


-
挥发性有机化合物(volatile organic compounds,VOCs)来源广泛,对大气环境和人体健康均有不利影响[1-3]。常见VOCs控制技术有吸附[4]、吸收[5]、冷凝[6]等回收技术和热氧化[7]、低温等离子体[8]、生物法[9]等销毁技术。工业中较为常用的技术为吸附和热氧化(包括蓄热燃烧、催化燃烧等)[10]。然而,吸附法仅仅将污染物进行了转移,后期存在吸附剂再生问题。而热氧化主要用于高浓度VOCs处理[11],且存在能量效率低的问题。相比之下,低温等离子体因其具有可快速启动和关闭、能耗低、净化效率高等特点而广受研究者关注[12-14]。
低温等离子体的产生方法有介质阻挡放电(DBD)、电晕放电、滑动弧放电、辉光放电等[15]。其中DBD是一种最为常见的低温等离子体产生方法,其放电均匀且稳定[16]。传统的DBD反应器是在高压电极和接地极之间嵌入一层绝缘介质,然而单一的介质阻挡放电低温等离子体在降解VOCs时存在副产物多、能量效率低等问题[17]。有研究[18-19]发现,若在两电极之间加入双层介质,在外加电压下,形成双介质阻挡放电(DDBD),可以有效提高VOCs的去除率并抑制副产物的产生。ZHANG等[18]比较了单双介质阻挡放电对苯乙烯降解的影响,发现DDBD反应器比DBD反应器的CO和CO2选择性提高了40%,同时DBD反应器中生成大量油状有机副产物,而DDBD反应器中则没有。TANG等[19]对比了单双介质阻挡放电反应器对NO的去除,发现DDBD反应器中产生的放电更加均匀稳定,使能量可以得到高效利用,而DBD反应器中的放电强度较大,有利于NO的去除。但李云霞等[20]的研究发现,当外加电压(4.0~6.5 kV)较低时,单介质反应器的CS2去除率高于双介质反应器。因此,双介质阻挡放电反应器对于某种污染物的去除效果并非绝对的优于单介质反应器,而要视具体条件而定。目前,关于双介质和单介质阻挡放电低温等离子体在不同条件下降解VOCs的系统比较研究仍然较少。因此,比较2种反应器在不同条件下的VOCs降解效果,可为实际应用过程中反应器的合理选择提供参考。
本研究首先对比分析了单介质和双介质反应器的放电特征,随后以甲苯为目标污染物,以甲苯去除率、矿化率、CO2选择性为指标,分析了2种反应器在不同电压、不同浓度下对甲苯的去除效果,并对副产物O3、N2O以及反应器的能量效率进行了比较分析。
-
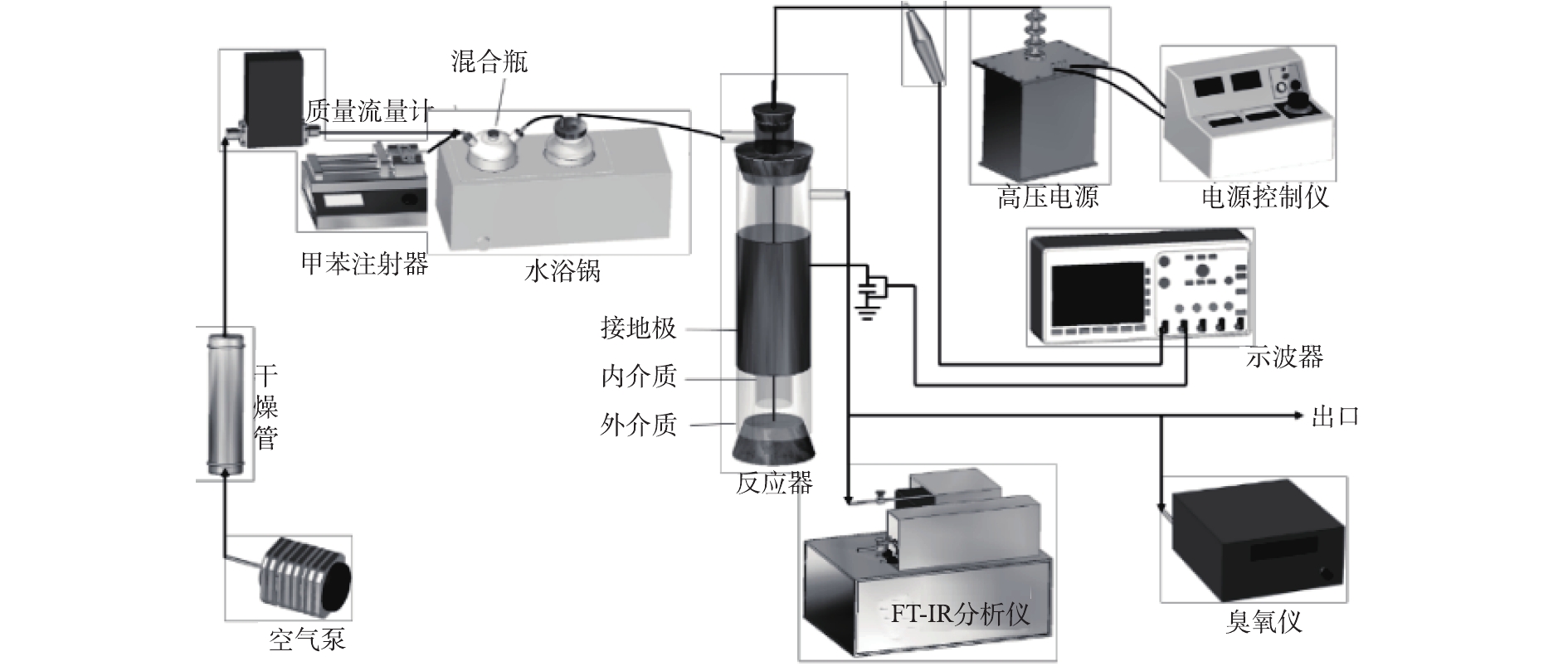
所有实验均在常温常压下进行,实验装置如图1所示。将注射泵注入的液态甲苯与经过干燥后的空气在混合瓶中混合,用来模拟甲苯废气,混合瓶置于恒温水浴锅中以保证甲苯能持续挥发。由质量流量计控制气体流量为2 L·min−1,通过改变注射器的推进速度来调整甲苯浓度,本实验中甲苯浓度分别为616、1 027、1 848 mg·m−3。低温等离子体在自制的线筒式介质阻挡放电反应器中产生,反应管为石英玻璃材质,高压电极为直径1 mm的不锈钢丝,接地极为缠绕在玻璃管外壁的铜皮(厚0.05 mm,长100 mm)。单介质反应器管内径为30 mm,厚1.5 mm,双介质反应器外管尺寸与单介质相同,内管内径为15 mm,厚1 mm。实验所用电源为50 Hz高压交流电源(GJTK-0.01/30K,上海南罡电除尘器有限公司)。使用示波器(DPO3054,Tektronix)通过高压探头(P6015A,Tektronix)测定电流电压波形图。甲苯及其降解过程中产生的H2O、CO2、CO、N2O的浓度由傅里叶变换红外光谱仪(Nicolet Antaris IGS,Thermo Scientific Company)测定,O3浓度通过臭氧检测仪(2B Technologies Model 106-M)测得。
-
反应器的输入功率计算方法[21]见式(1),甲苯的降解效果以甲苯去除率、矿化率、CO2选择性和能量效率作为评价指标,计算方法见式(2)~式(5)。
式中:P为输入功率,W;η为甲苯去除率;MR为矿化率;
SCO2 为CO2选择性;f为频率,取值50 Hz;EF为能量效率,g·(kWh)−1;C为电容,取值0.47 μF;A为示波器所测李萨如图的面积;Cin、Cout为反应器进、出口甲苯浓度,mg·m−3;CCO2 、CCO为反应器出口CO2、CO浓度,mg·m−3;Q为甲苯气体流量,取值2 L·min−1。 -
图2为单介质和双介质反应器分别在14 kV和24 kV电压下一个正负变换周期内的电流波形图。随着电压的升高,微放电的数量和强度明显增加。在同一电压下,双介质反应器中的微放电数量更少,这是由于双介质反应器中的内层介质阻碍电子的运动,微放电到达内层介质后不易继续向外层介质传播,导致检测到的微放电数量较少。
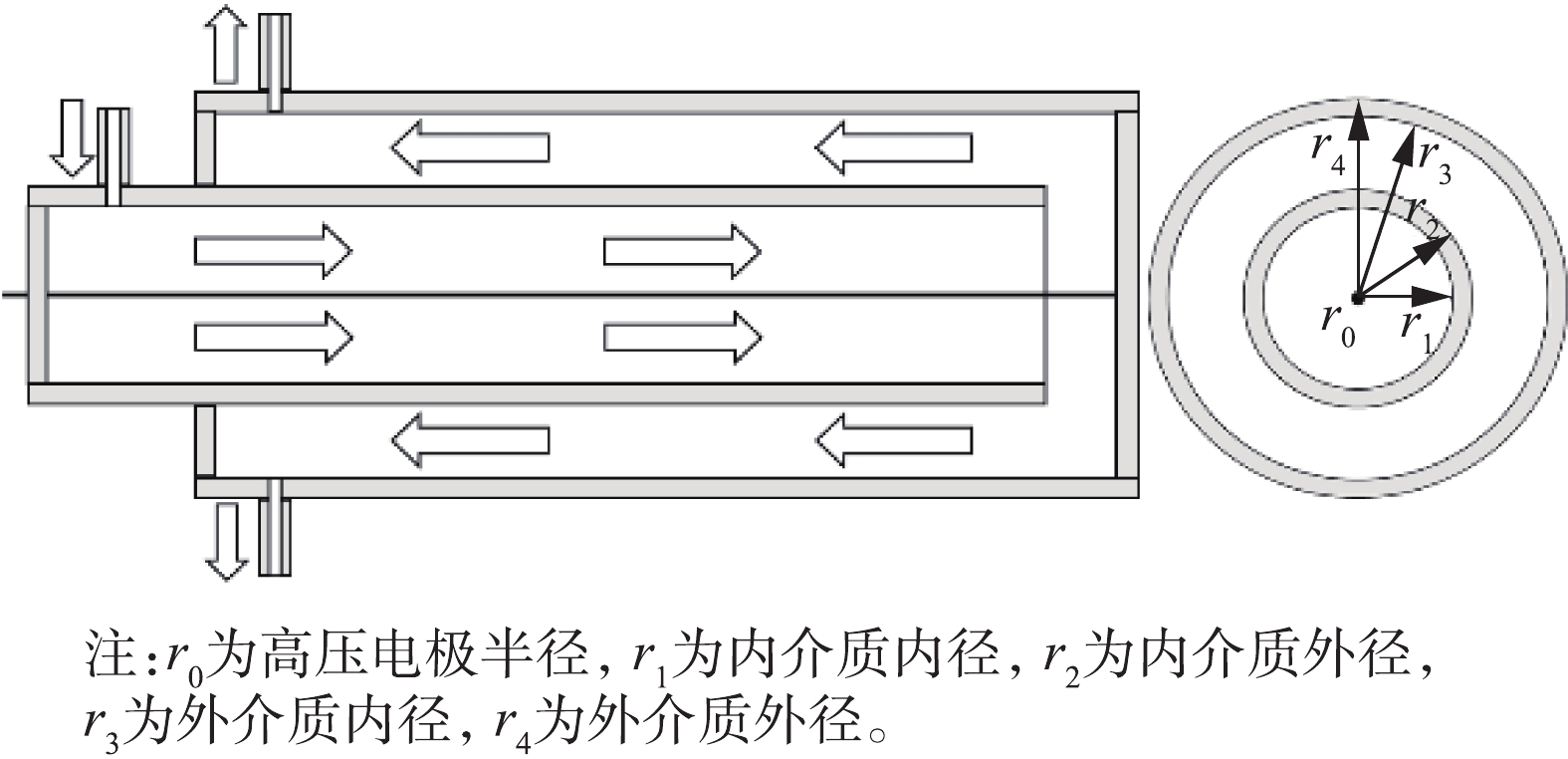
双介质反应器的结构及气流方向如图3所示。通过理论计算的方法估算2种反应器电场强度,计算方法[22-23]见式(6)和式(7)。
式中:Eg,双和Eg,单分别为双介质和单介质反应器放电区域的电场强度,kV·cm−1;V为放电电压,kV;εd为介质的相对介电常数,取值为4;εg为空气的相对介电常数,取值为1;r为反应器中放电间隙据轴心的距离,cm;r0=0.05 cm;r1=0.75 cm;r2=0.85 cm;r3=1.5 cm;r4=1.65 cm。
不同反应器的平均电场强度和输入功率比较结果见图4。在相同电压下,双介质反应器的平均电场强度略大于单介质反应器,而双介质反应器的输入功率远小于单介质反应器,与类似研究中的结果[23-24]一致。由于内管的屏蔽作用,在高压电极附近产生的高能电子只有一部分能够运动到外管附近的区域,从而产生更小的电流,以致相同电压下的输入功率更小,这与电流电压波形图(图2)中双介质反应器的微放电数量少于单介质反应器这一现象是一致的。
-
在不同的浓度和电压下,甲苯在2种反应器中的去除率见图5。甲苯浓度分别为616、1 027、1 848 mg·m−3,电压为14~24 kV时,双介质反应器中的甲苯去除率为9.4%~100%、7.4%~99%、5.1%~64%,单介质为67%~98%、46%~90%、26%~59%。随电压的升高和甲苯浓度的降低,甲苯去除率升高。电压升高,放电间隙电场强度增大,电子运动速度加快,使得反应器中的活性粒子数量增加、能量增大,与甲苯分子的碰撞概率增加,使得更多的甲苯被分解[25]。反应器入口浓度升高,甲苯去除率下降。在相同电压下,活性粒子数量和能量一定,浓度升高意味着更多的甲苯分子进入反应器,因此,每一个甲苯分子所能接触到的活性粒子数量减少,导致去除率下降[26]。可以看出,双介质反应器的去除率-电压曲线斜率高于单介质反应器,即双介质反应器的甲苯去除率随电压升高变化更快。电压为20 kV、浓度为616 mg·m−3时,双介质反应器对甲苯的去除率达到了94.51%。李云霞等[20]比较了单双介质反应器对CS2的去除效果,得到了相同的结论。这可能与2种反应器的气体击穿电压和放电间隙电场强度有关。同时,双介质反应器对甲苯去除率高于单介质反应器时的电压的结果并不是固定的,这一电压随甲苯浓度的升高而增加。甲苯在低温等离子体中降解后并非完全生成CO2和H2O,还会生成有机副产物和CO[27]。因此,还须通过矿化率和CO2选择性来评价反应器中的甲苯降解效果。图6和图7反映了不同条件下2种反应器中的甲苯矿化率和CO2选择性。2种反应器的甲苯矿化率均随电压的升高和甲苯浓度的降低而升高,说明高电压、低浓度更有利于甲苯转化为COx。当浓度和放电电压相同时,单介质反应器的矿化率高于双介质反应器,这是由单介质反应器的输入功率更大而导致的。当浓度为616 mg·m−3时,双介质反应器的矿化率为16.66%~40.22%,单介质的矿化率为20.21%~40.27%。
随甲苯浓度的升高,2个反应器中的CO2选择性均降低,说明浓度升高不仅导致矿化率降低,而且使CO2在COx中所占比率下降。在高浓度下,每个甲苯分子所能接触到的活性粒子数目减少,不仅导致其降解率下降,还会发生不充分氧化而生成CO。在单介质反应器中,CO2选择性随电压的升高而降低,在浓度为616、1 027、1 848 mg·m−3时,分别为52.11%~49.28%、50.23%~46.58%、45.75%~43.19%。这说明,随电压的升高,CO对矿化率的贡献逐渐增大。在双介质反应器中,CO2选择性随电压的升高,先降低后升高。这可能是因为电压小于20 kV时,甲苯的降解主要发生在内管中,随电压的升高,外管中有足够的场强促进甲苯的降解和CO的氧化。在相同条件下,双介质反应器的CO2选择性高于单介质。这是因为甲苯在内管中降解时产生的CO随气流进入外管后,可以进一步与臭氧或氧自由基反应生成CO2。而单介质反应器中只有一个放电区域,在各处发生的反应大致相同,不存在类似于双介质中的CO进一步氧化的过程。
-
在低温等离子体降解甲苯时,不可避免地会出现O3、N2O等产物[28]。图8和图9反映了2种反应器在不同条件下的O3浓度和N2O浓度。甲苯浓度越高,O3和N2O的浓度越低。在一定放电条件下,反应器中的活性粒子数目和能量一定,甲苯浓度较高时,甲苯分子与活性粒子碰撞的概率增大,更多的活性粒子参与甲苯的降解而非O3和N2O的形成。在双介质反应器中,O3及N2O的浓度均随电压升高而增大。电压升高产生更多的活性粒子,与O2和N2发生一系列反应,生成更多的O3和N2O。在单介质反应器中,随电压的升高,O3浓度先升高后降低。当外加电压过高时,单介质反应器中可能出现火花放电,系统温度升高,使部分臭氧分解[29]。不同于O3,N2O不会因高温而发生分解,其浓度随电压的升高而增大。在同一电压下,单介质反应器具有更高的输入功率,产生更多的活性粒子,因此,N2O浓度高于双介质反应器。双介质反应器的击穿电压更高,不存在单介质反应器中O3因高温而分解的情况,因此,电压超过20 kV后,双介质反应器中的O3浓度更高。相对于N2O,O3很容易被分解,因此,可以在反应器中加入合适的催化剂,来抑制O3的产生[30-31]。
-
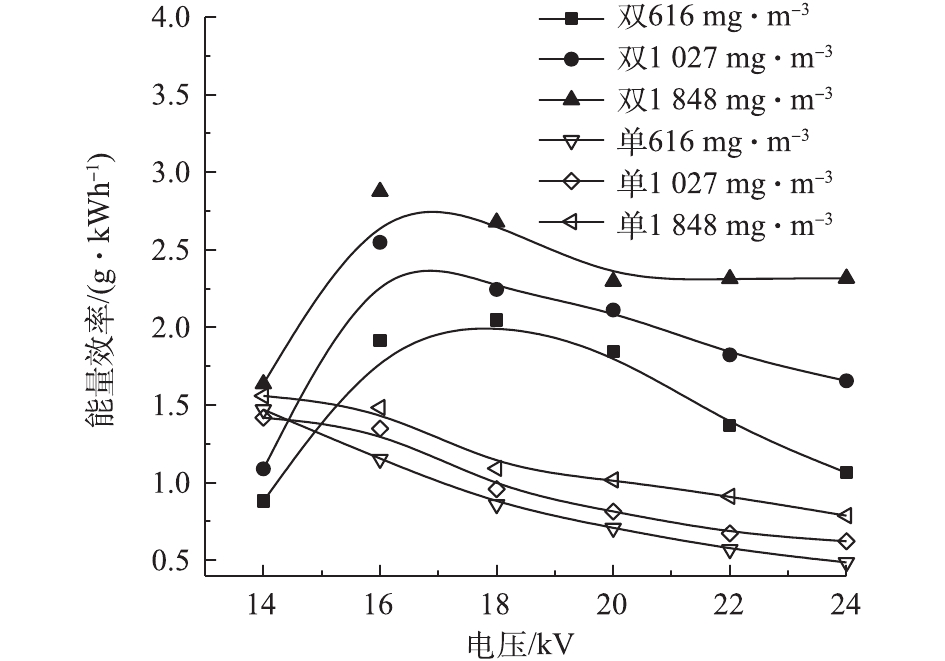
图10反映了2种反应器在不同条件下的能量效率。当甲苯浓度较高时,能量效率较高。虽然高浓度下甲苯的去除率较低,但系统的能量能够被更加有效地利用。随电压的升高,单介质反应器的能量效率逐渐降低[32],而双介质反应器则先升高后下降[33]。电压为14 kV时,双介质反应器对甲苯的去除率不到10%,只有很少的一部分甲苯被降解,导致能量效率较低。电压在16~18 kV时,双介质反应器有最佳的能量效率;电压超过16 kV时,双介质反应器的能量效率高于单介质反应器。这说明甲苯的去除率高并不代表其能量效率也高,因为在相同电压下,2个反应器的输入功率不同。
-
1)在相同电压下,双介质反应器具有更高的平均电场强度,单介质反应器输入功率更高。
2)电压升高、浓度降低,甲苯去除率增加。在浓度为616 mg·m−3,电压为14~20 kV时,单介质反应器对甲苯去除率高;电压为20~24 kV时,双介质反应器对甲苯去除率高。随甲苯浓度的变化,这一电压范围也会发生改变。
3)甲苯矿化率随电压的升高和浓度的降低而升高。在同一条件下,单介质反应器的矿化率高于双介质反应器。单介质反应器的CO2选择性随电压的升高而降低,双介质反应器的CO2选择性随电压先降低后升高。在相同条件下,双介质反应器的CO2选择性更高。
4)在双介质反应器中,O3和N2O浓度均随电压的升高和甲苯浓度的降低而升高。在单介质反应器中,O3浓度随电压的升高先升高后降低,N2O浓度随电压的升高而升高。电压低于20 kV时,单介质反应器中O3浓度较高。双介质反应器中的N2O浓度低于单介质反应器。
5)单介质反应器的能量效率随电压的升高而降低,双介质反应器在电压为16~18 kV时,具有最佳能量效率;电压大于16 kV时,双介质反应器的能量效率高于单介质反应器。
单介质和双介质阻挡放电低温等离子体降解甲苯的比较
Comparison of single and double dielectric barrier discharge non-thermal plasma for toluene removal
-
摘要: 为研究介质阻挡放电(DBD)反应器结构对低温等离子体降解甲苯的影响,设计了具有单层介质和双层介质的DBD反应器。对2种反应器的放电特征、甲苯去除率、矿化率、CO2选择性和能量效率进行了比较,并对施加电压和初始浓度对甲苯降解效果的影响进行了分析。结果表明:在相同电压下,双介质反应器(DDBD)具有更高的电场强度,而单介质反应器(SDBD)的输入功率更高;当甲苯浓度和电压分别为616、1 027、1 848 mg·m−3和14~24 kV时,双介质中的甲苯去除率为9.4%~100%、7.4%~99%、5.1%~64%,单介质为67%~98%、46%~90%、26%~59%。这说明低电压下单介质反应器的甲苯去除率更高,而高电压下则相反,并且,浓度降低、电压升高有利于甲苯的降解。单介质反应器的能量效率随电压升高而降低,双介质反应器则先升高后下降,且双介质反应器的能量效率高于单介质反应器(16~24 kV)。以上研究可为介质阻挡放电在VOCs去除方面的应用提供参考。Abstract: In order to investigate the effect of dielectric barrier discharge (DBD) reactor structure on toluene degradation in non-thermal plasma, reactors with single and double barrier were designed. The discharge characteristics, toluene removal efficiency, mineralization rate, CO2 selectivity and energy efficiency of the two rectors were compared, and the effects of voltage and initial concentration on toluene degradation were analyzed. The results indicated that DDBD reactor had higher electric field strength and SDBD reactor had higher power under the same applied voltage. The removal efficiency of toluene in the DDBD reactor ranged from 9.4%~100%, 7.4%~99%, 5.1%~64% and that in the SDBD reactor ranged from 67%~98%, 46%~90%, 26%~59%, when the initial toluene concentration and the applied voltage were 616, 1 027, 1 848 mg·m−3 and were 14~24 kV, respectively. It showed that SDBD has a higher toluene removal efficiency at low applied voltage, while the removal efficiency is higher in DDBD at a higher applied voltage. Moreover, the decrease of toluene concentration and the increase of applied voltage were favorable for toluene degradation. In SDBD, the energy efficiency decreased with the increase of applied voltage, while that of the DDBD reactor first increased and then decreased. The energy efficiency of the DDBD reactor was higher than that of the SDBD reactor at 16~24 kV. The present study could provide reference for the application of DBD in VOCs abatement.
-
Feammox是在厌氧的环境条件下以及微生物的催化作用下,能够实现以Fe(Ⅲ)为电子受体,NH4+为电子供体,发生类似于Anammox的氨氧化反应,且最终实现Fe(Ⅲ)还原和NH4+的氧化过程,也称之为厌氧铁氨氧化过程。Feammox作为一种新型的生物脱氮反应,其自然反应广泛存在于湿地、河流及湖泊底泥、稻田、旱地土壤等不同环境体系中[1-4]。Feammox反应过程中NH4+被氧化生成NO2−、NO3−和N2后,氮的损失可通过反硝化或Anammox进行[5],且其可直接产生N2的途径使得氮循环路径极大程度的缩短,并导致生态系统氮的损失[4]。此过程不但对陆地生态系统和水生生态系统具有重大意义,对整个自然界间氮的循环机制产生更深层次的认知和理解。
Feammox是由微生物驱使的生物脱氮过程,Feammox菌是Feammox反应的驱动者。Huang等[5]在2015年首次分离出了能够同时发生铵氧化和铁还原的嗜酸微生物科细菌A6(Acidimicrobiaceae bacterium A6)。Huang等[6]研究发现,A6能够以无机碳(CO2)作为碳源将NH4+氧化的同时还原Fe(Ⅲ),对NH4+的去除率可达52%。A6对重金属也具有一定的耐受性,Gilson等[7]研究发现在铀(U)污染的湿地沉积物中依然存在A6,并且其在厌氧状态下能将U(VI)作为电子受体还原和氧化NH4+。相比于氨氧化细菌,Feammox菌在厌氧条件下反应,以Fe(Ⅲ)替代氧作为电子受体,因此不需供氧且产生较少的N2O[8]。根据Feammox的脱氮特点,相比于传统的硝化反硝化脱氮,Feammox工艺应用于污水中可实现自养脱氮,不需要外加碳源和曝气,减少污泥量的生成,对解决低碳氮比(C/N)废水反硝化过程碳源不足问题,以及含重金属废水的处理具有一定的可行性和理论依据。
本文综述了Feammox反应机理以及Fe(Ⅲ)还原过程中电子传递机制,并探讨Feammox对自然界中氮损失的贡献程度及对生态环境的影响,最后分析了Feammox在污水脱氮领域的探索和应用,对其未来发展趋势进行了总结和归纳。
1. 铁氨氧化研究进展(Research progress of Feammox)
1.1 铁氨氧化发展历程
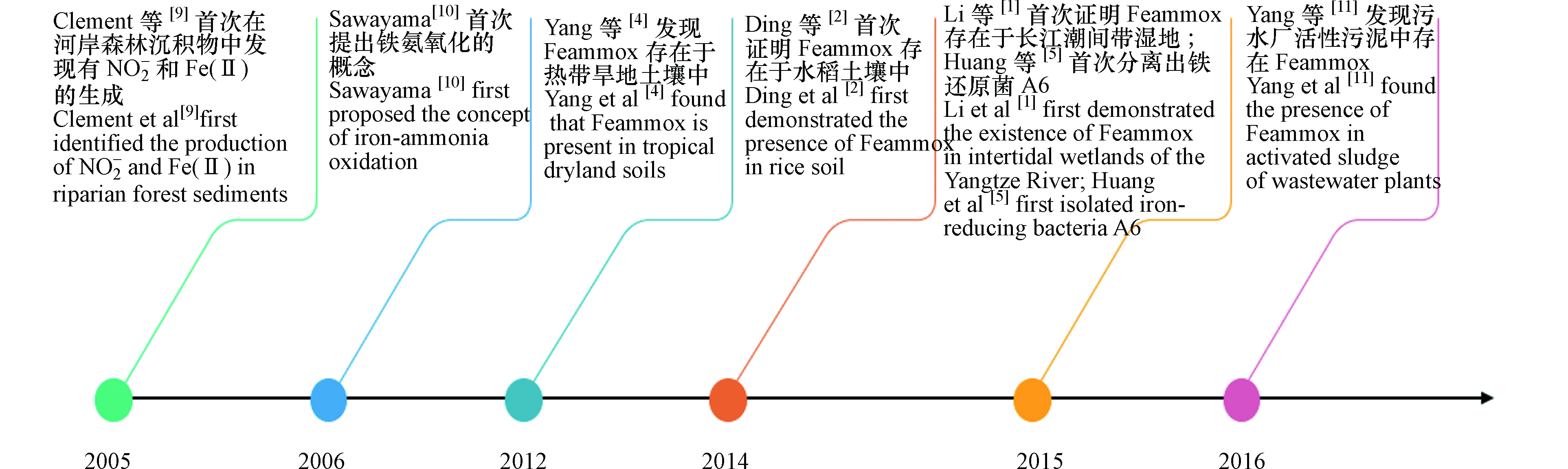
Feammox作为新发现的氮素转化机制,近几年来对其不断深入研究取得较多进展,Feammox主要研究历史节点如图1。Feammox反应最早发现在2005年,Clément等[9]在探索河岸森林沉积物中铁和氮循环的动力学时,发现在厌氧条件下,测定的沉积物样品中有NO2−和Fe(Ⅱ)的意外产生。因此猜测有一生物过程使用Fe(Ⅲ)作为电子受体,同时氧化NH4+生成NO2−,Fe(Ⅲ)被还原为Fe(Ⅱ)。在2006年,Sawayama通过实验研究,在厌氧的条件下含有NH4+、乙二胺四乙酸铁钠(EDTA-Fe)和碳酸氢钠(NaHCO3)的固定床反应器流出物中有NO2−生成,且流出的NO2−浓度与添加的NaHCO3和EDTA-Fe的浓度呈正比,证明铁还原微生物以NaHCO3为无机碳源将NH4+转化为NO2−,Fe(Ⅲ)还原为Fe(Ⅱ),并将此反应命名为Feammox过程[10]。2012年,Yang等[4]使用同位素标记的铵和Fe(Ⅲ)培养热带旱地土壤泥浆,研究发现Feammox在热带旱地土壤中能产生N2、NO2−或NO3−,且Feammox的主要途径是产生N2。2014年,Ding等[2]使用15N标记的铵基同位素示踪和乙炔抑制技术,首次证明Feammox存在于水稻土壤中,且Feammox是造成水稻土壤中氮损失的一个重要途径。2015年,Li等[1]在长江潮间带湿地沉积物中检测到Feammox,其潜在速率为0.24—0.36 µg·g−1·d−1 。2017年,Yang等[11]对污水厂产生的污泥进行厌氧消化,通过添加磁铁矿、Fe2O3和Fe(OH)3的3种不同类型的Fe(Ⅲ)源可诱导Feammox发生,其中Fe(OH)3效果最佳且对总氮的去除率达到了20.1%。添加三价铁化合物使得活性污泥中含有的铁能够改善和强化污泥的絮体结构[12],不仅可减少污泥体积和增加甲烷产量也可诱导Feammox产生去除污泥中的铵,减轻活性污泥对环境的危害[11],给水厂污泥固体废弃物的处置提供了较好的方法和新颖的思路。
1.2 铁氨氧化反应原理
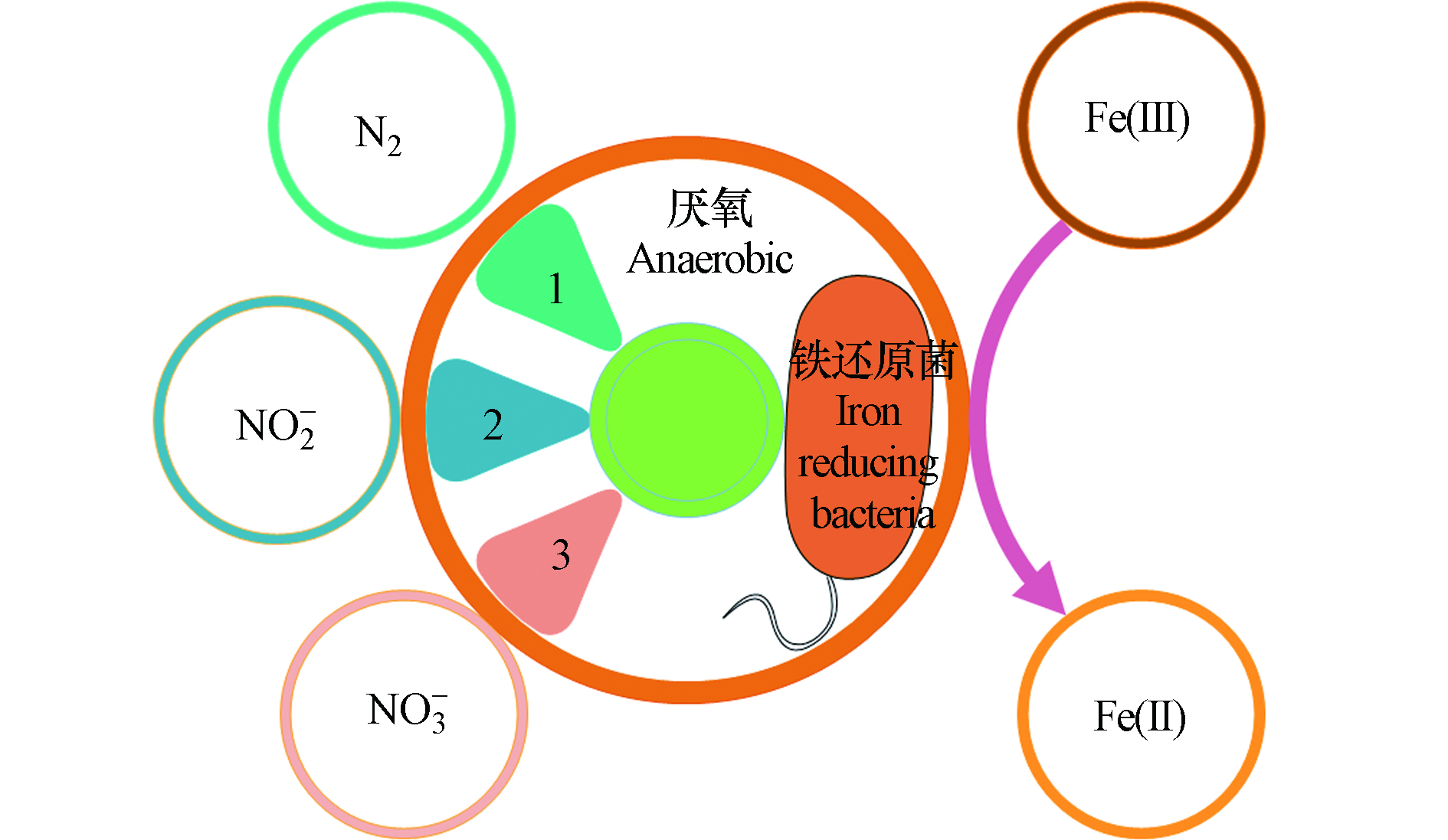
Feammox反应是在厌氧条件下,以及铁还原菌的驱使作用下,以三价铁为最终电子受体,氨氮为电子供体发生的生化反应,其反应产物有N2、NO2−、NO3−的3种不同形态的氮素(图2)。
Feammox反应机制极为复杂,致使Feammox产物氮素形态差异的原因尚不明确[13-14]。不同形态下的三价铁化合物作为电子供体Feammox的反应方程式及吉布斯自由能(ΔG)如表1。
表 1 Feammox反应方程式及吉布斯自由能Table 1. Feammox reaction equation and Gibbs free energy电子受体Electron acceptor 化学反应方程式Chemical reaction equation 吉布斯自由能ΔG/( kJ·mol−1 )Gibbs free energy 参考文献Reference FeOOH 6FeOOH+10H++NH4+—6Fe2++8H++NO2− −30.9 [9] 3Fe(OH)3+5H++NH4+—3Fe2++9H2O+0.5N2 −245 [4] Fe(OH)3 6Fe(OH)3+10H++NH4+—6Fe2++16H2O+NO2− −164 8Fe(OH)3+14H++NH4+—8Fe2++ 21H2O+ NO3− −207 Fe2O3·0.5H2O 3Fe2O3·0.5H2O+10H++NH4+—6Fe2++8.5H2O+NO2− ≤−145.08 [5] | Show Table DownLoad:
CSV
DownLoad:
CSV
不同形态的三价铁化合物作为电子受体,以及产物的不同其ΔG均有所差异。3种不同形态的铁化合物的ΔG均小于零,其反应均可自发进行。从热力学角度分析,电子受体为Fe(OH)3时,通过测定生成物为N2的反应ΔG为−245 kJ·mol−1 ,小于生成物为NO2−和NO3−的ΔG。另外生成N2的反应在相同条件下氧化NH4+所需Fe(Ⅲ)的量更少且产生能量更多,反应进程中消耗的铁氮比(Fe/N)为3∶1,而生成物为NO2−和NO3−的反应所消耗的Fe/N值分别为6∶1和8∶1。该反应又能够适应于较宽范围的pH,而NO2−和NO3−的生成要求pH值小于6.5,因此相比之下N2更易生成[4,15]。Yang等[4]研究证明了旱地土壤中N2的产生是Feammox主要反应途径。而Shrestha等[16]研究发现在没有任何NO2−或NO3−的湿地土壤中,并在厌氧和铁还原条件下,NH4+被氧化成了NO2−。在生成物为NO2−时,Fe(OH)3作为电子供体更易促进反应的进行,其反应ΔG为−164 kJ·mol−1,而FeOOH和Fe2O3·0.5H2O作为电子供体的Feammox反应ΔG为−30.90 kJ·mol−1和小于−145.08 kJ·mol−1,这也可为Feammox研究电子受体材料的选取提供一定理论依据。
1.3 铁氨氧化电子传递机理
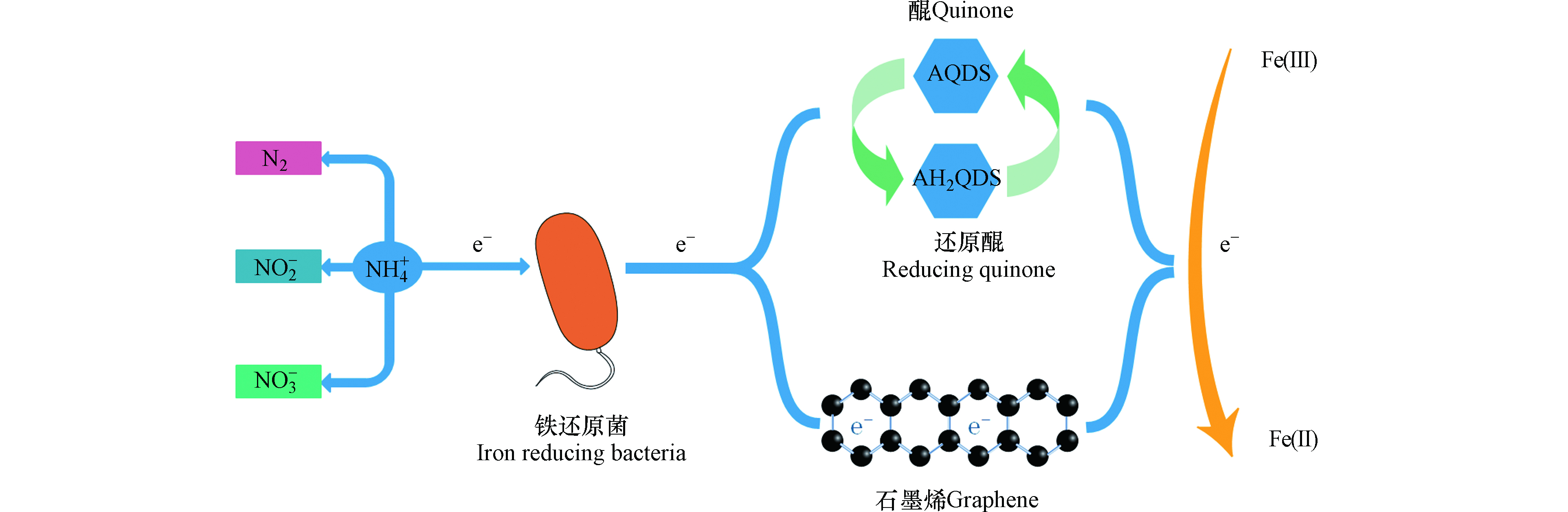
电子穿梭体是一种可溶性的小分子有机物或无机物,可通过自身的氧化还原作用加速微生物向细胞外部进行电子传递,间接电子传递和直接电子传递是胞外电子传递的两种主要方式,而穿梭体介导的电子传递属于间接电子传递[17-19]。电子穿梭体的来源可分为内生电子穿梭体(如黄素类化合物、黑色素、吩嗪化合物、醌类物质等)和外生电子穿梭体(如腐殖质、蒽醌、萘醌、半胱氨酸、硫单质、维生素等)[20-23],其中微生物自身能分泌的物质称为内生电子穿梭体,而能利用天然存在或人工合成的某些物质为外生电子穿梭体[24]。目前研究发现铁还原菌能够利用蒽醌、腐殖质和生物炭等作为电子穿梭体以其为电子载体向Fe(Ⅲ)输送电子(图3)[6,15,24]。Yang等[25]也通过实验证明AQDS和AH2QDS可作为铵与Fe2O3之间的电子传递体,能够成功地促进NH4+的氧化提升脱氮效率,并通过对比发现,添加AQDS的脱氮效率达到82.6%,而未添加AQDS的脱氮效率仅为64.3%。廖宏燕等[26]对螯合铁介导的Feammox脱氮效果进行了研究,研究发现腐殖酸铁介导的反应脱氮效果较好,总氮和氨氮去除率达到83.32%和83.68%,并推测N2是螯合铁介导Feammox反应的主要产物。腐殖酸的电子穿梭作用关键在于其所含有的醌基基团,而醌基基团可接受还原腐殖质的电子,并且研究发现腐殖质中醌基自由基的含量越高其电子接受能力也越高[27-28]。
通过固体导电介质的电子传导与电子穿梭体的分子扩散有一定的区别,固体材料的电子传导响应于电压差促使电子迁移,而水溶性的电子穿梭体在于浓度梯度产生的扩散[29]。生物炭作为一种固态材料,形态上虽区别于其它的水溶性电子穿梭体,其仍可以起到电子穿梭体的作用。Zhou等[30]研究了不同粒径的生物炭对异化铁还原菌(DIRB)和产甲烷菌的影响,发现在水铁矿富集物中加入生物炭后,DIRB菌和产甲烷菌均得到了富集,尺寸较小的粉末状生物炭对Fe(Ⅲ)还原效果更好。由于生物炭具有的芳香结构和氧化还原活性的醌类化合物有助于微生物细胞和不溶性矿物质之间的胞外电子转移,因此促进了Fe(Ⅲ)的还原效率[30-31]。另外Song等[32]研究发现活性炭表面的氧化还原活性含氧官能团对微生物还原Fe(Ⅲ)有促进作用,通过实验证明,经由HNO3改性处理的活性炭增加了表面官能团醌与氢醌的数量,并在兼性细菌MR-1的作用下Fe(Ⅲ)的还原率将近100%,与原始活性炭相比,Fe(Ⅲ)在还原率和还原程度上都有极大提高,活性炭具有的良好的氧化还原特性,使得其电子接受和电子供给能力发挥了极大作用。但是通过生物炭的电子传输不全归因于醌部分,Chen等[29]认为通过生物炭的电子传递也归因于生物炭自身材料的导电特性,并且发现附着在生物炭上的金属还原菌和巴氏杆菌可通过生物炭进行两个物种之间的电子转移,而不是通过细胞间的电子转移。显然生物炭能成为电子传递的通道,使得电子不通过细胞间的接触进行传递。另外Guan等[33]在红树林沉积物中加入石墨烯和9,10-蒽醌-2-磺酸酯(AQS),使得Feammox速率分别提高了31%和56%,但石墨烯参与的Feammox的Fe(Ⅲ)还原仅占总Fe(Ⅲ)还原的1.5%—4.9%,可能石墨烯具有的导电性能促使了部分电子转移给了Fe(Ⅲ),使得Fe(Ⅲ)得到还原。
石墨烯和生物炭作为固性材料均具有导电性能,但石墨烯不具有氢醌结构又能够促进Fe(Ⅲ)还原和Feammox反应速率,这也表明石墨烯和生物炭一样能够起到电子传递的桥梁作用,并与其它水溶性电子穿梭体的传递机制显著不同。目前对电子穿梭机制并没有完全的认知和了解,但随着对电子穿梭体传递电子机制的不断探究对Feammox的反应机理研究有极大帮助。
1.4 铁氨氧化参与的氮损失
在土壤环境体系中最常见的氮去除是有机氮矿化,其次是硝化作用,然后是反硝化作用[34]。但Feammox在陆地生态系统中的显著活动,造成了土壤中一定量的氮损失(表2),大量的NH4+被直接或间接氧化生成N2,也在一定程度下缓轻了陆地生态系统生境破坏和水生生态系统中水体富营养化的现象[35]。
表 2 铁氨氧化在自然界产生的氮损失Table 2. Nitrogen loss in nature due to Feammox样品Sample 来源地点Source location 铁氨氧化速率/(µg·g−1·d−1)Feammox rate 氮损失/(kg·ha-1·a-1)Nitrogen loss pH 文献Reference 热带旱地土壤 美国卢基洛山脉 0.19—0.45 — 4.25—6.15 [4] 丘陵水稻土壤 中国江西省金县(28°10'—28°45'N,116°1'—116°34'E) 0.17—0.59 7.8—61(占国内常规施氮肥3.9%—31%) 4.69—5.72 [2] 水稻土壤 江苏省金坛市(31°39'49.42”N , 119°28'4.12”E) 0.047—0.319 3.64(施肥土壤),15.97—24.91(不施肥土壤),(占国内常规施氮肥7.99%—12.45%) 6.25—6.76 [12] 潮间带湿地 中国上海崇明(31°30'N,121°59'E) 0.24—0.36 115—180(占长江湿地无机氮的3.1%—4.9%) 春潮(8.32—8.46),小潮(8.48—8.75) [1] 太湖底泥 太湖梅梁湾(31°31'39— 31°32' 30 N,120°10' 26—120°11' 70 E) 0.29(无藻区底泥),0.01—0.05(聚藻区底泥) — 7.26—7.43 [3] 河岸带土壤 太湖贡湖湾(31°27'32—31°28'01N, 120°19'27—120°21'18E) 0.25—0.29 — 6.87—7.42 [13] 菜地土壤 太湖流域宛山荡(31°34'32—31°36'30N, 120°30'12—120°32'24E) 0.06—0.23 — (占产生氮气的7.3%—12.4%) 4.50—5.21 [44] 农田土壤 太湖万山地区(31°35′15—31°35′27 N,120°28′43—120°30′32 E) 0.12—0.18 17.8(占总氮损失的4.2%) 7.23—7.43 [36] | Show TableDownLoad:
CSV
不同类型的土壤中Feammox速率和对氮损失的贡献占比略有差异,这与土壤的pH、总有机碳(TOC)、土壤Fe(Ⅲ)含量、土壤水分和铁还原微生物的丰度差异等诸多因素有关[2,5,36-37]。另外,Feammox速率也受季节和空间的影响。Ding等[38]对太湖流域万山地区的农田土壤、河岸土壤和河流沉积物在夏冬两季的Feammox速率以及铁还原菌的多样性和丰度进行了研究,结果发现,在夏季Feammox速率为0.05—0.19 µg·g−1·d−1大于冬季的0.02—0.09 µg·g−1·d−1,农田土壤中的Feammox速率最大,其后依次为河岸土壤和河流沉积物,土壤水分、Fe(Ⅲ)含量和TOC是影响Feammox速率和铁还原菌多样性的重要因素。而在水稻土壤中,由于其富含有Fe(Ⅲ)和高浓度的NH4+的特征,以及在水淹没状态下产生的厌氧环境,可能为Feammox发生过程及其相关的微生物提供了良好条件[39]。Li等[39]利用同15N同位素示踪技术证实了Feammox在我国南方水稻土中的存在,发现水稻土壤中约6.91%的施用氮肥的流失是由Feammox造成的,并对Feammox相关的微生物群落进行了研究,地杆菌属、GOUTA 19、 Nitrososphaeraceae和假单胞菌属为Feammox发生的潜在驱动力,而pH、土壤粒径、NH4+、C/N和TN是影响这些微生物组成的重要因素。Qin等[40]对位于太湖沿岸不同深度之间的水稻土的Feammox速率进行了研究,结果发现,Feammox在稻田深度为20—30 cm的区域活性最强,其速率为0.313—0.427 µg·g−1·d−1,在水稻表层土壤0—10 cm处,94.72%—96.89%的氮素流失是由反硝化造成的,反硝化作用是造成水稻表层土壤氮损失的最主要原因,而10 cm以下深层土壤的氮损失则由Feammox主导。Yi等[41]研究了施肥对水稻土中Feammox的影响,研究表明施肥可以丰富土壤中Fe(Ⅲ)和有机碳的含量,进而加速电子传递过程,刺激Feammox反应进程,并有助于微生物群落的转移,改变水稻土中微生物的氮转化过程。施肥能够导致反应底物NH4+和 NO3−含量升高,改变土壤理化性质,进而影响土壤微生物群落结构和功能微生物数量,尤其是有机肥对土壤中Feammox菌(A6)数量提升具有促进作用[42-43]。丁帮璟等[44]通过测定Feammox在菜地土壤中的反应速率,Feammox脱氮贡献占比为7.3%—12.4%,且Feammox速率与土壤Fe(Ⅲ)和TOC含量呈正相关性,约85%的N2生成是由反硝化和Anammox所贡献。在各项研究中可知,反硝化在土壤氮损失中起到最主要作用,而Feammox和Anammox等其它过程对土壤氮损失也占有一定比例,在土壤中Feammox和Anammox以及反硝化在各自反应过程中可能存在一定的联系。Ding等[36]研究发现在太湖河口地区Feammox、Anammox和反硝化分别约占总氮损失的3.5%—4.2%、2.8%—3.9%和92.6%—93.1%,并且发现Feammox速率和反硝化速率之间存在显著相关性,两者之间可能存在协同作用,但Feammox和Anammox之间并未发现具有明显的相关性。Feammox和Anammox可能会发生耦合作用,但是两者对NH4+的利用也会存在竞争[1,36],Feammox和反硝化存在关联可能是因为Feammox还原生成的Fe(Ⅱ)供给了反硝化利用[45],或者Feammox产生的NO3− 或NO2−被反硝化利用促进N2产生。
Feammox的发现和相关研究目前多在于陆地生态系统,其在海洋生态系统中的相关研究和报道较于陆地生态系统相对较少。海洋生态系统中的氮循环除了固氮和反硝化两大过程能够改变氮形态并起到关键的转化过程之外,氨化、硝化、Anammox和异化硝酸盐还原为铵(DNRA)等过程也是海洋生态系统氮循环和造成氮损失的重要途径[46]。此外,Toro等[47]在海洋沉积物中发现Anammox与依赖硫化物的反硝化之间的耦合作用,可同时去除与NO2−还原相关的铵和硫化物,这种耦合作用能力也可能会造成海洋中的氮损失。Emilia等[48]研究发现海洋沉积物中普遍存在依赖硫酸盐的厌氧氨氧化(Sulfammox)和Feammox反应过程,而沉积物中的Feammox速率达到2 µg·g−1·d−1N。这也表明了在极其复杂的海洋氮循环中,Feammox过程是参与氮循环和氮损失不可忽视的组成部分。Laufer等[49]在沿海海岸沉积物中研究发现存在自养的硝酸盐还原Fe(Ⅱ)氧化细菌,这种依赖于Fe(Ⅱ)的反硝化作用同样可能成为造成海洋氮损失的重要途径。另外,依赖型Fe(Ⅱ)反硝化的Fe(Ⅱ)源也可能来源于Feammox反应过程中还原产生的Fe(Ⅱ),在陆地土壤和海洋沉积物中,这两种反应极都有可能存在耦合作用。
不管Feammox存在于何种环境体系,其参与的氮转换途径已经成为计算全球氮损失不可缺少的一部分计算过程,并占有重要比例。Feammox过程的出现会改变以往反硝化和厌氧氨氧化等其它脱氮过程对自然界中氮去除的贡献格局,从而形成新的脱氮贡献格局,对氮污染的去除具有重大意义[50]。在实际环境体系中的复杂氮循环过程中,Feammox和其它各种复杂的反应过程之间存在怎样的关联,又如何通过相互之间的作用影响氮的损失,则需要进一步的探索和研究。
2. 铁氨氧化在污水脱氮中的应用探索(Application of feammox in wastewater denitrification)
2.1 铁氨氧化耦合脱氮理论基础
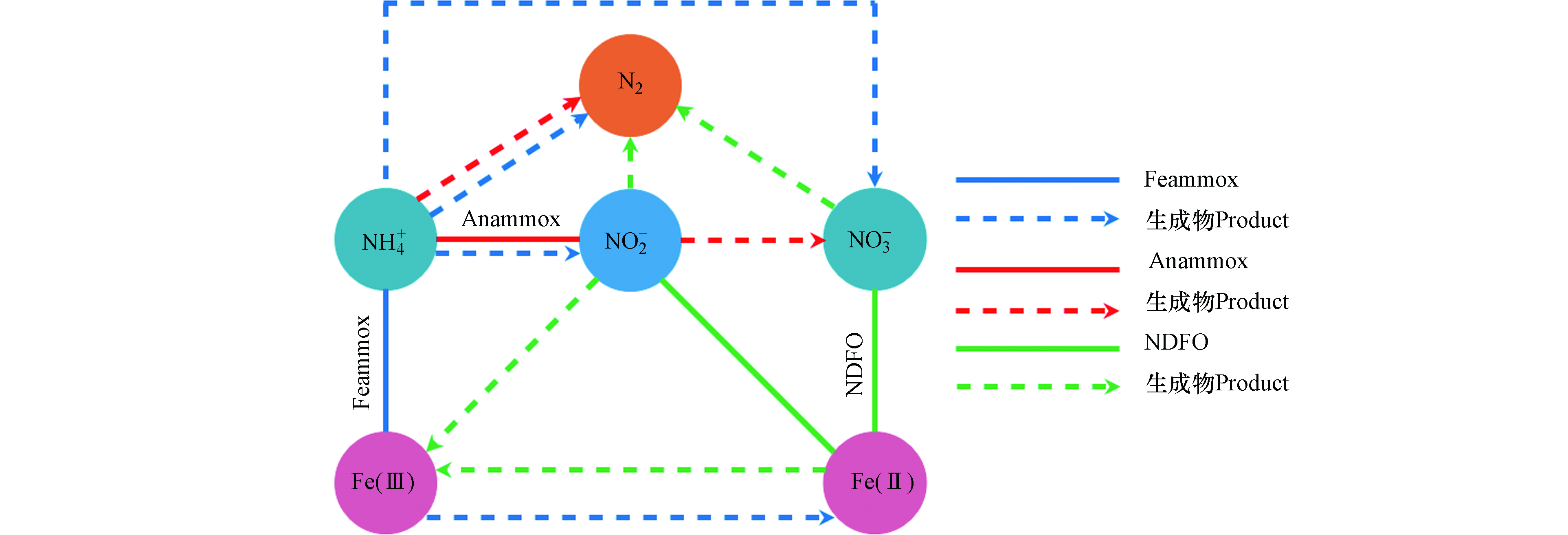
在污水处理中,氮素不同形态之间的转换是一个较为复杂的过程,微生物驱使作用下的污水生物脱氮主要包含有氨化、同化、硝化、反硝化、完全氨氧化(Comammox)、Anammox、Sulfammox、Feammox、亚硝酸盐/硝酸盐型厌氧甲烷氧化(n-DAMO)和DNRA等反应过程[51]。在污水生物脱氮过程中,Feammox反应生成的NO2−会被Anammox菌利用与NH4+反应生成N2,Anammox会伴随着Feammox反应进行而发生耦合作用(图4)。Li等[52]对Anammox与Feammox耦合的可行性及特点进行了研究,在含有Anammox的活性污泥的反应器中,以Fe(Ⅲ)作为电子受体进行厌氧反应,在反应器流出物中检测出有NO2−、NO3−和Fe(Ⅱ)的生成,且污泥中保留有Anammox菌,表明在Anammox污泥中存在Fe(Ⅲ)还原和NH4+氧化反应。另外,Park等[53]以养猪废水处理厂的厌氧污泥作为培养物的反应器中发现NH4+的氧化去除不仅促进了Fe(Ⅲ)的还原,而且有助于Anammox反应。刘恒蔚等[54]利用微生物技术对Anammox反应器和接种了Anammox污泥的Feammox反应器中的功能微生物进行了对比分析,发现Anammox菌均占主导地位,但Feammox反应器中微生物群落结构与Anammox反应器相比具明显的差异性。以上研究表明,以接种Anammox污泥启动的Feammox反应系统中,存在以NO2−或Fe(Ⅲ)为电子受体的氨氧化脱氮过程[55],但当NO2−与Fe(Ⅲ)同时作为电子受体反应时,Anammox优先于Feammox反应[56],其中Anammox脱氮占有重要比例,这也是Feammox与Anammox能够进行耦合反应的重要前提基础。另外值得注意的是,在反应系统中除了Feammox反应能够将Fe(Ⅲ)还原为Fe(Ⅱ)外,Anammox菌也具有还原Fe(Ⅲ)的能力,有研究报道称,Anammox菌能以有机物作为电子供体将Fe(Ⅲ)还原为Fe(Ⅱ),且其还原Fe(Ⅲ)的活性受pH、电子供体和受体种类的影响[57]。Zhao等[57]研究发现以甲酸盐和Fe(Ⅲ)-NTA分别作为电子受体和供体,在pH 7的条件下,Anammox菌具有较高的铁还原活性。在实际反应过程中,系统中会存在一定浓度的Fe(Ⅲ)和Fe(Ⅱ),有关研究表明,适当的Fe(Ⅱ)浓度能够促进Anammox菌生长速率,显著提高Anammox活性[58]。Fe(Ⅱ)能够通过提高Anammox菌细胞内的总铁、血红素C和联氨脱氢酶活性,从而更好地刺激Anammox菌生长,不仅对其丰度有促进作用,还能够提升Anammox脱氮效率和诱导更多基因表达[59-62],并根据这一特点,有些研究者便通过投加适量的Fe(Ⅱ)来缩短Anammox反应器的启动时间[60]。同样研究发现,适量的Fe(Ⅲ)也能够提高Anammox菌16S rRNA及功能基因hzsB的丰度,并提升对氮的去除率[63],但过高的Fe(Ⅱ)和Fe(Ⅲ)浓度则均会抑制Anammox菌活性。
值得思考和研究的问题是,在Anammox与Feammox耦合反应过程中,NH4+作为电子供体,反应进程中是否对NH4+存在显著竞争,系统中存在的Fe(Ⅲ)和Feammox还原生成的Fe(Ⅱ),以及在合适条件下Anammox菌自身拥有的Fe(Ⅲ)还原能力所还原生成的Fe(Ⅱ),是否会对Anammox菌起到促进作用,以致使其更好的利用Feammox反应产生的NO2−,并促进Feammox和Anammox的耦合反应,这些可能受Fe(Ⅲ)和Fe(Ⅱ)形态的存留时间及相应浓度,系统反应条件等综合因素的影响。
Feammox和铁型反硝化(NDFO)也存在耦合反应(图4),在Feammox反应进程中通过氧化NH4+而还原生成的Fe(Ⅱ)可为亚铁氧化细菌还原NO3−提供电子供体,氧化生成的Fe(Ⅲ)可再次被铁还原菌利用[8,64-65]。Li等[66]将Feammox污泥接种在含有Fe(Ⅲ)、NH4+和NO3−的反应器中,通过同位素和微生物群落分析,证明了反应器中存在Feammox、Anammox和NDFO过程,在运行62d后发现,67.6%的NH4+和58.8%的NO3−能够同时进行转化去除,并且成功实现了Fe(Ⅲ)和Fe(Ⅱ)的循环转换。吴悦溪等[56]利用二沉池普通活性污泥培养Feammox,并分析了Feammox反应系统中存在的脱氮途径及其对NH4+的去除的贡献程度,结果发现,反应系统中是由Feammox主导并存在与Anammox和NDFO的耦合作用,57.7%的NH4+是由Feammox反应去除,而Anammox对NH4+的去除占42.3%。Feammox和NDFO的耦合反应,理论上可实现Fe(Ⅲ)和Fe(Ⅱ)的再生循环转换,但在实际反应中除了受各种复杂影响因素之外,值得注意的是Feammox的产物并不全是NOx−。因此,NDFO生成的Fe(Ⅲ)的量具有一定的局限性,Fe(Ⅲ)和Fe(Ⅱ)并不以等量的方式进行循环再生,需要补充额外的NO3−来氧化Fe(Ⅱ)产生Fe(Ⅲ)[67]。Yang等[67]便通过添加NaNO3提供NO3−,利用Feammox和NDFO的耦合作用触发Fe(Ⅲ)和Fe(Ⅱ)循环,进而去除NH4+。结果发现总氮的去除率达到了90.1%,而未提供NO3−的对照组总氮去除率仅为41.6%。NO3−的加入极大提升了脱氮效率,且NO3− 还原产生的NO2−可以进一步氧化Fe(Ⅱ)。但较高的NO2−浓度可能会对NO3−的还原起到抑制作用[68],由于Fe(Ⅱ)会被NO2−氧化生成Fe的氢氧化物,这种Fe的氢氧化物会附着在细胞表面造成细胞结壳,使得细胞的代谢和活性受到抑制而导致NO3−无法还原[69]。另外,有关研究者发现当使用螯合铁Fe(Ⅱ)-EDTA时,Fe(Ⅱ)的氧化则是由微生物催化,而不会被NO2−氧化致使细胞产生结壳现象[70]。Zhou等[68]利用可将硝酸盐还原和Fe(Ⅱ)氧化的NDFO菌株W5接种到海绵铁填充的生物滤池中,当进水NO2−浓度为10 mg·L−1时,NO3−的去除率达到最低为50.35%。另外以FeSO4作为Fe(Ⅱ)源,其浓度从800 mg·L−1增加到1500 mg·L−1时,细胞结壳严重NO3−去除率降低,而以Fe(Ⅱ)-EDTA做为Fe(Ⅱ)源且浓度为1100 mg·L−1时,脱氮效率可达到90%。NO3−和NO2−均能够将Fe(Ⅱ)氧化生成Fe(Ⅲ),除了通过补充NO3−之外,也可直接补充NO2−,这也是直接弥补系统中Fe(Ⅲ)量不足的有效方式,也避免通过直接投加Fe(Ⅲ)造成铁泥污染现象的发生。Yang等[71]便通过向Feammox反应系统间歇的添加NO2−,NO2−能够氧化Fe(Ⅱ),Fe(Ⅱ)和Fe(Ⅲ)的浓度在NO2−的添加下分别上升和减少,并随着NO2−的消耗以及Feammox的作用下分别下降和上升。NO2−的添加诱导了Fe(Ⅲ)和Fe(Ⅱ)之间的循环转换,并在Feammox的作用下,NH4+的去除率达到96%,且反应过程没有检测到Anammox菌,系统中没有发生Anammox反应。
Feammox和NDFO的耦合作用机制是触发系统中Fe(Ⅲ)和Fe(Ⅱ)循环的有利前提,也是通过利用Fe(Ⅲ)和Fe(Ⅱ)之间的循环转换而去除氮的主要启示来源,利用外来投加适量的NO3−和NO2−作为末端电子受体,保持系统中Fe(Ⅲ)和Fe(Ⅱ)循环平衡进而脱氮,其所具有的可行性使得Feammox运用于污水脱氮具有一定的理论和实践基础。Feammox与Anammox和NDFO的耦合作用能够强化脱氮过程,提升脱氮效率,但其作用机制及影响因素较为复杂,因此分析耦合作用影响因素,研究多变量因素之间的协同作用,确定最佳可控反应条件,以及深入研究各反应对氮去除的贡献程度,可为未来实际污水脱氮应用提供重要理论依据并具有重大实际意义。
2.2 铁氨氧化在污水脱氮中的探索研究
目前,Feammox在污水脱氮中的应用仍处于初级探索阶段[72],利用Feammox处理实际废水的研究比较少,并且Feammox反应主要是NH4+和Fe(Ⅲ)之间发生的反应,因此对模拟废水中的NH4+去除研究较多。目前Feammox反应启动及对氮去除的主要技术手段有:(1)向反应器添加从土壤中富集培养出的Feammox菌液;(2)利用Anammox污泥培养驯化出Feammox污泥来启动Feammox反应;(3)利用微生物固定化技术将Feammox菌固定化。实验选取的反应器类型主要有ASBR、MBR和血清瓶等装置(表3)。
表 3 不同反应装置和接种污泥下的氨氮转化效果Table 3. Transformation effect of NH4+-N under different reactor and inoculated sludge反应器类型Reactor type 废水类型Wastewater type 启动方式Start mode Fe(Ⅲ) Fe(Ⅲ)/(mg·L−1) pH 转化率/%Removal rate 文献Reference ASBR 模拟废水 接种二沉池污泥 FeCl3 27—33 7.4—7.6 53.8 [56] ASBR 模拟废水 接种Anammox污泥 FeCl3 12—24 7.4—7.6 52.73 [73] MBR 模拟废水 添加Feammox菌液 Fe(OH)3 573.2 4.5—5 41.49 [74] 生活污水 添加Feammox菌液 — 0.43—0.49 7.32 40.05 血清瓶 小榄江水 添加Feammox菌液 — 0.06—0.08 7.12 12.90 [75] 硫铁矿污水 添加Feammox菌液 — 13.43—13.55 4.5 44.64 | Show TableDownLoad:
CSV
李海晖等[75]对实际污水进行了研究,通过向城市生活污水、和矿山废水中添加使用NH4+和Fe(Ⅲ)富集培养出的Feammox菌液,来探讨Feammox对实际废水中NH4+的去除效果。结果发现,经过10d的反应,城市生活污水中的NH4+浓度由初始203.94 mg·L−1降低至122.26 mg·L−1,去除率为40.05%,矿山废水中的NH4+去除率达到了44.64%。表明Feammox对实际废水中的NH4+去除起到了良好的效果。吴胤等[74]通过配置不同NH4+浓度的模拟废水来探讨对Feammox反应所产生的影响,向NH4+浓度分别为75 mg·L−1、150 mg·L−1和400 mg·L−1的模拟废水中加入Feammox菌液并进行厌氧反应。结果发现,反应15d后,初始NH4+浓度为75 mg·L−1和150 mg·L−1的反应器中,NH4+去除率分别为41.49%和7.45%,而在NH4+浓度为400 mg·L−1的模拟废水中Feammox反应受到抑制,表明在低氨氮废水中Feammox能起到较好的反应。姚海楠等[73]考虑到Feammox菌对重金属具有耐受性,便利用处理垃圾渗滤液的Anammox污泥作为接种污泥启动Feammox反应,并同样探究了不同NH4+浓度对Feammox反应的影响。当进水NH4+浓度为400 mg·L−1时,经过12 d反应,NH4+的转换量为40.69 mg·L−1,也同样证明了Feammox反应在高氨氮废水中对NH4+的去除率小于在低氨氮废水中,另外还发现反应器中的NO3−的生成量与NH4+的转化量差异不大,反应器中的NH4+主要转化为了NO3−。Feammox菌对重金属具有较强的耐受性,能对重金属起到固定效应并可降低其在污水中的毒性[73-74],这也可能是Feammox能够在NH4+浓度为400 mg·L−1和Fe(Ⅲ)浓度为500 mg·L−1的高浓度废水中仍能发生反应的重要原因[73]。此外,Feammox处理垃圾渗滤液过程也受pH的影响,Wang等[76]利用Feammox处理垃圾渗滤液的过程中研究发现,在pH 4.5时,垃圾渗滤液的反硝化效果最好,而在pH 3.5时的酸性环境中Feammox菌大量死亡。大量研究表明Feammox反应偏向于酸性环境,但过酸环境会抑制其反应,并导致微生物死亡。刘志文等[77]利用微生物固定化技术,使用磁性壳聚糖凝胶球(MCHBs)将Feammox菌固定化,并探讨其与游离菌液对NH4+的去除效果及差异。结果发现,在进水NH4+ 浓度为60 mg·L−1运行16 d后,粒径为1—2 mm的MCHBs 对NH4+的去除比游离细菌高出17.39%,达到了53.62%,实验过程中NH4+ 主要转换为了NO3−,而NO2−与N2只有少量的生成。Feammox反应过程中,由于NH4+作为电子供体,也是最佳的研究去除对象和目标污染物,对于低氨氮废水Feammox反应对NH4+的去除具有一定效果,但并不显著,因此对于高氨氮废水仍需求有效的解决方式。Yang等[78]向含有高NH4+的Feammox反应器中间歇的充气,在充气条件下,O2作为末端电子受体将Feammox还原生成的Fe(Ⅱ)氧化为Fe(Ⅲ),实现Fe(Ⅲ)和Fe(Ⅱ)循环,进而持续发生Feammox反应进行连续脱氮,最终总氮的去除率达到98.5%。与以往添加NO2−和NO3−实现铁循环进行脱氮不同的是,此充气方式简单且来源易得,可避免二次污染,对未来实际高氨氮废水的去除具有更简便的方式和广泛的适用性。虽然Feammox是自养脱氮过程,但Le等[79]研究了在有机碳源存在下的Feammox的氨氧化过程,研究发现Feammox能够与异养条件产生耦合作用,在进水COD与NH4+浓度分别为250 mg·L−1和200 mg·L−1,确定并控制两者最佳浓度比值为1.4,pH为中性的条件下,COD与NH4+的去除率达到了98.3%和58.8%,且反应最终产物为N2。此研究在有机碳的存在下显著提升了NH4+的去除,彻底实现了由NH4+转化为N2,并具有较高的去除率,对于一些NH4+和COD含量高的废水具有巨大的应用潜力。Feammox反应过程需要Fe(Ⅲ)源,铁的氧化物形态多样且廉价易得,Zhu等[80]通过添加不同的Fe(Ⅲ)化合物,以探索铁氧化物和碳源在废水处理中对NH4+的去除影响,结果表明,添加Fe2O3的反应器中引发的Feammox反应对NH4+去除效果最好,其去除负荷高达0.06 g·m−3·d−1。而与Le研究不同的是,在无COD的情况下NH4+的去除率为53%,比有COD的反应器高出23%,在反应器存在有机碳源的情况下,有机物抑制了Fe(Ⅲ)还原,也可能如Le的研究所示,当COD与NH4+浓度存在合适的比值时,才能对NH4+进行高效去除,但在有机碳源存在的情况下,其浓度大小如何影响对NH4+的去除需要进一步研究。
根据目前的研究现状,Feammox在污水处理方面的研究还相对单薄,且其所涉及的相关性功能微生物目前尚未统一[51]。除了目前已知的与Feammox相关的微小杆菌(Exiguobacterium spp)、A6菌、铁还原菌、以及利用Anammox污泥培养Feammox获得的Anammox菌之外,在污水处理领域中尚未分离出纯的Feammox菌种[10,24,52]。Feammox菌作为厌氧自养细菌,具有独特的代谢机制,在反应的过程中具有无需提供氧气和碳源并产生较少N2O的特点[66],有利于与其他脱氮反应联合应用于低碳氮比污水的处理[81]。Zhu等[82]便将Feammox与生物电化学系统相结合,两者的耦合反应对NH4+的去除率比单独Feammox反应高出38.8%,且此系统具有较低的能耗和运行成本。因此,一旦Feammox工艺成功运行于实际污水处理中,将会大幅减小污水厂曝气量和外投加碳源的经济成本,并减少污泥产量和温室气体排放[83],这也是Feammox应用于工程上所具有的独特优势和潜能。
3. 结语(Conclusion)
Feammox作为新型的氮循环转换途径,在陆地和海洋沉积物中均有Feammox作用的发生,并参与其所在环境体系氮的损失,也是构成全球自然生态系统中氮损失的重要组成部分。经过近些年来的不断探索研究,Feammox在污水脱氮领域具有巨大的应用潜力,尤其是对废水中NH4+ 的去除具有较好的效果。另外,Feammox能够与Anammox和NDFO进行耦合脱氮,为污水中氮素的去除开辟了新的方法和途径,因此Feammox工艺脱氮也是继Anammox工艺脱氮之后备受瞩目的研究热点。
然而Feammox污水脱氮仍处于初步探索阶段,应用于实际工程中,仍具有一定的距离和面临着诸多挑战。在未来的发展中Feammox应注重以下研究:(1)目前发现的Feammox菌的种类比较少,与Feammox相关的功能性微生物没有统一,现已知的A6菌是唯一能起到纯Feammox作用的菌种。应从微生物宏基因和蛋白质组学领域作为出发点,研究Feammox的发生情景和其所处环境微生物的多样性,通过分析特定功能微生物及其所受影响因子,进一步剖析Feammox的作用机制。(2)Feammox反应的产物含有NO2−和NO3−,通过深入研究Feammox与Anammox和NDFO的耦合作用机制,确定耦合过程最佳反应条件,给污水中氮素的高效去除和精确的反应控制提供可靠的参考方案。有效地与NDFO反应进行结合,通过实现Fe(Ⅲ)和Fe(Ⅱ)的循环转换对氮素进行脱除,实现更加节约资源的绿色环保脱氮。(3)深入探究铁还原菌作用下的胞外电子传递机制,不仅对实现铁循环和加快Feammox反应速率具有重大意义,也对污水高效脱氮和Feammox作用机理的研究提供有力依据。
-

图 2 单介质和双介质反应器电流波形图
Figure 2. Current waveform of single and double dielectric barrier reactor
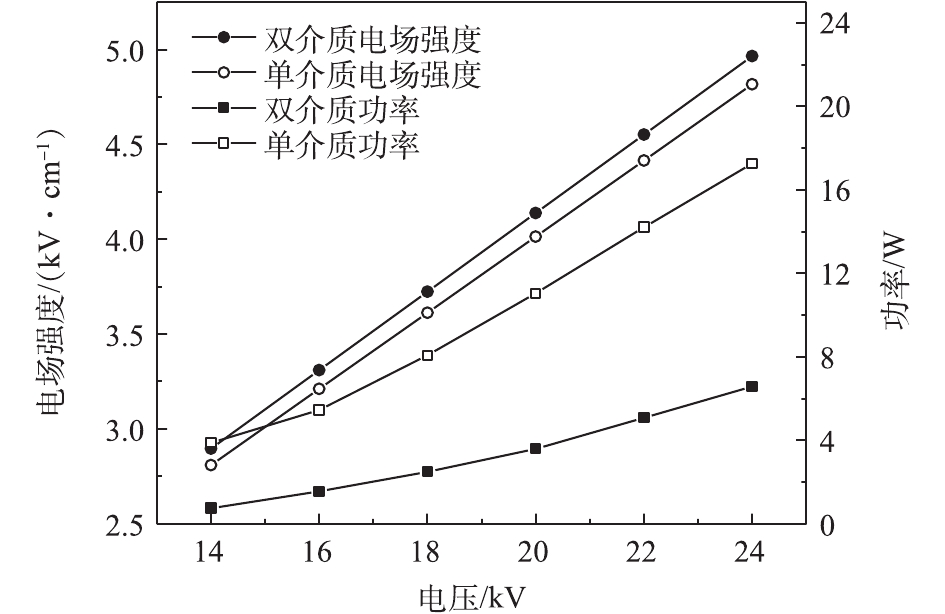
图 4 不同反应器的平均电场强度和输入功率
Figure 4. Average electric field and input power of different reactors
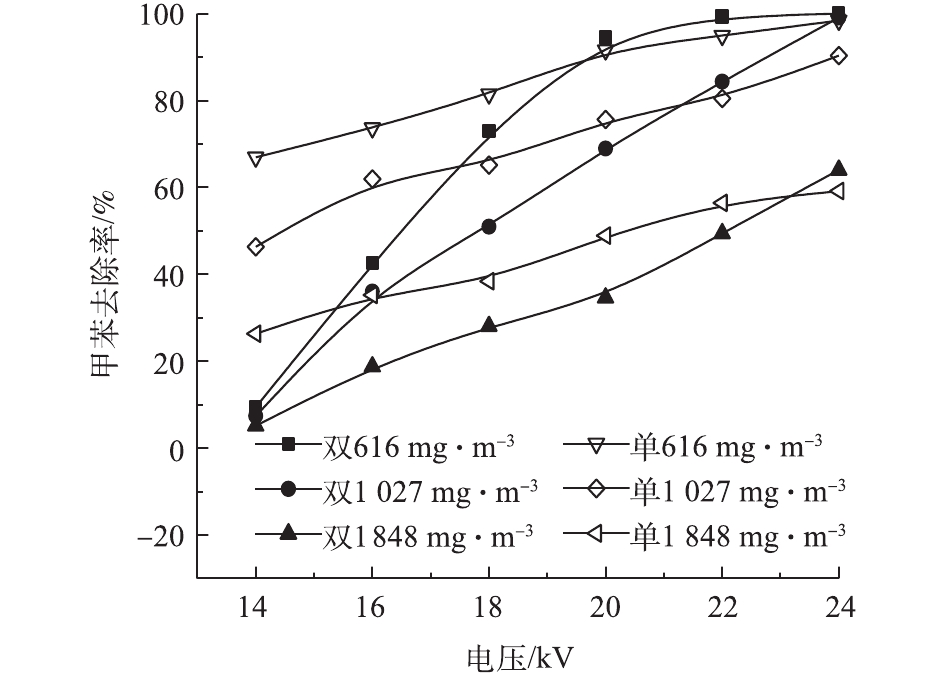
图 5 不同反应器中浓度和电压对甲苯去除率的影响
Figure 5. Effect of concentration and voltage on the toluene removal efficiency in different reactors
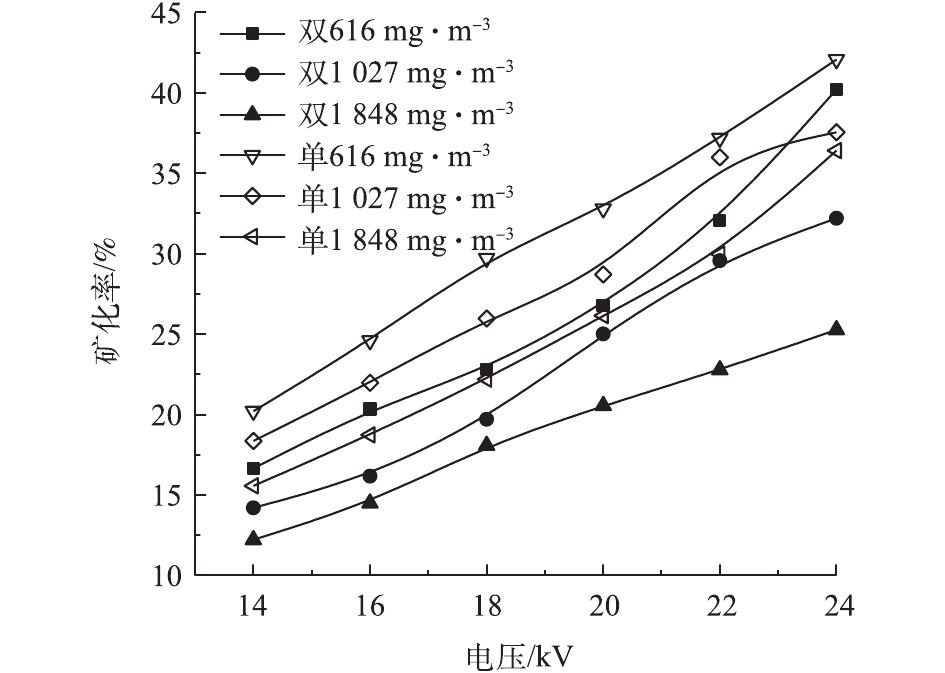
图 6 不同反应器中浓度和电压对矿化率的影响
Figure 6. Effect of concentration and voltage on the mineralization rate in different reactors

图 7 不同反应器中浓度和电压对CO2选择性的影响
Figure 7. Effect of concentration and voltage on the CO2 selectivity in different reactors

图 8 不同反应器中甲苯浓度和电压对O3浓度的影响
Figure 8. Effect of toluene concentration and voltage on O3 concentration in different reactors
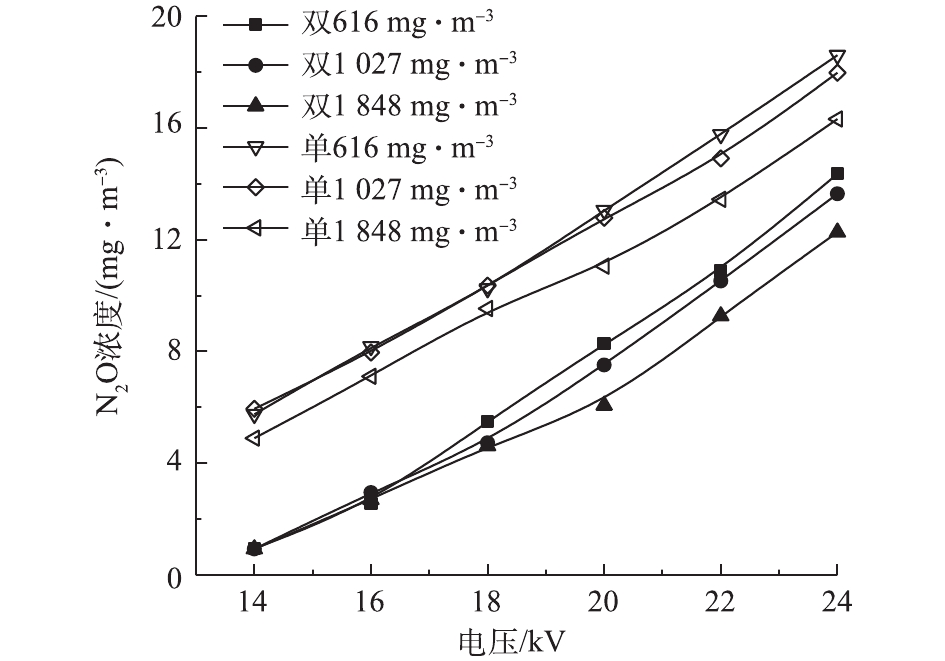
图 9 不同反应器中甲苯浓度和电压对N2O浓度的影响
Figure 9. Effect of toluene concentration and voltage on N2O concentration in different reactors
-
[1] ZHANG J N, XIAO J F, CHEN X F, et al. Allowance and allocation of industrial volatile organic compounds emission in China for year 2020 and 2030[J]. Journal of Environmental Science, 2018, 69: 155-165. doi: 10.1016/j.jes.2017.10.003 [2] HUI L R, LIU X G, TAN Q W, et al. VOC characteristics, sources and contributions to SOA formation during haze events in Wuhan, Central China[J]. Science of the Total Environment, 2019, 650: 2624-2639. doi: 10.1016/j.scitotenv.2018.10.029 [3] HU R Y, LIU G J, ZHANG H, et al. Levels, characteristics and health risk assessment of VOCs in different functional zones of Hefei[J]. Ecotoxicology and Environmental Safety, 2018, 160: 301-307. doi: 10.1016/j.ecoenv.2018.05.056 [4] ZHANG X Y, GAO B, CREAMER A E, et al. Adsorption of VOCs onto engineered carbon materials: A review[J]. Journal of Hazardous Materials, 2017, 338: 102-123. doi: 10.1016/j.jhazmat.2017.05.013 [5] HARIZ R, RIOSANZ J I, MERCIER C, et al. Absorption of toluene by vegetable oil-water emulsion in scrubbing tower: Experiments and modeling[J]. Chemical Engineering Science, 2017, 157: 264-271. doi: 10.1016/j.ces.2016.06.008 [6] BELAISSAOUI B, MOULLEC Y L, FAVRE E. Energy efficiency of a hybrid membrane/condensation process for VOC (volatile organic compounds) recovery from air: A generic approach[J]. Energy, 2016, 95: 291-302. doi: 10.1016/j.energy.2015.12.006 [7] MUHAMMAD S K, SHAIKH A R, MOHAMMAD M H. Catalytic oxidation of volatile organic compounds (VOCs): A review[J]. Atmospheric Environment, 2016, 140: 117-134. doi: 10.1016/j.atmosenv.2016.05.031 [8] SAVITA K P, VEERAPANDIAN C L, NATHALIE D G, et al. Abatement of VOCs using packed bed non-thermal plasma reactors: A review[J]. Catalysts, 2017, 7: 1-33. [9] ZHANG S H, YOU J P, CHRISTIAN K. Current advances of VOCs degradation by bioelectrochemical systems: A review[J]. Chemical Engineering Journal, 2018, 334: 2625-2637. doi: 10.1016/j.cej.2017.11.014 [10] 席劲瑛, 武俊良, 胡洪营, 等. 工业VOCs气体处理技术应用状况调查分析[J]. 中国环境科学, 2012, 32(11): 1955-1960. doi: 10.3969/j.issn.1000-6923.2012.11.005 [11] 栾志强, 郝郑平, 王喜芹. 工业固定源VOCs治理技术分析评估[J]. 环境科学, 2011, 32(12): 2216-2227. [12] QIN C H, GUO H, LIU P, et al. Toluene abatement through adsorption and plasma oxidation using ZSM-5 mixed with γ-Al2O3, TiO2 or BaTiO3[J]. Journal of Industrial and Engineering Chemistry, 2018, 63: 449-455. doi: 10.1016/j.jiec.2018.03.005 [13] AKIRA M. Generation of non-thermal plasma combined with catalysts and their application in environmental technology[J]. Catalysis Today, 2013, 211: 2-8. doi: 10.1016/j.cattod.2013.03.029 [14] QIN C H, GUO H, BAI W W, et al. Kinetics study on non-thermal plasma mineralization of adsorbed toluene over γ-Al2O3 hybrid with zeolite[J]. Journal of Hazardous Materials, 2019, 369: 430-438. doi: 10.1016/j.jhazmat.2019.01.098 [15] ARNE M V, MORENT R, NATHALIE D G, et al. Non-thermal plasmas for non-catalytic and catalytic VOC abatement[J]. Journal of Hazardous Materials, 2011, 195: 30-54. doi: 10.1016/j.jhazmat.2011.08.060 [16] 王保伟, 王超, 徐艳, 等. 介质阻挡放电等离子体反应器降解盐酸四环素[J]. 化工学报, 2018, 69(4): 1687-1694. [17] KIM H H, YOSHIYUKI T, NOBUAKI N, et al. A multidisciplinary approach to understand the interactions of nonthermal plasma and catalyst: A review[J]. Catalysis Today, 2015, 256: 13-22. doi: 10.1016/j.cattod.2015.04.009 [18] ZHANG H B, LI K, SHU C H, et al. Enhancement of styrene removal using a novel double-tube dielectric barrier discharge (DDBD) reactor[J]. Chemical Engineering Journal, 2014, 256: 107-118. doi: 10.1016/j.cej.2014.06.105 [19] TANG X L, GAO F Y, WANG J G, et al. Comparative study between single- and double-dielectric barrier discharge reactor for nitric oxide removal[J]. Industrial & Engineering Chemistry Research, 2014, 53: 6197-6203. [20] 李云霞, 朱承驻, 陈天虎, 等. 介质阻挡放电反应器中的二硫化碳降解特性[J]. 环境科学研究, 2013, 26(2): 188-193. [21] LEE B, KIM D W, PARK D W. Dielectric barrier discharge reactor with the segmented electrodes for decomposition of toluene adsorbed on bare-zeolite[J]. Chemical Engineering Journal, 2019, 357: 188-197. doi: 10.1016/j.cej.2018.09.104 [22] 赵卫东, 蔡忆昔, 韩文赫, 等. 同轴圆柱结构DBD装置放电功率的模拟计算及实验研究[J]. 高压电器, 2010, 46(6): 25-28. [23] 马天鹏, 钟方川. 估算线筒式介质阻挡放电场强和电子平均动能的方法[J]. 核聚变与等离子体物理, 2017, 37(4): 399-403. [24] 侯世英, 曾鹏, 刘坤, 等. 单介质与双介质结构介质阻挡放电水处理性能的比较[J]. 高电压技术, 2012, 38(7): 1562-1567. [25] YAO X H, ZHANG J, LIANG X H, et al. Plasma-catalytic removal of toluene over the supported manganese oxides in DBD reactor: Effect of the structure of zeolites support[J]. Chemosphere, 2018, 208: 922-930. doi: 10.1016/j.chemosphere.2018.06.064 [26] WANG B W, CHI C M, XU M, et al. Plasma-catalytic removal of toluene over CeO2-MnOx catalysts in an atmosphere dielectric barrier discharge[J]. Chemical Engineering Journal, 2017, 322: 679-692. doi: 10.1016/j.cej.2017.03.153 [27] OSMAN K, MARC A D. A comparative study of dilute VOCs treatment in a non-thermal plasma reactor[J]. Chemical Engineering Journal, 2016, 201: 308-315. [28] KIM H H, OGATA A, SHIGERU F. Oxygen partial pressure-dependent behavior of various catalysts for the total oxidation of VOCs using cycled system of adsorption and oxygen plasma[J]. Applied Catalysis B: Environmental, 2008, 79: 356-367. doi: 10.1016/j.apcatb.2007.10.038 [29] ZHANG H B, LI K, SUN T H, et al. The removal of styrene using a dielectric barrier discharge (DBD) reactor and the analysis of the by-products and intermediates[J]. Research on Chemical Intermediates, 2013, 39(3): 1021-1035. doi: 10.1007/s11164-012-0664-0 [30] ZHENG B, HAN H, YANG S L, et al. Vertically-oriented graphenes supported Mn3O4 as advanced catalysts in post plasma-catalysis for toluene decomposition[J]. Applied Surface Science, 2018, 436: 570-578. doi: 10.1016/j.apsusc.2017.12.081 [31] ZHU R Y, MAO Y B, JIANG L Y, et al. Performance of chlorobenzene removal in a nonthermal plasma catalysis reactor and evaluation of its byproducts[J]. Chemical Engineering Journal, 2015, 279: 463-471. doi: 10.1016/j.cej.2015.05.043 [32] CHANG T, SHEN Z X, HUANG Y, et al. Post-plasma-catalytic removal of toluene using MnO2-Co3O4 catalysts and their synergistic mechanism[J]. Chemical Engineering Journal, 2018, 348: 15-25. doi: 10.1016/j.cej.2018.04.186 [33] 吴萧, 刘盛余, 何廷宇, 等. 介质阻挡放电低温等离子体技术处理3种代表性VOC[J]. 环境工程学报, 2017, 11(10): 5502-5508. doi: 10.12030/j.cjee.201612064 期刊类型引用(9)
1. 陈梦杰,王世宇,王瑞雪,孔令策,李晓森. 近五年基于高级氧化技术降解罗丹明B的研究进展. 化学试剂. 2024(09): 1-11 .  百度学术
百度学术
2. 王迎辉,郭秀荣,张昊男,杜丹丰,亓占丰. 低温等离子木质钙钛矿柴油机尾气净化器设计与试验. 农业工程学报. 2024(16): 52-63 . 百度学术
3. 侯浩,党小庆,李世杰,于欣,王鹏勇,孟祥康. 低温等离子体耦合Co-Mn双金属催化剂降解三氯乙烯. 环境化学. 2023(04): 1185-1195 . 百度学术
4. 张瑜,米俊锋,杜胜男. 介质阻挡放电技术应用于NO_x脱除的研究进展. 科技导报. 2023(11): 52-60 . 百度学术
5. 余康,李民,孙高攀,周鹏,谭金浪,王斌,王涛,穆晓亮,赵璐,房克功. 介质阻挡放电等离子体转化H_2S-CO_2酸气制合成气的影响因素研究. 燃料化学学报(中英文). 2023(12): 1782-1790 . 百度学术
6. 刘建奇,刘鑫,陈佳尧,钟方川. MnFe/海泡石后置协同低温等离子体降解甲苯的研究. 安全与环境工程. 2022(03): 161-167 . 百度学术
7. 韩丰磊,朱一凡,贾继磊,刘晓琳,季纯洁,司佩壮,张宇鹏. 低温等离子体耦合催化剂降解VOCs的研究进展. 油气田环境保护. 2022(06): 6-12 . 百度学术
8. 叶凯,刘香华,姜月,于颖,赵亚飞,庄烨,郑进保,陈秉辉. 低温等离子体协同CeO_2/13X催化降解甲苯. 化工学报. 2021(07): 3706-3715 . 百度学术
9. 刘建奇,刘鑫,陈佳尧,钟方川. Fe/海泡石黏土协同等离子体降解甲苯的研究. 环境污染与防治. 2021(11): 1357-1363+1370 . 百度学术
其他类型引用(9)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 7283
- HTML全文浏览数: 7283
- PDF下载数: 101
- 施引文献: 18
