


-
聚酯(PET)是目前最常用的聚合物之一,它被广泛用于制造纤维、织物及其他产品,如饮料瓶、照相胶片等[1-3]。然而,随着聚酯使用量的增加,其不可生物降解的特性使得其回收利用成为了环境保护的一大关注点[4]。在PET的回收方法中,乙二醇醇解所得产物能用于生产各类产品,如疏水性纺织染料[2]、聚氨酯泡沫[5]、醇酸树脂[6-7]等,已被科学研究及工业生产广泛采用。有研究[8-12]发现,乙二醇醇解PET的主要产物是对苯二甲酸乙二醇酯(BHET)。尽管BHET用途广泛,但聚酯面料中含有的染料会残留在BHET晶体中,使其应用受到了限制,如将其用于生产再生聚酯纤维时,BHET中所含染料会直接影响再生聚酯的颜色。目前,国内外鲜有对聚酯降解产物脱色的报道,工业上主要使用纯度高及颜色浅的聚酯瓶片来生产再生聚酯产品,因此,找到一种经济的方法,来实现对聚酯面料降解产物的有效脱色是聚酯循环再利用中亟待解决问题。
目前,常用的染料废水脱色技术主要包括臭氧氧化法、超滤法、氧化法、光化学降解法和吸附法[13-16]。在这些方法中,吸附是最有效的方法之一,并已成功用于去除废水中的染料[17]。作为多孔材料,活性炭(AC)通常具有良好的内部孔结构、高比表面积以及位于外表面和内表面的一定量的化学官能团[18-19]。因此,它在分离多种化合物方面是非常有效的,并且已被广泛用作废水处理中的吸附剂[20-22]。但是,它在生产过程中形成的非极性特征限制了其应用[23]。已经有研究[24]采取各种处理方法来使AC成为极性更强的材料,包括酸碱改性,离子改性和微波改性等。其中,离子改性已被用于在AC上引入金属化合物和金属颗粒等,以提高其对水中氟化物,氰化物,和重金属砷等的吸附容量。
为实现对BHET脱色的同时不破坏其分子结构,本研究选用活性炭对其进行吸附脱色,并采用FeSO4·7H2O对活性炭进行了离子改性,以提高其对BHET中所含染料的吸附性能;在探究离子浓度、超声时间、煅烧温度对活性炭物理及化学性能的影响的基础上,通过中心组合法设计2因素3水平的响应面实验,以优化活性炭的改性工艺,从而提高其对染料的脱色效率;最后,通过吸附热力学与动力学实验,分析了染料在AC上吸附的机理及吸附容量变化,以实现活性炭在BHET脱色中的最大化利用。
-
无色聚酯纤维、分散红60(DR60)、活性炭(AC)、七水合硫酸亚铁(FeSO4·7H2O)、95%乙醇、乙二醇(EG)、醋酸锌(ZnAC2)均为分析纯。
-
紫外可见光分光光度计(UV-1800型,上海美谱达仪器有限公司);热失重分析仪(TGA Q-500型,美国TA Instruments公司);物理吸附仪(TriStar 3020型,美国Micromeritics公司);管式煅烧炉(GSL 1600X型,合肥科晶材料技术有限公司);高功率数控超声波清洗器(KH-400KDE型,昆山禾创超声仪器有限公司);循环水式多用真空泵(SHB型,郑州长城科工贸有限公司);电子天平(AR1530/C型,豪斯国际贸易有限公司)。
-
以过量乙二醇作为解聚剂,醋酸锌作为催化剂,将聚酯高分子降解成单体分子对苯二甲酸乙二醇酯(BHET),其反应如式(1)所示,醇解温度为196 ℃,反应时间为3 h,采用文献中的方法[9-10]进行降解。实验中使用分散红60(DR60)和无色聚酯纱线来模拟有色废弃聚酯,以保证BHET中只含单一染料。
 (1)
(1)
为计算活性炭样品吸附DR60的量,可使用DR60的标准曲线来计算BHET溶液中DR60浓度。配制浓度分别为4、8、10、20 mg·L−1的DR60溶液,其溶剂为95%的乙醇和水的混合溶剂(醇∶水的体积比为1∶2),使用UV-1800分光光度计扫描溶液在400~650 nm区域内的紫外吸收谱。如图1(a)所示,DR60溶液分别在553 nm和515 nm处有2个特征吸收峰。对不同浓度的DR60在553 nm处的吸光度进行线性拟合,即得到DR60在553 nm处的吸光度标准曲线,如图1(b)所示。
采用FeSO4·7H2O对活性炭进行改性。称取10 g活性炭,加入到100 mL的FeSO4·7H2O溶液中,将反应溶液置于100 Hz超声波清洗器中,分散一定时间,并于常温条件下浸渍24 h。反应结束后,将反应混合液过滤分离,并用去离子水将活性炭反复冲洗,至中性后,放入50 ℃鼓风干燥烘箱,烘干至恒重,装入密封袋备用。为探究改性条件对活性炭性质的影响,共进行3组单因素实验,具体实验条件如表1所示。
实验组1为不同浓度FeSO4·7H2O对活性炭的改性,该组改性活性炭分别定义为AC-Fe2+-0.2 mol·L−1、AC-Fe2+-0.4 mol·L−1、AC-Fe2+-0.8 mol·L−1、AC-Fe2+-1.6 mol·L−1;实验组2为不同超声时间下对活性炭的改性,该组改性活性炭分别定义为AC-Fe2+-1 h、AC-Fe2+-2 h、AC-Fe2+-4 h、AC-Fe2+-8 h;实验组3为不同煅烧温度下对活性炭的改性,将烘干得到的样品在管式炉中煅烧,该组改性活性炭分别定义为AC-Fe2+-350 ℃、AC-Fe2+-550 ℃、AC-Fe2+-750 ℃、AC-Fe2+-950 ℃。
为探究离子浓度、超声时间及煅烧温度这3个因素如何影响改性AC对BHET中DR60的吸附性能,设置了3组对比吸附实验。每组实验均设置1个对照组和4个实验组,对照组未改性AC与实验组改性AC均采用100 mL初始浓度为10 g·L−1的BHET溶液作为脱色样本,每个样本中活性炭的用量均为1 g,整个吸附过程保持在90 ℃恒温水浴中进行,取样间隔为10 min,脱色时间为120 min,120 min内的平均脱色率为12个取样点的脱色率平均值。为保证正确计算出溶液中DR60的浓度,取样时溶剂与DR60标准曲线绘制时保持一致,即取2 mL样品溶于1 mL 95%乙醇溶液中,以防止BHET溶液在常温下部分结晶析出。脱色过程中样品在t时刻的染料浓度根据DR60的标准曲线计算得到,染料脱除率由式(2)计算得到。
式中:Y为脱色率;C0为溶液中染料初始浓度,mg·L−1;Ct为t时刻溶液中染料浓度,mg·L−1。
采用中心组合法设计了包括离子浓度及超声时间2个因素在内的2因素3水平响应面分析实验(13组)。具体实验设计及实验结果如表2所示。AC样品120 min内平均脱色率实验方法和计算方法与吸附实验相同。
把响应面实验优化得到的最佳改性AC定义为AC-Fe2+-max。配制系列浓度为10、20、30、40和50 g·L−1的DR60 BHET溶液,量取100 mL置于250 mL锥形瓶中,分别加入0.02 g AC和样品,密封瓶口,置于90 ℃下吸附12 h,取2 mL样品溶于1 mL 95%的乙醇溶液中,测定吸光度,代入DR60标准曲线拟合方程中计算溶液中DR60的浓度Ce,利用式(3)计算吸附量qe。
式中:qe为平衡吸附容量,mg·g−1;C0为溶液中染料初始浓度,mg·L−1;Ce为吸附平衡时溶液中染料浓度,mg·L−1。根据qe计算结果,可作出AC对DR60的吸附等温线。常用的等温吸附模型为Langmuir和Freundlich等温吸附方程,如式(4)和式(5)所示。
式中:C0为溶液中染料初始浓度,mg·L−1;Ce为吸附质的平衡浓度,mg·L−1;Q为单层吸附容量,mg·g−1;b为朗格缪尔常数;qe为平衡吸附量,mg·g−1;kf为吸附平衡常数;n是吸附强度的指标[25]。
选择AC、AC-Fe2+-max以探究未改性及改性活性炭的动力学过程。配制初始浓度为50 g·L−1的DR60 BHET溶液,量取100 mL置于250 mL锥形瓶中,分别加入0.25 g AC及AC-Fe2+-max,密封瓶口,置于90 ℃下吸附12 h,每隔0.5 h取2 mL样品,溶于1 mL 95%的乙醇溶液中,测定吸光度,代入DR60标准曲线拟合方程中计算t时刻溶液中DR60的浓度Ct,利用式(3)计算t时刻的吸附量qt。根据计算结果,可作出活性炭对DR60的吸附动力学模型曲线。常用的吸附动力学模型为Lagergren拟一级动力学和拟二级动力学,分别如式(6)和式(7)所示。
式中:qe为吸附平衡时AC对染料的吸附量,mg·g−1;qt为t时刻AC对染料的吸附量,mg·g−1;k1是一级动力学吸附的速率常数,min−1;k2为二级动力学吸附的速率常数,g·(mg·min)−1。
-
采用比表面积分析仪测定改性前后活性炭的比表面积和孔径分布;采用热重分析仪测试改性前后活性炭的表面化学性质。
-
1)改性对活性炭孔结构的影响。不同浓度Fe2+改性AC的N2吸附-脱附曲线及孔径分布如图2所示,其孔径结构参数如表3所示。由图2可知,AC、AC-Fe2+-0.2 mol·L−1、AC-Fe2+-0.4 mol·L−1、AC-Fe2+-0.8 mol·L−1、AC-Fe2+-1.6 mol·L−1符合IV型吸附等温线的特征,表现出了中孔材料的吸附特性[26]。结合表3中参数可知,随着改性时Fe2+浓度的增加,孔径结构参数并没有呈现出明显的变化规律,参数之间差距较小,故可推测FeSO4改性会破坏AC的孔结构。但在Fe2+浓度为0.2~1.6 mol·L−1的范围内,FeSO4对AC的孔结构破坏作用不会因其浓度的变化而产生显著差异。图2(b)为不同浓度下改性AC的孔径分布曲线,可看出改性AC的孔径均集中在2~4 nm,符合中孔(2~50 nm)的孔径特征,这与吸附等温线的分析结果一致。不同超声时间下Fe2+改性AC的N2吸附-脱附曲线及孔径分布图如图3所示,其孔径结构参数如表4所示。由图3可知,AC及改性AC符合IV型吸附等温线的特征,改性后AC的吸附容量与未改性AC相比有所下降,但依然符合中孔材料的吸附特性。结合表4中参数可知,随着超声时间的增加,比表面积呈现出先上升后下降的趋势,在超声4 h时,改性AC的比表面积达到最大值,为1 229.154 8 m2·g−1,与未改性AC的比表面积仅相差26 m2·g−1。根据以上结果可推测,FeSO4会在一定程度上破坏AC的孔结构,使其比表面积降低,但同时作用的超声振荡又会使得AC表面产生新的孔隙结构,从而增加AC的比表面积。另外,AC-Fe2+-4 h的中孔表面积最大,这说明超声可加速FeSO4对AC表面微孔的腐蚀,使其变成中孔结构。图3(b)为不同时间下改性AC的孔径分布曲线,可看出改性AC的孔径均集中在2~4 nm,符合中孔的孔径(2~50 nm)特征,这与吸附等温线的分析结果一致。
在不同煅烧温度下,Fe2+改性AC的N2吸附-脱附曲线及孔径分布如图4所示,其孔径结构参数如表5所示。由图4可知,AC及改性AC均符合IV型吸附等温线的特征,改性后AC的吸附容量与未改性AC相比明显下降。结合表5中参数可知,煅烧后样品的比表面积均下降,4组改性样品的微孔表面积、中孔表面积、微孔孔容、中孔孔容等数据均呈现出与比表面积相同的变化趋势。这说明煅烧会破坏AC的孔结构,且温度越高,破坏越明显。图4(b)为不同煅烧温度下改性AC的孔径分布曲线,可看出改性AC的孔径均集中在2~4 nm左右,符合中孔(2~50 nm)的孔径特征,与吸附等温线的分析结果一致。
2)改性对活性炭表面官能团的影响。图5为不同Fe2+浓度下改性AC的DTG曲线,可以看出,不同浓度FeSO4改性AC的DTG曲线在150~900 ℃共有4个明显的失重峰,而未改性AC只在500~800 ℃有1个失重峰。在150~800 ℃形成的其他3个峰为升温过程中AC样品分解释放CO2和CO所导致的,其中在150~500 ℃的2个失重峰为CO2的解吸峰,表示AC表面羧基、内酯基和内酰胺基团的分解[27-30],且这2个峰强度随着FeSO4浓度的升高而增加。曲线中500~800 ℃的峰对应于羰基和酚基分解释放CO而造成的失重,未改性AC与改性AC在此位置均产生了相应的失重峰,这说明AC在改性前表面就含有一定数量的羰基和酚基,在经过改性之后,有部分的羰基和酚基分别被氧化为羧基和醌基,故改性AC在该温度范围内的失重峰相比于未改性AC稍有减弱。改性AC在800~900 ℃的失重峰尖锐,这说明改性AC在此处有一个很快的失重过程,根据FeSO4在温度高于680 ℃时即受热分解成Fe2O3、SO2和SO3等物质的性质,如式(8)和式(9)所示,可推测此处的失重峰为FeSO4受热分解所致。
图6为Fe2+浓度为0.8 mol·L1时,不同超声时间下改性AC的DTG曲线。与不同浓度FeSO4的改性结果相似,不同超声时间下改性AC在150~900 ℃共有4个失重峰,其中150~500 ℃的2个失重峰为AC表面羧基、内酯基和内酰胺基团分解产生CO2而形成的失重峰,600~800 ℃的失重峰为AC表面羰基和酚基分解产生CO而形成的失重峰,在800~900 ℃的失重峰为FeSO4受热分解产生Fe2O3、SO2和SO3而形成的失重峰。150~700 ℃的3个峰强度随着超声时间的不同并未有明显差别,但观察图中800~900 ℃的分解峰可看出,该组改性样在800~900 ℃的峰强随着超声时间的增长而增大,可推测超声时间越长,AC中所含的FeSO4的量也越多。
图7为Fe2+浓度为0.8 mol·L−1、超声时间为4 h时不同煅烧温度下改性AC的DTG曲线。与前2组FeSO4的改性结果不同,不同煅烧温度下改性AC在150~900 ℃只有2个失重峰,其中在600~800 ℃的失重峰为AC表面羰基和酚基分解产生CO而形成的失重峰,800~900 ℃的失重峰为FeSO4受热分解产生Fe2O3、SO2和SO3而形成的失重峰。150~500 ℃因羧基、内酯基和内酰胺基团分解而释放CO2的失重峰基本消失,这说明在煅烧过程中,这些峰已经基本分解。观察600~800 ℃的失重峰可发现,在温度高于550 ℃时,该处的峰值随着煅烧温度的升高而逐渐降低,这说明羰基和酚基在不同程度上的分解。当煅烧温度为750 ℃和950 ℃时,在煅烧后的样品表面观察到了红色物质,这证实了高于680 ℃的煅烧温度会使得FeSO4分解为Fe2O3。同时,从DTG曲线中可看出,尽管AC-Fe2+-350 ℃, AC-Fe2+-550 ℃的煅烧温度未达到680 ℃,位于800~900 ℃的峰代表FeSO4的受热分解峰,与未煅烧样品AC-Fe2+-4 h相比,峰值仍有所下降,且在该组实验设置的温度范围内,FeSO4分解峰的值随着煅烧温度的升高而降低。
-
1)离子浓度对改性AC吸附性能的影响。图8(a)为不同浓度下离子改性AC及未改性AC在120 min内对BHET溶液中DR60的吸附曲线。从图8(a)中可看出,4个浓度梯度下改性AC的脱色率值均大于未改性AC,这说明Fe2+改性能提高AC对DR60的脱色速率,且随着改性时Fe2+浓度的增大,改性AC在120 min内的脱色率呈现出先上升后下降的趋势(图8(b))。
2)超声时间对改性AC吸附性能的影响。图9(a)为不同超声时间下离子改性AC及未改性AC在120 min内对BHET溶液中DR60的吸附影响。从图9(a)可看出,改性AC对DR60的脱色率均大于未改性AC。图9(b)为超声时间对改性AC平均脱色率的影响。从图9(b)中可看出,改性AC对DR60的脱色率随改性时超声时间的增加呈现出了先增大后减小的趋势,AC-Fe2+-4 h对DR60平均脱色率最高,且脱色率平均值变化规律为AC-Fe2+-1 h < AC-Fe2+-2 h < AC-Fe2+-8 h < AC-Fe2+-4 h,这与4个样品的比表面积变化趋势一致,这说明与表面官能团及负载的离子量相比,比表面积在吸附中的影响仍然占据着主导作用。
3)煅烧温度对改性AC吸附性能的影响。图10(a)为不同煅烧温度下离子改性AC及未改性AC在120 min内对BHET溶液中DR60的吸附曲线。可以看出,除AC-Fe2+-750 ℃的脱色率曲线基本与AC重合之外,其他3个样品的脱色率曲线均位于AC之下,这是由样品的比表面积和表面官能团的共同作用的结果,煅烧后AC的表面官能团在不同程度上被破坏,同时其孔结构在浸渍改性过程中被破坏,这是造成该组改性样品吸附性能差于未改性AC的原因。与相同改性条件下只浸渍不煅烧的样品AC-Fe2+-4 h相比,该组煅烧样品性质的主要差别体现在表面官能团的数量与种类有所减少,说明改性官能团的存在与否及数量多少会对AC的吸附性能造成一定程度的影响。煅烧温度对改性AC吸附DR60的影响如图10(b)所示,随着改性时煅烧温度的增加,改性AC对DR60的吸附并未呈现出明显的变化规律。
-
在design expert 8.0中采用逐步回归的方法对表2中的实验结果进行方差分析,结果如表6所示。根据表中统计分析结果,可拟合得到平均脱色率与2个实验变量值之间的二次多项响应面方程,其响应面方程如式(10)所示。P≤0.01,回归方程显著,P>0.05,非正常误差不显著,这说明本次拟合得到的响应面方程可用。X1和X2的P值分别为0.306 5和0.210 7,这说明超声时间对AC平均脱色率的影响大于Fe2+浓度,
X21 和X22 项的P值均小于0.01,说明其对Y值,即AC平均脱色率的影响极显著。模型中不存在X1X2二次项,说明Fe2+浓度和超声时间之间不存在明显的交互影响。式中:Y为120 min内的平均脱色率;X1为Fe2+浓度,mol·L−1;X2代表超声时间,min。
在Design expert 8.0中利用得到的响应面方程对Y值进行最优化,在Fe2+浓度为1.224 mol·L−1和超声时间为4.93 h时,AC的平均脱色率可达最大值,为94.273 7%。
为验证上述预测值的准确性,在该条件下,对AC进行3次重复改性,并将得到的改性样品用于BHET的脱色实验中,3个样品120 min内的平均脱色率值分别为95.40%、92.73%、92.32%,3次吸附实验平均脱色率的均值为93.48%,与预测值94.27%十分接近,证明该模型具有实际意义。
-
1)离子改性活性炭吸附热力学分析。图11为AC及AC-Fe2+-max的Langmuir和Freundlich等温线拟合曲线,表7为根据拟合方程计算所得的各项参数汇总。根据表中的数据可知,在90 ℃时,Langmuir等温方程对AC和AC-Fe2+-max的决定系数分别为0.990 5和0997 1,Freundlich等温方程的决定系数分别为0.900 1和0.946 9,这说明Langmuir等温方程可以更好地描述DR60在AC及AC-Fe2+-max上的吸附过程。Langmuir等温曲线表示吸附质在吸附剂表面有限的吸附位点上为单层吸附,吸附质在吸附剂表面平面内无迁移,这说明DR60在AC及AC-Fe2+-max上的吸附为均一单层分布,这与硝酸改性AC所得的热力学实验结果相同[31]。
2)离子改性活性炭吸附动力学分析。图12为AC及AC-Fe2+-max的拟一级动力学和拟二级动力学曲线。如图12(a)所示,以ln(qe − qt)对t作出的曲线与一级动力学模型的前半段拟合性较好,后半段差别较大,对AC与AC-Fe2+-max,其决定系数R2分别为0.948 4,0.935 7。而以t/q对t所作出的曲线与其各自的二级动力学拟合曲线基本重合,t/q对t呈现出良好的线性关系,对AC与AC-Fe2+-max,其决定系数分别为0.999 3、0.999 7。这说明BHET中的DR60在AC与AC-Fe2+-max上的吸附过程更符合二级动力学方程。利用拟合方程的斜率和截距以计算表8中所示的动力学速率常数k1、k2及平衡吸附量qe1、qe2。从表8中可看出,通过二级动力学模型拟合曲线计算所得的qe值与实验所得的qe值十分接近。结合图12所示的结果,可以得出AC及AC-Fe2+-max对BHET中DR60的吸附均符合二级动力学模型,一些相关文献中也报道了相似的结果[31-32]。这表明,吸附过程中发生了化学反应[32],即在DR60吸附到AC及AC-Fe2+-max表面的过程中,发生了电子共享或电子交换的化学反应过程[31, 33]。
根据表8中的结果,AC及AC-Fe2+-max对DR60根据二级动力学计算所得的吸附容量分别为45.683 mg·g−1及46.468 mg·g−1,二者对DR60的实际吸附容量分别为42.289 mg·g−1及43.907 mg·g−1。结合上文中脱色速率,AC-Fe2+-max对DR60平均脱色率可达93.48%,这远高于未改性AC的82.04%,同时其吸附容量也比未改性的AC高出约1.8 mg·g−1,故从吸附速率和吸附容量来看,AC-Fe2+-max的吸附性能都优于AC。
-
1) AC表面羧基及内酯基的含量会随着改性时FeSO4浓度的增加而增加,而AC中FeSO4的含量会随着超声时间的增加而增加;同时,改性AC的比表面积随着超声时间的增加呈现出先上升后下降的趋势,在超声4 h改性条件下其比表面积与未改性AC仅相差26 m2·g−1;高温煅烧会使得AC表面各种官能团在不同程度上分解,包括负载在AC表面的FeSO4,煅烧同时也会破坏AC的孔结构,使其比表面积等各项参数值下降。
2)在3组改性AC对BHET脱色实验中,120 min内平均脱色率随着改性浓度和超声时间的增加均呈现出先上升后下降的趋势,在AC-Fe2+-0.8 mol·L−1和AC-Fe2+-4 h条件下脱色率可达到最大值,且未煅烧的2组改性AC在120 min内平均脱色率均高于未改性AC,煅烧后改性AC的脱色性能下降到低于未改性AC的水平。
3)在Fe2+浓度为1.224 mol·L−1,超声时间为4.93 h,且无煅烧的条件下,改性AC对DR60的120 min内平均脱色率可达93.48%,可实现对BHET的完全脱色。
4) AC及AC-Fe2+-max的吸附热力学与吸附动力学实验结果表明,二者的吸附特征均更符合Langmuir吸附等温模型及二级动力学模型,DR60在AC及AC-Fe2+-max上的吸附为均一单层分布,吸附过程中包含化学反应,AC-Fe2+-max吸附容量相较于未改性AC提高了1.8 mg·g−1。
Fe2+改性活性炭的制备及其对聚酯降解产物的脱色性能分析
Preparation of Fe2+ modified activated carbon and its performance analysis of decolorization of polyester degradation products
-
摘要: 针对聚酯降解产物的回收再利用问题,采用Fe2+对活性炭进行浸渍改性,并用于聚酯降解产物的脱色。通过比表面积测定、TG分析对改性前后的活性炭进行了表征;研究了改性时Fe2+浓度、超声时间及煅烧温度对活性炭的孔结构、表面官能团以及吸附性能的影响,并以此为基础,通过响应面实验优化了Fe2+对活性炭的改性工艺。结果表明,在Fe2+浓度为1.224 mol·L−1,超声时间为4.93 h,且无煅烧的条件下,改性活性炭对染料的平均脱色率最高,可达93.483 5%,可在2 h内实现对聚酯降解产物的完全脱色,且其吸附容量比未改性活性炭提高了1.8 mg·g−1。对改性前后的活性炭进行吸附热力学与吸附动力学实验,发现二者的吸附特征符合Langmuir吸附等温模型及拟二级动力学模型,其决定系数分别为0.990 5、0.997 1及0.999 3、0.999 7。这说明染料在活性炭上的吸附为均一单层分布,吸附过程中包含化学反应。使用Fe2+对活性炭进行浸渍改性后再对聚酯降解产物进行脱色,不仅能提高活性炭对染料的脱色效率,还能提高对其对染料的吸附容量。Abstract: In order to reuse the polyester degradation products effectively, Fe2+ was used to impregnating the activated carbon for decolorization of polyester degradation products. The fresh and modified activated carbon was characterized by specific surface area test and TGA analysis. The effects of Fe2+ concentration, ultrasonic time and calcination temperature on the pore structure, surface function groups and adsorption performance of the activated carbon were studied during the modification, and the response surface experiment was conducted to optimize activated carbon modification process by Fe2+. The results showed that he average decolorization rate of the modified activated carbon reached the highest value of 93.484% under the conditions of Fe2+ concentration of 1.224 mol·L−1, ultrasonic time of 4.93 h and without calcination. The complete decorization occurred for the polyester degradation products within 2 h, and the adsorption capacity of modified activated carbon increased by 1.8 mg·g−1 compared with the fresh activated carbon. The adsorption thermodynamics and kinetics experiments of activated carbon before and after modification were conducted. Their adsorption characteristics were in accordance with the Langmuir adsorption isotherm model and the pseudo second-order kinetic model, and the corresponding fitting correlation coefficients were 0.990 5, 0.997 1 and 0.999 3, 0.999 7, respectively. This indicated that the adsorption of the dye on activated carbon was a uniform single layer distribution, and the chemical reaction was contained in the adsorption process. Decolorization of polyester degradation products using Fe2+ modified activated carbon can not only improve the dyes decolorization efficiency, but also the adsorption capacity of dyes.
-
聚酯(PET)是目前最常用的聚合物之一,它被广泛用于制造纤维、织物及其他产品,如饮料瓶、照相胶片等[1-3]。然而,随着聚酯使用量的增加,其不可生物降解的特性使得其回收利用成为了环境保护的一大关注点[4]。在PET的回收方法中,乙二醇醇解所得产物能用于生产各类产品,如疏水性纺织染料[2]、聚氨酯泡沫[5]、醇酸树脂[6-7]等,已被科学研究及工业生产广泛采用。有研究[8-12]发现,乙二醇醇解PET的主要产物是对苯二甲酸乙二醇酯(BHET)。尽管BHET用途广泛,但聚酯面料中含有的染料会残留在BHET晶体中,使其应用受到了限制,如将其用于生产再生聚酯纤维时,BHET中所含染料会直接影响再生聚酯的颜色。目前,国内外鲜有对聚酯降解产物脱色的报道,工业上主要使用纯度高及颜色浅的聚酯瓶片来生产再生聚酯产品,因此,找到一种经济的方法,来实现对聚酯面料降解产物的有效脱色是聚酯循环再利用中亟待解决问题。
目前,常用的染料废水脱色技术主要包括臭氧氧化法、超滤法、氧化法、光化学降解法和吸附法[13-16]。在这些方法中,吸附是最有效的方法之一,并已成功用于去除废水中的染料[17]。作为多孔材料,活性炭(AC)通常具有良好的内部孔结构、高比表面积以及位于外表面和内表面的一定量的化学官能团[18-19]。因此,它在分离多种化合物方面是非常有效的,并且已被广泛用作废水处理中的吸附剂[20-22]。但是,它在生产过程中形成的非极性特征限制了其应用[23]。已经有研究[24]采取各种处理方法来使AC成为极性更强的材料,包括酸碱改性,离子改性和微波改性等。其中,离子改性已被用于在AC上引入金属化合物和金属颗粒等,以提高其对水中氟化物,氰化物,和重金属砷等的吸附容量。
为实现对BHET脱色的同时不破坏其分子结构,本研究选用活性炭对其进行吸附脱色,并采用FeSO4·7H2O对活性炭进行了离子改性,以提高其对BHET中所含染料的吸附性能;在探究离子浓度、超声时间、煅烧温度对活性炭物理及化学性能的影响的基础上,通过中心组合法设计2因素3水平的响应面实验,以优化活性炭的改性工艺,从而提高其对染料的脱色效率;最后,通过吸附热力学与动力学实验,分析了染料在AC上吸附的机理及吸附容量变化,以实现活性炭在BHET脱色中的最大化利用。
1. 材料与方法
1.1 实验试剂
无色聚酯纤维、分散红60(DR60)、活性炭(AC)、七水合硫酸亚铁(FeSO4·7H2O)、95%乙醇、乙二醇(EG)、醋酸锌(ZnAC2)均为分析纯。
1.2 实验仪器
紫外可见光分光光度计(UV-1800型,上海美谱达仪器有限公司);热失重分析仪(TGA Q-500型,美国TA Instruments公司);物理吸附仪(TriStar 3020型,美国Micromeritics公司);管式煅烧炉(GSL 1600X型,合肥科晶材料技术有限公司);高功率数控超声波清洗器(KH-400KDE型,昆山禾创超声仪器有限公司);循环水式多用真空泵(SHB型,郑州长城科工贸有限公司);电子天平(AR1530/C型,豪斯国际贸易有限公司)。
1.3 实验方法
以过量乙二醇作为解聚剂,醋酸锌作为催化剂,将聚酯高分子降解成单体分子对苯二甲酸乙二醇酯(BHET),其反应如式(1)所示,醇解温度为196 ℃,反应时间为3 h,采用文献中的方法[9-10]进行降解。实验中使用分散红60(DR60)和无色聚酯纱线来模拟有色废弃聚酯,以保证BHET中只含单一染料。
 (1)
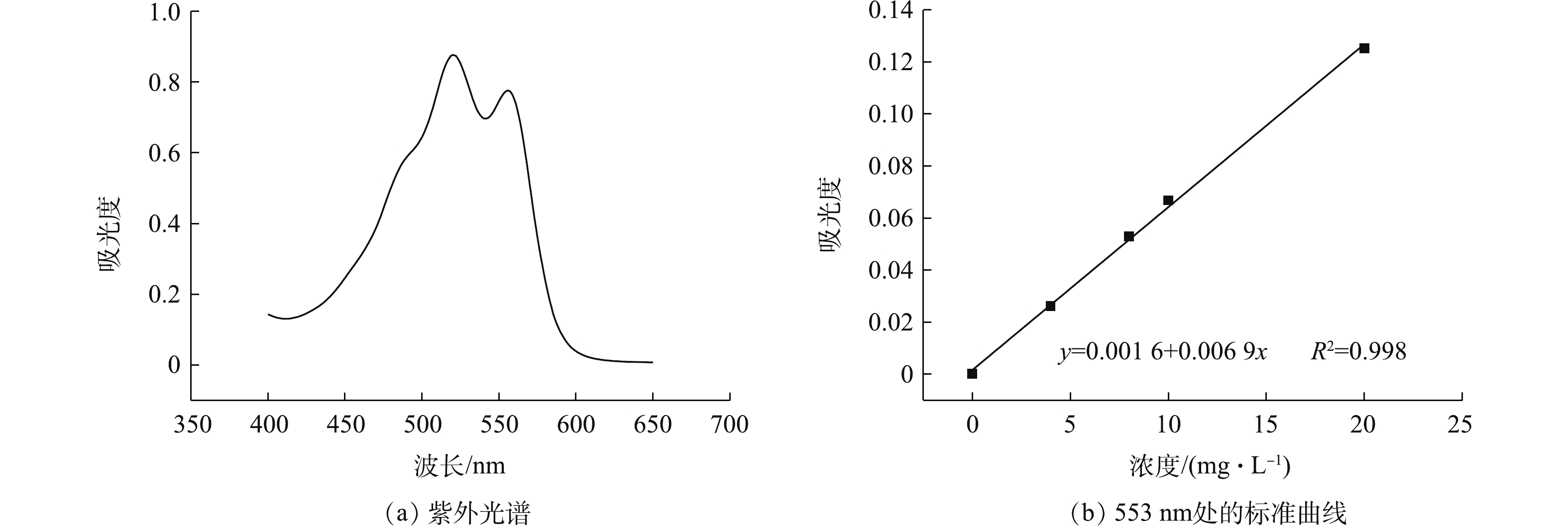
(1)为计算活性炭样品吸附DR60的量,可使用DR60的标准曲线来计算BHET溶液中DR60浓度。配制浓度分别为4、8、10、20 mg·L−1的DR60溶液,其溶剂为95%的乙醇和水的混合溶剂(醇∶水的体积比为1∶2),使用UV-1800分光光度计扫描溶液在400~650 nm区域内的紫外吸收谱。如图1(a)所示,DR60溶液分别在553 nm和515 nm处有2个特征吸收峰。对不同浓度的DR60在553 nm处的吸光度进行线性拟合,即得到DR60在553 nm处的吸光度标准曲线,如图1(b)所示。
采用FeSO4·7H2O对活性炭进行改性。称取10 g活性炭,加入到100 mL的FeSO4·7H2O溶液中,将反应溶液置于100 Hz超声波清洗器中,分散一定时间,并于常温条件下浸渍24 h。反应结束后,将反应混合液过滤分离,并用去离子水将活性炭反复冲洗,至中性后,放入50 ℃鼓风干燥烘箱,烘干至恒重,装入密封袋备用。为探究改性条件对活性炭性质的影响,共进行3组单因素实验,具体实验条件如表1所示。
表 1 活性炭改性实验条件Table 1. Modification conditions of AC实验组 Fe2+浓度/(mol·L−1) 超声时间/h 煅烧温度/℃ 1 0.2、0.4、0.8、1.6 0.5 无煅烧 2 0.8 1、2、4、8 无煅烧 3 0.8 4 350、550、750、950 | Show Table DownLoad:
CSV
DownLoad:
CSV
实验组1为不同浓度FeSO4·7H2O对活性炭的改性,该组改性活性炭分别定义为AC-Fe2+-0.2 mol·L−1、AC-Fe2+-0.4 mol·L−1、AC-Fe2+-0.8 mol·L−1、AC-Fe2+-1.6 mol·L−1;实验组2为不同超声时间下对活性炭的改性,该组改性活性炭分别定义为AC-Fe2+-1 h、AC-Fe2+-2 h、AC-Fe2+-4 h、AC-Fe2+-8 h;实验组3为不同煅烧温度下对活性炭的改性,将烘干得到的样品在管式炉中煅烧,该组改性活性炭分别定义为AC-Fe2+-350 ℃、AC-Fe2+-550 ℃、AC-Fe2+-750 ℃、AC-Fe2+-950 ℃。
为探究离子浓度、超声时间及煅烧温度这3个因素如何影响改性AC对BHET中DR60的吸附性能,设置了3组对比吸附实验。每组实验均设置1个对照组和4个实验组,对照组未改性AC与实验组改性AC均采用100 mL初始浓度为10 g·L−1的BHET溶液作为脱色样本,每个样本中活性炭的用量均为1 g,整个吸附过程保持在90 ℃恒温水浴中进行,取样间隔为10 min,脱色时间为120 min,120 min内的平均脱色率为12个取样点的脱色率平均值。为保证正确计算出溶液中DR60的浓度,取样时溶剂与DR60标准曲线绘制时保持一致,即取2 mL样品溶于1 mL 95%乙醇溶液中,以防止BHET溶液在常温下部分结晶析出。脱色过程中样品在t时刻的染料浓度根据DR60的标准曲线计算得到,染料脱除率由式(2)计算得到。
Y=(C0−Ct)C0×100% (2) 式中:Y为脱色率;C0为溶液中染料初始浓度,mg·L−1;Ct为t时刻溶液中染料浓度,mg·L−1。
采用中心组合法设计了包括离子浓度及超声时间2个因素在内的2因素3水平响应面分析实验(13组)。具体实验设计及实验结果如表2所示。AC样品120 min内平均脱色率实验方法和计算方法与吸附实验相同。
表 2 响应面实验设计及实验结果汇总Table 2. Design and results of response surface experiment实验编号 编码(实验)水平 120 min平均脱色率/% 离子浓度/(mol·L−1) 超声时间/h 1 −1(0.8) −1(4) 87.318 6 2 −1(0.8) 1(6) 84.916 0 3 1(1.6) −1(4) 88.316 8 4 1(1.6) 1(6) 87.542 1 5 0(1.2) −1.41(3.59) 86.032 3 6 0(1.2) 1.41(6.41) 85.190 2 7 −1.41(0.636) 0(5) 85.845 3 8 1.41(1.764) 0(5) 85.765 6 9 0(1.2) 0(5) 95.429 6 10 0(1.2) 0(5) 94.546 4 11 0(1.2) 0(5) 93.686 2 12 0(1.2) 0(5) 92.843 5 13 0(1.2) 0(5) 94.705 7 | Show TableDownLoad:
CSV
把响应面实验优化得到的最佳改性AC定义为AC-Fe2+-max。配制系列浓度为10、20、30、40和50 g·L−1的DR60 BHET溶液,量取100 mL置于250 mL锥形瓶中,分别加入0.02 g AC和样品,密封瓶口,置于90 ℃下吸附12 h,取2 mL样品溶于1 mL 95%的乙醇溶液中,测定吸光度,代入DR60标准曲线拟合方程中计算溶液中DR60的浓度Ce,利用式(3)计算吸附量qe。
qe=(C0−Ce)VW (3) 式中:qe为平衡吸附容量,mg·g−1;C0为溶液中染料初始浓度,mg·L−1;Ce为吸附平衡时溶液中染料浓度,mg·L−1。根据qe计算结果,可作出AC对DR60的吸附等温线。常用的等温吸附模型为Langmuir和Freundlich等温吸附方程,如式(4)和式(5)所示。
Ceqe=1Qb+CeQ (4) lnqe=lnkf+nlnCe (5) 式中:C0为溶液中染料初始浓度,mg·L−1;Ce为吸附质的平衡浓度,mg·L−1;Q为单层吸附容量,mg·g−1;b为朗格缪尔常数;qe为平衡吸附量,mg·g−1;kf为吸附平衡常数;n是吸附强度的指标[25]。
选择AC、AC-Fe2+-max以探究未改性及改性活性炭的动力学过程。配制初始浓度为50 g·L−1的DR60 BHET溶液,量取100 mL置于250 mL锥形瓶中,分别加入0.25 g AC及AC-Fe2+-max,密封瓶口,置于90 ℃下吸附12 h,每隔0.5 h取2 mL样品,溶于1 mL 95%的乙醇溶液中,测定吸光度,代入DR60标准曲线拟合方程中计算t时刻溶液中DR60的浓度Ct,利用式(3)计算t时刻的吸附量qt。根据计算结果,可作出活性炭对DR60的吸附动力学模型曲线。常用的吸附动力学模型为Lagergren拟一级动力学和拟二级动力学,分别如式(6)和式(7)所示。
ln(qe−qt)=lnqe−k1t (6) tq=1k2qe2+tqe (7) 式中:qe为吸附平衡时AC对染料的吸附量,mg·g−1;qt为t时刻AC对染料的吸附量,mg·g−1;k1是一级动力学吸附的速率常数,min−1;k2为二级动力学吸附的速率常数,g·(mg·min)−1。
1.4 分析方法
采用比表面积分析仪测定改性前后活性炭的比表面积和孔径分布;采用热重分析仪测试改性前后活性炭的表面化学性质。
2. 结果与讨论
2.1 改性对活性炭物理及化学性质的影响
1)改性对活性炭孔结构的影响。不同浓度Fe2+改性AC的N2吸附-脱附曲线及孔径分布如图2所示,其孔径结构参数如表3所示。由图2可知,AC、AC-Fe2+-0.2 mol·L−1、AC-Fe2+-0.4 mol·L−1、AC-Fe2+-0.8 mol·L−1、AC-Fe2+-1.6 mol·L−1符合IV型吸附等温线的特征,表现出了中孔材料的吸附特性[26]。结合表3中参数可知,随着改性时Fe2+浓度的增加,孔径结构参数并没有呈现出明显的变化规律,参数之间差距较小,故可推测FeSO4改性会破坏AC的孔结构。但在Fe2+浓度为0.2~1.6 mol·L−1的范围内,FeSO4对AC的孔结构破坏作用不会因其浓度的变化而产生显著差异。图2(b)为不同浓度下改性AC的孔径分布曲线,可看出改性AC的孔径均集中在2~4 nm,符合中孔(2~50 nm)的孔径特征,这与吸附等温线的分析结果一致。不同超声时间下Fe2+改性AC的N2吸附-脱附曲线及孔径分布图如图3所示,其孔径结构参数如表4所示。由图3可知,AC及改性AC符合IV型吸附等温线的特征,改性后AC的吸附容量与未改性AC相比有所下降,但依然符合中孔材料的吸附特性。结合表4中参数可知,随着超声时间的增加,比表面积呈现出先上升后下降的趋势,在超声4 h时,改性AC的比表面积达到最大值,为1 229.154 8 m2·g−1,与未改性AC的比表面积仅相差26 m2·g−1。根据以上结果可推测,FeSO4会在一定程度上破坏AC的孔结构,使其比表面积降低,但同时作用的超声振荡又会使得AC表面产生新的孔隙结构,从而增加AC的比表面积。另外,AC-Fe2+-4 h的中孔表面积最大,这说明超声可加速FeSO4对AC表面微孔的腐蚀,使其变成中孔结构。图3(b)为不同时间下改性AC的孔径分布曲线,可看出改性AC的孔径均集中在2~4 nm,符合中孔的孔径(2~50 nm)特征,这与吸附等温线的分析结果一致。
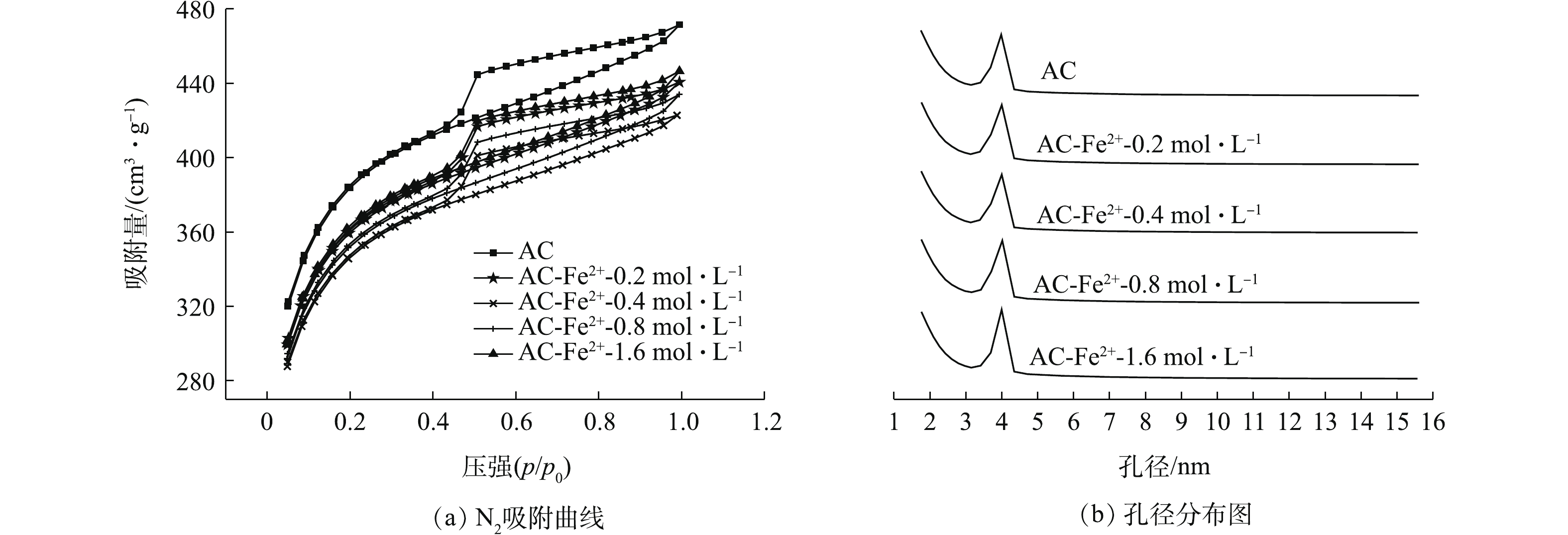 图 2 不同浓度Fe2+改性AC的N2吸附等温线及孔径分布图Figure 2. Adsorption isotherms and pore size distribution of AC modified by Fe2+ with different concentrations表 3 不同浓度Fe2+离子改性AC孔径结构数据Table 3. Data of pore structure of ACs modified by Fe2+ with different concentrations
图 2 不同浓度Fe2+改性AC的N2吸附等温线及孔径分布图Figure 2. Adsorption isotherms and pore size distribution of AC modified by Fe2+ with different concentrations表 3 不同浓度Fe2+离子改性AC孔径结构数据Table 3. Data of pore structure of ACs modified by Fe2+ with different concentrations样品 比表面积/(m2·g−1) 微孔表面积/(m2·g−1) 中孔表面积/(m2·g−1) 总孔容/(cm2·g−1) 微孔孔容/(cm2·g−1) AC 1 255.144 9 620.843 4 634.301 5 0.392 7 0.312 2 AC-Fe2+-0.2 mol·L−1 1 152.140 9 552.206 6 599.934 2 0.379 6 0.290 4 AC-Fe2+-0.4 mol·L−1 1 113.631 7 527.849 6 585.728 1 0.363 0 0.275 9 AC-Fe2+-0.8 mol·L−1 1 132.991 7 540.272 5 592.719 2 0.368 2 0.282 0 AC-Fe2+-1.6 mol·L−1 1 166.269 5 551.380 0 614.889 5 0.374 7 0.287 6 | Show TableDownLoad:
CSV
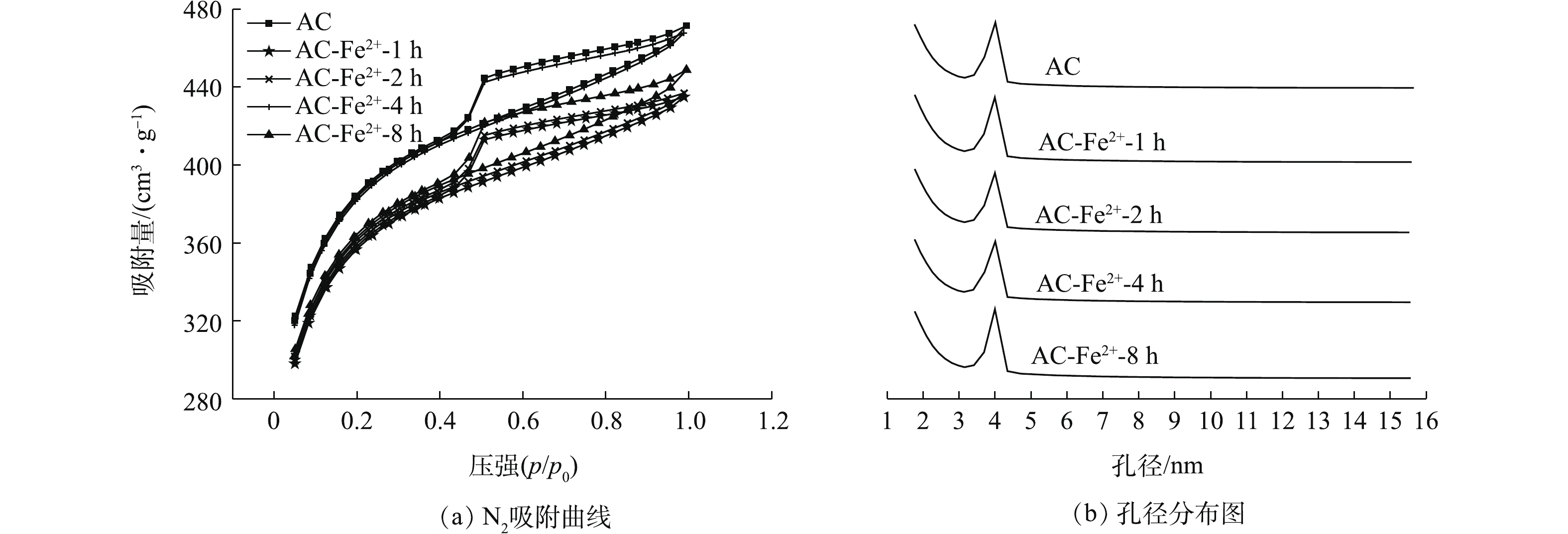 图 3 不同超声时间下改性AC的N2吸附等温线及孔径分布图Figure 3. N2 adsorption isotherms and pore size distribution of modified AC at different ultrasonic time表 4 不同超声时间下Fe2+改性AC孔径结构数据Table 4. Data of pore structure of modified ACs at different ultrasonic time
图 3 不同超声时间下改性AC的N2吸附等温线及孔径分布图Figure 3. N2 adsorption isotherms and pore size distribution of modified AC at different ultrasonic time表 4 不同超声时间下Fe2+改性AC孔径结构数据Table 4. Data of pore structure of modified ACs at different ultrasonic time样品 比表面积/(m2·g−1) 微孔表面积/(m2·g−1) 中孔表面积/(m2·g−1) 总孔容/(cm2·g−1) 微孔孔容/(cm2·g−1) AC 1 255.144 9 620.843 4 634.301 5 0.392 7 0.312 2 AC-Fe2+-1 h 1 143.085 0 551.982 8 591.102 2 0.372 0 0.290 2 AC-Fe2+-2 h 1 178.305 9 587.138 6 591.167 3 0.361 2 0.294 5 AC-Fe2+-4 h 1 229.154 8 583.239 3 645.915 5 0.397 1 0.304 9 AC-Fe2+-8 h 1 163.558 3 556.831 4 606.726 9 0.385 0 0.292 7 | Show TableDownLoad:
CSV
在不同煅烧温度下,Fe2+改性AC的N2吸附-脱附曲线及孔径分布如图4所示,其孔径结构参数如表5所示。由图4可知,AC及改性AC均符合IV型吸附等温线的特征,改性后AC的吸附容量与未改性AC相比明显下降。结合表5中参数可知,煅烧后样品的比表面积均下降,4组改性样品的微孔表面积、中孔表面积、微孔孔容、中孔孔容等数据均呈现出与比表面积相同的变化趋势。这说明煅烧会破坏AC的孔结构,且温度越高,破坏越明显。图4(b)为不同煅烧温度下改性AC的孔径分布曲线,可看出改性AC的孔径均集中在2~4 nm左右,符合中孔(2~50 nm)的孔径特征,与吸附等温线的分析结果一致。
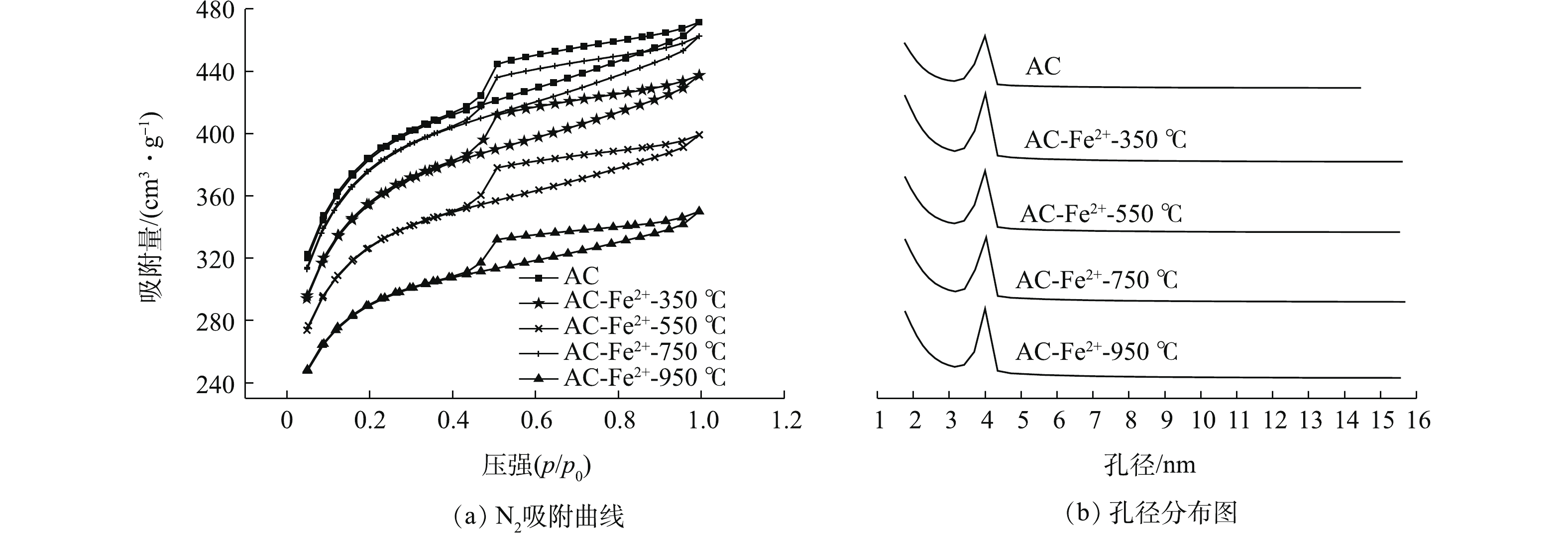 图 4 不同煅烧温度下改性AC的N2吸附等温线及孔径分布图Figure 4. N2 adsorption isotherms and pore size distribution of modified AC at different calcination temperatures表 5 不同煅烧温度Fe2+改性AC孔径结构数据Table 5. Data of pore structure of modified ACs at different calcination temperatures
图 4 不同煅烧温度下改性AC的N2吸附等温线及孔径分布图Figure 4. N2 adsorption isotherms and pore size distribution of modified AC at different calcination temperatures表 5 不同煅烧温度Fe2+改性AC孔径结构数据Table 5. Data of pore structure of modified ACs at different calcination temperatures样品 比表面积/(m2·g−1) 微孔表面积/(m2·g−1) 中孔表面积/(m2·g−1) 总孔容/(cm2·g−1) 微孔孔容/(cm2·g−1) AC 1 255.144 9 620.843 4 634.301 5 0.392 7 0.312 2 AC-Fe2+-350 ℃ 1 134.926 7 540.431 2 594.495 5 0.373 6 0.285 3 AC-Fe2+-550 ℃ 1 047.606 2 525.750 4 521.855 9 0.320 3 0.274 6 AC-Fe2+-750 ℃ 1 210.382 9 576.059 4 634.323 4 0.392 9 0.301 0 AC-Fe2+-950 ℃ 920.449 4 507.856 8 412.592 6 0.250 9 0.265 4 | Show TableDownLoad:
CSV
2)改性对活性炭表面官能团的影响。图5为不同Fe2+浓度下改性AC的DTG曲线,可以看出,不同浓度FeSO4改性AC的DTG曲线在150~900 ℃共有4个明显的失重峰,而未改性AC只在500~800 ℃有1个失重峰。在150~800 ℃形成的其他3个峰为升温过程中AC样品分解释放CO2和CO所导致的,其中在150~500 ℃的2个失重峰为CO2的解吸峰,表示AC表面羧基、内酯基和内酰胺基团的分解[27-30],且这2个峰强度随着FeSO4浓度的升高而增加。曲线中500~800 ℃的峰对应于羰基和酚基分解释放CO而造成的失重,未改性AC与改性AC在此位置均产生了相应的失重峰,这说明AC在改性前表面就含有一定数量的羰基和酚基,在经过改性之后,有部分的羰基和酚基分别被氧化为羧基和醌基,故改性AC在该温度范围内的失重峰相比于未改性AC稍有减弱。改性AC在800~900 ℃的失重峰尖锐,这说明改性AC在此处有一个很快的失重过程,根据FeSO4在温度高于680 ℃时即受热分解成Fe2O3、SO2和SO3等物质的性质,如式(8)和式(9)所示,可推测此处的失重峰为FeSO4受热分解所致。
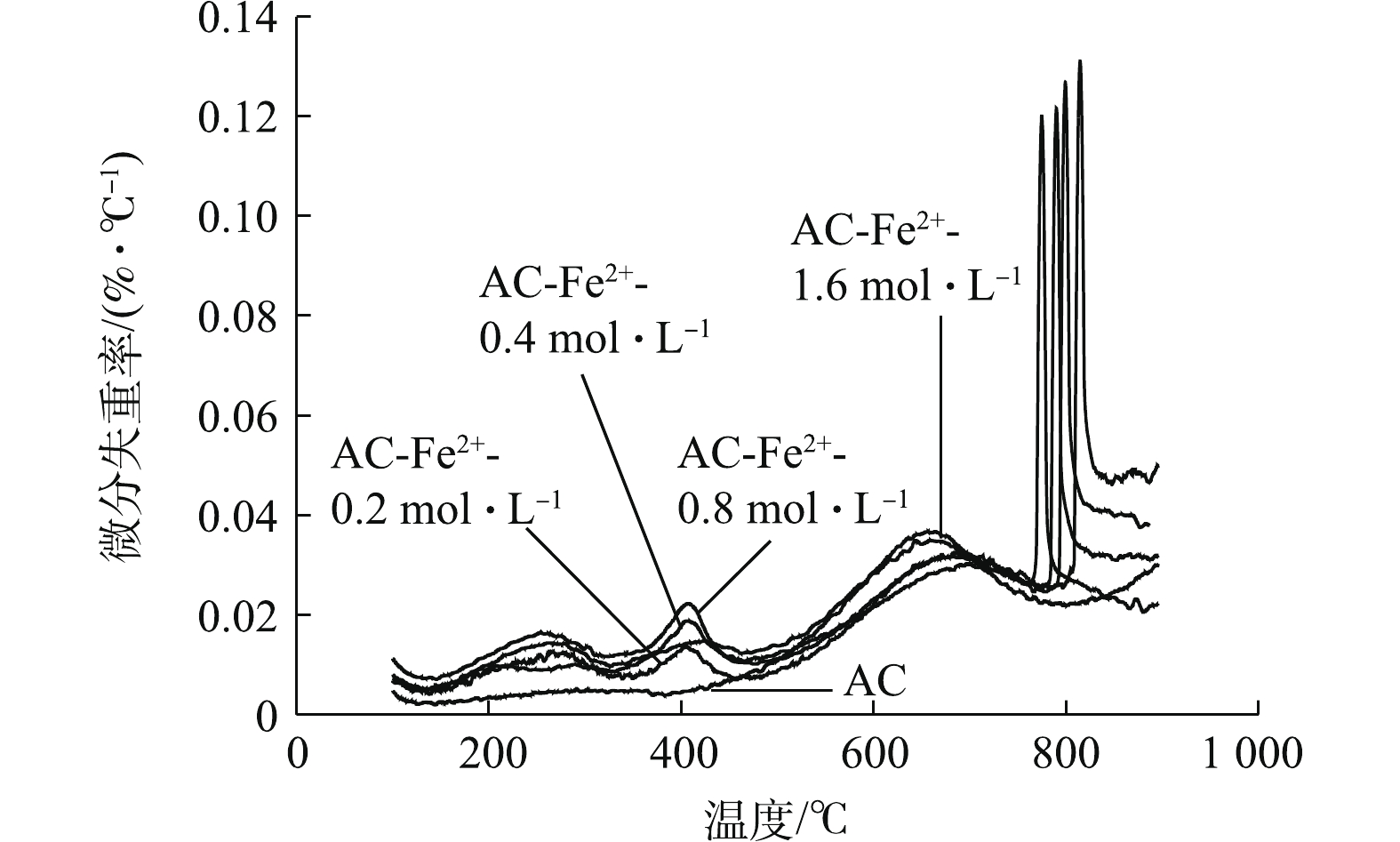 图 5 不同Fe2+浓度下改性AC的DTG曲线Figure 5. DTG curves of modified ACs by Fe2+ at different concentrations
图 5 不同Fe2+浓度下改性AC的DTG曲线Figure 5. DTG curves of modified ACs by Fe2+ at different concentrationsFeSO4=Fe2O3+SO2↑+SO3↑+14H2O↑ (8) 2SO3=2SO2+O2 (9) 图6为Fe2+浓度为0.8 mol·L1时,不同超声时间下改性AC的DTG曲线。与不同浓度FeSO4的改性结果相似,不同超声时间下改性AC在150~900 ℃共有4个失重峰,其中150~500 ℃的2个失重峰为AC表面羧基、内酯基和内酰胺基团分解产生CO2而形成的失重峰,600~800 ℃的失重峰为AC表面羰基和酚基分解产生CO而形成的失重峰,在800~900 ℃的失重峰为FeSO4受热分解产生Fe2O3、SO2和SO3而形成的失重峰。150~700 ℃的3个峰强度随着超声时间的不同并未有明显差别,但观察图中800~900 ℃的分解峰可看出,该组改性样在800~900 ℃的峰强随着超声时间的增长而增大,可推测超声时间越长,AC中所含的FeSO4的量也越多。
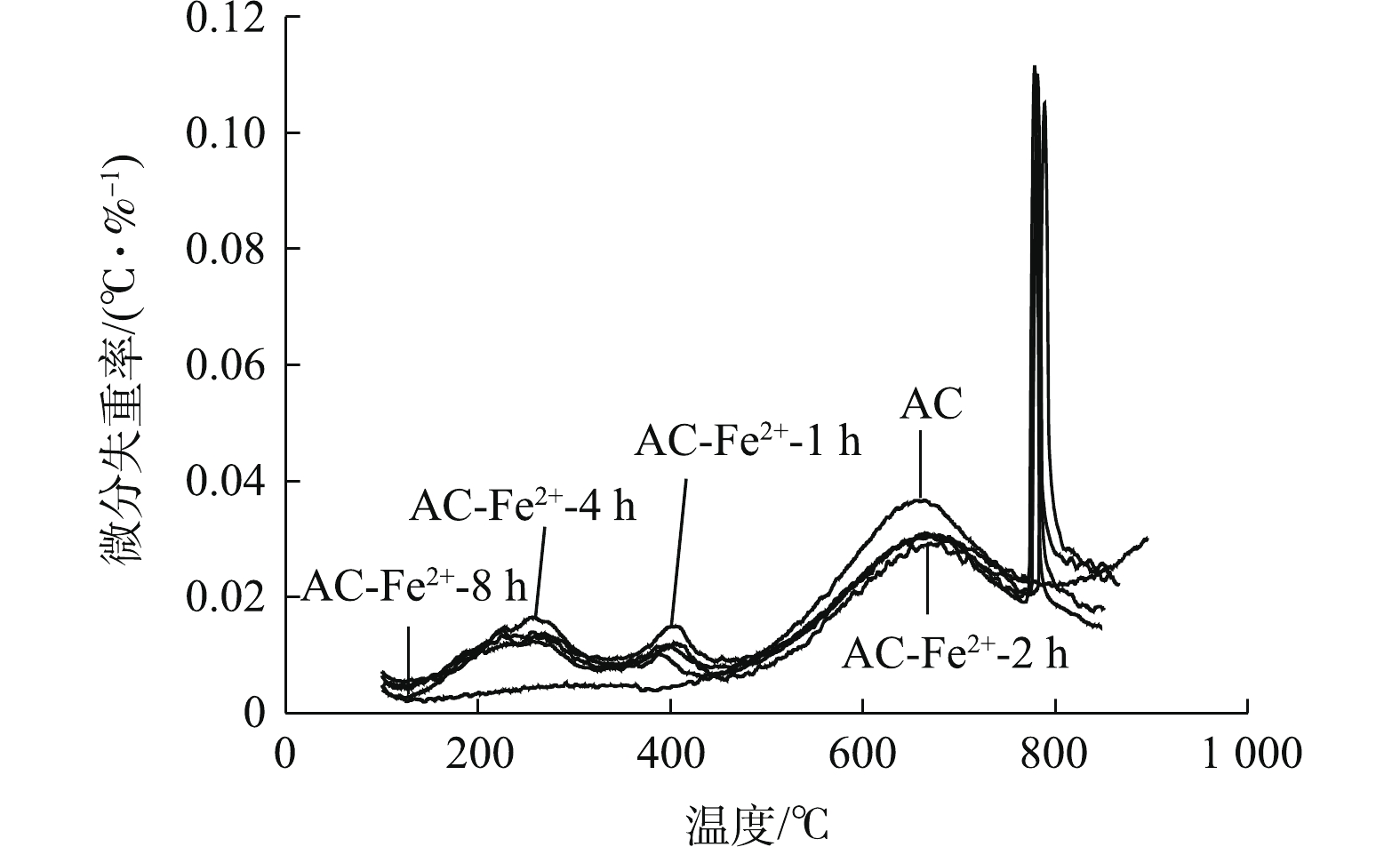 图 6 不同超声时间下改性AC的DTG曲线Figure 6. DTG curves of modified ACs at different ultrasonic times
图 6 不同超声时间下改性AC的DTG曲线Figure 6. DTG curves of modified ACs at different ultrasonic times图7为Fe2+浓度为0.8 mol·L−1、超声时间为4 h时不同煅烧温度下改性AC的DTG曲线。与前2组FeSO4的改性结果不同,不同煅烧温度下改性AC在150~900 ℃只有2个失重峰,其中在600~800 ℃的失重峰为AC表面羰基和酚基分解产生CO而形成的失重峰,800~900 ℃的失重峰为FeSO4受热分解产生Fe2O3、SO2和SO3而形成的失重峰。150~500 ℃因羧基、内酯基和内酰胺基团分解而释放CO2的失重峰基本消失,这说明在煅烧过程中,这些峰已经基本分解。观察600~800 ℃的失重峰可发现,在温度高于550 ℃时,该处的峰值随着煅烧温度的升高而逐渐降低,这说明羰基和酚基在不同程度上的分解。当煅烧温度为750 ℃和950 ℃时,在煅烧后的样品表面观察到了红色物质,这证实了高于680 ℃的煅烧温度会使得FeSO4分解为Fe2O3。同时,从DTG曲线中可看出,尽管AC-Fe2+-350 ℃, AC-Fe2+-550 ℃的煅烧温度未达到680 ℃,位于800~900 ℃的峰代表FeSO4的受热分解峰,与未煅烧样品AC-Fe2+-4 h相比,峰值仍有所下降,且在该组实验设置的温度范围内,FeSO4分解峰的值随着煅烧温度的升高而降低。
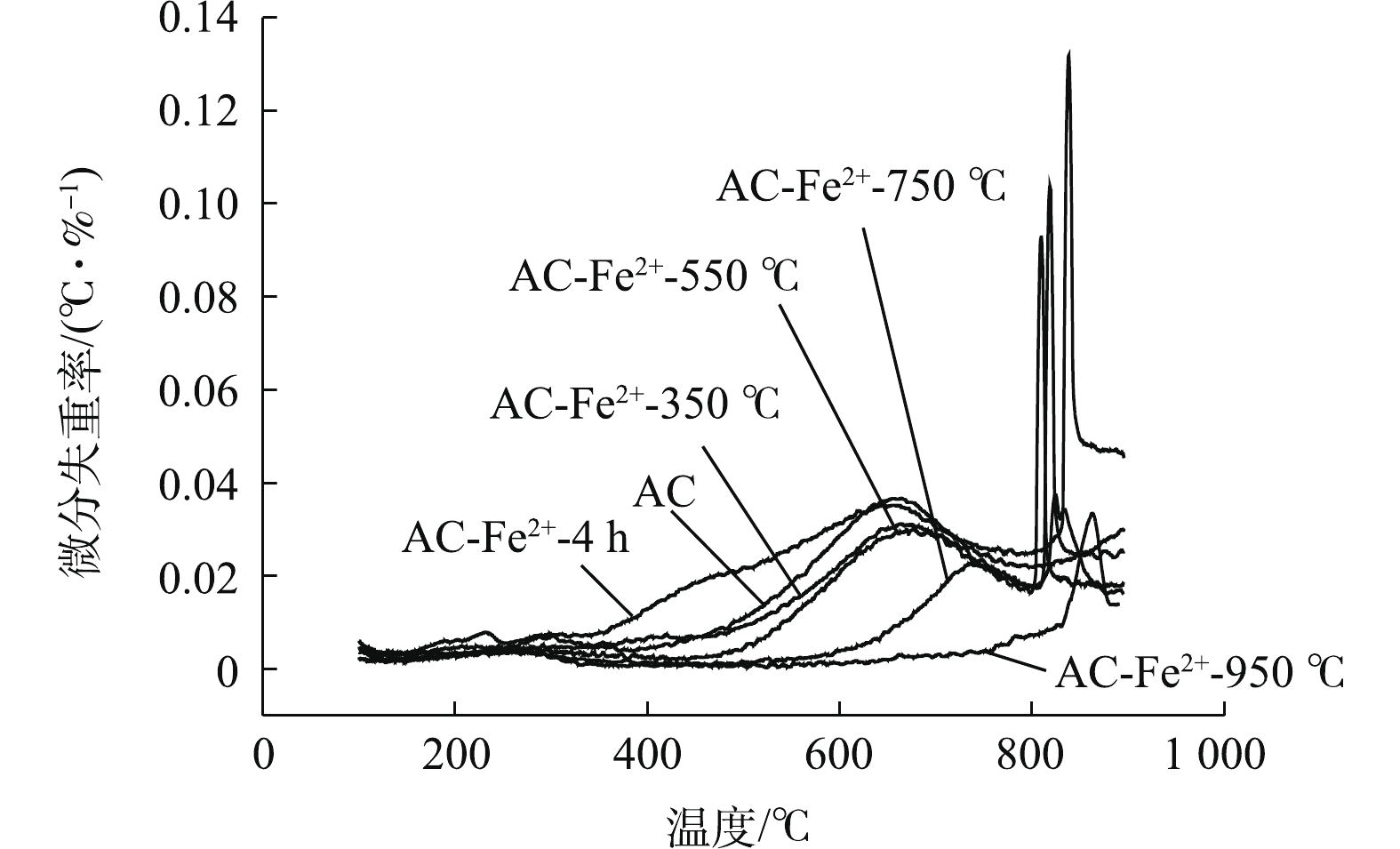 图 7 不同煅烧温度下改性AC的DTG曲线Figure 7. DTG curves of modified ACs at different calcination temperatures
图 7 不同煅烧温度下改性AC的DTG曲线Figure 7. DTG curves of modified ACs at different calcination temperatures2.2 单因素对改性活性炭吸附DR60的影响
1)离子浓度对改性AC吸附性能的影响。图8(a)为不同浓度下离子改性AC及未改性AC在120 min内对BHET溶液中DR60的吸附曲线。从图8(a)中可看出,4个浓度梯度下改性AC的脱色率值均大于未改性AC,这说明Fe2+改性能提高AC对DR60的脱色速率,且随着改性时Fe2+浓度的增大,改性AC在120 min内的脱色率呈现出先上升后下降的趋势(图8(b))。
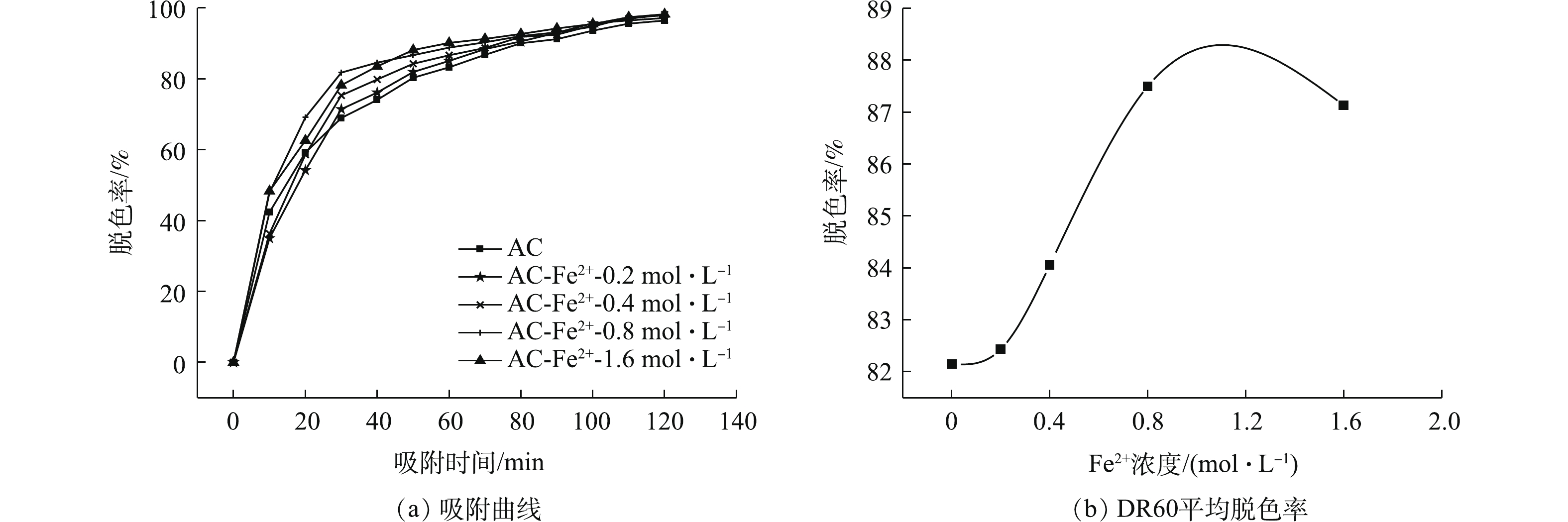 图 8 改性AC对DR60的吸附曲线及Fe2+浓度对平均脱色率的影响Figure 8. Adsorption curve of modified AC for DR60 and effect of Fe2+ concentration on the average decolorization rate
图 8 改性AC对DR60的吸附曲线及Fe2+浓度对平均脱色率的影响Figure 8. Adsorption curve of modified AC for DR60 and effect of Fe2+ concentration on the average decolorization rate2)超声时间对改性AC吸附性能的影响。图9(a)为不同超声时间下离子改性AC及未改性AC在120 min内对BHET溶液中DR60的吸附影响。从图9(a)可看出,改性AC对DR60的脱色率均大于未改性AC。图9(b)为超声时间对改性AC平均脱色率的影响。从图9(b)中可看出,改性AC对DR60的脱色率随改性时超声时间的增加呈现出了先增大后减小的趋势,AC-Fe2+-4 h对DR60平均脱色率最高,且脱色率平均值变化规律为AC-Fe2+-1 h < AC-Fe2+-2 h < AC-Fe2+-8 h < AC-Fe2+-4 h,这与4个样品的比表面积变化趋势一致,这说明与表面官能团及负载的离子量相比,比表面积在吸附中的影响仍然占据着主导作用。
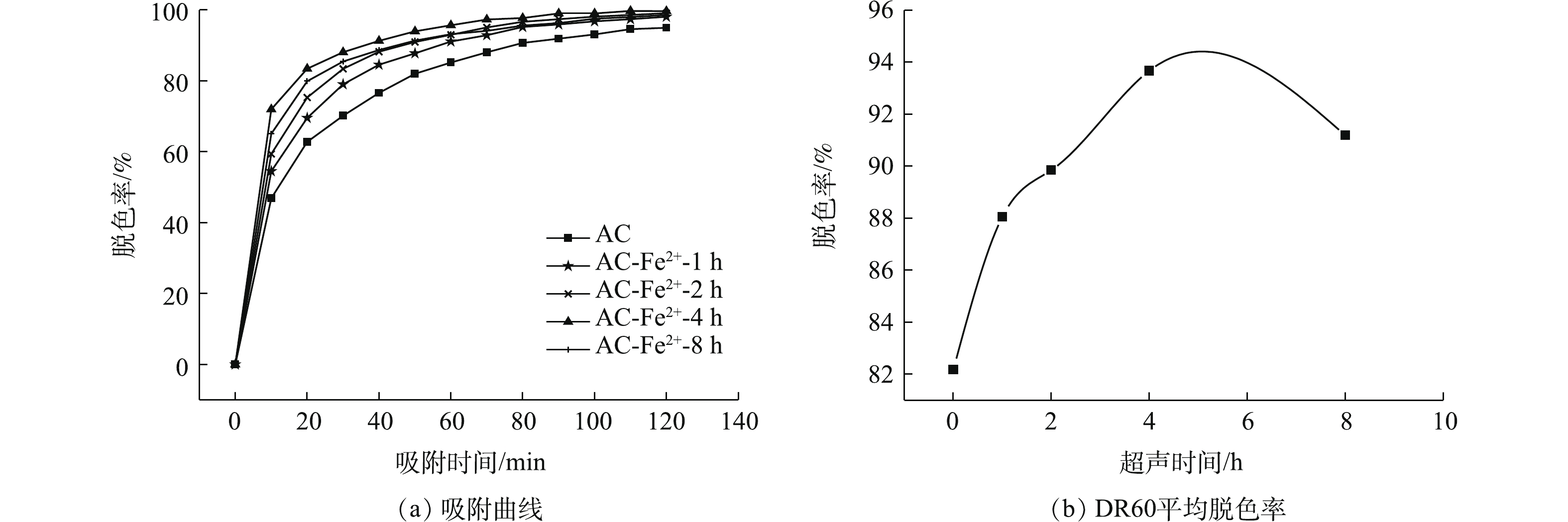 图 9 改性AC对DR60的吸附曲线及超声时间对平均脱色率的影响Figure 9. Adsorption curve of modified AC for DR60 and effect of ultrasonic time on the average decolorization rate
图 9 改性AC对DR60的吸附曲线及超声时间对平均脱色率的影响Figure 9. Adsorption curve of modified AC for DR60 and effect of ultrasonic time on the average decolorization rate3)煅烧温度对改性AC吸附性能的影响。图10(a)为不同煅烧温度下离子改性AC及未改性AC在120 min内对BHET溶液中DR60的吸附曲线。可以看出,除AC-Fe2+-750 ℃的脱色率曲线基本与AC重合之外,其他3个样品的脱色率曲线均位于AC之下,这是由样品的比表面积和表面官能团的共同作用的结果,煅烧后AC的表面官能团在不同程度上被破坏,同时其孔结构在浸渍改性过程中被破坏,这是造成该组改性样品吸附性能差于未改性AC的原因。与相同改性条件下只浸渍不煅烧的样品AC-Fe2+-4 h相比,该组煅烧样品性质的主要差别体现在表面官能团的数量与种类有所减少,说明改性官能团的存在与否及数量多少会对AC的吸附性能造成一定程度的影响。煅烧温度对改性AC吸附DR60的影响如图10(b)所示,随着改性时煅烧温度的增加,改性AC对DR60的吸附并未呈现出明显的变化规律。
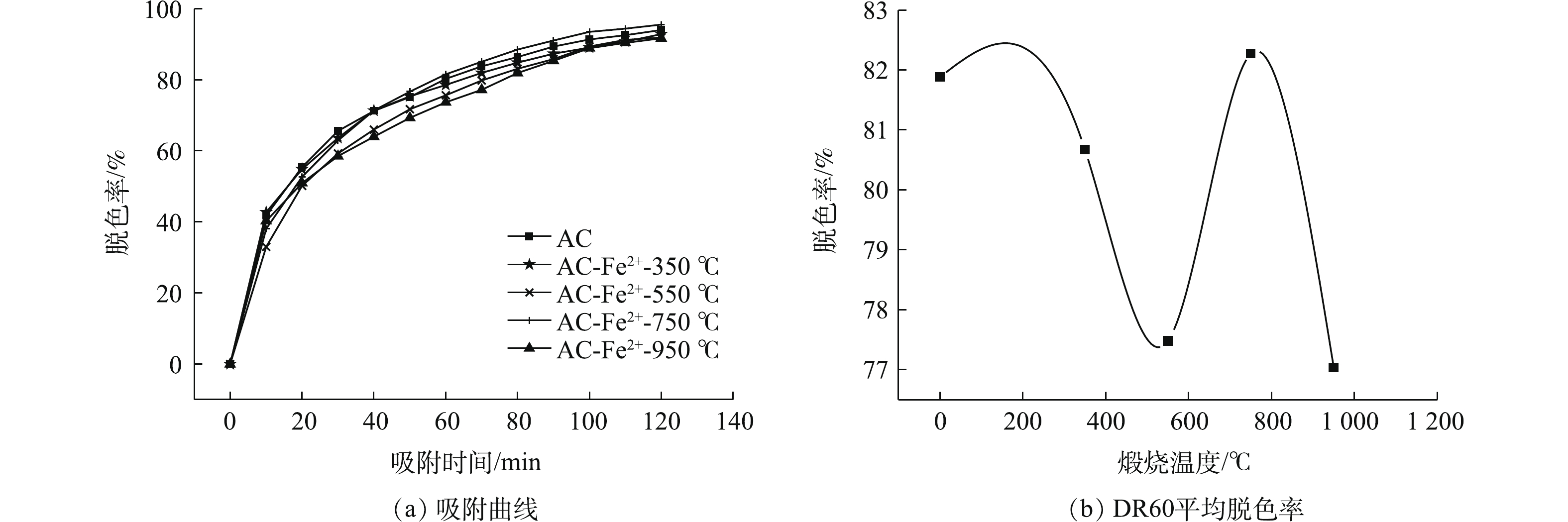 图 10 改性AC对DR60的吸附曲线及煅烧温度对平均脱色率的影响Figure 10. Adsorption curve of modified AC for DR60 and effect of calcination temperature on the average decolorization rate
图 10 改性AC对DR60的吸附曲线及煅烧温度对平均脱色率的影响Figure 10. Adsorption curve of modified AC for DR60 and effect of calcination temperature on the average decolorization rate2.3 响应面实验结果分析
在design expert 8.0中采用逐步回归的方法对表2中的实验结果进行方差分析,结果如表6所示。根据表中统计分析结果,可拟合得到平均脱色率与2个实验变量值之间的二次多项响应面方程,其响应面方程如式(10)所示。P≤0.01,回归方程显著,P>0.05,非正常误差不显著,这说明本次拟合得到的响应面方程可用。X1和X2的P值分别为0.306 5和0.210 7,这说明超声时间对AC平均脱色率的影响大于Fe2+浓度,
X21 和X22 项的P值均小于0.01,说明其对Y值,即AC平均脱色率的影响极显著。模型中不存在X1X2二次项,说明Fe2+浓度和超声时间之间不存在明显的交互影响。表 6 响应面法优化Fe2+改性活性炭方差分析结果Table 6. Variance analysis result of optimization of modified activated carbon by response surface methodology变异来源 平方和 自由度 均方 F P 模型 194.83 4 48.71 37.70 <0.000 1(显著) X1 1.54 1 1.54 1.19 0.306 5 X2 2.39 1 2.39 1.85 0.210 7 
105.26 1 105.26 81.48 <0.000 1 
110.53 1 110.53 85.56 <0.000 1 残差 10.33 8 1.29 失拟项 6.35 4 1.59 1.59 0.331 1(不显著) 纯误差 3.98 4 1.00 总值 205.17 12 | Show TableDownLoad:
CSV
Y=−4.01X21−4.11X22+0.44X1−0.55X2+94.49(R2=0.9496) (10) 式中:Y为120 min内的平均脱色率;X1为Fe2+浓度,mol·L−1;X2代表超声时间,min。
在Design expert 8.0中利用得到的响应面方程对Y值进行最优化,在Fe2+浓度为1.224 mol·L−1和超声时间为4.93 h时,AC的平均脱色率可达最大值,为94.273 7%。
为验证上述预测值的准确性,在该条件下,对AC进行3次重复改性,并将得到的改性样品用于BHET的脱色实验中,3个样品120 min内的平均脱色率值分别为95.40%、92.73%、92.32%,3次吸附实验平均脱色率的均值为93.48%,与预测值94.27%十分接近,证明该模型具有实际意义。
2.4 改性活性炭的吸附热力学与动力学
1)离子改性活性炭吸附热力学分析。图11为AC及AC-Fe2+-max的Langmuir和Freundlich等温线拟合曲线,表7为根据拟合方程计算所得的各项参数汇总。根据表中的数据可知,在90 ℃时,Langmuir等温方程对AC和AC-Fe2+-max的决定系数分别为0.990 5和0997 1,Freundlich等温方程的决定系数分别为0.900 1和0.946 9,这说明Langmuir等温方程可以更好地描述DR60在AC及AC-Fe2+-max上的吸附过程。Langmuir等温曲线表示吸附质在吸附剂表面有限的吸附位点上为单层吸附,吸附质在吸附剂表面平面内无迁移,这说明DR60在AC及AC-Fe2+-max上的吸附为均一单层分布,这与硝酸改性AC所得的热力学实验结果相同[31]。
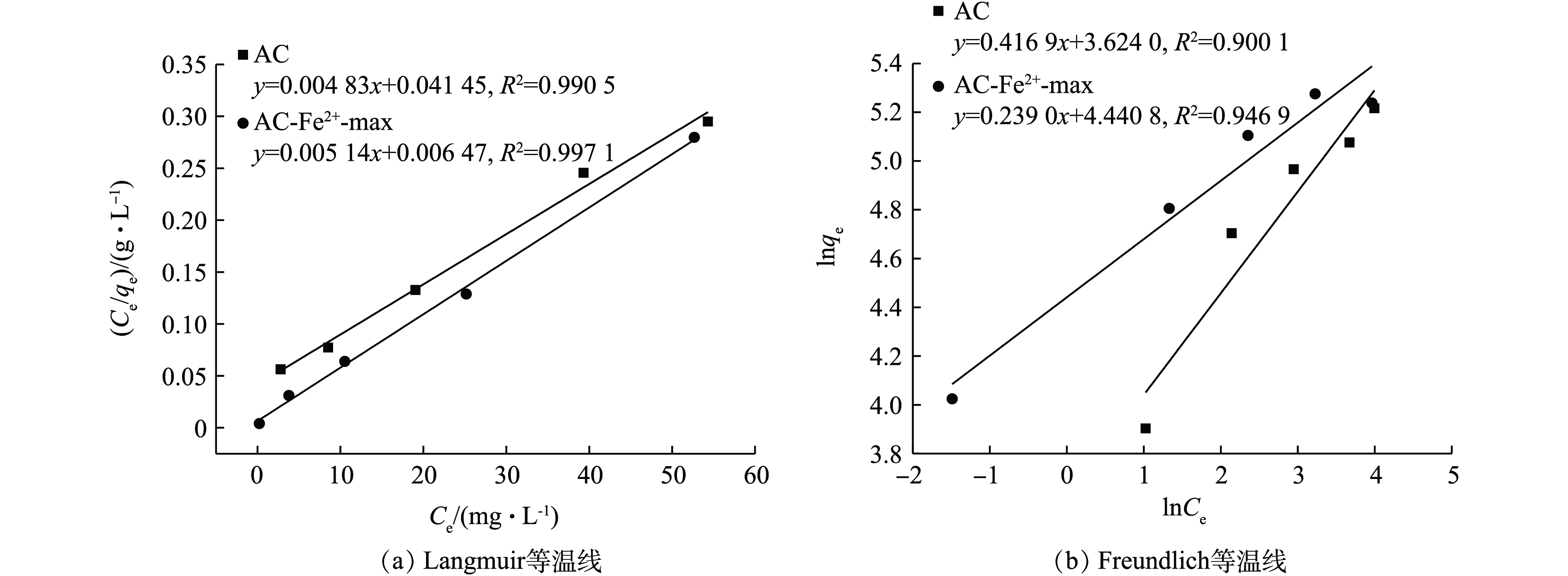 图 11 Langmuir和Freundlich等温线拟合Figure 11. Fitting liners of Langmuir and Freundlich models表 7 Langmuir模型和Freundlich模型拟合结果Table 7. Fitting results of Langmuir and Freundlich models
图 11 Langmuir和Freundlich等温线拟合Figure 11. Fitting liners of Langmuir and Freundlich models表 7 Langmuir模型和Freundlich模型拟合结果Table 7. Fitting results of Langmuir and Freundlich models样品 Langmuir Freundlich R2 Q/(mg·g−1) b/(L·mg−1) R2 n kf AC 0.990 5 208.333 3 0.115 7 0.900 1 0.416 9 39.026 5 AC-Fe2+-max 0.997 1 196.078 4 0.784 6 0.946 9 0.239 0 84.842 8 | Show TableDownLoad:
CSV
2)离子改性活性炭吸附动力学分析。图12为AC及AC-Fe2+-max的拟一级动力学和拟二级动力学曲线。如图12(a)所示,以ln(qe − qt)对t作出的曲线与一级动力学模型的前半段拟合性较好,后半段差别较大,对AC与AC-Fe2+-max,其决定系数R2分别为0.948 4,0.935 7。而以t/q对t所作出的曲线与其各自的二级动力学拟合曲线基本重合,t/q对t呈现出良好的线性关系,对AC与AC-Fe2+-max,其决定系数分别为0.999 3、0.999 7。这说明BHET中的DR60在AC与AC-Fe2+-max上的吸附过程更符合二级动力学方程。利用拟合方程的斜率和截距以计算表8中所示的动力学速率常数k1、k2及平衡吸附量qe1、qe2。从表8中可看出,通过二级动力学模型拟合曲线计算所得的qe值与实验所得的qe值十分接近。结合图12所示的结果,可以得出AC及AC-Fe2+-max对BHET中DR60的吸附均符合二级动力学模型,一些相关文献中也报道了相似的结果[31-32]。这表明,吸附过程中发生了化学反应[32],即在DR60吸附到AC及AC-Fe2+-max表面的过程中,发生了电子共享或电子交换的化学反应过程[31, 33]。
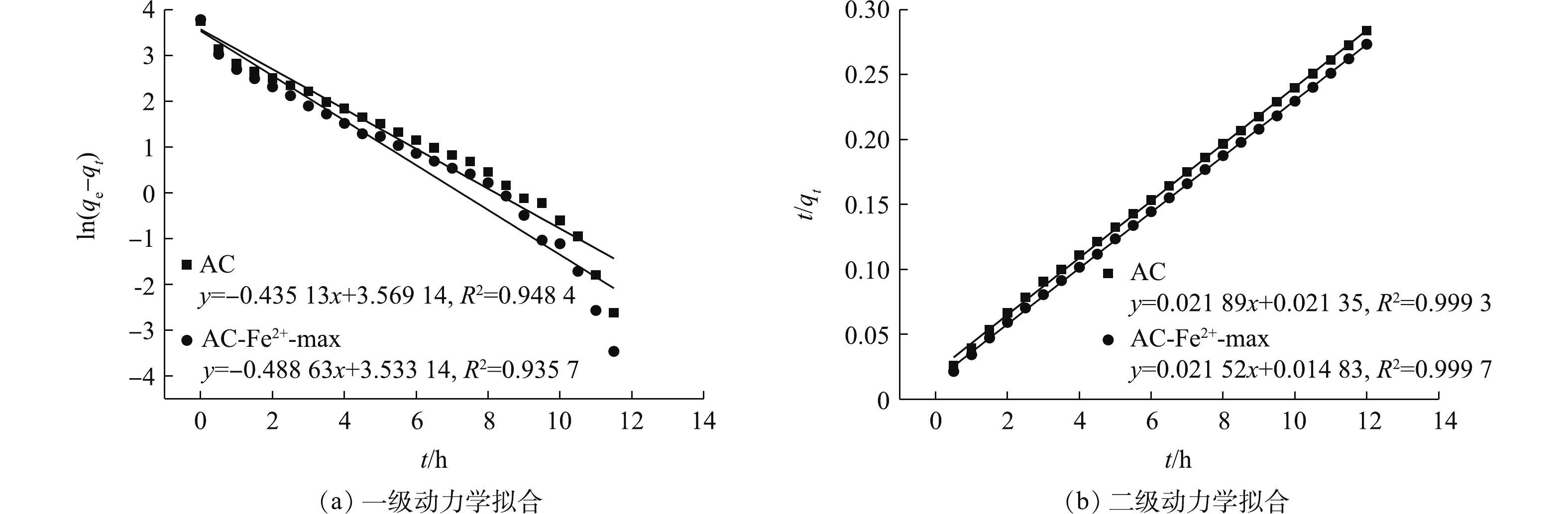 图 12 拟一级动力学曲线和拟二级动力学曲线Figure 12. Fitting liners of pseudo-first order and pseudo-secondary order kinetics表 8 拟一级动力学和拟二级动力学拟合结果Table 8. Fitting results of pseudo-first order and pseudo-secondary order kinetics
图 12 拟一级动力学曲线和拟二级动力学曲线Figure 12. Fitting liners of pseudo-first order and pseudo-secondary order kinetics表 8 拟一级动力学和拟二级动力学拟合结果Table 8. Fitting results of pseudo-first order and pseudo-secondary order kinetics样品 qe,exp/(mg·g−1) 拟一级动力学 拟二级动力学 k1/h−1 qe,cal/(mg·g−1) R2 Δq/% k2/(g·mg·h−1) qe,cal/(mg·g−1) R2 Δq/% AC 42.289 0.435 35.486 0.948 4 16.560 0.021 45.683 0.999 3 8.026 AC-Fe-max 43.907 0.489 34.231 0.935 7 22.037 0.031 46.468 0.999 7 5.833 | Show TableDownLoad:
CSV
根据表8中的结果,AC及AC-Fe2+-max对DR60根据二级动力学计算所得的吸附容量分别为45.683 mg·g−1及46.468 mg·g−1,二者对DR60的实际吸附容量分别为42.289 mg·g−1及43.907 mg·g−1。结合上文中脱色速率,AC-Fe2+-max对DR60平均脱色率可达93.48%,这远高于未改性AC的82.04%,同时其吸附容量也比未改性的AC高出约1.8 mg·g−1,故从吸附速率和吸附容量来看,AC-Fe2+-max的吸附性能都优于AC。
3. 结论
1) AC表面羧基及内酯基的含量会随着改性时FeSO4浓度的增加而增加,而AC中FeSO4的含量会随着超声时间的增加而增加;同时,改性AC的比表面积随着超声时间的增加呈现出先上升后下降的趋势,在超声4 h改性条件下其比表面积与未改性AC仅相差26 m2·g−1;高温煅烧会使得AC表面各种官能团在不同程度上分解,包括负载在AC表面的FeSO4,煅烧同时也会破坏AC的孔结构,使其比表面积等各项参数值下降。
2)在3组改性AC对BHET脱色实验中,120 min内平均脱色率随着改性浓度和超声时间的增加均呈现出先上升后下降的趋势,在AC-Fe2+-0.8 mol·L−1和AC-Fe2+-4 h条件下脱色率可达到最大值,且未煅烧的2组改性AC在120 min内平均脱色率均高于未改性AC,煅烧后改性AC的脱色性能下降到低于未改性AC的水平。
3)在Fe2+浓度为1.224 mol·L−1,超声时间为4.93 h,且无煅烧的条件下,改性AC对DR60的120 min内平均脱色率可达93.48%,可实现对BHET的完全脱色。
4) AC及AC-Fe2+-max的吸附热力学与吸附动力学实验结果表明,二者的吸附特征均更符合Langmuir吸附等温模型及二级动力学模型,DR60在AC及AC-Fe2+-max上的吸附为均一单层分布,吸附过程中包含化学反应,AC-Fe2+-max吸附容量相较于未改性AC提高了1.8 mg·g−1。
-
图 2 不同浓度Fe2+改性AC的N2吸附等温线及孔径分布图
Figure 2. Adsorption isotherms and pore size distribution of AC modified by Fe2+ with different concentrations
图 3 不同超声时间下改性AC的N2吸附等温线及孔径分布图
Figure 3. N2 adsorption isotherms and pore size distribution of modified AC at different ultrasonic time
图 4 不同煅烧温度下改性AC的N2吸附等温线及孔径分布图
Figure 4. N2 adsorption isotherms and pore size distribution of modified AC at different calcination temperatures
图 5 不同Fe2+浓度下改性AC的DTG曲线
Figure 5. DTG curves of modified ACs by Fe2+ at different concentrations
图 6 不同超声时间下改性AC的DTG曲线
Figure 6. DTG curves of modified ACs at different ultrasonic times
图 7 不同煅烧温度下改性AC的DTG曲线
Figure 7. DTG curves of modified ACs at different calcination temperatures
图 8 改性AC对DR60的吸附曲线及Fe2+浓度对平均脱色率的影响
Figure 8. Adsorption curve of modified AC for DR60 and effect of Fe2+ concentration on the average decolorization rate
图 9 改性AC对DR60的吸附曲线及超声时间对平均脱色率的影响
Figure 9. Adsorption curve of modified AC for DR60 and effect of ultrasonic time on the average decolorization rate
图 10 改性AC对DR60的吸附曲线及煅烧温度对平均脱色率的影响
Figure 10. Adsorption curve of modified AC for DR60 and effect of calcination temperature on the average decolorization rate
图 11 Langmuir和Freundlich等温线拟合
Figure 11. Fitting liners of Langmuir and Freundlich models
图 12 拟一级动力学曲线和拟二级动力学曲线
Figure 12. Fitting liners of pseudo-first order and pseudo-secondary order kinetics
表 1 活性炭改性实验条件
Table 1. Modification conditions of AC
实验组 Fe2+浓度/(mol·L−1) 超声时间/h 煅烧温度/℃ 1 0.2、0.4、0.8、1.6 0.5 无煅烧 2 0.8 1、2、4、8 无煅烧 3 0.8 4 350、550、750、950
下载: 导出CSV
表 2 响应面实验设计及实验结果汇总
Table 2. Design and results of response surface experiment
实验编号 编码(实验)水平 120 min平均脱色率/% 离子浓度/(mol·L−1) 超声时间/h 1 −1(0.8) −1(4) 87.318 6 2 −1(0.8) 1(6) 84.916 0 3 1(1.6) −1(4) 88.316 8 4 1(1.6) 1(6) 87.542 1 5 0(1.2) −1.41(3.59) 86.032 3 6 0(1.2) 1.41(6.41) 85.190 2 7 −1.41(0.636) 0(5) 85.845 3 8 1.41(1.764) 0(5) 85.765 6 9 0(1.2) 0(5) 95.429 6 10 0(1.2) 0(5) 94.546 4 11 0(1.2) 0(5) 93.686 2 12 0(1.2) 0(5) 92.843 5 13 0(1.2) 0(5) 94.705 7
下载: 导出CSV
表 3 不同浓度Fe2+离子改性AC孔径结构数据
Table 3. Data of pore structure of ACs modified by Fe2+ with different concentrations
样品 比表面积/(m2·g−1) 微孔表面积/(m2·g−1) 中孔表面积/(m2·g−1) 总孔容/(cm2·g−1) 微孔孔容/(cm2·g−1) AC 1 255.144 9 620.843 4 634.301 5 0.392 7 0.312 2 AC-Fe2+-0.2 mol·L−1 1 152.140 9 552.206 6 599.934 2 0.379 6 0.290 4 AC-Fe2+-0.4 mol·L−1 1 113.631 7 527.849 6 585.728 1 0.363 0 0.275 9 AC-Fe2+-0.8 mol·L−1 1 132.991 7 540.272 5 592.719 2 0.368 2 0.282 0 AC-Fe2+-1.6 mol·L−1 1 166.269 5 551.380 0 614.889 5 0.374 7 0.287 6
下载: 导出CSV
表 4 不同超声时间下Fe2+改性AC孔径结构数据
Table 4. Data of pore structure of modified ACs at different ultrasonic time
样品 比表面积/(m2·g−1) 微孔表面积/(m2·g−1) 中孔表面积/(m2·g−1) 总孔容/(cm2·g−1) 微孔孔容/(cm2·g−1) AC 1 255.144 9 620.843 4 634.301 5 0.392 7 0.312 2 AC-Fe2+-1 h 1 143.085 0 551.982 8 591.102 2 0.372 0 0.290 2 AC-Fe2+-2 h 1 178.305 9 587.138 6 591.167 3 0.361 2 0.294 5 AC-Fe2+-4 h 1 229.154 8 583.239 3 645.915 5 0.397 1 0.304 9 AC-Fe2+-8 h 1 163.558 3 556.831 4 606.726 9 0.385 0 0.292 7
下载: 导出CSV
表 5 不同煅烧温度Fe2+改性AC孔径结构数据
Table 5. Data of pore structure of modified ACs at different calcination temperatures
样品 比表面积/(m2·g−1) 微孔表面积/(m2·g−1) 中孔表面积/(m2·g−1) 总孔容/(cm2·g−1) 微孔孔容/(cm2·g−1) AC 1 255.144 9 620.843 4 634.301 5 0.392 7 0.312 2 AC-Fe2+-350 ℃ 1 134.926 7 540.431 2 594.495 5 0.373 6 0.285 3 AC-Fe2+-550 ℃ 1 047.606 2 525.750 4 521.855 9 0.320 3 0.274 6 AC-Fe2+-750 ℃ 1 210.382 9 576.059 4 634.323 4 0.392 9 0.301 0 AC-Fe2+-950 ℃ 920.449 4 507.856 8 412.592 6 0.250 9 0.265 4
下载: 导出CSV
表 6 响应面法优化Fe2+改性活性炭方差分析结果
Table 6. Variance analysis result of optimization of modified activated carbon by response surface methodology
变异来源 平方和 自由度 均方 F P 模型 194.83 4 48.71 37.70 <0.000 1(显著) X1 1.54 1 1.54 1.19 0.306 5 X2 2.39 1 2.39 1.85 0.210 7 X21 </td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><i>X</i><sub>1</sub></td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.54</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.54</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.19</td> <td align="center" valign="middle" style="padding-top:1.5pt;">0.306 5</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><i>X</i><sub>2</sub></td> <td align="center" valign="middle" style="padding-top:1.5pt;">2.39</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">2.39</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.85</td> <td align="center" valign="middle" style="padding-top:1.5pt;">0.210 7</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><inline-formula>$X_1^2$<img class="inline-formula" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead> <tr> <td class="table_top_border" align="center" valign="middle">变异来源</td> <td class="table_top_border" align="center" valign="middle">平方和</td> <td class="table_top_border" align="center" valign="middle">自由度</td> <td class="table_top_border" align="center" valign="middle">均方</td> <td class="table_top_border" align="center" valign="middle"><i>F</i></td> <td class="table_top_border" align="center" valign="middle"><i>P</i></td> </tr> </thead>
<tbody> <tr> <td class="table_top_border2" align="center" valign="middle">模型</td> <td class="table_top_border2" align="center" valign="middle">194.83</td> <td class="table_top_border2" align="center" valign="middle">4</td> <td class="table_top_border2" align="center" valign="middle">48.71</td> <td class="table_top_border2" align="center" valign="middle">37.70</td> <td class="table_top_border2" align="center" valign="middle">&lt;0.000 1(显著)</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><i>X</i><sub>1</sub></td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.54</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.54</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.19</td> <td align="center" valign="middle" style="padding-top:1.5pt;">0.306 5</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><i>X</i><sub>2</sub></td> <td align="center" valign="middle" style="padding-top:1.5pt;">2.39</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">2.39</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.85</td> <td align="center" valign="middle" style="padding-top:1.5pt;">0.210 7</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><inline-formula>$X_1^2$<alternatives><img class="graphic" src="201811127_M3.jpg"><img class="graphic" src="201811127_M3.png"></alternatives></inline-formula></td> <td align="center" valign="middle" style="padding-top:1.5pt;">105.26</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">105.26</td> <td align="center" valign="middle" style="padding-top:1.5pt;">81.48</td> <td align="center" valign="middle" style="padding-top:1.5pt;">&lt;0.000 1</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><inline-formula>$ X_2^2 $<alternatives><img class="graphic" src="201811127_M4.jpg"><img class="graphic" src="201811127_M4.png"></alternatives></inline-formula></td> <td align="center" valign="middle" style="padding-top:1.5pt;">110.53</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">110.53</td> <td align="center" valign="middle" style="padding-top:1.5pt;">85.56</td> <td align="center" valign="middle" style="padding-top:1.5pt;">&lt;0.000 1</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;">残差</td> <td align="center" valign="middle" style="padding-top:1.5pt;">10.33</td> <td align="center" valign="middle" style="padding-top:1.5pt;">8</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.29</td> <td align="center" valign="middle"> </td><td align="center" valign="middle"> </td></tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;">失拟项</td> <td align="center" valign="middle" style="padding-top:1.5pt;">6.35</td> <td align="center" valign="middle" style="padding-top:1.5pt;">4</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.59</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.59</td> <td align="center" valign="middle" style="padding-top:1.5pt;">0.331 1(不显著)</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;">纯误差</td> <td align="center" valign="middle" style="padding-top:1.5pt;">3.98</td> <td align="center" valign="middle" style="padding-top:1.5pt;">4</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.00</td> <td align="center" valign="middle"> </td><td align="center" valign="middle"> </td></tr> <tr> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle">总值</td> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle">205.17</td> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle">12</td> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle"> </td><td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle"> </td><td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle"> </td></tr> </tbody>
</table></div></foreignObject></svg>"></inline-formula></td> <td align="center" valign="middle" style="padding-top:1.5pt;">105.26</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">105.26</td> <td align="center" valign="middle" style="padding-top:1.5pt;">81.48</td> <td align="center" valign="middle" style="padding-top:1.5pt;"><0.000 1</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><inline-formula>$ X_2^2 $<img class="inline-formula" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead> <tr> <td class="table_top_border" align="center" valign="middle">变异来源</td> <td class="table_top_border" align="center" valign="middle">平方和</td> <td class="table_top_border" align="center" valign="middle">自由度</td> <td class="table_top_border" align="center" valign="middle">均方</td> <td class="table_top_border" align="center" valign="middle"><i>F</i></td> <td class="table_top_border" align="center" valign="middle"><i>P</i></td> </tr> </thead>
<tbody> <tr> <td class="table_top_border2" align="center" valign="middle">模型</td> <td class="table_top_border2" align="center" valign="middle">194.83</td> <td class="table_top_border2" align="center" valign="middle">4</td> <td class="table_top_border2" align="center" valign="middle">48.71</td> <td class="table_top_border2" align="center" valign="middle">37.70</td> <td class="table_top_border2" align="center" valign="middle">&lt;0.000 1(显著)</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><i>X</i><sub>1</sub></td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.54</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.54</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.19</td> <td align="center" valign="middle" style="padding-top:1.5pt;">0.306 5</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><i>X</i><sub>2</sub></td> <td align="center" valign="middle" style="padding-top:1.5pt;">2.39</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">2.39</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.85</td> <td align="center" valign="middle" style="padding-top:1.5pt;">0.210 7</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><inline-formula>$X_1^2$<alternatives><img class="graphic" src="201811127_M3.jpg"><img class="graphic" src="201811127_M3.png"></alternatives></inline-formula></td> <td align="center" valign="middle" style="padding-top:1.5pt;">105.26</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">105.26</td> <td align="center" valign="middle" style="padding-top:1.5pt;">81.48</td> <td align="center" valign="middle" style="padding-top:1.5pt;">&lt;0.000 1</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;"><inline-formula>$ X_2^2 $<alternatives><img class="graphic" src="201811127_M4.jpg"><img class="graphic" src="201811127_M4.png"></alternatives></inline-formula></td> <td align="center" valign="middle" style="padding-top:1.5pt;">110.53</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">110.53</td> <td align="center" valign="middle" style="padding-top:1.5pt;">85.56</td> <td align="center" valign="middle" style="padding-top:1.5pt;">&lt;0.000 1</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;">残差</td> <td align="center" valign="middle" style="padding-top:1.5pt;">10.33</td> <td align="center" valign="middle" style="padding-top:1.5pt;">8</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.29</td> <td align="center" valign="middle"> </td><td align="center" valign="middle"> </td></tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;">失拟项</td> <td align="center" valign="middle" style="padding-top:1.5pt;">6.35</td> <td align="center" valign="middle" style="padding-top:1.5pt;">4</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.59</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.59</td> <td align="center" valign="middle" style="padding-top:1.5pt;">0.331 1(不显著)</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;">纯误差</td> <td align="center" valign="middle" style="padding-top:1.5pt;">3.98</td> <td align="center" valign="middle" style="padding-top:1.5pt;">4</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.00</td> <td align="center" valign="middle"> </td><td align="center" valign="middle"> </td></tr> <tr> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle">总值</td> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle">205.17</td> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle">12</td> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle"> </td><td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle"> </td><td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle"> </td></tr> </tbody>
</table></div></foreignObject></svg>"></inline-formula></td> <td align="center" valign="middle" style="padding-top:1.5pt;">110.53</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1</td> <td align="center" valign="middle" style="padding-top:1.5pt;">110.53</td> <td align="center" valign="middle" style="padding-top:1.5pt;">85.56</td> <td align="center" valign="middle" style="padding-top:1.5pt;"><0.000 1</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;">残差</td> <td align="center" valign="middle" style="padding-top:1.5pt;">10.33</td> <td align="center" valign="middle" style="padding-top:1.5pt;">8</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.29</td> <td align="center" valign="middle"> </td><td align="center" valign="middle"> </td></tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;">失拟项</td> <td align="center" valign="middle" style="padding-top:1.5pt;">6.35</td> <td align="center" valign="middle" style="padding-top:1.5pt;">4</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.59</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.59</td> <td align="center" valign="middle" style="padding-top:1.5pt;">0.331 1(不显著)</td> </tr> <tr> <td align="center" valign="middle" style="padding-top:1.5pt;">纯误差</td> <td align="center" valign="middle" style="padding-top:1.5pt;">3.98</td> <td align="center" valign="middle" style="padding-top:1.5pt;">4</td> <td align="center" valign="middle" style="padding-top:1.5pt;">1.00</td> <td align="center" valign="middle"> </td><td align="center" valign="middle"> </td></tr> <tr> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle">总值</td> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle">205.17</td> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle">12</td> <td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle"> </td><td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle"> </td><td class="table_bottom_border" style="padding-top:1.5pt;" align="center" valign="middle"> </td></tr> </tbody>
</table></div></foreignObject></svg>)
105.26 1 105.26 81.48 <0.000 1 X22 110.53 1 110.53 85.56 <0.000 1 残差 10.33 8 1.29 失拟项 6.35 4 1.59 1.59 0.331 1(不显著) 纯误差 3.98 4 1.00 总值 205.17 12
下载: 导出CSV
表 7 Langmuir模型和Freundlich模型拟合结果
Table 7. Fitting results of Langmuir and Freundlich models
样品 Langmuir Freundlich R2 Q/(mg·g−1) b/(L·mg−1) R2 n kf AC 0.990 5 208.333 3 0.115 7 0.900 1 0.416 9 39.026 5 AC-Fe2+-max 0.997 1 196.078 4 0.784 6 0.946 9 0.239 0 84.842 8
下载: 导出CSV
表 8 拟一级动力学和拟二级动力学拟合结果
Table 8. Fitting results of pseudo-first order and pseudo-secondary order kinetics
样品 qe,exp/(mg·g−1) 拟一级动力学 拟二级动力学 k1/h−1 qe,cal/(mg·g−1) R2 Δq/% k2/(g·mg·h−1) qe,cal/(mg·g−1) R2 Δq/% AC 42.289 0.435 35.486 0.948 4 16.560 0.021 45.683 0.999 3 8.026 AC-Fe-max 43.907 0.489 34.231 0.935 7 22.037 0.031 46.468 0.999 7 5.833
下载: 导出CSV
-
[1] DUQUE-INGUNZA I, LOPEZ-FONSECA R, RIVAS B D, et al. Process optimization for catalytic glycolysis of post-consumer pet wastes[J]. Journal of Chemical Technology & Biotechnology, 2014, 89(1): 97-103. [2] SHUKLA S R, HARAD A M, JAWALE L S. Chemical recycling of pet waste into hydrophobic textile dyestuffs[J]. Polymer Degradation & Stability, 2009, 94(4): 604-609. [3] WELLE F. Twenty years of pet bottle to bottle recycling: An overview[J]. Resources Conservation & Recycling, 2011, 55(11): 865-875. [4] SHUKLA S R, HARAD A M, JAWALE L S. Recycling of waste pet into useful textile auxiliaries[J]. Waste Management, 2008, 28(1): 51-56. doi: 10.1016/j.wasman.2006.11.002 [5] KACPERSKI M, SPYCHAJ T. Rigid polyurethane foams with poly(ethylene terephthalate)/triethanolamine recycling products[J]. Polymers for Advanced Technologies, 2015, 10(10): 620-624. [6] DULLIUS J, RUECKER C, OLIVEIRA V, et al. Chemical recycling of post-consumer pet: Alkyd resins synthesis[J]. Progress in Organic Coatings, 2006, 57(2): 123-127. doi: 10.1016/j.porgcoat.2006.07.004 [7] PHUANGNGAMPHAN P, THONGPIN C. Synthesize of polydiacetylene containing copolyurethane using polyol product derived from chemical recycling of pet bottles[J]. Energy Procedia, 2014, 56: 326-333. [8] XI G, LU M, SUN C. Study on depolymerization of waste polyethylene terephthalate into monomer of bis(2-hydroxyethyl terephthalate)[J]. Polymer Degradation & Stability, 2005, 87(1): 117-120. [9] LI M J, HUANG Y H, JU A Q, et al. Synthesis and characterization of azo dyestuff based on bis(2-hydroxyethyl) terephthalate derived from depolymerized waste poly(ethylene terephthalate) fibers[J]. Chinese Chemical Letters, 2014, 25(12): 1550-1554. doi: 10.1016/j.cclet.2014.09.022 [10] LI M J, HUANG Y H, YU T S, et al. Chemical recycling of waste poly(ethylene terephthalate) fibers into azo disperse dyestuffs[J]. RSC Advances, 2014, 87(4): 46476-46480. doi: 10.1039/C4RA07608G [11] LI M J, LUO J, HUANG Y H, et al. Recycling of waste poly(ethylene terephthalate) into flame-retardant rigid polyurethane foams[J]. Journal of Applied Polymer Science, 2014, 131(19): 5829-5836. [12] LU J, LI M J, LI Y Y, et al. Synthesis and sizing performances of water-soluble polyester based on bis(2-hydroxyethyl) terephthalate derived from depolymerized waste poly(ethylene terephthalate) fabrics[J]. Textile Research Journal, 2019, 89(4): 572-579. [13] ELASS K, LAACHACH A, ALAOUI A, et al. Removal of methyl violet from aqueous solution using a stevensite-rich clay from morocco[J]. Applied Clay Science, 2011, 54(1): 90-96. doi: 10.1016/j.clay.2011.07.019 [14] KARAGOZOGLU B, TASDEMIR M, DEMIRBAS E, et al. The adsorption of basic dye (astrazon blue fgrl) from aqueous solutions onto sepiolite, fly ash and apricot shell activated carbon: kinetic and equilibrium studies[J]. Journal of Hazardous Materials, 2007, 147(1/2): 297-306. [15] AHMAD F, WAN M A W D, AHMAD M A, et al. Cocoa (theobroma cacao) shell-based activated carbon by CO2, activation in removing of cationic dye from aqueous solution: kinetics and equilibrium studies[J]. Chemical Engineering Research & Design, 2012, 90(10): 1480-1490. [16] MOHAMADI N, KHANI H, GUPTA V K, et al. Adsorption process of methyl orange dye onto mesoporous carbon material-kinetic and thermodynamic studies[J]. Journal of Colloid & Interface Science, 2011, 362(2): 457-642. [17] CHIOU M S, CHUANG G S. Competitive adsorption of dye metanil yellow and RB15 in acid solutions on chemically cross-linked chitosan beads[J]. Chemosphere, 2006, 62(5): 731-740. doi: 10.1016/j.chemosphere.2005.04.068 [18] JIANG Z, YAN L, SUN X, et al. Activated carbons chemically modified by concentrated H2SO4 for the adsorption of the pollutants from wastewater and the dibenzothiophene from fuel oils[J]. Langmuir, 2003, 19(3): 731-736. doi: 10.1021/la020670d [19] WANG S, ZHU Z H, COOMES A, et al. The physical and surface chemical characteristics of activated carbons and the adsorption of methylene blue from wastewater[J]. Journal of Colloid & Inion on the Structure Aterface Science, 2005, 284(2): 440-446. [20] CHAN L S, CHEUNG W H, ALLEN S J, et al. Separation of acid-dyes mixture by bamboo derived active carbon[J]. Separation & Purification Technology, 2009, 67(2): 166-172. [21] NJOKU V O, FOO K Y, HAMEED B H. Microwave-assisted preparation of pumpkin seed hull activated carbon and its application for the adsorptive removal of 2,4-dichlorophenoxyacetic acid[J]. Chemical Engineering Journal, 2013, 215(3): 383-388. [22] CHAN L S, CHEUNG W H, MCKAY G. Adsorption of acid dyes by bamboo derived activated carbon[J]. Desalination, 2008, 218(1): 304-312. [23] GOKCE Y, AKTAS Z. Nitric acid modification of activated carbon produced from waste tea and adsorption of methylene blue and phenol[J]. Applied Surface Science, 2014, 313: 352-359. doi: 10.1016/j.apsusc.2014.05.214 [24] BHATNAGAR A, HOGLAND W, MARQUES M, et al. An overview of the modification methods of activated carbon for its water treatment applications[J]. Chemical Engineering Journal, 2013, 219(3): 499-511. [25] CAO Y, GU Y, WANG K, et al. Adsorption of creatinine on active carbons with nitric acid hydrothermal modification[J]. Journal of the Taiwan Institute of Chemical Engineers, 2016, 66: 347-356. doi: 10.1016/j.jtice.2016.06.008 [26] SING K S W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (recommendations 1984)[J]. Pure and Applied Chemistry, 1985, 57(4): 603-619. doi: 10.1351/pac198557040603 [27] FIGUEIREDO J L, PEREIRA M F R, FREITAS M M A, et al. Modification of the surface chemistry of activated carbons[J]. Carbon, 1999, 37(9): 1379-1389. doi: 10.1016/S0008-6223(98)00333-9 [28] YU C, FAN X, YU L, et al. Adsorptive removal of thiophenic compounds from oils by activated carbon modified with concentrated nitric acid[J]. Energy & Fuels, 2013, 27(3): 1499-1505. [29] ZIELKE U, HUTTINGER K J, HOFFMAN W P. Surface-oxidized carbon fibers: Surface structure and chemistry[J]. Carbon, 1996, 34(8): 983-998. doi: 10.1016/0008-6223(96)00032-2 [30] EL-HENDAWY A N A. Influence of HNO3 oxidation on the structure and adsorptive properties of corncob-based activated carbon[J]. Carbon, 2003, 41(4): 713-722. doi: 10.1016/S0008-6223(03)00029-0 [31] DJILANI C, ZAGHDOUDI R, DJAZI F, et al. Adsorption of dyes on activated carbon prepared from apricot stones and commercial activated carbon[J]. Journal of the Taiwan Institute of Chemical Engineers, 2015, 53: 112. doi: 10.1016/j.jtice.2015.02.025 [32] BRUNAUER S, DEMING L S, DEMING W E, et al. On a theory of the van der wals adsorption of gases[J]. Journal of the American Chemical Society, 1940, 62(7): 1723-1732. doi: 10.1021/ja01864a025 [33] FOO K Y, HAMEED B H. Microwave-assisted preparation and adsorption performance of activated carbon from biodiesel industry solid reside: influence of operational parameters[J]. Bioresource Technology, 2012, 103(1): 398-404. doi: 10.1016/j.biortech.2011.09.116 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2749
- HTML全文浏览数: 2749
- PDF下载数: 49
- 施引文献: 0
