





-
随着社会经济的发展,柴油的使用量增加,但是柴油在生产、运输、装卸、加工及使用过程中的泄露会对土壤环境造成一定的污染,直接或间接地危害人类的生命与健康[1-2]。因此,解决柴油污染土壤问题已成为世界各国所共同面临的问题[3]。
目前,针对柴油污染土壤修复的方法主要包括机械、物理、化学和生物修复方法等[4]。其中,机械、物理、化学修复方法具有费用高、容易产生二次污染等不足[5-7]。而生物修复技术是一种高效、环境友好、低成本的技术,能够将柴油等污染物通过微生物代谢转化成无毒的终产物[8-9],因而被广泛应用于修复柴油污染土壤之中[10]。刘沙沙等[11]已成功利用醋酸钙不动杆菌降解柴油以及污染物,经过62 d的生物修复实验,柴油去除率为69.8%。然而,柴油组成的复杂性决定了其降解需要有不同菌株的参与[12],TAO等[13]研究了土著细菌联合体与外源芽孢杆菌(Bacillus subtilis)共同培养降解原油的实验,细菌群落分析结果表明,在确定的共培养条件下,细菌多样性降低,降解效率提高,同时证明芽孢杆菌对长链烷烃有很好的降解效果。
大量的研究证明,微生物在修复有机物污染土壤的过程中具有良好的应用前景,但目前对于构建微生物菌群的研究较少,本研究从柴油污染土壤中筛选、分离出能够降解柴油污染物的微生物,采用组合实验构建优势菌群,探究了其柴油生物降解特性,研究分析了该菌群中各菌种之间的互作机制,为构建降解柴油的菌群提供参考。
-
紫外分光光度计(UV-2102C,中国上海),GC-MS(Agilent6890/5975I,安捷伦),台式高速冷冻离心机(H-2050R,中国长沙),恒温振荡培养箱(HZQ-X160,中国太仓)。实验所用试剂均为分析纯。
柴油污染土壤取自上海金山卫金山大道城河路;柴油为市售0#柴油(密度:0.84 kg·L−1);菌种:实验所用菌种均为从柴油污染土壤样品中筛选分离得到。
-
1)微生物菌种生物量的测定方法。将菌落接种于灭菌的种子培养基中,每2 h取样,采用分光光度计在波长600 nm下测定吸光度值,绘制柴油降解细菌的生长曲线。
2)微生物菌种对柴油降解能力的测定方法。残余柴油浓度采用分光光度法[14]进行测定。菌株对柴油的降解能力采用柴油降解率表示。降解率计算公式如式(1)所示。
式中:R为柴油降解率;C0为柴油的初始浓度,mg·mL−1;Ct为柴油的降解过程测定浓度,mg·mL−1。
3)微生物高效降解柴油的条件优化实验。以构建好的柴油污染物降解菌群为研究对象,考察了初始pH(5.0、6.0、7.0、8.0、9.0)、初始柴油浓度(1.0、3.0、5.0、7.0、9.0 mL·L−1)、初始接种量(体积比5.0%、10.0%、15.0%)对柴油降解率和细菌生物量的影响结果。在30 ℃,150 r·min−1条件下培养5 d,定时取样并测定其中的柴油降解率以及生物量的变化,每组实验重复3次,取其平均值。
4)微生物多样性测试。将培养24 h的菌群混合液按10%的接种量接种到无机盐培养基中,柴油浓度为7.0 mL·L−1,pH=7.0,在30 ℃,150 r·min−1条件下培养,14 d后,将混合菌进行收集,离心弃上清液,收集菌体,测试微生物多样性,该工作由上海美吉医药科技有限公司完成。
5)微生物降解柴油产物的检测。将筛选获得的高效单菌种分别接种到种子培养基中,培养24 h后,离心收集菌体,稀释使其OD600=1.50,并按照最佳的体积比混合后接种于无机盐培养基中,以不加细菌为对照组,柴油浓度为7.0 mL·L−1,在30 ℃、150 r·min−1的恒温振荡培养箱中培养14 d。然后将培养基取出加入1∶1(体积比)硫酸5.0 mL酸化水样,继续加入0.2 g·L−1的氯化钠破乳[15],然后加入石油醚20.0 mL(60~90 ℃)超声10 min。将上述溶液8 000 r·min−1离心10 min,将上清液转移至另一干净的三角瓶中,下层溶液倒入原三角瓶并用石油醚重新提取1次。合并2次提取液,过膜,利用GC-MS测定培养基内降解产物组成及含量。
6)微生物降解十五烷产物的检测。将本研究所构建的混合菌群接种于添加了十五烷(7.0 mL·L−1)的无机盐培养基中,在30 ℃、150 r·min−1的振荡箱里培养14 d,分别取降解3、6、14 d的培养液,按照上述方法处理并检测。
-
从柴油污染土壤中筛选获得4株具有较强柴油降解能力的菌株,其菌落形态和柴油降解能力结果见表1。
-
微生物初步鉴定结果如表2所示。通过16S rRNA测序结果可知,1#、2#、3#、4#菌株分别与Bacillus sp. VOC18、Enterococcus faecalis、Lysinibacillus、Rhodococcus equi有97%、98%、99%、99%的相似性。本研究分别对1#、2#、3#、4#菌株命名为Bacillus sp. VOC18-L1,Enterococcus faecalis-L2,Lysinibacillus-L3,Rhodococcus equi-L4(简称L1,L2,L3和L4)。
-
每种菌的生长曲线如图1所示。由图1可知,4种细菌在24 h均进入指数生长阶段,在该阶段,微生物生长速度快,活性较强,因此,后续实验均采用培养24 h的菌液为接种液。
-
菌群构建的结果如表3所示。由表3可知,编号11的菌群组合在5 d内对柴油的降解率最高,可达到39.6%,这可能是由于不同菌种之间的协同作用,使得混合培养的菌群对于柴油污染物的降解效果要优于单菌,因此,选用该混合菌群作为最佳降解菌群进行后续的研究。
-
1)采用正交实验法确定柴油降解菌群的最佳菌种混合比例。菌群中各菌种的相对含量对柴油等有机物的降解效率有显著的影响。因此,本研究通过正交实验研究了不同混合比例的菌种对柴油降解效率的影响关系,结果如表4所示。由表4可知,降解效果最好的菌种比例为L1∶L2∶L3∶L4=3∶1∶3∶4,在5 d时,降解柴油效率为52.5%,本研究将此比例的微生物组合命名为OCDL-3134。由于该菌群中的每种细菌都具有特定的作用,因此,柴油降解效率明显得到提高[16-17]。然而,将培养了14 d的微生物进行多样性分析结果如图2所示,由图2可以看出,OCDL-3134经过14 d的培养后,菌种之间的比例变为2∶6∶5∶1,这说明在降解过程中,4种菌根据环境的变化发挥着协同作用,并自行调整他们之间的相对丰度,以实现充分利用柴油污染物的目的。此时L2和L3菌种变为优势菌种,说明在培养后期,L2和L3菌种发挥了重要作用,这与其自身的功能是相一致的。同时也表明确定各种微生物的初始接种比例的菌群构建方案具有一定的合理性。有研究[18]表明,一旦长链烷烃耗尽,就会缺乏碳源和能量用于其生长,而由长链烷烃降解形成的短链烃类化合物则被其他菌种继续代谢利用而进一步降解。
2)初始pH对OCDL-3134菌体生长和柴油降解效率的影响。环境pH可引起细胞膜电荷的变化,从而影响微生物对营养物质的吸收,影响代谢过程中酶的活性,改变营养物质的可给性和有害物质的毒性。本研究探讨了初始pH对OCDL-3134菌体生长和柴油降解效率的影响。由图3(a)可知,当环境pH过高或过低时,会对微生物产生抑制,这可能是由于此条件严重影响了细菌利用柴油的能量和物质代谢进程,从而影响菌体生长,因此会显著降低柴油的生物降解效率。当pH为7.0时,OCDL-3134对柴油降解的效率达到最佳,此时生物量也达到最高,如图3(b)所示。因此,OCDL-3134对柴油的降解效率和生物量的最佳初始pH均为7.0。
3)柴油浓度对OCDL-3134菌体生长和柴油降解效率的影响。柴油浓度是微生物代谢过程的一个重要因素,对微生物降解性能有一定影响[19]。柴油浓度较低时,碳源不足,细菌生长缓慢,对柴油降解效果不佳。随着柴油浓度的增加,碳源可以满足微生物生长,微生物的降解效果也随之增大。但随着柴油浓度继续增加,柴油降解菌的活性受到抑制,同时培养基表面形成一层油膜,使得溶液内的溶解氧浓度降低,抑制微生物的生长繁殖,从而影响对柴油的降解。如图4(a)所示,随着柴油浓度的增大,微生物的柴油降解率呈先上升后下降的趋势,在柴油浓度为7.0 mL·L−1时,降解效果最好,达到50.0%以上。同时,图4(b)为初始柴油浓度对生物量的影响结果,随着柴油初始浓度的增加,生物量呈先增后降的趋势,结果表明OCDL-3134在初始柴油浓度为7.0 mL·L−1时生物量最佳。
4)接种量对OCDL-3134菌体生长和柴油降解效率的影响。如图5所示,接种量在体积比为10.0%时,微生物对柴油的降解率较高。此后,再增加接种量反而导致新增细胞减少,进而使菌株整体活性下降,降解柴油的后劲不足。15.0%的接种量虽然生物量最多,但由于微生物大量繁殖,造成菌株集中,短时间内消耗了培养基中大量营养成分,不利于新菌株的持续生长,从而影响了菌株对柴油的降解率,因此,根据实验结果最佳接种量为10.0%。
-
1)菌群OCDL-3134降解柴油过程中的产物分析。各单菌种和OCDL-3134对柴油降解的产物结果如图6所示。图6(a)是原始柴油的组分,由此可知,原始柴油的组分非常复杂,主要包括C13~C24的烷烃。图6(b)是L1培养14 d后的产物,通过与图6(a)对比可发现,L1对短链烷烃降解效果好,推测可能是由于该菌在生长代谢过程中产生了相应的表面活性剂[13],促进了微生物与柴油的接触,从而对短链烷烃具有较好的降解效果;图6(c)是L2培养14 d后的产物,通过与图6(a)对比可发现,整体烃类的含量降低。该菌株具有利用该有机物代谢产酸的能力[20],可以将柴油降解的一些产物分解成小分子的酸,因此,在柴油降解过程中发挥着重要作用;图6(d)是L3培养14 d后的产物。与图6(a)对比可发现,短链以及长链烷烃的含量菌有所减少。该菌能够在好氧条件下代谢简单的碳水化合物,因此,在小分子烃类物质的降解过程具有重要作用,同时在柴油降解的后期可能会具有重要贡献,这一点与图3的结果是一致的;图6(e)是L4培养14 d后产物,与图6(a)对比可发现,整体的烃类含量降低,而环苜蓿烯的含量也有明显的下降。有研究[20]表明,该菌株的主要特性是能够有效降解芳香烃,因此,对于柴油中芳香烃的降解具有重要作用。
综上所述,L1、L2、L3、L4对柴油污染物中的有机烃类物质具有一定的降解效果,并且不同的菌种对不同链长的烃类物质降解效果也有显著差异。然而,L1、L2、L3、L4却不能将柴油完全降解。而L1、L2、L3、L4混合形成的菌群OCDL-3134对柴油降解效果显著,如图6(f)所示,相同时间内,柴油几乎被完全降解,转化成无毒无害的酸类小分子、二氧化碳和水。这表明混合菌对柴油的降解效率显著优于单菌株。
2)菌群OCDL-3134降解十五烷过程中的产物分析。使用GC-MS检测了以柴油作为碳源的微生物降解的产物,实验结果表明,混合菌群对柴油的降解效果显著优于单菌种的效果。对此已有大量研究[21]证明了微生物对柴油等有机污染物的降解作用,但是生物降解长链烷烃的机理研究报道并不多见,而且对其降解途径也缺乏了解。由于柴油属于混合物,且主要的烷烃为十五烷和十六烷,因此,本研究选择十五烷作为研究对象,初步探讨微生物降解十五烷的机理,如图7所示。由图7(a)可知,第3天样品中的主要成分是十五烷,这表明在前3 d菌种要先适应新环境,降解效率低。而到第6天,OCDL-3134中的各菌种在协同作用下将十五烷降解为C13H28、C13H26O2等化合物,如图7(b)所示。据报道[15],微生物对直链烷烃最常见的降解途径为烷烃末端氧化,微生物攻击直链烷烃的末端甲基,由加氧酶、脱氢酶、水化酶等混合功能氧化酶催化,生成伯醇,再进一步氧化为醛和脂肪酸,脂肪酸接着通过氧化进一步代谢,被彻底氧化成二氧化碳和水。如图7(c)所示,在第15天,十五烷以及产物被OCDL-3134彻底降解为水和二氧化碳等小分子物质。
3) OCDL-3134降解柴油过程中细菌代谢功能的预测。图8是从KEGG数据库中获得的代谢丰度图,碳水化合物代谢的丰度最好,表明微生物一开始是对柴油的代谢,而氨基酸的代谢功能丰度是其次的,可能发挥的作用是对中间代谢产物脱氨基,从而进一步代谢成醛、酮、酸等小分子物质,因此,细菌首先利用柴油,并将柴油分解成中间代谢产物,然后经过代谢途径分解成酸类小分子物质。其他的代谢途径如能量代谢、辅因子和维生素的代谢、核酸代谢等也参与进来,最终一起合作完成降解柴油的任务。而且可以发现,4种菌种的异质降解和代谢丰度较好,这一代谢丰度说明微生物具有增强降解柴油的能力,这与TAO等[13]的研究结果相一致,但由于是混合菌,故不能判断是哪一种菌种产生的作用。代谢功能预测进一步证明了混合菌在柴油完全降解方面优于单种菌种。
-
1)通过排列组合的方式将筛选出的微生物菌种进行组合,得出高效柴油降解菌群OCDL-3134,通过正交实验得出它们之间接种量最优比例为3∶1∶3∶4,同时对混合菌柴油降解性能进行优化,实验测得微生物降解柴油的最优条件为pH=7.0,初始柴油的浓度为7.0 mL·L−1,初始接种量为10.0%,在此优化条件下,测得第14天柴油的最佳降解率为89.0%,这说明所筛选的混合菌种具有较高的应用价值。
2)通过GC-MS检测和微生物多样性功能预测分析证明了微生物对柴油以及十五烷的协同作用高于单菌株的降解效果,4种菌种之间存在协同作用,能够将长链烷烃降解为短链烷烃和小分子物质,并获得自身生长与代谢的能源和碳源。KEGG数据库中获得的代谢丰度图也进一步证明了混合菌在柴油完全降解方面优单种菌种。
柴油降解菌的筛选、菌群构建及其对柴油和十五烷的降解机理
Screening and community construction of diesel oil degrading bacteria and their degradation mechanism of diesel oil and pentadecane
-
摘要: 针对柴油污染土壤生物修复技术效率低的问题,通过构建高效降解菌群修复柴油污染的土壤,采用组合优化和正交实验构建最佳组合与接种比例的菌群,并研究其柴油降解特性。结果表明,通过筛选、鉴定并命名的4株柴油降解菌为Bacillus sp. VOC18-L1、Enterococcus faecalis-L2、Lysinibacillus-L3、Rhodococcus equi-L4;当4株菌接种比例为3∶1∶3∶4,pH = 7.0,30 ℃,转速150 r·min−1时,柴油降解的效果最佳,14 d对7.0 mL·L−1的柴油降解率达到89.0%。通过气相色谱质谱联用仪(GC-MS)检测柴油降解产物,发现该混合菌株能将柴油中的烷烃降解为短链烷烃,最终转化为小分子物质。同时利用KEGG数据库获得代谢丰度图并初步预测每种菌的功能,根据微生物多样性测试结果,进一步证明了混合菌对柴油完全降解的效果优于单种菌种。通过人工构建的微生物菌群可以有效地应用于柴油污染土壤的修复。Abstract: In view of the current low efficiency of bioremediation technology for diesel oil-contaminated soil, a highly efficient flora was constructed to remediate the polluted soil. A combinatorial optimization and orthogonal design were used to construct the flora with the optimal combination and inoculation ratios, and their degradation characteristics of the diesel oil were also studied. The results showed that four diesel degrading bacteria were screened, identified and named as Bacillus sp. VOC18-L1、Enterococcus faecalis-L2、Lysinibacillus-L3、Rhodococcus equi-L4. The optimal degradation efficiency was obtained at their inoculation ratios of 3∶1∶3∶4, pH=7.0, 30 ℃ and the rotational speed of 150 r·min−1. After 14 days of incubation, the degradation efficiency of 7.0 mL·L−1 diesel oil reached 89.0%. The degradation products of diesel oil were detected using GC-MS, which indicated that these mixed strains could first degrade the long-chain alkanes in diesel oil into short-chain alkanes and finally converted them into some small molecular organics. The metabolic abundance maps obtained from KEGG database were used to predict the functions of each strain. According to the results of microbial diversity test, it was further proven that the mixed flora was superior to pure strain in the complete degradation of diesel oil. The artificially optimized and constructed microbial flora could effectively degrade diesel oil in contaminated soil.
-
砷是煤中普遍存在、毒性较强且易挥发的痕量元素之一,在煤燃烧过程中,砷可随烟气被释放进入环境导致污染,是重要的人为大气砷排放源[1-4]。烟气中的砷除一部分以气态形式存在,大部分可在灰尘等颗粒物中富集[5-10]。无论是气态还是颗粒态砷最终均进入环境并发生迁移转化[11-12],对人体构成健康风险[13-17]。
将烟气砷污染控制技术按实施位置分为燃烧前、燃烧中和燃烧后三类控制技术[18]。燃烧前控制技术的关键是采用物理或化学前处理方法减少煤中的砷含量。煤中砷酸盐和有机砷较少,黄铁矿形态砷居多[19]。连续化学浸提法分析表明,砷以硫化物结合态为主,其次为有机物结合态,其它形态砷含量与煤种相关[20]。燃烧中砷污染控制技术的核心是通过混煤燃烧、添加化学药剂将挥发性强的气态砷转化为不易挥发的砷酸盐等,然后与粉煤灰发生凝并或被吸附而被除尘器捕集[21-22],以达到炉膛出口烟气砷含量降低的目标。燃烧后脱除主要针对末端烟气中的砷,通过吸附剂对其进行物理、化学吸附,实现气态砷在吸附剂表面的固化和稳定化,从而减少烟气中的气态砷含量。
本文综述了近年来烟气砷污染控制领域的主要研究进展,按燃烧前、燃烧中和燃烧后三类控制技术展开论述[18],对比分析了不同控制技术的原理、效果和优缺点。
1. 烟气砷排放现状(The situation of arsenic emission from flue gas)
燃煤烟气中的砷来源于煤炭,煤中砷含量与地质形成过程密切相关。因此,不同国家和地区燃煤中砷含量差异明显,不同煤种砷含量也不相同。表1至表2分别列出了不同国家、地区和煤种中砷的含量[8, 15, 23-26]。世界上煤中砷平均含量为8.3 mg·kg−1。我国煤中砷平均含量为3.18 mg·kg−1,略低于世界平均水平。我国燃煤发电仍然是最主要的能源利用方式,2019年我国燃煤机组发电量为50465 GWh[27],占总发电量约69%。据统计,2020年我国煤炭产量为39亿t[28-29],其中45%以上的煤炭被用于燃煤发电[8].
表 1 不同国家和地区煤砷含量Table 1. The average contents of arsenic in coal from different countries and areas国家和地区Country and Areas 砷丰度/(mg·kg−1)As 样本数Number of samples 参考文献Ref. 中国 3.18 737 [8] 印度 0.1 4 [26] 美国 1.5 — [30] 俄国 7.8 59 [31] 世界 8.3 119 [32] 中国贵州 3.9 140 [15] Donets Basin 9.4 — [23] Danville 12.7 33 [24] Springfield 9.4 64 [24] 注: —表示文献中未给出, 下同. Note:— Indicates that it is not given in the literature, the same below. | Show Table DownLoad:
CSV
表 2 不同煤种砷的含量Table 2. The average contents of arsenic in different ranks of coal| Show TableDownLoad:
CSV
DownLoad:
CSV
表 2 不同煤种砷的含量Table 2. The average contents of arsenic in different ranks of coal| Show TableDownLoad:
CSV
煤中砷以不同结合形态存在。绝大部分有机结合态砷可被燃烧释放并发生形态转化,粉煤灰中几乎检测不到有机结合态砷;燃烧温度越高,煤中残渣态砷越趋于向可交换态与酸溶态转化,粉煤灰中残渣态比例降低,气态砷所占比例升高[34-35]。热力学平衡计算表明,在900—1200 K氧化气氛条件下,砷主要以As2O3(g)存在[34];煤燃烧后,气态砷占总砷含量的24.5%,大多数砷吸附在颗粒物上[36]。部分不能被脱除的颗粒态、气态砷仍通过末端烟气排放进入大气[8, 37-40]。2003—2006年,我国工业砷排放逐年增加,2006年达到了902.95 t,其中燃煤电厂砷排放量达到了522.13 t,占总排放量半数以上[8]。平均每发电1 MWh将产生大气砷排放1.36—9.07 mg[36]。2000—2006年,燃煤电厂大气砷排放量由354.01 t逐步上升至615.70 t[38]。2020年中国燃煤电厂的大气砷排放量为216—257 t[38]。据统计,我国部分地区大气砷平均浓度超过了《国家环境空气质量标准》和世界卫生组织规定的参考值6.0 ng·m−3和6.6 ng·m−3[41]。燃煤电厂持续产生的大气砷排放不容忽视。此外,烟气砷污染控制还可以减缓SCR催化剂中毒[42-43],降低电厂的运行成本。
2. 燃烧前控制(Pre-combustion removal)
较早用于燃烧前砷控制的技术是通过煤洗选去除矸石进而达到降低砷含量的目的[44-45]。根据水溶性强弱,煤中砷可分为硫化物态(36%)、有机结合态(26%)、砷酸盐(17%)、硅酸盐(16%)、水溶解态与可交换态(5%)[46]。根据砷元素在煤中的主要赋存状态与浸出特征,黄铁矿形态的砷含量较高[19, 47]。煤矸石中含有大量的黄铁矿[48-51],洗选煤技术通过去除煤矸石实现有毒元素协同脱除[52-53]。洗选煤技术可分为湿法洗选煤技术与干法洗选煤技术[45]。干法选煤技术在空气中进行选煤,利用煤与矸石的物理性质差别,通过外力,如气流、振动、摇动与它们组合作用进行筛选,比如风选、跳汰选、电磁选和空气重介质流化床选煤等[44]。湿法选煤技术用液态流体作为分选介质,煤矸石密度比煤大,经过流体时,煤矸石被截留后与煤分离[45],同时一些有毒元素被协同脱除。湿法选煤包括重介质法与浮选法等[54]。湿法洗选是我国现行的主流技术。王文峰等[52]的研究表明,基于重介质的物理洗选煤的砷平均脱除率为62.1%,最低为42.5%,最高为84.3%。表3中总结了一些文献中洗选煤技术的砷脱除效果,部分文献研究发现,洗选煤技术不同程度上降低了煤中的砷含量。
表 3 煤洗选砷脱除效果Table 3. The arsenic removal efficiency of coal washing分类Category 方法Method 最低效率/%Min 平均效率/%Avg 最大效率/%Max 样本数N 文献Ref. 干式 重介质法 42.5 62.1 84.3 6 [52] 干式与湿式 重介质和浮选法 45.3 63.4 84.2 6 [58] 湿式 浮选法 12.5 52.4 89.6 10 [59] 干式与湿式 重介质、跳汰选和浮选法 — 16.5 — 47 [60] | Show TableDownLoad:
CSV
脱除率主要与砷赋存状态密切相关。但也受煤级、粒度以及洗选工艺影响,因此不同样品在不同的洗选工艺中差异较大。褐煤与有机砷含量较高的煤无法通过洗选脱砷,甚至发生严重的砷富集现象。王明仕等[55]的研究中有4个煤样洗选后砷发生富集,它们的平均富集率为82.4%。这是因为煤中的硫与砷具有很强的相关性,但部分煤中硫可能以有机硫或细分散的矿物存在[46, 56],导致砷与硫的脱洗率为负。洗选煤的砷脱除效率还与煤的总砷含量与煤粉粒度有关。研究发现,煤中砷含量为0—0.55 mg·kg−1时,砷主要以有机结合态为主,溶解性差、密度小,洗选过程可表现出一定程度的富集;含量为5.55—8.00 mg·kg−1时,煤中的硫化物态砷含量最高,平均脱除率为67.30%;含量大于8.00 mg·kg−1时平均脱除率为49.82%[46]。此外,有研究发现精末、精小粒、精中粒、精大粒洗选后砷脱除率表现为先变高后降低的趋势,表明它们的脱除程度在精中粒中相对较高[57]。
洗选煤技术降低了煤炭中所含的灰份和硫份,减少很多因燃煤产生的环境问题。通过洗选煤筛分出煤矸石同时将砷含量降低,但不同研究中砷洗选脱除效率相差较大,存在不确定性,脱除效率受到多种条件的影响。另外,洗选煤会产生大量高砷煤泥等废物[46, 52, 57-62],存在较高二次污染风险。
3. 燃烧中控制(Removal during combustion)
燃烧中砷污染控制技术是在煤燃烧过程中通过混煤、添加化学添加剂等方式,使其不易转化为气态砷或富集在细微飞灰颗粒上,而是以固态砷酸盐存在或吸附在粗飞灰颗粒上,从而与底灰飞灰等共同被APCDs捕集。研究证明,炉膛烟气中的As2O3(g)与CaO和CaCO3反应,生成以砷酸钙(Ca3(AsO4)2)、砷酸铁(FeAsO4)为代表的易团聚、热稳定性强、挥发性弱的化合物,减少气态砷释放[15, 63-65]。
3.1 混煤
混煤燃烧是一种洁净煤燃烧技术,混煤在一定程度上解决锅炉结渣问题,还能降低氮、硫氧化物与重金属的排放[66-67]。研究表明,Heshan烟煤与Huolinhe褐煤1:1混燃后,细颗粒物中的砷向粗颗粒物中转移,同时与Heshan烟煤燃烧后相比,PM10中砷降低了33%[66]。经分析,Huolinhe褐煤中含有较多矿物质,混煤燃烧降低了灰熔融温度,促进了铁、钙与铝硅酸盐的反应,抑制细颗粒物产生,而粗颗粒物中的砷更易被脱除。此外,煤中矿物质与砷相互反应产生砷酸盐,抑制砷的挥发。但有研究发现不同配比下,混煤中砷的挥发率均高于两种原煤砷挥发率的加权平均值,导致砷挥发率增加[66-69]。主要原因是褐煤中的高挥发分促进了混煤中的焦炭燃烧,进而加速了硫化物结合态砷分解生成气态砷,因此混煤的砷挥发特性更接近于单一褐煤[69]。目前混煤法进行砷脱除具有一定局限性,煤种、煤质以及矿物含量对脱除效果影响显著,应用范围较窄。
3.2 化学添加剂
郭胜利等[65]通过改性碳酸钙混燃技术,实现了燃煤有毒元素有效控制。硫酸铝(Al2(SO4)3)改性碳酸钙与无烟煤混合燃烧,砷脱除效率达到46.15%。经分析,Al3+半径小于Ca2+,它与碳酸根中氧成键的能量更低。Al3+取代Ca2+的位置,形成点缺陷[70],碳酸钙晶体内部重新排列,Ca2+被活化后利用率增加。Zhao等[15]研究了氧化钙混燃技术,向煤粉中掺入0.3% wt. CaO,燃烧后,PM1中砷降低了56%,PM10中砷降低了6.8%。在该控制技术应用过程中,在900 ℃时主要发生化学吸附且产物为热稳定性很强的砷酸钙。研究发现,气态砷富集在PM1中,氧化钙加入后,大部分砷被吸附剂表面的活化阳离子捕获,气态砷含量明显降低,但炉内仍有部分氧化钙团聚成大于PM1的颗粒,并与气态砷发生反应,导致PM10中砷含量下降不明显。燃烧中砷污染控制不需要对现有APCDs进行改造也无需增加新设备,砷脱除成本低。
4. 燃烧后控制(Post-combustion removal)
燃烧后烟气砷污染控制技术,是利用原有的空气污染控制设备与吸附剂将烟气砷固定并脱除,降低砷排放量。研究发现,APCDs对砷具有协同控制作用[71-75],包括选择性催化还原脱硝(SCR)、静电除尘器(ESP)、布袋除尘器(FF)与湿法烟气脱硫(WFGD),它们的砷脱除效果见表4。
表 4 APCDs的砷脱除效果总结Table 4. Summary of performance of APCDs for arsenic removal from flue gas控制设备Control device 平均脱除效率/% Average removal efficiency 样本数N 文献Ref. SCR 6.4 5 [73] ESP 83.0 4 [71] ESP 96.1 5 [73] ESP 98.3 1 [74] ESP+FF 99.8 1 [75] WFGD 61.0 4 [71] WFGD 48.2 1 [75] WFGD 75.0 1 [74] ESP+WFGD 97.3 — [72] ESP+WFGD 99.6 1 [74] | Show TableDownLoad:
CSV
2020年,燃煤电厂全部完成超低排放改造(ULE)[76]。某电厂改造后,空气污染控制设备(APCDs)对砷的协同脱除效率由约95%提高至约97%[77]。即使完成了ULE改造,2020年中国燃煤电厂的大气砷排放量仍达到216—257 t[38]。,SCR对砷的脱除效果差,ESP最好,WFGD次之。APCDs的砷平均脱除效率已经很高,但现有APCDs无法有效脱除气态砷,尤其是水溶性很低的As2O3(g),而它的毒性极大,加上电厂烟气排放量巨大,这部分砷对生态环境的影响不容忽视。因此燃烧后的烟气还需要添加额外的吸附剂将气态砷固定并脱除。
除尘器可有效捕集到Ca/Fe/Al的砷酸盐[3],其热稳定性强,沸点高不易挥发,环境化学性质较为稳定。因此,将As2O3(g)转化为稳定性强的砷酸盐可以实现气态砷的固化[15, 63-64, 76,78]。CaO、CaSiO3、Fe2O3、γ-Al2O3及其改性材料可以被用于吸附脱除烟气砷。按照文献中的吸附剂组成特点,将其区分为碳基、钙基、铁基、铝基吸附剂、复合金属吸附剂以及其他吸附剂。
4.1 碳基吸附剂
常见的碳基吸附剂包括各种活性炭[36, 79-81]、石墨烯[82]、C60[83]和纳米碳管[84]等。碳基吸附剂具有比表面积大、孔容积大、孔隙率高等特点,因此具有很强的吸附能力[79-81]。Wu等[85]通过密度泛函理论(DFT)模拟了以六元碳环椅形、之字形吸附剂在分子水平上吸附As2O3(g)的过程,认为As2O3分子可以被水平或垂直形式吸附在碳原子表面,6种可能的吸附形式中吸附能最大为− 45.47 kJ·mol−1,最小为− 497.74 kJ·mol−1,这证明活性炭吸附剂与气态砷可发生化学吸附。在模拟烟气条件下,低浓度SO2降低了相邻碳吸附活性位点的静电势,促进了其对As2O3(g)的吸附。由于竞争吸附效应,SO2浓度较高时可抑制其对As2O3(g)的吸附。Charpenteau等[79]研究了商品活性炭、废旧轮胎热解活性炭和黑炭对气态砷的脱除作用,200 ℃时3种吸附剂的砷脱除效率分别为69%、50%和72%;400 ℃时分别为72%、51%和38%。可见不同碳基吸附剂在不同温度时的吸附效果存在明显差异。Marczak等[36]采用商品活性炭将烟煤燃烧烟气的砷浓度由146.2 μg·m−2降低至17.3 μg·m−2,脱除效率达到88.2%。López-Antón等[80]模拟了煤燃烧过程中砷和硒的挥发过程,并采用3种活性炭进行吸附实验,砷吸附容量分别为0.30 mg·g−1、0.35 mg·g−1和0.56 mg·g−1。提高活性炭的用量可以改善气态砷的吸附效果。Player等[81]通过提高活性炭的用量并优化吸附条件,最终实现99.94%的As2O3脱除效率。碳基吸附剂对烟气中气态砷具有较强的吸附效果,但选择性较差,易受烟气组分影响,应用成本相对较高,在实际应用中存在一定难度。另外,碳基吸附剂的热稳定性差,吸附效果受温度影响明显。一般地,超过400 ℃时,砷吸附效果会显著下降[81]。
4.2 钙基吸附剂
钙基吸附剂主要包括氧化钙、碳酸钙、硫酸钙和硅酸钙等。钙基吸附剂对As2O3(g)有较好的脱除效果,可与As2O3(g)发生如下(1)—(7)反应[86-92]:
CaO(s)+As2O3(g)→CaO⋅As2O[88]3 (1) CaO(s)+1/3As2O3+1/3O2→1/3Ca3(AsO4)[86,88−92]2 (2) CaCO3+1/3As2O3+1/3O2→1/3Ca3(AsO4)2+CO[91]2 (3) CaSO4+1/3As2O3→1/3Ca3(AsO4)2+SO2+1/6O[65,88]2 (4) CaSO4+1/2As2O3→1/2Ca2As2O7+SO[87]2 (5) CaSiO3+1/3As2O3+1/3O2→1/3Ca3(AsO4)2+SiO[89]2 (6) 2CaSiO3+As2O3+O2→Ca2As2O7⋅2SiO[87]2 (7) 3Ca2As2O7→2Ca3(AsO4)2+As2O3+O[72]2 (8) 温度较低时钙基吸附剂与As2O3(g)发生物理吸附,类似反应(1),高温时则几乎为化学吸附。研究发现,450 ℃以下,CaO与As2O3以物理吸附为主,750 ℃以上发生化学吸附(反应2)[88]。As2O3(g)与CaO和CaCO3可发生反应(2)、(3)后生成砷酸钙Ca3(AsO4)2[86, 88-92],与CaSO4和CaSiO3反应产物为Ca2As2O7和Ca3(AsO4)2(反应4—7),600 ℃主要产物为单斜晶型的Ca3(AsO4)2,800—1000 ℃时逐步转化为菱面晶型[91],1000 ℃以上时生成的Ca2As2O7则再次分解为Ca3(AsO4)2(反应8),因此CaSO4或CaSiO3与As2O3(g)的最终产物同样也为Ca3(AsO4)2[72]。
从反应(2)—(7)可以看出,除CaSO4外,CaO、CaCO3和CaSiO3与As2O3反应均有O2参与,因此O2可以促进砷的化学吸附[86-92]。As2O3吸附转化过程中CaO晶体表面的晶格氧与表面吸附O2同时对As2O3起到氧化作用[88],促进砷的脱除。此外,研究表明CO2也可促进CaO对砷的脱除效果,但具体机理还需要进一步研究[91]。研究显示,在1300 ℃以下CaO和CaSiO3对砷的吸附容量随温度升高而升高[87, 89-90]。然而,CaSO4的吸附容量却随温度升高而降低。高浓度SO2(5.721 g·m−3)显著抑制了CaO对As2O3的吸附作用,但900 ℃以上时,CaO与SO2反应生成了CaSO4,同样对As2O3(g)具有一定吸附能力,减弱了SO2的抑制效果[88],见化学反应(4)和(5)。SO2浓度较低时(2.002 g·m−3)CaO的吸附容量几乎不受影响[91]。NO对CaO脱除As2O3(g)的影响较小[87, 90]。表5中列出了不同温度与时间下的钙基吸附剂的吸附效果。钙基吸附剂适于高温烟气或混燃过程中气态砷的产生抑制和吸附脱除,在价格成本方面表现出优势。
表 5 钙基吸附剂的砷脱除效果总结Table 5. Summary of performance of calcium-based sorbents for arsenic removal from flue gas吸附剂Sorbent 模拟烟气Simulated flue gas 温度/℃Temp. 吸附时间/minTime 吸附容量/(mg·g−1)Absorption capacity 文献Ref. CaSiO3 N2/O2 1200 10 4.98 [87] CaSiO3 N2/O2/NO/SO2 1200 10 3.50 [87] CaO N2/O2/H2O 750 5 3.75 [88] CaO N2/O2/H2O/SO2 750 5 8.69 [88] CaO N2/O2 800 30 11.82 [91] CaO N2/O2/SO2 800 30 11.51 [91] CaO N2/O2/NO/SO2 1000 10 1.65 [90] CaO N2/O2/NO/SO2 1300 10 1.94 [90] CaSO4 N2/O2 1000 10 3.79 [90] | Show TableDownLoad:
CSV
4.3 铁基吸附剂
Fu等[93]研究发现富砷烟煤燃烧过程中,含铁矿物在砷捕获和形态转化过程中发挥着关键作用。鉴于此,将Fe2O3、Fe3O4或含铁矿物及氧化物称为铁基吸附剂。研究表明,Fe2O3的表面晶格氧将As2O3(g)氧化为As2O5(s),晶格氧可以通过化学吸附O2再生,且As2O5(s)与Fe2O3进一步反应生成砷酸铁(FeAsO4)[80, 92, 94]。DFT研究结果也表明,As2O3(g)可在Fe2O3的(001)面形成8种稳定的吸附结构,吸附能最低为− 275.52 kJ·mol−1[95]。此外,在Fe3O4的(111)面形成四种稳定的吸附结构,均为化学吸附,其中吸附能最低为− 197.96 kJ·mol−1[93]。研究证明,FeAsO4通过下列化学反应(9)生成,该反应的分步反应为(10)—(12)[80]。此外,Fe3O4、铁磁珠与As2O3发生反应(13),可以生成FeAsO4与砷酸亚铁(Fe3(AsO4)2)[96]。
As2O3(g)+Fe2O3+O2(g)→FeAsO4 (9) As2O3(g)↔As2O3(ads) (10) As2O3(ads)+FexOy↔As2O5(ads)+FexOy−2 (11) As2O5(ads)+Fe2O3→2FeAsO4 (12) 4As2O3(g)+3Fe3O4+4O2(g)→6FeAsO4+Fe3(AsO4)2 (13) 表6中列出了不同温度与时间下的铁基吸附剂的吸附效果。在600—900 ℃的温度区间,Fe2O3的砷脱除效率随温度升高由57.02%降低至43.38%[92];在150—900 ℃的温度区间内,Fe2O3/γ-Al2O3的砷脱除效率先升高后降低,600 ℃时效率最高为68%[94]。O2可促进铁基吸附剂对砷的吸附脱除。As2O3(g)转化为FeAsO4会消耗Fe2O3的晶格氧,而O2可以有效补充表面晶格氧[80, 92, 94]。在一定浓度范围内,SO2有利于砷的脱除,可在吸附剂表面与O2、H2O在Fe(Ⅲ)催化作用下生成
HSO−4 SO2−4 NO−3 NO−3 表 6 烟气组分和温度对铁基吸附剂的砷脱除效果Table 6. The arsenic removal efficiencies of iron-based adsorbents in different gas components and reaction temperatures吸附剂Sorbent 模拟烟气Simulated flue gas 温度/℃Temp. 吸附时间/minTime 脱除效率/%Removal efficiency 吸附容量/(mg·g−1)Absorption capacity 文献Ref. Fe2O3 N2/O2 600 90 57.02 1.38 [92] Fe2O3 N2/O2 900 90 43.38 1.08 [92] 铁磁珠 N2/O2/SO2 600 60 42.75 0.53 [96] Fe2O3/γ-Al2O3 N2/O2/SO2/NO 600 — 68.00 — [94] Fe2O3/γ-Al2O3 N2/O2/SO2/NO 900 — 46.00 — [94] | Show TableDownLoad:
CSV
4.4 铝基吸附剂
Hu等[97]通过DFT理论计算,研究了γ-Al2O3(001)表面吸附As2O3(g)的吸附结构和过渡态。结果表明,吸附能最低达到了− 409.13 kJ·mol−1,研究认为As2O3(g)在γ-Al2O3表面存在如图1所示的4种吸附方式[98]。不同烟气组分和温度下铝基吸附剂吸附效果见表7。
 图 1 (a)As3+物理吸附;(b)As3+弱化学吸附;(c)As3+强化学吸附;(d)As5+强化学吸附Figure 1. (a) As3+ physical adsorption; (b) As3+ weak chemical adsorption; (c) As3+ strong chemical adsorption; (d) As5+ strong chemical adsorption(图片借鉴自文献[98])表 7 不同烟气组分和温度下铝基吸附剂的砷脱除效果Table 7. The arsenic removal efficiencies of aluminum-based adsorbents in different gas components and reaction temperatures
图 1 (a)As3+物理吸附;(b)As3+弱化学吸附;(c)As3+强化学吸附;(d)As5+强化学吸附Figure 1. (a) As3+ physical adsorption; (b) As3+ weak chemical adsorption; (c) As3+ strong chemical adsorption; (d) As5+ strong chemical adsorption(图片借鉴自文献[98])表 7 不同烟气组分和温度下铝基吸附剂的砷脱除效果Table 7. The arsenic removal efficiencies of aluminum-based adsorbents in different gas components and reaction temperatures吸附剂Sorbent 模拟烟气Simulated flue gas 温度/℃Temp 吸附时间/minTime 吸附容量/(mg·g−1)Absorption capacity 文献Ref. γ-Al2O3 N2/O2/H2O 300 60 66.62 [99] γ-Al2O3 N2/O2/H2O 400 60 52.30 [99] γ-Al2O3 N2/O2/H2O/SO2 300 60 56.92 [99] γ-Al2O3 N2/O2/H2O/SO2 400 60 46.49 [99] γ-Al2O3 N2/O2/H2O 750 90 9.27 [98] γ-Al2O3 N2/O2/H2O/HCl/SO2 750 90 8.44 [98] γ-Al2O3 N2 400 60 0.99 [100] γ-Al2O3 N2 600 60 1.48 [100] γ-Al2O3 N2/O2 400 60 1.55 [100] γ-Al2O3 N2/O2 600 60 2.17 [100] | Show TableDownLoad:
CSV
模拟烟气条件下,砷脱除效果随温度升高而降低[99]。在较高温度条件下,γ-Al2O3比表面积显著下降并开始向θ-Al2O3转变,导致脱除效果变差[98]。由于竞争吸附效应,SO2一定程度上抑制了γ-Al2O3对As2O3(g)的吸附。NO显著抑制铝基吸附剂对砷的脱除效果,NO与As2O3(g)在γ-Al2O3表面铝原子上发生竞争吸附,同时竞争晶格氧,NO则被氧化为NO2,而As2O3(g)也被转化为氧化形态。尽管晶格氧可以通过O2进行不断补充,NO仍然会表现出抑制效果。SO2与NO同时存在时,它们可在γ-Al2O3上的Al-OH位生成(Al-SO3NO)中间体,随后在O2存在条件下,转化为(Al-SO4)和(NO2),在一定程度上避免了晶格氧耗竭从而减弱了NO对As2O3(g)的吸附抑制效应[99]。铝基吸附剂的适宜脱除温度低于钙基吸附剂,价格相对低廉,脱除效果较好。
4.5 复合金属吸附剂
单金属氧化物吸附剂对砷吸附虽然在As2O3(g)浓度较低时具有较高的脱除效率,但吸附容量有限,较难满足高浓度As2O3(g)或复杂烟气条件下的需求,且γ-Al2O3与CaO高温时会出现比表面积下降[88]或晶型转化[98]等问题。为了提高吸附剂的吸附容量和稳定性,改善吸附效果,研究提出了多种复合吸附剂用于烟气砷污染控制。表8列出了不同复合吸附剂对砷的脱除效果,其中复合吸附剂主要包括α-Al2O3、γ-Al2O3负载Pt吸附剂[101-103]、Fe-Mn双元氧化物(FMBO)[104]、氧化锰改性凹凸棒土(Mn(Ⅳ)/ATP)[105]等。
表 8 不同烟气组分和温度下复合吸附剂的砷脱除效果Table 8. The arsenic removal efficiencies of aluminum-based adsorbents in different gas components and reaction temperatures吸附剂Sorbent 模拟烟气Simulated flue gas 温度/℃Temp. 吸附时间/minTime 吸附容量/(mg·g−1)Absorption capacity 文献Ref. Pd/α-Al2O3 N2/H2/CO/CO2/H2S 204 150 4.74 [101] Pd/γ-Al2O3 N2/H2/CO2 200 300 70.00 [102] FMBO N2/O2/CO2/H2O/NO/SO2 300 30 17.98 [104] FMBO N2/O2/CO2/H2O/NO/SO2 600 30 21.65 [104] FMBO N2/O2/CO2/H2O/NO/SO2 700 30 8.22 [104] Mn(Ⅳ)/ATP N2/O2/CO2/H2O/NO/SO2 600 30 6.66 [105] Mn(Ⅳ)/ATP N2/O2/CO2/H2O/NO/SO2 600 60 10.98 [105] Mn(Ⅳ)/ATP N2/O2/CO2/H2O/NO/SO2 600 180 25.01 [105] | Show TableDownLoad:
CSV
As2O3(g)与Pd/γ-Al2O3发生化学吸附,砷吸附容量达到70 mg·g−1[102],吸附效率达到63.81%[103],吸附容量明显优于单金属氧化物吸附剂。吸附产物为As3Pd8与AsPd2[101-102]。当As/Pd原子比提高时,As3Pd8可向AsPd2转化[101]。在初始较短时间内,反应产物为As3Pd8[101],随着反应时间的延长,产物逐渐转化为AsPd2[102]。
CaO、Fe2O3和γ-Al2O3脱除As2O3(g)时需要晶格氧或吸附剂表面的化学吸附氧将As2O3(g)氧化为As2O5(s),再形成稳定的产物。MnO2可将As2O3(g)氧化为As2O5(s),但无法与As2O5(s)形成稳定的产物,MnO2与Fe2O3制成复合吸附剂(FMBO)后,MnO2将As2O3(g)氧化为As2O5(s),而Fe2O3不再需要消耗晶格氧,便可将As2O5(s)固化在FMBO表面生成FeAsO4。最佳条件下,FMBO的砷吸附容量达到21.65 mg·g−1。凹凸棒土(ATP)是一种含有Fe2O3的硅铝酸盐,具有高比表面积与较大的吸附容量,且天然矿物的成本低、性质稳定。ATP可作为复合材料载体制备复合吸附剂[104-106]。He等[105]制作了氧化锰改性凹凸棒土(Mn(Ⅳ)/ATP)用于脱除烟气砷,脱除机理与FMBO类似,最佳条件时的砷吸附容量为25.01 mg·g−1。
FMBO吸附容量受温度影响明显。在600 ℃以下,温度越高FMBO与Mn(Ⅳ)/ATP的砷吸附容量越高,600 ℃时吸附容量最大,但温度达到700 ℃以上时,由于MnO2发生团聚或分解,比表面积下降,吸附容量下降明显[104-105]。CO2会产生明显的吸附抑制作用,CO2通过占据FMBO与Mn(Ⅳ)/ATP表面的活性位点,浓度高时,表面的铁被还原为二价[107],导致吸附效果下降。FMBO与Mn(Ⅳ)/ATP的吸附机理与实验结论都证实了O2对吸附效果产生促进作用,此处不再赘述。随着NO浓度增大,FMBO与Mn(Ⅳ)/ATP的砷吸附容量先升高后降低,NO可以在它们的表面形成NO+、NO2+和NO2,这些官能团可以将As2O3(g)氧化为As2O5(s),但NO浓度过高会导致晶格氧与化学吸附氧过度消耗,使砷脱除效率下降。SO2对FMBO与Mn(Ⅳ)/ATP的影响规律不同,SO2为1.144—5.721 g·m−3时,浓度越高Mn(Ⅳ)/ATP的砷吸附容量越高。SO2可在H2O和O2存在下生成HSO4−或SO42-。Mn氧化物与ATP结合后更容易吸附H2O并生成羟基,SO2生成双齿表面配合物并与表面羟基结合后稳定存在,而锰氧化物还可加速SO2氧化为硫酸盐的过程。SO2达到5.721 g·m−3时FMBO的吸附容量略有降低,而Mn(Ⅳ)/ATP的吸附容量未受抑制。
Mn(Ⅳ)/ATP与铁锰双元氧化物吸附剂的最佳砷脱除温度为600 ℃,高于SCR的最佳工作温度,但将砷脱除设备置于SCR前,吸附剂可耐受较高SO2、NO与颗粒物的环境。Pd/γ-Al2O3在204 ℃时的砷吸附容量很大,可放置在SCR后或除尘器之后,此时烟气中NOx与颗粒物含量较低。
4.6 其他脱除技术
除了4.2—4.5节中提到的砷脱除技术,还有一些脱除方法包括粉煤灰吸附法、异相凝并技术与液相氧化脱除技术。粉煤灰中含有大量的Ca、Si、Al与Fe的氧化物[108-110],因此利用粉煤灰也可以对烟气砷实现控制。Li等[65]研究了3种粉煤灰回注技术在进行砷原位固定的可行性,在900 ℃,模拟烟气环境中,3种粉煤灰砷吸附容量分别为5.97、8.33、5.54 mg·g−1。Wang等[66]在某电厂实际工况下研究了改性粉煤灰在SCR出口回注技术协同脱除砷等重金属,烟气中总砷浓度降低了78.1%。SO2与NO会抑制粉煤灰的砷脱除效果[65-66]。
异相凝并是一种新兴的烟气砷脱除技术,它向SCR与ESP的烟道间喷入羧甲基纤维素钠、聚丙烯酰胺、磺胺树脂与羟甲基纤维素等凝并剂,使颗粒态砷通过电荷中和、桥架等方式互相团聚成更大的颗粒,且更易以镶嵌的形式与液滴发生吸附,从而形成更大的团聚体。异相凝并吸附剂喷入烟道后,其汽化降低了烟道温度,促进了气态砷的非均相冷凝、成核作用,从而更易被固定[111-112]。凝并剂能够促使PM1长大至1—10 μm;在脱硫石膏中,凝并后砷含量降低67.6%。与未凝并工况对比,气态与颗粒态砷向10 μm以上颗粒转移,最终排放至大气的砷降低69.3%[111]。研究发现,凝并粉煤灰的砷批淋滤浸出浓度降低,在纯净水中的浸出量降低了50%,吸附在凝并飞灰上的砷迁移转化能力减弱[112]。
除了固体吸附材料,也有研究探索了液相氧化剂对砷的脱除效果。As2O5比As2O3的毒性低50倍,且As2O5的溶解性好,将As(Ⅲ)氧化为As(Ⅴ)后不仅毒性降低,也利于溶解吸收。一些氧化剂溶液可以在实验室条件下实现对气态砷的吸收,包括KMnO4、Na2S2O8/H2O2、芬顿试剂、NaClO、NaClO/NaClO2和CH3COOOH/H2O2[113-118]。最佳条件下,吸收效率可接近100%。其中KMnO4的砷脱除效率受SO2影响最大,SO2超过4.290 g·m−3时,砷脱除效率便降低至不到60%;其他氧化剂在SO2超过11.441 g·m−3时仍有超过50%的效率。NO明显抑制砷脱除效率[113-116, 118],这是由于NO持续消耗氧化剂,NO被氧化为硝酸根与亚硝酸根。此外,NO与NaClO2反应产生ClNO和ClNO2中间体,这可能导致NaClO2与NaClO的持续消耗。但Na2S2O8/H2O2氧化剂却非如此[117],随着NO浓度增加,砷脱除效率出现先升高后降低的趋势,这可能是因为NO较低时与As2O3竞争氧化剂分子,当NO较高时,NO的氧化产物NO2对As2O3仍有氧化作用。除NaClO/NaClO2外[118],CO2均对砷脱除起到了抑制作用,而CO2对NaClO/NaClO2氧化剂几乎无影响。除Na2S2O8/H2O2/Ca(OH)2外[117],O2对以上氧化剂也可以产生抑制作用。砷液相氧化脱除技术的最佳温度为50—60 ℃,已经接近电厂烟气出口温度,但在实际条件下需要增加新设备,成本较高,且存在二次污染风险。
4.7 小结
活性炭等碳基吸附剂的优点是吸附容量较大,当气体成分复杂时,吸附剂特异性差,再加上成本高等原因,少有工业应用。单金属氧化物吸附剂的研究众多,包括氧化钙、氧化铝、氧化铁,这些吸附剂具有一定的抵抗酸性气体能力,具有一定的吸附特异性。这类吸附剂能适应更宽的温度,钙基吸附剂能适应超过1000 ℃的高温,而氧化铝、氧化铁的吸附温度范围略低,但SCR的最佳工作温度仅为400 ℃左右,未达到其最佳吸附温度。在实际工况下,单金属氧化物吸附剂的吸附容量仍有待提升,可以通过金属氧化物吸附剂进行修饰、负载、多元复合等方式实现吸附容量的提升,如Pd/γ-Al2O3、FMBO和Mn(Ⅳ)/ATP。Pd/α-Al2O3与Pd/γ-Al2O3吸附容量更大,吸附选择性强,最适应吸附温度与电厂烟气更接近,但缺点是吸附剂的成本极高,工业应用价值低。FMBO与Mn(Ⅳ)/ATP的吸附选择性与抗酸性气体能力更强,成本略高于单金属氧化物吸附剂,具有很好的工业应用潜力。目前已知的燃烧后砷污染控制技术中,粉煤灰、改性粉煤灰与异相凝并技术已经有实际的工业应用,其中粉煤灰吸附剂兼顾了固体废弃物资源化,应用成本较低,但粉煤灰的组成往往随煤种与电厂的工况发生变化,不同粉煤灰的脱除效果差距大。目前异相凝并技术的工业应用效果最好,成本适中,可以进行大规模工业应用。氧化脱除技术在很宽的NO与SO2浓度范围内具有良好的适用性,且脱除效率高。烟气中的砷浓度已经很低,但量大流速快并需要进行长时间吸收。在上述这些研究中,最长反应时间皆未超过30 min,但在实际生产中则是长时间反应;另外,由于烟气中其他还原性物质消耗氧化剂,使氧化剂频繁添加,所产生的还原产物可能随烟气带出,导致二次污染。
5. 总结与展望(Summary and prospect)
考虑到我国能源结构的特点,在工业生产过程中,燃煤产生的砷排放不容忽视。燃煤烟气砷污染控制不仅可以减少大气砷污染,同时也可以最大限度避免SCR催化剂失活,降低烟气治理成本。该部分从燃烧前、燃烧中和燃烧后控制技术进行了总结与展望。
燃烧前控制技术主要是采用洗选方式将燃煤中砷含量很高的煤矸石去除,使原煤中砷含量降低。降低洗选煤的耗水量与二次污染风险,提高其效果的稳定性是该技术重点研究之处。
燃烧中控制技术主要是在煤燃烧过程中通过化学添加剂或混煤等方式,将砷转化为化学性质稳定不易挥发的砷酸盐,主要以颗粒物为载体固定在底灰、飞灰中。受限于燃煤电厂实际情况与现实条件,目前通过吸附剂与煤粉混燃脱砷的相关研究较少,且研究处于有限的工况下,很难代表持续运行情况。例如以下几个方面需要进行深入研究:(1)实际燃煤烟气中吸附剂脱砷的关键影响因素有哪些?如何进行性能、空燃比,过量空气系数等参数的调控;(2)吸附剂在炉膛中的喷射方式,流动与分布情况是否影响其脱除效果,如改变喷射角度、喷射位置、混和与喷入的先后顺序,利用模型与实际工况实验进行详细研究。
燃烧后控制技术在烟气燃烧区域后用吸附剂进行烟气砷脱除。研究表明,金属氧化物进行修饰、负载制备多元复合物后,吸附性能和稳定性得到提升。目前,燃烧后控制技术几乎都在固定床脱砷实验台进行,这些实验装置与燃煤电厂的实际工况差距较大,有必要开展中试规模实验。未来燃烧后砷污染控制的方向和趋势应着眼以下几点:(1)提高废弃吸附剂中砷的热稳定性并降低其生物有效性,降低废旧吸附剂的二次污染风险。(2)研究可循环利用的吸附剂,减少资源浪费;(3)拓展吸附剂的工作温度宽度,使其适应于多APCDs的工况,便于升级改造。将吸附剂进行化学改性,若得到化学性质更稳定的吸附剂,其生物有效性便可能降低;若稳定性适中,可再通过一些方法脱附,便可能进行吸附剂再生后循环利用;若改性后,不同温度时的吸附效果变化,其工作温度便得到一定程度的拓展。
-
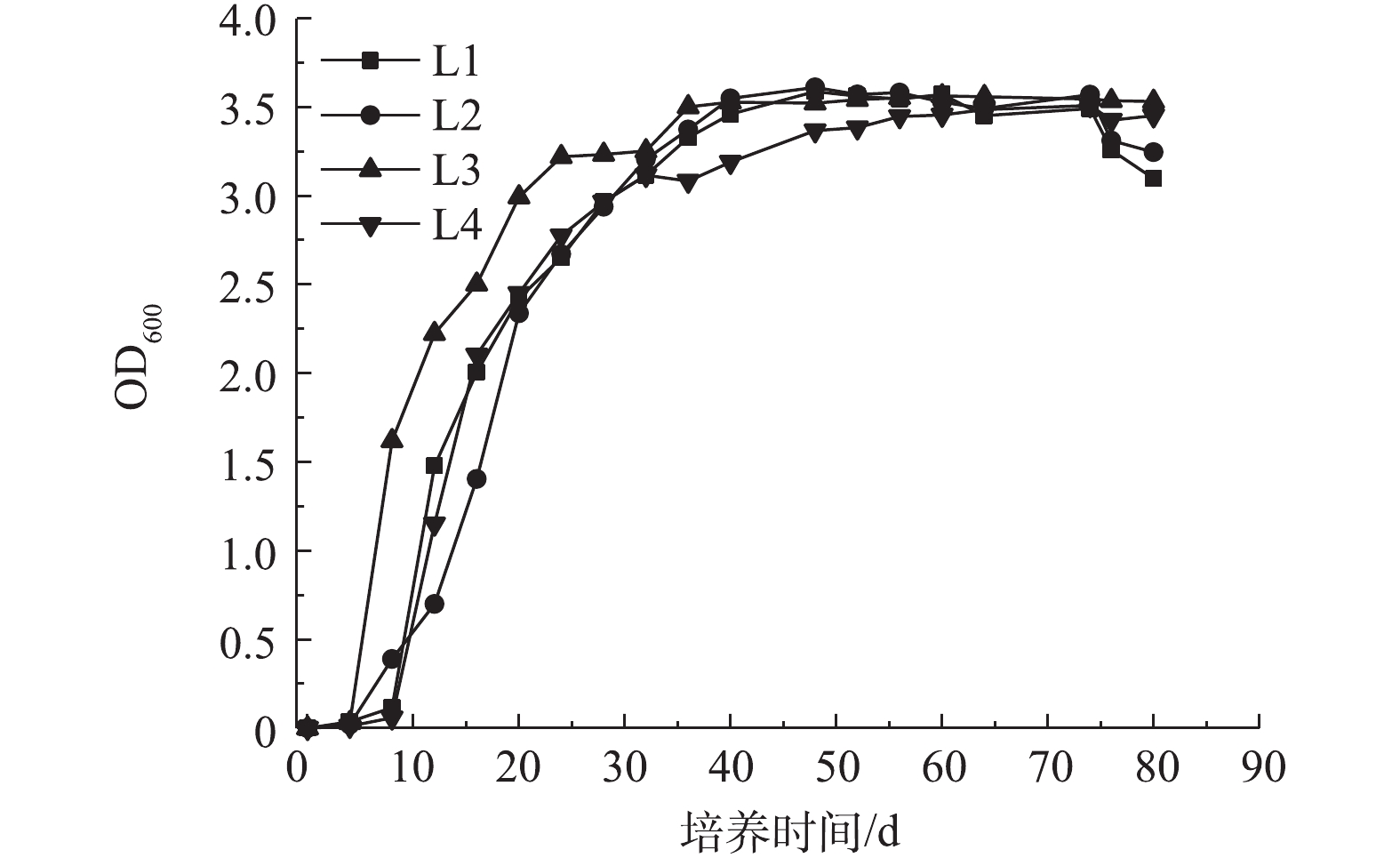
图 1 柴油降解菌L1、L2、L3和L4的生长曲线
Figure 1. Growth curves of diesel oil degrading bacteria L1, L2, L3 and L4
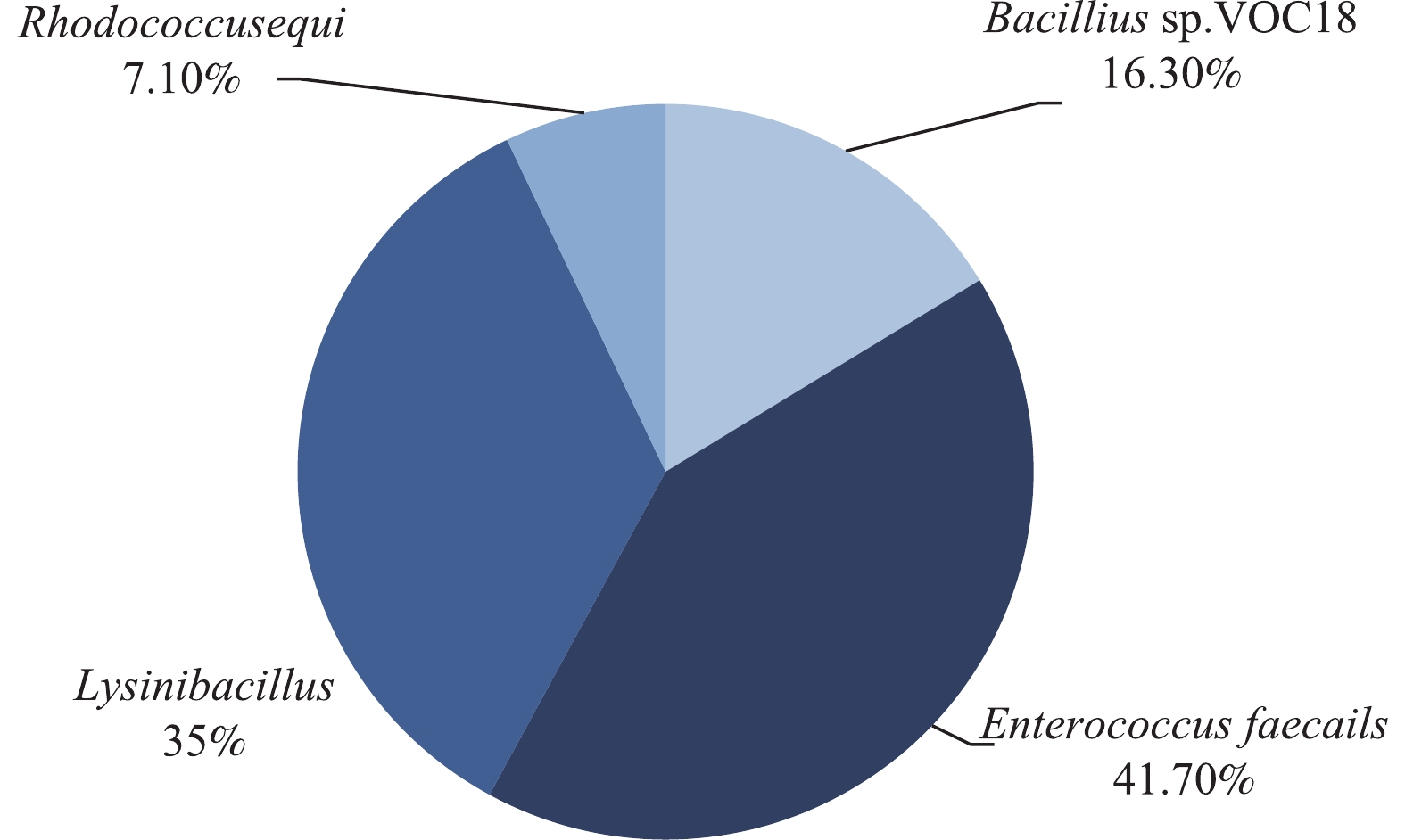
图 2 OCDL-3134降解柴油14 d后的菌群结构示意图
Figure 2. Analysis of microbial community structure of OCDL-3134 for diesel oil degradation after 14 d

图 3 初始pH对OCDL-3134的柴油降解效率和细菌生物量影响
Figure 3. Effect of initial pH on the degradation efficiency of diesel oil by OCDL-3134 and bacteria biomass
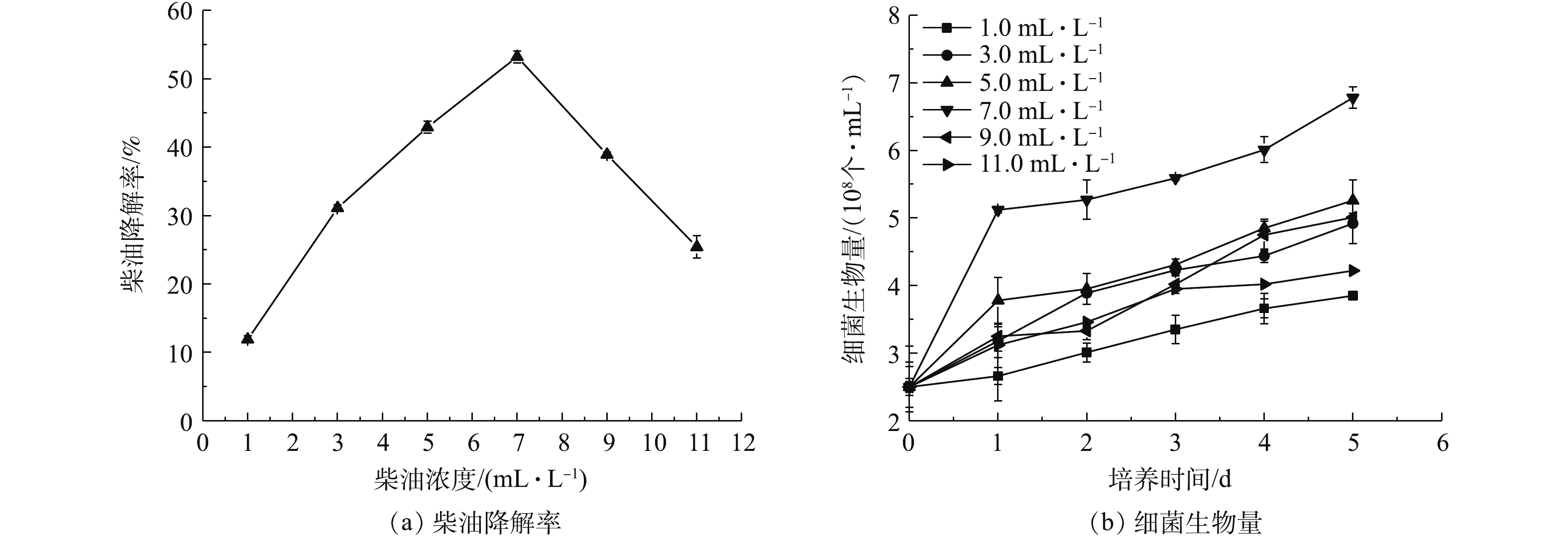
图 4 初始柴油浓度对OCDL-3134降解柴油效率及细菌生物量的影响
Figure 4. Effect of initial diesel oil concentration on the degradation efficiency of diesel oil by OCDL-3134 and bacteria biomass
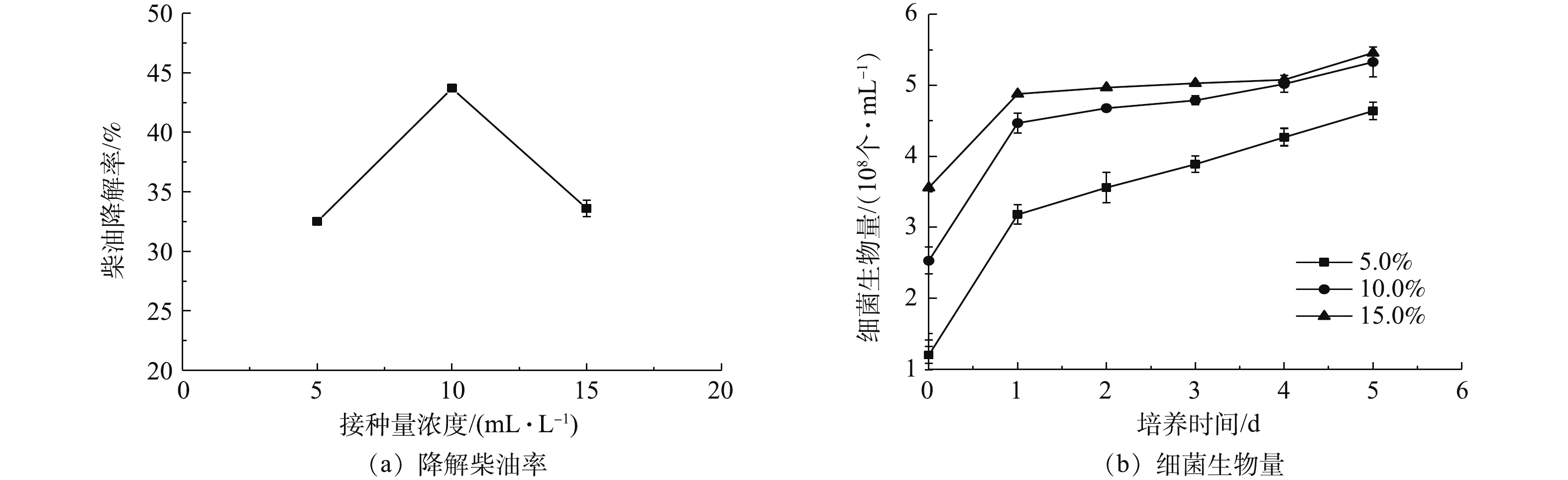
图 5 接种量对OCDL-3134降解柴油效率及细菌生物量的影响
Figure 5. Effect of inoculation on the degradation efficiency of diesel oil by OCDL-3134and bacteria biomass
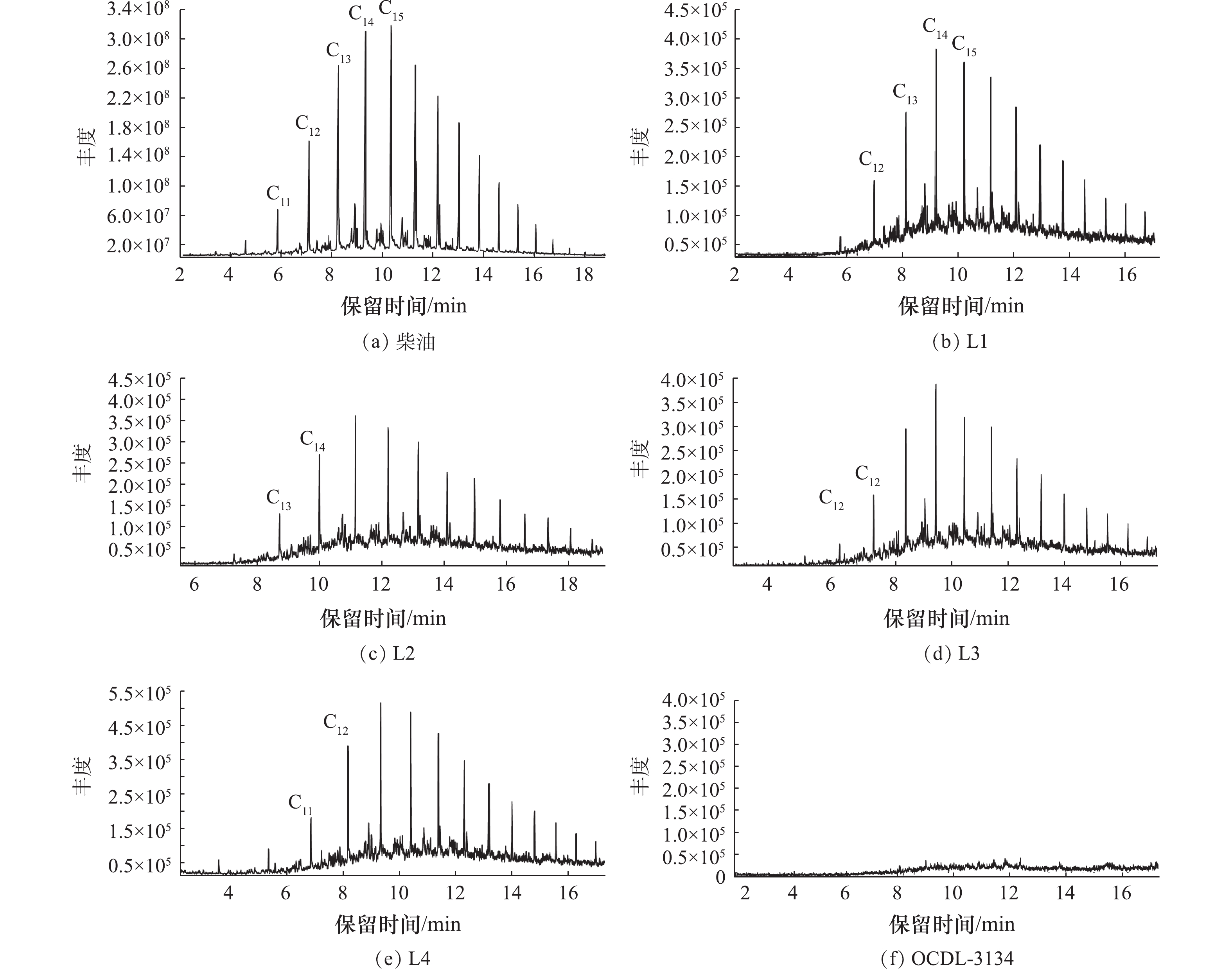
图 6 L1、L2、L3、L4和OCDL-3134降解柴油的产物气相色谱图
Figure 6. Gas chromatographic charts of products from diesel oil degradation by L1, L2, L3, L4 and OCDL-3134
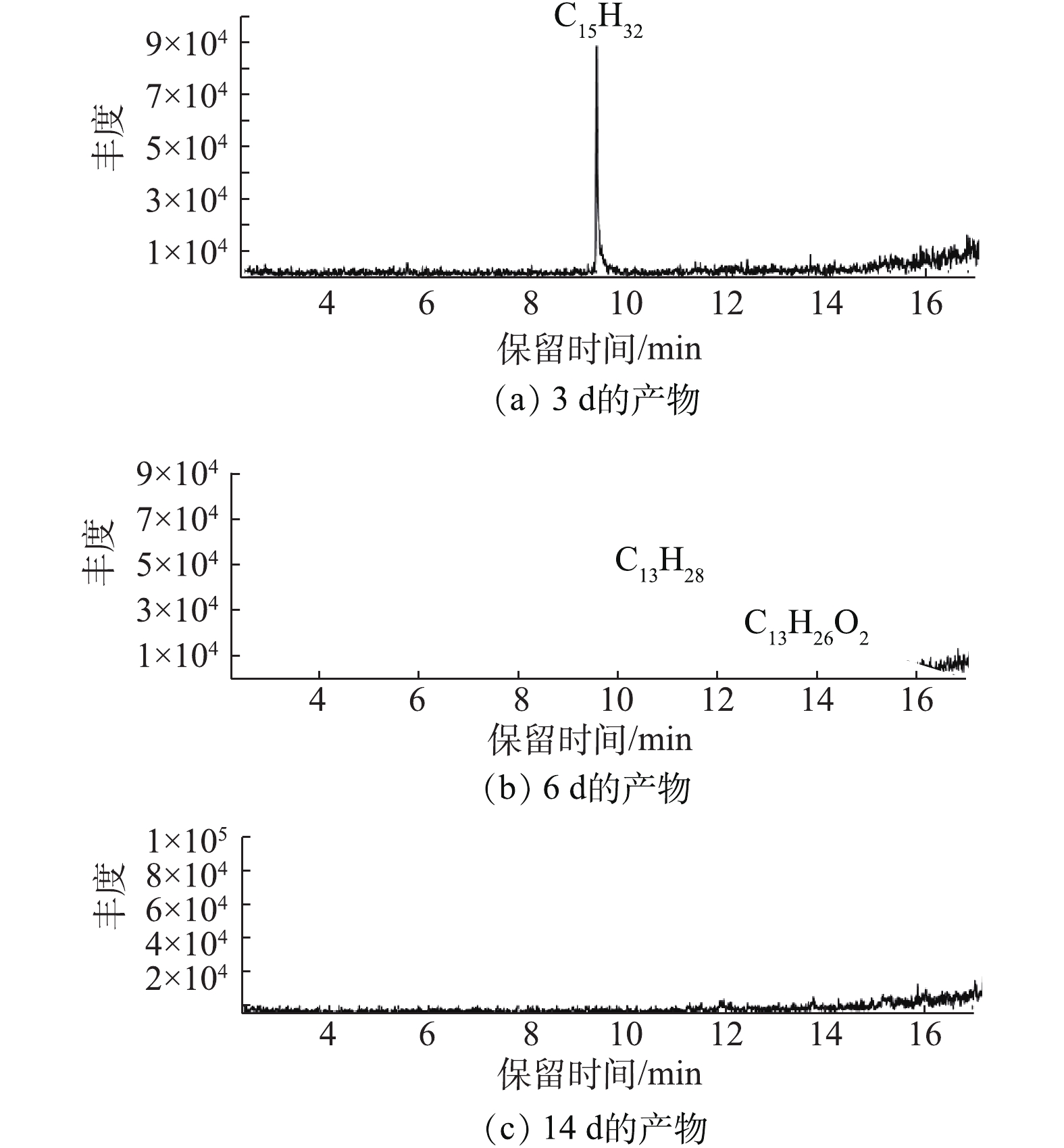
图 7 OCDL-3134降解十五烷在不同时间的产物气相色谱图
Figure 7. Gas chromatographic charts of degradation of pentadecane by OCDL-3134 at different time
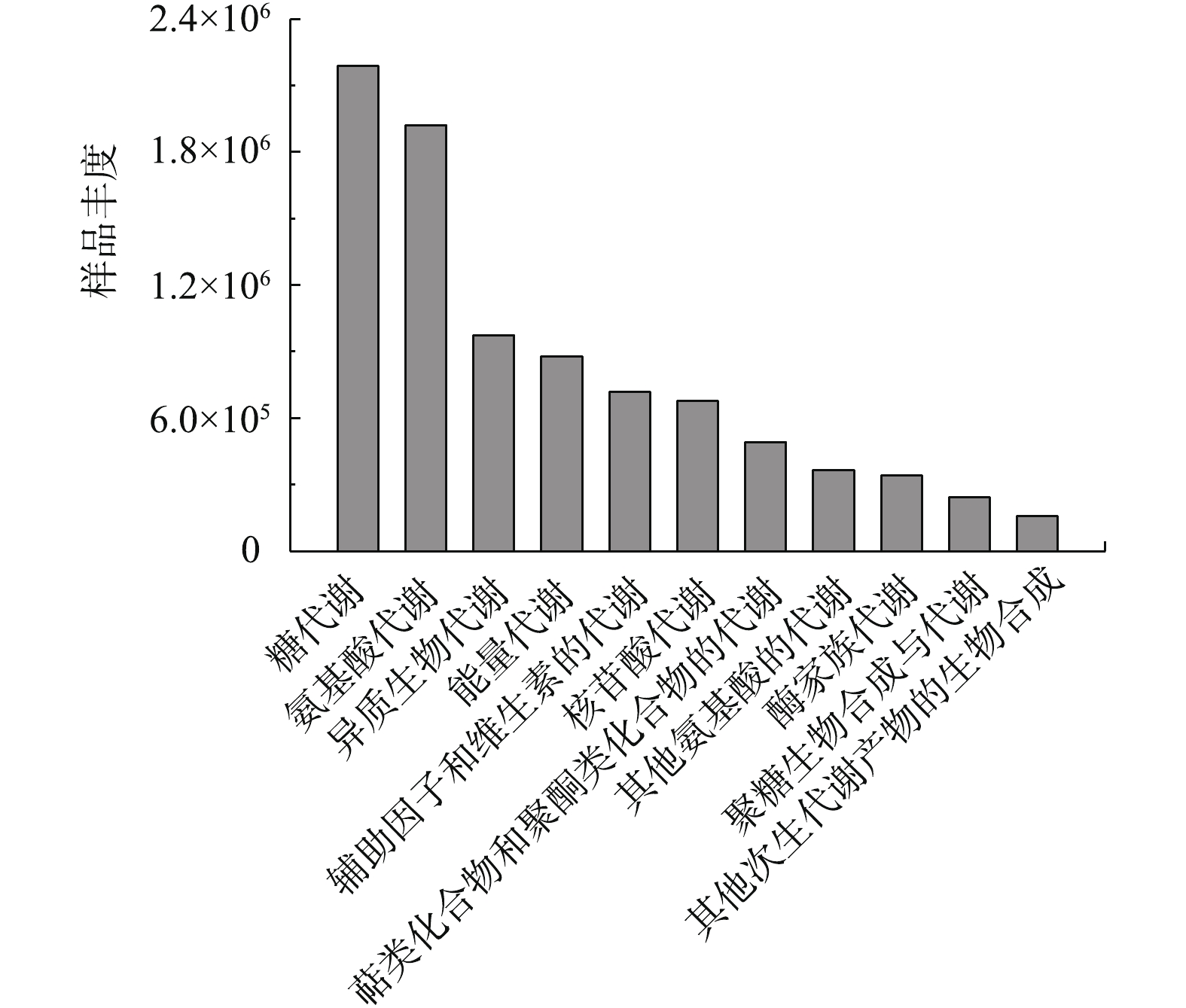
图 8 OCDL-3134代谢柴油的功能预测
Figure 8. Metabolic function prediction of OCDL-3134 metabolizing diesel
表 1 菌株的菌落形状及柴油降解能力
Table 1. Colony shape and diesel degradability of strains
菌株号 菌落形态 菌体形态 菌落颜色 降解率/% 1# 菌落为扁平、边缘不整齐、表面粗糙皱褶 杆状 白色 27.0 2# 菌落透明、光滑、有光泽 球形 白色 29.0 3# 菌落微黄、表面光滑、边缘整齐 杆状 微黄色 32.0 4# 菌落淡红色、湿润、不规则 杆状 淡红色 35.0
下载: 导出CSV
表 2 4种柴油降解菌株的生理生化实验结果
Table 2. Physiological and biochemical characteristics of four diesel degrading bacteria
实验类型 菌株号 1# 2# 3# 4# 淀粉水解实验 − − − − 明胶实验 + − + + 尿素实验 − + − − 甲基红实验 + − + + V-P实验 + − + − 吲哚实验 − − − − 柠檬酸盐实验 − − − − 硫化氢实验 − − − + 触酶实验 − + − + 葡萄糖发酵实验 + − + + 乳糖发酵实验 − − − − 木糖发酵实验 − − − − 麦芽糖发酵实验 + − + + 蔗糖发酵实验 + − − − 注:“+”表示显阳性,“−”表示显阴性。
下载: 导出CSV
表 3 菌种组合对柴油降解效率的实验结果
Table 3. Experimental results of degradation efficiency of diesel oil by species strain combination
编号 组合 降解率/% 1 L1+L2 20.9 2 L1+L3 26.2 3 L1+L4 26.1 4 L2+L3 25.8 5 L2+L4 30.9 6 L3+L4 29.3 7 L1+L2+L3 23.5 8 L1+L2+L4 31.5 9 L1+L3+L4 25.0 10 L2+L3+L4 21.1 11 L1+L2+L3+L4 39.6 12 空白 10.9
下载: 导出CSV
表 4 4种柴油降解菌的接种比例和对应的柴油降解效率表
Table 4. Inoculation ratio of four diesel oil degrading bacteria and their diesel oil biodegradation efficiency
接种比例(L1∶L2∶L3∶L4) 降解率/% 接种比例(L1∶L2∶L3∶L4) 降解率/% 3∶3∶1∶2 16.5 2∶4∶3∶2 14.8 1∶1∶1∶1 39.3 4∶1∶4∶2 18.5 2∶1∶2∶3 15.3 2∶2∶1∶4 15.7 4∶4∶1∶3 21.4 4∶3∶2∶4 19.1 3∶4∶2∶1 15.6 1∶2∶2∶2 25.5 2∶3∶4∶1 13.7 3∶1∶3∶4 52.5 1∶4∶4∶4 22.3 3∶2∶4∶3 13.2 1∶3∶3∶3 13.1 4∶2∶3∶1 11.7
下载: 导出CSV
-
[1] LI S, ZHANG S, DONG H, et al. Presence of aliphatic and polycyclic aromatic hydrocarbons in near-surface sediments of an oil spill area in Bohai Sea[J]. Marine Pollution Bulletin, 2015, 100(1): 169-175. doi: 10.1016/j.marpolbul.2015.09.009 [2] LECKLIN T, RY M R, KUIKKA S. A bayesian network for analyzing biological acute and long-term impacts of an oil spill in the Gulf of Finland[J]. Marine Pollution Bulletin, 2011, 62(12): 2822-2835. doi: 10.1016/j.marpolbul.2011.08.045 [3] 徐志霞, 张颖, 金显敏, 等. 高效石油降解菌株的筛选及菌群的构建[J]. 海南师范大学学报(自然科学版), 2015, 28(4): 421-424. [4] CHANDRAN P, DAS N. Degradation of diesel oil by immobilized Candida tropicalis and biofilm formed on gravels[J]. Biodegradation, 2011, 22(6): 1181-1189. doi: 10.1007/s10532-011-9473-1 [5] 陆秀君, 郭书海, 孙清, 等. 石油污染土壤的修复技术研究现状及展望[J]. 沈阳农业大学学报, 2003, 34(1): 64-68. [6] 张树才, 牟桂芹. 石油污染地的土壤修复技术[J]. 安全、健康和环境, 2009, 9(8): 29-31. doi: 10.3969/j.issn.1672-7932.2009.08.015 [7] 姜昌亮. 石油污染土壤的物理化学处理-生物修复工艺与技术研究[D]. 北京: 中国科学院研究生院, 2001. [8] RADWAN S S, DASHTI N, ELNEMR I M. Enhancing the growth of Vicia faba plants by microbial inoculation to improve their phytoremediation potential for oily desert areas[J]. International Journal of Phytoremediation, 2005, 7(1): 19-32. doi: 10.1080/16226510590915783 [9] ALARC N A, DAVIES J F, AUTENRIETH R L, et al. Arbuscular mycorrhiza and petroleum-degrading microorganisms enhance phytoremediation of petroleum-contaminated soil[J]. International Journal of Phytoremediation, 2008, 10(4): 251-263. doi: 10.1080/15226510802096002 [10] LU J. Marine oil spill detection, statistics and mapping with ERS SAR imagery in south-east Asia[J]. International Journal of Remote Sensing, 2003, 24(15): 3013-3032. doi: 10.1080/01431160110076216 [11] 刘沙沙, 陈志良, 董家华, 等. 柴油降解菌的分离鉴定及降解特性研究[J]. 土壤通报, 2013, 44(6): 1440-1444. [12] 何丽媛, 党志, 唐霞, 等. 混合菌对原油的降解及其降解性能的研究[J]. 环境科学学报, 2010, 30(6): 1220-1227. [13] TAO K, LIU X, CHEN X, et al. Biodegradation of crude oil by a defined co-culture of indigenous bacterial consortium and exogenous Bacillus subtilis[J]. Bioresource Technology, 2017, 224: 327-332. doi: 10.1016/j.biortech.2016.10.073 [14] 刘海华. 紫外分光光度法测量土壤中柴油的含量[J]. 黑龙江科技信息, 2008(18): 44-44. doi: 10.3969/j.issn.1673-1328.2008.18.044 [15] 杨丽芹, 蒋继辉. 微生物对石油烃类的降解机理[J]. 油气田环境保护, 2011, 21(2): 24-26. doi: 10.3969/j.issn.1005-3158.2011.02.009 [16] YUKI K, YOH T, TOSHIHIRO H, et al. Physiological and molecular characterization of a microbial community established in unsaturated, petroleum-contaminated soil[J]. Environmental Microbiology, 2010, 7(6): 806-818. [17] YUSTE L, CORBELLA M A E, TURI GANO M A J, et al. Characterization of bacterial strains able to grow on high molecular mass residues from crude oil processing[J]. FEMS Microbiology Ecology, 2000, 32(1): 69-75. doi: 10.1111/fem.2000.32.issue-1 [18] A H, D G, E R. Sequential growth of bacteria on crude oil[J]. Applied Microbiology, 1975, 30(1): 10-19. [19] 申圆圆. 土壤中石油污染物行为特征及植物根际修复研究[D]. 西安: 长安大学, 2012. [20] BOONTAWAN A. Isolation and characterization of Jatropha oil-degradation by Enterococcus faecalis and Burkholderia cenocepacia W-1 under anaerobic condition[J]. African Journal of Biotechnology, 2011, 10(63): 13841-13851. doi: 10.5897/AJB [21] PRINCE R C, MCFARLIN K M, BUTLER J D, et al. The primary biodegradation of dispersed crude oil in the sea[J]. Chemosphere, 2013, 90(2): 521-526. doi: 10.1016/j.chemosphere.2012.08.020 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5098
- HTML全文浏览数: 5098
- PDF下载数: 72
- 施引文献: 0
