


 下载:
下载:



 百度学术
百度学术


-
半干法脱硫工艺具有占地面积小、投资少、无废水产生、脱硫产物为干态、便于处理运输等优点[1-2],越来越多的企业选择该类脱硫工艺,进而产生大量的脱硫灰。预计到2020年,中国干法-半干法脱硫灰每年产生量稳定在2×107 t左右。目前,对此类脱硫灰国内外还没有成熟的工业化处理技术,从而导致大量的脱硫灰没有得到合理的处理利用[3-4],大量露天堆放的脱硫灰会产生一定的环境风险。大量无法利用的脱硫灰不能露天堆放,很多企业会选择填埋处置[5-7],而填埋处置又会造成大量资金和资源的浪费。因此,脱硫灰已经成为限制干法-半干法脱硫发展的一项难题。
脱硫灰的粒径主要在0.004~0.200 mm,钢厂脱硫灰的粒径较小一些,中位粒径(D50)为5~7 μm;而电厂的脱硫灰则粒径较大些,中位粒径为170~200 μm[8-10]。脱硫灰粒度相对较小,比表面积相对较大,具有较高的活性并适合用于生产较高品质的建材。脱硫灰的主要组分有CaSO3,CaCO3,CaSO4,Ca(OH)2,含量其次相对较多的是Al2O3和SiO2,此外,还含有少量的MgO、Fe2O3、Na2O等[11-14],其组成比较复杂。脱硫灰的热重分析结果[15-17]表明:脱硫灰中亚硫酸钙发生高效氧化的温度为400~560 ℃,在该区间内存在明显的放热现象。
脱硫灰无法大量应用的主要原因是其中含有大量的亚硫酸钙,亚硫酸钙在潮湿的环境下可以与氧气发生缓慢的氧化反应,产生二水硫酸钙,从而造成体积膨胀,使建材的强度和稳定性下降,进而严重影响其在建筑材料中的应用[18]。亚硫酸钙在水泥中水化较慢,不会很快凝结硬化,会造成水泥和建材的缓凝[19]。当温度高于800 ℃或脱硫灰处于强酸环境时,亚硫酸钙则会分解释放出二氧化硫,造成二次污染。要想实现脱硫灰的资源化利用,就必须将脱硫灰中的亚硫酸钙氧化成硫酸钙,变成脱硫石膏,用脱硫石膏代替天然石膏应用于建材中,最终实现脱硫灰的资源化利用。通过加热使脱硫灰中亚硫酸钙氧化为硫酸钙是最直接、最有可能快速进入生产实践阶段的处理方法。本研究以应用最为广泛的循环流化床工艺的脱硫灰为实验材料,对主要的热氧化影响因素进行了探究,为脱硫灰热氧化提供了准确的最优工艺参数,为脱硫灰氧化的工业化生产提供参考。
全文HTML
-
本实验脱硫灰取自于中国河北省某钢厂的烧结烟气循环流化床脱硫灰,为干燥的细小粉末,其颜色为淡黄色。实验试剂:碘、碘化钾、碘酸钾、硫代硫酸钠、无水碳酸钠、醋酸钠、磷酸、醋酸、10 g·L−1淀粉溶液等,除碘酸钾为优级纯外,其他均为分析纯。
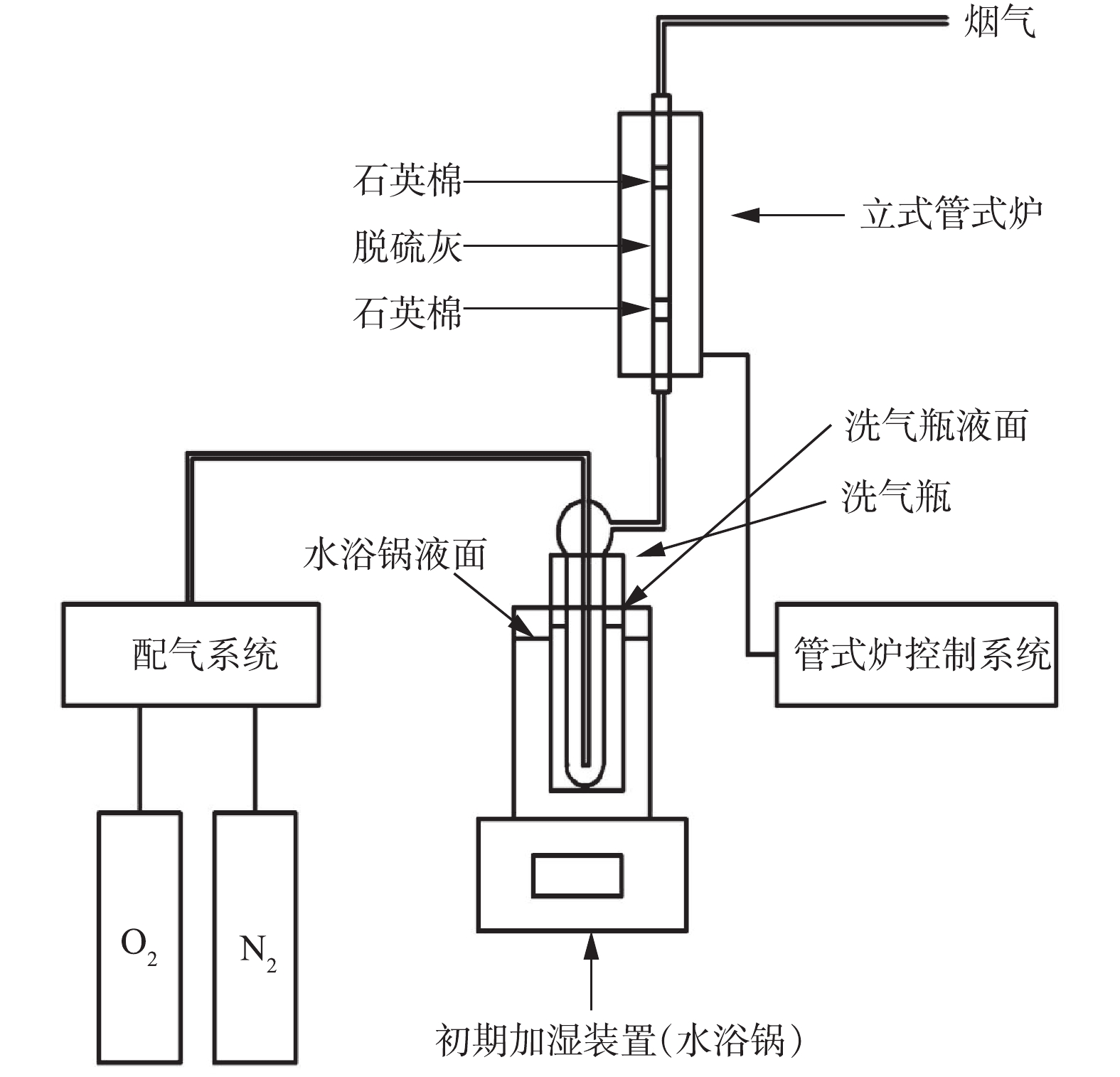
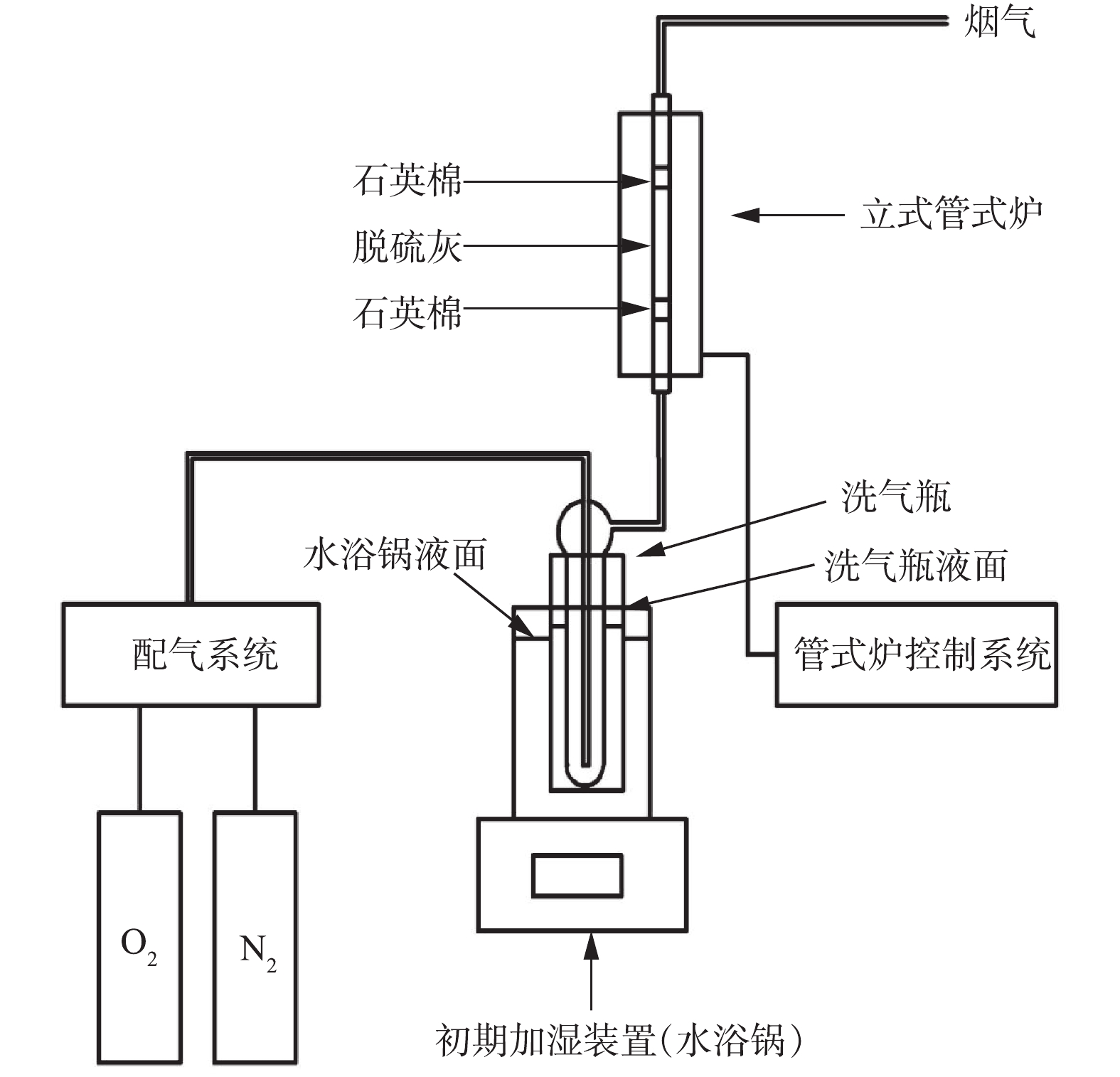
利用X射线荧光光谱仪(XRF)、X射线衍射仪(XRD)、激光粒度分布仪、热重-质谱分析(TG-MAS)、扫描电子显微镜(SEM)对脱硫灰样品进行了多种分析,多角度了解脱硫灰的理化特性。实验装置如图1所示。
-
实验材料化学分析方法采用的是GB/T 5484-2012中的碘量法和在詹鹏等[20]的脱硫灰分析方法基础上改进的方法,实验证明改进后的碘量法稳定可靠,误差范围更小。
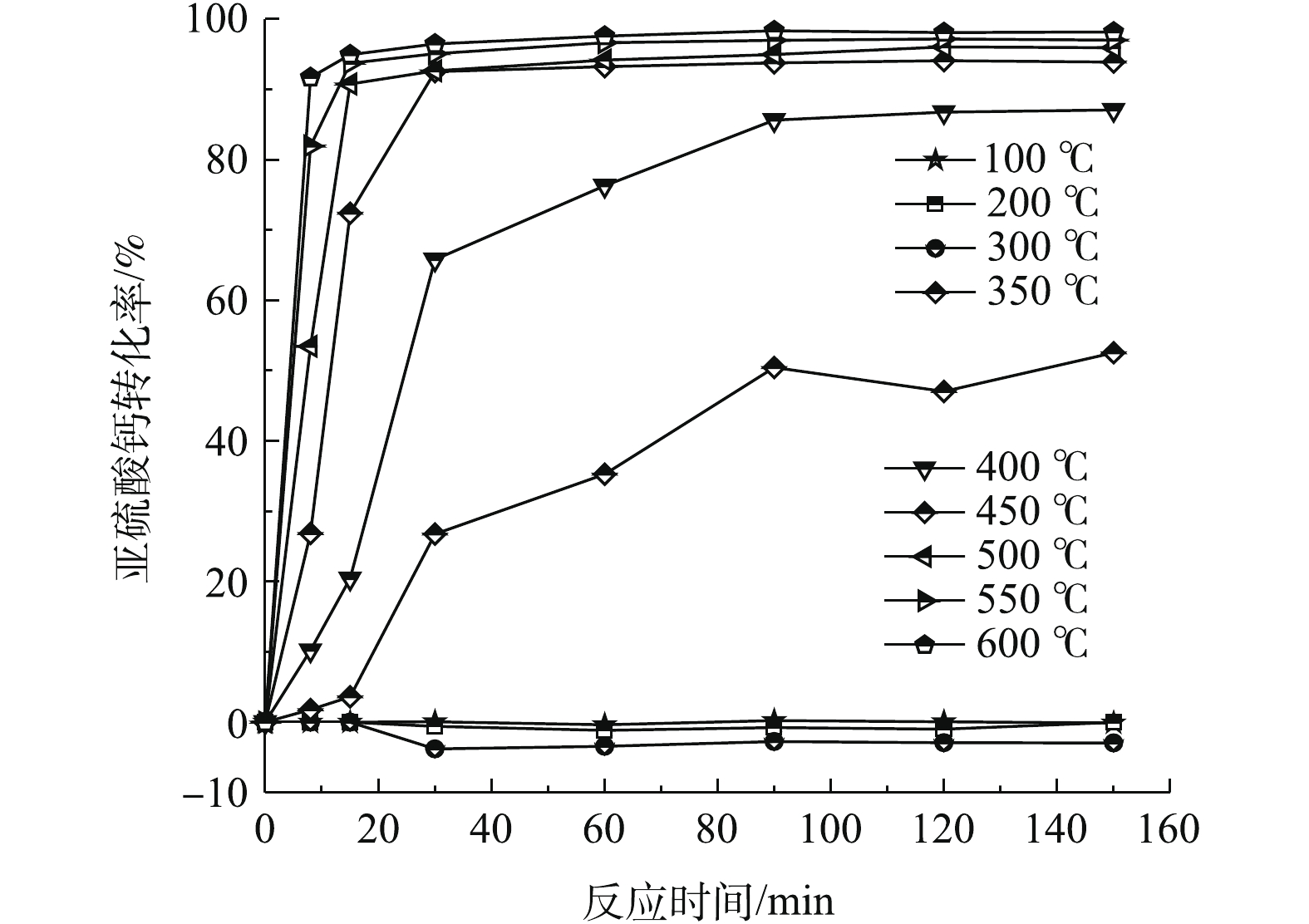
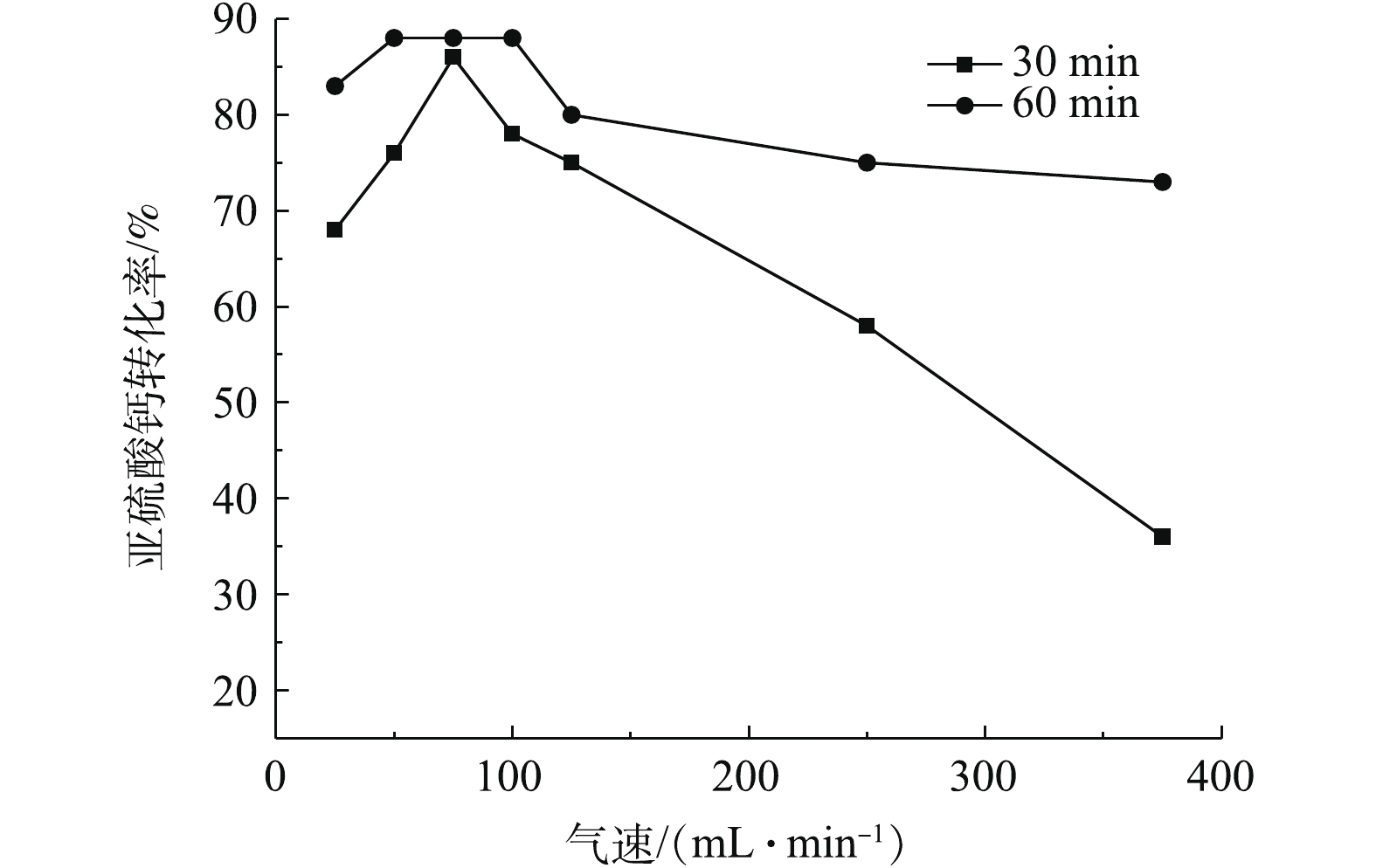
实验考察了不同氧化状态对脱硫灰氧化的影响。①静固态:实验主要考察了温度对亚硫酸钙氧化的影响,并为后期优化实验提供数据支撑。分别设定反应温度为100、200、300、350、400、450、500、550和600 ℃,每批次共5组,每隔30 min取出1组。②膨化态:实验主要考察了不同膨化程度(即反应气体流速)对脱硫灰氧化的影响。设定氮氧比为4∶1,气速分别为25、50、75、100、125、250、375、500 mL·min−1,反应温度为350 ℃,反应时间分别为30 min和60 min。
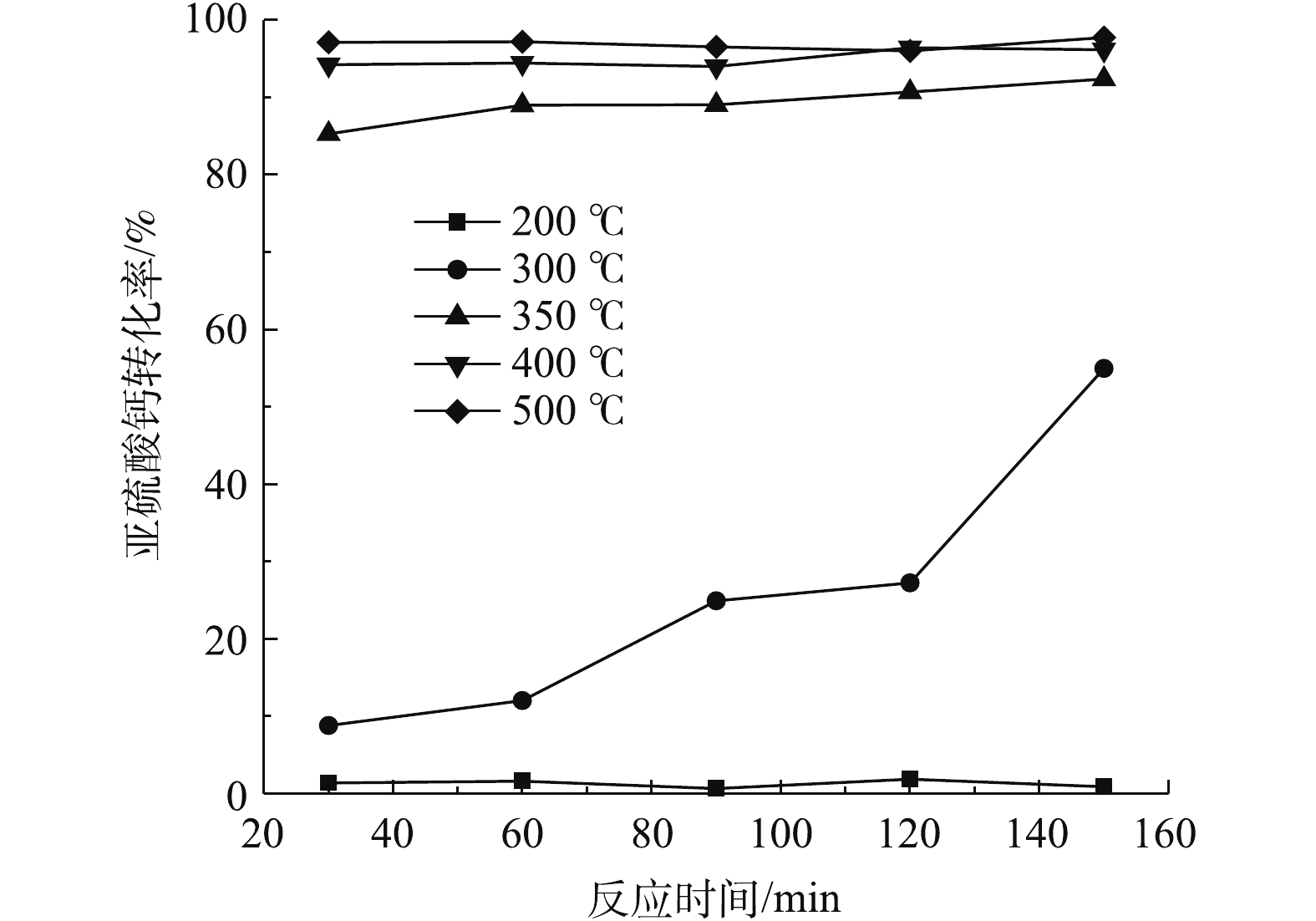
在进行不同温度对脱硫灰中亚硫酸钙氧化的影响研究时,设定氮氧比=3∶2,气速为250 mL·min−1,反应温度分别为200、300、350、400、450 ℃,反应时间为30 min。
在进行不同含氧量对脱硫灰中亚硫酸钙氧化的影响研究时,主要考察了400 ℃和450 ℃条件下氧含量对亚硫酸钙氧化的影响。设定氮氧比(常温常压下反应气中氮气与氧气的比值)为3∶2、3.5∶1、4∶1和4.5∶0.5,气速为250 mL·min−1,反应时间为30 min。
实验考察了水汽含量对脱硫灰中亚硫酸钙氧化的影响。①在进行水汽量恒定、气速不同的影响实验时,设定反应温度为400 ℃,氮氧比为4∶1,气速分别为25、50、75、100、125、250、375、500 mL·min−1,反应时间为30 min,让气体通过装有恒温80 ℃蒸馏水的洗气瓶,使气体中带有少量且恒定的水汽。②在进行气速恒定、水汽量不同的影响实验时,设定反应温度为400 ℃,氮氧比为4∶1,气速为75 mL·min−1,反应时间为30 min。让气体通过定量加水的汽化加热管,使气体中带有一定的水汽。
根据脱硫灰的热重曲线规律可知,脱硫灰随温度的升高,失重越大,且不同脱硫灰含水率存在一定差异。为简化计算,本研究忽略了不同温度对脱硫灰减重的影响,用氧化转化前后样本中亚硫酸钙含量变化率(简称转化率或氧化率)来表示脱硫灰的氧化程度,根据式(1)进行计算。
式中:C为脱硫灰中亚硫酸钙的转化率;C1为反应前脱硫灰中亚硫酸钙的质量分数;C2为反应后脱硫灰中亚硫酸钙的质量分数。
1.1. 实验试剂与仪器
1.2. 实验方法
-
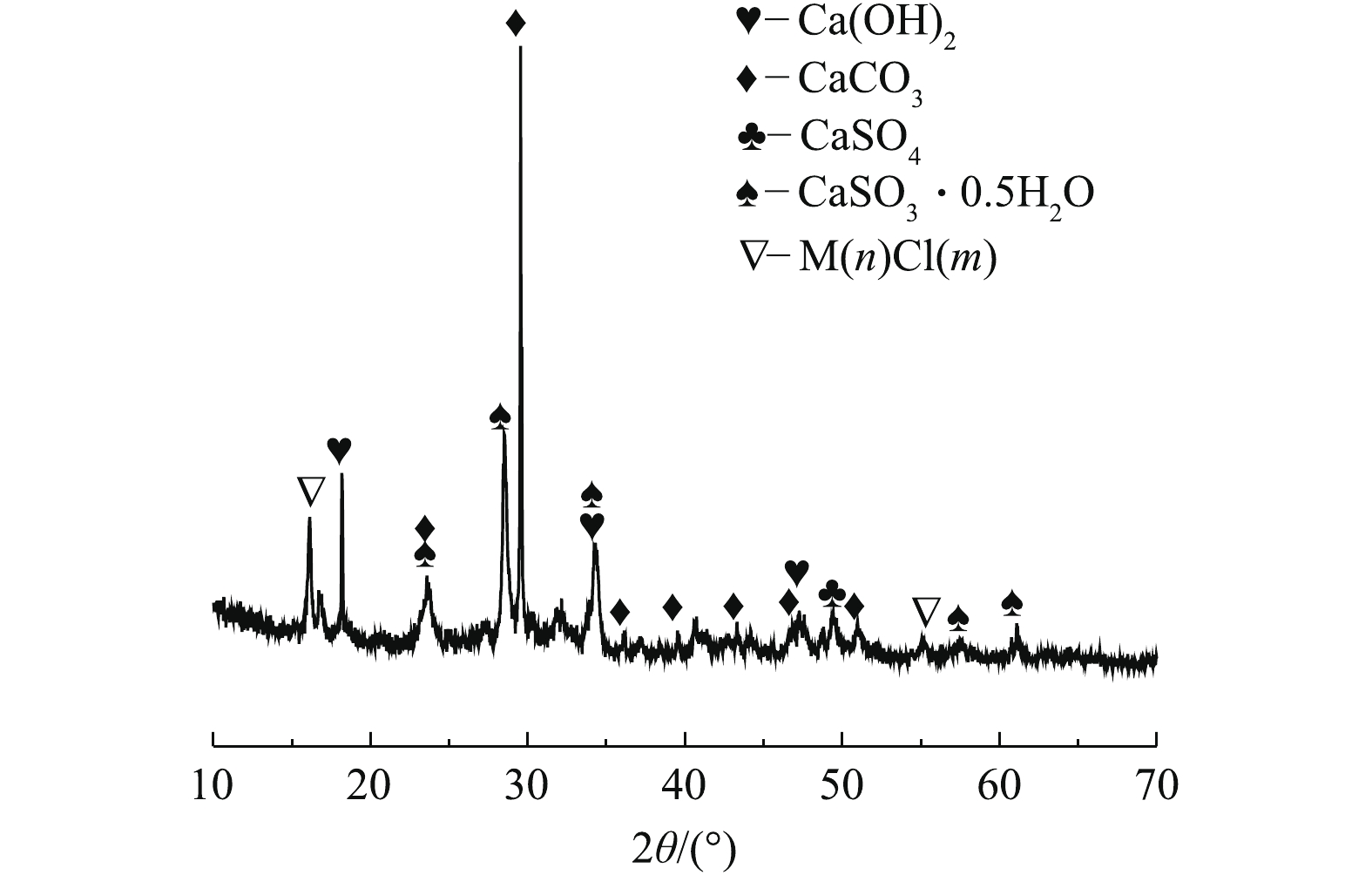
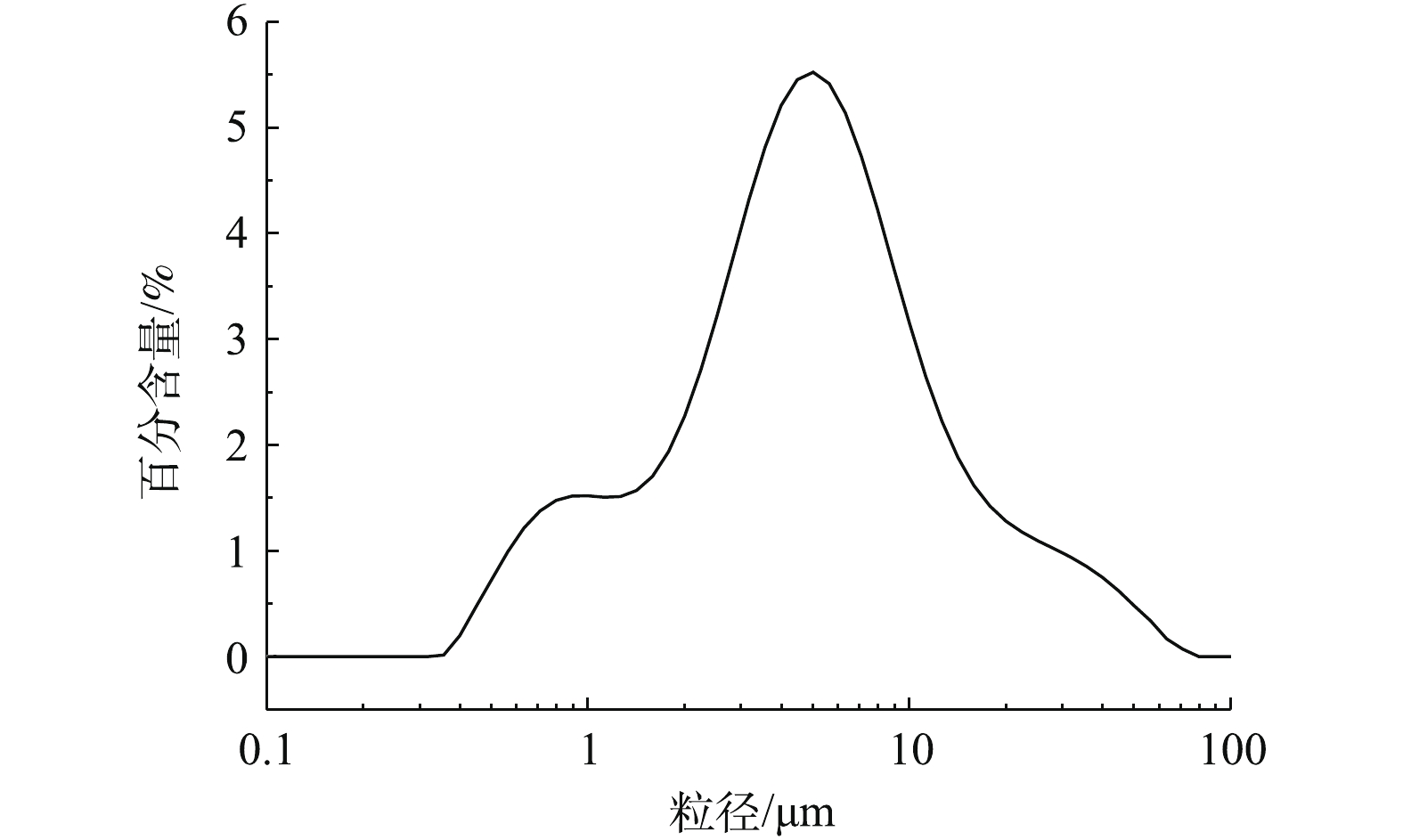
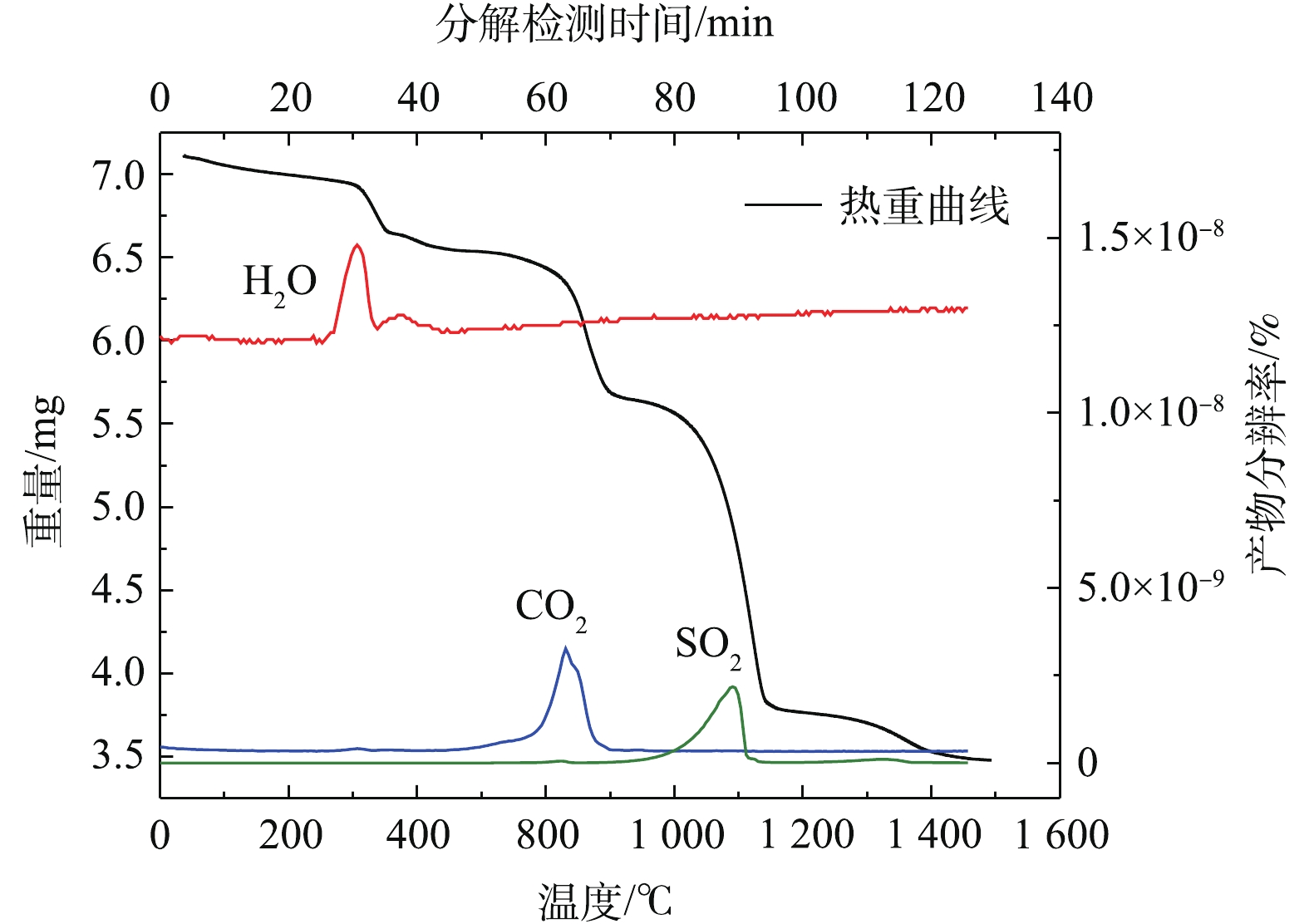
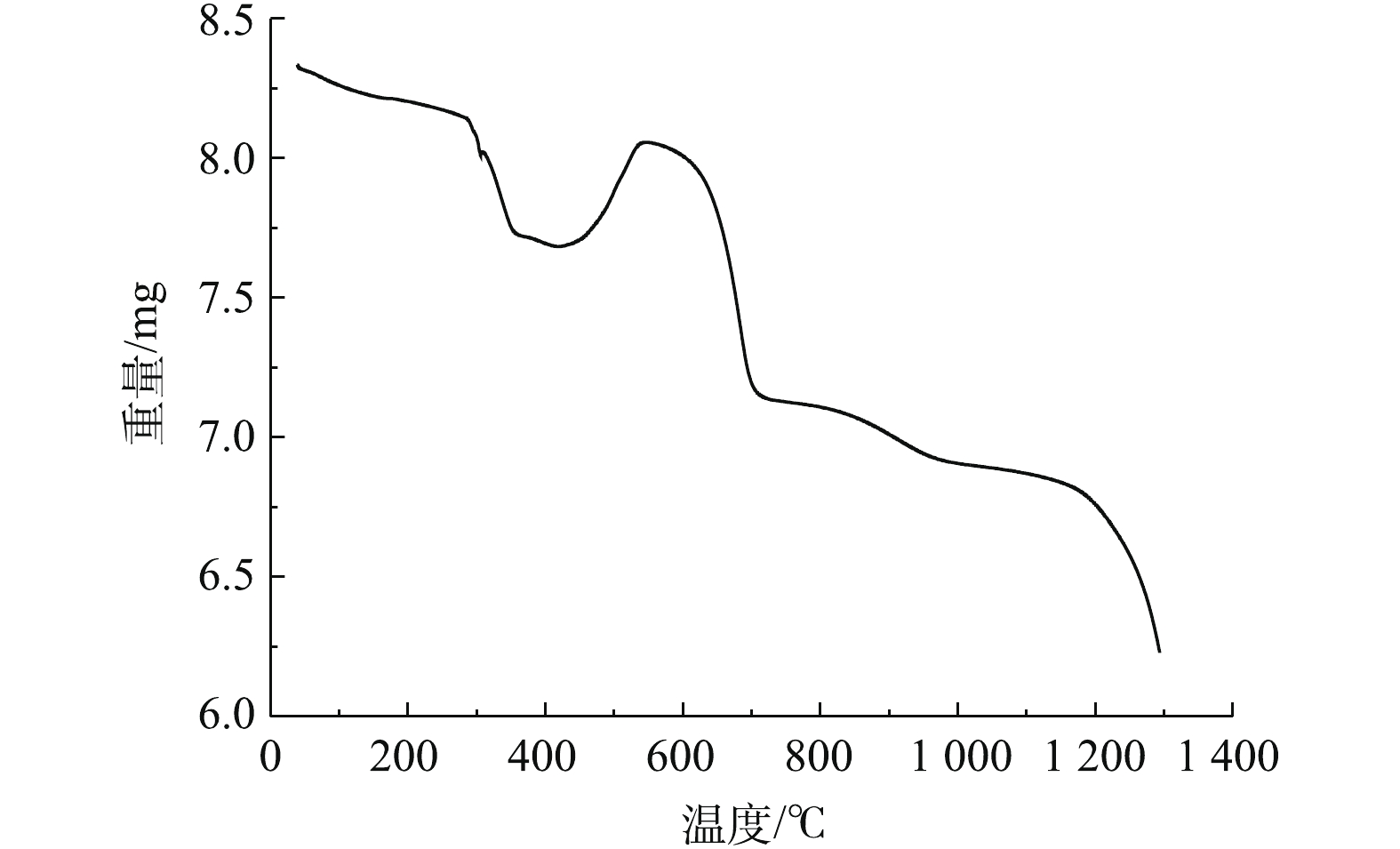
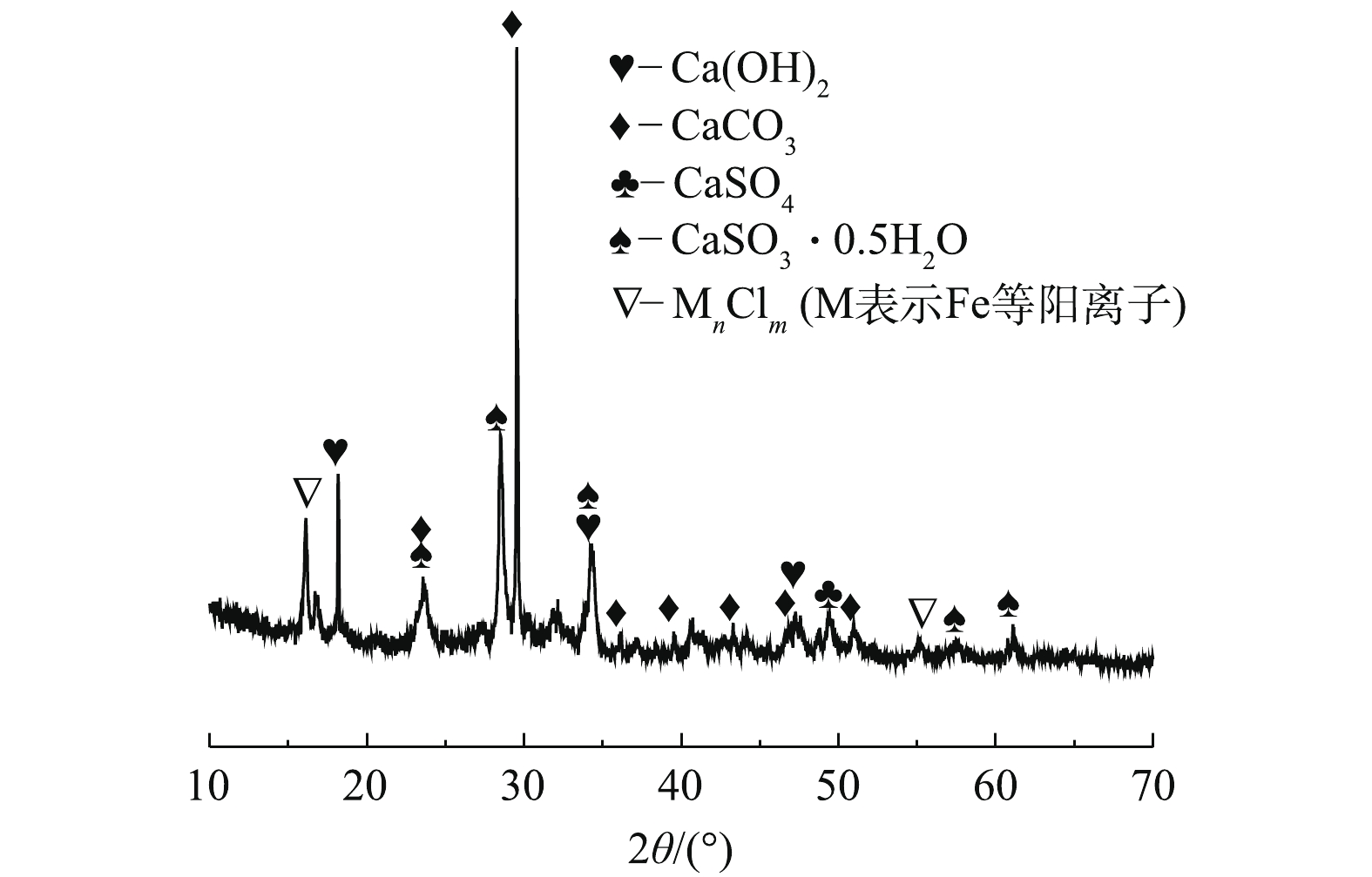
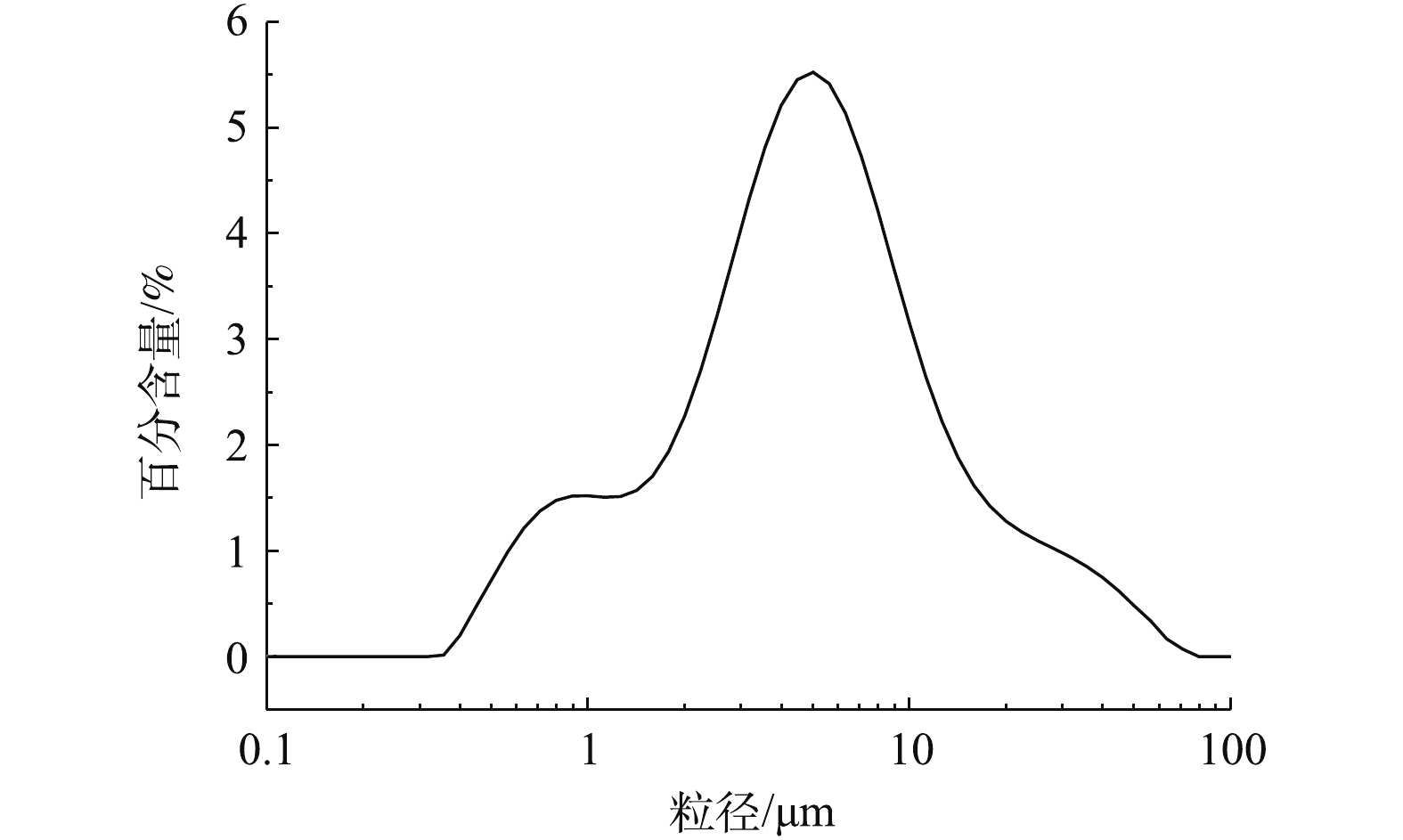
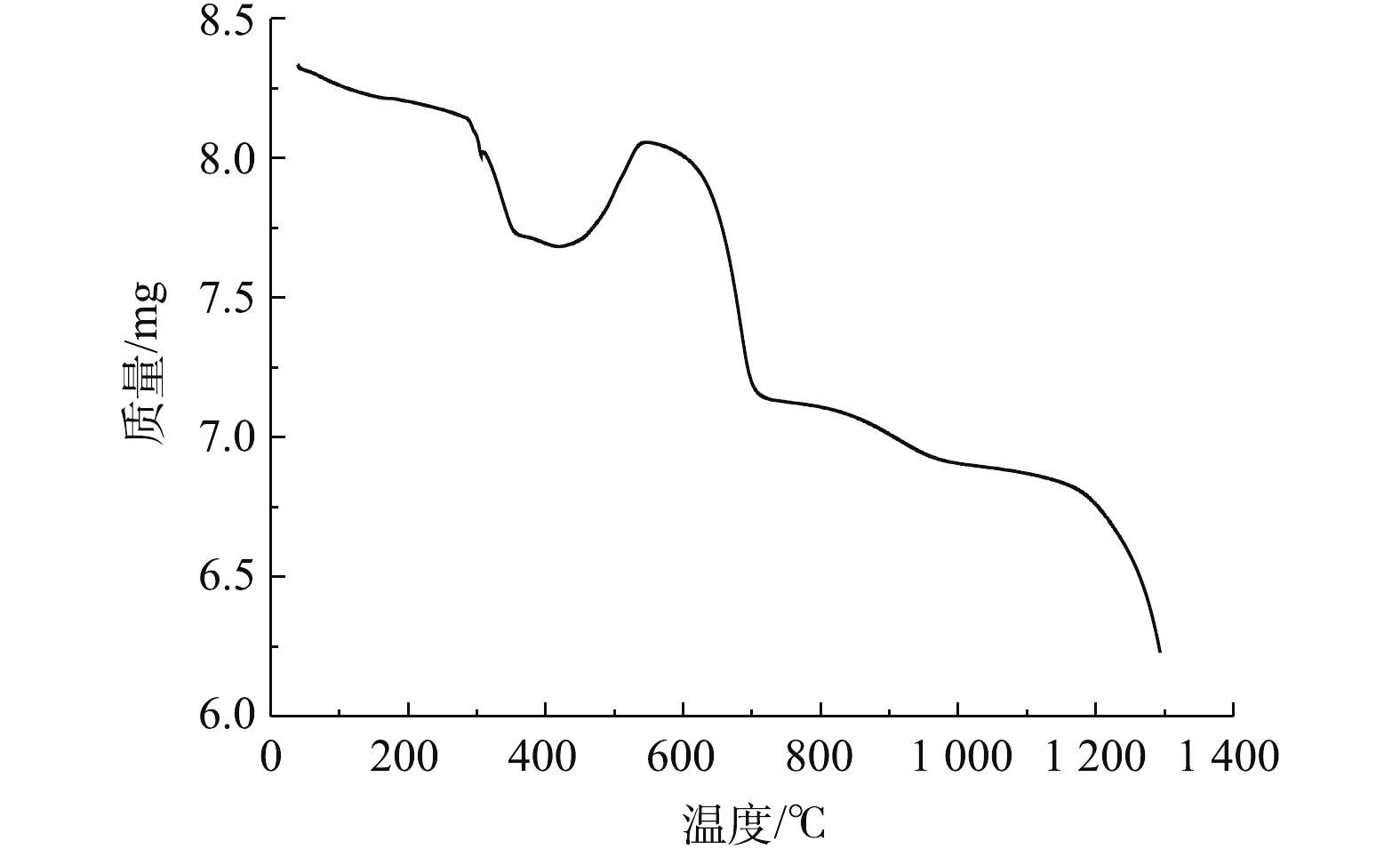
根据表1和图2可知,脱硫灰的主要成分为半水合亚硫酸钙、碳酸钙、氢氧化钙和硫酸钙。由图3可知,脱硫灰平均粒径为7.252 μm,中位粒径(D50)为4.521 μm,80%的粒子粒径为1.039~16.162 μm。根据实验升温速率为10 ℃·min−1的热重-质谱图(图4)可知:300~400 ℃为CaSO3·0.5H2O失水和Ca(OH)2分解的失重区间;550~700 ℃为CaCO3分解失重区间;750~900 ℃为CaSO3分解失重区间;1 100~1 200 ℃为CaSO4分解区间。在600 ℃后边有一个微弱的SO2的峰,这部分变化可能是由于脱硫灰中少量的硫酸亚铁分解造成的。根据空气氛围热重曲线(图5)可知:当小于300 ℃时,主要为脱硫灰中的吸附水和部分结合水蒸发失重;300~450 ℃为CaSO3·0.5H2O失去结晶水和Ca(OH)2分解的失重区间;450~550 ℃为CaSO3氧化增重区间;550~700 ℃为CaCO3分解失重区间;1 100~1 300 ℃为CaSO4分解区间。但对于脱硫灰氧化而言,在450~550 ℃内,只是氧化增量大于失重量,即为氧化速率较大的温度区间,但脱硫灰开始明显氧化的温度肯定小于450 ℃。由脱硫灰的SEM表征结果(图6)可以看出,由脱硫灰大小不一,形状极不规则,但表面很光滑。脱硫灰粒子有很多孔道,孔道直径在0.1~0.2 μm左右。细小而多孔的脱硫灰有较大的比表面积,从而可以与气体接触更加充分,故有较强的表面活性。
-
膨化态是介于静固态和流化态之间的一种形态。该状态下固体之间空隙较大,气固接触相对比较充分,不需要大量的气体使之保持该种状态,即废气量小。通入反应气体的流速越大,气固接触就越充分。静固态实验是在马弗炉中空气氛围下完成的。膨化态实验是模拟工业无预热氧化条件在立式管式炉中进行的,即反应气体进入反应器前的温度为室温。
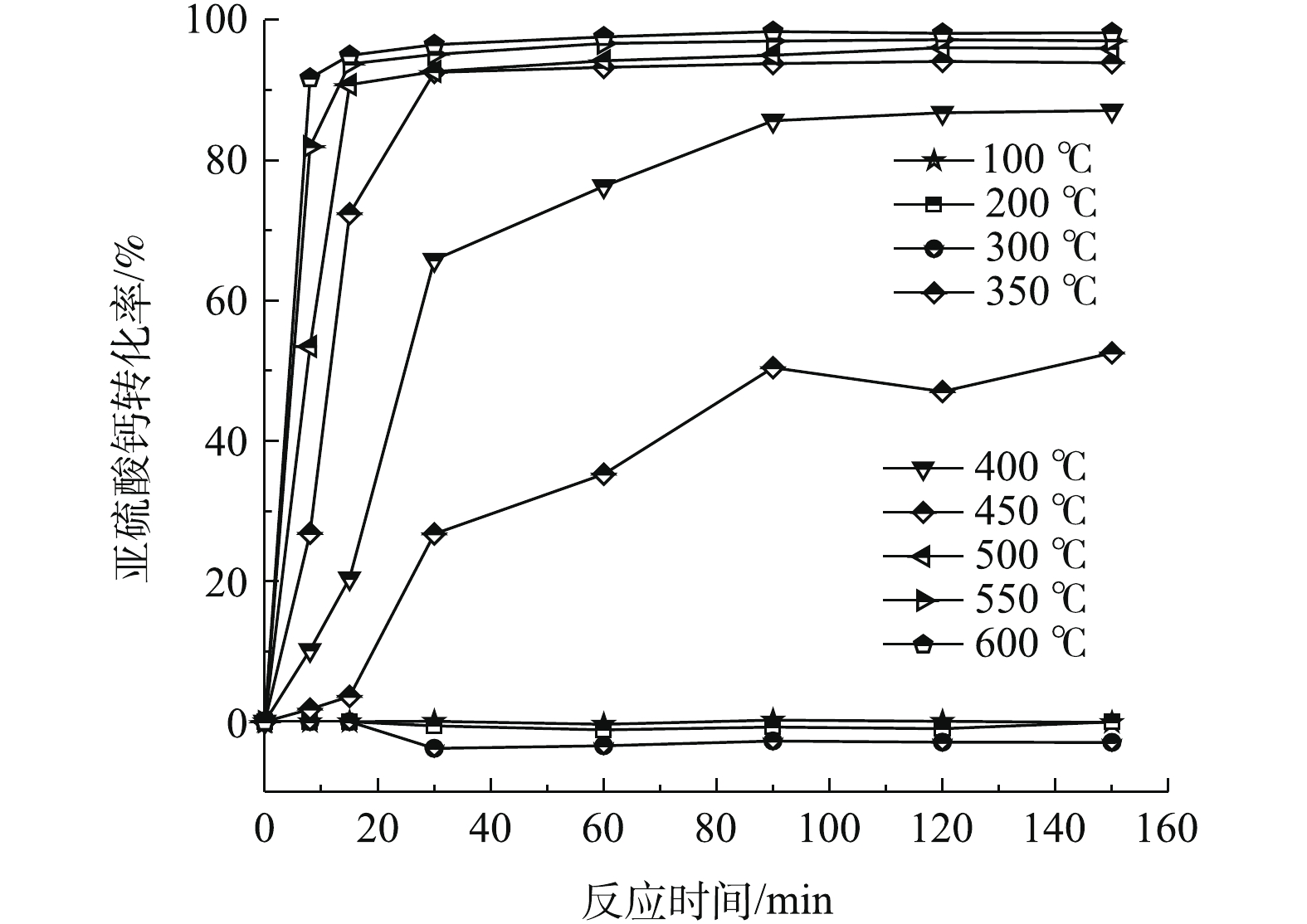
在静固态时,由图7可知:当温度小于300 ℃时,亚硫酸钙氧化增重远远小于该温度下的失重,表现出转化率小于零的现象;当温度高于350 ℃时,脱硫灰发生了明显的氧化反应,但反应速率并没有达到最大;温度在350~400 ℃时,在30~90 min内以恒定速率进行氧化反应,90 min以后反应趋于饱和。产生该现象可能的原因是温度升高时脱硫灰发生了团聚反应[21-22]。此过程中脱硫灰整体体积变小、密度增大、孔隙减小、且反应速率比较慢,导致内部亚硫酸钙无法与氧气充分地接触,故其不能被继续氧化。当温度高于450 ℃时,氧化速率足够大,在脱硫灰团聚完成之前就已经完成氧化,在30 min内,反应趋于相对完全,此时氧化转化率在92%以上。根据不同温度下的氧化转化率对比可知,温度越高,氧化速率提升越快。
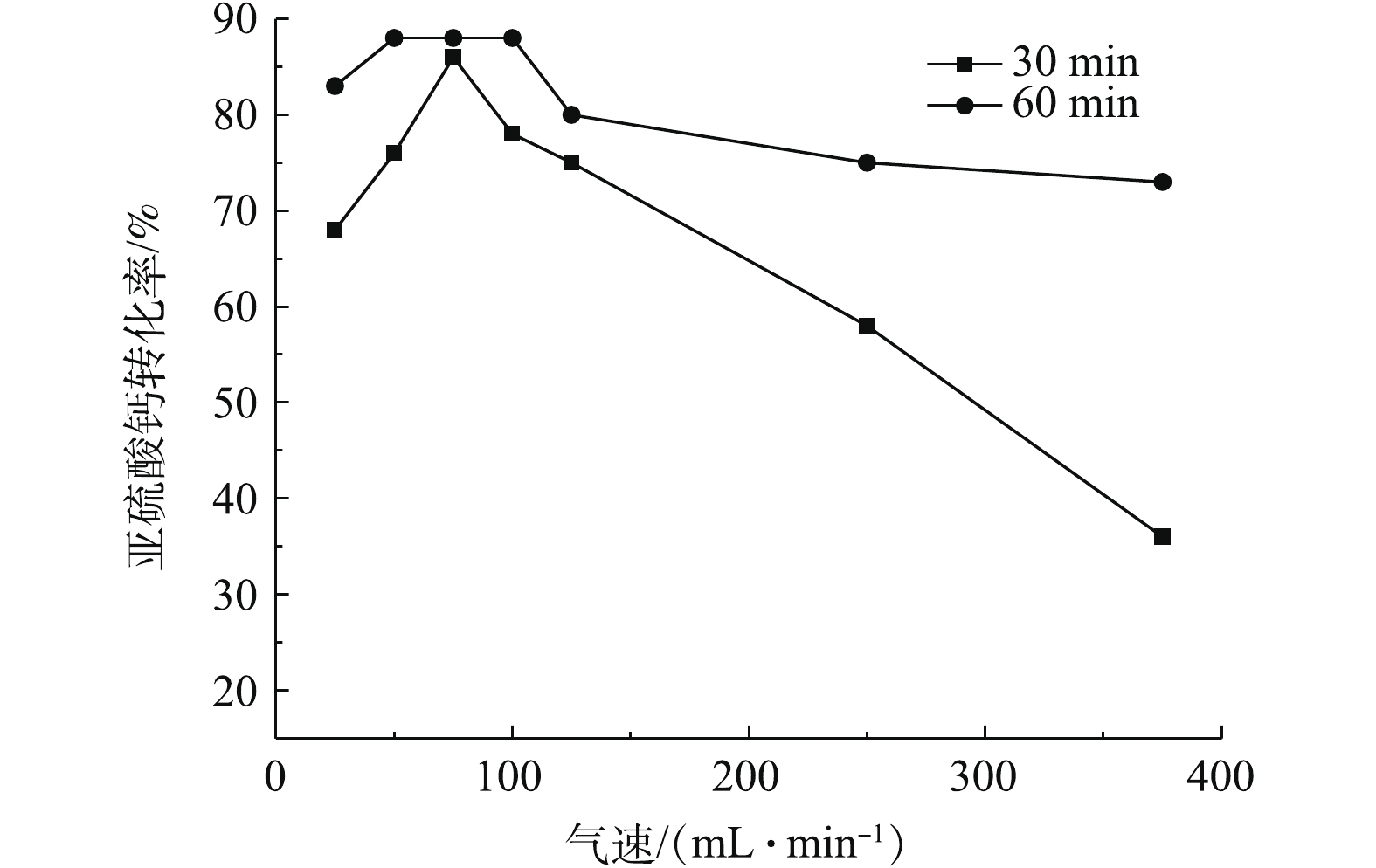
在膨化态时,由图8中30 min对应的曲线可知,当流速在75 mL·min−1时反应速率最大,氧化转化率为86%,而由60 min对应的曲线可知,在50~100 mL·min−1时,反应速率接近,氧化转化率为88%。造成这种现象的原因可能有2点:其一是该条件下的最大氧化效率为88%,虽然在不同气速下实验前30 min氧化速率有所不同,但在反应60 min后均达到了最大值,其二是在该条件下气固接触更加充分,且氧化放热能使大部分亚硫酸钙完成热氧化。当气速大于250 mL·min−1时,脱硫灰呈现沸腾状态,气固接触更加充分。但室温空气进入反应器后会使反应器内温度略有降低,并带走亚硫酸钙的氧化热,从而致使氧化效率明显降低。当气速小于250 mL·min−1时,曲线呈现为倒U或V型,其原因是:气速较小时反应器内的氧含量达不到氧化反应的需氧要求,且气固接触程度不高,从而致使氧化效率较低;当气速大于75 mL·min−1时,石英管内的脱硫灰被气流分成若干份,每份脱硫灰内部的亚硫酸钙接触到氧气的概率明显降低,即气固完全接触的程度有所降低,从而导致氧化效率降低。
通过对同一温度下的实验结果对比可知,流化态氧化效果比静固态的氧化效果更好,且在流化态较低速时,转化率的提升幅度为2~3倍。综上所述,适当提高气固接触程度对氧化反应具有较大的影响。在室温反应气体进入反应器时,并不是接触程度越高越好,而是存在1个最佳的气速值,根据以上研究结果,本研究的最佳气速为75 mL·min−1。
-
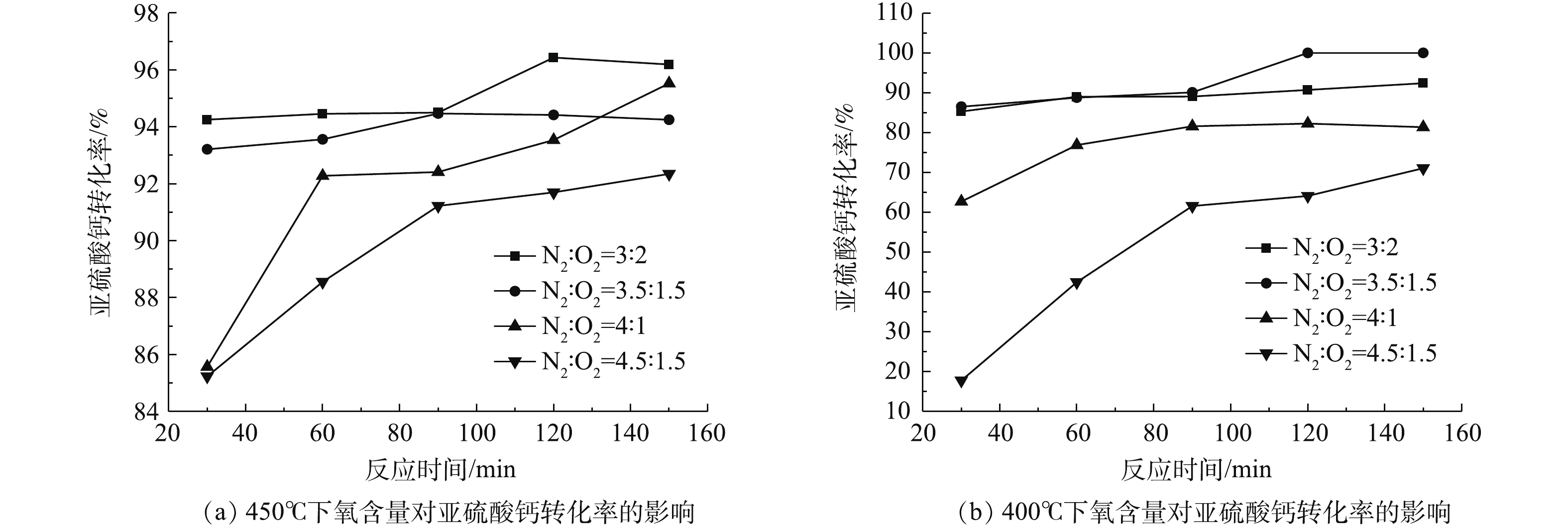
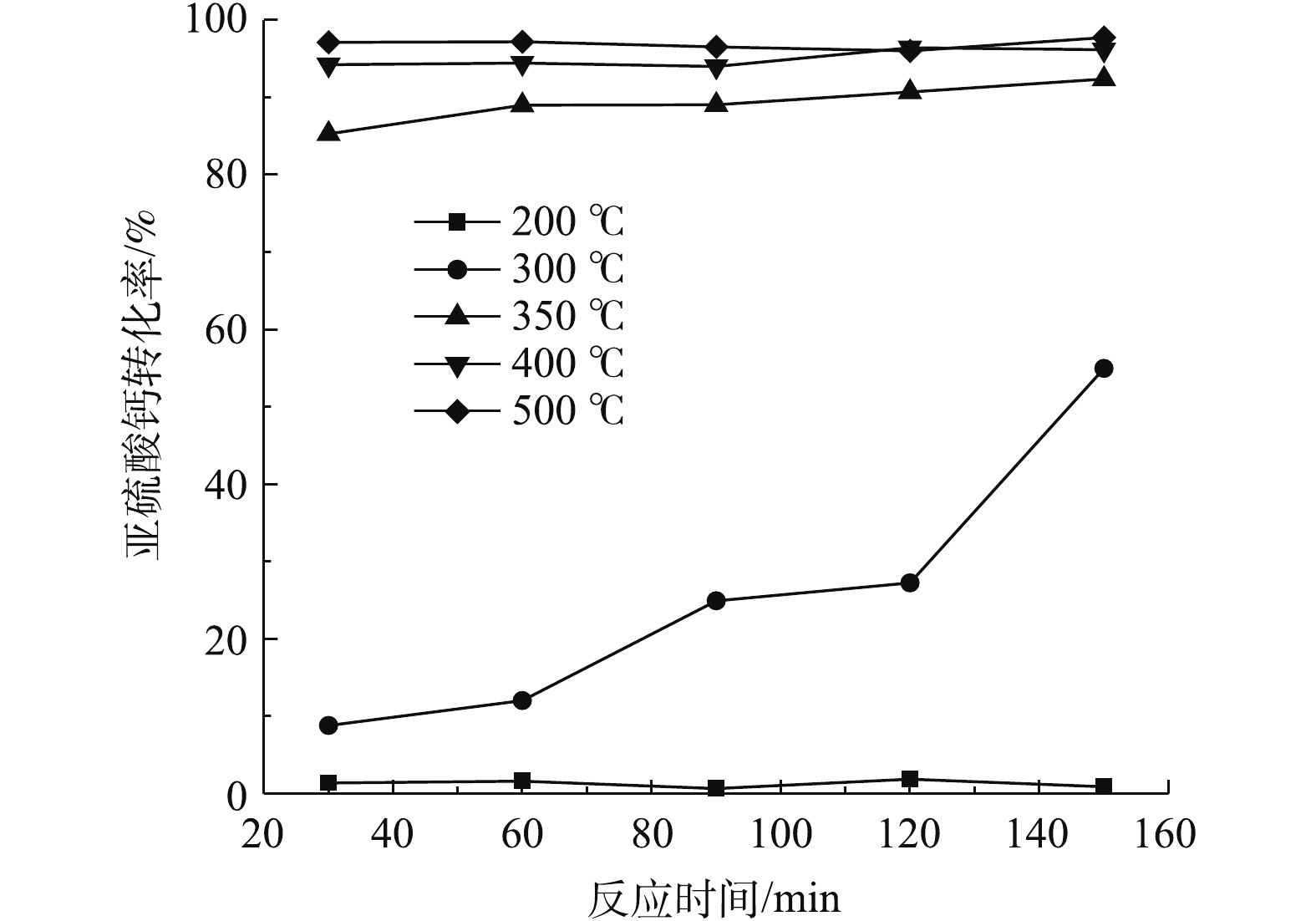
该部分实验条件是在40%氧含量(高氧)的条件下进行的,旨在探究该条件下亚硫酸钙氧化反应对温度的响应情况。由图9可知,提高氧含量可降低脱硫灰初始氧化温度,可整体提高氧化效率。300 ℃时静态固体态氧化反应不明显。当氧含量提升至空气的2倍后,300 ℃时即发生了明显的氧化反应,表明初始氧化温度有所降低。反应在120~150 min时,氧化转化率可提升至55%,这是由于脉冲进气带来的振动效果在较长时间的积累导致气固接触更加充分造成的。当温度为350 ℃时,30 min氧化转化率为85%,与静固态实验相比氧化转化率提升了近60%;当反应时间大于60 min时,转化率略有提高,但提高的效果并不显著,氧化转化率在90%~92%,此时与静固态450 ℃处理效果相近;当温度高于400 ℃时,氧化转化率可达94%;在400~450 ℃时,最大氧化转化率在96%~97%,此时反应达到最大转化率。
-
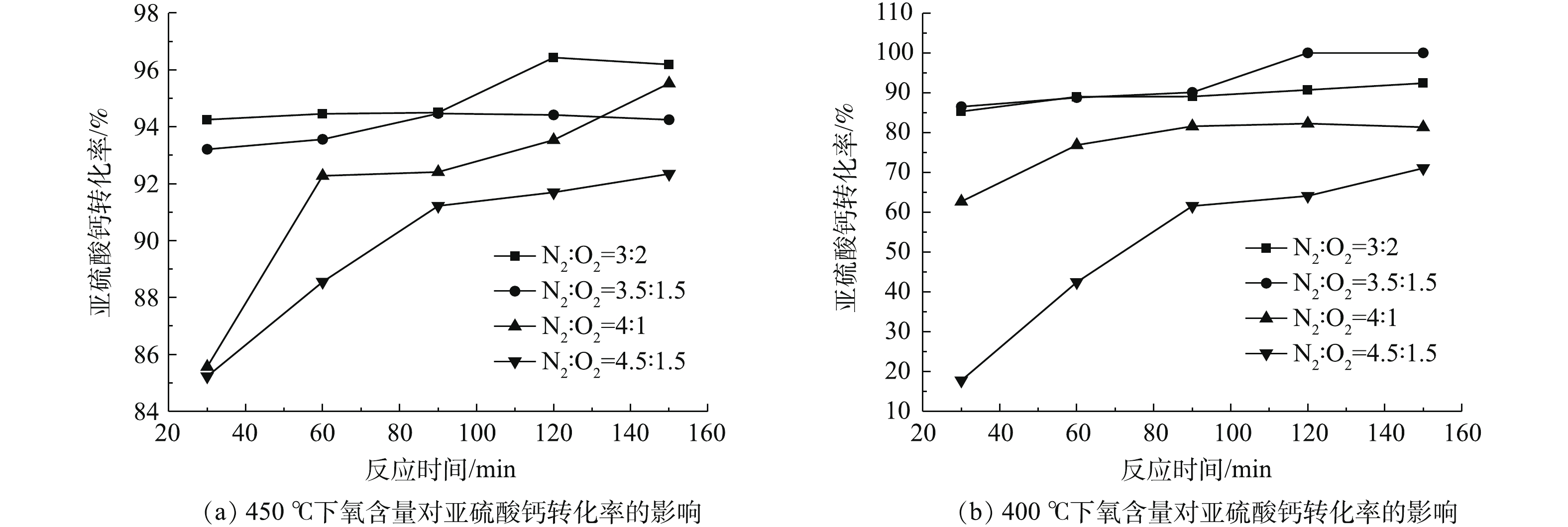
氧含量的增加有利于反应的进行,但温度对反应的影响更大一些。由图10可知,随着氧含量的提高,反应效率均得到了不同程度的提高。当温度为400 ℃时,氧含量的提高对反应转化率的影响更大一些。氮氧比在4.5∶0.5时,30 min的亚硫酸钙转化率在16%左右;当氮氧比提升至3.5∶1.5时,亚硫酸钙转化率提升至85%左右,亚硫酸钙转化率大约提升了69%,反应速率得到了显著提升。当温度为450 ℃,氮氧比为4.5∶0.5时,30 min的亚硫酸钙转化率在85%左右;当氮氧比提升至3.5∶1.5时,亚硫酸钙转化率提升至93%左右;此时氮氧比的提升同样使得转化率得到一定的提升,但亚硫酸钙转化率仅提升了8%左右,提升的显著程度并没有像400 ℃时那样大。将图10(a)和图10(b)中任意对应点进行比较可知:450 ℃的转化率恒大于等于400 ℃的转化率,当氮氧比在3∶2时,温度的提升致使转化率提升了8%左右;当氮氧比在3.5∶1.5时,温度的提升致使转化率提升也是8%左右。由此可知:当氮氧比小于3.5∶1.5时,温度从400 ℃提升至450 ℃时对转化率的影响是一定的,且转化率提高在8%左右;当氮氧比大于4∶1时,温度对转化率的影响十分显著;氮氧比为4.5∶0.5时,450 ℃的最低转化率为85%,而400 ℃的最低转化率为17%,最高为71%。以上结果充分表明,当氧含量较低时,亚硫酸钙充分氧化所需的温度则需更高。
-
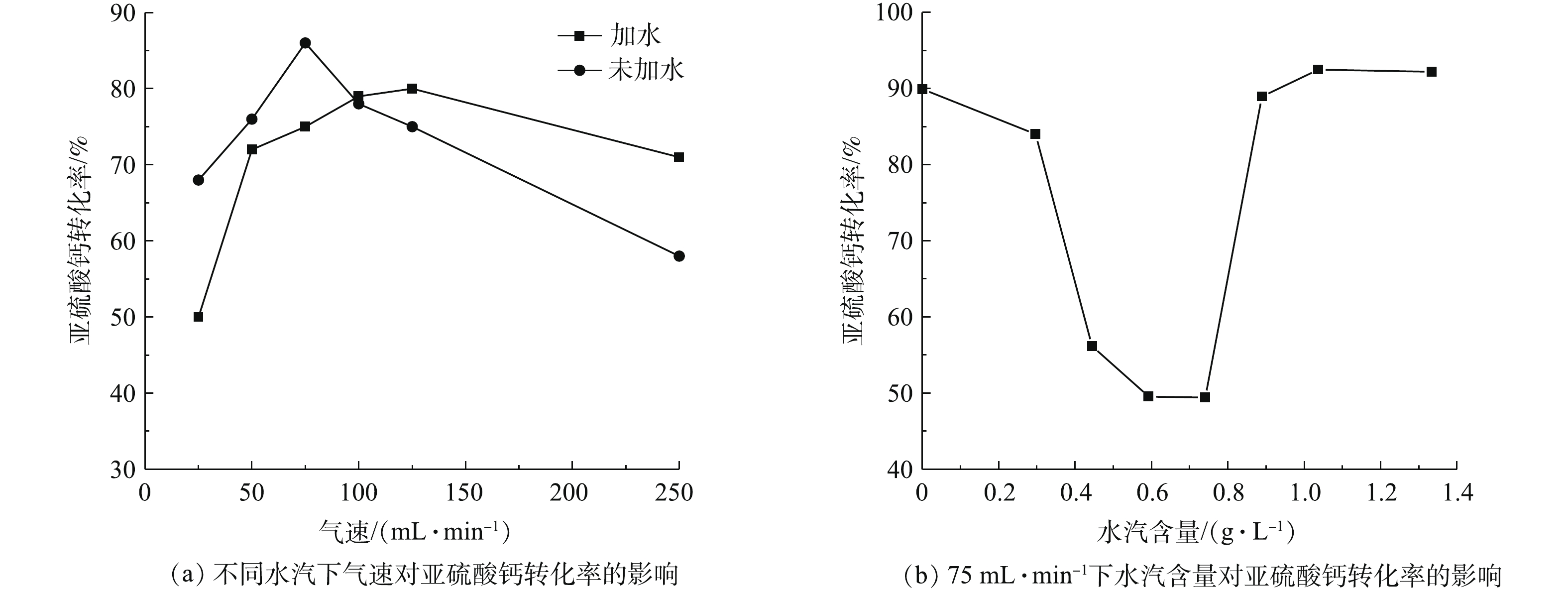
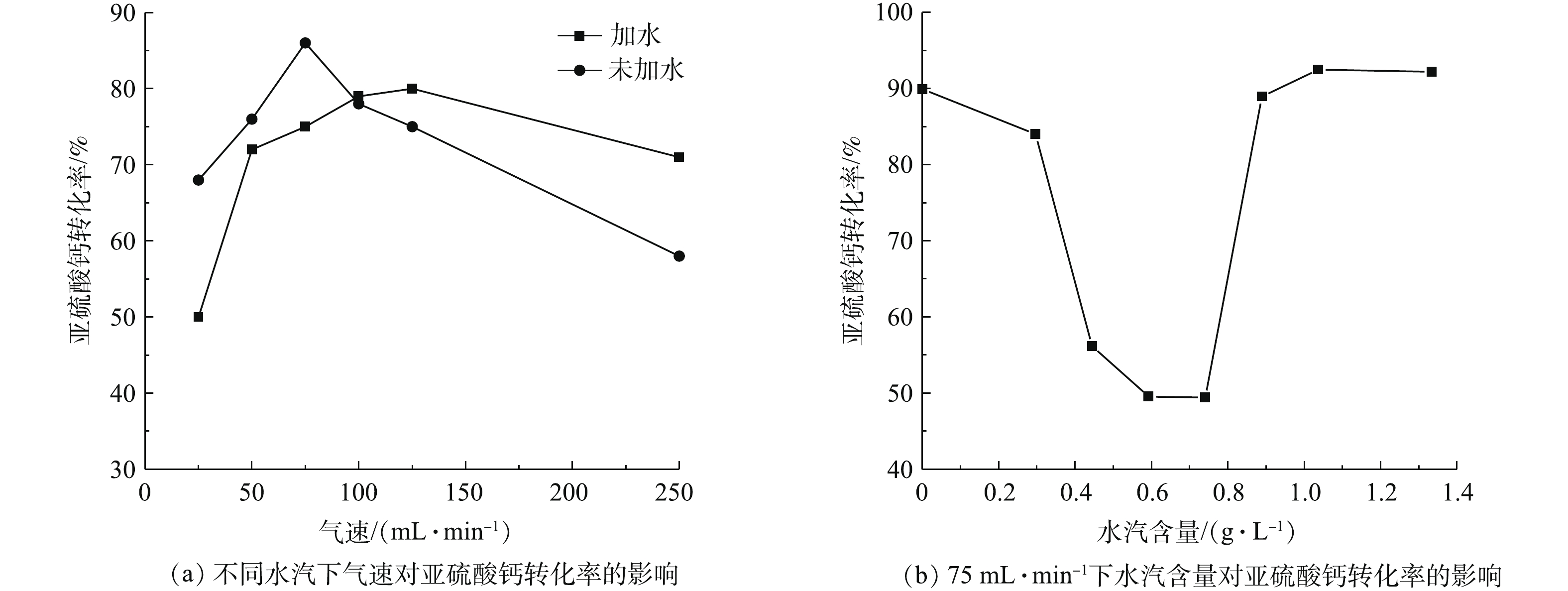
为方便实验的进行和数据的分析,本研究中水汽含量为常温常压下每升配气中加入蒸馏水的质量。由图11(a)可知,当气体流速小于100 mL·min−1时,水汽的加入降低了氧化反应的效率;而气体流速大于100 mL·min−1,水汽的加入提高了氧化反应的效率。造成这种现象的原因是:当气流较小时,带入水汽的量很少,而有限的水汽在该条件下与脱硫灰中的吸水性物质优先发生了反应,从而抑制了亚硫酸钙的氧化;当气体流速增大,带入的水汽足够多时,未进行上述反应的水汽通过某种特殊的反应过程促进了亚硫酸钙和氧气的反应。为证明该猜测,对实验进行了一定改进,从而使少量气体能够带有大量的水汽,此时气速为75 mL·min−1。由图11(b)可知:当水汽量小于0.88 g·L−1时,转化率随水汽量的增加而降低,最低转化率为49.42%,转化率明显降低了;当水汽量为1.03 g·L−1和1.33 g·L−1时,转化率分别为92.01%和92.47%。该实验结果不仅证明了上述观点的正确性,而且还可以说明,当水汽量较多时,水汽可以促进氧化反应的进行,如果继续增加水汽含量,转化率的改变并不会太大。产生这种结果的原因是:一方面大量水汽的加入会使气速增加,气固接触更加充分;另一方面则使得亚硫酸钙稳定性发生变化,进而使氧化反应变得更加容易进行。
2.1. 脱硫灰理化特性
2.2. 氧化状态
2.3. 温度的影响
2.4. 氧含量的影响
2.5. 水汽含量的影响
-
1)脱硫灰中亚硫酸钙的氧化转化率随温度的增加而增加,气固接触越充分,氧化转化率相对越高。气体速度通过对气固接触状态和反应器中温度的影响,进而影响亚硫酸钙的氧化。脱硫灰中亚硫酸钙氧化的最优工艺条件:温度为400 ℃、氮氧比为4∶1、气速为75 mL·min−1。
2)当氧化温度较低时,氧含量的增加可以较大程度地提高氧化效率,提升幅度在6%左右;在较高的温度下,氧含量对反应的影响较弱。
3)水汽对亚硫酸钙氧化反应的影响具有双面性。当水汽含量<0.88 g·L−1时,随水汽含量的增加,转化效率逐渐降低;当水蒸气量>0.88 g·L−1时,水汽的加入使得转化效率略有提高,但水汽含量的增加对转化效率增加的影响是有限的。
