

-
剩余污泥是采用活性污泥法的污水处理厂主要的副产物[1]。目前,针对剩余污泥有多种处理处置方法,主要包括填埋、焚烧、厌氧消化、好氧堆肥等。其中,厌氧消化作为一种成本较低、稳定有机物、减少污泥体积、产出甲烷能源的技术,吸引了越来越多的关注[2]。然而,由于污泥在厌氧消化过程中具有缓慢的水解速率和较弱的产甲烷潜力,使该技术的应用受到了较大的限制[3-4]。为改善污泥的厌氧消化性能,常采用热水解[5]、超声波[6]、碱解法[4]等预处理技术对污泥中的细胞和胞外聚合物进行破解,从而释放有机物进入液相,达到改善污泥的水解速率和产甲烷潜力的目的。在众多预处理技术中,超声波破解效率快、破解程度高且无化学添加,使超声波破解污泥技术成为了热门的研究方向之一[6-8]。然而,目前大部分超声波破解污泥的研究仅限于实验室阶段,距离工业化应用仍有较大差距。
超声波反应器分探头式和槽式超声波反应器2种[9]。近年来,一些研究人员对2种类型的超声波技术进行了工业化应用的尝试。NICKEL等[10]在研究中报道了一种管道式超声波反应器,最大功率为3.6 kW,体积为1.3 L,利用该反应器破解污泥后,厌氧消化一级反应动力学速率常数由0.26 d−1提高至0.52 d−1。此外,NICKEL等[10]还开发了一种工业化规模探头式超声波反应器,体积为29 L,共计5个频率20 kHz、功率2 kW的超声波换能器,停留时间为30 s,在连续塞流式超声波处理污泥情况下,70 Wh·L−1能量输入条件下可获得50%的COD溶出率。GOGATE等[11-12]先后研发了双频率、三频率多探头超声波槽式反应器,但只是应用于废水处理,并未在污泥处理中取得应用。
上述研究对2种类型的超声波反应器均进行了优化并进行了工业化应用尝试。有研究[11-14]表明,同等功率下低功率多换能器槽式超声波反应器相比传统大功率单换能器探头式超声波反应器的声场强度更大,声场分布更均匀,能量利用效率更高。然而,目前对于工业化规模低功率多探头槽式超声波反应器破解污泥的研究还未见报道。本研究利用1台250 L的低功率多探头槽式超声波反应器,建立工业化规模超声波破解污泥实验,并探究了工业化规模超声波对不同浓度固体(TS)污泥物理化学特性及后续厌氧消化性能的改善效果。
-
实验所用污泥为天津某污水处理厂的剩余污泥,该厂采用A2/O活性污泥处理工艺,处理能力为300 000 m3·d−1。污泥取自于污泥脱水车间的脱水污泥,固体浓度为20%。
-
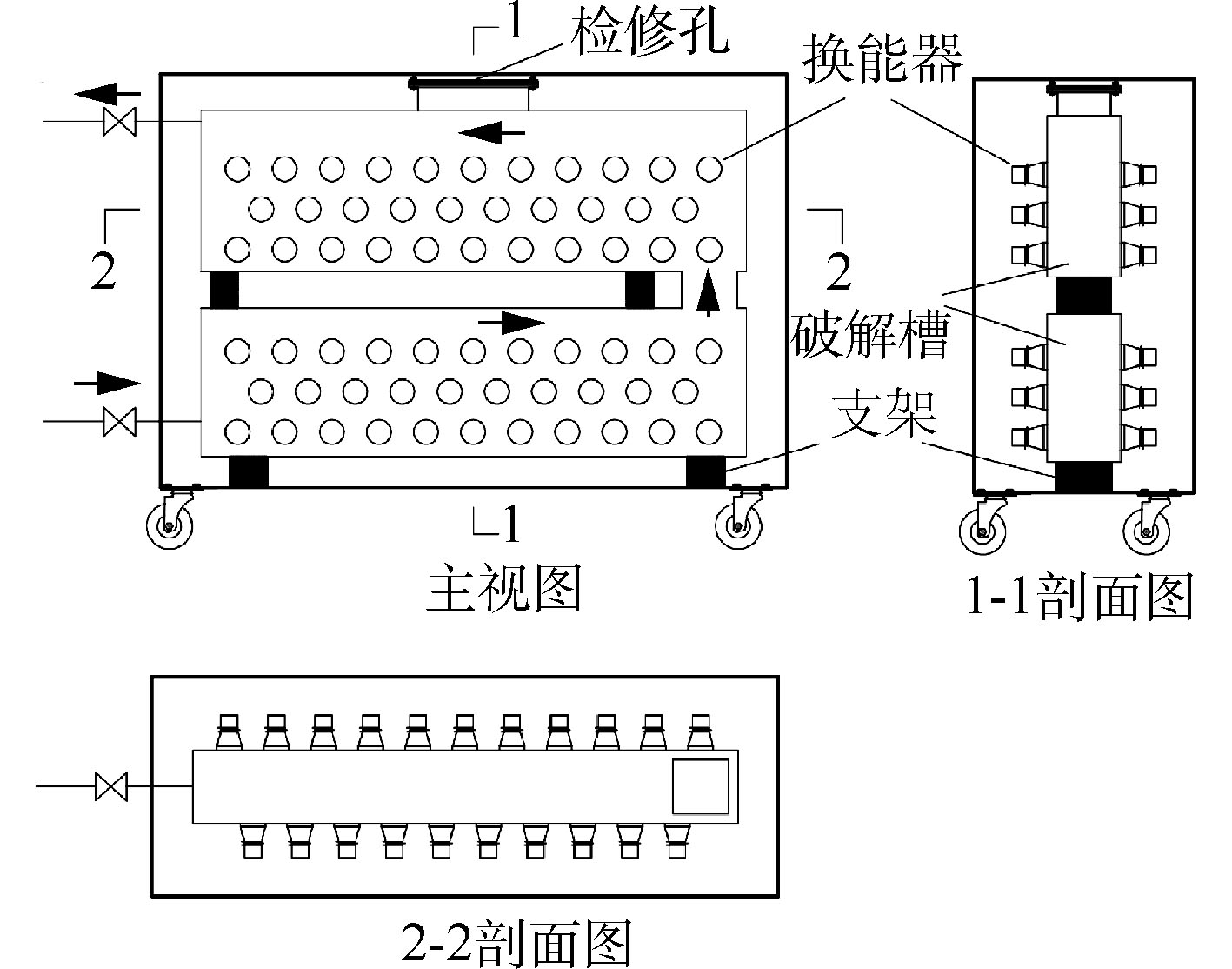
多探头槽式超声波反应器如图1所示。该超声波预处理设备主要由超声波换能器探头、超声波破解槽、超声波反应器箱体、超声波发生器、超声波发生器机柜和电缆线路系统组成。超声波破解槽体容积为250 L,总功率为10 kW。超声波换能器探头参数为20 kHz,100 W,按需求均匀错位布置于破解槽体两侧壁,反应器箱体设隔音棉环绕破解槽四周,破解槽和反应器箱体材料为304不锈钢,超声波发生器电源2台,分别使用电缆连接、控制左右两侧壁的探头,进行开闭,发生器电源功率数字显示实时超声波功率。
-
污泥在搅拌池内稀释搅拌TS至2%、4%、6%、8%、10%,之后泵入超声反应器内,工作时调整总输入功率为10 kW,设计4个污泥破解时间分别为15、30、45和60 min。破解方式为序批式,污泥加满反应器后,关闭进泥泵及反应器进出阀门,开启超声反应器,在运行15、30、45和60 min时取样监测。
-
厌氧消化实验采用全自动甲烷潜力测试系统(AMPTSⅡ,碧普(瑞典)有限公司),反应瓶体积650 mL,有效容积400 mL,混合投加泥按照基质和接种泥VS 2∶1的比例进行配比。投加污泥后,对瓶内通入氮气5 min以排出氧气。发酵温度稳定设置在37.5 ℃。沼气中CO2被100 mL瓶内80 mL 3 mol·L−1的NaOH所吸收,剩余的气体进入自动气体计量装置,每天的产气量数据会自动储存在自动化系统中,随时可以调出。
-
TS、VS、SCOD采用美国公共卫生协会推荐的标准方法[15]进行测定;溶解性多糖和总多糖采用蒽酮比色法[16-17]测定;溶解性蛋白质和总蛋白质采用Lowry法[17-18]测定;利用马尔文激光粒度仪(MasterSizer 2000,英国马尔文仪器有限公司)测定污泥粒径分布及平均粒径。
-
当超声破解污泥时,可以通过观测污泥中固体颗粒平均粒径的变化来分析其对污泥的破解效果。图2为不同TS污泥平均粒径随超声时间的变化情况。由图2可知,相比对照组污泥,60 min超声波破解时间条件下,2% TS和4% TS污泥的平均粒径分别由52.82 μm和55.17 μm降低至17.16 μm和22.39 μm,而6%、8%和10% TS的污泥降低幅度较小,分别由60.62、58.86和65.22 μm降低至37.66、50.22和52.14 μm。污泥粒径变小证实了超声波对污泥中的固体微粒进行了破解。在本研究中,低固体浓度和高固体浓度的污泥的粒径都有一定降低,不同的粒径变化反映了污泥不同的破解程度。对于TS在6%以上的污泥,超声波的大部分声能被固体物质所吸收,不能对污泥进行有效的破解;对于2% TS和4% TS的污泥,超声波可以对其污泥进行有效的破解,污泥中的絮体、微生物、固体颗粒和菌胶团被打散,有机物溶出,固体污泥粒径变小。
-
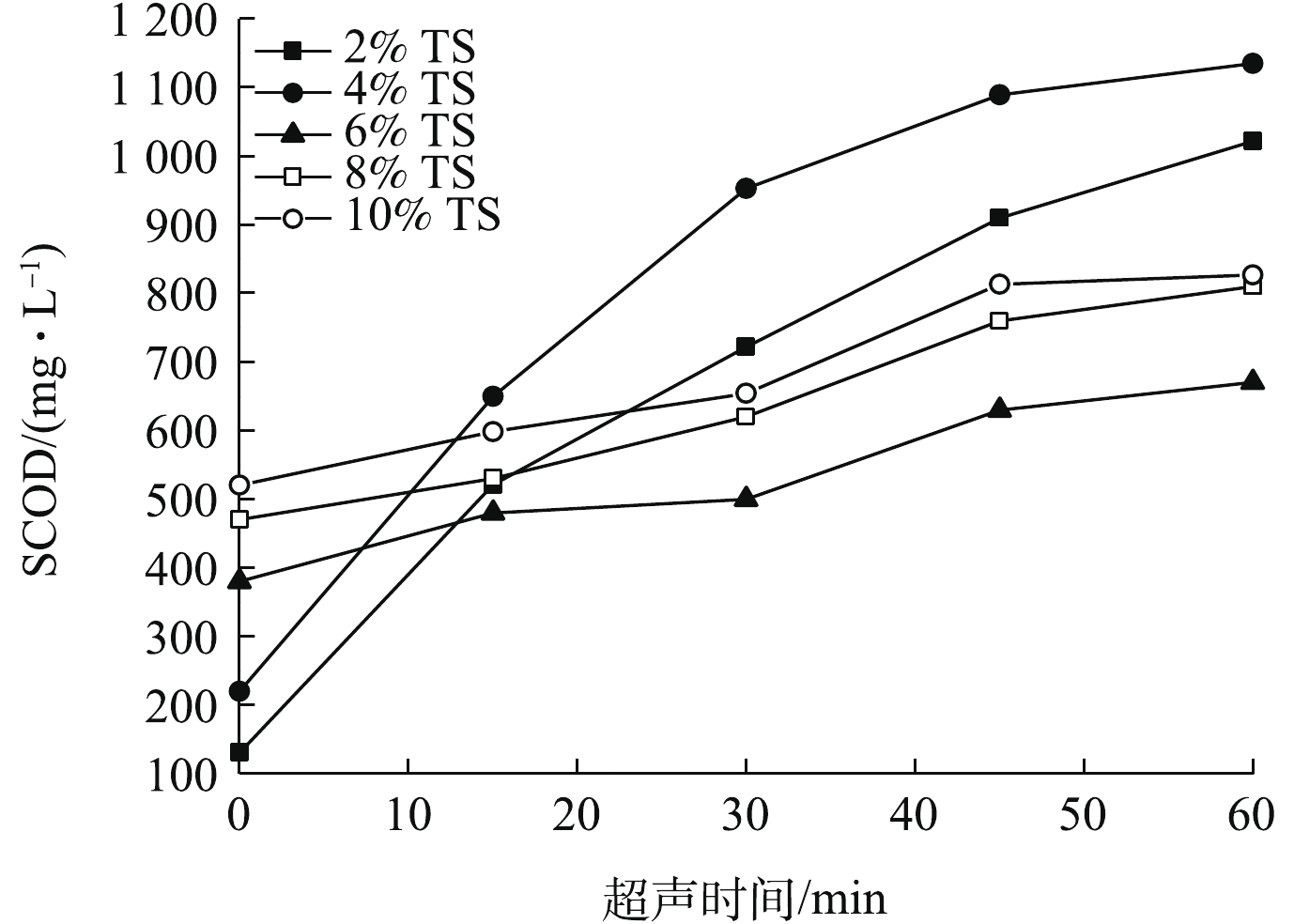
为进一步研究超声对污泥的破解程度,需要对污泥中耗氧类有机化合物(以COD计)、蛋白质、多糖的溶出情况进行测定分析。不同TS污泥超声波破解后,污泥SCOD随时间的变化如图3所示。由图3可知,随着污泥破解时间的延长,各不同比例TS下污泥SCOD均逐渐增加。当TS为2%和4%时,超声破解时间为30 min时,SCOD分别从130 mg·L−1和220 mg·L−1增加到722 mg·L−1和953 mg·L−1;超声破解时间增加至60 min,SCOD分别增长到1 022 mg·L−1和1 135 mg·L−1。当TS为6%、8%和10%时,SCOD随超声时间的增加而增加,超声破解时间增加到60 min,SCOD浓度从380、470和520 mg·L−1分别增长到670、810和827 mg·L−1。上述结果说明,超声对TS浓度2%和4%的污泥进行了有效的破解,前30 min内,破解效率较高,30 min后,SCOD增长速度明显变缓;超声波对4% TS的破解效果优于对2% TS污泥的破解效果;超声波对TS为6%、8%和10%的污泥破解效果较差,SCOD随超声时间的增加保持微弱的增加趋势。超声波对低固体浓度污泥破解效率高,但对高固体浓度的污泥破解效率较低。其主要原因是:对于较高浓度的污泥,超声波中的能量大部分被固体物质所吸收,故空化作用被衰减,达不到有效破解污泥的目的[19]。因此,利用超声波破解污泥时,宜选用TS浓度不高于4%的污泥。
-
剩余污泥中的有机成分主要包括蛋白质、多糖和脂类,大部分的有机成分都被污泥中微生物细胞及胞外聚合物包裹,污泥被超声预处理以后,SCOD增加,这主要归因于蛋白质和多糖的溶出[7]。不同浓度的TS污泥被超声波破解后,溶解性蛋白质的浓度随超声时间的变化如图4所示。由图4可知,和SCOD溶出情况类似。当TS高于6%时,蛋白质溶出效果并不明显;当污泥TS为2%时,超声时间30 min和60 min条件下蛋白质浓度分别为302 mg·L−1和609 mg·L−1;当污泥TS为4%时,超声时间为30 min和60 min条件下蛋白质浓度分别为403 mg·L−1和653 mg·L−1。上述结果表明,超声波对6% TS或更高固体浓度污泥的蛋白质溶出效果不明显,当污泥TS在4%时,超声波对蛋白质破解效果优于TS为2%的污泥。
蛋白质是污泥有机物中的重要组成部分,在本研究中,对于TS浓度为2%和4%的污泥进行超声破解后,蛋白质溶出较明显。按照蛋白质的COD当量系数1.50 g·g−1[20]计算,60 min超声破解后,TS浓度为2%和4%的污泥溶出的蛋白质COD当量分别占溶出COD总量的89.3%和86.3%,以上结果证实了溶出物质的主要成分是蛋白质。
-
超声波破解不同浓度TS的污泥后,溶解性多糖浓度随超声时间的变化如图5所示。由图5可知,与COD及蛋白质的溶出情况不同,在30 min破解时间内,各浓度TS下的污泥溶解性多糖的溶出趋势较为相似,并无明显差别。在破解时间60 min后,2%、4%、6%、8%和10% TS污泥中溶解性多糖的浓度分别为312.4、251.1、222.7、251.2和266.3 mg·L−1。此结果表明,在本研究中,当超声波破解污泥时,超声波对污泥中多糖溶出效果有限。多糖溶出效果较差的原因可能是在本研究中所用的脱水污泥特性所致。
多糖同样是污泥有机物中的重要组成部分,在本研究中,按照多糖的COD当量系数1.07 g·g−1[20]计算,在60 min超声破解后,TS浓度2%、4%、6%、8%和10%污泥中溶解性多糖的COD当量分别占溶出COD总量的32.7%、23.7%、35.6%、33.2%和34.5%。因此,多糖也是污泥有机溶出物的主要成分,但在COD总量占比中,相比蛋白质较少。
-
不同超声波破解时间下TS浓度2%污泥厌氧消化累积甲烷产率的变化如图6所示。由图6可知,在27 d厌氧消化过程中,厌氧消化累积甲烷产率随超声时间的增加逐渐升高。相比对照组污泥,15、30、45和60 min超声波预处理后,污泥预处理的累积甲烷产率分别增加了7.5%、41.2%、44.6%和48.5%;15 min超声波的破解时间对污泥甲烷产率增加量较小(增加了7.5%),30 min超声波破解时间下甲烷产率明显提升了41.2%,而破解时间的进一步增加对甲烷产率增加则有限。
结合前面对污泥有机物的溶出效果的研究结果,TS浓度为2%的污泥COD在60 min内几乎始终保持线性增加的趋势。然而,在评价累积甲烷产率时,30 min是有效提升甲烷产率的超声破解时间,30 min之后仅对甲烷产率有微弱提升。上述结果说明,COD的增加和甲烷产率的增加并不是绝对相关的,这和KIM等[21]的研究结果是一致的。超声波破解污泥时,较长的时间虽然会使有机物溶出浓度有一定增加,但是溶出的有机物中并不全是易生物降解的有机物,因此,超声时间对厌氧消化性能的改善也需要进行综合评估。
不同超声时间下TS浓度为4%的污泥厌氧消化过程中的累积甲烷产率如图7所示。由图7可知,随超声时间的增加,厌氧消化累积甲烷产率逐渐增加。相比对照组污泥,15、30、45和60 min超声波预处理后,污泥预处理的累积甲烷产率分别增加了8.4%、30.2%、33.6%和34.4%;相比未处理污泥,15 min超声波的破解时间对污泥甲烷产率仅增加了8.4%,30 min超声波对甲烷产率显著增加了30.2%,破解时间的继续增加对甲烷产率作用不明显。
当超声对TS浓度为4%的污泥进行破解时,前30 min超声破解时间下溶解性有机污染物的溶出量占60 min超声破解时间下溶解性有机污染物溶出量的90%,这说明30 min内对污泥破解已经达到了较高的程度,而30~60 min的超声破解对污泥COD溶出较少。上述结果说明,在低功率密度超声破解污泥时,延长时间并不意味COD溶出率和甲烷产率的增加,仍存在有效的破解时间范围,在该范围内进行超声波破解可以促进COD溶出率和甲烷产率增加,当超过该时间范围后对污泥厌氧消化改善不明显。因此,在实际应用中,需要对破解时间进行优化,根据需要选择最合适的破解时间,以获得更高的能量利用效率。
不同超声时间下,6% TS(图8(a))、8% TS(图8(b)和10% TS(图8(c)污泥厌氧消化过程中的累积甲烷产率如图8所示。相比对照组,随超声时间的增加,各浓度的TS下甲烷产率未见明显升高。因此,当TS为6%时,不但对COD、蛋白质和多糖破解溶出有限,对厌氧消化累积甲烷产量也没有明显改善。
由上述结果可知,在利用超声波预处理污泥时,要对污泥浓度、破解时间进行优化,选取最优的污泥浓度和超声波破解时间,以获得更好的破解效果和更高的能量利用效率。该多探头槽式超声波反应器破解污泥的最佳固体浓度条件是不高于4%,超声波破解时间是不超过30 min。
-
不同浓度的TS下,污泥厌氧消化后VS去除率随超声时间变化情况如表1所示。由表1可知,随固体浓度的增加,VS去除率呈现降低的趋势;而随超声时间的增加,VS去除率呈现升高的趋势。固体浓度越高,污泥厌氧消化后有机物去除率越低,该结果与FERNÁNDEZ等[22]的研究结果是一致的;超声波破解污泥后,2% TS和4% TS污泥的VS去除率相比对照组有明显提升,而6%和更高浓度TS的污泥VS去除率相比对照组提升并不明显,这与甲烷产率的结果是对应的;2% TS和4% TS污泥的VS去除率在超声破解时间为15 min时分别提升了1.84%和1.5%,在超声破解30 min时VS去除率分别提升了6.34%和8.92%,30 min之后VS去除率未见明显升高。上述结果说明,30 min内对污泥的破解更有效,继续延长时间对VS去除率提升并不明显。
在厌氧消化过程中,有机物被微生物代谢降解,转化为甲烷,因此,甲烷产率和VS去除率基本是一致的,与甲烷产量的改善情况一样,30 min破解时间对污泥厌氧消化VS去除率最有效。
-
1)随污泥固体浓度的增加,多探头槽式超声波反应器破解污泥的程度逐渐降低,污泥粒径降低幅度逐渐减缓,污泥中有机物溶出效果逐渐减弱。该多探头槽式超声波反应器破解污泥的最佳固体浓度条件为不高于4%。
2)对于TS浓度为2%和4%的污泥,随超声破解时间的延长,多探头槽式超声波反应器破解污泥的程度逐渐增加,污泥中溶解性有机物浓度逐渐增加,在30 min破解时间条件下,污泥甲烷产率分别提升了41.2%和30.2%,30 min后延长超声破解时间对厌氧消化性能改善影响较小。
3)对于TS浓度为2%和4%的污泥,多探头槽式超声波超声破解30 min时,VS去除率相比对照组分别提升了6.34%和8.92%。本研究为超声波破解污泥技术的工业化应用提供了理论依据。
工业化规模超声波预处理对不同固体浓度污泥厌氧消化性能的影响
Effect of full-scale ultrasonic pretreatment on anaerobic digestion performance of sludge with different solid concentrations
-
摘要: 针对活性污泥厌氧消化水解速率慢的问题,通过工业化规模超声波反应器对不同固体浓度污泥开展了破解研究。采用粒径分析及溶解性COD、蛋白质和多糖浓度监测的方法研究了超声波破解前后污泥物理化学特性的变化;评估了超声波破解对污泥厌氧消化产甲烷潜力及有机物降解规律的影响。结果表明:工业化规模超声波破解不同固体浓度污泥后,污泥粒径均有所降低,而溶解性COD、蛋白质和多糖的浓度均有增加;超声波对污泥的破解程度与破解时间和固体浓度有关,其随破解时间增加而增加,随污泥固体浓度增加而减弱;超声波破解固体浓度2%和4%的污泥30 min后,累积甲烷产率分别提升41.2%和30.2%,当破解时间和固体浓度进一步增加时,污泥甲烷产率无明显变化。本研究结果可为超声波破解污泥技术的工业化应用提供参考。Abstract: To resolve the problem of low hydrolysis rate of activated sludge during anaerobic digestion process, an industrial scale ultrasonic reactor was used to disintegrate excess sludge under different solid concentrations. The physical and chemical characters of sludge before and after ultrasonic disintegration were evaluated in terms of particle size, and soluble COD, protein, carbohydrate. Effects of ultrasonic disintegration on cumulative methane yield and organism decomposition during anaerobic digestion process were studied. The results indicated that with the pretreatment of ultrasonic operation, the particle size of sludge was reduced, while an obvious increase of the concentrations of SCOD, soluble protein and carbohydrate occurred. The disintegration degree increased with sonication time extension, and decreased with the increase of solid concentrations. After 30 min ultrasonic pretreatment of 2% and 4% TS sludge, their methane yield increased up to 41.2% and 30.2%, respectively. However, there was no obvious increase of methane yield as ultrasonic time and sludge TS further increased. Through above research, the results provide technological support for ultrasonic pretreatment at industrial scale.
-
Key words:
- full-scale /
- ultrasonic pretreatment /
- sludge pretreatment /
- activated sludge /
- anaerobic digestion
-
近年来,基于过硫酸盐的高级氧化技术(persulfate−based advanced oxidation processes,PS−AOPs)作为去除水中难降解有机物的有效方法而受到广泛关注[1 − 3]. 研究表明,PS−AOPs主要依赖于体系中的硫酸根自由基(SO4·−)、羟基自由基(·OH)等作为活性氧化物质(reactive oxide species,ROS)发挥氧化作用,进而分解、矿化体系中的有机污染物. 含Co、Ag、Cu、Mn、Fe、Au、Ru等多种金属元素在内的多种非均相催化剂可有效激活过一硫酸盐(peroxymonosulfate,PMS)和过二硫酸盐(peroxydisulfate,PDS)去除体系中的有机污染物[4-5]. 非均相催化的显著优点在于固相催化剂可以通过过滤或离心等操作从反应后的体系中分离,能够有效控制产物中的金属残留、降低二次污染风险[6].
近年来,随着纳米催化科学的发展和表征技术的进步,金属催化剂表面锚定的活性粒子被缩小到原子水平,被定义为单原子催化剂(single−atom catalysts,SAC). SAC相较于常规纳米催化剂显示出以下几点突出优势[7 − 8]:(1)锚定在载体上的特定位点的金属单原子与载体阴离子之间将形成强化学键,将有效提高催化剂的反应活性和稳定性;(2)SAC表面金属原子理论分散度高达100%,具有理论上的最大原子利用效率;(3)活性位点结构及分散状态均一,可从原子甚至电子层次下研究催化体系中的构效关系. 基于以上显著优势,根据Web of Science(WOS)和China National Knowledge Infrastructure(CNKI)发文量统计数据(见图1)可以明显看出,SAC的开发和应用得到了越来越多研究者的关注,并逐渐成为非均相PS−AOPs技术研究的发展前沿[9 − 10]. 本文重点综述了SAC活化过硫酸盐产生氧化活性物质的驱动机制,对结构调控与催化性能的相关性进行了系统总结,深入了解结构−性能关系,指出了精确调整催化剂活性位点的策略,而后进一步讨论了当前的研究局限和未来的研究需求.
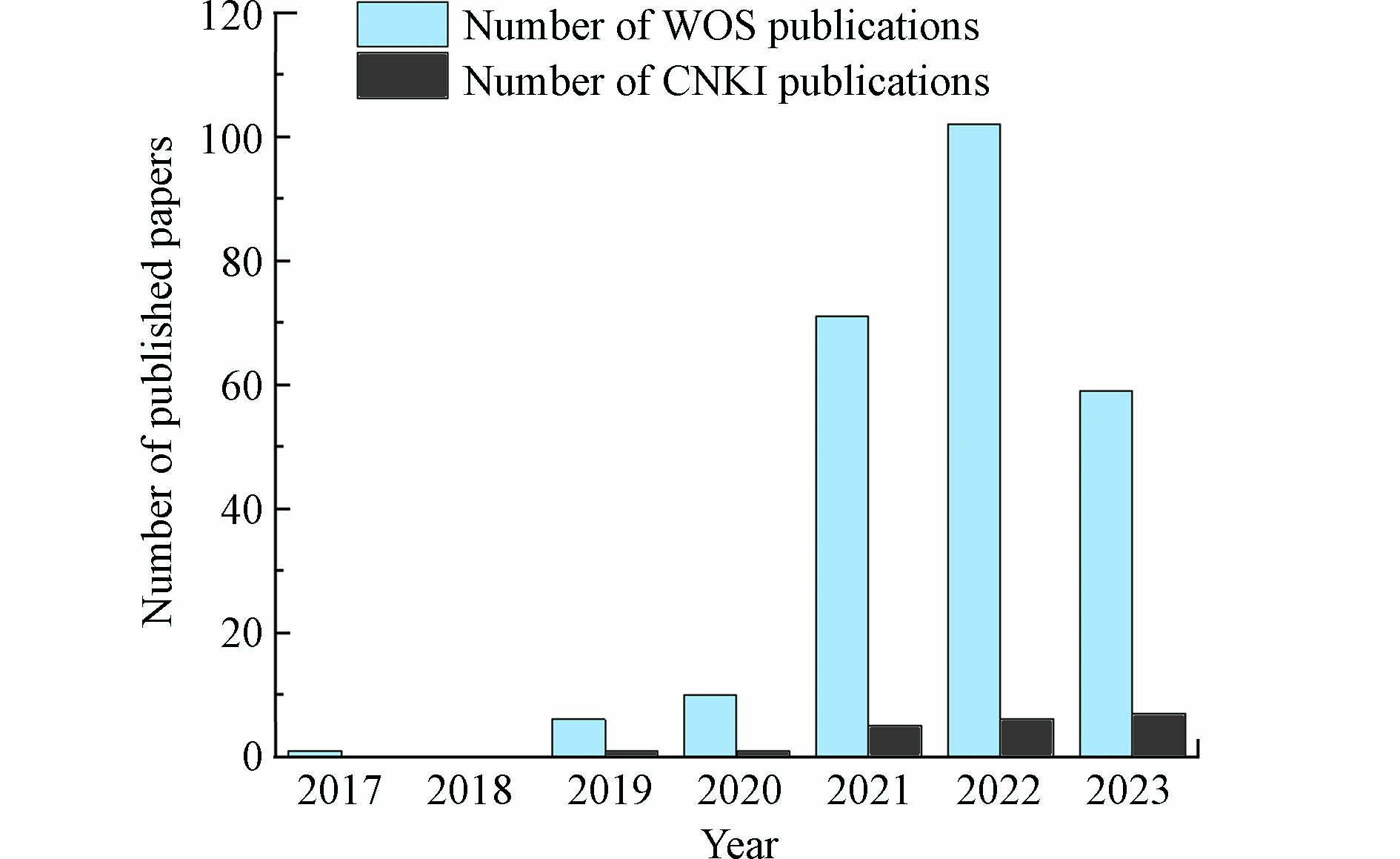 图 1 历年SAC活化过硫酸盐相关论文发布量统计图Figure 1. Statistical chart of papers published on SAC activation of persulfate over the years
图 1 历年SAC活化过硫酸盐相关论文发布量统计图Figure 1. Statistical chart of papers published on SAC activation of persulfate over the years1. 过硫酸盐的特点(Characteristics of persulfate)
通过能量转移或还原电子转移过程均可分解PMS和PDS中过氧键(O—O)并产生SO4·−和·OH. 但由于PMS中的O—O键不对称,与氢相连的O—O带部分正电荷,而PDS中O—O键的电荷分布对称,因而二者具有不同的反应特性(表1)[11 − 12]. PMS更易受到各种亲核试剂的攻击,包括X−、CN−、N3−和HCO3−[13 − 15]. PMS与卤化物离子的反应尤其值得关注,其反应过程中有可能不涉及过硫酸盐活化(即SO4·−的形成)而是形成卤素自由基,但在PDS中未观察到此反应[14 − 15]. 此外,尽管PDS的氧化还原电位高于PMS(E0(HSO5−/HSO4−)=+1.82 vs. NHE;E0(S2O82−/HSO4−)=+2.08 vs. NHE),由于在过氧化物连接体的两侧存在两个SO3基团而产生的空间位阻,也使得PMS比PDS相对选定有机物的反应性更高,例如,PMS与芳香族和脂肪族醛的反应更快[11].
表 1 PMS和PDS理化性质、反应性和主要氧化剂对比Table 1. Comparison of Physical–Chemical Properties, Reactivity, and Main Oxidants for PMS and PDSPMS PDS 离子式 HSO5− S2O82− 结构示意图 

标准还原电位(E0 vs NHE.) 1.82 V 2.08 V 双氧键解离能 377 kJ·mol−1 92 kJ·mol−1 248 nm处的摩尔吸收系数 19.1 L·mol−1·cm−1 27.5 L·mol−1·cm−1 解离常数(pKa) 9.3 −3.5 与亲核试剂的反应性 对亲核试剂(如X−和HCO3−)的有效氧原子转移反应将导致二级氧化剂的形成 可忽略(在过量背景阴离子下稳定) 与自由基的反应性 与pH相关pH < pKa2 = 9.3 (HSO5−)k(SO4·−) < 105 ( mol·L−1·s)−1k(·OH) = 1.7 × 107 (mol·L−1·s)−1pH > pKa2 = 9.3 (SO52−)k(SO4·−) < 105 (mol·L−1·s)−1k(·OH) = 2.1 × 109 (mol·L−1·s)−1 k(SO4·−) = 1.2 × 106 (mol·L−1·s)−1 首选活化方法 基于非均相催化剂的电子转移活化 基于能量转移的活化 | Show Table DownLoad:
CSV
DownLoad:
CSV
上述PMS和PDS的结构差异导致了具体活化过程的差异. 例如,由于离子结构不对称,PMS比PDS更容易被过渡金属(如Co2+、CuFe2O4、Fe2O3)激活生成SO4·−[16 − 17]. 在碳质材料和贵金属催化剂通过电子转移机制氧化有机物方面,PMS也比PDS更有效[18 − 19]. 相比于PMS,因为PDS键解离能较低,能量转移过程(即光解、热分解)激活PDS效果更明显[20 − 21]. 然而,相对处理效率取决于水基质. 由于水体背景成分的存在,如Cl−和HCO3−,使PMS氧化体系净水效果优于PDS. PMS与天然亲核试剂的反应提高了SO4·−的生成率,并产生二级非自由基氧化剂(如HOCl),而无机杂质在PDS过程中往往会淬灭自由基使其失去氧化活性[22 − 23]. 将pH值提高到PMS的pKa2以上(即9.3)会使摩尔吸收系数增加10倍(从13.8 (mol·L−1)−1·cm−1增加到149.5 (mol·L−1)−1·cm−1),从而提高UV/PMS过程的自由基生成效率[24]. 而在碱性条件下,UV/PDS体系中的主要物质从SO4·−转变为·OH,而摩尔吸收率和相关自由基生成率没有变化. 注意,由于pKa值极低,PDS在较宽的pH范围内不会发生质子化[25].
近年来,非自由基途径因其对复杂水体中痕量有机污染物的高效降解而在废水净化领域受到广泛关注. 具体来说,该反应途径对具有富电子基团的有机污染物具有更高的选择性,从而对多种无机离子和有机质等水环境背景物质具有更强的抵抗力[26]. 目前,对于非均相反应体系中的非自由基过程,提出了3种可能的反应途径:电子转移过程(固相催化剂作为电子传导媒介)、高价态金属氧化和单线态氧氧化[4]. 一般来说,过硫酸盐与催化剂表面金属物种之间的电子转移是ROS生成的主要过程. 因此,SAC中高度分散的单金属原子使催化中心数量和过硫酸盐活化的金属利用效率最大化[27 − 28],并且SAC中独特的电子结构使其表现出明显由于同类金属颗粒催化剂的过硫酸盐活化性能[29]. 与此同时,在有关于PS−AOPs的研究报道中,SAC中单金属原子的载体主要是各种碳材料,包括石墨烯[30]、还原氧化石墨烯[31]、碳纳米管[32]、非金属原子(如N、S、B等等)掺杂的石墨烯[33]、石墨氮[34]等等,其中往往含有丰富的潜在活性位点,如空穴结构、氧官能团和非金属原子等,可以作为过硫酸盐活化位点[35],或是作为污染物吸附位点促进体系中目标污染物的快速去除[36]. 其他载体材料,如金属氧化物(例如,MgO、FeOx、ZnO、WO3、CuO、Co3O4、Al2O3、CeO2)[37 − 38]和金属纳米颗粒(Pd、Pt、Rh、Ni、Ru、Cu、Ag、Au等)[39 − 40]也可作为SAC载体,此类SAC已被应用于多领域研究,而有关于PS−AOPs的研究报道有MXene[41]、Mn3O4[42]和In2O3[43]. 综上所述,SAC往往具有种类丰富、数量可观的活性位点,能够有效活化过硫酸盐产生自由基和非自由基氧化作用,实现目标污染物的高效去除.
2. 单原子催化剂在过硫酸盐活化体系中的研究进展(Research progress of SAC in persulfate activation systems)
近年来,一系列SAC作为过硫酸盐活化剂被用于降解各种有机微污染物(表2),SAC均表现出远高于相应的具有纳米尺度金属位点的催化剂的催化性能[44]. 从催化机理看,纳米粒子结构复杂,其中组成金属原子可能位于不同的化学环境中发挥催化作用,因而活性位点种类繁杂,难以建立结构−性能关系. 一般而言,SAC的催化活性是由金属活性位点与载体类型、配位环境原子排布方式来协同决定的,如图2所示[45 − 46]. 相关研究从原子甚至电子层次下研究催化体系中的构效关系,揭示了过硫酸盐在催化剂的表面的反应机理、提出了相应的材料性能优化策略.
表 2 单原子催化剂/过硫酸盐降解有机污染物概况Table 2. Summary of degradation of organic pollutants by single atom catalyst/persulfate催化剂Catalyst 单原子位点Single atomic site 反应条件Reaction condition 催化性能Catalytic performance 金属离子溶出Metal ion leaching concentration Fe−g−C3N4 [47] Fe [亚甲基蓝]0 = 10 mg·L−1[PMS]0 = 0.5 mmol·L−1[催化剂]0 = 50 mg·L−1反应时间 = 30 min 100%(第一次循环)~85%(第四次循环)100%(再生处理后第五次循环) [Fe] = 0.022 mg·L−1(第1次循环)[Fe] = 0.017 mg·L−1(第2次循环)[Fe] = 0.011 mg·L−1(第3次循环) Fe−NC[28] Fe [污染物]0 = 22 mg·L−1[PMS]0 = 0.5 mmol·L−1[催化剂]0 = 100 mg·L−1反应时间 = 160 s 100%(双酚A)~80%(磺胺甲恶唑)100%(2−氯苯酚)~35%(马卡西平)100%(4−氯苯酚) [Fe] = 0.391 mg·L−1 Co−NC[48] Co [污染物]0 = 50 µmol·L−1[PMS]0 = 0.24 mmol·L−1[催化剂]0 = 20 mg·L−1反应时间 = 15 min 100%(苯甲酸)100%(布洛芬)100%(对羟基苯甲酸)100%(双酚A)100%(对氯苯酚) [Co] = 0.048 mg·L−1(第1次循环)[Co] = 0.039 mg·L−1(第5次循环) Co−g−C3N4[34] Co [污染物]0 = 5 mg·L−1[PMS]0 = 1.0 mmol·L−1[催化剂]0 = 100 mg·L−1反应时间 = 20 min 100%(环丙沙星)100%(磺胺甲恶唑)100%(四环素)100%(苯酚) — Mn−NC[49] Mn [磷酸氯喹]0 = 25 mg·L−1[PMS]0 = 2.0 mmol·L−1[催化剂]0 = 1000 mg·L−1反应时间 = 30 min ~90%(第一次循环)~55%(第二次循环)~55%(第三次循环) [Mn] = 0.018 mg·L−1 Mn−NC[50] Mn [污染物]0 = 20 mg·L−1[PMS]0 = 0.2 g·L−1[催化剂]0 = 200 mg·L−1反应时间 = 6 min 100%(双酚A)100%(亚甲基橙)100%(苯酚)97.8%(环丙沙星)100%(氧氟沙星) [Mn] = 2.097 mg·L−1 CuSA−NC[51] Cu [2,4 −二氯苯酚]0 = 100 µmol·L−1[PDS]0 = 0.5 mmol·L−1[催化剂]0 = 40 mg·L−1反应时间 = 30 min ~95% [Cu] = 0.013 mg·L−1 Cu−N4/C−B[33] Cu [双酚A]0 = 20 mg·L−1[PMS]0 = 0.2 g·L−1[催化剂]0 = 100 mg·L−1反应时间 = 5 min ~95% — Ni−NC[52] Ni [苯酚]0 = 10 mg·L−1[PMS]0 = 0.1 g·L−1[催化剂]0 = 100 mg·L−1反应时间 = 60 min 94.8%(第1次循环)86.8%(第2次循环)81.9%(第3次循环) [Ni] = 0.0291 mg·L−1(第1次循环)[Ni] = 0.0111 mg·L−1(第2次循环)[Ni] = 0.0058 mg·L−1(第3次循环) Fe/Cu–N–C[53] Fe和Cu [氯霉素]0 = 20 mg·L−1[PDS]0 =5.0 mmol·L−1[催化剂]0 = 100 mg·L−1反应时间 = 60 min 82.4%(第1次循环)~70%(第5次循环) — Cu−SA/MXene[41] Cu [双酚A]0 = 10 mg·L−1[PMS]0 = 2.0 mmol·L−1[催化剂]0 = 500 mg·L−1反应时间 = 10 min >90% (5次循环降解) [Cu] = 6.8×10−4 mg·L−1 pH=3.0[Cu] = 4.5×10−4 mg·L−1 pH=5.0[Cu] = 2.1×10−4 mg·L−1 pH=7.0[Cu] = 1.6×10−4 mg·L−1 pH=9.0[Cu] = 0.8×10−4 mg·L−1 pH=11.0 Co SAC/Mn3O4[42] Co [磺胺甲恶唑]0 = 2 mg·L−1[PMS]0 = 0.2 mmol·L−1[催化剂]0 = 200 mg·L−1反应时间 = 30 min >90% (6次循环降解) [Co] = ~0.055 mg·L−1(第1次循环)[Co] = ~0 mg·L−1(第6次循环) Cu−In2O3/Ov Cu [四环素]0 = 20 mg·L−1[PMS]0 = 1 mmol·L−1[催化剂]0 = 500 mg·L−1反应时间 = 20 min >90% (5次循环降解) [Cu] = 0.032 mg·L−1 | Show TableDownLoad:
CSV
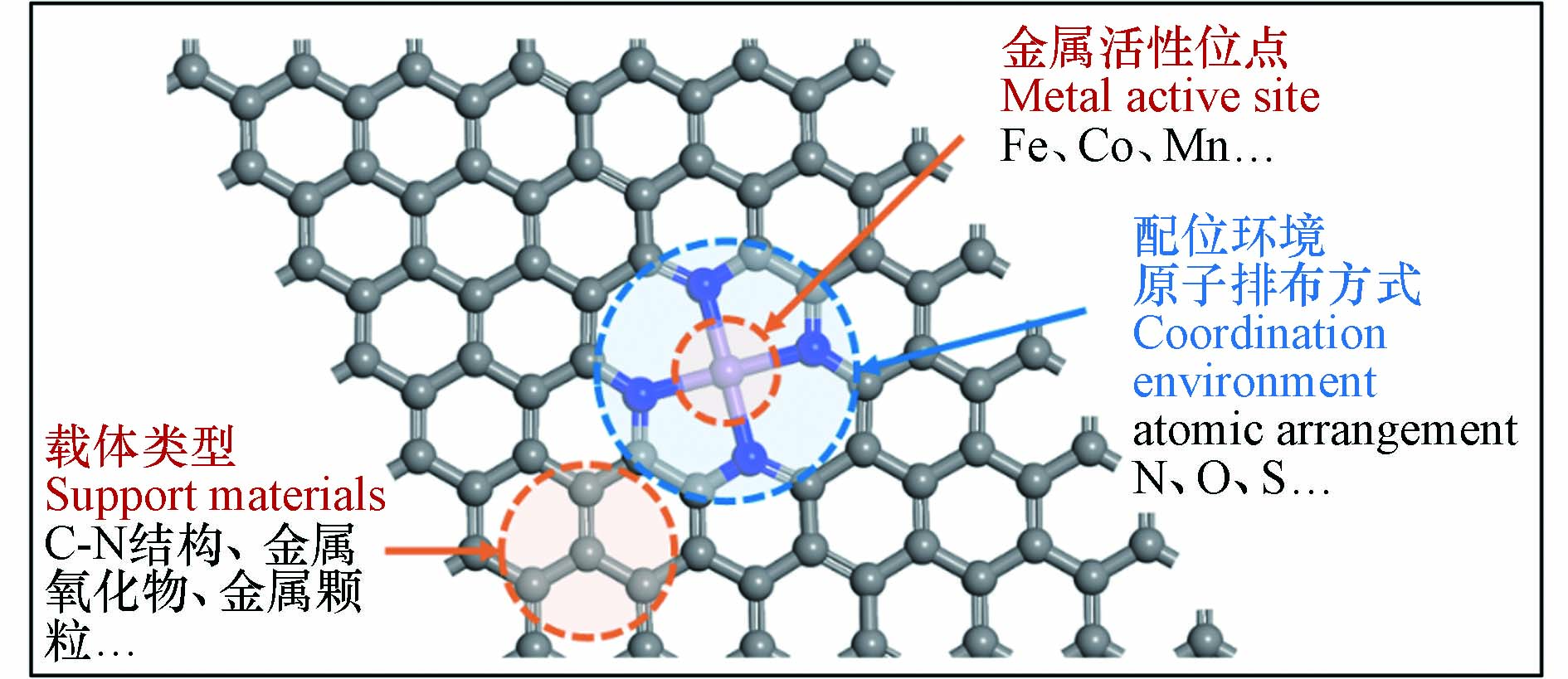 图 2 标记单原子催化剂的金属活性位点、载体类型和配位环境原子排布方式示意图Figure 2. Schematic of SAC, highlighting center active metal, support type and coordination environment atomic arrangement
图 2 标记单原子催化剂的金属活性位点、载体类型和配位环境原子排布方式示意图Figure 2. Schematic of SAC, highlighting center active metal, support type and coordination environment atomic arrangement2.1 单原子催化剂活化过一硫酸盐
金属中心及其载体种类是影响氧化活性物质生成和氧化途径发生的关键性决定因素[46]. 相关研究表明,在保持金属原子负载量相近、配位环境相类似的情况下,具有不同的金属原子位点的SAC活化PMS将导致不同的ROS主导污染物的降解,污染物去除效果差异明显(图3a)[54]. Gao等[44]从Co−NPs中制备Co−SAC作为PMS催化剂,制备过程如图3b所示. Co−SAC比Co−NPs和其他Co基催化剂表现出显著提高的污染物分解活性(图3c). 研究者将Co−SAC的优异性能归因于单一金属原子的高密度和超细分散,进而最大限度地增加催化中心的数量和PMS活化反应中的金属利用效率,同时碳载体特性(石墨度、杂化和组成)也显著改变了SAC的电荷再分配,从而调节了催化剂对PMS的催化活性. 相似的结论在其他研究报告中也有提及[9, 55 − 56]. 目前大部分SAC是以具有C−N−金属配位的杂原子掺杂碳为载体,MXene的通式为Mn+1XnTx(M:过渡金属,X:C或N;Tx:表面官能团,如—OH、—O、—Cl或−F;n = 1、2或3),是一种过渡金属碳化物和碳氮化物,被广泛认定为一种优异的催化剂载体材料[57 − 59]. 将Cu单原子锚定在Ti3C2Tx MXene纳米片上的复合催化剂(Cu−SA/MXene−900)活化PMS降解污染物的效果明显优于其他含Cu催化剂,包括其同类型纳米催化剂(Cu−NP/MXene−900),如图3d中所示. 实验与密度泛函理论(density functional theory,DFT)计算结果表明,Cu−SA/MXene−900将PMS转化为1O2的效率高达99.71%[41],如图3d中所示,Cu−SA/MXene活化PMS时将选择性吸附PMS的末端O,促进SO5·−的产生进而转化为1O2. 因此,不同的金属活性位点和载体类型对于氧化活性物质和氧化途径的驱动机制有所差别.
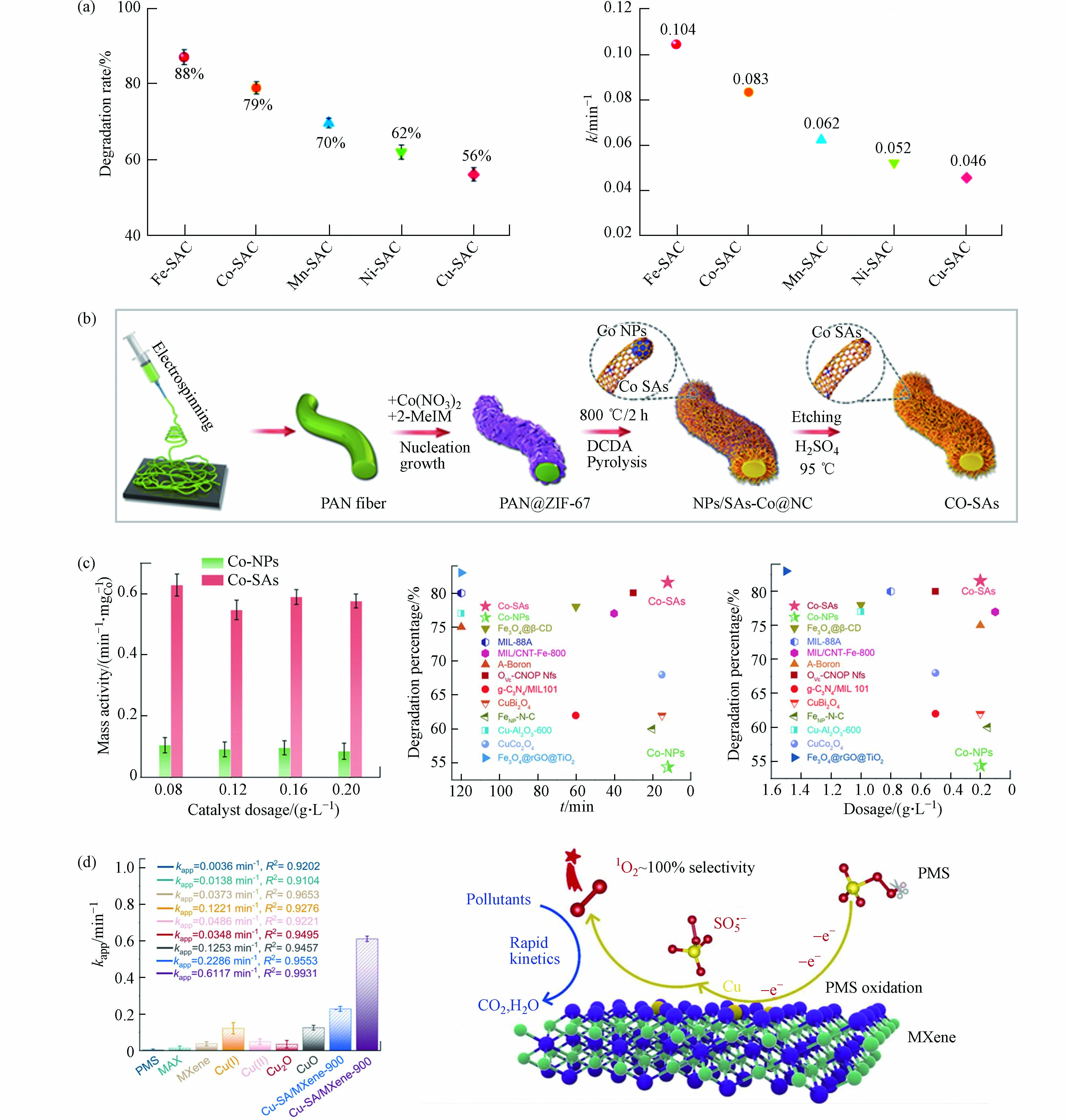 图 3 (a)M−SACs在30min内对BPA的降解曲线及降解速率;(b)由IF−67包覆PAN纳米纤维制备Co−NPs和Co−SAC的方案;(c)不同体系中按Co重量归一化的反应速率常数,该体系和其他研究中提及的催化剂对BPA降解百分比(%)(反应时间和催化剂用量)[44];(d)双酚A在不同催化剂活化PMS作用下的速率常数,Cu−SA/MXene活化PMS高效转化生成1O2过程示意图[41]Figure 3. (a) Degradation curves and degradation rate of BPA catalyzed by M−SACs within 30 min; (b) Preparation of Co−NPs and Co−SAC by IF−67 coated PAN nanofibers; (c) Reaction rate constants normalized by Co weight in different systems, and the percentage (%) of BPA degradation by catalysts mentioned in this system and other studies (reaction time and catalyst dosage)[44]; (d) Apparent rate constant of BPA in the presence of various catalysts/PMS and the schematic diagram of the efficient conversion process of Cu−SA/MXene activated PMS to generate 1O2 [41]
图 3 (a)M−SACs在30min内对BPA的降解曲线及降解速率;(b)由IF−67包覆PAN纳米纤维制备Co−NPs和Co−SAC的方案;(c)不同体系中按Co重量归一化的反应速率常数,该体系和其他研究中提及的催化剂对BPA降解百分比(%)(反应时间和催化剂用量)[44];(d)双酚A在不同催化剂活化PMS作用下的速率常数,Cu−SA/MXene活化PMS高效转化生成1O2过程示意图[41]Figure 3. (a) Degradation curves and degradation rate of BPA catalyzed by M−SACs within 30 min; (b) Preparation of Co−NPs and Co−SAC by IF−67 coated PAN nanofibers; (c) Reaction rate constants normalized by Co weight in different systems, and the percentage (%) of BPA degradation by catalysts mentioned in this system and other studies (reaction time and catalyst dosage)[44]; (d) Apparent rate constant of BPA in the presence of various catalysts/PMS and the schematic diagram of the efficient conversion process of Cu−SA/MXene activated PMS to generate 1O2 [41]结合以上论述可知,合理的金属活性位点和载体类型组合有可能激发自由基和非自由基协同作用体系,有助于去除催化剂上的中间体,提高催化剂的稳定性和可重复使用性[60]. Li等[36]提出了Co−SAC激活PMS的双反应点催化机理,包括“供体−受体复合物(donor–acceptor complex)”机制和基于1O2的非自由基途径. Co−SAC由FeCo−PBA纳米球煅烧衍生制备(图4a),该催化剂活化PMS在6 min内对BPA的降解效率达100%,而不含钴的催化剂对BPA的去除率为22%,证明原子分散的Co对PMS的分解起着重要的催化作用. 通过理论计算验证了几种可能的活性位点与PMS之间的吸附能(Eads),结果表明Co−SAC中的吡咯N(CoN4)的吸附能最高. 因此,PMS更倾向与其结合并活化PMS分解生成1O2,与自由基淬灭和电子顺磁共振谱图(electron paramagnetic resonance,EPR)结果相一致. 同时,CoN4作为电子供体,而BPA中的−OH基团作为电子受体,两者之间遵循“供体−受体复合物”机制(图4b). 因此,基于CoN4位点上同步发生的PMS活化与污染物吸附过程,大大降低了ROS的迁移距离,原位生成的1O2与相邻的BPA在Co−SAC表面快速反应,进而大大提高了污染物降解速率. 马军院士课题组[61]近期研究报告中指出,在Co−SAC活化PMS体系中·OH和SO4·−主要来自催化剂表面CoNx位点与PMS之间的作用过程,与此同时,Co−SAC中N−C骨架将催化PMS诱导出现直接电子转移的非自由基氧化途径,而这种自由基氧化与非自由基氧化相结合的混合氧化途径使PMS氧化工艺在去除复杂水污染物方面具有广阔的应用前景. 相应地,实验结果证明Co−SAC活化PMS氧化体系在60 h的连续流反应中一直保持较高的污染物去除率. 因此,活性位点的催化机制及其周围ROS转化规律有所差异,明确的材料设计思路有助于开发自由基和非自由基协同作用体系.
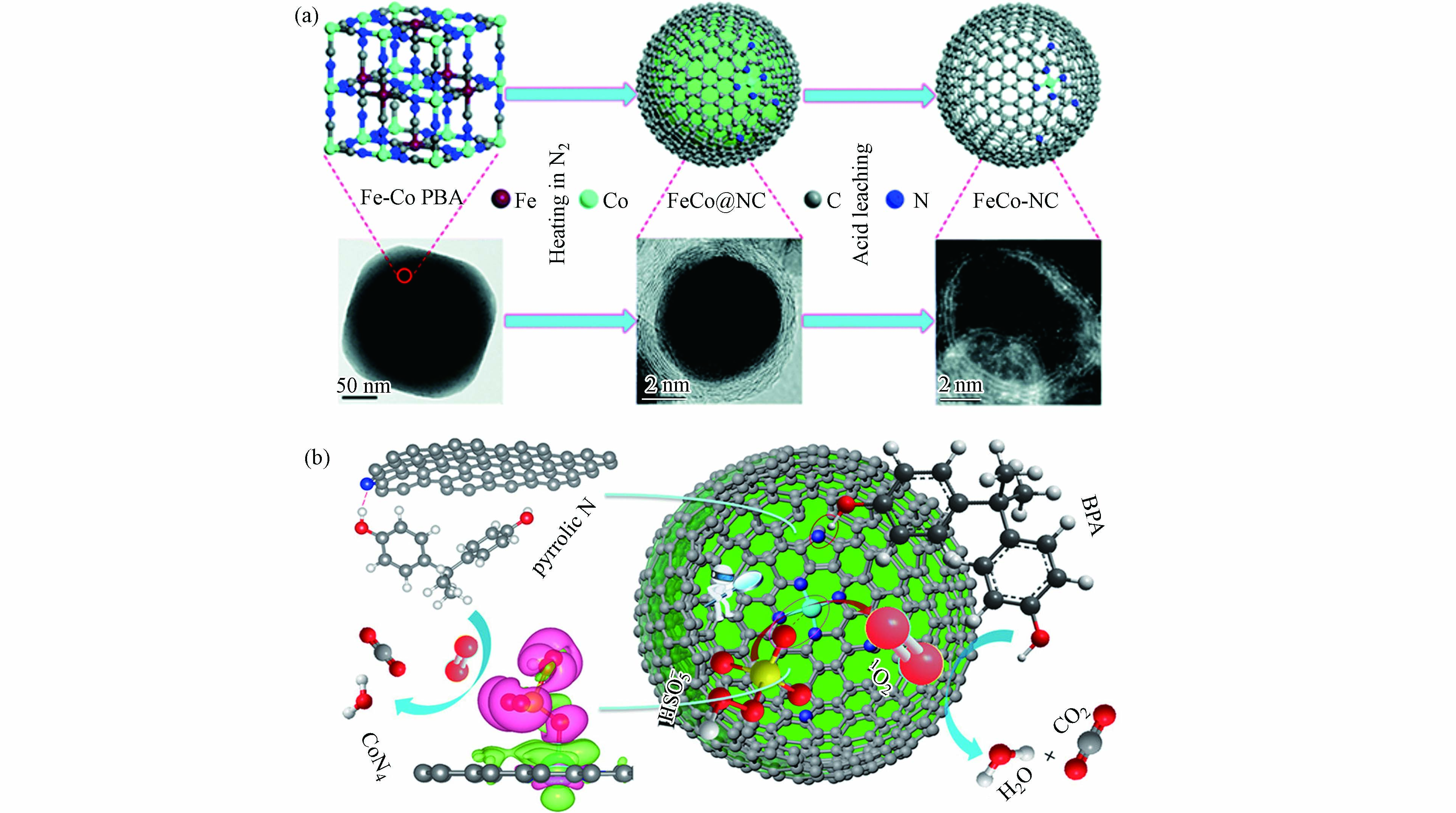 图 4 (a)Co−SAC的制备路线及相关的电镜图像,(b)Co−SAC活化PMS的双反应点机理示意图(紫色和绿色分别代表电子积累和电子消耗)[36]Figure 4. (a) Preparation of Co−SAC and related electron microscope images. (b) Schematic diagram of the mechanism of the double reaction sites of Co−SAC activation of PMS (purple and green represent electron accumulation and electron consumption, respectively) [36]
图 4 (a)Co−SAC的制备路线及相关的电镜图像,(b)Co−SAC活化PMS的双反应点机理示意图(紫色和绿色分别代表电子积累和电子消耗)[36]Figure 4. (a) Preparation of Co−SAC and related electron microscope images. (b) Schematic diagram of the mechanism of the double reaction sites of Co−SAC activation of PMS (purple and green represent electron accumulation and electron consumption, respectively) [36]除了直接改变位点的金属原子种类与载体类型外,通过调控SAC中原子排布也可以有效改善催化剂活性. 相关研究从原子尺度直观地揭示结构−活性和反应机理之间的关系,为定制具有高活性催化剂提供关键依据. Qi等[56]以木质素为碳源制备了N配位Co SAC用于活化PMS降解有机污染物,通过实验与DFT计算发现,Co单原子位点诱导的直接电子转移是体系中目标污染物降解的主要机制. 因为该催化剂在Co原子位点与配位N(C)处的电荷积累较多,此催化活性位点将作为高效电子转移渠道,使得污染物中的电子转移到过硫酸盐形成硫酸根离子进而得以分解. 对于N配位Fe SAC,相关研究指明PMS在Fe原子位点将首先被激活形成Fe−N−C−PMS*(*表示吸附位点),然后与邻近的Fe原子协调形成一个复合物,随后生成FeⅣ=O[62]. 该研究中确定了FeⅣ=O产生的浓度阈值,指明Fe1−Fe1距离为0.4 nm的铁原子是PMS活化反应生成FeⅣ=O的活性位点(图5a),通过PMS的双位点吸附形成络合物是关键步骤(图5b). 通过有机污染物降解实验证明,以FeⅣ=O为主要氧化活性物质的类Fenton体系具有优异的污染物去除能力、pH耐受性和对单电子氧化电位(one−electron oxidation potential)较高的污染物选择性降解. 除了金属原子位点间距以外,金属的平均氧化状态也会影响Fe SAC−PMS中自由基途径与高价物种途径的调控[63]. 处于高旋转状态的Fe2+−N4倾向于激活PMS形成SO4·−和·OH,而Fe3+−N4则容易生成Fe5+=O. 另外相关研究报告了通过将缺电子硼(B)或富电子磷(P)原子掺入碳基底的调控策略对Cu−N4电子结构的影响及其相应的PMS活化性能(图5c)[33]. 研究发现缺电子型Cu−N4/C−B催化剂表现出优异的PMS活化性能,而富电子型Cu−N4/C−P催化剂的性能明显低于Cu−N4/C催化剂,因为与B原子的长程相互作用降低了Cu活性位点的电子密度,从而优化了催化剂对于PMS的吸附能. 该研究提供了一种通过调节原子排布精细控制金属活性位点电子结构的方法. 类似地,通过将电子接收型氧化S基团(oxidized S groups)或电子供给型噻吩类S物种(thiophene−like S species)引入SACs的碳载体中,也可有效调整单个原子位点在氧还原反应(oxygen reduction reaction,ORR)过程中的动力学活性[64]. 然而,有关于S掺杂的SAC在过硫酸盐AOPs中的研究尚无报道. 除了碳基SAC,相关研究报道了在Mn3O4表面构建具有配位结构不对称的Co原子位点,探究金属原子配位环境对于催化剂性能的影响[42]. 该研究通过DFT计算揭示了N1O2配位结构中的N导致Co位点上发生了明显的电子离域现象,从而促进了催化剂表面对PMS的吸附、裂解和CoⅣ=O的形成.
 图 5 (a)2Fe−N4的计算结构模型(粉色、蓝色和灰色的球分别代表Fe、N和C原子),(b)PMS在2Fe−N4位点上的吸附结构和相应的吸附能[62]; (c) Cu−N4/C−B和Cu−N4/C−P的制备策略示意图,色条表示Cu−N4位点的电子密度,富电子(蓝色)和缺电子(红色)[33]Figure 5. (a) The computational structure model of 2Fe−N4 (the pink, blue and gray spheres represent Fe, N and C atoms, respectively), (b) the adsorption structure and corresponding adsorption energy of PMS at the 2Fe−N4 site[62]; Schematic of the preparation strategy for Cu−N4/C−B and Cu−N4/C−P. The color bar indicates the electronic density of Cu−N4 site, electro−rich (blue) and electro−poor (red)[33]
图 5 (a)2Fe−N4的计算结构模型(粉色、蓝色和灰色的球分别代表Fe、N和C原子),(b)PMS在2Fe−N4位点上的吸附结构和相应的吸附能[62]; (c) Cu−N4/C−B和Cu−N4/C−P的制备策略示意图,色条表示Cu−N4位点的电子密度,富电子(蓝色)和缺电子(红色)[33]Figure 5. (a) The computational structure model of 2Fe−N4 (the pink, blue and gray spheres represent Fe, N and C atoms, respectively), (b) the adsorption structure and corresponding adsorption energy of PMS at the 2Fe−N4 site[62]; Schematic of the preparation strategy for Cu−N4/C−B and Cu−N4/C−P. The color bar indicates the electronic density of Cu−N4 site, electro−rich (blue) and electro−poor (red)[33]由此可见,不同的金属活性位点与载体类型、配位环境原子排布方式对于氧化活性物质和氧化途径的驱动机制有所差别. 大部分文献中报道了不同的单原子金属位点被锚定在各种碳基质(例如,多孔碳、石墨烯和C3N4)上,从而形成用于PMS活化的碳基SAC,这些碳基SAC的制备方法主要有有机小分子、聚合物、MOFs和生物质的热解. 此外,在难降解有机污染物的降解过程中,通过单原子位点和碳载体中的非金属活性物质,触发了自由基和非自由基途径. 综上所述,对于PMS激活过程中无金属催化位点与SAC位点的协同作用及相互关系还有待进一步研究[10],金属载体SAC与非金属原子掺杂载体SAC的催化活性及其反应特性研究较少、缺乏相应案例说明.
2.2 单原子催化剂活化过二硫酸盐
Zuo等[65]制备了锚定在双层C3N4−rGO载体中单原子Fe位点的催化剂(C3N4−Fe−rGO),表征结果证明Fe原子位于rGO和C3N4层之间,该碳基底具有优异的电子转移能力,有助于PDS被活化生成SO4·−(图6a). 特别地,双层结构显著抑制了铁原子的团聚和浸出,C3N4−Fe−rGO在0—14 pH范围内均表现出稳定的催化能力,这是目前非均相类Fenton体系所能达到的最宽pH范围(图6b). 然而,Jiang等[66]发现Fe单原子催化剂(Fe−N−C)活化PDS体系中没有自由基产生,而是生成了Fe(V),进而通过直接的双电子提取过程实现有机污染物的选择性降解. 此外,经热处理和酸浸处理后,酞菁铁和金属有机骨架合成了氮掺杂碳上的单原子铁(SAFe−N−C)(图6c)[67]. 原子分散的Fe−Nx位点的引入不仅提高了催化活性,而且调节了PDS活化的反应途径. 原子分散的Fe−Nx位点,产生更多的1O2,使反应途径以1O2为主(图6d). 因此,SAFe−N−C在较宽的pH范围内表现出较高的催化活性,并对无机阴离子具有良好的抗性. 基于以上分析可知,同种金属SAC活化PDS体系降解污染物反应过程中发挥主要作用的ROS不完全一致,说明活性位点ROS产生机制与其周围ROS转化规律有所差异,对此过程中发生的具体反应过程尚不明晰,影响污染物降解效果的关键因素有待阐明.
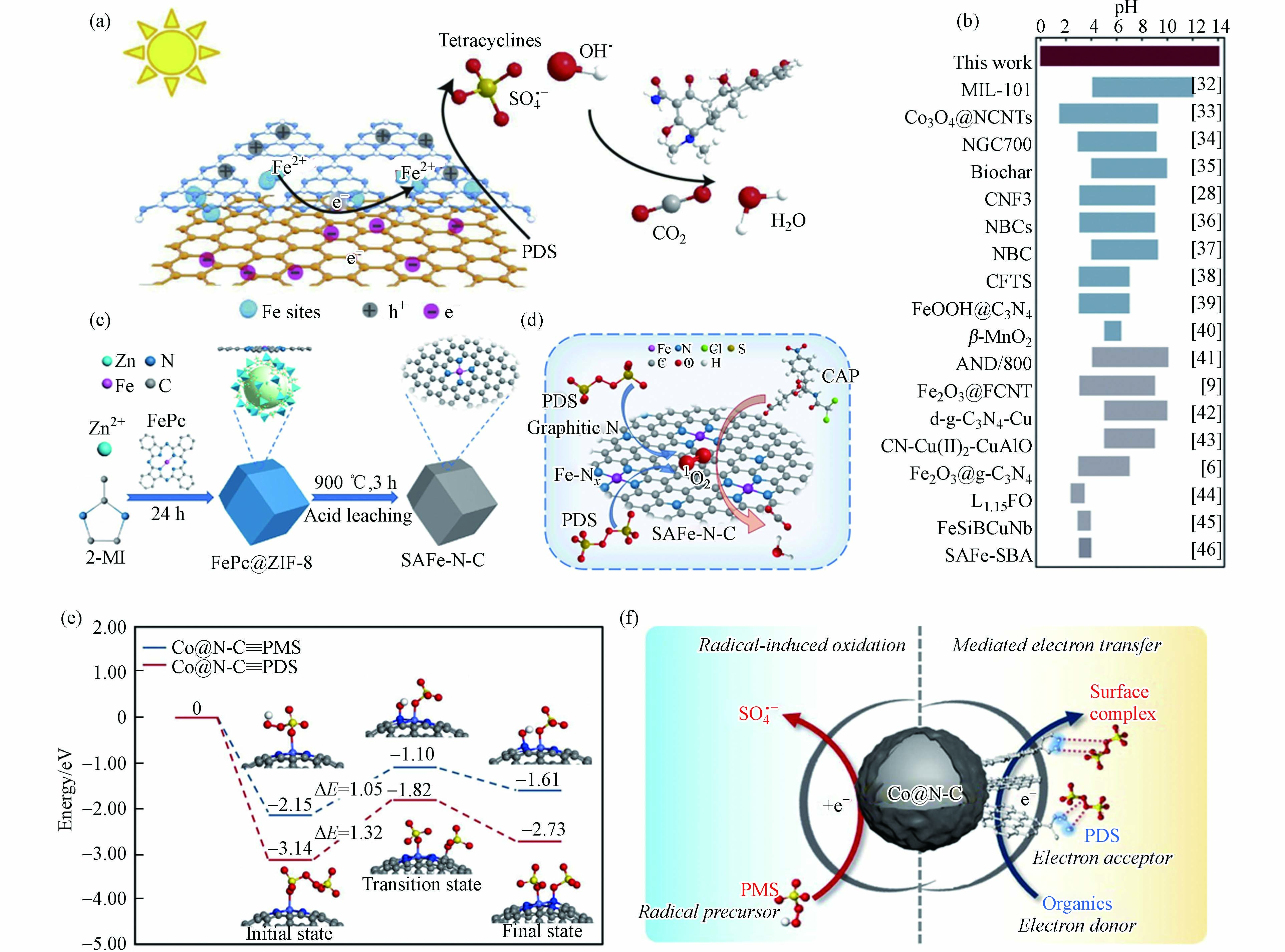 图 6 (a)C3N4−Fe−rGO活化PDS的反应机理示意图,(b)类Fenton反应催化剂的典型工作pH范围[65];(c)SAFe−N−C制备示意图,(d)PDS和SAFe−75−N−C体系的反应机理[67];(e)Co@N−C上PMS和PDS解离过程计算的势能分布和(f)催化剂表面反应机理示意图(浅蓝色、蓝色、灰色、红色、黄色和白色球体分别代表Co、N、C、O、S和H原子)[68]Figure 6. (a) Schematic diagram of reaction mechanism of C3N4−Fe−rGO activation of PDS, (b) Typical working pH range of Fenton−like catalysts[65]; (c) Schematic diagram of preparation of SAFe−N−C, (d) Reaction mechanism of PDS and SAFe−75−N−C system[67]; (e) Potential energy distribution calculated during the dissociation of PMS and PDS at Co@N−C and (f) Schematic diagram of catalyst surface reaction mechanism (light blue, blue, grey, red, yellow and white spheres represent Co, N, C, O, S and H atoms, respectively)[68]
图 6 (a)C3N4−Fe−rGO活化PDS的反应机理示意图,(b)类Fenton反应催化剂的典型工作pH范围[65];(c)SAFe−N−C制备示意图,(d)PDS和SAFe−75−N−C体系的反应机理[67];(e)Co@N−C上PMS和PDS解离过程计算的势能分布和(f)催化剂表面反应机理示意图(浅蓝色、蓝色、灰色、红色、黄色和白色球体分别代表Co、N、C、O、S和H原子)[68]Figure 6. (a) Schematic diagram of reaction mechanism of C3N4−Fe−rGO activation of PDS, (b) Typical working pH range of Fenton−like catalysts[65]; (c) Schematic diagram of preparation of SAFe−N−C, (d) Reaction mechanism of PDS and SAFe−75−N−C system[67]; (e) Potential energy distribution calculated during the dissociation of PMS and PDS at Co@N−C and (f) Schematic diagram of catalyst surface reaction mechanism (light blue, blue, grey, red, yellow and white spheres represent Co, N, C, O, S and H atoms, respectively)[68]3. 结论与展望(Conclusion and prospect)
总而言之,SAC活化过硫酸盐的研究近年来取得了迅速的发展. 研究者致力于探索催化剂的活性位点,揭示其在催化氧化过程中的作用机制. 同时,一系列的原位分析方法被开发,用于深入研究SAC活化过硫酸盐体系中的ROS演化规律和污染物降解机制. 然而,对于SAC活化过硫酸盐体系的环境修复机制和实际应用,仍存在巨大的挑战. 未来的研究应集中在:
(1) 通过理论模拟和原位表征技术加强基础研究,填补过硫酸盐与SAC相互作用机制的研究空白——进一步确定反应过程中SAC表面活化的PDS/PMS的存在状况,揭示不同金属原子与其周围配位结构对氧化活性物质生成和转化的影响、明确单一反应物种对有机污染物整体降解的贡献及其协同效应. 与此同时,确定体系中氧化活性物质去除难降解有机污染物的效果及其造成有毒产物二次污染的风险.
(2) 以环境应用为基础进行材料设计与其反应器开发,积极应对与工程实践和复杂水质相关的挑战——之前综述了有关与PS−AOPs中纳米催化剂的固定化方法和反应器设计开发的最新研究进展,指明通过合理的高效催化剂设计,并结合膜技术、固定床反应器或其他集成设备或连续反应器中,是实现非均相PS−AOPs走向实际应用的必经之路. 此外,为了确认PS−AOPs的经济效益和环境后果,需要对反应系统的处理效果进行合理评估,涉及运行成本、可持续性和环境适用性.
-
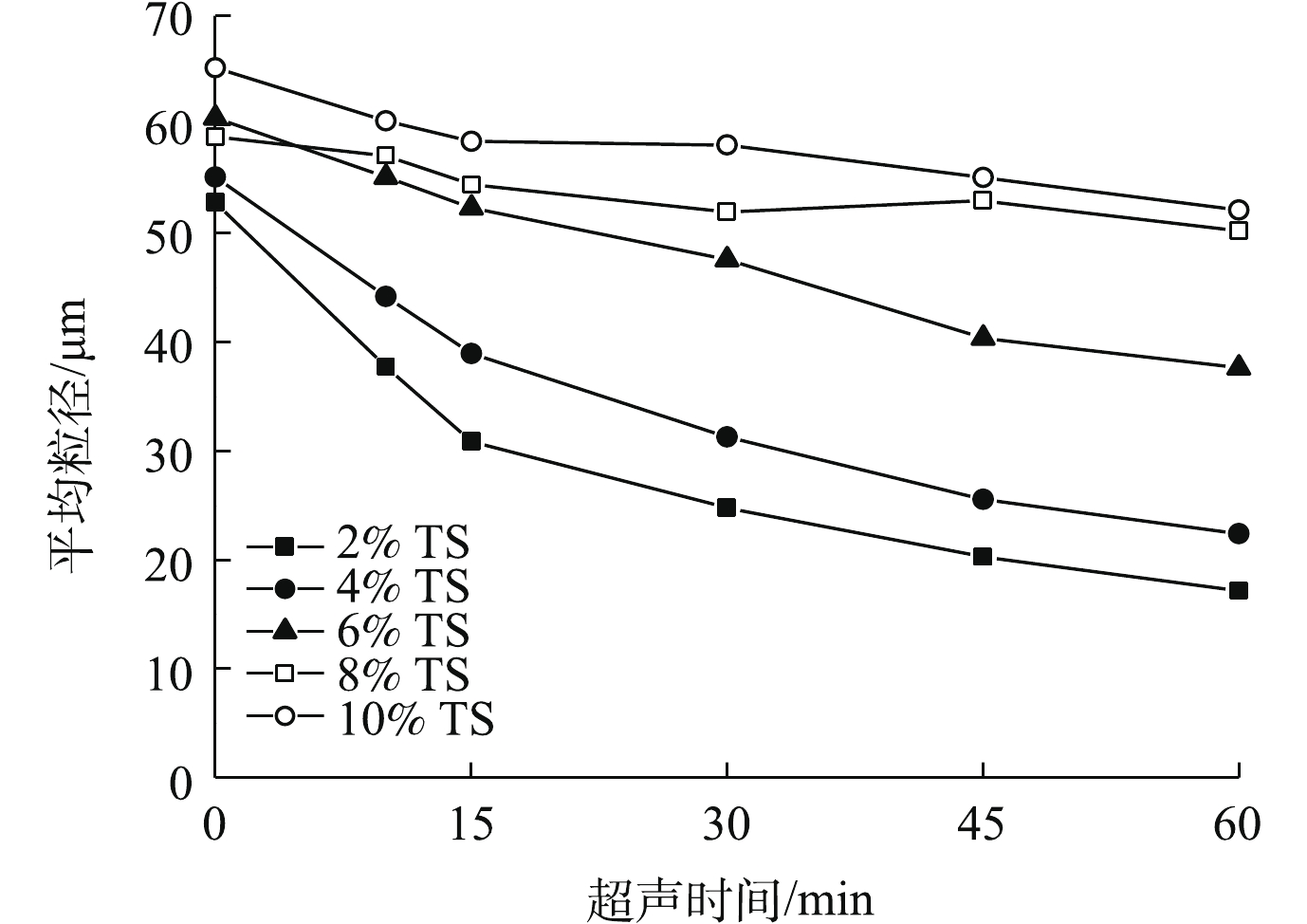
图 2 超声预处理后污泥平均粒径的变化
Figure 2. Mean particle size of sludge after ultrasonic pretreatment
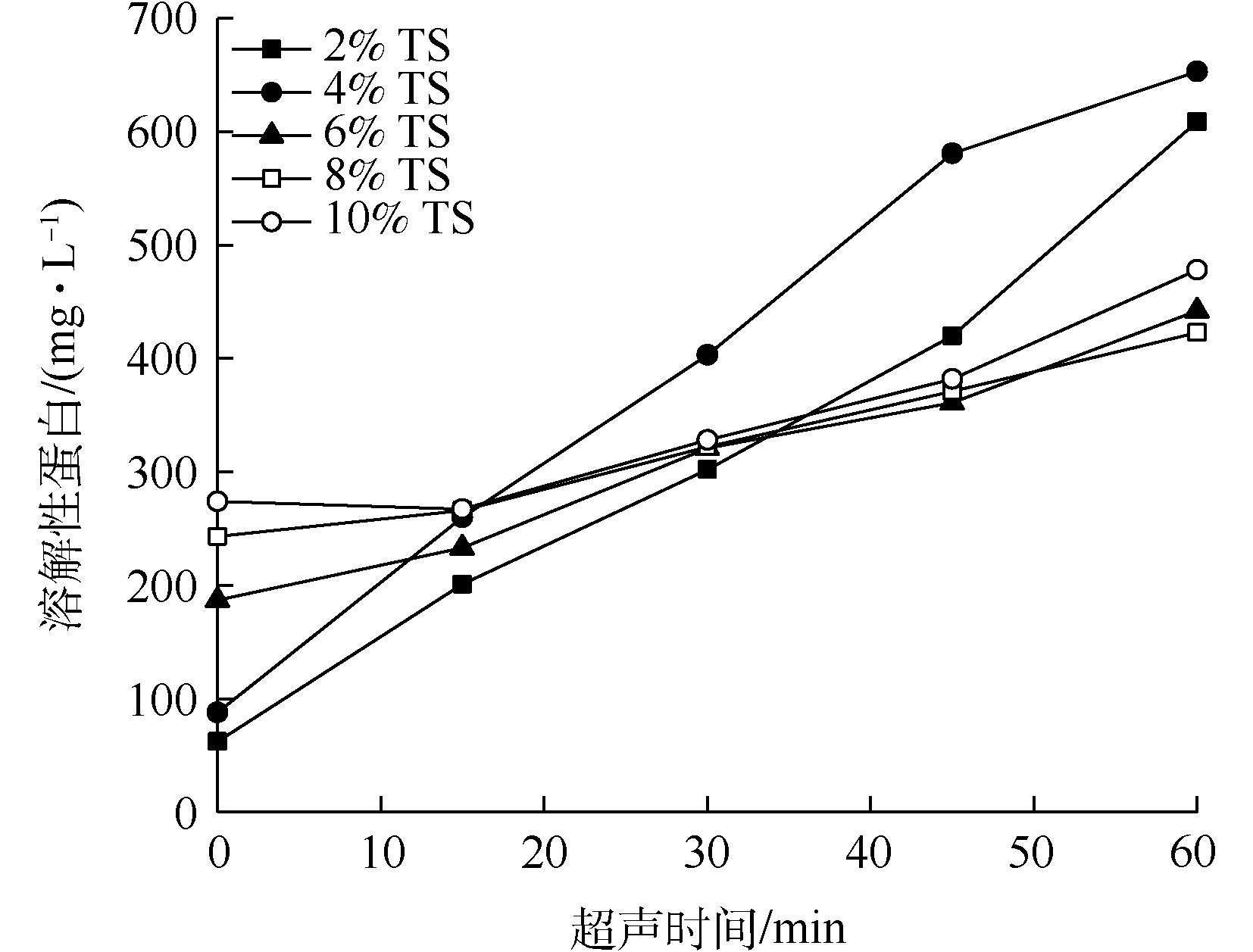
图 4 超声预处理后污泥中溶解性蛋白质浓度的变化
Figure 4. Changes in soluble protein concentration of sludge after ultrasonic pretreatment
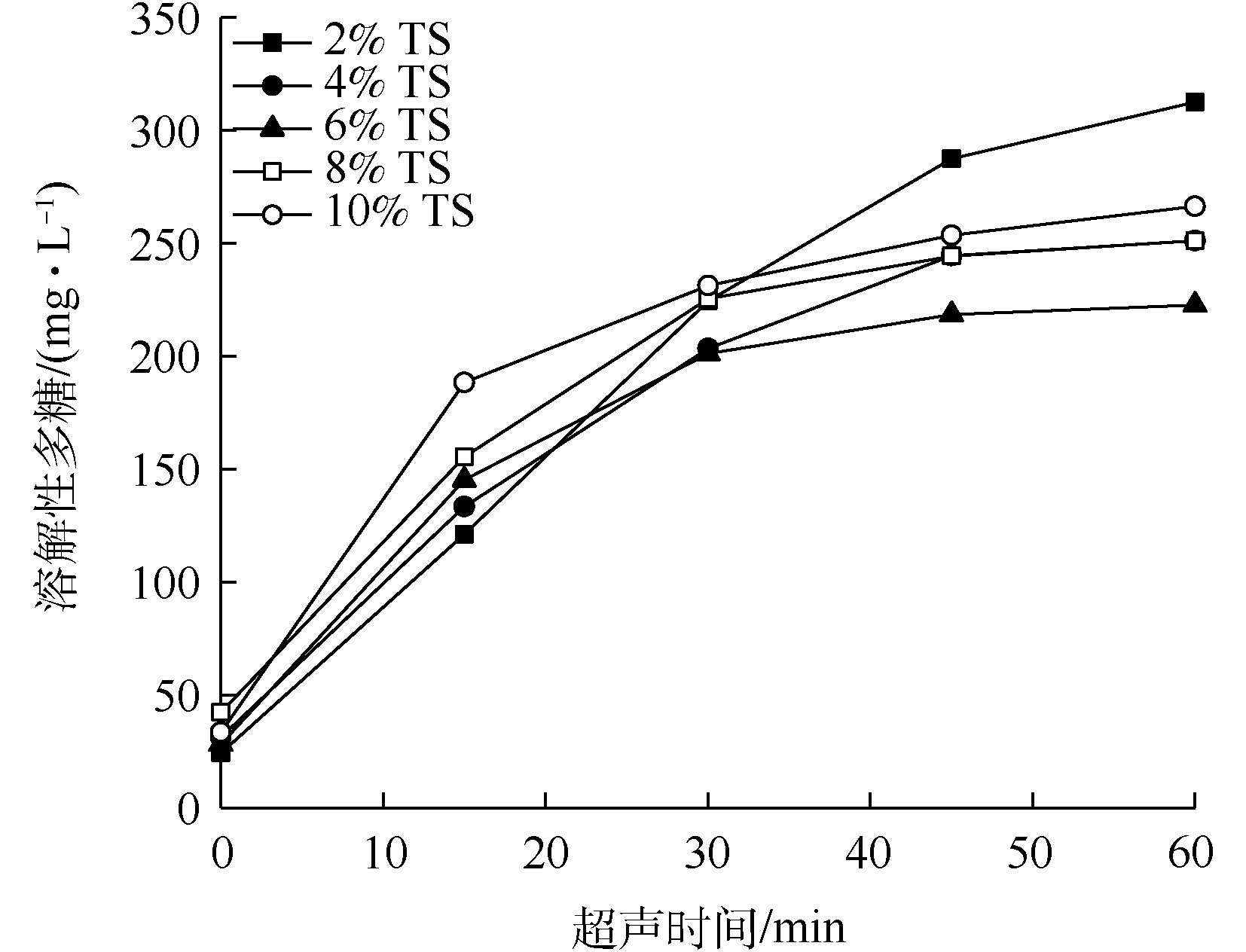
图 5 超声预处理后污泥中溶解性多糖浓度的变化
Figure 5. Changes in soluble carbohydrate concentration of sludge after ultrasonic pretreatment
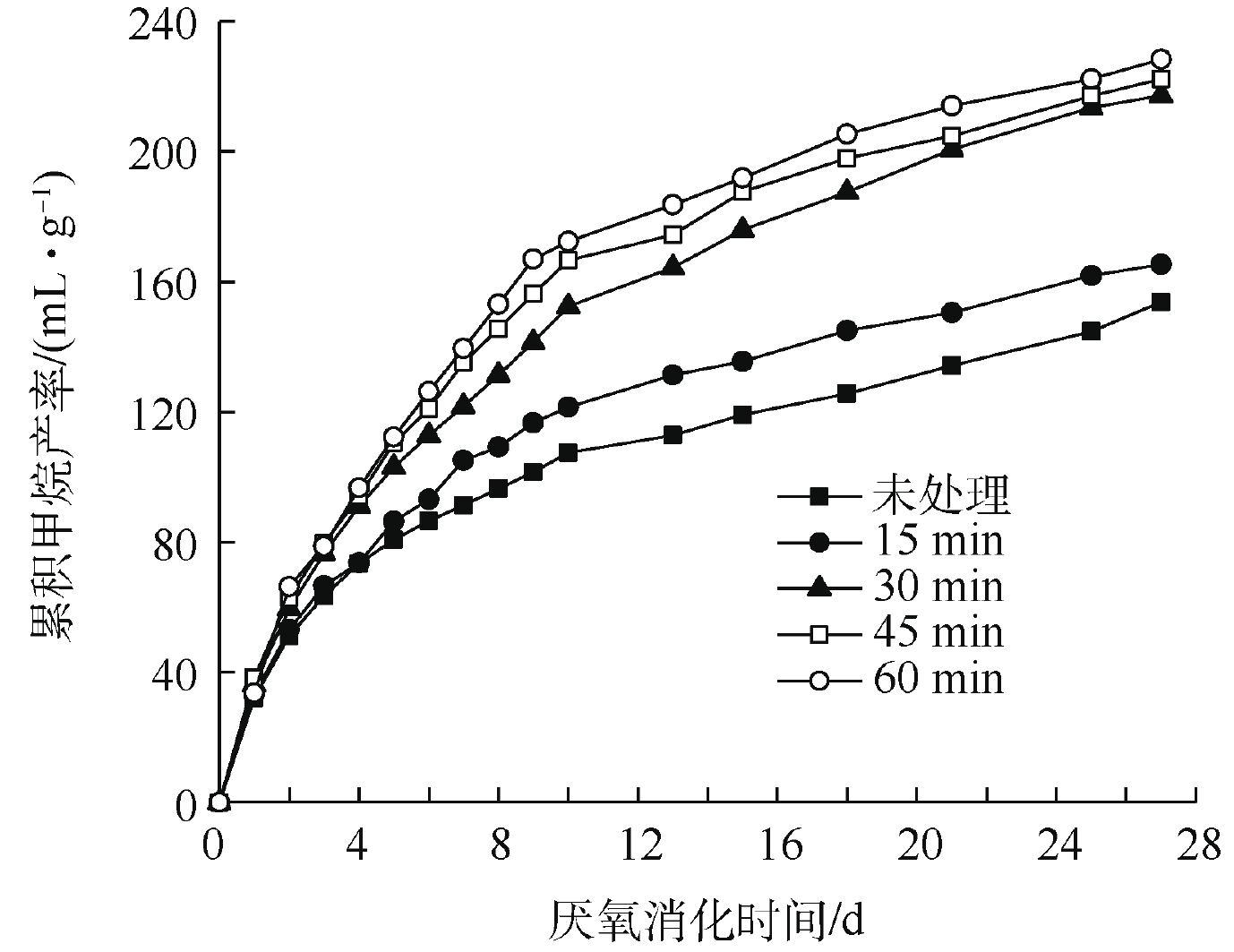
图 6 超声预处理2% TS污泥后累积甲烷产率的变化
Figure 6. Changes in cumulative methane yield of 2% TS sludge after ultrasonic pretreatment
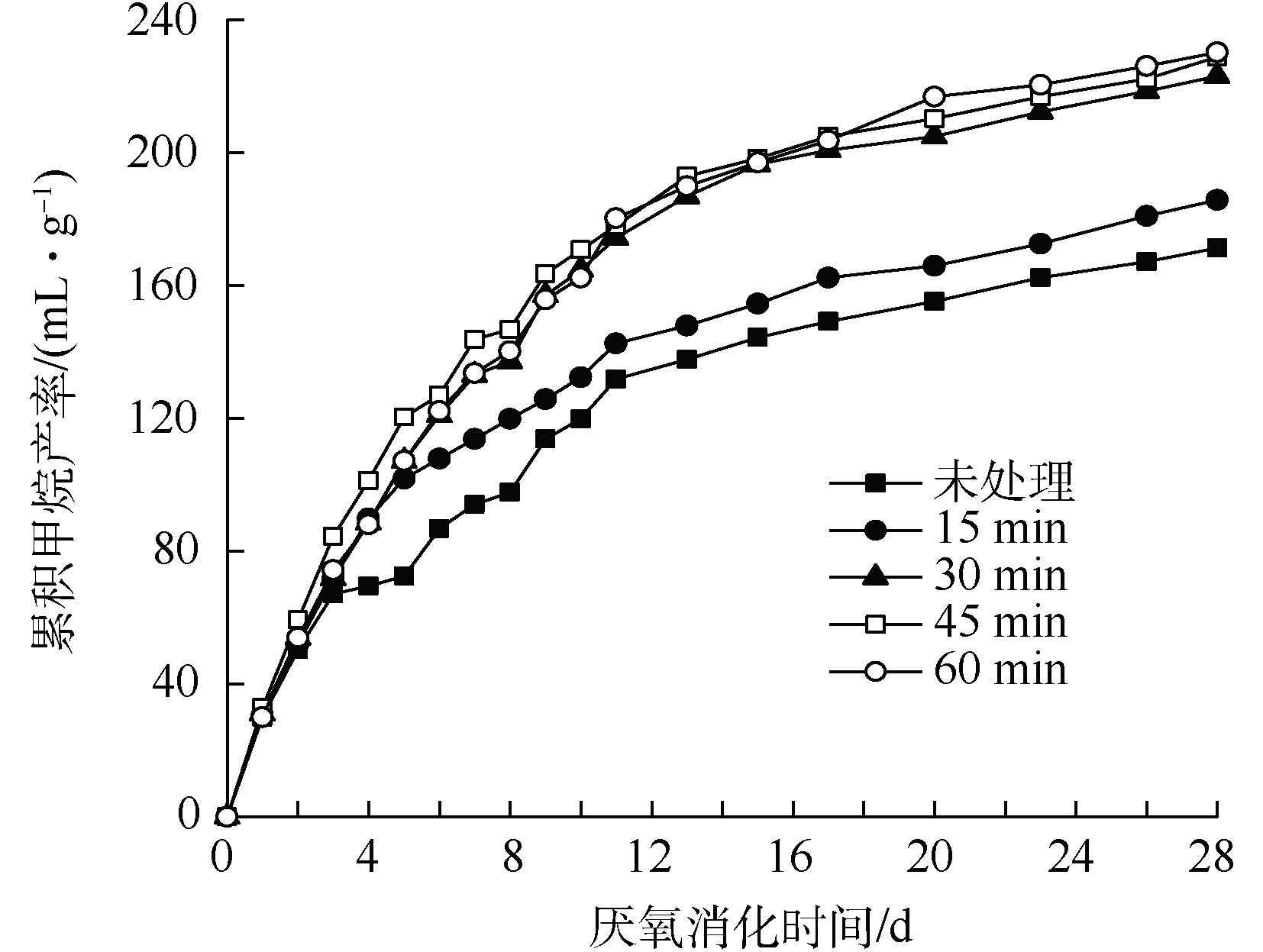
图 7 超声预处理后4% TS污泥累积甲烷产率的变化
Figure 7. Changes in cumulative methane yield of 4% TS sludge after ultrasonic pretreatment
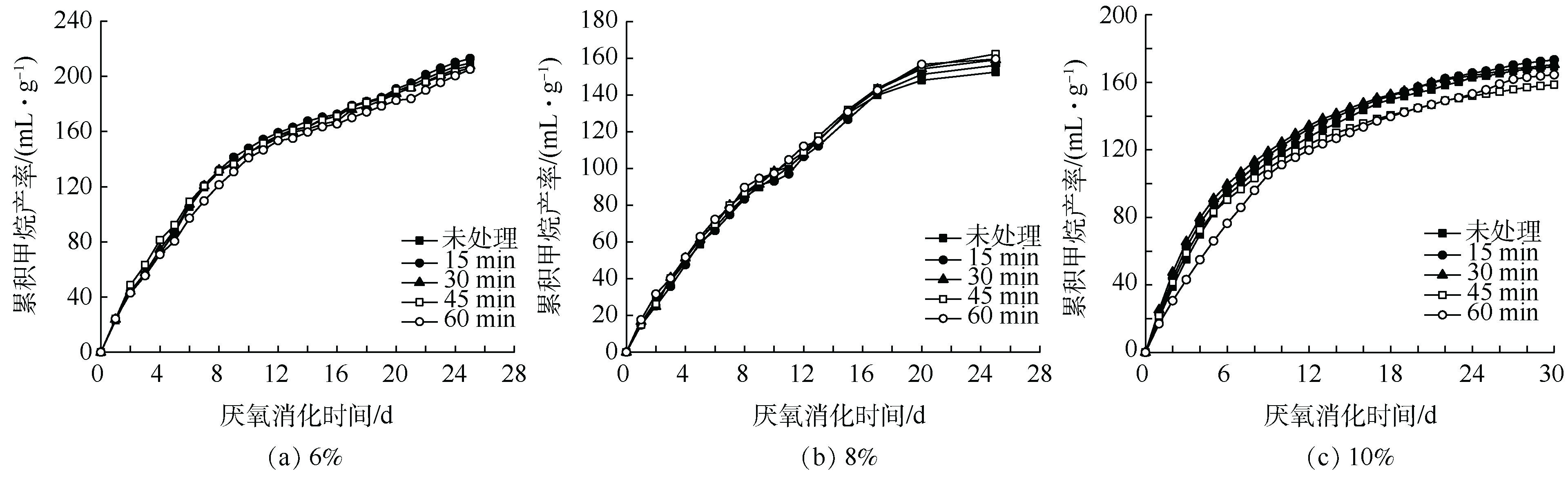
图 8 超声预处理后6%、8%和10%TS污泥累积甲烷产率的变化
Figure 8. Change in cumulative methane yields of 6%,8% and 10% TS sludge after ultrasonic pretreatment
表 1 超声预处理后污泥VS去除率的变化
Table 1. Changes in VS removal of sludge after ultrasonic pretreatment
TS浓度/% VS去除率(对照组)/% VS去除率(超声处理)/% 超声15 min 超声30 min 超声45 min 超声60 min 2 34.83 36.67 41.17 40.68 42.61 4 33.52 35.02 42.44 41.70 42.94 6 32.11 33.74 32.08 34.21 31.04 8 28.54 30.01 29.62 29.55 28.42 10 28.27 27.32 29.09 30.62 28.33
下载: 导出CSV
-
[1] ZHANG L, DUAN H, YE L, et al. Increasing capacity of an anaerobic sludge digester through FNA pre-treatment of thickened waste activated sludge[J]. Water Research, 2019, 149: 406-413. doi: 10.1016/j.watres.2018.11.008 [2] 晏发春, 汪恂, 张雷, 等. 高温热水解预处理厌氧消化技术实例分析[J]. 中国给水排水, 2016, 32(18): 35-37. [3] ŞAHINKAYA S, SEVIMLI M F. Synergistic effects of sono-alkaline pretreatment on anaerobic biodegradability of waste activated sludge[J]. Journal of Industrial and Engineering Chemistry, 2013, 19(1): 197-206. [4] TIAN X, WANG C, TRZCINSKI A P, et al. Insights on the solubilization products after combined alkaline and ultrasonic pre-treatment of sewage sludge[J]. Journal of Environmental Sciences, 2015, 29: 97-105. doi: 10.1016/j.jes.2014.07.024 [5] 王平. 热水解厌氧消化工艺的分析和应用探讨[J]. 给水排水, 2015, 3(1): 33-38. doi: 10.3969/j.issn.1002-8471.2015.01.008 [6] PILLI S, YAN S, TYAGI R D, et al. Anaerobic digestion of ultrasonicated sludge at different solids concentrations-computation of mass-energy balance and greenhouse gas emissions[J]. Journal of Environmental Management, 2016, 166: 374-386. [7] ZHANG B, JI M, WANG F, et al. Damage of EPS and cell structures and improvement of high-solid anaerobic digestion of sewage sludge by combined (Ca(OH)2 + multiple-transducer ultrasonic) pretreatment[J]. RSC Advances, 2017, 7(37): 22706-22714. doi: 10.1039/C7RA01060E [8] GONZALEZ A, HENDRIKS A, VAN L, et al. Pre-treatments to enhance the biodegradability of waste activated sludge: Elucidating the rate limiting step[J]. Biotechnology Advances, 2018, 36(5): 1434-1469. doi: 10.1016/j.biotechadv.2018.06.001 [9] PILLI S, YAN S, LEBLANC R, et al. Ultrasonic pretreatment of sludge: A review[J]. Ultrasonics Sonochemistry, 2011, 18(1): 1-18. doi: 10.1016/j.ultsonch.2010.02.014 [10] NICKEL K, NEIS U. Ultrasonic disintegration of biosolids for improved biodegradation[J]. Ultrasonics Sonochemistry, 2007, 14(4): 450-455. doi: 10.1016/j.ultsonch.2006.10.012 [11] GOGATE P, SIVAKUMAR M, PANDIT A. Destruction of rhodamine B using novel sonochemical reactor with capacity of 7.5 L[J]. Separation and Purification Technology, 2004, 34(1/2/3): 13-24. [12] GOGATE P, MUJUMDAR S, PANDIT A. Sonochemical reactors for waste water treatment: Comparison using formic acid degradation as a model reaction[J]. Advances in Environmental Research, 2003, 7(2): 283-299. doi: 10.1016/S1093-0191(01)00133-2 [13] GOGATE P, SUTKAR V, PANDIT A. Sonochemical reactors: Important design and scale up considerations with a special emphasis on heterogeneous systems[J]. Chemical Engineering Journal, 2011, 166(3): 1066-1082. doi: 10.1016/j.cej.2010.11.069 [14] ASGHARZADEHAHMADI S, ABDULRAMAN A, PARTHASARATHY R, et al. Sonochemical reactors: Review on features, advantages and limitations[J]. Renewable and Sustainable Energy Reviews, 2016, 63: 302-314. doi: 10.1016/j.rser.2016.05.030 [15] American Public Health Association. Standard Methods for the Examination of Water and Wastewater[M]. Washington D C: American Public Health Association, 1995. [16] GAUDY A F. Colorimetric determination of protein and carbohydrate[J]. Industrial Water Wastes, 1962, 7: 17-22. [17] JIMENEZ J, VEDRENNE F, DENIS C, et al. A statistical comparison of protein and carbohydrate characterisation methodology applied on sewage sludge samples[J]. Water Research, 2013, 47(5): 1751-1762. doi: 10.1016/j.watres.2012.11.052 [18] LOWRY O H, ROSEBROUGH N J, FARR A L, et al. Protein measurement with the Folin phenol reagent[J]. Journal of Biological Chemistry, 1951, 193(1): 265-275. [19] TYAGI V, LO S, APPELS L, et al. Ultrasonic treatment of waste sludge: A review on mechanisms and applications[J]. Critical Reviews in Environmental Science and Technology, 2014, 44(11): 1220-1288. doi: 10.1080/10643389.2013.763587 [20] JIANG S, CHEN Y, ZHOU Q. Effect of sodium dodecyl sulfate on waste activated sludge hydrolysis and acidification[J]. Chemical Engineering Journal, 2007, 132(1/2/3): 311-317. [21] KIM D, CHO S, LEE M, et al. Increased solubilization of excess sludge does not always result in enhanced anaerobic digestion efficiency[J]. Bioresource Technology, 2013, 143: 660-664. doi: 10.1016/j.biortech.2013.06.058 [22] FERNÁNDEZ J, PÉREZ M, ROMERO L I. Kinetics of mesophilic anaerobic digestion of the organic fraction of municipal solid waste: Influence of initial total solid concentration[J]. Bioresource Technology, 2010, 101(16): 6322-6328. doi: 10.1016/j.biortech.2010.03.046 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3085
- HTML全文浏览数: 3085
- PDF下载数: 55
- 施引文献: 0
