









-
城市污水处理厂进水碳源不足是一个普遍存在的问题,导致后续脱氮效率较低。目前,解决该问题的主要方法之一是外加部分碳源,如甲醇和乙酸钠等。添加的部分碳源还有毒性,而且药剂成本较高。如何以较低的成本提高脱氮效率是低碳氮比污水生物脱氮亟待解决的问题,因此,寻找合适的外加碳源成为目前关注的热点[1]。水解酸化是把污泥中的大分子有机物分解成小分子有机物,得到挥发性脂肪酸(VFAs)的过程。而VFAs中的乙酸和丙酸是增强生物脱氮的有利碳源,其反硝化速率比甲醇和乙醇更高[2]。
超磁分离水体净化工艺是近年来发展起来的一种物化水处理技术。磁分离技术借助外加磁场强化固液分离效率,较生物吸附技术处理效率高,较膜分离技术能耗低,能弥补现有碳源浓缩技术各自的劣势,满足节能降耗需求[3-5]。其能快速有效地去除生活污水中的大部分有机物,COD分离去除率约为75%,SCOD的分离去除率超过60%,TP去除率接近90%[6]。本研究所采用的超磁分离设备的进水为生化处理前的污水,所以超磁分离污泥类似于初沉污泥。而初沉污泥中含有大量的有机物,是很好的发酵底物[7]。目前,国内外有许多关于初沉污泥[7]、剩余污泥[8]以及两者混合污泥[9]的水解产酸的研究报道。但是对于超磁分离污泥与剩余污泥协同水解酸化的相关研究,还很少见。现有研究[10]发现,在不调控pH,温度为30 ℃的反应条件下,既可以为生化系统提供更多的SCOD,又可以避免系统过高的N、P负荷。
本研究在维持温度30 ℃,不调控pH条件下,选取了2种超磁分离后污泥(R1、R2)、剩余污泥(W1、W2),设置R1、W1为一组,设置R2、W2为另一组,进行了超磁分离污泥、混合污泥以及剩余污泥3种不同类型污泥水解酸化的对比研究,其中混合污泥为超磁分离污泥以及剩余污泥按不同比例混合后的污泥(5组)。探究了污泥性质的差异对水解酸化及酸化产物组分的影响,为污水厂通过污泥产酸发酵获得碳源进行污泥种类的选择提供参考。
-
R1、W1分别为污水处理厂停产前超磁分离污泥以及含水率为80%的脱水污泥;R2、W2分别为污水处理厂停产后超磁分离污泥以及某强化生物除磷(EBPR)中试工艺的二沉池中的剩余污泥。其中R1所用污水取自东坝污水处理厂细格栅之后,R2所用污水取自污水处理厂进水井(粗格栅之前)。实验前,将W1用蒸馏水稀释,将W2在4 ℃下浓缩24 h,然后排出上清液。以期达到与超磁分离污泥相似的挥发性固体(VSS)。实验前,取1 d内不同时段的污泥,混合后接种。4种污泥特征(至少经过3次重复测定取平均值)结果见表1。R1、W1、R2和W2的初始pH为7.55、7.68、6.85和6.91,含水率为0.984 7、0.982 2、0.968 3和0.977 2。投加比例见表2,1~7号投加的比例以剩余污泥的体积和VSS计,其中1号为超磁分离污泥,7号为剩余污泥,2~6号为投加了不同比例的剩余污泥。
-
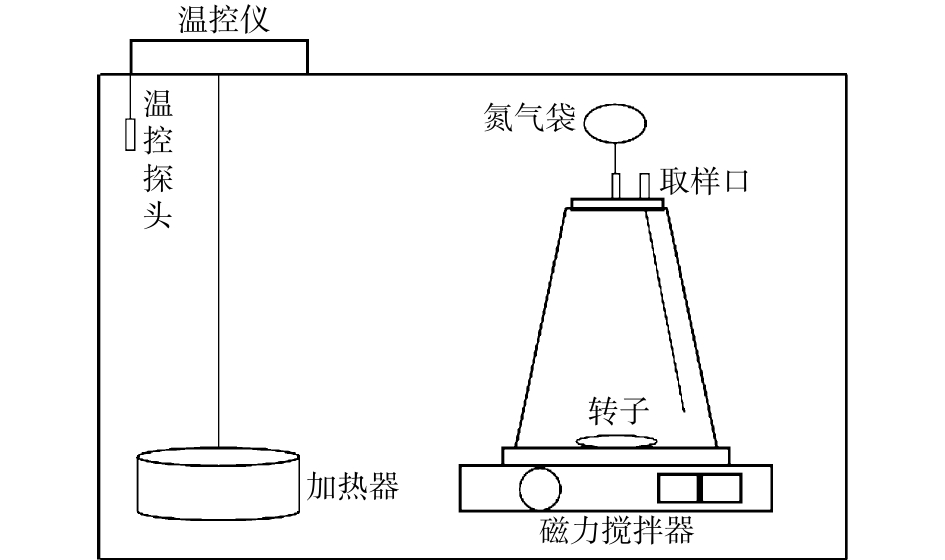
超磁分离污泥水解酸化的批次实验在恒温培养箱中进行,实验装置如图1所示,采用7个2 L的反应器,接种污泥体积为1.8 L。实验开始前,曝氮气3 min,以驱除反应器中的氧气,然后使用橡胶塞密封,橡胶塞上开2个孔,分别是氮气袋,以及取样口,反应器采用磁力搅拌器搅拌。
-
本研究在首创东坝污水处理厂现场进行,每天早晚各取反应器的出水进行相关指标的测定。由于水解消化后污泥脱水性能变差,因此,各指标测定前须对样品进行预处理。预处理主要包括离心及过滤2个过程。离心采用100 mL的离心管,设置转速为5 000 r·min−1,离心45 min。然后将上清液用0.45 μm的微孔滤膜过滤,去除上清液中小颗粒物质,避免阻塞测定仪器并确保测量精度。
常规分析参考文献中的方法[11],其中TCOD、SCOD采用重铬酸钾法,TN采用过硫酸钾氧化紫外分光光度法,TP采用过硫酸钾氧化钼酸铵分光光度法,SOP采用钼酸铵分光光度法,
NH+4 -N采用纳氏试剂光度法,VSS和SS采用重量法。pH采用HACH HQ40d测定仪测定。VFAs采用瑞士万通883型离子色谱仪测定。 -
污泥水解情况可以使用SCOD[9]来表示。2种剩余污泥在不同接种比例下对超磁分离污泥水解酸化的影响如图2所示。由图2(a)和图2(b)可见,2种超磁分离污泥(R1、R2)自然水解产生的SCOD均在第4天达到峰值,分别为1 118.68 mg·L−1和2 063.50 mg·L−1;虽然两者水解得到的SCOD不同,但是从图2(c)可以看出,其SCOD/VSS的变化规律是一致的,最高值均出现在第4天,为110 mg·g−1。说明2种超磁分离后的污泥水解产酸的效果基本是一致的。
剩余污泥(W1、W2)自然水解产生的SCOD均在第7天达到峰值,分别为1 599.88 mg·L−1和4 954.80 mg·L−1。由图2(a)可以看出,2号和3号的SCOD最大值均出现在第4天,分别为1 196.80 mg·L−1和1 248.40 mg·L−1;4号的SCOD最大值出现在第5天,为1 262.57 mg·L−1;5号、6号和7号的SCOD最大值均出现在第7天,分别为1 443.68、1 493.96和1 599.88 mg·L−1。随着剩余污泥比例的增加,不仅可以增加SCOD的析出量,还可以延长其达到最大值的时间;与R1、W1水解不同的是,由图2(b)可以看出,2~7号的SCOD最大值均在第7天,并且其随着接种比例的增加而增大,分别为2 435.30、2 622.70、2 668.80、3 151.00、3 423.20和4 954.80 mg·L−1。这与苏高强等[12]的研究结果相似。
W1、W2产SCOD出现如此大的差异,推测其原因是:一方面,W1为脱完水后的污泥,其中聚丙烯酰胺(PAM)的存在增加了分子间的团聚性,进而减少了发酵微生物与消化基质的接触[13],从而减少了SCOD的产量;另一方面,W2为某稳定运行的EBPR系统,污泥中微生物的含量较W1多,水解酸化菌通过对污泥中微生物细胞壁破坏从而促使细胞内容物释放[14]。
-
水解酸化过程中产生的VFAs主要是由发酵产酸菌对可溶性有机物的吸收转化。实验发现,3种污泥产生的酸主要是乙酸、丙酸、正丁酸、异丁酸和正戊酸,将其乘以相应的系数换算成COD后相加,其和为挥发性有机酸量[8]。实验选取R1、W1进行分析,污泥水解过程中VFAs的生成情况如图3所示。由图3可以看出,VFAs的变化规律与SCOD是一致的,均呈先增大后减少的趋势。1号(超磁分离污泥)自然水解VFAs的峰值出现在第4天,峰值为353.54 mg·L−1,与SCOD的变化趋势相同的是,混合污泥2~6号分别在第4、4、5、7和7天,水解液中产生的VFAs达到最大值,分别为399.98、436.52、449.03、520.05和556.97 mg·L−1,7号(剩余污泥)自然水解产生的VFAs的峰值出现在第7天,为477.52 mg·L−1。从图3中还来可以看出,接种剩余污泥能提高VFAs的产生量,并且随着接种剩余污泥的增加,也能延长其VFAs达到峰值的时间。
在初始阶段,污泥中易降解颗粒物质首先被水解酸化菌转化为VFAs,随着反应的进行,易降解物质被消耗完全,水解酸化菌开始利用较难降解的颗粒及大分子物质,这样导致VFAs的产速变慢[15]。由图3可以看出:混合污泥与超磁分离、剩余污泥比较,更易酸化产VFAs。这是因为一方面混合污泥吸附大量胶体和易降解有机物,水解酸化菌能被有效利用;另一方面,超磁分离污泥中虽然有机物含量很高,但多数属于慢速降解碳源;剩余污泥中的有机物主要存在其细胞内和胞外聚合物中,不经过有效预处理水解酸化菌难以利用。
-
SCOD向VFAs的转化率能直接用来反映污泥的产酸效果[16]。实验选取R1、W1进行分析,由图4可以看出,在前4 d,VFAs∶SCOD均逐渐变大,混合污泥VFAs∶SCOD比值一直领先超磁分离、剩余污泥。1~7号的VFAs∶SCOD分别在第4、4、4、5、7、7和7天达到最大值分别为0.316、0.334、0.350、0.360、0.361、0.373和0.299。因此,仅从VFAs∶SCOD来看:混合污泥较之于超磁分离具有较高的产酸优势;且剩余污泥接种量的增加也加快了水解酸化的速率,从而加深了酸化的程度。
ELEFSINIOTIS等[17]指出,反硝化优先利用乙酸,其次为丁酸(包括异丁酸和正丁酸)和丙酸,最后是戊酸(包括异戊酸和正戊酸)。CHEN等[18]发现,适宜作为除磷碳源的2种有机酸为乙酸和丙酸,从短期看,乙酸作为碳源除磷效果较好,而从长期看,丙酸作为碳源要比乙酸作为碳源的除磷效果好。可见SCFAs的组成情况对其作为碳源被利用具有重要的影响。
由于超磁分离污泥SCOD在第4天即达到最大值,此时选取R1、W1进行分析,结果如图5所示。实验中污泥水解酸化主要生成5种挥发性脂肪酸,分别为乙酸、丙酸、正丁酸、异丁酸和正戊酸。超磁分离污泥中5种酸的含量大小为乙酸>正戊酸>正丁酸>异丁酸>丙酸,而剩余污泥中5种酸的含量大小为乙酸>丙酸>正戊酸>正丁酸>异丁酸。混合污泥中随着剩余污泥占比的增加,丙酸和异丁酸的含量也有不同程度的增加,正丁酸出现了下降的趋势,而正戊酸的变化不大。从图5中易看出,各种污泥产VFAs中,乙酸均具有明显优势。这与苏高强等[9]、刘绍根等[1]、吴昌生等[19]的研究结果是一致的。之所以乙酸占比最高,其主要原因为:一方面,水解产物被产酸菌降解为乙酸,且乙酸可以直接从碳水化合物和蛋白质的水解酸化得到;另一方面,其他的有机酸(丙酸、丁酸或戊酸等)在某些胞内酶的作用下也可进一步生成乙酸[20]。
-
不同比例的剩余污泥对N元素的影响见图6。超磁分离污泥以及剩余污泥中含有大量的蛋白质,所以水解酸化过程中除了有VFAs、SCOD等有机物溶出以外,还会伴随着N元素的释放。本研究主要以
NH+4 -N和TN为考察对象。在以往对于污泥厌氧发酵的研究中,都出现了不同程度的N元素的释放[1, 9-10, 19]。对于R1、W1,由图6(a)可知,3种不同的污泥的NH+4 -N都呈现出逐渐增长的趋势。并且随着剩余污泥接种量的增加,NH+4 -N的增加量也越大。反应进行到第4天时,1~7号的增加量分别为78.79、85.97、91.11、94.68、97.28、115.32和115.91 mg·L−1。对于R2、W2,由图6(b)可知,3种不同的污泥呈现出与R1、W1一样的变化规律,不同于R1、W1的是,其
NH+4 -N的增加量更大。第4天,1~7号NH+4 -N的增加量分别为127.34、147.56、153.53、176.34、206.19、244.41和399.83 mg·L−1。由于剩余污泥主要是由一些活性生物絮体组成,因此,含有较多的蛋白质,蛋白质水解能释放出大量的氨氮。系统中的TN主要是以
NH+4 -N的形式存在,由图6(c)和图6(d)中可以看出,TN具有和NH+4 -N相似的变化规律。剩余污泥接种量的增加也加快了N元素的溶出,含有大量氮元素的水解酸化液若投加到脱氮系统中,势必增加系统的N负荷。因此,剩余污泥的接种量应该综合考虑氮元素的释放对于整个系统后续的脱氮除磷的影响。 -
在污泥的厌氧消化过程中,随着污泥的解体和细胞的破壁,会有大量的磷释放到水解酸化液中。如果将水解酸化液直接用于脱氮除磷的碳源,会增加后续处理的磷负荷。所以,在此之前都会进行前处理,对氮磷进行部分回收。因此,监测P的溶出情况很有必要[21]。
在以往对于污泥水解酸化的研究中,随着时间的延长,都在不同程度上伴随着磷元素的析出。吴昌生等[19]在对碱预处理絮凝污泥水解酸化影响的研究中发现:在25 ℃时,磷酸盐浓度在第480分钟达到峰值,为7.65 mg·L−1;在35 ℃时,在第480分钟达到峰值,为15.23 mg·L−1。苏高强等[9]发现混合污泥厌氧发酵在第6天时磷酸盐的释放量为120 mg·L−1。由于超磁分离在污水处理前端就已经去除了系统中绝大多数的磷酸盐,减轻了后续的处理压力,所以对于超磁分离污泥的水解酸化,并不希望有P元素的析出。
对比2种超磁分离污泥(R1、R2)P的释放情况,由图7可知,不管是TP还是SOP,其值较初始值都没有较大的变化,并没有P的析出。推测可能是由于超磁分离污泥中有PAC(聚合氯化铝),抑制了磷酸盐的释放。对比2种剩余污泥(W1、W2)的TP,由图7(b)可知,TP的浓度在前5 d逐渐升高,在第5天达到峰值,为24.15 mg·L−1,此后逐渐降低。由图7(a)可知,2~6号TP的浓度稳定在1~2 mg·L−1,并没有很明显的磷的析出;由图7(d)可以看出,TP的浓度在第3天即达到峰值,为385.11 mg·L−1,此后浓度稳定在390 mg·L−1左右,由图7(c)可知,2~6号TP的浓度在3 d后分别稳定在4.31、9.61、16.96、32.81、57.50 mg·L−1左右。2种剩余污泥释磷情况有巨大差异,推测其原因是:W1来源的东坝污水处理厂采用前端化学除磷工艺,所以污泥中几乎没有P的富集;而W2取自某稳定运行的EBPR中试实验的二沉池污泥,其出水能稳定满足北京市地标(DB 11/890-2012)B限值标准甚至北京市地标(DB 11/890-2012)A限值标准出水标准,因此,其二沉池中污泥富集了大量的磷酸盐,污泥水解酸化时,在厌氧条件下导致了剩余污泥中的聚磷菌的释磷。单从P元素的释放情况来看,W2显然不适合用作接种污泥。
-
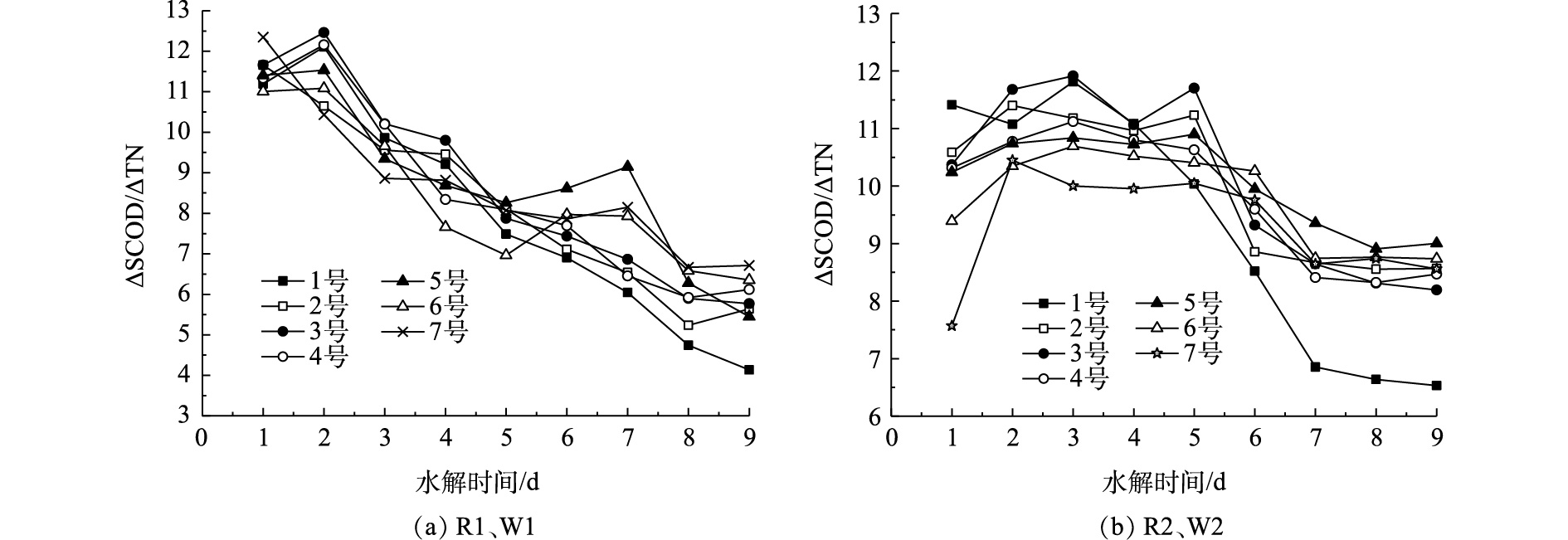
污泥水解酸化旨在获取较多可利用碳源,但同时也存在着氮元素的释放。较高的氮释放势必会增加系统的氮负荷,同时加剧对碳源的竞争,最终降低系统的脱氮效率[9, 22]。因此,在污泥水解酸化反应获得较多碳源的同时尽量减少总氮的释放,即达到较高的ΔSCOD/ΔTN值。由于超磁分离后的污泥水解产酸在第4天达到最大值,所以考察了第4天时各污泥的ΔSCOD/ΔTN值。由图8(a)可以看出,在第4天,3号的ΔSCOD/ΔTN值最大,为9.80,此时,剩余污泥的投加比例为12.2%。由图8(b)可以看出,在第4天,3号的ΔSCOD/ΔTN值最大,为9.86,此时,剩余污泥的投加比例为13.6%。由此可见,在只考虑N元素的影响时,虽然2种剩余污泥来源不同,但其在第4天达到最大值时的污泥接种比例是相近的。综合考虑剩余污泥对于超磁分离污泥水解酸化效果影响发现,当剩余污泥接种量W1为12.2%,W2为13.6%时,既可以为系统提供更多的SCOD,又可以避免过高的氮负荷。
-
1) 2种超磁分离污泥(R1、R2)自然水解产生的SCOD均在第4天达到峰值,剩余污泥(W1、W2)自然水解产生的SCOD均在第7天达到峰值,随着剩余污泥接种量的增加,混合污泥SCOD的析出量也逐渐增加。
2)对R1、W1进行产酸分析发现:剩余污泥接种量的增加促进了混合污泥VFAs的生成;各种污泥产VFAs中,乙酸均具有明显优势,并且会促进丙酸的累积。
3) VFAs∶SCOD值的分析结果表明,混合污泥较之于超磁分离、剩余污泥具有快速、高效的产酸优势,且剩余污泥接种量的增加也加快了水解酸化的速率并且加深了酸化的程度,但是会延长其达到峰值的时间。
4)污泥产酸发酵的同时,还存在着N元素的释放,且随着剩余污泥接种量的增加,N元素的释放更明显;对比2种剩余污泥(W1、W2),W1作为接种污泥时,并没有明显的P元素的释放,当W2作为接种污泥时,伴随着比较明显的P元素的释放。
5)综合考虑剩余污泥对于超磁分离污泥水解酸化效果影响发现,当剩余污泥接种量W1为12.2%,W2为13.6%时,既可以为系统提供更多的SCOD,又可以避免过高的氮负荷。
超磁分离污泥与剩余污泥协同水解酸化
Synergistic hydrolysis and acidification of ReCoMag sludge and excess sludge
-
摘要: 以超磁分离污泥作为研究对象,用2种不同的剩余污泥作为接种污泥,维持温度在30 ℃,探究了剩余污泥对超磁分离污泥厌氧水解酸化产物及产率的影响。结果表明:随着剩余污泥接种量的增加,混合污泥SCOD的析出量也逐渐增加;接种剩余污泥量的增加促进了混合污泥VFAs的生成;各种污泥产VFAs中,乙酸均具有明显优势,并会促进丙酸的累积;混合污泥较之于超磁分离和剩余污泥具有快速、高效的产酸优势,且随着剩余污泥接种量的增加,加快了水解酸化的速率并且加深了酸化的程度,但会延长其达到最大值的时间。污泥产酸发酵获得内碳源的同时,还存在着N元素的释放,且随着剩余污泥接种量的增加,这种伴随现象更明显。对比2种剩余污泥(W1、W2)发现,W1作为接种污泥时,并没有明显的P元素的释放;当W2作为接种污泥时,伴随着比较明显的P元素的释放。综合考虑剩余污泥对于超磁分离污泥水解酸化效果的影响发现,当剩余污泥接种量W1为12.2%,W2为13.6%时,既可以为系统提供更多的SCOD,又可以避免过高的氮负荷。Abstract: ReCoMag sludge was used as the research object, two different types of excess sludge were used as inoculated sludge, the effects of excess sludge on anaerobic hydrolysis acidification products and their yields of ReCoMag sludge were investigated when the temperature was maintained at 30 ℃. The experimental results show that the SCOD releasing amount from mixed sludge gradually increased with the increase of inoculum amount of excess sludge. The increase of the excess sludge inoculum amount promoted VFAs formation of mixed sludge; acetic acid has obvious advantages among various sludge-producing VFAs, and it could promote the accumulation of propionic acid. Mixed sludge had a fast and efficient acid-producing advantage over super-magnetic separation and excess sludge, and the increase of the inoculum amount of excess sludge accelerated hydrolysis acidification rate and deepened the acidification degree, but it extended the time reaching the maximum value. When the sludge was acidified and fermented to obtain the internal carbon source, N element release also occurred at the same time, and the more inoculation amount of excess sludge, the more obvious above concomitant phenomenon. Comparing two types of excess sludge (W1, W2), no obvious P element release occurred for W1 as inoculated sludge, while obvious P element release occurred for W2. Considering the effect of excess sludge on the hydrolysis and acidification effect of ReCoMag sludge, the inoculum amounts of 12.2% W1 and 13.6% W2 with ReCoMag sludge could produce more SCOD for the system and avoid excessively high N load.
-
Key words:
- ReCoMag sludge /
- excess sludges /
- hydrolysis acidification /
- internal carbon source
-
采用静电式空气净化器净化室外新风或室内循环空气,是改善室内空气品质的主要方法之一。一般来说,随着静电式空气净化器的电场强度增大,净化效率会提高,但臭氧产生量亦会增大[1-2]。臭氧是一种具有特殊气味和强氧化性的气体。人体长期暴露于高浓度臭氧会引起呼吸系统疾病,故应采取措施,防止其进入室内环境。臭氧控制措施主要包括两类:优化电极配置以抑制臭氧释放,借助热分解、吸附或催化之类后处理技术分解臭氧。在优化电极配置仍无法抑制臭氧产生的情况下,后者成为研究热点。热分解(含热催化分解)会带来明显的空气温升,吸附法则存在活性炭烧蚀等不利影响,故室温催化是最具实际应用前景的技术。
分解臭氧的催化剂主要活性组分包括贵金属(Ag、Au、Pd、Pt等)和过渡金属氧化物(Mn、Cu、Fe、Co、Ni等)[3]。过渡金属催化剂中,锰氧化物因其低毒性、高活性及优良的可调结构和物理化学性能,已被用作臭氧分解的活性组分[4-5]。锰氧化物存在多种价态和晶型结构。其中,α-MnO2因其具有开放的2×2孔道结构、较大的比表面积、较低的Mn平均氧化态及丰富的表面吸附氧物种,其催化分解臭氧的活性优于其他晶型结构MnO2 [6-7]。同时,α-MnO2中存在能有效捕获臭氧分子的氧空位及丰富的Mn3+/Mn4+氧化还原对,可通过替换孔道结构中部分阳离子或掺杂改性来提高α-MnO2的室温臭氧分解性能[8-9]。但在相对较高湿度下,锰氧化物的催化活性会明显降低。金属Ag价格较低,而且Ag和氧化锰间相互作用可产生更多晶格缺陷和氧空位,从而提高了锰氧化物的还原能力和氧的迁移率[10-11]。因此,采用Ag掺杂不仅可提高催化剂的抗水性,还可改善催化剂的催化活性。本课题组前期研究表明,MnO2或Ag/MnO2催化剂具有优异的室温催化分解臭氧性能[12-14]。
净化室外新风或室内循环空气的静电式空气净化器收尘区极板间距通常仅数毫米,且极板面积大。这意味着待净化空气流经电场空间时,会与极板接触充分,因此,极板涂覆臭氧分解催化材料,有望实现臭氧的原位分解。基于此,本研究采用铝合金板(常用作空气净化器的极板)表面涂覆MnO2或Ag/MnO2催化剂的方法,系统考察涂覆浆料构成(粘结剂的类型和含量、催化剂活性组分)、反应时间和环境条件(臭氧浓度和空气湿度)等因素对催化分解臭氧性能的影响,为优化静电式空气净化器性能以同时实现除尘提效、臭氧控制提供参考。
1. 实验部分
1.1 催化剂的制备与表征
MnO2或Ag/MnO2的制备参照文献[3]。催化剂晶体结构采用D8 Advance型X射线衍射仪(XRD, 德国 Bruker公司)测定;比表面积和孔结构采用ASAP 2020型全自动比表面积及微孔物理吸附仪(BET,美国 Micromeritics公司)测定;表面形貌采用ZEISS Sigma 500 扫描电子显微镜(SEM,德国 Bruker公司)检测。
1.2 催化剂涂覆步骤
1)表面粗化处理。实验采用表面光滑的铝合金板,其尺寸为100 mm ×55 mm×1 mm。为改善基体与涂层的结合力,需先对铝合金板进行化学粗化处理:先将铝合金板浸泡于40 ℃的混合溶液(NaOH质量浓度为30 g∙L−1,Na2CO3质量浓度为25 g∙L−1 )中约1.5 min,以去除表面的油脂和氧化膜;然后用去离子水冲洗去除多余的碱液;将铝合金板浸泡于混合酸液(将1.5 mol∙L−1 HCl和0.08 mol∙L−1 H2C2O4等体积混合得到)中刻蚀2 h,以便形成粗糙表面,然后用去离子水冲洗除去表面酸液;最后将其置于105 ℃鼓风干燥箱中烘干备用。
2)涂层浆料配制。分别采用复配比为5/7的羧甲基纤维/丁苯橡胶(carboxymethyl cellulose/styrene rubber,CMC/SBR)和海藻酸钠(sodium alginate,SA)作粘结剂配制涂层浆料。浆料配制方法:取一定量的粘结剂溶于去离子水中,少量多次添加MnO2或Ag/MnO2催化剂粉末,并用顶置式电子搅拌器以2 000 r·min−1转速搅拌20 min,得到混合均匀的催化剂浆料。
3)催化剂涂覆。将表面粗化的铝合金板固定在提拉涂膜机夹具上,以10 cm∙min−1的速度浸入催化剂浆料中,浸泡0.5 min后以相同速度提出铝合金板试片。将试片置于105 ℃鼓风干燥箱中干燥1 h初步去除水分,随后以2 ℃∙min−1的速率升温至300 ℃,并在此温度下焙烧20 min,最终获得涂覆催化剂的试片。
1.3 催化剂涂层结合性能评价
通过震荡法脱落涂层,以涂层脱落率
ϕ m脱落后 脱落率(%)=m涂覆后−m脱落后m涂覆后−m涂覆前×100% (1) 式中:
m涂覆前 m涂覆后 1.4 催化分解臭氧性能评价
催化剂分解臭氧性能在自行搭建的测试系统中进行(图1)。测试系统由方形石英管、轴流风机、气流均布板、石英试片插槽组成。其中,石英方管的外径尺寸为68 mm×68 mm、壁厚为3 mm、长度为1 000 mm,在距进出口各150 mm处设置1个直径为20 mm的检测口用于检测管道内气体的臭氧浓度、风速和湿度等。风量由微型调频风扇调变。试片插槽是尺寸为60 mm×30 mm×10 mm。试片插槽间距为分别为3、4和5 mm,槽宽1.1 mm、槽深4 mm,将涂覆好的试片嵌入插槽中可组成臭氧分解催化剂试片组进行实验。试片与气流方向平行放置,对应插槽间距为3、4 和5 mm的试片数量分别为18、14和11。模拟气体由臭氧和室内空气组成,臭氧由体积分数5%的 O2/N2合成气经臭氧发生器产生。气体湿度采用美国 Cole-Parmer仪器有限公司生产的7116-CP湿度计检测。
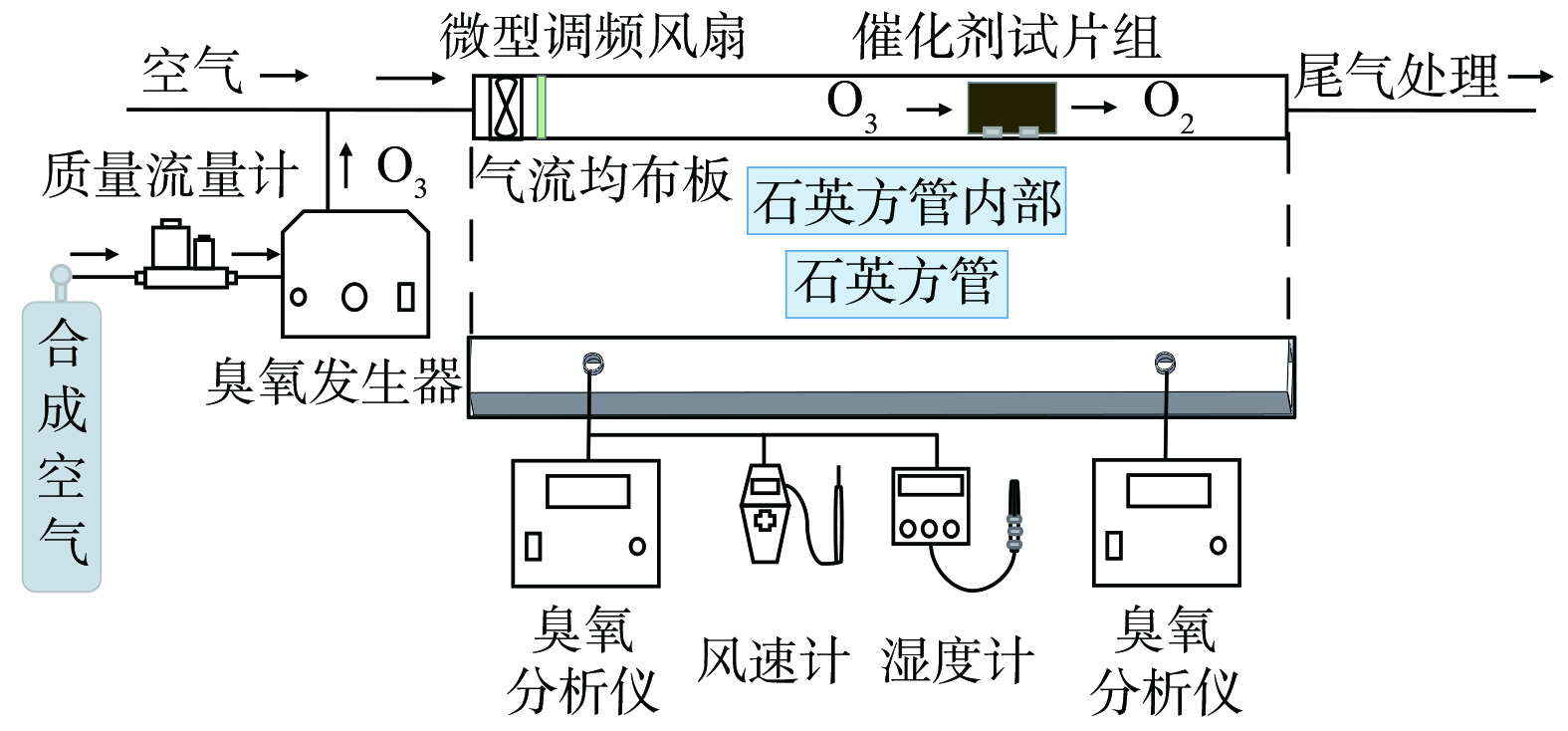 图 1 臭氧催化分解性能测试系统装置示意图Figure 1. Schematic diagram of ozone catalytic decomposition performance test system
图 1 臭氧催化分解性能测试系统装置示意图Figure 1. Schematic diagram of ozone catalytic decomposition performance test system臭氧分解率计算公式如式(2)所示。
臭氧分解率(%)=C入口−C出口C入口×100% (2) 式中:
C入口 C出口 2. 结果与讨论
2.1 催化剂表征
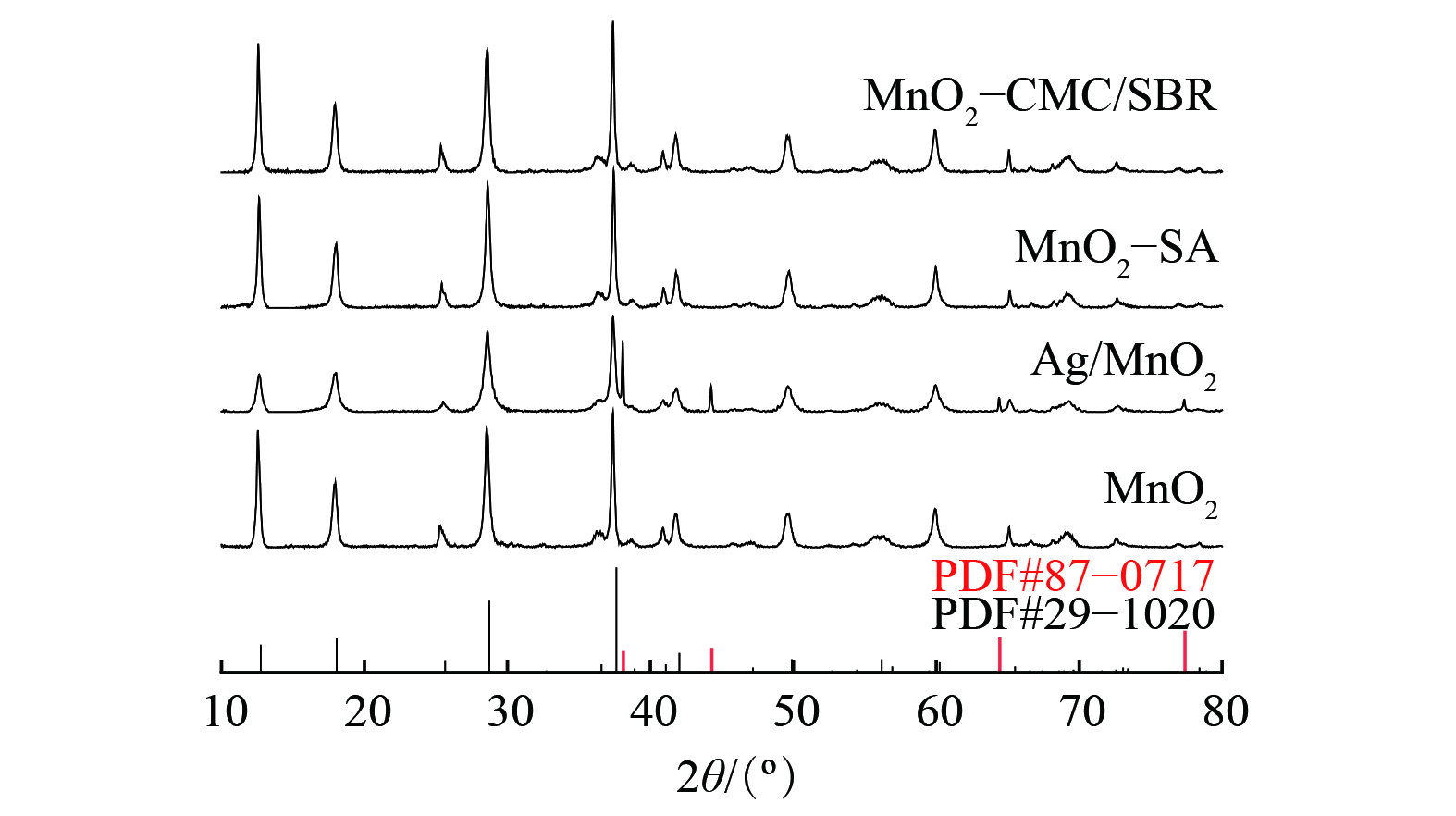
1) XRD、BET表征和脱落率。图2为MnO2和涂层的X射线衍射仪分析结果。MnO2和涂层样品的X射线衍射峰基本相同,皆在2θ角分别为12.7°、18.1°、28.7°、37.6°和60.2°处出现特征峰,与四方晶系锰钾矿MnO2的标准谱图(JCPDS 29-1020)相吻合。这表明添加CMC/SBR复配粘结剂和SA粘结剂均未改变或破坏催化剂的晶体结构。Ag/MnO2除含有锰钾矿MnO2的特征峰(JCPDS 29-1020)外,在2θ角分别为38.1°、44.3°、64.4°、77.4°处存在立方相Ag的特征峰(JCPDS 87-0717)。这表明在催化剂掺杂Ag的制备过程中,部分Ag未进入到MnO2隧道中,而在MnO2表面形成Ag纳米颗粒。
表1为MnO2和涂层的BET,以及脱落率测试结果。添加粘结剂会导致催化剂比表面积减小,且粘结剂含量越高,比表面积越小。平均孔径随粘结剂含量增加而增加,这是由于粘结剂对催化剂微孔产生了堵塞。同时,粘结团聚又在涂层中产生了新孔隙。当粘结剂含量相同时,添加SA粘结剂比添加CMC/SBR复配粘结剂的比表面积更大,这是由于SA热分解温度(220~280℃)低于SBR分解温度(>400℃),经300℃焙烧后SA大部分热分解,MnO2会更充分地暴露[15-16]。
表 1 催化剂的BET和脱落率测试结果Table 1. BET and the rate of bond failure test results of catalyst催化剂种类 BET/(m2∙g−1) 孔径/nm 孔体积/(cm3∙g−1) 脱落率/% MnO2 21.3 23.0 0.10 - MnO2-CMC/SBR0.5% 17.1 17.8 0.09 7.18 MnO2- CMC/SBR1.0% 14.3 21.4 0.09 2.15 MnO2- CMC/SBR1.5% 10.1 54.2 0.07 0.84 MnO2- SA0.5% 20.3 21.4 0.09 13.16 MnO2- SA1.0% 16.1 33.5 0.08 4.15 MnO2- SA1.5% 10.7 53.6 0.07 1.25 | Show Table DownLoad:
CSV
DownLoad:
CSV
催化剂涂层的脱落率随粘结剂含量的增加而减小。这是由于粘结剂含量越高,对MnO2的粘结强度越大。当粘结剂含量≥1.0%时,MnO2-CMC/SBR和MnO2-SA催化剂的脱落率均小于5%。而当粘结剂含量相同时,催化剂MnO2-CMC/SBR的脱落率小于MnO2-SA,这也同样说明了经300 ℃焙烧后MnO2-SA催化剂中SA粘结剂相比CMC/SBR粘结剂分解得更彻底,故粘结能力下降。
2) SEM表征。利用扫描电子显微镜分别对催化剂及涂层样品进行观察(图3)。图3(a)和(b)表明,MnO2和Ag/MnO2粉末的微观形貌相似,均呈现海胆球状的团簇结构,颗粒直径为1~50 μm。所有团簇体颗粒均为具有规则的长直纳米棒紧密交织团聚而成,纳米棒的长度为200~500 nm,平均直径约20 nm。
图3(c)和(d)表明,添加CMC/SBR复配粘结剂后MnO2纳米棒变短,长直边界线消失,表面出现少量SBR球状突起。纳米棒的平均直径增至40~60 nm后,催化剂出现一定程度团聚。这说明粘结剂对MnO2产生粘结和包覆,且随粘结剂含量的增加SBR球状突起增多,包覆作用增强。图3(e)和(f)表明,添加SA时涂层催化剂纳米棒变短,平均直径增至40~60 nm,无明显突起,相比添加CMC/SBR时纳米棒具有更清晰的边界。这说明催化剂受到SA包覆作用相对更弱些,与BET结果相对应。
2.2 催化分解臭氧性能评价
2.2.1 浆料构成对臭氧分解性能的影响
1) 粘结剂类型及含量的影响。图4为粘结剂类型及含量对臭氧分解性能的影响。图4(a)表明,当CMC/SBR粘结剂质量分数由0.5%增至1.5%时,臭氧分解率由37.9%降至21.7%。类似地,图4(b)表明,当SA粘结剂质量分数由0.5%增至1.5%时,臭氧分解率从53.0%降至39.1%。随着粘结剂含量的增加,催化剂对臭氧的分解性能降低。这可能归因于添加粘结剂对MnO2产生一定包覆和粘结作用,使其比表面积减小,活性组分MnO2不能充分暴露,从而减弱了催化剂与臭氧的接触。由于300 ℃焙烧时,添加的SA相对CMC/SBR热分解得更充分,从而导致添加SA后催化剂比表面积高于相同含量的CMC/SBR,且活性组分MnO2暴露更充分,因此,MnO2-SA比MnO2-CMC/SBR具有更高的臭氧分解率。MnO2-SA0.5%涂层的臭氧分解率为50.2%,而MnO2-SA1.0%涂层的臭氧分解率为46.8%,两者相差不大,而SA含量为0.5%和1.0%时涂层脱落率分别为13.16%和4.15%。因此,综合考虑催化剂涂层结合性能和臭氧分解性能,选取质量分数为1.0%的SA粘结剂进行后续实验。
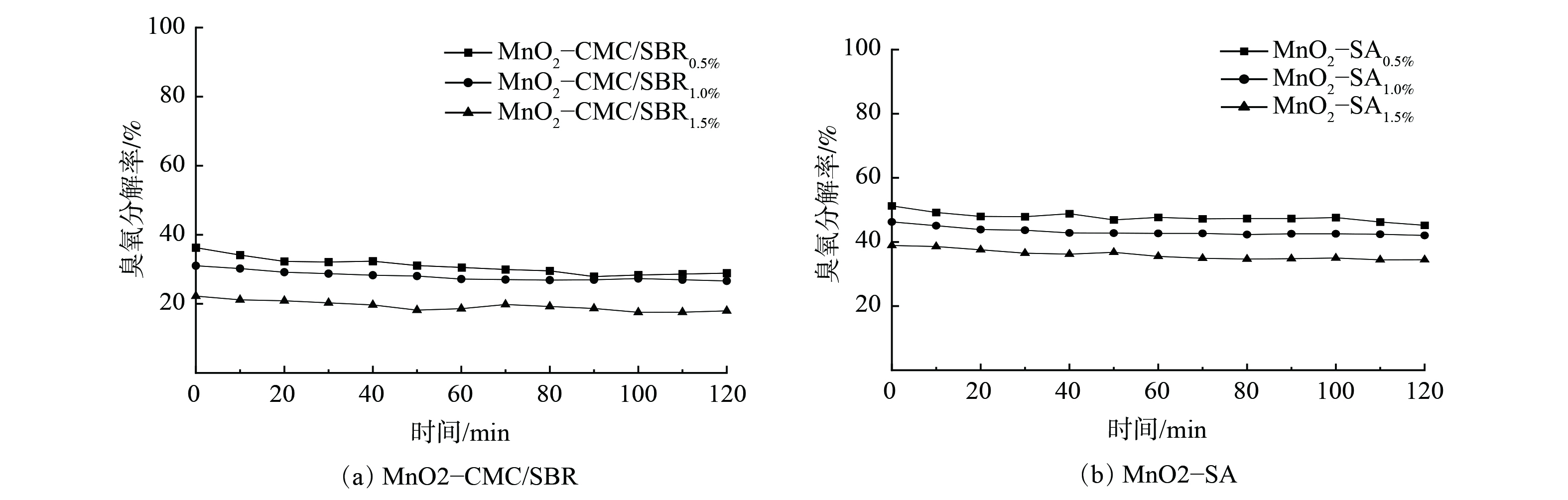 图 4 不同粘结剂及含量对臭氧分解性能的影响Figure 4. Effect of different binder and content on ozone decomposition注:测试条件为臭氧质量浓度2.1 mg∙m-3,RH=15%,风速1.5 m·s-1,板长100 mm,板间距5 mm,涂覆量70 g∙m-2。
图 4 不同粘结剂及含量对臭氧分解性能的影响Figure 4. Effect of different binder and content on ozone decomposition注:测试条件为臭氧质量浓度2.1 mg∙m-3,RH=15%,风速1.5 m·s-1,板长100 mm,板间距5 mm,涂覆量70 g∙m-2。2) 催化剂活性组分的影响。图5为Ag修饰前后MnO2催化剂的臭氧分解性能。催化剂Ag/MnO2-SA1.0%反应进行2 h后,臭氧分解率为64.4%,而MnO2- SA1.0%的臭氧分解率仅42.1%。Ag修饰可使MnO2催化分解臭氧效率提高约50%。氧化锰表面存在的氧空位是臭氧吸附和分解的主要活性中心。Ag掺杂MnO2后,一方面进入到MnO2隧道中的Ag不仅可更均匀分布在MnO2晶体中,还能显著降低Mn—O配位数,在晶体结构保持完整的同时促进催化剂生成更多的氧空位[9];另一方面,MnO2表面存在的Ag纳米颗粒对臭氧更易形成化学吸附,同时Ag纳米颗粒还与MnO2间形成协同效应,以提高催化剂表面的氧空位浓度,进而提高系统的催化活性[17]。
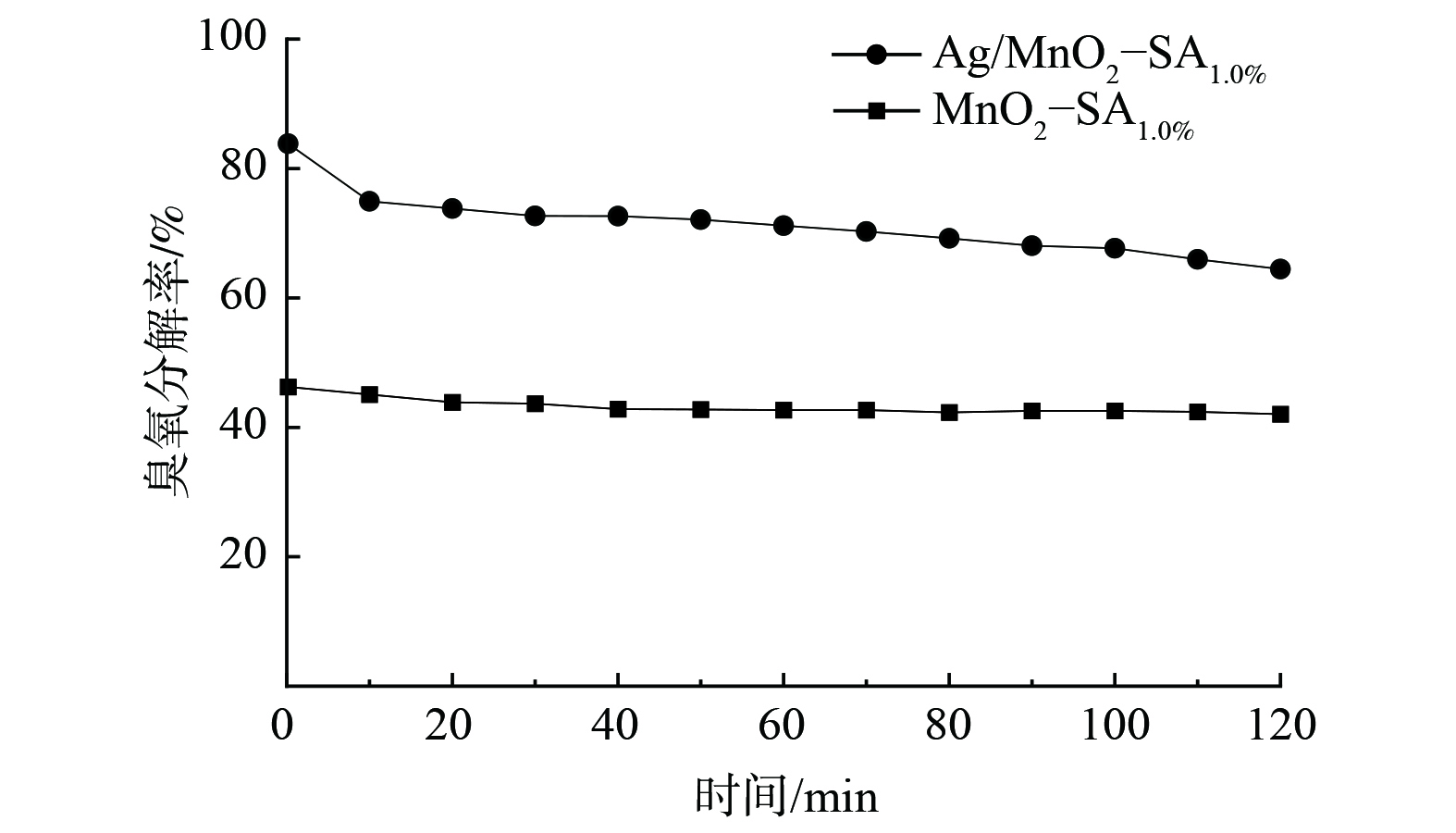 图 5 不同催化剂活性组分对臭氧分解性能的影响Figure 5. Effect of different active components on ozone decomposition注:测试条件为臭氧质量浓度2.1 mg∙m−3,RH=15%,风速1.5 m·s−1,板长100 mm,板间距5 mm,涂覆量70 g∙m−2。
图 5 不同催化剂活性组分对臭氧分解性能的影响Figure 5. Effect of different active components on ozone decomposition注:测试条件为臭氧质量浓度2.1 mg∙m−3,RH=15%,风速1.5 m·s−1,板长100 mm,板间距5 mm,涂覆量70 g∙m−2。2.2.2 反应时间对臭氧分解性能的影响
在相同板间距条件下,可通过改变极板长度及气流通过极板速度来调整臭氧分解的反应时间,结果如图6所示。图6(a)表明,极板长度由100 mm增至200 mm,对应反应时间由0.066 s增至0.133 s时,臭氧分解率由85.5%增至98%。图6(b)表明,气流风速从1.0 m·s−1增至2.5 m·s−1,对应的反应时间由0.200 s减至0.080 s时,臭氧分解率从100%逐渐降至86%,且反应0.1 s以上时,臭氧分解率均达到90%以上。
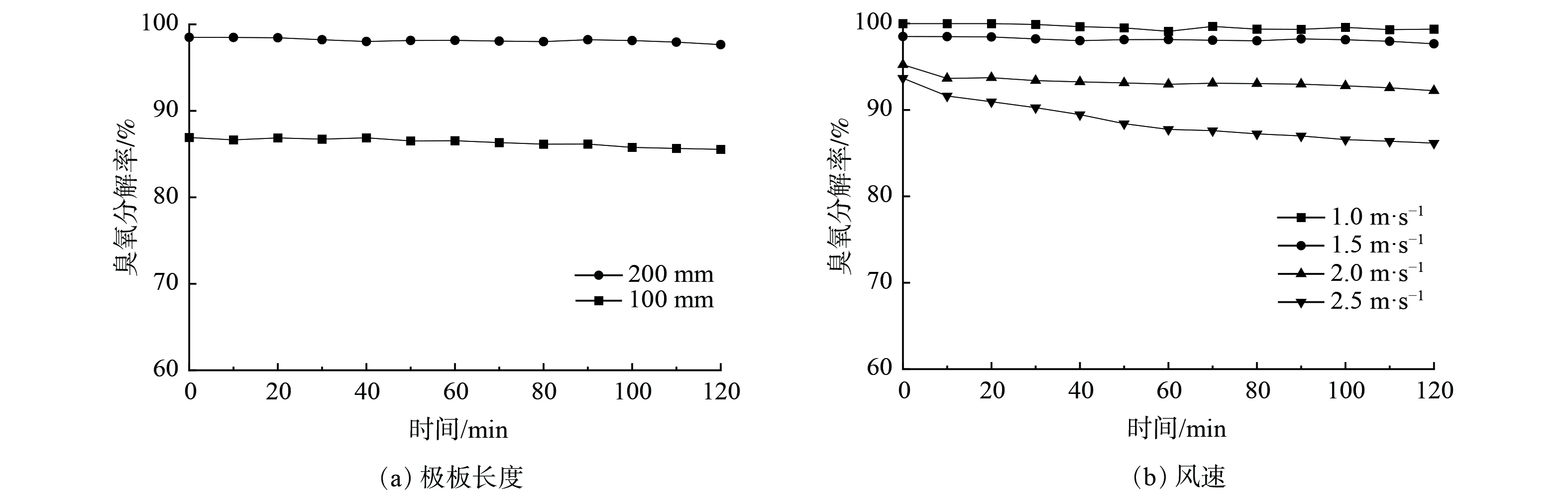 图 6 反应时间对臭氧分解性能的影响Figure 6. Effect of reaction time on ozone decomposition注:催化剂为Ag/MnO2-SA1.0 %,测试条件为臭氧质量浓度2.1 mg∙m-3,RH=15%,板间距3 mm,涂覆量70 g∙m-2。
图 6 反应时间对臭氧分解性能的影响Figure 6. Effect of reaction time on ozone decomposition注:催化剂为Ag/MnO2-SA1.0 %,测试条件为臭氧质量浓度2.1 mg∙m-3,RH=15%,板间距3 mm,涂覆量70 g∙m-2。2.2.3 环境条件对臭氧分解性能的影响
1)初始臭氧浓度的影响。图7为初始臭氧质量浓度分别为2.1、10.7和21.4 mg∙m−3时的臭氧分解率,其对应分解率分别为97.6%、92.5%和86.1%,即分解率随着臭氧质量浓度提高而降低。这是由于当臭氧质量浓度增大时,单位体积催化剂所接触的臭氧分子增多,使部分臭氧分子未能参与反应而使分解率下降。进一步地,还考察了初始臭氧质量浓度为2.1 mg∙m−3的臭氧分解稳定性。在24 h内,Ag/MnO2-SA1.0 %催化剂的臭氧分解效率维持在97%以上,以上结果说明该催化剂稳定性良好。
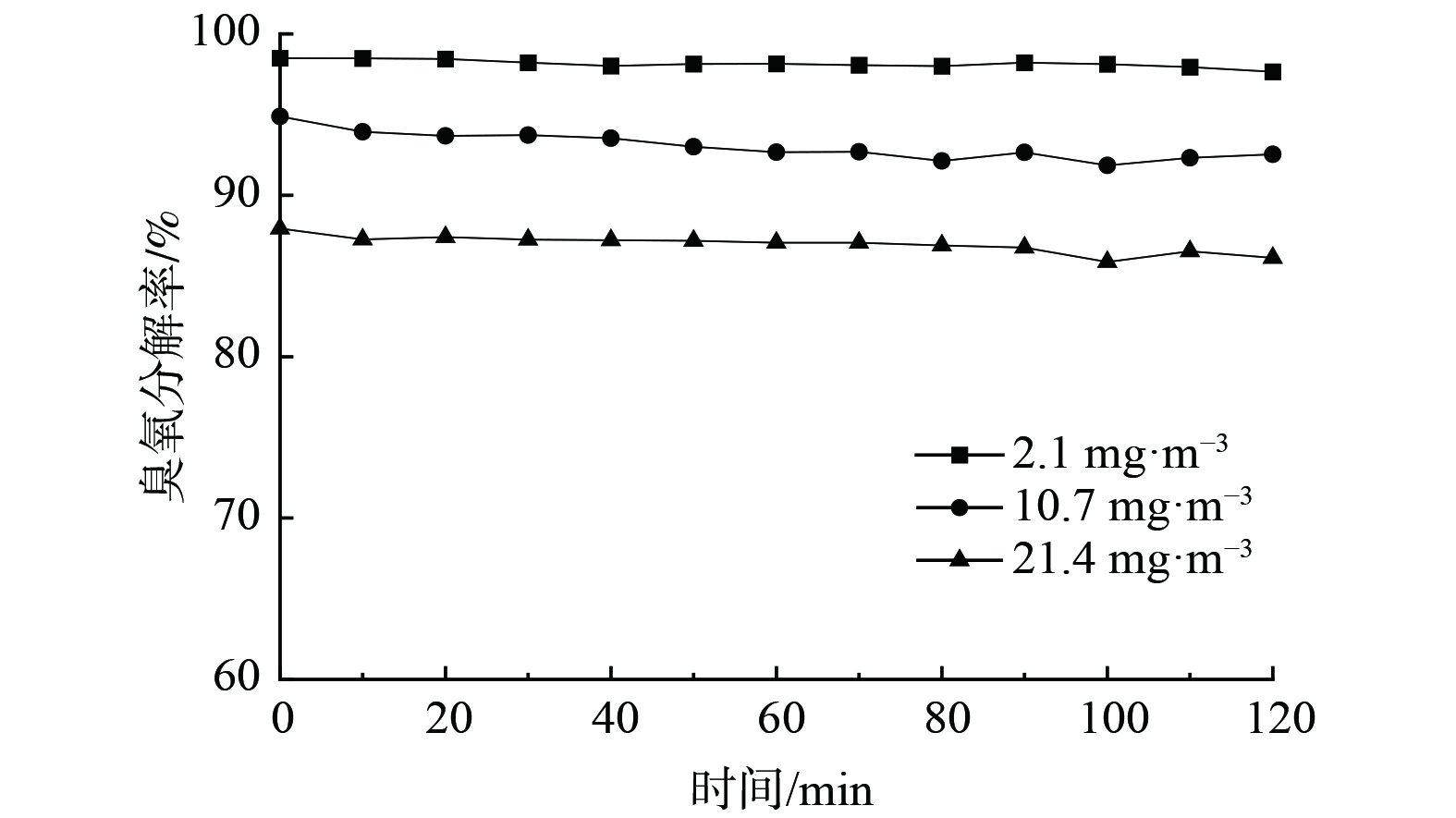 图 7 不同臭氧质量浓度对臭氧分解性能的影响Figure 7. Effect of different ozone concentration on ozone decomposition注:催化剂为Ag/MnO2-SA1.0 %,测试条件为RH=15%,风速1.5 m·s-1,板长200 mm,板间距3 mm,涂覆量70 g∙m-2。
图 7 不同臭氧质量浓度对臭氧分解性能的影响Figure 7. Effect of different ozone concentration on ozone decomposition注:催化剂为Ag/MnO2-SA1.0 %,测试条件为RH=15%,风速1.5 m·s-1,板长200 mm,板间距3 mm,涂覆量70 g∙m-2。2)空气湿度对臭氧分解性能的影响。空气湿度是影响催化剂催化分解臭氧性能的重要因素。在空气相对湿度分别为15%、30%和45%时,臭氧分解率分别为97.6%、96.1%和92.9%(图8)。催化剂的臭氧分解性能随相对湿度的增加有所下降,这是由于水分子和臭氧在催化剂表面形成了竞争吸附[18]。然而,即使湿度达到45%,仍能保持90%以上的分解率。这是由于掺杂的Ag可显著降低Ag/MnO2表面活性位与水分子间的亲和作用,从而提高Ag/MnO2催化剂的抗水性[9]。
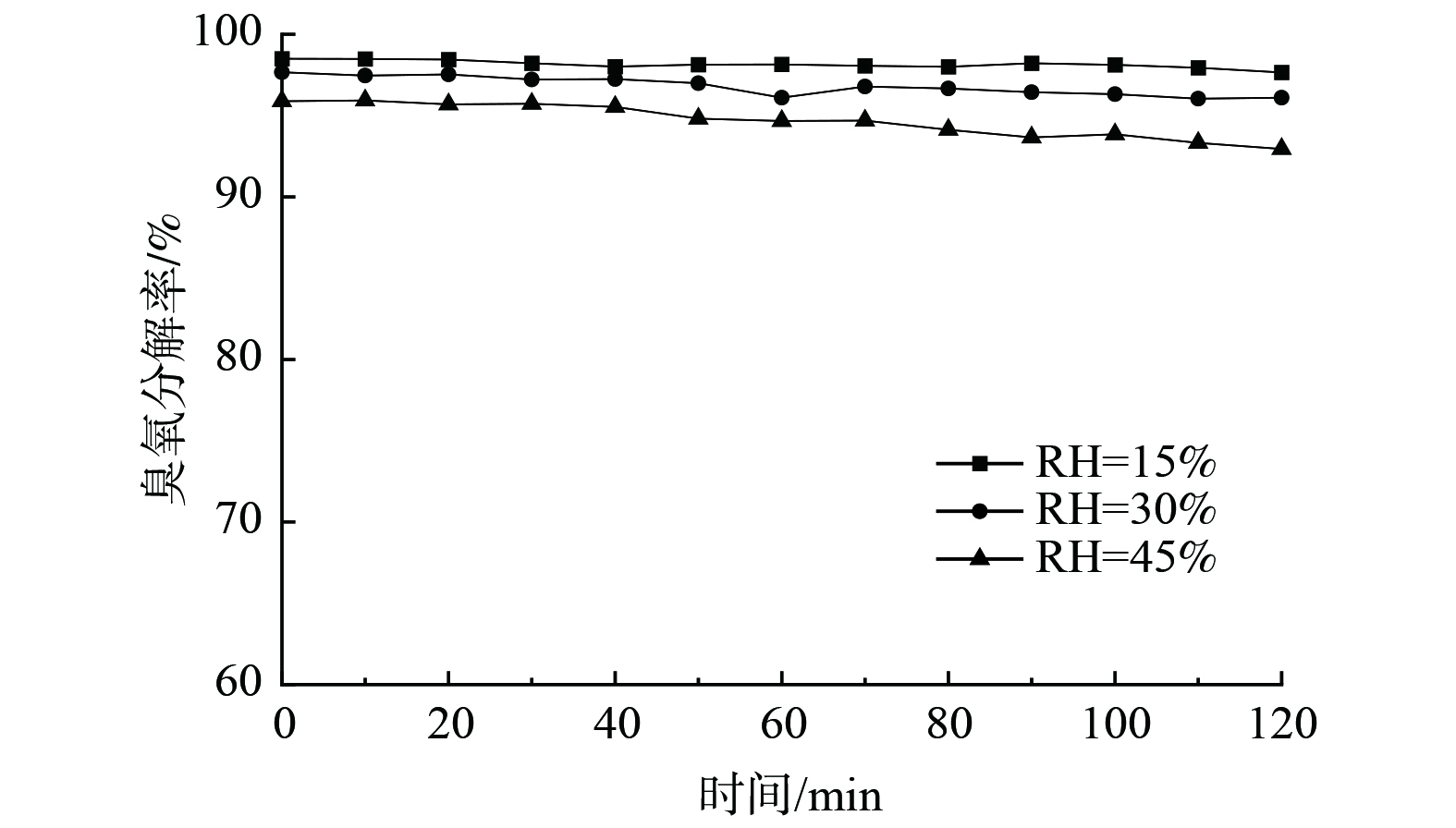 图 8 相对湿度对臭氧分解性能的影响Figure 8. Effect of relative humidity on ozone decomposition注:测试条件为Ag/MnO2-SA1.0 %,臭氧浓度2.1mg∙m-3,风速1.5 m·s-1, 板长200 mm,板间距3 mm,涂覆量70 g∙m-2。
图 8 相对湿度对臭氧分解性能的影响Figure 8. Effect of relative humidity on ozone decomposition注:测试条件为Ag/MnO2-SA1.0 %,臭氧浓度2.1mg∙m-3,风速1.5 m·s-1, 板长200 mm,板间距3 mm,涂覆量70 g∙m-2。3. 结论
1)极板表面涂覆臭氧分解催化剂可实现臭氧原位分解。入口臭氧质量浓度为2.1 mg·m−3、相对湿度为15%、反应时间大于0.1 s时,臭氧分解效率可达90%以上。
2)Ag修饰可使MnO2催化分解臭氧效率提高约50%,但催化剂浆液粘结剂的存在会导致催化分解臭氧效率一定程度下降,需根据催化剂的结合能力和臭氧分解性能合理选择粘结剂及其添加量。
3)在相同反应条件下,提高风速和臭氧浓度,臭氧分解效率下降,可通过增加极板的板长或降低净化器运行风速以获得较大的停留时间,从而使得臭氧高效分解。Ag掺杂可明显改善MnO2的抗水性。当湿度从15%增至45%时,臭氧分解效率从98%降至约92%,分解率仅降低6%。
-
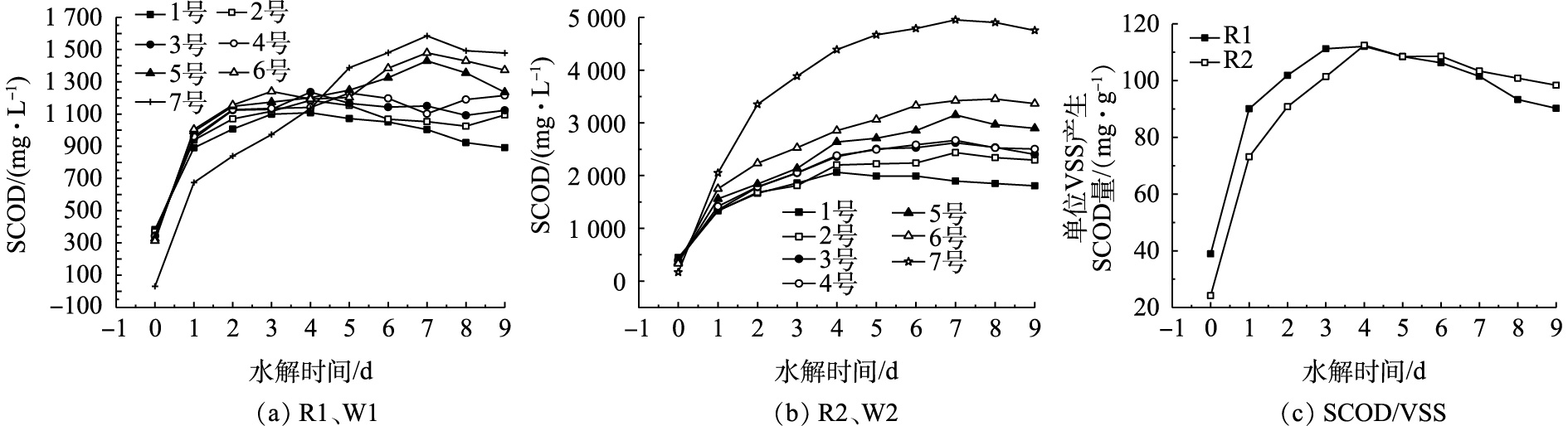
图 2 不同比例的剩余污泥对水解程度的影响
Figure 2. Effect of different proportions of excess sludge on hydrolysis
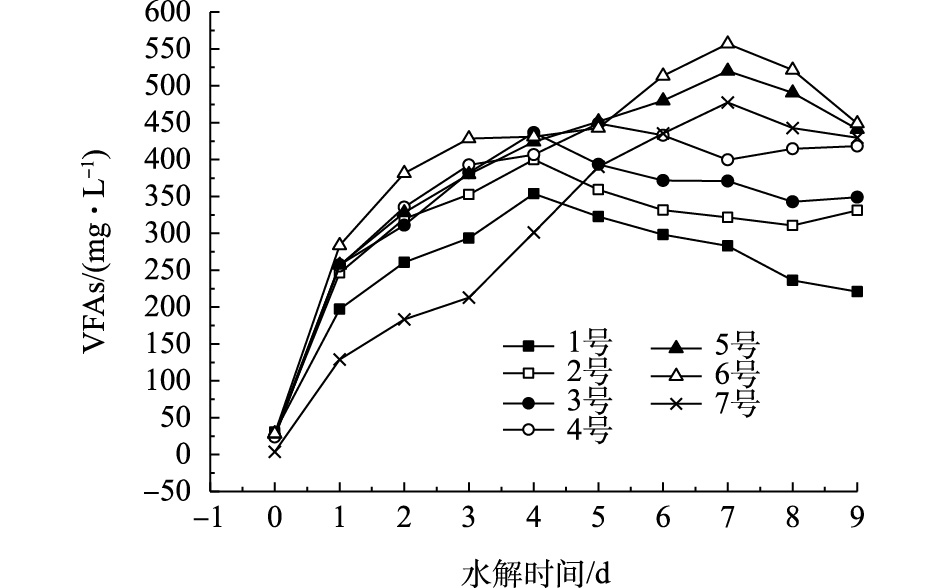
图 3 不同比例的剩余污泥对VFAs的影响
Figure 3. Effect of different proportions of excess sludge on VFAs
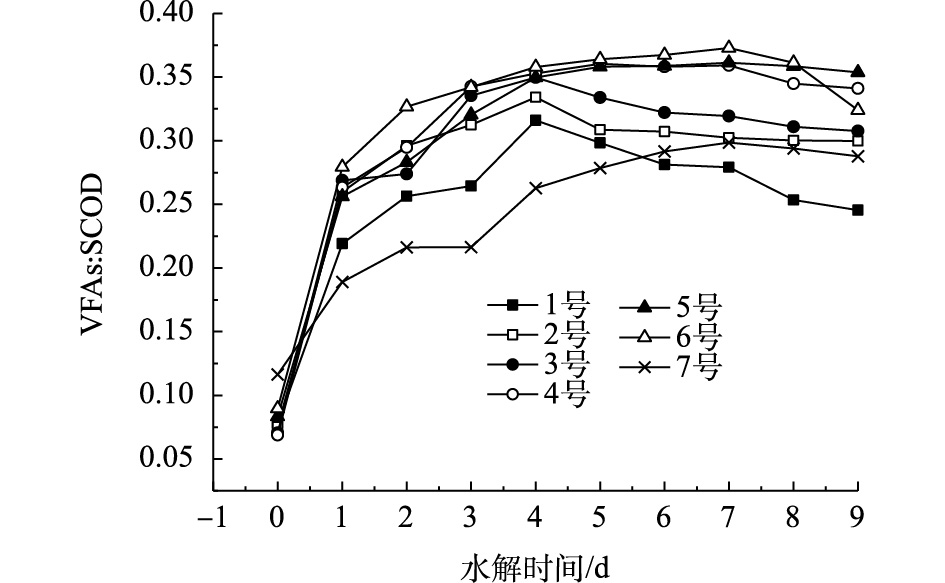
图 4 不同比例的剩余污泥对VFAs∶SCOD的影响
Figure 4. Effect of different proportions of excess sludge on VFAs∶SCOD
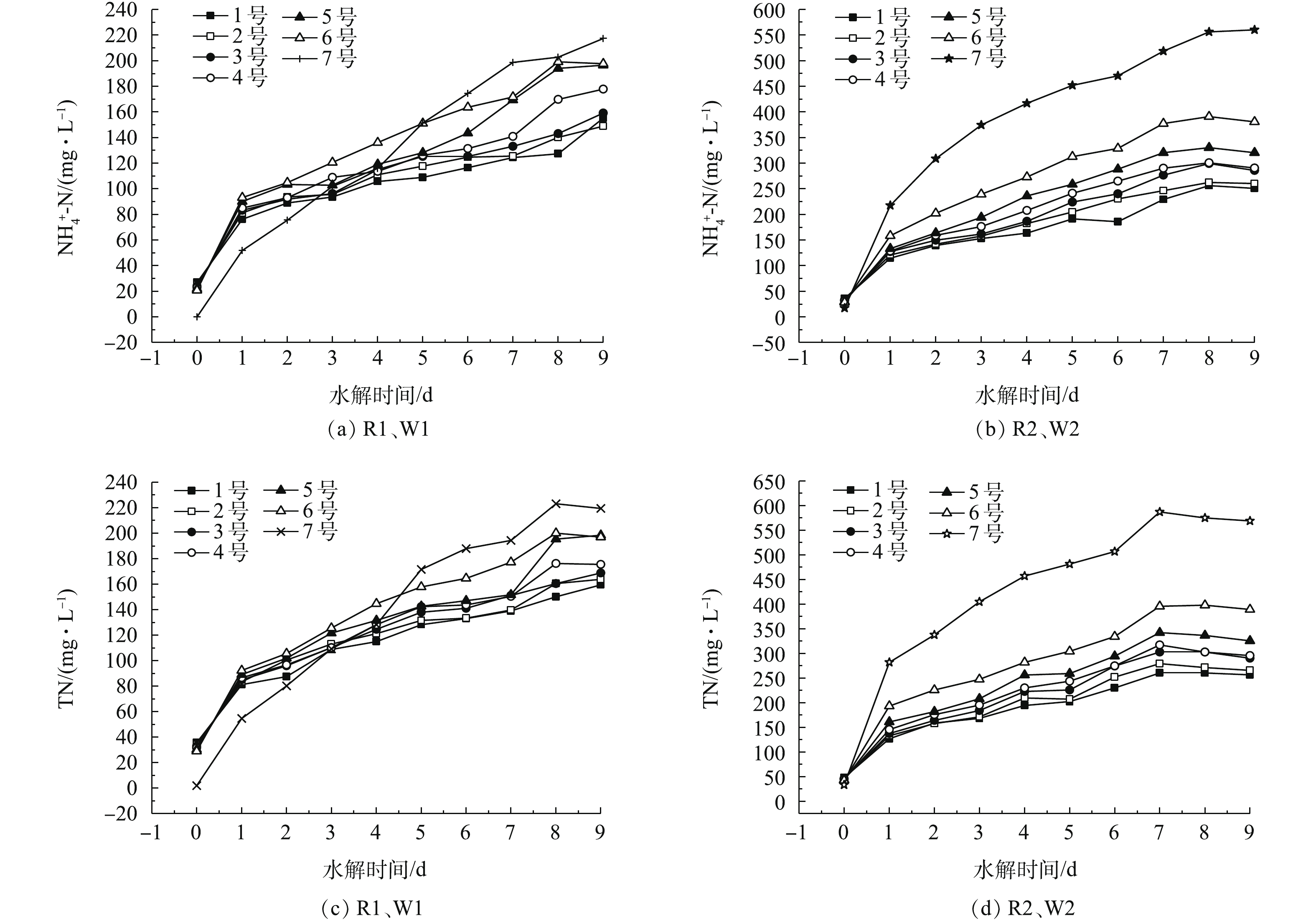
图 6 不同比例的剩余污泥对N元素的影响
Figure 6. Effect of different proportions of excess sludge on N element
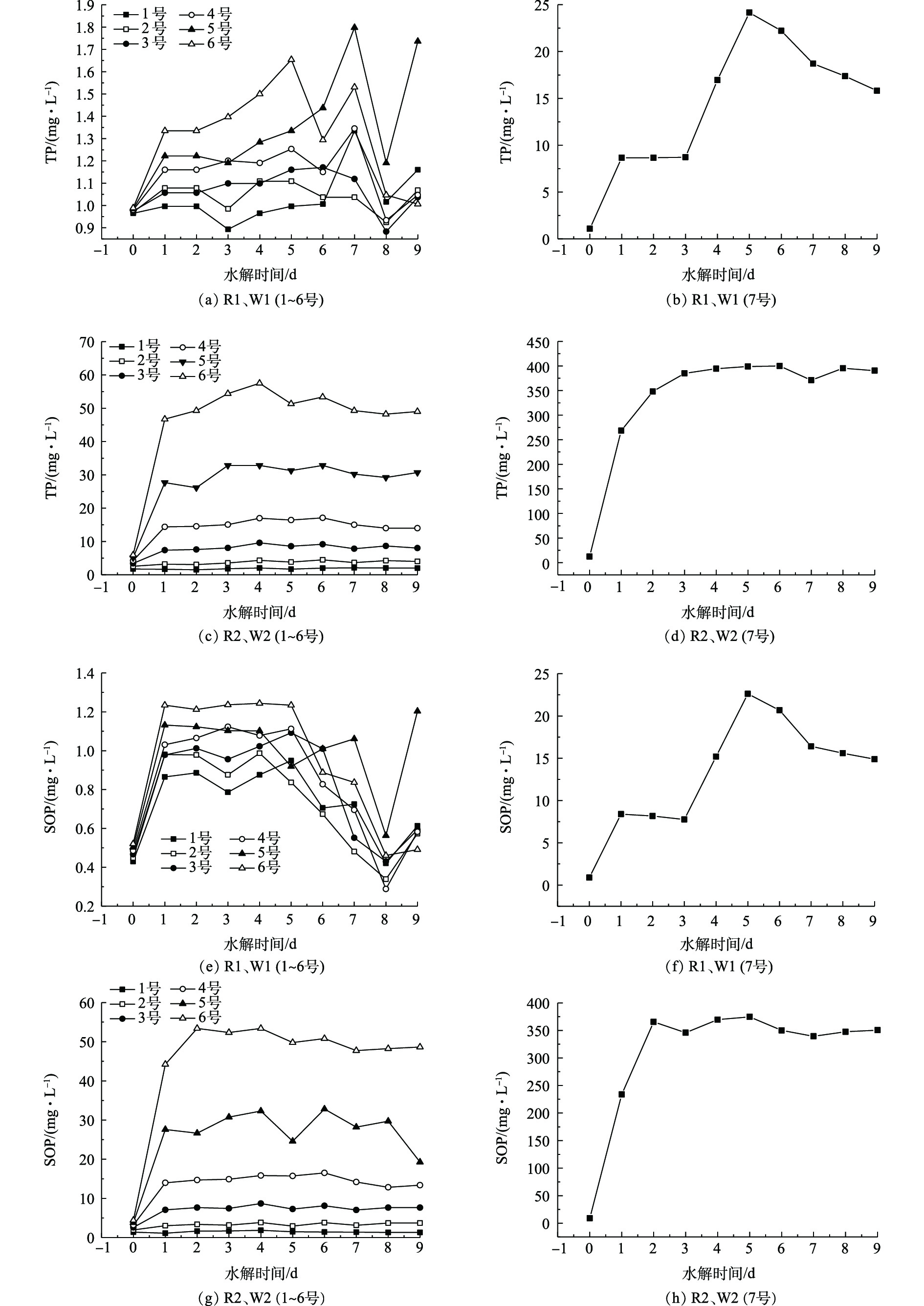
图 7 不同比例的剩余污泥对P元素的影响
Figure 7. Effect of different proportions of excess sludge on P element
表 1 4种污泥的主要理化指标
Table 1. Main physical and chemical indicators of four types of sludge
mg·L−1 污泥类型 TCOD SCOD SS VSS NH+4 -N
TN TP R1 14 004.3 388.63 16 280 9 980 26.86 35.55 0.97 W1 13 476.3 32.10 23 900 15 240 0 1.95 1.09 R2 36 270.3 444.42 32 480 18 350 36.025 48.33 1.74 W2 25 893.8 208.76 22 980 15 700 17.092 33.24 42.27
下载: 导出CSV
表 2 实验设计污泥投加量
Table 2. Experimental designed sludge dosages
% 污泥类型 1号 2号 3号 4号 5号 6号 7号 R1、W1(以体积计) 0 4 8 12 16 20 100 R1、W1(以VSS计) 0 6.1 12.2 18.3 24.4 30.5 100 R2、W2(以体积计) 0 8 16 24 32 40 100 R2、W2(以VSS计) 0 6.8 13.6 20.5 27.4 34.2 100
下载: 导出CSV
-
[1] 刘绍根, 徐锐, 胡星梅. 污泥性质对污泥水解酸化效果的影响[J]. 环境工程学报, 2015, 9(2): 572-578. doi: 10.12030/j.cjee.20150212 [2] 苏高强, 王淑莹, 郑冰玉, 等. 温度和污泥浓度对碱性条件下剩余污泥水解酸化的影响[J]. 环境工程学报, 2013, 7(4): 1231-1236. [3] 陈远炎, 郭苗芬. 磁絮凝的原理及其工业应用[J]. 有色金属(选矿部分), 1988(1): 42-47. [4] 李永泰. 永磁分离滚筒设计制造中的几个问题[J]. 铸造机械, 1973(5): 26-34. [5] DUANG D C, NATHAPORN A, KITIPHATMONTREE M. The effects of magnetic field on the removal of organic compounds and metals by coagulation and flocculation[J]. Physica Status Solidi, 2006, 3(9): 3201-3205. [6] 何秋杭, 金正宇, 宫徽, 等. 基于强化磁分离的市政污水碳源浓缩技术研究[J]. 水处理技术, 2018, 44(10): 114-118. [7] 李军, 任健, 王洪臣, 等. 初沉污泥水解酸化试验研究[J]. 北京工业大学学报, 2008, 12(12): 1304-1308. doi: 10.11936/bjutxb2008121304 [8] 李斯施, 刘东方, 赵乐军, 等. 臭氧预处理促进剩余污泥的水解酸化[J]. 环境工程学报, 2015, 9(7): 3426-3430. doi: 10.12030/j.cjee.20150757 [9] 苏高强, 彭永臻, 汪传新, 等. 污泥类型对污泥碱性发酵的影响[J]. 化工学报, 2011, 12(12): 3492-3497. doi: 10.3969/j.issn.0438-1157.2011.12.028 [10] 赵峰辉, 于德爽, 刘杰, 等. 温度对超磁分离初沉污泥水解酸化影响的研究[J]. 环境工程学报, 2019, 13(6): 1374-1381. doi: 10.12030/j.cjee.201812016 [11] 国家环境保护总局. 水和废水监测分析方法[M]. 4版. 北京: 中国环境科学出版社, 2002. [12] 苏高强, 汪传新, 郑冰玉, 等. pH对混合污泥水解酸化的影响[J]. 环境工程学报, 2012, 12(12): 4257-4262. [13] 雷彩虹, 孙颖, 杨英. 絮凝剂聚丙烯酰胺对高固体污泥厌氧消化的影响[J]. 工业安全与环保, 2018, 44(1): 24-26. doi: 10.3969/j.issn.1001-425X.2018.01.007 [14] 高永青, 张晶宇, 彭永臻, 等. pH值对剩余污泥水解酸化溶出物的影响[J]. 北京工业大学学报, 2011, 37(1): 139-145. doi: 10.11936/bjutxb2011010139 [15] YUAN Q, SPARLING R, OLESZKIEWICZ J A. VFA generation from waste activated sludge: Effect of temperature and mixing[J]. Chemosphere, 2011, 83(4): 603-607. [16] 刑光熹, 曹亚烃. 太湖地区水体氮的污染源和反硝化[J]. 中国科学(B辑), 2001, 31(2): 130-136. [17] ELEFSINIOTIS P, WAREHAM D G, SMITN M O. Use of volatile fatty acids from an acid-phase digester for denitrification[J]. Journal of Biotechnlogy, 2004, 114(3): 289-297. doi: 10.1016/j.jbiotec.2004.02.016 [18] CHEN Y G, ANDREW A R, TERRENCE M. The efficiency of enhanced biological phosphorus removal from real wastewater affected by different ratios of acetic to propionic acid[J]. Water Research, 2004, 38(1): 27-36. doi: 10.1016/j.watres.2003.08.025 [19] 吴昌生, 徐锐, 刘绍根, 等. 温度对碱预处理絮凝污泥水解酸化影响研究[J]. 安徽建筑大学学报, 2016, 24(1): 59-64. doi: 10.11921/j.issn.2095-8382.20160113 [20] FENG L, WANG H, CHEN Y, et al. Effect of solids retention time and temperature on waste activated sludge hydrolysis and short-chain fatty acids accumulation under alkaline conditions in continuous-flowreactors[J]. Bioresource Technology, 2009, 100(1): 44-49. doi: 10.1016/j.biortech.2008.05.028 [21] 郭京京. 强化污水处理厂剩余污泥微氧水解酸化的研究[D]. 北京: 北京工业大学, 2017. [22] 温沁雪, 薛莲, 陈志强. 污泥浓度对剩余污泥水解酸化过程的影响[J]. 中国给水排水, 2012, 28(21): 103-106. doi: 10.3969/j.issn.1000-4602.2012.21.030 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4467
- HTML全文浏览数: 4467
- PDF下载数: 65
- 施引文献: 0
