


-
随着排放法规的加严,后处理系统的系统集成是满足国六排放标准的必然要求,柴油车后处理主流技术路线为DOC(氧化型催化转化器)+DPF(柴油颗粒捕集器)+SCR(选择性催化还原转化器)+ASC(氨逃逸催化器)。其中,SCR技术是控制柴油机尾气中NOx排放最有效的技术之一,现已商业化用于移动污染源排放控制[1],催化剂是SCR技术的核心。CHA型小孔分子筛(SSZ-13、和SAPO-34)催化剂具备良好的NH3-SCR催化活性和抗水热老化能力,是满足国六阶段柴油车排放标准的必然选择[2-5],缺点是成本偏高,制备工艺复杂。ZSM-5分子筛价格低廉、制备工艺成熟、NH3-SCR效率较高,具有对CHA型分子筛催化剂进行低成本取代的潜力,一直是国内外研究的热点[6-8]。在NH3-SCR反应中,Cu/ZSM-5分子筛催化剂具有良好的NOx转化率和N2选择性,但其在700 ℃以上的高温水热条件下极易劣化[9-11]。在国六阶段,DPF在运行过程中须周期性地反复再生,再生过程中排气温度可达到700 ℃以上,加之尿素水解引入的大量水蒸气,严重制约了铜基ZSM-5分子筛催化剂的实际应用。
许多研究者[12-14]对ZSM-5分子筛的水热老化性能进行了研究。SHI等[15]发现,Fe-H-ZSM-5在新鲜时和在750 ℃、5%H2O水热老化后的NH3-SCR活性高于Fe-Na-ZSM-5,是由于Fe-H-ZSM-5和Fe-Na-ZSM-5的Fe种类分布和酸度不同。宋守强等[16]研究了磷改性的ZSM-5分子筛在甲醇制丙烯(MTP)反应中的水热稳定性,发现高硅铝比会明显加剧磷氧化物的迁移、缩合及与骨架铝的配位成键作用,提高其水热稳定性。DING等[17]合成了多级孔ZSM-5分子筛,发现通过引入介孔和磷改性均可提高ZSM-5的稳定性。
解决Cu/ZSM-5催化剂水热老化问题的关键在于探明催化剂在水热老化条件下的NH3-SCR失活机理。目前,对其失活机理尚无明确的认识,普遍被接受的水热老化失活机理主要包括Cu/ZSM-5催化剂的Brønsted酸位减少和活性中心数目损失机理[18]。尽管已有对ZSM-5水热老化后Brønsted酸减少和活性中心损失机理的研究,但仍无法系统解释不同水热条件下Cu/ZSM-5催化剂的失活现象。本研究针对水热老化过程中不同温度、H2O(g)含量处理后的Cu/ZSM-5催化剂,通过对老化前后的催化剂物理化学结构表征,对催化剂失活的构效关系进行了研究。
-
通过浸渍法制备Cu/ZSM-5催化剂,CuO的质量分数为3%。利用硝酸铜(Cu(NO3)2)制备前驱体溶液,将商业ZSM-5分子筛(Si/Al为25)加入前驱体溶液中,在110 ℃下水浴搅拌至蒸干,然后将样品120 ℃干燥10 h,550 ℃焙烧5 h,得到粉末样催化剂。将催化剂粉末压片并用40~60目筛网过筛,得到催化剂样品,标记为CZ5。
快速水热老化是评估催化剂高温水热稳定性的手段之一,老化条件:H2O(g)浓度为5%~15%,以N2作为H2O(g)载气,空气作为平衡气,老化温度为600~800 ℃,老化时间为12 h,将老化后的催化剂样品标记为CZ5-X-Y,其中X代表温度,Y代表H2O(g)含量。
-
样品的NH3-SCR催化活性和N2选择性评价在立式固定床石英反应器中进行。模拟反应气体组成如下:φ(NO)=0.05%、φ(NH3)=0.05%、φ(O2)=5%、N2作为平衡气、总流量为1 000 mL·min−1、空速30 000 h−1。通过FT-IR光谱仪(Thermo IS10,美国)分析NO、NO2、N2O和NH3的浓度。催化剂NO转化率和N2选择性[19-20]按式(1)和式(2)计算。
式中:RNO为NO转化率;φ(M)in为M气体的进口浓度;φ(N)out为N气体的出口浓度;S为N2选择性。
-
采用比表面积分析仪(Autosorb AS-1,美国)测定老化前后催化剂的比表面积和孔结构;采用X射线衍射仪(D/MAX-RB,日本)测试催化剂的晶体结构;采用化学吸附仪(ChemiSorb 2720,美国)评价催化剂的氧化还原性;采用扫描电子显微镜(XL-30-ESEM,瑞士)测试新鲜和水热老化催化剂的粒度和形貌;采用X射线光电子能谱分析(ESCALab 220i-XL,美国)测试催化剂表面元素价态。
-
图1(a)为不同温度老化条件下,各Cu/ZSM-5催化剂的NO转化率和N2选择性,老化条件如表1所示。从图1(a)中可以看出,不同温度(5%H2O)水热处理后,CZ5催化剂的NO转化率均有所降低,低温条件下(T<250 ℃),催化剂NO转化率显著降低;在高于400 ℃时,催化剂的NO转化率也略有下降。在CZ5-600-5%催化剂上NO起燃温度(NO转化率为50%)为151 ℃,温度窗口(NO转化率大于80%)为175~429 ℃。当老化温度提高至800 ℃,CZ5-800-5%催化剂上NO起燃温度为187 ℃,温度窗口为226~375 ℃。随着水热处理温度的升高,Cu/ZSM-5的NO转化率明显降低。不同老化条件的催化剂的N2选择性如图1(b)所示,不同温度(5%H2O)水热处理后的各CZ5催化剂表现出相近的变化规律,但随着老化温度的提高,N2选择性未表现出良好的规律性,且选择性均在95%以上,变化幅度较小。
图2(a)为不同H2O(g)含量(750 ℃)条件下,各Cu/ZSM-5催化剂上NO转化率和N2选择性,老化条件如表1所示。从图2(a)中可以看出,经过750 ℃,不同浓度H2O(g)条件老化后的各催化剂上NO起燃温度均向高温方向移动。CZ5-750-5%、CZ5-750-10%和CZ5-750-15%催化剂上NO起燃温度分别为160、167和181 ℃,窗口温度分别为195~450、190~450和230~461 ℃,与新鲜催化剂相比,催化剂性能均出现不同程度的劣化。随着H2O(g)浓度的增加,各Cu/ZSM-5催化剂上NO转化率表现出不同规律,在H2O(g)含量分别为5%和10%时,催化剂性能差异不大,与CZ5-750-5%相比,CZ5-750-10%样品上NO起燃温度提高幅度仅为5 ℃,温度窗口几乎没有变化;当水含量为15%时,CZ5-750-15%催化剂性能出现明显劣化,NO起燃温度提高至181 ℃,温度窗口明显向高温方向移动。N2选择性如图2(b)所示,不同催化剂的N2选择性同样未表现出良好的规律性。
催化剂水热老化的主要变量是温度和H2O(g)含量。提高H2O(g)含量对催化剂SCR活性的影响小于提高老化温度对催化剂NH3-SCR活性的影响。
-
1)水热老化对Cu/ZSM-5催化剂孔结构的影响。不同温度和不同H2O(g)含量老化后各催化剂的BET结果如表1所示。从表1中可以看出,老化后样品的比表面积整体呈现下降趋势。CZ5-600-5%、CZ5-670-5%和CZ5-750-5%催化剂的比表面积(SBET)分别为298、286和285 m2·g−1,催化剂比表面积没有表现出明显变化。当水热老化温度达到800 ℃时,CZ5-800-5%催化剂的SBET大幅减小为256 m2·g−1,与600 ℃老化样品相比,下降幅度达14%。而H2O(g)含量的提高对比表面积的影响不大,比表面积呈略下降趋势。老化温度的提高和H2O(g)含量的增加都会导致催化剂的比表面积下降,但温度的升高(特别是800 ℃以上的高温)对催化剂比表面积的影响更大,这是由于高温会导致催化剂活性组分聚集或载体结构坍塌,导致比表面积急剧下降,温度可能是影响催化剂比表面积变化的主要原因。
CZ5催化剂的孔容为0.095 1 cm3·g−1,随着水热老化温度的提高孔容逐渐增加,当水热处理温度达到750 ℃和800 ℃时,孔容分别为0.211 4 cm3·g−1和0.476 5 cm3·g−1,水热老化温度越高,催化剂孔容增加越快。因此,随着水热老化温度的升高,Cu/ZSM-5催化剂的结构逐渐被破坏,孔容变大,孔径提高,特别是在老化温度高于750 ℃后,ZSM-5孔道坍塌,这和催化剂比表面积减少结果相一致。但水热处理过程中H2O(g)浓度的增加对催化剂孔容孔径的影响很小。
2)水热老化对Cu/ZSM-5晶体结构的影响。新鲜和水热老化后的Cu/ZSM-5催化剂的XRD图谱如图3所示,从图3(a)和图3(c)中可以看出,水热老化前后各Cu/ZSM-5催化剂样品均具有典型的MFI结构衍射峰,且衍射峰尖锐,具有良好的结晶度[15],这表明水热老化后各催化剂保持了完整的ZSM-5晶相结构。随着水热老化温度的升高,特征峰的强度降低,这表明ZSM-5的结晶度降低。同时,在所有XRD图谱中均未显示CuO的衍射峰,这表明活性组分CuO在催化剂表面得到了良好的分散。
将衍射峰放大后,如图3(b)所示,随着老化温度的升高,衍射峰均不同程度地向高衍射角移动。与新鲜催化剂相比,CZ5-750-5%、CZ5-750-10%和CZ5-750-15%催化剂的衍射峰由于晶粒高温聚集向高衍射角移动,而在图3(d)中H2O(g)含量的增加不会改变衍射峰位置。由布拉格方程(见式(3))可以看出,当n和λ不变时,晶面间距d会变小,θ增加。高温会导致催化剂晶体团聚,晶面间距d减小,衍射峰向高衍射角移动。综上所述,水热老化温度对ZSM-5结构的影响大于水蒸汽浓度变化的影响。
式中:d为晶面间距;θ为入射光线、反射线和反射晶面之间的角度;λ为波长,n为反射次数。
3)水热老化对Cu/ZSM-5催化剂氧化还原性的影响。H2消耗信号与催化剂表面的铜活性物种的氧化还原现象有关,新鲜和水热老化各Cu/ZSM-5催化剂上H2-TPR如图4(a)和图4(b)所示。结果表明,各Cu/ZSM-5催化剂样品均能检测到至少2个还原峰。低温峰(200~400 ℃)归因于Cu2+还原为Cu+[21-24],而高温峰(430~570 ℃)归因于氧化铜还原[25-27],而Cu+还原的峰一般出现在600 ℃以后。CZ5-670和CZ5-750在400~450 ℃的肩峰归因于氧化铜微晶的还原[28]。
CZ5催化剂在275 ℃和436 ℃分别出现2个还原峰。对比发现,随着老化温度的提高,上述2个还原峰均向高温方向移动,这说明催化剂的还原性降低,这也是水热老化后各Cu/ZSM-5催化剂活性降低的原因之一。CZ5-670-5%和CZ5-750-5%催化剂在温度400~450 ℃出现的肩峰归属于氧化铜微晶的还原峰[29]。对比各催化剂发现,CZ5-670-5%样品中421 ℃和505 ℃的2个还原峰随着老化温度的提高还原峰温度逐渐向高温方向移动,进而CZ5-800-5%样品中在471 ℃形成一个典型的还原峰。上述结果表明,随着老化温度的升高,ZSM-5分子筛笼中的活性铜离子逐渐转变成铜微晶并最终变成铜氧化物,这可能也是催化剂氧化还原性降低的原因,随之催化剂活性出现明显下降。
750 ℃不同H2O(g)含量条件下,水热老化各Cu/ZSM-5催化剂的H2-TPR结果如图4(b)所示。与CZ5-750-5%对比,随着H2O(g)含量的增加,低温峰(<350 ℃)位置基本不变,且强度很低,随着H2O(g)的增加,436 ℃位置的峰面积逐渐增大。结果表明,随着H2O(g)含量的增加,部分Cu2+和水结合,形成更多的氧化铜微晶。总之,水热老化增加水蒸汽含量对催化剂的还原性能影响要小于老化温度的影响。这和两者对催化剂活性的影响规律保持一致。
4)水热老化对Cu/ZSM-5催化剂表面形貌的影响。不同温度水热老化前后各Cu/ZSM-5催化剂形貌如图5所示。在800 ℃水热老化后,CZ5-800-5%催化剂可以观察到颗粒团聚,而随着H2O(g)的增加,未能观察到明显的团聚现象。新鲜催化剂具有尖锐的边缘,经600 ℃水热老化后,CZ5-600-5%催化剂颗粒略微破碎成小块,直径约为1~10 μm。随着老化温度的进一步升高,这种现象变得更为明显。在750 ℃和800 ℃水热老化后,CZ5-750-5%和CZ5-800-5%催化剂颗粒的小块重新团聚成大颗粒,这与BET结果中比表面积大幅降低相一致。水热老化过程中随着H2O(g)含量的增加未观察到明显的团聚现象发生。水热老化过程中温度的提高对催化剂表面形貌的影响较为显著。
5)水热老化对Cu/ZSM-5催化剂表面元素价态的影响。水热老化前后各Cu/ZSM-5催化剂XPS能谱如图6(a)和图6(b)所示。Cu2p3/2的峰位于930~934 eV,位于933.1 eV附近的峰归属于Cu2+[30-31],图6中并没有观察到Cu+的信号峰,这可能是由于催化剂表面Cu+含量较低或XPS灵敏度较低[32]。Cu2p3/2信号可以拟合为2个峰,峰I (930~931 eV)和峰II (932~934 eV)分别为不同的Cu2+化学态。其中,前者归属于CuO活性物种信号峰;后者归属于与分子筛的表面氧原子配位的Cu2+离子[33]信号峰。水热老化前后各Cu/ZSM-5催化剂均可观察到峰I和峰II,不同温度下,5%H2O水热老化催化剂对应的峰I强度基本保持不变,随着水热老化温度的升高,峰II的强度明显下降,这表明与分子筛的表面氧原子配位的Cu2+转化为CuO微晶或者CuO物质,这与H2-TPR结果一致。另一方面,750 ℃不同H2O(g)含量水热处理各催化剂XPS结果变化不大,峰II强度略有升高,这说明水热老化过程中,H2O(g)含量的变化不是影响Cu2+变化的主因,H2O(g)含量小于老化温度对催化剂中Cu2+变化的影响。
-
1)随着水热老化温度的升高,Cu/ZSM-5催化剂的NH3-SCR活性显著下降,比表面积减小,孔容增大,催化剂发生团聚现象,催化剂的表面氧原子配位的Cu2+转化为CuO微晶或者CuO物质,活性Cu2+离子减少,导致催化剂低温活性下降。水热老化温度是影响催化剂NH3-SCR性能变化的主因。
2)随着水热老化H2O(g)含量的升高,Cu/ZSM-5催化剂上NH3-SCR性能无明显劣化,当H2O(g)含量增加至15%时,催化剂低温活性显著下降,H2O(g)含量的增加对Cu/ZSM-5催化剂结构没有显著的影响;水热老化后SCR的低温性能(<300 ℃)的下降主要是由于老化温度提高所致。
3)对Cu/ZSM-5催化剂,优化催化剂的抗高温能力是提高其水热稳定性的最直接有效的方式。提高ZSM-5分子筛结构稳定性或减少催化剂活性Cu2+迁移可能是优化Cu/ZSM-5的抗水热能力的有效途径。
水热老化条件对Cu/ZSM-5催化剂NH3-SCR反应的影响
Effect of hydrothermal aging conditions on NH3-SCR reaction of Cu/ZSM-5 catalysts
-
摘要: 针对Cu/ZSM-5高温水热失活的问题,通过浸渍法合成了Cu/ZSM-5催化剂,并对该催化剂进行了不同温度和不同H2O(g)含量的水热老化。采用比表面积分析、SEM观察、X射线衍射分析、H2-程序升温还原、X射线光电子能谱分析对Cu/ZSM-5催化剂的理化性能进行了表征。分别研究了不同水热老化条件下Cu/ZSM-5催化剂的NH3-SCR性能和水热失活机理。结果表明,经水热处理后,各Cu/ZSM-5催化剂的NH3-SCR性能均有所降低。随着老化温度的提高,催化剂的分子筛载体出现结构坍塌,比表面积减小,孔容积增大,但仍保持MFI结构,老化温度的提高同样使催化剂活性Cu2+减少并一部分转化为CuO微晶,而H2O(g)含量的变化对催化剂的物理化学结构的影响较小。在高温水热老化过程中,温度对催化性能劣化的影响大于水蒸汽含量,是催化剂失活的主要原因。Abstract: Aiming at the hydrothermal deactivation of Cu/ZSM-5 catalysts under high temperature, they were synthesized by impregnation method and then the hydrothermal aging treatment of these catalysts were carried out under different temperatures and water vapor contents. The physicochemical properties of the catalysts were characterized by BET, SEM, XRD, H2-TPR and XPS. The NH3-SCR performance and hydrothermal deactivation mechanism of the Cu/ZSM-5 catalysts under different hydrothermal aging conditions were studied. The results showed that the NH3-SCR performance of each Cu/ZSM-5 catalyst was reduced after hydrothermal treatment. With the increase of temperature of hydrothermal aging, the zeolite supports of Cu/ZSM-5 collapsed, the specific surface area decreased and the pore volume increased while the MFI structure of Cu/ZSM-5 remained unchanged, even the isolated active Cu2+ in it decreased and partly transformed to CuO microcrystal. However, the change in water vapor content had slight effect on the physicochemical structure of the catalyst. High temperature hydrothermal deactivation study found that the effect of temperature on catalytic performance degradation was greater than that of water vapor contents. High temperature was the main reason for catalyst deactivation.
-
Key words:
- molecular sieve ofzeolites /
- NH3-SCR /
- ZSM-5 /
- hydrothermal stability
-
随着排放法规的加严,后处理系统的系统集成是满足国六排放标准的必然要求,柴油车后处理主流技术路线为DOC(氧化型催化转化器)+DPF(柴油颗粒捕集器)+SCR(选择性催化还原转化器)+ASC(氨逃逸催化器)。其中,SCR技术是控制柴油机尾气中NOx排放最有效的技术之一,现已商业化用于移动污染源排放控制[1],催化剂是SCR技术的核心。CHA型小孔分子筛(SSZ-13、和SAPO-34)催化剂具备良好的NH3-SCR催化活性和抗水热老化能力,是满足国六阶段柴油车排放标准的必然选择[2-5],缺点是成本偏高,制备工艺复杂。ZSM-5分子筛价格低廉、制备工艺成熟、NH3-SCR效率较高,具有对CHA型分子筛催化剂进行低成本取代的潜力,一直是国内外研究的热点[6-8]。在NH3-SCR反应中,Cu/ZSM-5分子筛催化剂具有良好的NOx转化率和N2选择性,但其在700 ℃以上的高温水热条件下极易劣化[9-11]。在国六阶段,DPF在运行过程中须周期性地反复再生,再生过程中排气温度可达到700 ℃以上,加之尿素水解引入的大量水蒸气,严重制约了铜基ZSM-5分子筛催化剂的实际应用。
许多研究者[12-14]对ZSM-5分子筛的水热老化性能进行了研究。SHI等[15]发现,Fe-H-ZSM-5在新鲜时和在750 ℃、5%H2O水热老化后的NH3-SCR活性高于Fe-Na-ZSM-5,是由于Fe-H-ZSM-5和Fe-Na-ZSM-5的Fe种类分布和酸度不同。宋守强等[16]研究了磷改性的ZSM-5分子筛在甲醇制丙烯(MTP)反应中的水热稳定性,发现高硅铝比会明显加剧磷氧化物的迁移、缩合及与骨架铝的配位成键作用,提高其水热稳定性。DING等[17]合成了多级孔ZSM-5分子筛,发现通过引入介孔和磷改性均可提高ZSM-5的稳定性。
解决Cu/ZSM-5催化剂水热老化问题的关键在于探明催化剂在水热老化条件下的NH3-SCR失活机理。目前,对其失活机理尚无明确的认识,普遍被接受的水热老化失活机理主要包括Cu/ZSM-5催化剂的Brønsted酸位减少和活性中心数目损失机理[18]。尽管已有对ZSM-5水热老化后Brønsted酸减少和活性中心损失机理的研究,但仍无法系统解释不同水热条件下Cu/ZSM-5催化剂的失活现象。本研究针对水热老化过程中不同温度、H2O(g)含量处理后的Cu/ZSM-5催化剂,通过对老化前后的催化剂物理化学结构表征,对催化剂失活的构效关系进行了研究。
1. 材料与方法
1.1 催化剂制备及老化
通过浸渍法制备Cu/ZSM-5催化剂,CuO的质量分数为3%。利用硝酸铜(Cu(NO3)2)制备前驱体溶液,将商业ZSM-5分子筛(Si/Al为25)加入前驱体溶液中,在110 ℃下水浴搅拌至蒸干,然后将样品120 ℃干燥10 h,550 ℃焙烧5 h,得到粉末样催化剂。将催化剂粉末压片并用40~60目筛网过筛,得到催化剂样品,标记为CZ5。
快速水热老化是评估催化剂高温水热稳定性的手段之一,老化条件:H2O(g)浓度为5%~15%,以N2作为H2O(g)载气,空气作为平衡气,老化温度为600~800 ℃,老化时间为12 h,将老化后的催化剂样品标记为CZ5-X-Y,其中X代表温度,Y代表H2O(g)含量。
1.2 催化剂活性测试
样品的NH3-SCR催化活性和N2选择性评价在立式固定床石英反应器中进行。模拟反应气体组成如下:φ(NO)=0.05%、φ(NH3)=0.05%、φ(O2)=5%、N2作为平衡气、总流量为1 000 mL·min−1、空速30 000 h−1。通过FT-IR光谱仪(Thermo IS10,美国)分析NO、NO2、N2O和NH3的浓度。催化剂NO转化率和N2选择性[19-20]按式(1)和式(2)计算。
RNO=φ(NO)in−φ(NO)outφ(NO)in×100% (1) S=φ(NO)in−φ(NO)out+φ(NH3)in−φ(NH3)out−φ(NO2)out−2φ(N2O)outφ(NO)in−φ(NO)out+φ(NH3)in−φ(NH3)out×100% (2) 式中:RNO为NO转化率;φ(M)in为M气体的进口浓度;φ(N)out为N气体的出口浓度;S为N2选择性。
1.3 催化剂表征
采用比表面积分析仪(Autosorb AS-1,美国)测定老化前后催化剂的比表面积和孔结构;采用X射线衍射仪(D/MAX-RB,日本)测试催化剂的晶体结构;采用化学吸附仪(ChemiSorb 2720,美国)评价催化剂的氧化还原性;采用扫描电子显微镜(XL-30-ESEM,瑞士)测试新鲜和水热老化催化剂的粒度和形貌;采用X射线光电子能谱分析(ESCALab 220i-XL,美国)测试催化剂表面元素价态。
2. 结果与讨论
2.1 水热老化对Cu/ZSM-5 催化剂NH3-SCR性能的影响
图1(a)为不同温度老化条件下,各Cu/ZSM-5催化剂的NO转化率和N2选择性,老化条件如表1所示。从图1(a)中可以看出,不同温度(5%H2O)水热处理后,CZ5催化剂的NO转化率均有所降低,低温条件下(T<250 ℃),催化剂NO转化率显著降低;在高于400 ℃时,催化剂的NO转化率也略有下降。在CZ5-600-5%催化剂上NO起燃温度(NO转化率为50%)为151 ℃,温度窗口(NO转化率大于80%)为175~429 ℃。当老化温度提高至800 ℃,CZ5-800-5%催化剂上NO起燃温度为187 ℃,温度窗口为226~375 ℃。随着水热处理温度的升高,Cu/ZSM-5的NO转化率明显降低。不同老化条件的催化剂的N2选择性如图1(b)所示,不同温度(5%H2O)水热处理后的各CZ5催化剂表现出相近的变化规律,但随着老化温度的提高,N2选择性未表现出良好的规律性,且选择性均在95%以上,变化幅度较小。
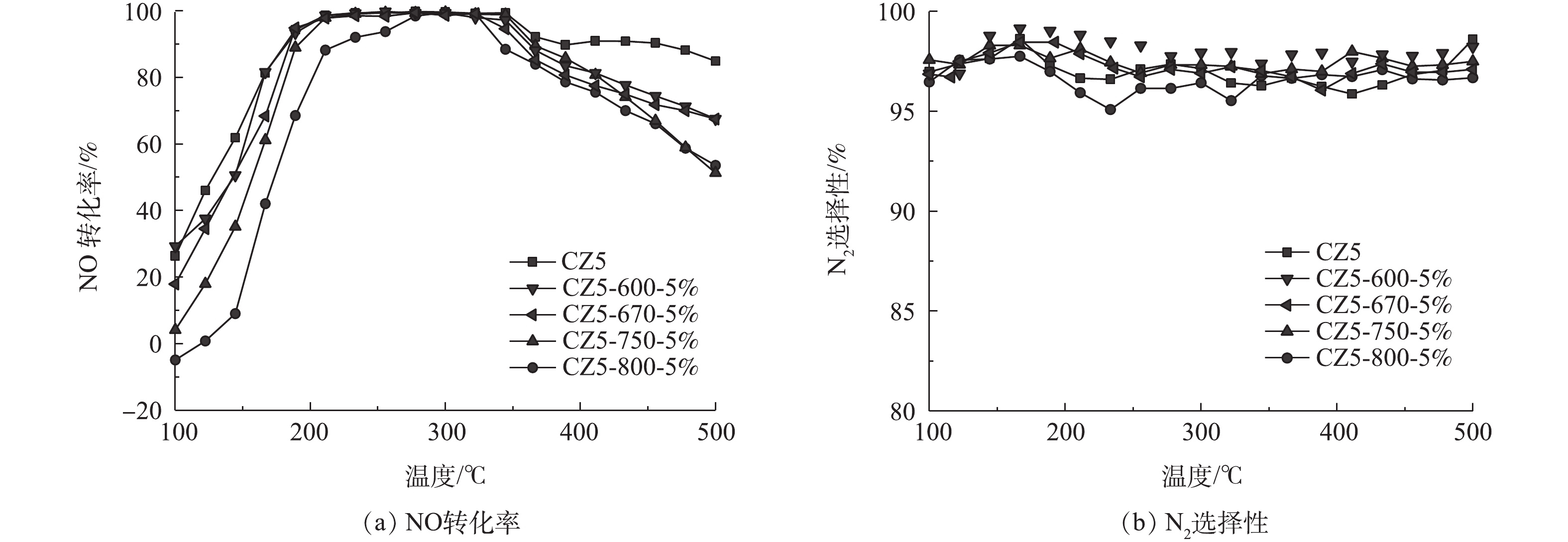 图 1 不同温度水热(5%H2O)老化各Cu/ZSM-5催化剂上NH3-SCR性能Figure 1. Performance comparison of NH3-SCR activity of Cu/ZSM-5 catalysts over different temperature at 5%H2O表 1 不同催化剂的质构参数Table 1. Structure parameters of different catalysts
图 1 不同温度水热(5%H2O)老化各Cu/ZSM-5催化剂上NH3-SCR性能Figure 1. Performance comparison of NH3-SCR activity of Cu/ZSM-5 catalysts over different temperature at 5%H2O表 1 不同催化剂的质构参数Table 1. Structure parameters of different catalysts催化剂 BET比表面积/(m2·g−1) 单点总孔容/(10−2cm3·g−1) 吸附平均孔径/nm CZ5 316 9.51 28.94 CZ5-600-5% 298 14.82 29.17 CZ5-670-5% 286 15.59 30.99 CZ5-750-5% 285 21.14 31.18 CZ5-800-5% 256 47.65 44.64 CZ5-750-10% 276 26.53 34.55 CZ5-750-15% 270 26.59 34.63 | Show Table DownLoad:
CSV
DownLoad:
CSV
图2(a)为不同H2O(g)含量(750 ℃)条件下,各Cu/ZSM-5催化剂上NO转化率和N2选择性,老化条件如表1所示。从图2(a)中可以看出,经过750 ℃,不同浓度H2O(g)条件老化后的各催化剂上NO起燃温度均向高温方向移动。CZ5-750-5%、CZ5-750-10%和CZ5-750-15%催化剂上NO起燃温度分别为160、167和181 ℃,窗口温度分别为195~450、190~450和230~461 ℃,与新鲜催化剂相比,催化剂性能均出现不同程度的劣化。随着H2O(g)浓度的增加,各Cu/ZSM-5催化剂上NO转化率表现出不同规律,在H2O(g)含量分别为5%和10%时,催化剂性能差异不大,与CZ5-750-5%相比,CZ5-750-10%样品上NO起燃温度提高幅度仅为5 ℃,温度窗口几乎没有变化;当水含量为15%时,CZ5-750-15%催化剂性能出现明显劣化,NO起燃温度提高至181 ℃,温度窗口明显向高温方向移动。N2选择性如图2(b)所示,不同催化剂的N2选择性同样未表现出良好的规律性。
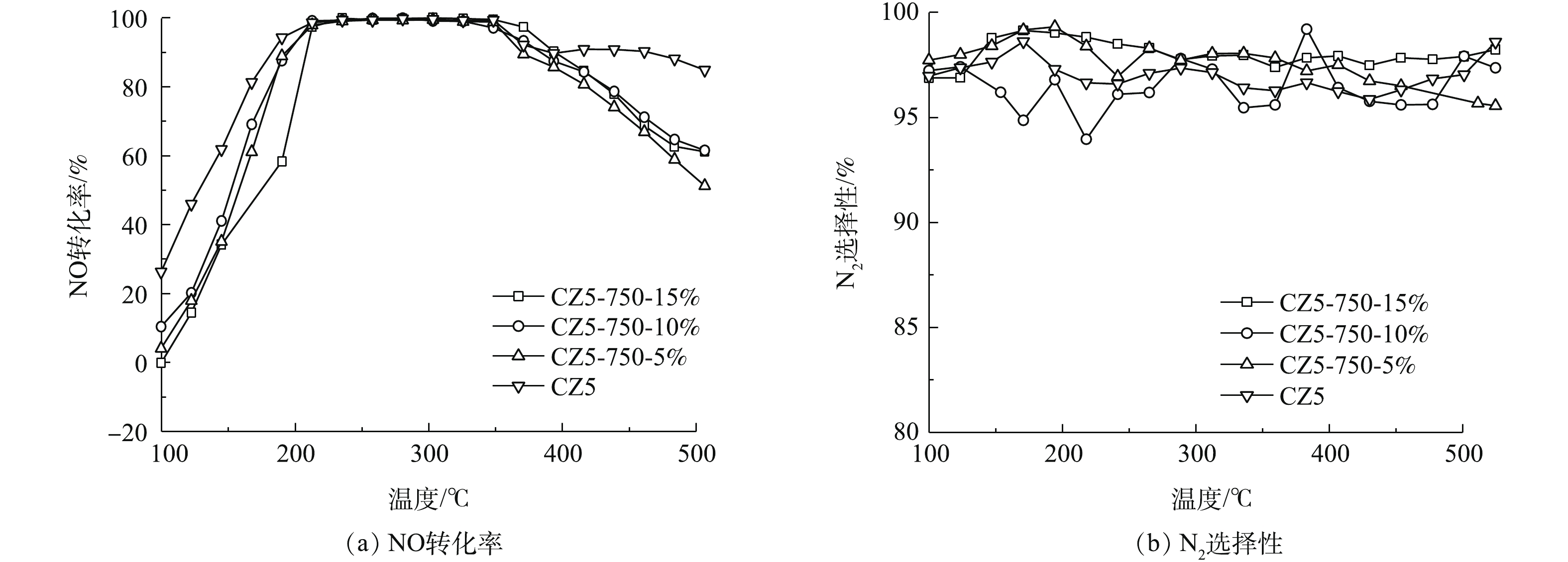 图 2 不同H2O(g)含量水热(750 ℃)老化Cu/ZSM-5催化剂上NH3-SCR性能Figure 2. Performance comparison of NH3-SCR activity of Cu/ZSM-5 catalysts over different H2O contents at 750 ℃
图 2 不同H2O(g)含量水热(750 ℃)老化Cu/ZSM-5催化剂上NH3-SCR性能Figure 2. Performance comparison of NH3-SCR activity of Cu/ZSM-5 catalysts over different H2O contents at 750 ℃催化剂水热老化的主要变量是温度和H2O(g)含量。提高H2O(g)含量对催化剂SCR活性的影响小于提高老化温度对催化剂NH3-SCR活性的影响。
2.2 催化剂表征结果
1)水热老化对Cu/ZSM-5催化剂孔结构的影响。不同温度和不同H2O(g)含量老化后各催化剂的BET结果如表1所示。从表1中可以看出,老化后样品的比表面积整体呈现下降趋势。CZ5-600-5%、CZ5-670-5%和CZ5-750-5%催化剂的比表面积(SBET)分别为298、286和285 m2·g−1,催化剂比表面积没有表现出明显变化。当水热老化温度达到800 ℃时,CZ5-800-5%催化剂的SBET大幅减小为256 m2·g−1,与600 ℃老化样品相比,下降幅度达14%。而H2O(g)含量的提高对比表面积的影响不大,比表面积呈略下降趋势。老化温度的提高和H2O(g)含量的增加都会导致催化剂的比表面积下降,但温度的升高(特别是800 ℃以上的高温)对催化剂比表面积的影响更大,这是由于高温会导致催化剂活性组分聚集或载体结构坍塌,导致比表面积急剧下降,温度可能是影响催化剂比表面积变化的主要原因。
CZ5催化剂的孔容为0.095 1 cm3·g−1,随着水热老化温度的提高孔容逐渐增加,当水热处理温度达到750 ℃和800 ℃时,孔容分别为0.211 4 cm3·g−1和0.476 5 cm3·g−1,水热老化温度越高,催化剂孔容增加越快。因此,随着水热老化温度的升高,Cu/ZSM-5催化剂的结构逐渐被破坏,孔容变大,孔径提高,特别是在老化温度高于750 ℃后,ZSM-5孔道坍塌,这和催化剂比表面积减少结果相一致。但水热处理过程中H2O(g)浓度的增加对催化剂孔容孔径的影响很小。
2)水热老化对Cu/ZSM-5晶体结构的影响。新鲜和水热老化后的Cu/ZSM-5催化剂的XRD图谱如图3所示,从图3(a)和图3(c)中可以看出,水热老化前后各Cu/ZSM-5催化剂样品均具有典型的MFI结构衍射峰,且衍射峰尖锐,具有良好的结晶度[15],这表明水热老化后各催化剂保持了完整的ZSM-5晶相结构。随着水热老化温度的升高,特征峰的强度降低,这表明ZSM-5的结晶度降低。同时,在所有XRD图谱中均未显示CuO的衍射峰,这表明活性组分CuO在催化剂表面得到了良好的分散。
 图 3 不同条件下水热老化Cu/ZSM-5催化剂XRD图谱Figure 3. X-ray diffraction patterns of Cu/ZSM-5 with different conditions of hydrothermal aging
图 3 不同条件下水热老化Cu/ZSM-5催化剂XRD图谱Figure 3. X-ray diffraction patterns of Cu/ZSM-5 with different conditions of hydrothermal aging将衍射峰放大后,如图3(b)所示,随着老化温度的升高,衍射峰均不同程度地向高衍射角移动。与新鲜催化剂相比,CZ5-750-5%、CZ5-750-10%和CZ5-750-15%催化剂的衍射峰由于晶粒高温聚集向高衍射角移动,而在图3(d)中H2O(g)含量的增加不会改变衍射峰位置。由布拉格方程(见式(3))可以看出,当n和λ不变时,晶面间距d会变小,θ增加。高温会导致催化剂晶体团聚,晶面间距d减小,衍射峰向高衍射角移动。综上所述,水热老化温度对ZSM-5结构的影响大于水蒸汽浓度变化的影响。
2dsinθ=nλ (3) 式中:d为晶面间距;θ为入射光线、反射线和反射晶面之间的角度;λ为波长,n为反射次数。
3)水热老化对Cu/ZSM-5催化剂氧化还原性的影响。H2消耗信号与催化剂表面的铜活性物种的氧化还原现象有关,新鲜和水热老化各Cu/ZSM-5催化剂上H2-TPR如图4(a)和图4(b)所示。结果表明,各Cu/ZSM-5催化剂样品均能检测到至少2个还原峰。低温峰(200~400 ℃)归因于Cu2+还原为Cu+[21-24],而高温峰(430~570 ℃)归因于氧化铜还原[25-27],而Cu+还原的峰一般出现在600 ℃以后。CZ5-670和CZ5-750在400~450 ℃的肩峰归因于氧化铜微晶的还原[28]。
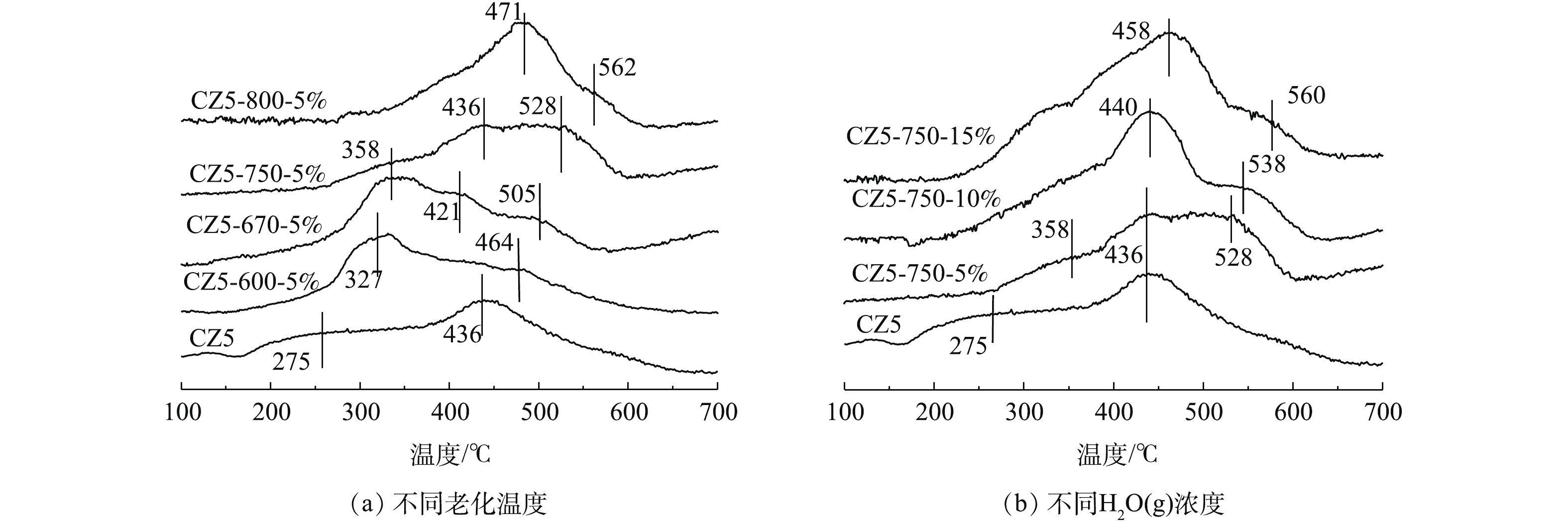 图 4 不同条件下水热老化Cu/ZSM-5催化剂H2-TPR图谱Figure 4. H2-TPR of Cu/ZSM-5 with different conditions of hydrothermal aging
图 4 不同条件下水热老化Cu/ZSM-5催化剂H2-TPR图谱Figure 4. H2-TPR of Cu/ZSM-5 with different conditions of hydrothermal agingCZ5催化剂在275 ℃和436 ℃分别出现2个还原峰。对比发现,随着老化温度的提高,上述2个还原峰均向高温方向移动,这说明催化剂的还原性降低,这也是水热老化后各Cu/ZSM-5催化剂活性降低的原因之一。CZ5-670-5%和CZ5-750-5%催化剂在温度400~450 ℃出现的肩峰归属于氧化铜微晶的还原峰[29]。对比各催化剂发现,CZ5-670-5%样品中421 ℃和505 ℃的2个还原峰随着老化温度的提高还原峰温度逐渐向高温方向移动,进而CZ5-800-5%样品中在471 ℃形成一个典型的还原峰。上述结果表明,随着老化温度的升高,ZSM-5分子筛笼中的活性铜离子逐渐转变成铜微晶并最终变成铜氧化物,这可能也是催化剂氧化还原性降低的原因,随之催化剂活性出现明显下降。
750 ℃不同H2O(g)含量条件下,水热老化各Cu/ZSM-5催化剂的H2-TPR结果如图4(b)所示。与CZ5-750-5%对比,随着H2O(g)含量的增加,低温峰(<350 ℃)位置基本不变,且强度很低,随着H2O(g)的增加,436 ℃位置的峰面积逐渐增大。结果表明,随着H2O(g)含量的增加,部分Cu2+和水结合,形成更多的氧化铜微晶。总之,水热老化增加水蒸汽含量对催化剂的还原性能影响要小于老化温度的影响。这和两者对催化剂活性的影响规律保持一致。
4)水热老化对Cu/ZSM-5催化剂表面形貌的影响。不同温度水热老化前后各Cu/ZSM-5催化剂形貌如图5所示。在800 ℃水热老化后,CZ5-800-5%催化剂可以观察到颗粒团聚,而随着H2O(g)的增加,未能观察到明显的团聚现象。新鲜催化剂具有尖锐的边缘,经600 ℃水热老化后,CZ5-600-5%催化剂颗粒略微破碎成小块,直径约为1~10 μm。随着老化温度的进一步升高,这种现象变得更为明显。在750 ℃和800 ℃水热老化后,CZ5-750-5%和CZ5-800-5%催化剂颗粒的小块重新团聚成大颗粒,这与BET结果中比表面积大幅降低相一致。水热老化过程中随着H2O(g)含量的增加未观察到明显的团聚现象发生。水热老化过程中温度的提高对催化剂表面形貌的影响较为显著。
 图 5 不同条件水热老化Cu/ZSM-5催化剂SEM图Figure 5. SEM images of Cu/ZSM-5 with different conditions of hydrothermal aging
图 5 不同条件水热老化Cu/ZSM-5催化剂SEM图Figure 5. SEM images of Cu/ZSM-5 with different conditions of hydrothermal aging5)水热老化对Cu/ZSM-5催化剂表面元素价态的影响。水热老化前后各Cu/ZSM-5催化剂XPS能谱如图6(a)和图6(b)所示。Cu2p3/2的峰位于930~934 eV,位于933.1 eV附近的峰归属于Cu2+[30-31],图6中并没有观察到Cu+的信号峰,这可能是由于催化剂表面Cu+含量较低或XPS灵敏度较低[32]。Cu2p3/2信号可以拟合为2个峰,峰I (930~931 eV)和峰II (932~934 eV)分别为不同的Cu2+化学态。其中,前者归属于CuO活性物种信号峰;后者归属于与分子筛的表面氧原子配位的Cu2+离子[33]信号峰。水热老化前后各Cu/ZSM-5催化剂均可观察到峰I和峰II,不同温度下,5%H2O水热老化催化剂对应的峰I强度基本保持不变,随着水热老化温度的升高,峰II的强度明显下降,这表明与分子筛的表面氧原子配位的Cu2+转化为CuO微晶或者CuO物质,这与H2-TPR结果一致。另一方面,750 ℃不同H2O(g)含量水热处理各催化剂XPS结果变化不大,峰II强度略有升高,这说明水热老化过程中,H2O(g)含量的变化不是影响Cu2+变化的主因,H2O(g)含量小于老化温度对催化剂中Cu2+变化的影响。
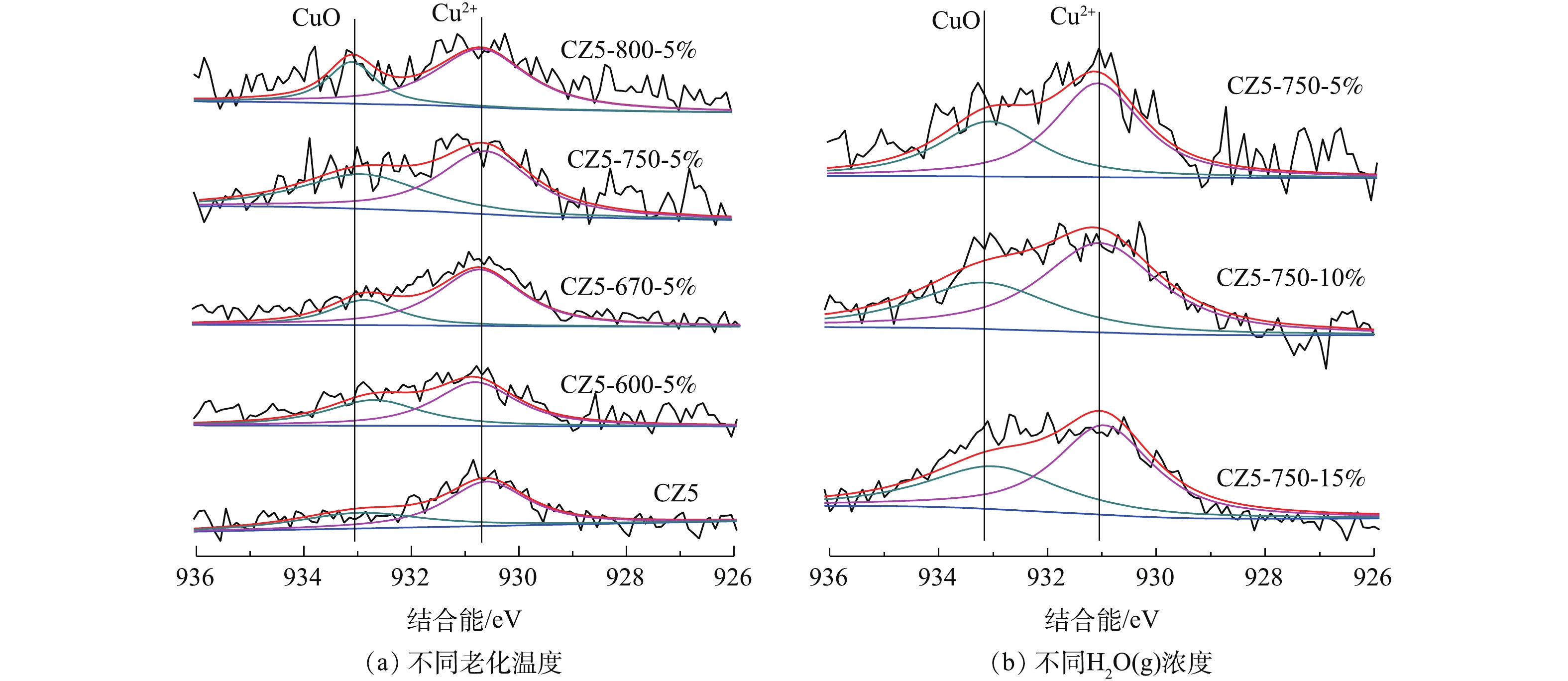 图 6 不同条件水热老化Cu/ZSM-5催化剂XPS图谱Figure 6. XPS spectra of Cu/ZSM-5 with different conditions of hydrothermal aging
图 6 不同条件水热老化Cu/ZSM-5催化剂XPS图谱Figure 6. XPS spectra of Cu/ZSM-5 with different conditions of hydrothermal aging3. 结论
1)随着水热老化温度的升高,Cu/ZSM-5催化剂的NH3-SCR活性显著下降,比表面积减小,孔容增大,催化剂发生团聚现象,催化剂的表面氧原子配位的Cu2+转化为CuO微晶或者CuO物质,活性Cu2+离子减少,导致催化剂低温活性下降。水热老化温度是影响催化剂NH3-SCR性能变化的主因。
2)随着水热老化H2O(g)含量的升高,Cu/ZSM-5催化剂上NH3-SCR性能无明显劣化,当H2O(g)含量增加至15%时,催化剂低温活性显著下降,H2O(g)含量的增加对Cu/ZSM-5催化剂结构没有显著的影响;水热老化后SCR的低温性能(<300 ℃)的下降主要是由于老化温度提高所致。
3)对Cu/ZSM-5催化剂,优化催化剂的抗高温能力是提高其水热稳定性的最直接有效的方式。提高ZSM-5分子筛结构稳定性或减少催化剂活性Cu2+迁移可能是优化Cu/ZSM-5的抗水热能力的有效途径。
-
图 1 不同温度水热(5%H2O)老化各Cu/ZSM-5催化剂上NH3-SCR性能
Figure 1. Performance comparison of NH3-SCR activity of Cu/ZSM-5 catalysts over different temperature at 5%H2O
图 2 不同H2O(g)含量水热(750 ℃)老化Cu/ZSM-5催化剂上NH3-SCR性能
Figure 2. Performance comparison of NH3-SCR activity of Cu/ZSM-5 catalysts over different H2O contents at 750 ℃
图 3 不同条件下水热老化Cu/ZSM-5催化剂XRD图谱
Figure 3. X-ray diffraction patterns of Cu/ZSM-5 with different conditions of hydrothermal aging
图 4 不同条件下水热老化Cu/ZSM-5催化剂H2-TPR图谱
Figure 4. H2-TPR of Cu/ZSM-5 with different conditions of hydrothermal aging
图 5 不同条件水热老化Cu/ZSM-5催化剂SEM图
Figure 5. SEM images of Cu/ZSM-5 with different conditions of hydrothermal aging
图 6 不同条件水热老化Cu/ZSM-5催化剂XPS图谱
Figure 6. XPS spectra of Cu/ZSM-5 with different conditions of hydrothermal aging
表 1 不同催化剂的质构参数
Table 1. Structure parameters of different catalysts
催化剂 BET比表面积/(m2·g−1) 单点总孔容/(10−2cm3·g−1) 吸附平均孔径/nm CZ5 316 9.51 28.94 CZ5-600-5% 298 14.82 29.17 CZ5-670-5% 286 15.59 30.99 CZ5-750-5% 285 21.14 31.18 CZ5-800-5% 256 47.65 44.64 CZ5-750-10% 276 26.53 34.55 CZ5-750-15% 270 26.59 34.63
下载: 导出CSV
-
[1] FAHAMI A R, NOVA I, TRONCONI E. A kinetic modeling study of NO oxidation over a commercial Cu-CHA SCR catalyst for diesel exhaust aftertreatment[J]. Catalysis Today, 2017, 297(15): 10-16. [2] KARAMITROS D, KOLTSAKIS G. Model-based optimization of catalyst zoning on SCR-coated particulate filters[J]. Chemical Engineering Science, 2017, 173(14): 514-524. [3] FICKEL D W, D′ADDIO E, LAUTERBACH J A, et al. The ammonia selective catalytic reduction activity of copper-exchanged small-pore zeolites[J]. Applied Catalysis B: Environmental, 2011, 102(3/4): 441-448. [4] KWAK J H, TONKYN R G, KIM D H, et al. Excellent activity and selectivity of Cu-SSZ-13 in the selective catalytic reduction of NOx with NH3[J]. Journal of Catalysis, 2010, 275(2): 187-190. doi: 10.1016/j.jcat.2010.07.031 [5] FICKEL D W, FEDEYKO J M, LOBO R F. Copper coordination in Cu-SSZ-13 and Cu-SSZ-16 investigated by variable-temperature XRD[J]. Journal of Physical Chemistry C, 2010, 114(3): 1633-1640. doi: 10.1021/jp9105025 [6] WILKEN N, KAMASAMUDRAM K, CURRIER N W. Heat of adsorption for NH3, NO2 and NO on Cu-Beta zeolite using microcalorimeter for NH3 SCR applications[J]. Catalysis Today, 2010, 151(3/4): 237-243. [7] 张惠, 王喜芹, 栾志强, 等. 铜-胺改性ZSM-5吸附剂的制备及其对NOx的净化机理[J]. 环境工程学报, 2013, 7(12): 4887-4890. [8] 李富霞, 任晓光, 李鹏, 等. 焙烧条件对CuO/ZSM-5催化剂脱硫脱硝性能的影响[J]. 环境工程学报, 2013, 7(8): 3117-3122. [9] 石晓燕, 刘福东, 单文坡, 等. 水热老化对不同方法制备的Fe-ZSM-5用于NH3选择性催化还原NOx的影响[J]. 催化学报, 2012, 33(3): 454-464. [10] GOMEZ S A, CAMPERO A, MARTINEZ-HERNANDEZ A. Changes in Cu2+ environment upon wet deactivation of Cu-ZSM-5 deNOx catalysts[J]. Applied Catalysis A: General, 2000, 197(1): 157-164. doi: 10.1016/S0926-860X(99)00546-3 [11] PARK J H, PARK H J, BAIK J H. Hydrothermal stability of Cu-ZSM-5 catalyst in reducing NO by NH3 for the urea selective catalytic reduction process[J]. Journal of Catalysis, 2006, 240(1): 47-57. doi: 10.1016/j.jcat.2006.03.001 [12] 任爱玲, 刘卉, 张硕, 等. Ce-Mn/ZSM-5催化剂的制备及其低温脱硝性能分析[J]. 现代化工, 2018, 38(6): 73-77. [13] 杨晓初, 肖海平, 万震天, 等. 煅烧温度影响MNOx/ZSM-5催化NO氧化性能研究[J]. 热能动力工程, 2018, 33(4): 56-62. [14] 杜蒙蒙, 温正城, 康普滋, 等. ZSM-5负载Ce-Co催化氧化NO的机理研究[J]. 热能动力工程, 2018, 33(3): 93-99. [15] SHI X Y, HE H, XIE L J. The effect of Fe species distribution and acidity of Fe-ZSM-5 on the hydrothermal stability and SO2 and hydrocarbons durability in NH3-SCR reaction[J]. Chinese Journal of Catalysis, 2015, 36(4): 649-656. doi: 10.1016/S1872-2067(14)60268-0 [16] 宋守强, 李明罡, 李黎声, 等. 磷改性ZSM-5分子筛的水热稳定性[J]. 石油学报(石油加工), 2014, 30(2): 194-203. doi: 10.3969/j.issn.1001-8719.2014.02.002 [17] DING J, XUE T, WU H H, et al. One-step post-synthesis treatment for preparing hydrothermally stable hierarchically porous ZSM-5[J]. Chinese Journal of Catalysis, 2017, 38(1): 48-57. doi: 10.1016/S1872-2067(16)62549-4 [18] IWASAKI M, YAMAZAKI K, BANNO K, et al. Characterization of Fe/ZSM-5 DeNOx catalysts prepared by different methods: Relationships between active Fe sites and NH3-SCR performance[J]. Journal of Catalysis, 2008, 260(2): 205-216. doi: 10.1016/j.jcat.2008.10.009 [19] 李振国, 马杰, 刘双喜, 等. 黏结剂对柴油车用V2O5-WO3/TiO2催化剂选择性催化还原性能的影响[J]. 工业催化, 2011, 19(11): 60-63. doi: 10.3969/j.issn.1008-1143.2011.11.011 [20] 李振国, 马杰, 王务林, 等. 制备条件对柴油车用V2O5-WO3/TiO2催化剂催化性能的影响[J]. 工业催化, 2011, 19(5): 30-33. doi: 10.3969/j.issn.1008-1143.2011.05.006 [21] BEUTEL T, SARKANY J, LEI G D, et al. Redox chemistry of Cu/ZSM-5[J]. Journal of Physical Chemistry, 1996, 100(2): 845-851. doi: 10.1021/jp952455u [22] PRALIAUD H, MIKHAILENKO S, CHAJAR Z, et al. Surface and bulk properties of Cu-ZSM-5 and Cu/Al2O3 solids during redox treatments. Correlation with the selective reduction of nitric oxide by hydrocarbons[J]. Applied Catalysis B: Environmental, 1998, 16(4): 359-374. doi: 10.1016/S0926-3373(97)00093-3 [23] RICHTER M, FAIT M J G, ECKELT R, et al. Oxidative gas phase carbonylation of methanol to dimethyl carbonate over chloride-free Cu-impregnated zeolite Y catalysts at elevated pressure[J]. Applied Catalysis B: Environmental, 2006, 73(3/4): 269-281. [24] SULTANA A, NANBA T, HANEDA M, et al. Influence of co-cations on the formation of Cu+ species in Cu/ZSM-5 and its effect on selective catalytic reduction of NOx with NH3[J]. Applied Catalysis B: Environmental, 2010, 101(1/2): 61-67. [25] NANBA T, MASUKAWA S, OGATA A, et al. Active sites of Cu-ZSM-5 for the decomposition of acrylonitrile[J]. Applied Catalysis B: Environmental, 2005, 61(3/4): 288-296. [26] RICHTER M, FAIT M J G, ECKELT R, et al. Gas-phase carbonylation of methanol to dimethyl carbonate on chloride-free Cu-precipitated zeolite Y at normal pressure[J]. Journal of Catalysis, 2007, 245(1): 11-24. doi: 10.1016/j.jcat.2006.09.009 [27] KEFIROV R, PENKOVA A, HADJIIVANOV K, et al. Stabilization of Cu+ ions in BEA zeolite: Study by FTIR spectroscopy of adsorbed CO and TPR[J]. Microporous and Mesoporous Materials, 2008, 116(1/2/3): 180-187. [28] RUTKOWSKA M, PACIA I, BASĄG S, et al. Catalytic performance of commercial Cu-ZSM-5 zeolite modified by desilication in NH3-SCR and NH3-SCO processes[J]. Microporous and Mesoporous Materials, 2017, 246: 193-206. doi: 10.1016/j.micromeso.2017.03.017 [29] 吕刚, 范啸天, 宋崇林, 等. Cu/ZSM-5催化剂制备及SCR催化性能研究[J]. 工程热物理学报, 2015, 36(10): 2276-2281. [30] LI Z G, CHEN X Y, LI J H, et al. Synthesis and evaluation of mesopore structured ZSM-5 and a CuZSM-5 catalyst for NH3-SCR reaction: Studies of simulated exhaust and engine bench testing[J]. RSC Advances, 2016, 6: 102570-102581. doi: 10.1039/C6RA20237C [31] SARMA D D, RAO C N R. XPES studies of oxides of second- and third-row transition metals including rare earths[J]. Journal of Electron Spectroscopy and Related Phenomena, 1980, 20(1): 25-45. doi: 10.1016/0368-2048(80)85003-1 [32] WANG L, GAUDET J R, LI W, et al. Migration of Cu species in Cu/SAPO-34 during hydrothermal aging[J]. Journal of Catalysis, 2013, 306: 68-77. doi: 10.1016/j.jcat.2013.06.010 [33] 郑昌坤, 韩帅, 叶青. 铜源对Cu/ZSM-5催化剂氨选择性催化还原NO的影响[J]. 化学工程, 2018, 46(9): 23-27. doi: 10.3969/j.issn.1005-9954.2018.09.005 期刊类型引用(2)
1. 陈栋栋,彭丽娟,高慎勇,李云华,孙晓宇. 基于OBD监测的国六柴油机NO_x排放方法研究. 内燃机与动力装置. 2022(04): 48-53 .  百度学术
百度学术
2. 朱自如,黄鹏,王素梅,吕立群,葛蕴珊. 柴油车SCR装置实际使用过程劣化机理研究. 重庆理工大学学报(自然科学). 2022(12): 216-222 . 百度学术
其他类型引用(4)
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 6974
- HTML全文浏览数: 6974
- PDF下载数: 83
- 施引文献: 6
