试剂有限公司)与2 g γ-Al2O3(球形,国药集团化学试剂有限公司)分散在含有分散剂聚乙烯吡咯烷酮(质量分数2%)(AR,国药集团化学试剂有限公司)、乙醇(质量分数12%)(AR,国药集团化学试剂有限公司)和去离子水的混合溶液中。其中,Mn(NO3)2的量取决于Mn元素与γ-Al2O3的质量比。将混合物超声分散1 h后转移进容积为100 mL的聚四氟乙烯瓶中,在140 ℃条件下放置6 h[20-21]。混合物冷却至室温后,用去离子水洗涤3次,并在60 ℃下干燥,最后在马弗炉中以500 ℃煅烧产物6 h以获得催化剂。
1.3 数据的统计分析
VOCs废气的降解效果通常使用降解率与碳平衡进行表征。其中,VOCs的降解率(degradation rate, DR)由式(1)计算得到[22]。
DR=cin−coutcin×100% (1) 式中:
cin cout VOCs降解后的碳平衡(carbon balance, CB)可通过式(2)计算得到。
CB=nCO+nCO27×nT+4×nE+3×nA (2) 式中:
nCO nCO2 nT nE nA DDBD放电腔通过高压电源(CTP-2000K,南京苏曼电子有限公司)驱动放电,电源频率为10 kHz;放电腔两端的电压和电流分别通过高压探头( P6015A, Tektronix)及电流探头(5315, ETA)检测,并使用数字示波器记录(MDO3032, Tektronix)记录其放电波形。本课题组前期研究发现,调制脉冲电源可改善DDBD等离子体降解VOCs的能量效率。因此,在本实验中,电源通过一个矩形脉冲来调制一个中心频率为10 kHz 的正弦波形,并将高压电源的占空比和调制频率固定为20%与150 Hz。调制后的DDBD放电典型电流电压波形如图2所示。
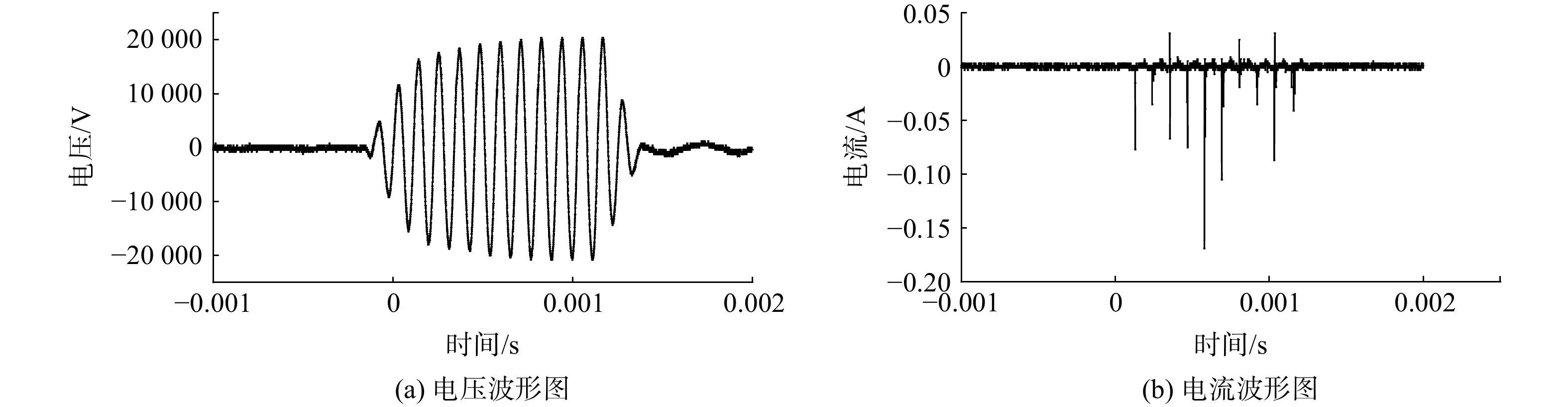 图 2 脉冲调制后DDBD放电典型电流电压波形图Figure 2. Typical current and voltage waveform of modulated DDBD discharge
图 2 脉冲调制后DDBD放电典型电流电压波形图Figure 2. Typical current and voltage waveform of modulated DDBD discharge反应器放电功率P可通过式(3)计算得到[23]。
P=f∫T0U(t)I(t)dt (3) 式中:T为脉冲电源的脉冲宽度,s;f为调制脉冲频率,Hz; U(t)为高压探头测得的放电电压,V;I(t)为电流探针测得的放电电流,A。
进而可通过式(4)计算得到低温等离子体降解VOCs过程中的特定输入能量(specific input energy,SIE)。SIE是低温等离子体降解VOCs效果评价的重要参数之一[24]。
SIE=PQ×60 (4) 式中:Q为模拟VOCs废气的流速,L ∙ min−1。
1.4 催化剂表征
催化剂的元素含量通过Prodigy ICP装置(利曼,美国)上的电感耦合等离子体(inductively coupled plasma, ICP)光电发射光谱进行测量。氮气吸附-脱附等温线在ASAP-2460分析仪上获得的。使用传统brunauer-emmett-teller(BET)和barrett-joyner-halenda(BJH)方程中的吸附数据确定催化剂的比表面积、孔径分布和孔体积;使用DD Max-2550PC型18 kW转靶X射线衍射仪(里加库,日本)记录催化剂粉末X射线衍射(X-ray diffraction, XRD)图;催化剂的X射线光电子能谱(X-ray photoelectron spectroscopy, XPS)由Thermo Escalab 250Xi型X射线光电子能谱仪(ThermoFisher,美国)在Al-K(1486.6 eV,150 W)辐射下获得;通过扫描电镜(scanning electron microscope, SEM)(JEOL 7800 F,日本)研究催化剂的形态。使用高分辨率透射电镜(high resolution transmission electron microscope, HR-TEM)(FEI Tecnai G2F30,美国)测定了催化剂的结构和元素图。
1.5 DFT计算方法
为分析催化剂的催化机理,使用密度泛函理论(density functional theory, DFT)模型计算了臭氧在Mn2O3晶体上的吸附过程。Mn2O3采用了最常见的(222)晶面,切面时,将其厚度设为1。单晶面包含43个单元,其中氧原子27个、锰原子16个。为避免表面间的原子相互作用,添加了2.4 nm的真空层。最终产生Mn2O3(222)晶面的模型,其晶格三维长度分别为a=1.330 77 nm, b=1.330 77 nm, c=2.50 nm,其3×3×1的超晶胞如图3所示。
在DFT计算过程中,采用原子PAW_PBE泛函,布里渊区k值设定为k=2×2×1。每一步运算都通过VASP 5.4.1 for Linux软件进行结构优化计算。运算采用的超算服务器,CPU为Intel Xeon Platinum单节点96核。
2. 结果与讨论
2.1 催化剂表征
实验制备MnOx/γ-Al2O3的XRD如图4(a)所示。在2θ为23°、33°、38°和55°处出现了较强的衍射峰,这4个衍射峰可较好地对应Mn2O3晶体立方结构(PDF 002-0896)的(211)、(222)、(400)和(440)晶面。其中,2θ为33°和55°是Mn2O3的主峰。这表明Mn2O3在γ-Al2O3上具有良好的分散性[25]。
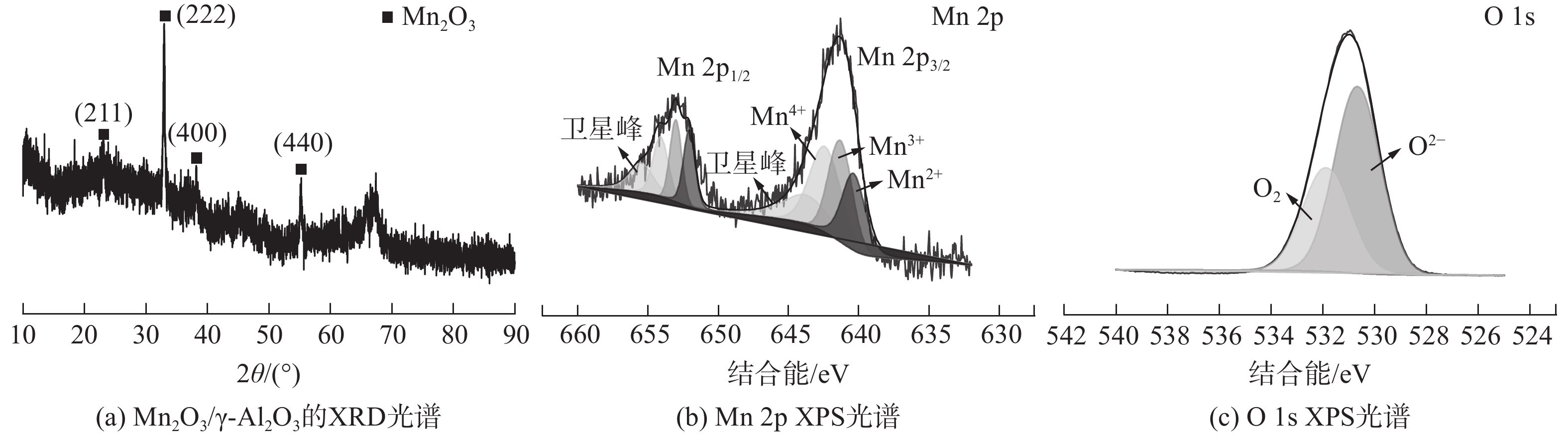 图 4 Mn2O3/γ-Al2O3催化剂的XRD光谱、Mn 2p XPS光谱和O 1s XPS光谱Figure 4. (a) XRD spectrum, Mn 2p XPS spectrum and O 1s XPS spectrum of Mn2O3/γ-Al2O3 catalyst
图 4 Mn2O3/γ-Al2O3催化剂的XRD光谱、Mn 2p XPS光谱和O 1s XPS光谱Figure 4. (a) XRD spectrum, Mn 2p XPS spectrum and O 1s XPS spectrum of Mn2O3/γ-Al2O3 catalyst图4(b)为Mn2p的XPS图谱,其中2个分别位于641.7 eV和653.4 eV的主峰与文献中的Mn2O3所对应的峰值相匹配。对XPS图谱进行高斯拟合后,位于642.5 eV、641.5 eV和640.4 eV处的3个峰值分别对应于Mn4+、Mn3+和Mn2+。在643.8 eV处的最低峰值是卫星峰值,这是由于电荷从外层电子壳层转移到能量较高的空轨道所致。O1s的XPS图谱如图4(c)所示。位于530.7 eV处的峰可归因于晶格氧(O2-)与Mn的结合,而位于531.9 eV处的峰可归因于表面吸附氧(O2)。
图5为Mn2O3/γ-Al2O3的SEM图像、TEM图像及选区电子衍射(selected area electron diffraction, SAED)图像。Mn2O3主要在γ-Al2O3表面以球形颗粒形式存在,且均匀分散在γ- Al2O3表面。Mn2O3的粒径约为10~100 nm。表面高度分散的Mn2O3晶体可促进催化过程中VOCs分子与催化剂间的接触。这可能会促进催化反应,最终促进VOCs的降解[26]。Mn2O3晶体呈立方结构与XRD结果一致。通过选区电子衍射分析获得Mn2O3的米勒指数为(211)、(222)、(400)和(440),与XRD分析中提到的一致。在图5(c)中截取的区域可观察到图3(b)中Mn2p的XPS光谱中2个主峰对应的2个晶面:(211)和(222)晶面,其晶面间距分别为0.386 nm和0.272 nm。
 图 5 Mn2O3/γ-Al2O3和Mn2O3纳米颗粒的电子显微镜图像Figure 5. SEM image, TEM image and SAED image of Mn2O3/γ-Al2O3 catalyst
图 5 Mn2O3/γ-Al2O3和Mn2O3纳米颗粒的电子显微镜图像Figure 5. SEM image, TEM image and SAED image of Mn2O3/γ-Al2O3 catalyst2.2 VOCs降解实验结果
2.2.1 VOCs单独降解与混合VOCs降解的降解率对比
各降解条件下VOCs降解率如图6所示。甲苯、丙酮和乙酸乙酯的降解率均随SIE上升而上升,这与已有研究的结果一致。这是由于3种VOCs的分子电离能和分子结构不同所决定的[13]。对比有无催化剂条件下VOC单独降解与混合气中VOCs降解的降解率(具体数值见表1),可发现混合气中甲苯的降解率相较甲苯单独降解时的降解率有明显提升。当SIE为700 J ∙ L−1时,甲苯单独降解时降解率为61%,而混合气中的甲苯降解率为84.7%,提升率为38.9%;而混合气中乙酸乙酯的降解率相较乙酸乙酯单独降解时的降解率也有所提升,同等SIE下提升率约为12.6%。不同的是,混合气中丙酮的降解率相较丙酮单独降解时的降解率发生了明显下降。在SIE为700 J ∙ L−1条件下,丙酮单独降解时降解率为50.1%,而混合气中丙酮降解率为31.1%,降低了37.9%。其原因可能是3种VOCs的分解产物之间存在协同效应。当等离子体中存在多种VOCs时,会比单种VOCs产生更多活性物种,如自由基等。这可能会更有效地促进VOCs分解,从而导致相对容易降解的甲苯和乙酸乙酯的降解率得到提升[27]。然而,如图7所示,除了丙酮本身较难降解外,其还是甲苯降解的有机副产物之一[28]。在混合气中,甲苯的降解率相比甲苯单独降解有了极大提升的同时,也导致其有机副产物中丙酮体积分数上升,最终导致混合气中丙酮降解率出现下降。
表 1 在SIE为700 J ∙ L−1时,各VOCs的降解率及其提升率Table 1. Degradation rate and improvement rate of VOCs at SIE of 700 J ∙ L−1指标 甲苯 乙酸乙酯 丙酮 单独 混合 单独 混合 单独 混合 无催化时的降解率 61% 84.7% 59.6% 67.1% 50.1% 31.1% 有催化时的降解率 72.6% 91.1% 70.2% 79.1% 58.4% 45.3% 提升率 19% 7.5% 17.9% 18% 16.7% 45.8% | Show Table DownLoad:
CSV
DownLoad:
CSV
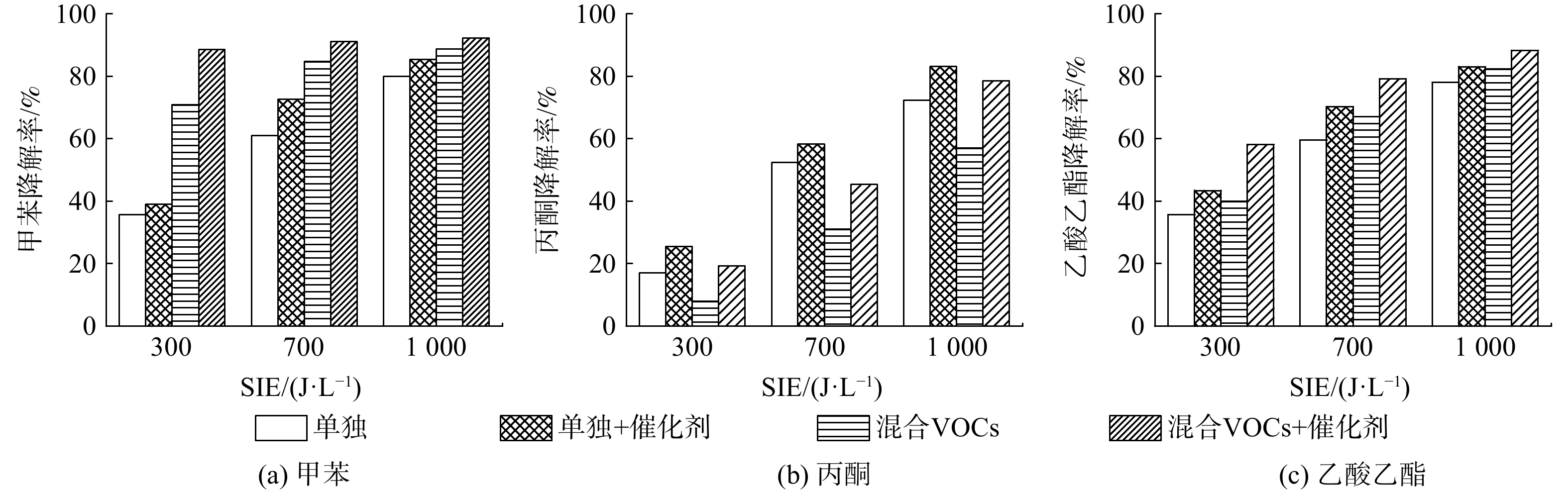 图 6 VOCs单独降解及在混合VOCs中降解时的降解率:Figure 6. Degradation rates of VOCs’ degradation alone and in mixed VOCs: (a) toluene; (b) acetone; (c) ethyl acetate
图 6 VOCs单独降解及在混合VOCs中降解时的降解率:Figure 6. Degradation rates of VOCs’ degradation alone and in mixed VOCs: (a) toluene; (b) acetone; (c) ethyl acetate随着Mn2O3/γ-Al2O3催化剂的引入,无论是单独或是混合状态,各VOCs的降解率均得以显著提升。当SIE为700 J ∙ L−1时,甲苯、乙酸乙酯及丙酮单独降解的降解率分别为61%、59.6%及50.1%;而在催化剂作用下,甲苯、乙酸乙酯以及丙酮单独降解的降解率分别提升至72.6%、70.2%及58.4%,此时催化剂对其降解率的提升量分别为19%、17.9%及16.7%。而在混合气中,同等SIE下甲苯、乙酸乙酯及丙酮的降解率分别为84.7%、67.1%及31.1%;在催化剂作用下,混合气中甲苯降解率被提升至91.1%,提升率约为7.5%;乙酸乙酯降解率被提升至79.2%,提升率约为18%;而丙酮的降解率被提升至45.3%,提升率为45.8%。此外,根据同等SIE下混合气中各VOCs的降解率可发现3种VOCs在混合气中的降解难度存在较大差距,甲苯、乙酸乙酯及丙酮的降解难度呈降序排列。这与前面单独降解的情况一致,表明混合和催化剂均不会改变VOCs的降解难易程度,从而说明电离能和分子结构是影响降解效率的重要因素。而Mn2O3/γ-Al2O3催化剂对混合气中甲苯、乙酸乙酯及丙酮降解率的提升效果随VOCs降解难度的上升而更加显著。
2.2.2 VOCs单独降解与混合VOCs降解的臭氧产量对比
甲苯、丙酮、乙酸乙酯单独降解,以及混合VOCs降解过程中的臭氧产量如图8所示。在各种条件下,VOCs降解过程中的臭氧产量均随SIE上升呈先升后降趋势。如式(5)~(6)所示,臭氧的形成可分为2部分:高能电子与氧分子发生非弹性碰撞,形成氧原子;氧原子和氧分子在第三体的参与下生成臭氧[29]。
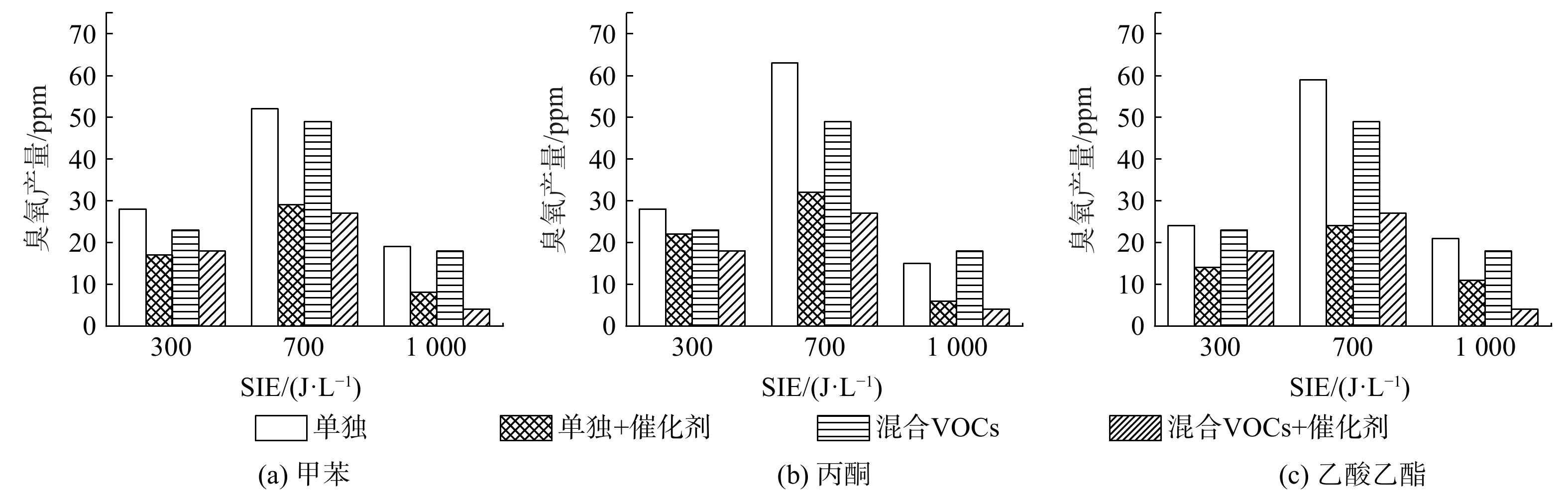 图 8 VOCs单独降解及在混合VOCs中降解时的臭氧产量Figure 8. Ozone production of VOCs’ degradation alone and in mixed VOCs: (a) toluene; (b) acetone; (c) ethyl acetate
图 8 VOCs单独降解及在混合VOCs中降解时的臭氧产量Figure 8. Ozone production of VOCs’ degradation alone and in mixed VOCs: (a) toluene; (b) acetone; (c) ethyl acetatee+O2→2O+e (5) O+O2+M→O3+M (6) 随着SIE上升,等离子体的电子密度和电子能量都随之增加,氧气分子与高能电子发生碰撞的几率随之上升,从而导致更多氧原子的产生,进而导致臭氧产量的上升。
然而,随着SIE的进一步上升,反应器腔体的温度也随之升高。STANISLAV等[30]发现反应器腔体温度的上升会导致臭氧产量的降低。随着Mn2O3/γ-Al2O3催化剂的引入,各条件下臭氧产量均出现明显降低。MnOx催化剂对臭氧生成有明显抑制作用[31],在混合VOCs中,这一抑制作用同样表现出色,并未因待降解气体成分的改变而表现异常。同时,混合VOCs中的臭氧产量相较3种VOCs单独降解时均有微弱下降。混合VOCs中VOCs总浓度的上升,将使更多氧原子参与VOCs及其中间产物的降解,从而使参加与O2发应生成臭氧的氧原子减少,即臭氧浓度比单种VOC降解时更少[4]。另外,VOCs体积分数的上升也会导致降解中间产物的增多,部分臭氧在深度氧化这些中间产物的过程中被消耗。
2.2.3 VOCs单独降解与混合VOCs降解的碳平衡对比
甲苯、丙酮、乙酸乙酯单独降解及混合VOCs降解的碳平衡情况如图9所示。随着SIE的上升,各条件下VOCs降解的碳平衡均呈上升趋势。此时,电场强度被增强,电子能量和密度也随之增强,进而提升了其与VOCs分子及VOCs分子降解中间产物碰撞的几率,从而导致碳平衡上升。此外,混合VOCs的碳平衡相较VOCs单独降解时的碳平衡均有一定程度下降。如,在SIE为700 J ∙ L−1时,甲苯单独降解的碳平衡为69.6%,丙酮单独降解的碳平衡为68.8%,乙酸乙酯单独降解的碳平衡为69.5%。而混合VOCs降解的碳平衡为67.1%,略有下降。相比VOCs单独降解,在混合VOCs中,由于VOCs体积分数的上升,部分等离子体放电产生的高能电子被用于甲苯、丙酮、乙酸乙酯分子的降解,被用于深度降解中间产物的高能电子数量则相应减少。最终导致混合VOCs降解的碳平衡较VOCs单独降解时有所降低。
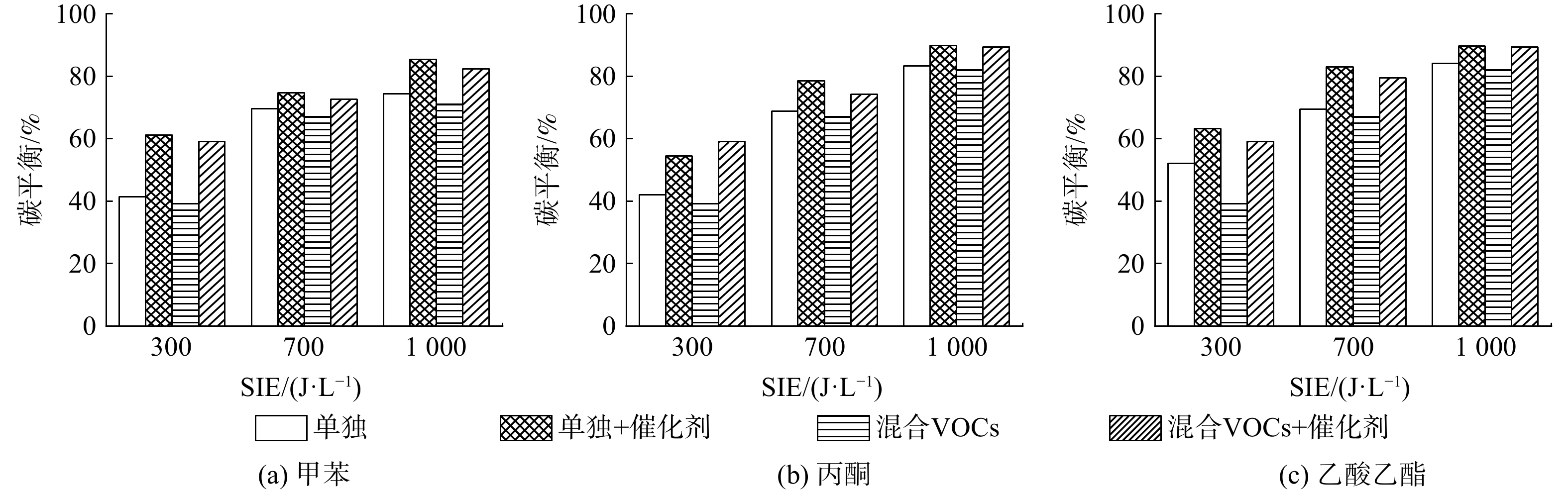 图 9 VOCs单独降解及在混合VOCs中降解时的碳平衡Figure 9. Carbon balance of VOCs’ degradation alone and in mixed VOCs: (a) toluene; (b) acetone; (c)ethyl acetate
图 9 VOCs单独降解及在混合VOCs中降解时的碳平衡Figure 9. Carbon balance of VOCs’ degradation alone and in mixed VOCs: (a) toluene; (b) acetone; (c)ethyl acetate随着Mn2O3/γ-Al2O3催化剂的引入,各条件下VOCs降解的碳平衡均得以提升。臭氧在催化剂表面可分解为氧分子和具有强氧化性的氧原子。氧原子除了可降解等离子体阶段中未降解的一部分VOCs外,还可将等离子体降解VOCs的中间产物深度氧化为COx和H2O。最终导致催化剂引入后的碳平衡得以提升。
2.2.4 臭氧在Mn2O3晶体上的吸附过程
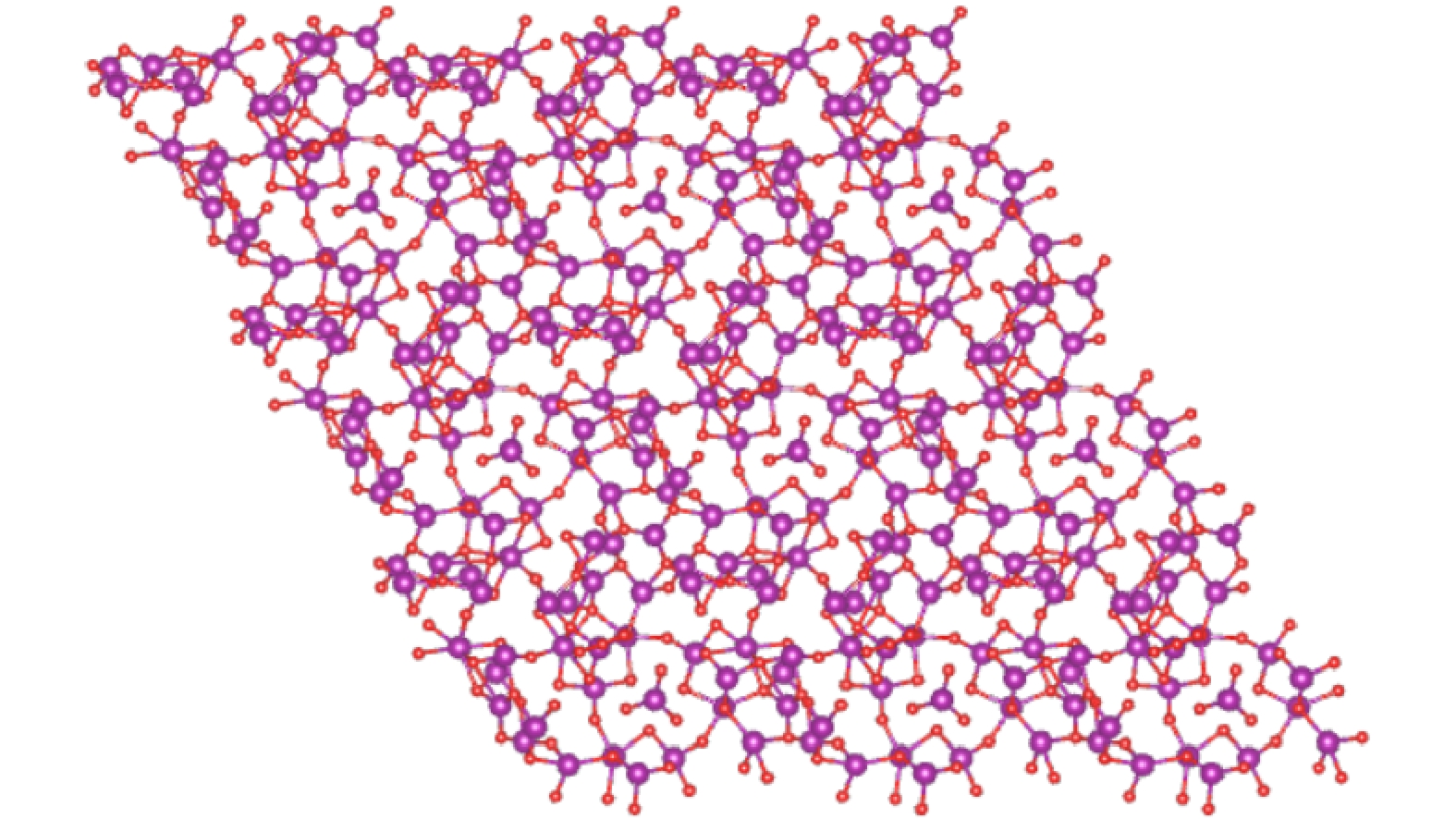
计算结果表明,O3中的2个O—O键长分别为0.128 9 nm、0.128 8 nm,键角为:118.1°。与实验结果得到的0.127 8 nm、0.127 8 nm、116.8°相比,误差分别为0.84%、0.80%、1.13%。误差极小表明选取的计算参数进行结构优化后,得到的参数处于可接受范围。对Mn2O3催化剂吸附O3的情形进行DFT计算,从而对O—Mn原子连接的方式进行研究。首先将优化后的O3分子模型和Mn2O3(222)晶面模型进行合并,并将O3分子置于晶面中一个Mn原子的正上方(图10)。
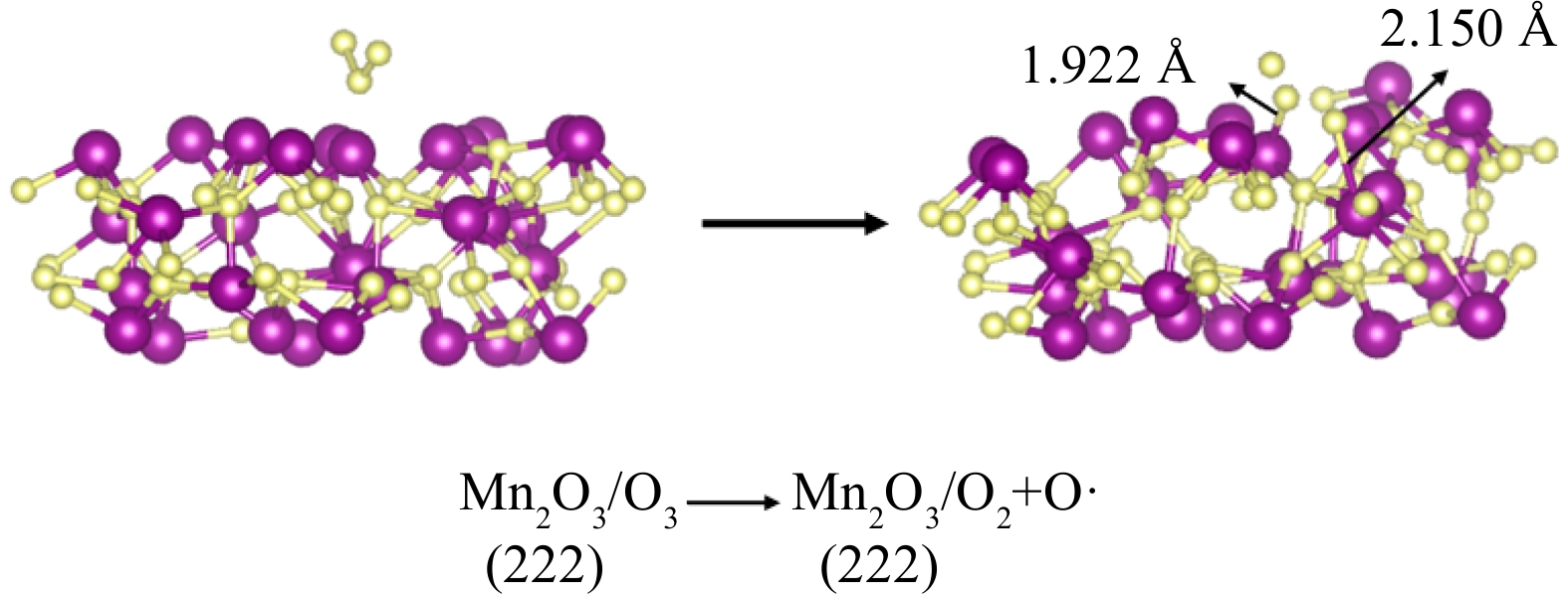 图 10 优化后的O3分子模型和Mn2O3(222)晶面模型Figure 10. Optimized O3 molecular model and Mn2O3 (222) crystal plane model
图 10 优化后的O3分子模型和Mn2O3(222)晶面模型Figure 10. Optimized O3 molecular model and Mn2O3 (222) crystal plane model通过计算得到其吸附能为−16.64 eV。O3中的2个O原子分别吸附在了2个Mn原子上,其中O—Mn键的长度为0.192 2、0.215 0 nm。同时,3个O原子间的距离增加,分别为0.201 0 nm、0.194 4 nm。另一个O原子形成孤立离子形态,使其氧化性大大增强。在吸附的2个Mn原子附近Mn—O键分别从(0.185 9 nm、0.186 9 nm、0.187 0 nm)、(0.186 6 nm、0.193 0 nm、0.189 2 nm、0.211 0 nm)变为了(0.187 1 nm、0.196 9 nm、0.204 8 nm)、(0.194 5 nm、0.222 7 nm、0.202 8 nm、0.258 6 nm),其中1个O原子的距离为0.258 6 nm,可视为强氧化性的孤立氧原子。这也表明O3的吸附对Mn2O3晶面的表面结构产生了影响,吸附属于化学吸附。
另外,本研究还计算了O3分子的马利肯电荷(Maliken charge),其吸附前后的电荷如表2所示。在O3吸附于Mn2O3晶面的过程中,3个原子分别获得1.03 e、1.14 e、1.09 e的电子。因此,为使电负性平衡,O原子将被作为电子供体,其氧化性大大增强。O3吸附于Mn2O3催化剂表面,对催化剂活性有很大影响。在反应过程中,O3吸附于Mn2O3的222切面中,通常是O原子与Mn原子进行相互连接,吸附于Mn原子表面,从而改变了原切面的结构,从而增强其催化作用。
表 2 O3分子的马利肯电荷Table 2. Maliken charge of O3 molecule原子种类 吸附前/e 吸附后/e O1 4.94 5.97 O2 4.94 6.09 O3 4.94 6.03 | Show TableDownLoad:
CSV
3. 结论
1) 对比单种VOCs与混合VOCs降解的降解率,可发现混合VOCs中甲苯的降解率相较单纯甲苯降解时的降解率有明显提升。乙酸乙酯的降解率相较单纯乙酸乙酯降解时的降解率略有提升。而丙酮的降解率相较单纯丙酮降解时的降解率却发生了明显下降。
2) VOCs降解过程中的臭氧产量均随SIE上升呈先升后降的趋势。同时,混合VOCs中的臭氧产量相较3种VOCs单独降解时均有微弱下降。Mn2O3/γ-Al2O3催化剂对于臭氧的生成有明显抑制作用。
3) 混合VOCs降解相较单种VOCs降解时的碳平衡均有一定程度下降。Mn2O3/γ-Al2O3催化剂的引入使得各条件下VOCs降解的碳平衡均得以提升。
4) Mn2O3/γ-Al2O3催化剂在协同低温等离子体降解多组分VOCs气体过程中,对混合VOCs中甲苯、乙酸乙酯及丙酮降解率的提升效果随VOCs种类降解难度的上升而更显著。
5) 通过DFT计算了O3在Mn2O3催化剂表面的吸附。O3吸附于Mn2O3的222切面中,通常是O原子与Mn原子进行相互连接,吸附于Mn原子表面,从而改变了原切面结构,增强了其催化作用。
-

图 1 实验期间各实验组表层沉积物中环境DNA片段浓度的变化
Figure 1. Variation of eDNA content in surface sediment during experimental period
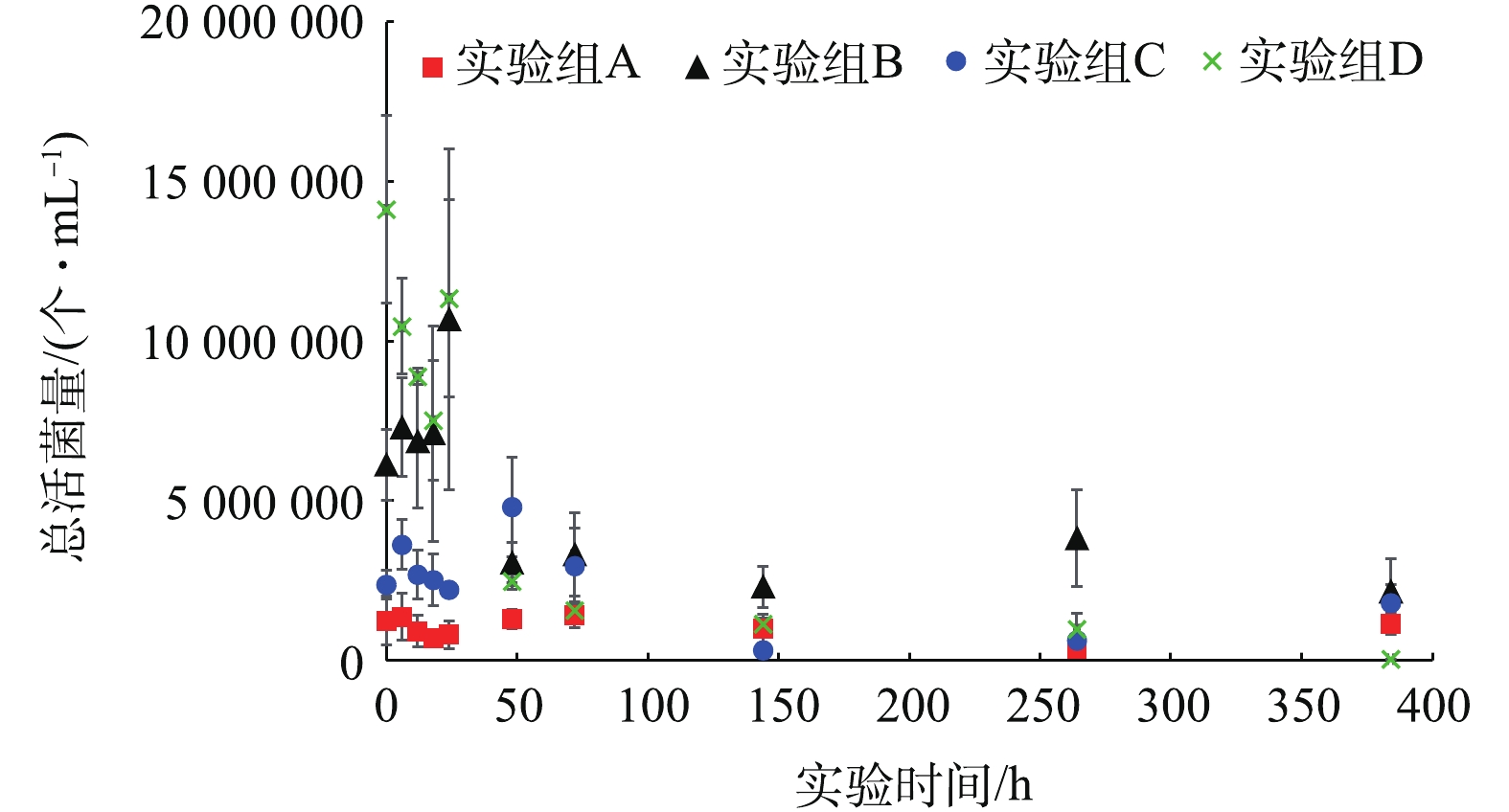
图 2 实验期间各实验组水样中活菌数的变化
Figure 2. Variation of viable bacteria count in water sample during experimental period
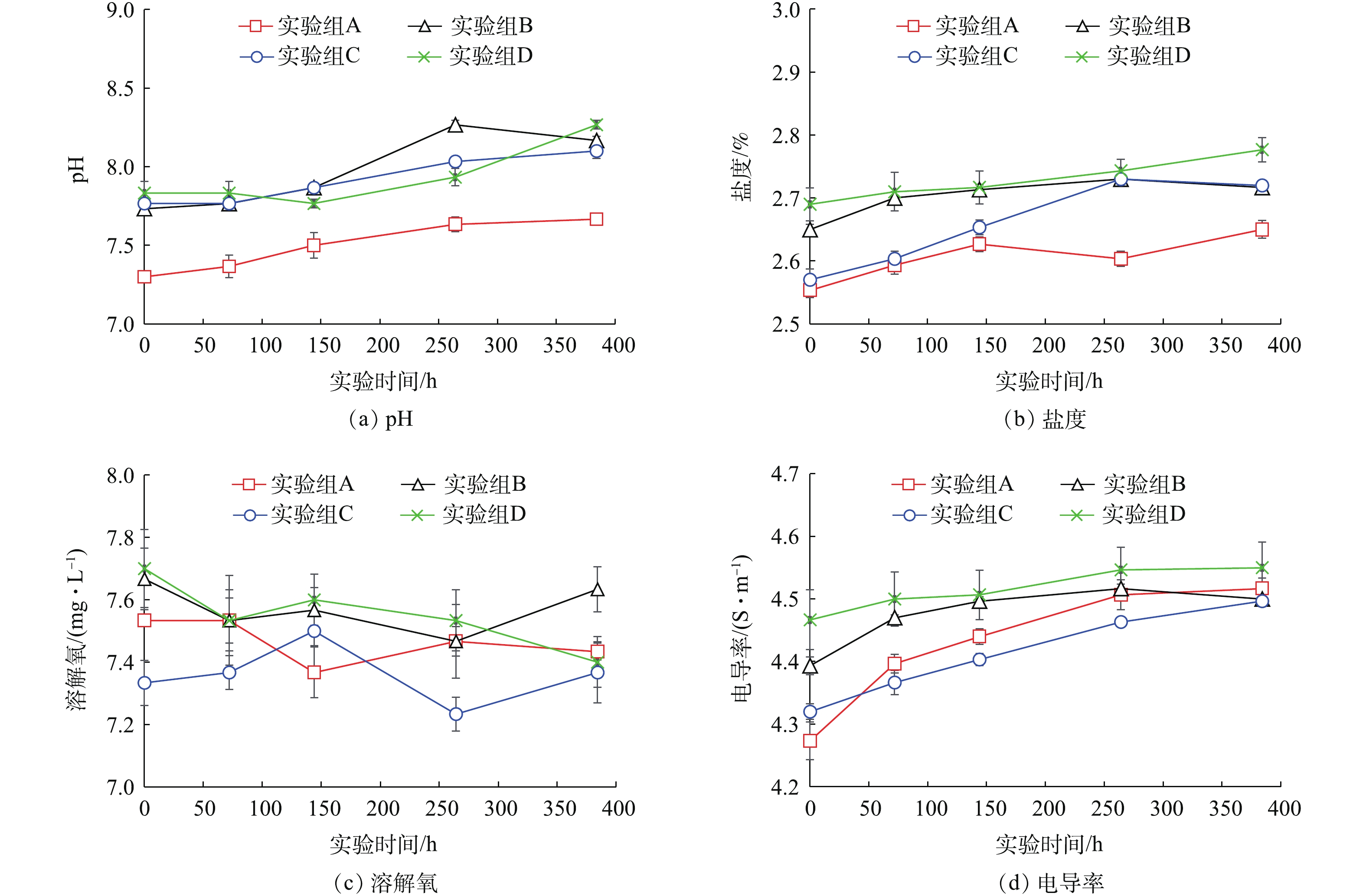
图 3 实验期间各实验组水样中pH、盐度、溶解氧、电导率变化
Figure 3. Variation of pH, salinity, dissolved oxygen, and conductivity in water sample during experimental period
表 1 小试实验条件设置
Table 1. Setup of lab-scale experiment
实验组 被测生物数量/只 石英砂/g 人造海水/mL 平行样数量/个 A 10 70 180 3 B 20 70 180 3 C 30 70 180 3 D 50 70 180 3 空白样 0 70 180 4
下载: 导出CSV
表 2 引物序列及PCR退火温度
Table 2. Nucleotide sequence of primers and PCR annealing temperature
引物名称 核苷酸序列 (5′~3′) 产物长度/bp 扩增目标区 退火温度/℃ 126F GTTTTAGGTGCTTGGGCCAG 126 线粒体COI基因 60 126R AGCATGCGCTGTTACTGAGA 413F CTTCGTTTTAGGTGCTTGGGC 413 线粒体COI基因 55 413R AGGAGGCCCCTGCTAAATGA
下载: 导出CSV
表 3 环境DNA片段降解速率与环境变量广义线性回归结果
Table 3. Results of general linear regression between eDNA decay rate and environmental variables
自变量 回归系数 标准差 t值 P值 生物丰度 3.34 4.02 1.830 0.41 总活菌量 0.000 006 0.000 017 0.366 0.72 pH 830.2 387.2 2.144 0.04 盐度 −6 022 1 549 −3.888 0.000 5 溶解氧 300 601 0.500 0.62
下载: 导出CSV
-
[1] TABERLET P, COISSAC E, HAJIBABAEI M, et al. Environmental DNA[J]. Molecular Ecology, 2012, 21(8): 1789-1793. doi: 10.1111/j.1365-294X.2012.05542.x [2] THOMSEN P F, WILLERSLEV E. Environmental DNA: An emerging tool in conservation for monitoring past and present biodiversity[J]. Biological Conservation, 2015, 18: 34-18. [3] 于水强, 王文娟. 环境DNA技术在地下生态学中的应用[J]. 生态学报, 2015, 35(15): 4968-4976. [4] 马鸿娟, 马利民, 任文伟, 等. 环境DNA及其在水生生态系统保护中的应用[J]. 生态学杂志, 2016, 35(2): 516-523. [5] RONDON M R, AUGUST P R, BETTERMANN A D, et al. Cloning the soil metagenome: A strategy for accessing the genetic and functional diversity of uncultured microorganisms[J]. Applied and Environmental Microbiology, 2000, 66(6): 2541-2547. doi: 10.1128/AEM.66.6.2541-2547.2000 [6] FICETOLA G F, MIAUD C, POMPANON F, et al. Species detection using environmental DNA from water samples[J]. Biology Letters, 2008, 4(4): 423-425. doi: 10.1098/rsbl.2008.0118 [7] KELLY R P, PORT J A, YAMAHARA K M, et al. Environmental monitoring: Harnessing DNA to improve environmental management[J]. Science, 2014, 344(6191): 1455-1456. doi: 10.1126/science.1251156 [8] WEI N, NAKAJIMA F, TOBINO T. Effects of treated sample weight and DNA marker length on sediment eDNA based detection of a benthic invertebrate[J]. Ecological Indicators, 2018, 93: 267-273. doi: 10.1016/j.ecolind.2018.04.063 [9] WEI N, NAKAJIMA F, TOBINO T. A microcosm study of surface sediment environmental DNA: Decay observation, abundance estimation, and fragment length comparison[J]. Environmental Science & Technology, 2018, 52(21): 12428-12435. [10] EICHMILLER J J, BEST S E, SORENSEN P W. Effects of temperature and trophic state on degradation of environmental DNA in lake water[J]. Environmental Science & Technology, 2016, 50(4): 1859-1867. [11] TSUJI S, USHIO M, SAKURAI S, et al. Water temperature-dependent degradation of environmental DNA and its relation to bacterial abundance[J]. Plos One, 2017, 12(4): e0176608. doi: 10.1371/journal.pone.0176608 [12] STRICKLER K M, FREMIER A K, GOLDBERG C S. Quantifying effects of UV-B, temperature, and pH on eDNA degradation in aquatic microcosms[J]. Biological Conservation, 2015, 183: 85-92. doi: 10.1016/j.biocon.2014.11.038 [13] US EPA. Methods for assessing the toxicity of sediment-associated contaminants with estuarine and marine amphipods[R]. United States, 1994. [14] YE J, COULOURIS G, ZARETSKAYA I, et al. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction[J]. BMC Bioinformatics, 2012, 13(1): 134. doi: 10.1186/1471-2105-13-134 [15] ZHU F, MASSANA R, NOT F, et al. Mapping of picoeucaryotes in marine ecosystems with quantitative PCR of the 18S rRNA gene[J]. FEMS Microbiology Ecology, 2005, 52(1): 79-92. doi: 10.1016/j.femsec.2004.10.006 [16] TEAM R. R: A language and environment for statistical computing[EB/OL]. [2019-10-01]. R Foundation for Statistical Computing, Vienna, Austria, 2017. https://www.r-project.org. [17] TEAM R. RStudio: integrated development for R[EB/OL]. [2019-10-01]. RStudio, Inc., Boston, MA, 2015. https://rstudio.com. [18] THOMSEN P F, KIELGAST J, IVERSEN L L, et al. Detection of a diverse marine fish fauna using environmental DNA from seawater samples[J]. Plos One, 2012, 7(8): e41732. doi: 10.1371/journal.pone.0041732 [19] BARNES M A, TURNER C R, JERDE C L, et al. Environmental conditions influence eDNA persistence in aquatic systems[J]. Environmental Science & Technology, 2014, 48(3): 1819-1827. [20] PIAGGIO A J, ENGEMAN R M, HOPKEN M W, et al. Detecting an elusive invasive species: A diagnostic PCR to detect Burmese python in Florida waters and an assessment of persistence of environmental DNA[J]. Molecular Ecology Resources, 2014, 14(2): 374-380. doi: 10.1111/1755-0998.12180 [21] WILLERSLEV E, HANSEN A J, CHRISTENSEN B, et al. Diversity of Holocene life forms in fossil glacier ice[J]. Proceedings of the National Academy of Sciences of the United States of America, 1999, 96(14): 8017-8021. doi: 10.1073/pnas.96.14.8017 [22] SEERSHOLM F V, PEDERSEN M W, SØE M J, et al. DNA evidence of bowhead whale exploitation by Greenlandic Paleo-Inuit 4000 years ago[J]. Nature Communications, 2016, 7: 13389. doi: 10.1038/ncomms13389 [23] GIGUET-COVEX C, PANSU J, ARNAUD F, et al. Long livestock farming history and human landscape shaping revealed by lake sediment DNA[J]. Nature Communications, 2014, 5: 3211. doi: 10.1038/ncomms4211 [24] PEDERSEN M W, OVERBALLE-PETERSEN S, ERMINI L, et al. Ancient and modern environmental DNA[J]. Biological Sciences, 2015, 370(1660): 20130383. doi: 10.1098/rstb.2013.0383 [25] PEDERSEN M W, RUTER A, SCHWEGER C, et al. Postglacial viability and colonization in North America’s ice-free corridor[J]. Nature, 2016, 537(7618): 45. doi: 10.1038/nature19085 [26] TAKAHARA T, MINAMOTO T, YAMANAKA H, et al. Estimation of fish biomass using environmental DNA[J]. Plos One, 2012, 7(4): e35868. doi: 10.1371/journal.pone.0035868 [27] PILLIOD D S, GOLDBERG C S, ARKLE R S, et al. Estimating occupancy and abundance of stream amphibians using environmental DNA from filtered water samples[J]. Canadian Journal of Fisheries and Aquatic Sciences, 2013, 70(8): 1123-1130. doi: 10.1139/cjfas-2013-0047 [28] DOI H, UCHII K, TAKAHARA T, et al. Use of droplet digital PCR for estimation of fish abundance and biomass in environmental DNA surveys[J]. Plos One, 2015, 10(3): e0122763. doi: 10.1371/journal.pone.0122763 [29] KLYMUS K E, RICHTER C A, CHAPMAN D C, et al. Quantification of eDNA shedding rates from invasive bighead carp Hypophthalmichthys nobilis and silver carp Hypophthalmichthys molitrix[J]. Biological Conservation, 2015, 183: 77-84. doi: 10.1016/j.biocon.2014.11.020 [30] LACOURSIÈRE-ROUSSEL A, ROSABAL M, BERNATCHEZ L. Estimating fish abundance and biomass from eDNA concentrations: Variability among capture methods and environmental conditions[J]. Molecular Ecology Resources, 2016, 16(6): 1401-1414. doi: 10.1111/1755-0998.12522 [31] TILLOTSON M D, KELLY R P, DUDA J J, et al. Concentrations of environmental DNA (eDNA) reflect spawning salmon abundance at fine spatial and temporal scales[J]. Biological Conservation, 2018, 220: 1-11. doi: 10.1016/j.biocon.2018.01.030 [32] JO T, MURAKAMI H, MASUDA R, et al. Rapid degradation of longer DNA fragments enables the improved estimation of distribution and biomass using environmental DNA[J]. Molecular Ecology Resources, 2017, 17(6): 25-33. [33] WEI N, NAKAJIMA F, TOBINO T. Variation of environmental DNA in sediment at different temporal scales in nearshore area of Tokyo Bay[J]. Journal of Water and Environment Technology, 2019, 17(3): 153-162. doi: 10.2965/jwet.18-047 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5534
- HTML全文浏览数: 5534
- PDF下载数: 83
- 施引文献: 0
