

-
国内外的饮用水中均检测出医药品污染物,其浓度通常处于ng·L−1或更低水平. 风险评价研究显示,饮用水中微量的医药品污染物对于健康的成年人影响较小,但是敏感性人群特别是孕妇及新生儿暴露于环境中的风险需要加以考虑[1]. 布洛芬(IBU)属于抗炎药物,据估计全球年产量达数千吨,其在地表水和废水中检测到的浓度范围为ng·L−1至低μg·L−1水平[2];传统的水处理技术无法对持久性污染物IBU有效去除,研究发现IBU能对淡水环境中的无尾类动物的胚胎发育产生影响[3]. 非均相催化臭氧氧化技术是一种能够在室温和常压下,将那些难以通过臭氧单独氧化的有机污染物高效分解的方法[4-5]. 臭氧在催化剂表面有效分解产生羟基自由基等活性氧来降解水中有机污染物[6-7]. 由于不同催化剂的属性差异较大,其促进臭氧有效分解的机理还不清楚,需要深入研究催化臭氧氧化水中有机污染物的作用机制[8-9].
本文采用固体酸γ-Al2O3和多价态的钛活性组份负载法制备了TiO2/γ-Al2O3介孔催化剂. 由于TiO2的Lewis酸性强于Al2O3,通过调节钛的负载量来控制γ-Al2O3的酸量,从而更深入的研究Lewis酸性位对布洛芬去除的影响,并通过吡啶红外光谱、电子自旋共振和原位激光显微拉曼光谱等测试手段来探究催化反应机理.
-
布洛芬(IBU)从东京化成工业株式会社购买. 自由基捕捉剂BMPO从东仁化学科技(上海)有限公司购买. 实验用超纯水(电阻率为18.2 MΩ·cm)是用Milli-Q超纯水仪制备的. 异丙醇铝(Al[OCH(CH3)2]3)购买于Sigma-Aldrich公司. 葡萄糖、钛酸四正丁酯(TBOT)、盐酸(HCl)、硝酸(HNO3)和氢氧化钠(NaOH)在国药集团化学试剂北京有限公司采购. 全部化学试剂均是分析纯. 溶液pH由2 mol·L−1盐酸或者氢氧化钠溶液进行调节.
-
γ-Al2O3制备[10]. 称取16.8 g异丙醇铝和14.4 g葡萄糖溶于216 mL水中,在35 ℃恒温条件下搅拌6 h,用硝酸溶液(质量分数为10%)将pH调到5.5,继续搅拌24 h,将其在100 ℃下烘干,然后用马弗炉在600 ℃下煅烧6 h得到γ-Al2O3.
TiO2/γ-Al2O3制备. 称取6 g γ-Al2O3和适量TBOT(Al/Ti = 75)溶于200 mL水中,在35 ℃条件下搅拌24 h后在100 ℃烘干,然后用马弗炉600 ℃煅烧6 h得到TiO2/γ-Al2O3. 经前期实验优化,当Al/Ti = 75时,TiO2/γ-Al2O3催化臭氧氧化水中布洛芬具有最佳催化活性.
-
催化剂晶相结构由Scintag-XDS-2000型X射线衍射仪(XRD)测试分析,用Cu Kα(λ = 0.154 nm)辐射为激发源,操作电压为40 kV,操作电流为100 mA. 催化剂的比表面积、孔径和孔容采用美国Micromeritics公司的ASAP-2020比表面积仪测定. 孔隙度是基于氮气吸附-解吸等温线及Barrett-Joyner-Halenda(BJH)法获得的. 利用日本Hitachi公司 SU8020 FESEM电子扫描电镜对催化剂形貌和结构进行了观察. 用马尔文Nano-ZS90型Zeta电位计测定了催化剂的表面零电荷点(pHpzc). 利用Nicolet 6700红外光谱(Pyridine-FTIR)对吡啶吸附的催化剂酸性位进行了表征. 具体方法:压片后的催化剂放入红外的样品池中,将样品在常温真空处理30 min,获得样品背景谱图,该仪器的分辨率为4 cm−1. 再分别于20 ℃,150 ℃和350 ℃解吸得到样品的吡啶红外谱图. 因红外谱图峰高正比于催化剂的酸量. 依此,可对催化剂的表面酸性作定性或半定量的分析[11].
-
催化臭氧氧化试验是在一个1.2 L鼓泡式玻璃反应器内进行的. 利用高纯度氧气作为气源,通过臭氧发生器(购于北京同林科技有限公司)进行放电生成臭氧,流量计测量后送入玻璃反应器底部微孔砂心布气头,经过磁力搅拌器搅拌后臭氧可均匀的分布于水中. 具体实验参数:在20 ℃条件下,将1 L浓度为10 mg·L−1的布洛芬水溶液为模型化合物和前期优化确定的1.5 g催化剂加入到反应器中,利用盐酸或氢氧化钠调节pH到7.0,然后通入臭氧浓度为30 mg·L−1的O3/O2混合气体,气体的流速为200 mL·min−1. 反应后残余的臭氧用碘化钾溶液吸收. 每隔一段时间采样1次,所取样品立即加几滴0.1 mol·L−1硫代硫酸钠以终止溶液中残余臭氧,然后用0.45 μm醋酸纤维滤膜滤出样品待测.
-
水溶液中布洛芬的浓度采用高效液相色谱(1200 series;Agilent,Santa Clara,CA)测定,色谱柱为Eclipse XDB-C18 column(5 μm,4.6 mm×150 mm;Agilent). 分析条件为:流动相为60:40(V/V)乙腈:磷酸盐缓冲液(20 mmol·L−1,pH = 2.5)溶液,柱温40 ℃,进样量20 μL,流量为1 mL·min−1,紫外检测波长220 nm. 总有机碳分析(TOC)采用岛津TOC-VCPH型总有机碳分析仪测定. 利用电感耦合等离子体原子发射光谱分析(ICP-OES)分析溶液中金属离子浓度. 气相臭氧浓度采用IDEAL-2000型臭氧浓度检测仪进行检测. 用靛蓝法测定了水中溶解臭氧的浓度[12]. 利用BMPO作为自由基捕捉获剂,利用德国布鲁克公司ESP300E型电子顺磁共振波谱仪(EPR)对反应溶液中的羟基自由基及超氧自由基进行直接测定,微波频率为9.79 GHz,功率为5.05 mW. 原位激光显微拉曼光谱分析利用Lab RAM HR Evolution型共聚焦激光显微拉曼光谱仪进行测定. 激光激发波长633 nm、分辨率为1 cm−1、持续时间为100 s. 样品制备方法:将0.1 g催化剂与2 mL超纯水或者饱和臭氧水摇匀后,快速滴入载玻片,调整视野开始扫描测定.
-
图1是γ-Al2O3和TiO2/γ-Al2O3的小角和广角XRD图谱. 在小角范围,γ-Al2O3和TiO2/γ-Al2O3在1o附近都显示出一个很强的衍射峰,说明这两种催化剂都具有介孔结构;在广角范围,Al2O3显示出典型的γ相(JCPD no. 00-046-1131)[10, 13]. TiO2/γ-Al2O3样品除了出现γ-Al2O3的特征峰,同时在25.3o出现强的TiO2的特征峰,说明TiO2负载于γ-Al2O3表面.
γ-Al2O3和TiO2/γ-Al2O3的扫描电镜照片见图2,γ-Al2O3显示出海绵结构,而对于TiO2/γ-Al2O3大的TiO2颗粒出现在γ-Al2O3表面,证实了TiO2负载于γ-Al2O3表面.
图3是γ-Al2O3和TiO2/γ-Al2O3的氮气吸附脱附等温线和孔径、孔容分布图. 所有样品都符合第Ⅳ类吸附等温线特征,说明这两种催化剂都有介孔结构,都表现出集中的孔径分布. 另外从表1可以看出,与γ-Al2O3相比,TiO2/γ-Al2O3的比表面积、孔径、孔容下降,这是由于TiO2负载于γ-Al2O3表面.
利用吡啶-红外光谱技术,对γ-Al2O3和TiO2/γ-Al2O3催化剂表面的酸性位进行了分析. 图4为催化剂在20 ℃、150 ℃和350 ℃脱附的吡啶-红外光谱. 在1200—1800 cm−1范围的吡啶-红外光谱代表吡啶环υ(C—N)和υ(C—C)振动[14]. 在20 ℃条件下脱附,γ-Al2O3和TiO2/γ-Al2O3催化剂在1221、1308、1448、1492、1578、1595、1614 cm−1位置都有吸收,TiO2/γ-Al2O3具有比γ-Al2O3更强的吸收强度. 吡啶-红外光谱在1221、1308、1448、1492、1614 cm−1处的吸收峰,代表吡啶被催化剂Lewis酸性位所吸附[15]. 150 ℃解吸时,1595 cm−1处的吸收峰消失表明吡啶和催化剂表面之间的弱相互作用,代表吡啶在催化剂表面上吸附氢键的位置. 350 ℃脱附过程中,1578 cm−1吸收峰逐渐消失,表明吡啶在中强的Lewis酸性位上发生了吸附. 随脱附温度升高,吡啶的吸收峰迁移到较高频位,表明吡啶在强Lewis酸性位发生了吸附. 基于不同温度条件下催化剂脱附曲线,进一步定量分析了γ-Al2O3和TiO2/γ-Al2O3表面的Lewis酸量[11]. 从表2可见,TiO2/γ-Al2O3与γ-Al2O3相比Lewis酸量略有增加. 这些结果说明钛负载于γ-Al2O3表面能够增大γ-Al2O3的Lewis酸量.
-
图5是不同工艺对布洛芬的催化性能. 由图5可知,TiO2/γ-Al2O3/O2、γ-Al2O3/O2和TiO2/O2过程对布洛芬的吸附去除率分别为6%、4%和2%;催化臭氧氧化比单独臭氧氧化更有利于布洛芬降解;TiO2/γ-Al2O3/O3工艺具有最高的催化活性. 反应30 min时,不同工艺过程均将布洛芬完全降解,催化臭氧氧化过程可极大提高布洛芬的矿化率. 反应60 min,TiO2/γ-Al2O3对布洛芬的TOC去除率达到80%,γ-Al2O3、TiO2及单独臭氧氧化过程中TOC去除率分别达62%、32%及26%. 由于布洛芬在TiO2/γ-Al2O3表面的吸附去除率只有6%,表明TiO2/γ-Al2O3与臭氧之间具有良好的协同作用. 通过对比表2和图5,可以发现催化剂表面的Lewis酸量与其催化活性呈正相关,TiO2/γ-Al2O3最大Lewis酸量使其具备最高的催化活性.
-
催化臭氧氧化过程,催化剂的表面属性与活性位共同决定了臭氧的吸附与高效分解. 文献报道臭氧可以在Lewis酸性位替代羟基基团与水分子结合,进行有效分解成活性氧物种[16]. 利用电子顺磁共振光谱对反应体系产生的羟基自由基及超氧自由基进行了考察. 由图6可见,在γ-Al2O3和TiO2/γ-Al2O3的臭氧悬浆,羟基自由基和超氧自由基的信号比单独臭氧溶液的要低,说明吸附于γ-Al2O3和TiO2/γ-Al2O3催化剂表面的臭氧没有分解产生羟基自由基和超氧自由基.
原位激光显微拉曼光谱用于研究γ-Al2O3及TiO2/γ-Al2O3催化剂上形成的活性氧物种(图7). γ-Al2O3的臭氧悬浆中,γ-Al2O3的拉曼光谱在945 cm−1有一个新的吸附峰,表明γ-Al2O3催化剂上有活性原子氧生成(≡Al—*O)[16-17];TiO2/γ-Al2O3的臭氧悬浆在892 cm−1和954 cm−1均出现了一个新吸收峰,表明TiO2/γ-Al2O3催化剂上既生成了过氧物种(≡Ti4+—*O2)又生成了活性原子氧(≡Al3+—*O). 通过与γ-Al2O3催化剂的原位激光显微拉曼光谱进行比较,发现TiO2/γ-Al2O3的拉曼光谱中活性氧物种吸收峰的位置有所偏移. 这是由于钛负载于γ-Al2O3表面改变了钛原子和铝原子对表面吸附的活性氧原子和过氧基团的键强度引起的[18-20]. 原位激光显微拉曼光谱证实了活性原子氧和过氧物种是TiO2/γ-Al2O3催化臭氧氧化反应的活性氧物种. 综合以上研究结果可得:TiO2/γ-Al2O3催化剂表面的Lewis酸性位为臭氧分解的活性位,当臭氧在γ-Ti-Al2O3催化剂表面分解时,会生成相对稳定的活性原子氧和过氧物种,这有助于有机物的吸附矿化.
-
图8是TiO2/γ-Al2O3催化剂的活性与稳定性实验. 催化剂重复使用6次后,布洛芬的降解速率几乎不变,TOC的去除率重复3次后就基本稳定,仅下降6%,并且反应后溶液中未检测到Ti、Al的溶出,这说明TiO2/γ-Al2O3催化剂具有很好的活性与稳定性.
-
成功制备了钛负载γ-Al2O3介孔分子筛催化剂. XRD、SEM、氮气吸附脱附和吡啶红外光谱表征结果表明,TiO2/γ-Al2O3催化剂具有大的比表面积,更多的Lewis酸量,二氧化钛高度分散于γ-Al2O3表面. TiO2/γ-Al2O3催化臭氧氧化布洛芬结果表明,该催化剂具有良好的催化活性与稳定性,反应60 min时,TOC的去除率为80%. 单独臭氧氧化过程中TOC的去除率仅为26%. 电子顺磁共振和原位激光显微拉曼光谱实验结果表明,TiO2/γ-Al2O3催化剂表面的Lewis酸性位是催化臭氧氧化反应的活性位,臭氧分解产生活性原子氧和过氧物种有利于有机物的矿化,从而表现出最高的催化活性. TiO2/γ-Al2O3是一种有前景的臭氧化催化剂.
TiO2/γ-Al2O3催化臭氧氧化水中布洛芬
Catalytic ozonation of ibuprofen by TiO2/γ-Al2O3 in water
-
摘要: 以葡萄糖为模板,采用蒸发诱导自组装法合成了γ-Al2O3介孔催化剂,以钛酸四正丁酯(TBOT)为原料制备TiO2/γ-Al2O3催化剂,并将其用于催化臭氧氧化水中布洛芬(IBU). X射线衍射、扫描电子显微镜、氮气吸附-脱附和吡啶红外光谱表征结果表明,TiO2均匀负载于γ-Al2O3表面,保持γ-Al2O3有序的介孔结构,具有较大的比表面积和更多的Lewis酸性位. 不同催化剂活性评价结果表明,TiO2的负载显著提高γ-Al2O3催化臭氧氧化水中布洛芬的活性,反应60 min,TiO2/γ-Al2O3催化剂的TOC去除率达到80%,而γ-Al2O3、TiO2及单独臭氧氧化过程中TOC去除率则分别达到62%、32%及26%. 电子顺磁共振和原位激光显微拉曼光谱实验结果表明,Lewis酸性位为臭氧高效分解的活性位,活性原子氧和过氧物种是TiO2/γ-Al2O3催化臭氧氧化反应的活性氧物种,有利于有机物的矿化,从而表现出最高的催化活性. 重复利用实验结果表明,TiO2/γ-Al2O3催化剂具有较长的使用寿命.
-
关键词:
- TiO2/γ-Al2O3 /
- 催化臭氧氧化 /
- 活性原子氧 /
- 布洛芬.
Abstract: The γ-Al2O3 mesoporous catalyst was synthesized by evaporation-induced self-assembly method using glucose as template. TiO2/γ-Al2O3 catalyst was prepared from tetrabn-butyl titanate (TBOT) as raw material, and was used to catalytic ozonation of ibuprofen (IBU) in water. The results of X-ray diffraction, scanning electron microscopy, nitrogen adsorption-desorption and pyridine infrared spectroscopy showed that titanium dioxide was uniformly loaded on the surface of γ-Al2O3, maintaining the ordered mesoporous structure of γ-Al2O3, with larger specific surface area and more Lewis acidic sites. The evaluation results of different catalyst activities showed that TiO2 support significantly improved the activity of γ-Al2O3 catalytic ozonation of ibuprofen in water, and the TOC removal rate of TiO2/γ-Al2O3 catalyst reached 80% after 60 min reaction. The TOC removal rates of γ-Al2O3, TiO2 and ozonation alone reached 62%, 32% and 26%, respectively. The results of electron paramagnetic resonance (EPR) and in-situ laser micro-Raman spectroscopy showed that Lewis acid sites were the active site for effective decomposition of ozone, while active atomic oxygen and peroxy species were the active oxygen species of TiO2/γ-Al2O3 catalytic ozonation reaction, which was conducive to the mineralization of organic matter and showed the highest catalytic activity. The reuse experiment results showed that the TiO2/γ-Al2O3 catalyst had a long service life.-
Key words:
- TiO2/γ-Al2O3 /
- catalytic ozonation /
- active atomic oxygen /
- ibuprofen.
-
铁是一种生物必须的营养元素,直接影响浮游植物的光合作用和碳水化合物形成,由于高含氧量和无机态铁的低溶解性,铁通常是制约HNLC海区(High nutrition low chlorophyll)初级生产力的关键微量元素[1-2]。大规模海洋施铁实验表明,水生态系统中生物可利用铁的增加可以显著提高浮游植物的生物量和光合作用率,从而提高初级生产力,并促使浮游植物的群落结构发生变化[3-7]。以往研究表明,光自养微生物对碳循环和全球气候起关键作用[8-9]。初级生产力的提高,深刻地影响着全球尺度的二氧化碳固定,对温室气体的控制具有重要意义。
水生态系统中,99%溶解性铁(dissolved iron,DFe)与有机配体结合。尽管大部分有机络合态铁不能直接被藻类利用,但通过一些地球化学转化过程,可转变为生物可利用铁[10-11]。Blazevic等[12]研究发现,海洋中腐殖酸结合态铁可以发生光还原反应,进而提高铁的生物可利用性。沼泽性河流是海洋DFe的重要来源[13]。沼泽性河流中大量存在的溶解性有机碳(DOC, dissolved organic carbon)与铁离子形成有机络合物,使水中保持较高浓度DFe。有机质中羧基和酚羟基是与铁络合的主要官能团。泥炭源中的酚酸类物质,含有稳定的芳香环结构。部分酚酸与铁有较高的配合能力,这类物质的存在保护了长距离迁移的DFe,保证了陆源DFe向水生态系统的有效输出[14]。
泥炭沼泽中存在多种类型酚酸,前人在金川泥炭中检测出了9种酚酸,包括对-羟基苯甲酸、丁香酸、香草酸、阿魏酸、对-香豆酸、没食子酸、原儿茶酸和咖啡酸,许多泥炭沼泽中都有这些酚酸的存在[14]。研究证实,酚酸等有机质可以和铁形成较为稳定的配合物,使其可以在淡水运输过程中迁移更长的距离[15]。其中,具有儿茶酚或者没食子酰基结构的原儿茶酸、没食子酸以及咖啡酸可以和Fe(Ⅱ)形成较为稳定的络合物,使得Fe(Ⅱ)在极易被氧化的碱性条件下也可以保存较长时间。而咖啡酸、没食子酸、原儿茶酸以及龙胆酸还对Fe(Ⅲ)有着明显的还原作用,同样有助于这两种铁形态之间的平衡[16]。
植物或微生物分泌代谢物质对环境中其他植物或微生物体产生不利或有利的影响,这种作用称为化感作用。在化感作用过程中分泌的物质即被称为化感物质,自然界的化感物质种类非常丰富,主要包括酚酸类、苯醌类、倍半萜类、黄酮类等几大类物质[17]。迄今发现的化感物质几乎都是植物的次生代谢物质,分子量较小,结构简单,其中酚酸类物质是一类重要的次生代谢产物,也是研究较多,被证实是化感活性较强的一类物质[18]。酚酸具有一定生物毒性。目前对于酚酸抑藻的机制还不十分清楚,其抑制作用可能通过多种方式实现。研究表明,酚酸与蛋白质分子易遵循疏水键-氢键多点键合理论结合。在酚酸存在的情况下,藻细胞的胞外磷酸酶活性受到抑制,碱性磷酸酶活性的抑制使藻利用磷的能力下降。酚酸与细胞膜蛋白的结合,会破坏生物体细胞膜结构,使植物多酚物质进一步穿过细胞膜,进入细胞体内,从而改变微生物细胞酶活性,减少藻类对外源性蛋白质的利用,并通过对细胞外酶的抑制达到抑藻的目的[19]。另外,如果酚酸进入细胞体后,通过与金属离子发生络合反应,形成沉淀而破坏微生物的正常新陈代谢也是植物多酚抑藻的原因所在[20]。尽管酚酸存在生物毒性,但适量前提下,对藻类生长有积极作用[21]。泥炭源典型酚酸与铁的络合物是否对藻类利用铁有显著影响尚待进一步研究。因此,探究酚-铁配合物络合稳定性及其生物可利用性有助于进一步了解生物对铁的吸收,更好地理解全球铁碳耦合循环。
铜绿微囊藻(microcystic aeruginosa) 是中国湖泊、水库及其他水域生态系统水体富营养化蓝藻水华的代表性藻类。本文铜绿微囊藻为培养对象,利用泥炭源典型酚酸及泥炭溶解有机质(DOM)开展了一系列培养试验,以期了解泥炭沼泽源酚酸以及酚-铁络合物对铜绿微囊藻生长的影响。
1. 材料与方法(Materials and methods)
1.1 实验材料
试验所用铜绿微囊藻藻源,由中国科学院水生生物研究所提供,采用BG-11培养基培养。
4种酚酸的配制:以1.7 g·L−1的浓度配制BG-11培养基,然后将对羟基苯甲酸、对香豆酸、水杨酸、咖啡酸加入,分别配制4份浓度为0.24 g·L−1的酚酸溶液。用0.22 μm滤膜在超净台中过滤,并用紫外光照射30 min,消除微生物的影响,现配现用。
藻种培养条件:实验前5天,将铜绿微囊藻进行扩大培养。光照强度4000 lx;光暗比24 h∶0 h;温度(25±1)°C;每天摇动培养瓶5次,使藻类生长进入对数生长期。进入对数增长期后,取铜绿微囊藻各300 mL加入到1 L锥形瓶,再加入BG-11培养基100 mL进行驯化培养。
1.2 实验设计
1.2.1 单一酚酸对铜绿微囊藻的抑制作用
由于对羟基苯甲酸和对香豆酸在泥炭中含量相对较高,水杨酸和咖啡酸和铁离子可以络合,实验选择这4种酚酸进行实验。
使用细胞计数仪确定当前藻液浓度,并根据藻液浓度取一定量的处于对数生长期的藻液加入250 mL锥形瓶内,其中分别添加稀释了不同倍数的酚酸溶液,最后用培养基补足,使得锥形瓶内的液体总体积达到150 mL。每个锥形瓶内藻的初始密度为105 cell·mL−1,酚酸的最终浓度梯度分别为0、10、20、40、60、80 mg·L−1,每组3个平行,置于光照培养箱内。培养温度为(20±1)℃,光照强度为4000 lx,24 h光照,每天震荡3—5次。
1.2.2 酚络合态铁对铜绿微囊藻生长的影响
选用水杨酸和咖啡酸与铁形成络合物,探究酚铁对藻类生长的影响。实验选择的酚酸浓度为5×10−5 mol·L−1,铁浓度为1×10−6 mol·L−1,在此条件下酚酸浓度为铁浓度的50倍,可以有效保护体系中的二价铁。此外,由于泥炭沼泽中也普遍存在草酸、柠檬酸、酒石酸、乙酸等无苯环的小分子有机酸,所以实验选择草酸、柠檬酸、乙酸作为干扰物质加入到酚铁体系中进行藻类的培养实验。
藻类的培养实验分为10组,每组添加的物质如下:A.水杨酸+硫酸亚铁;B.咖啡酸+硫酸亚铁;C.水杨酸+草酸+硫酸亚铁;D.水杨酸+乙酸+硫酸亚铁;E.咖啡酸+草酸+硫酸亚铁;F.咖啡酸+乙酸+硫酸亚铁;G.水杨酸+柠檬酸+硫酸亚铁;H.咖啡酸+柠檬酸+硫酸亚铁;I.不添加酸和铁的对照组;J.只添加铁的对照组。
以上试验均为期15 d,每隔48 h取样1次,记录藻细胞数量的变化,以及藻存活情况的变化和pH值的变化。培养周期结束后分别取10 mL和5 mL样品,测量样品中叶绿素a的含量和叶绿素荧光参数Fv/Fm(最大光能转化效率)。其中Fv/Fm常用来表征叶绿素PsⅡ(低铁环境藻类光系统Ⅱ)反应中心内禀光能转换效率,反映当时所有的PsⅡ反应中心均处于开放态时最大光量子产量。
1.2.3 生理指标的测定方法:
为测藻细胞存活率及藻细胞数量,使用5-CFDA染色,具体操作方法如下:
(1)用DMSO(二甲基亚砜)5-CFDA稀释至10 mmol·L−1。将99 μL已经配制好的BG-11培养基与1 μL的5-CFDA混合作为A液,摇晃10 s混匀;
(2)将50 μL样品与50 μL A液混匀,用移液枪至少吹打10次;
(3)在25℃条件下将上一步准备好的样品避光放置30 min;
(4)用移液枪将待测样品吹打10次或摇晃,使藻细胞分散,然后用移液枪取20 μL加入计数板内。在计数板插入仪器之前,稳定1 min,使样品在其中稳定下来。
将细胞染色后,使用细胞计数仪计数,并观察细胞的存活状态。
测量样品叶绿素a的含量:
(1)取藻液10 mL,4500 r·min-1离心15 min,去掉上层清液,将样品在4℃冰箱中放置1 d;
(2)取出后迅速加入5 mL的90%热乙醇(80℃)于80℃的热水浴萃取2 min,再用超声处理10 min,放在暗处萃取4 h后,用0.22 μm的滤头过滤。用酶标仪于波长665 nm和750 nm处测吸光值,然后滴加1滴1 mol·L−1的盐酸酸化,于波长665 nm和750 nm处再测吸光值。计算公式为:
Chla乙醇=27.9×[(E665−E750)−A665+A750)]×V乙醇/V样品 其中,Chla乙醇为热乙醇法测定的叶绿素a含量(μg·L−1);E665是乙醇萃取液于波长665 nm的吸光值;E750是乙醇萃取液于波长750 nm的吸光值;A665为乙醇萃取液酸化后在665 nm处的吸光值;A750为乙醇萃取液在750 nm处的吸光值;V乙醇为乙醇萃取液的体积(mL),V样品为所取样品的体积(mL)。
抑制率计算公式:IR=(1-N/N0×100%)抑制率为负值则有促进效果,抑制率为正则抑制。
1.2.4 泥炭沼泽源酚络合态铁对铜绿微囊藻生长的影响
培养前准备:
(1)提取泥炭中的DOM;
(2)将样品通过H型阳离子柱交换柱,去除样品中存在的金属离子;然后将DOM样品按照<1 KDa,1—3 KDa,>3 KDa分成3份;
(3)将3份样品中加入FeSO4,待稳定一段时间后,取10 mL加入培养基中,加入后Fe的浓度为5×10−6 mol·L−1。
铜绿微囊藻培养实验分为5组,每组3份平行,每组添加的物质如下:A.无添加;B.Fe,浓度为5×10−6 mol·L−1;C.DOM-Fe(<1 KDa),Fe浓度为5×10−6 mol·L−1;D.DOM-Fe(1—3 KDa),Fe浓度为5×10−6 mol·L−1;E.DOM-Fe(>3 KDa),Fe浓度为5×10−6 mol·L−1。
实验均为期15 d,每隔48 h取样1次,记录藻细胞数量的变化,以及藻存活情况的变化。
2. 结果与讨论(Results and discussion)
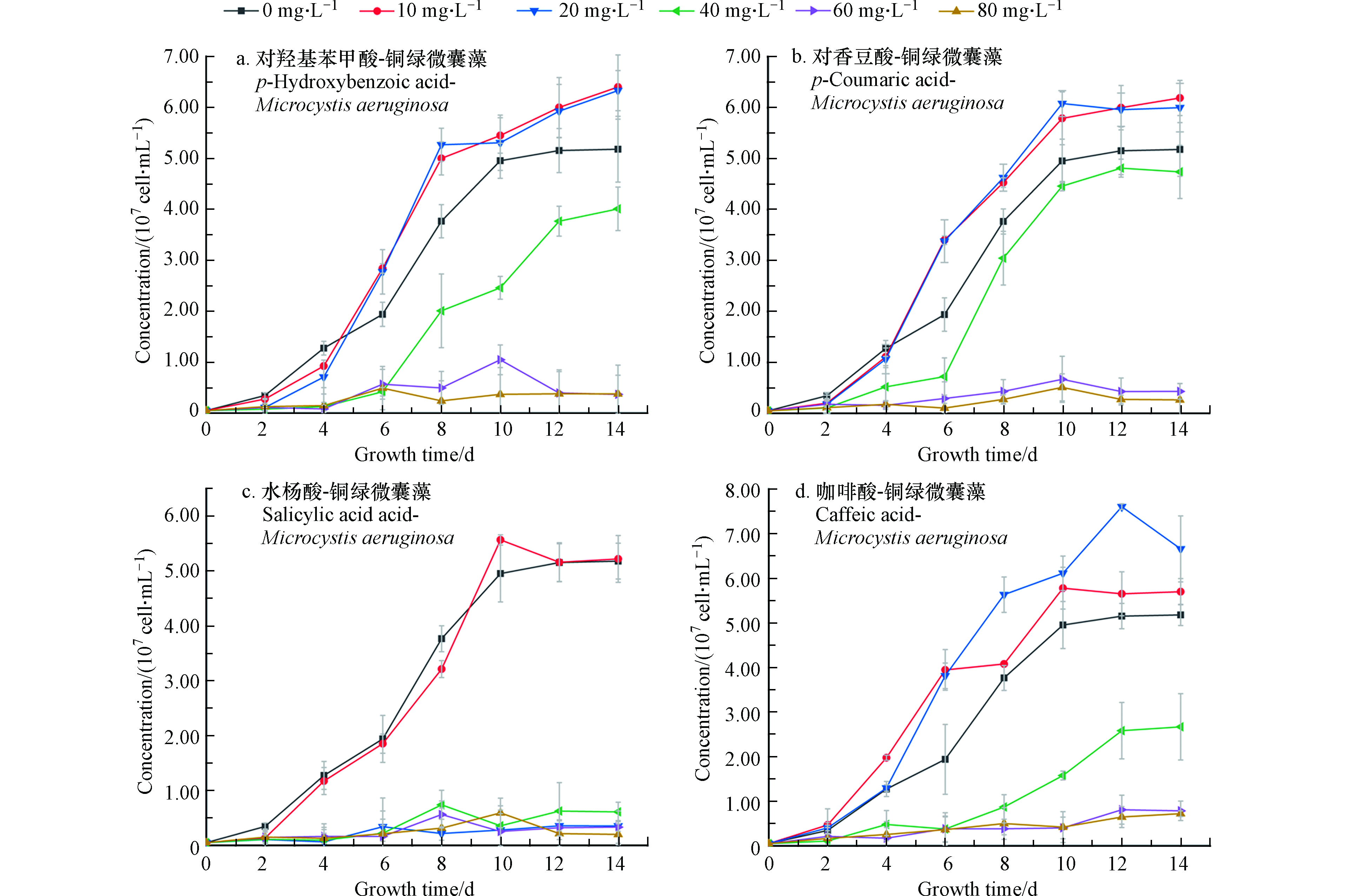
2.1 不同酚酸影响微囊藻生长的浓度效应
在微囊藻培养体系中分别加入不同浓度四种酚酸溶液,经过14 d培养和检测,得到微囊藻-酚酸的生长曲线如图1所示。图1中A、B为不同浓度的对羟基苯甲酸和对香豆酸对微囊藻生长情况的影响。可以看出,当酚酸浓度为10 mg·L−1和20 mg·L−1时,微囊藻的生长速率和最终达到的终点浓度明显高于控制组(0)抑制率为-23.5%—18%。从微囊藻浓度上来看,当酚酸浓度为10 mg·L−1和20 mg·L−1时,生长情况比较接近,说明在此浓度下,这两种酚酸对微囊藻的生长有一定的刺激作用。在A、B组中当酚酸浓度超过20 mg·L−1时,藻液浓度和藻的生长速率明显低于控制组;当浓度增加到40 mg·L−1时,这两种酚酸对微囊藻的生长起到了明显的抑制作用,而对羟基苯甲酸的对微囊藻生长的抑制作用更加明显;浓度继续增加到60—80 mg·L−1时,微囊藻前4—5天略有增长,然后基本停止了增长,保持在3×106 cell·mL−1左右。对香豆酸60 mg·L−1组的藻数量略高于80 mg·L−1组。
在C组水杨酸-微囊藻的实验中,水杨酸浓度为10 mg·L−1时,增长的速度与控制组接近,在8—10 d快速增长后,藻数量和控制组趋于一致。而当浓度大于10 mg·L−1时,都出现了明显的抑藻效果,抑制率约为80%。
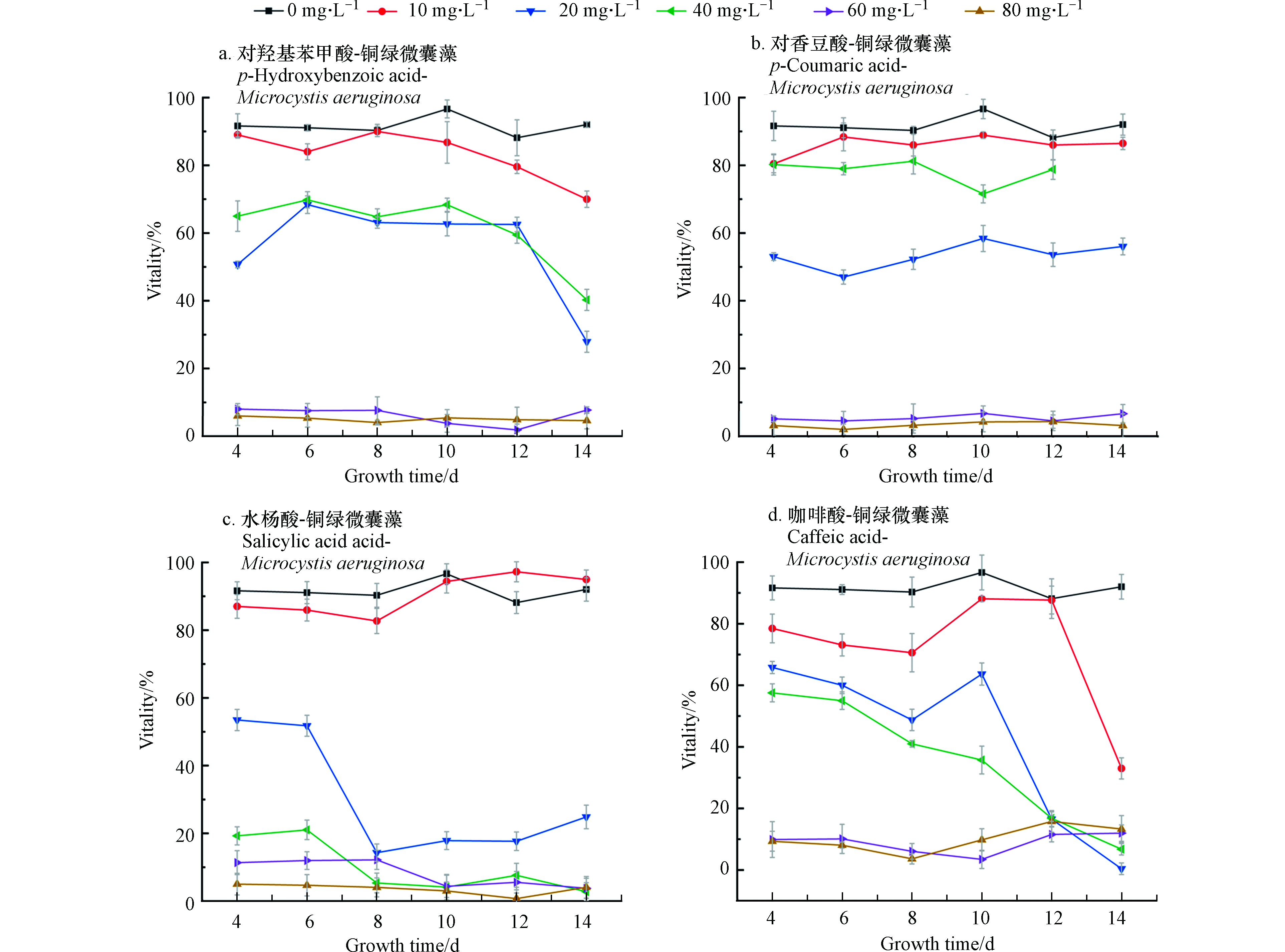
结合图2中存活率来看(从培养的第4天开始测微囊藻的存活率)当浓度为10 mg·L−1时,微囊藻的存活率都略低于控制组,差值在10%左右浮动,当浓度为20—40 mg·L−1,在开始计数时,藻类的存活率就已不同程度地低于控制组,并且A、C、D存活率在4—14 d整体处于下降的趋势,可以看出,水杨酸对微囊藻的抑制作用最强。
表1是各组样品的Fv/Fm,最大光能转化效率Fv/Fm常用来表征叶绿素PsII反应中心内禀光能转换效率,反映当时所有的PsII反应中心均处于开放态时最大光量子产量。
在非胁迫环境下,植物叶片叶绿素荧光参数Fv/Fm变化极小,表现出稳定的特点,但在胁迫条件下,该参数明显下降[22]。Fv/Fm可作为植物受环境胁迫的响应指标[23]。控制组的Fv/Fm为0.308。一般情况下,微囊藻的Fv/Fm在0.3左右,当Fv/Fm过低表明藻类受到环境胁迫,PSII中心受到损伤进而降低光合作用效率。由表1可以看出,当水杨酸浓度大于20 mg·L−1时会对微囊藻的光合作用产生明显抑制,当各组酚酸浓度超过60 mg·L−1时,藻类基本停止了光合作用,这和前文中藻类的生物量变化和存活率相吻合。表2是各个组叶绿素含量的均值,数据表明:各组中相对低浓度的酚酸,不仅对藻类数量的增长有促进作用,也促进了叶绿素含量的增加。
表 1 铜绿微囊藻的Fv/FmTable 1. Fv/Fm of Microcystis aeruginosa对羟基苯甲酸P-hydroxybenzoic acid 对香豆酸P-coumaric acid 水杨酸Salicylic acid 咖啡酸Caffeic acid 010×10−620×10−640×10−660×10−680×10−6 0.3080.3100.2920.3020.0390.000 0.3080.3100.3140.3240.0490.000 0.3080.3090.0230.0140.0000.000 0.3080.3310.2760.3600.0380.000 注:0—80×10−6分别对应添加的4种酚酸浓度,表中数据是在微囊藻培养期结束时测得的Fv/Fm。 Note: 0—80×10−6 respectively correspond to the four added phenolic acid concentrations. The data in the table are the Fv/Fm measured at the end of the Microcystis culture period. | Show Table DownLoad:
CSV
表 2 铜绿微囊藻叶绿素含量(g·L-1)Table 2. Chlorophyll content of Microcystis aeruginosa
DownLoad:
CSV
表 2 铜绿微囊藻叶绿素含量(g·L-1)Table 2. Chlorophyll content of Microcystis aeruginosa对羟基苯甲酸P-hydroxybenzoic acid 对香豆酸P-coumaric acid 水杨酸Salicylic acid 咖啡酸Caffeic acid 010×10−620×10−640×10−660×10−680×10−6 0.5681.0141.1380.7750.0000.000 0.5680.7370.8620.5680.0000.000 0.5680.7960.0090.0000.0000.000 0.5682.0932.2011.3590.0000.000 注:0—80×10−6分别对应添加的4种酚酸浓度,表中数据是微囊藻培养期结束时测得的叶绿素含量。 Note: 0—80×10−6 respectively correspond to the four phenolic acid concentrations added. The data in the table is the chlorophyll content measured at the end of the Microcystis culture period. | Show TableDownLoad:
CSV
2.2 酚络合态铁的生物可利用性
根据上述试验结果可以得出,当这4种典型泥炭源酚酸的浓度达10 mg·L−1时,从生物量、存活率以及光合作用强度来说对微囊藻的生长没有明显抑制作用。普遍认为,藻类吸收Fe主要是离子形态,而不是有机络合态Fe[24]。目前,仅观察到在产生铁载体的微生物中存在铁载体复合物的吸收及其在细胞中的还原[25]。已有研究发现,有机络合态铁中铁的释放途径不同。其中包括简单的配体-金属平衡(Ligand-Metal balance),平衡在初级生产者消耗铁后发生变化,促进铁从络合物中分离。另一个途径是基于配体的降解,这也导致了Fe和配体的分离。第三种可能是通过有机态铁的还原,降低配体对铁的亲和力,并导致Fe的释放。这个过程可能是由于络合物的自还原/氧化而发生的,这意味着配体氧化同时也释放产生Fe[26]。尤其是光还原分解对铁的生物利用度有很大影响,这一释放铁途径被称为AHS(aquatic humic substances)机制[12]。
图3是加入不同酸和Fe2+的微囊藻生长曲线。施加和不加Fe2+的对照试验表明,在培养1—9 d,两组生长速度以及生物量大致相同。在第9天后,施加Fe2+组的生长速度放缓,最终的藻密度低于对照组。图3表明在没有配体存在的情况下,加入一定量的铁对微囊藻生长促进作用不明显。同时,观察发现,在藻类指数生长阶段出现了较高的pH值(高达10.5),这与二氧化碳生物需求增加有关。在指数生长结束和平台期开始后,pH值略下降。在藻类生长平缓或生长不良的样品中,pH值无显著变化,pH值大多保持在6—8。
能促进藻类生长的酚酸应当与三价铁有较高的亲和力,与二价铁有较低的亲和力[27]。试验表明,相对其他3种酚酸铁配合物体系,水杨酸铁不能有效地为微囊藻提供生物可利用性铁,这可能与水杨酸的稳定性较强有关[27]。对照表明,用咖啡酸处理的微囊藻生长良好,最高浓度达到2.19×107 cell·mL−1。这可能是咖啡酸中的儿茶酚基结构所引起的,并且有更高的氧化还原潜力,更容易将Fe从配合物中释放。Santana等 [28]的研究也证实了在生物条件下还原络合物的可能性。总体上,酚酸的加入提高了藻类对Fe的生物可利用率。
图2显示藻类存活率从第10天开始明显下降,藻类计数可能包括了死藻,因此选择第11天数据进行显著性分析。结果表明(表3),在0.05的置信水平下,咖啡酸的加入对微囊藻生长有显著促进作用,而水杨酸在0.05的置信水平下,对微囊藻生长无显著促进作用。
 图 3 微囊藻生长曲线Figure 3. Microcystis growth curveA.水杨酸+硫酸亚铁 B.咖啡酸+硫酸亚铁 C.水杨酸+草酸+硫酸亚铁 D.水杨酸+乙酸+硫酸亚铁 E.咖啡酸+草酸+硫酸亚铁 F.咖啡酸+乙酸+硫酸亚铁 G.水杨酸+柠檬酸+硫酸亚铁 H.咖啡酸+柠檬酸+硫酸亚铁 1_3.不添加酸和铁的对照组 Fe1_3硫酸亚铁A. Salicylic acid+Ferrous sulfate B. Caffeic acid+Ferrous sulfate C. Salicylic acid+Oxalic acid+Ferrous sulfate D. Salicylic acid+Acetic acid+Ferrous sulfate E. Caffeic acid+Oxalic acid+Ferrous sulfate F. Caffeic acid+Acetic acid+Ferrous sulfate G. Salicylic acid+Citric acid+Ferrous sulfate H. Caffeic acid+Citric acid+Ferrous sulfate 1_3. Control group without acid and iron Fe1_3. Ferrous sulfate表 3 第11天不同试验组微囊藻浓度变化的相关性矩阵Table 3. Correlation matrix of changes in the concentration of Microcystis in different test groups on the 11th day
图 3 微囊藻生长曲线Figure 3. Microcystis growth curveA.水杨酸+硫酸亚铁 B.咖啡酸+硫酸亚铁 C.水杨酸+草酸+硫酸亚铁 D.水杨酸+乙酸+硫酸亚铁 E.咖啡酸+草酸+硫酸亚铁 F.咖啡酸+乙酸+硫酸亚铁 G.水杨酸+柠檬酸+硫酸亚铁 H.咖啡酸+柠檬酸+硫酸亚铁 1_3.不添加酸和铁的对照组 Fe1_3硫酸亚铁A. Salicylic acid+Ferrous sulfate B. Caffeic acid+Ferrous sulfate C. Salicylic acid+Oxalic acid+Ferrous sulfate D. Salicylic acid+Acetic acid+Ferrous sulfate E. Caffeic acid+Oxalic acid+Ferrous sulfate F. Caffeic acid+Acetic acid+Ferrous sulfate G. Salicylic acid+Citric acid+Ferrous sulfate H. Caffeic acid+Citric acid+Ferrous sulfate 1_3. Control group without acid and iron Fe1_3. Ferrous sulfate表 3 第11天不同试验组微囊藻浓度变化的相关性矩阵Table 3. Correlation matrix of changes in the concentration of Microcystis in different test groups on the 11th day1_3 Fe1_3 A B C D E F G H 1_3 1 0.963 0.971 −0.358 0.474 0.657 0.246 0.461 0.983 0.491 Fe1_3 0.963 1.00 1.000* −0.095 0.221 0.835 −0.022 0.682 0.996 0.707 A 0.971 1.000* 1.00 −0.126 0.251 0.818 0.008 0.659 0.999* 0.685 B −0.358 −0.095 −0.126 1.00 −0.992 0.468 −0.993 0.663 −0.179 0.637 C 0.474 0.221 0.251 −0.992 1.00 −0.352 0.970 −0.563 0.302 −0.534 D 0.657 0.835 0.818 0.468 −0.352 1.00 −0.569 0.972 0.785 0.979 E 0.246 −0.022 0.008 −0.993 0.970 −0.569 1.00 −0.746 0.062 −0.723 F 0.461 0.682 0.659 0.663 0.663 0.972 −0.746 1.00 0.618 0.999* G 0.983 0.996 0.999* −0.179 0.302 0.785 0.062 0.618 1.00 0.645 H 0.491 0.707 0.685 0.637 −0.534 0.979 −0.723 0.999* 0.645 1.00 注:*. 在 0.05 水平(双侧)上显著相关。Notes:*. Significant correlation at 0.05(bilateral) level. | Show TableDownLoad:
CSV
2.3 泥炭沼泽源DOM-Fe的藻类可利用性
许多泥炭源有机质同时含有酚羟基和羧基,具有酚酸性质。但由于泥炭有机质组成复杂多样,现有技术尚不能有效分离不同性质有机化合物。因此,利用不同分子量段有机质与铁的络合物,开展藻类培养试验有助于客观评估泥炭源DOM-Fe的藻类可利用性。利用不同分子量段DOM-Fe,进行培养试验,铜绿微囊藻的生长情况如图4。表4 结果表明,在0.05 的置信水平下,不同组之间差异不具统计显著性,但图4 还是反映出有机态铁对藻类生长的促进趋势.
结果显示(图3和图4),不同分子量结合态Fe均促进了铜绿微囊藻的生长,但影响程度不同。添加Fe后,铜绿微囊藻的生物量和控制组相比均有增加,微囊藻生长得到促进,最终达到107 cell·mL−1。其中,微囊藻在7—11 d增长最快;其次,对比生长终点可以发现不同DOM-Fe促进效果存在差异:E组>D组>C组>B组>A组>无Fe组;第三,添加DOM显著促进了藻的生长。研究表明,相对于Fe3+,藻类更倾向于利用Fe2+[29]。这是由于具有一定还原能力的DOM可以减缓二价铁的氧化[30],从而提高了藻类对铁的利用率。此外,不同分子量段DOM与Fe的络合稳定常数略有不同,较高分子量的DOM(>3 kD)与Fe的络合稳定常数较小[30],在光照或者其他条件下容易发生解离,产生易被藻类利用的Fe。而泥炭源低分子量DOM(<1 KD)络合态铁,由于其络合稳定的常数相对较高,在培养体系中更加稳定,相对不易被藻类利用。
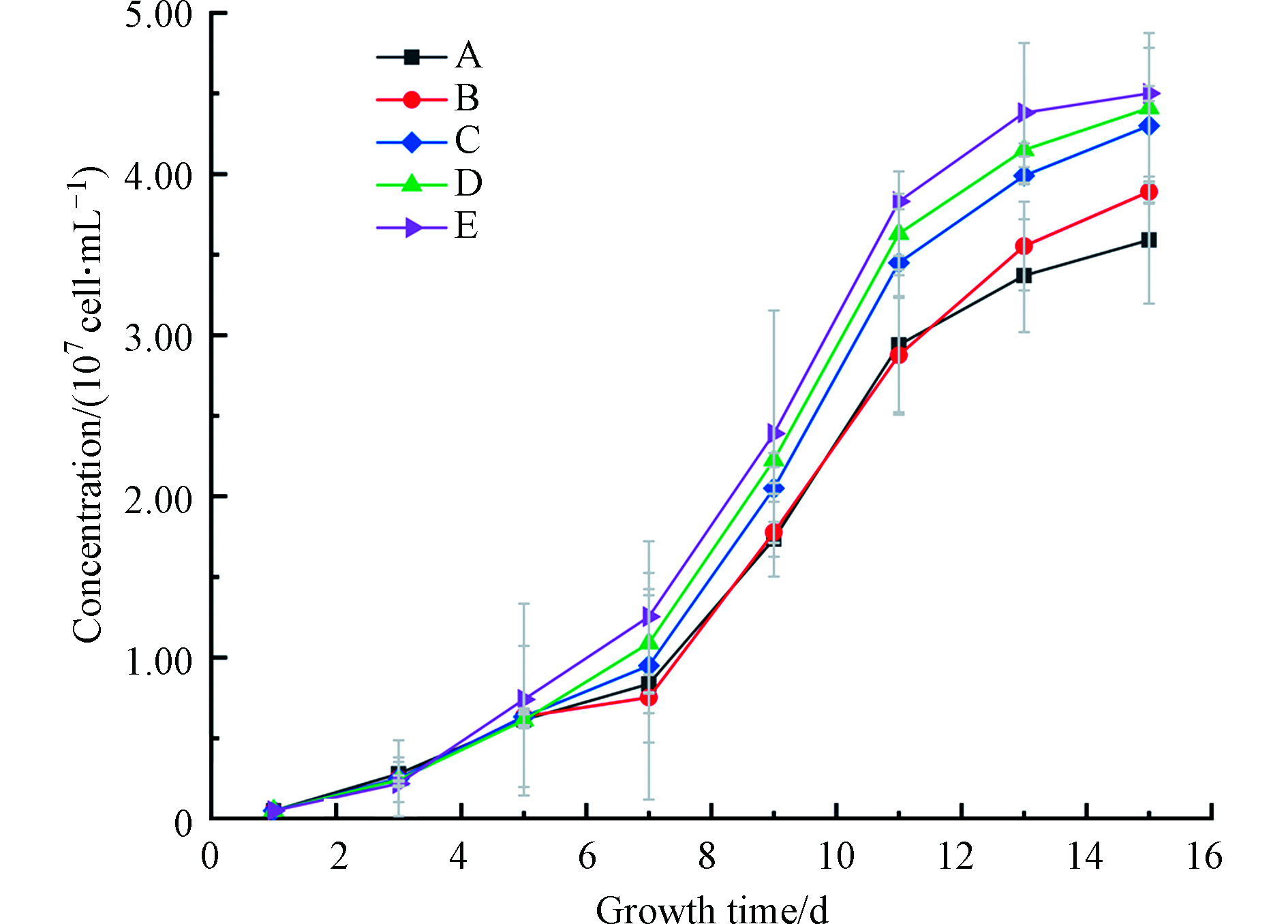 图 4 铜绿微囊藻生长曲线A.无添加 B.硫酸亚铁 C.DOM-Fe(<1KD ) D.DOM-Fe(1-3 KD) E.DOM-Fe(> 3KD)Figure 4. Microcystis aeruginosa growth curveA. No addition B. Ferrous sulfate C.DOM-Fe(<1KD) D.DOM-Fe(1-3KD) E.DOM-Fe(>3KD)表 4 第11天不同试验组铜绿微囊藻生长浓度变化的相关性矩阵Table 4. Correlation matrix of growth concentration changes of Microcystis aeruginosa in different test groups on the 11th day
图 4 铜绿微囊藻生长曲线A.无添加 B.硫酸亚铁 C.DOM-Fe(<1KD ) D.DOM-Fe(1-3 KD) E.DOM-Fe(> 3KD)Figure 4. Microcystis aeruginosa growth curveA. No addition B. Ferrous sulfate C.DOM-Fe(<1KD) D.DOM-Fe(1-3KD) E.DOM-Fe(>3KD)表 4 第11天不同试验组铜绿微囊藻生长浓度变化的相关性矩阵Table 4. Correlation matrix of growth concentration changes of Microcystis aeruginosa in different test groups on the 11th dayA B C D E A 1 −.751 .350 −.167 .770 B −.751 1 −.881 .776 −.158 C .350 −.881 1 −.982 −.327 D −.167 .776 −.982 1 .500 E .770 −.158 −.327 .500 1 注:*. 在 0.05 水平(双侧)上显著相关. Notes:*. Significant correlation at 0.05(bilateral) level. | Show TableDownLoad:
CSV
3. 结论(Conclusion)
(1)4种酚酸对藻类生长的影响均呈现“低促高抑”的规律。从藻类生物量和叶绿素含量来看,抑藻效果从高到低:水杨酸>对羟基苯甲酸>对香豆酸>咖啡酸;结合藻类的存活率,虽然低浓度酚酸刺激了藻类生物量的增长,但是也对藻类的生存产生了一定的负面影响:在添加10 mg·L−1酚酸的几组样品中,微囊藻的存活率都略低于控制组。
(2)添加酚酸的藻类样品中,当水杨酸浓度达到20 mg·L−1时,Fv/Fm明显降低(0.3降低到0.02左右),而其它3种酚酸浓度达到60 mg·L−1才出现抑制,说明水杨酸抑制作用最强。
(3)不同酚铁络合物的生物可利用性存在差异:相对咖啡酸和水杨酸,水杨酸络合态铁更难被藻类利用,除酚毒性效应外,还与其较高的络合稳定性有关。
(4)泥炭源不同分子段DOM-Fe对藻类生长的促进作用从高到低依次为:>3 KD,1—3 KD,<1 KD。高分子段DOM(>3 kD)与Fe的络合稳定常数最小,在光照或者其他条件下容易发生解离,更易释放Fe而被藻类利用;泥炭源低分子量DOM(<1 KD)络合态铁,因其络合稳定常数相对较高,相对不易被藻类利用。
致谢:感谢中国科学院水生生物研究所的大力支持。
-

图 1 催化剂的小角(A)和广角(B)XRD图
Figure 1. Small angle (A) and wide angle (B) XRD diagrams of catalysts

图 2 (A1、A2)γ-Al2O3和(B1、B2)TiO2/γ-Al2O3样品的扫描电镜图
Figure 2. TEM images of (A1, A2) γ-Al2O3 and (B1, B2) TiO2/γ-Al2O3 samples
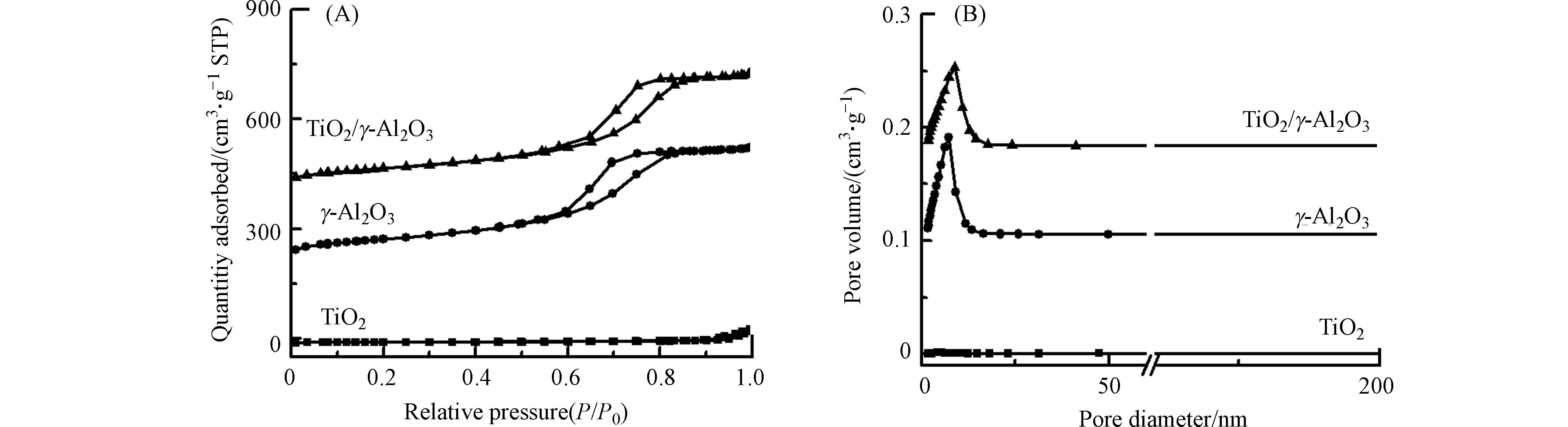
图 3 催化剂的氮气吸附脱附等温线(A)和孔径分布曲线(B)
Figure 3. Nitrogen adsorption and desorption isotherms (A) and pore size distribution curves (B) of catalysts
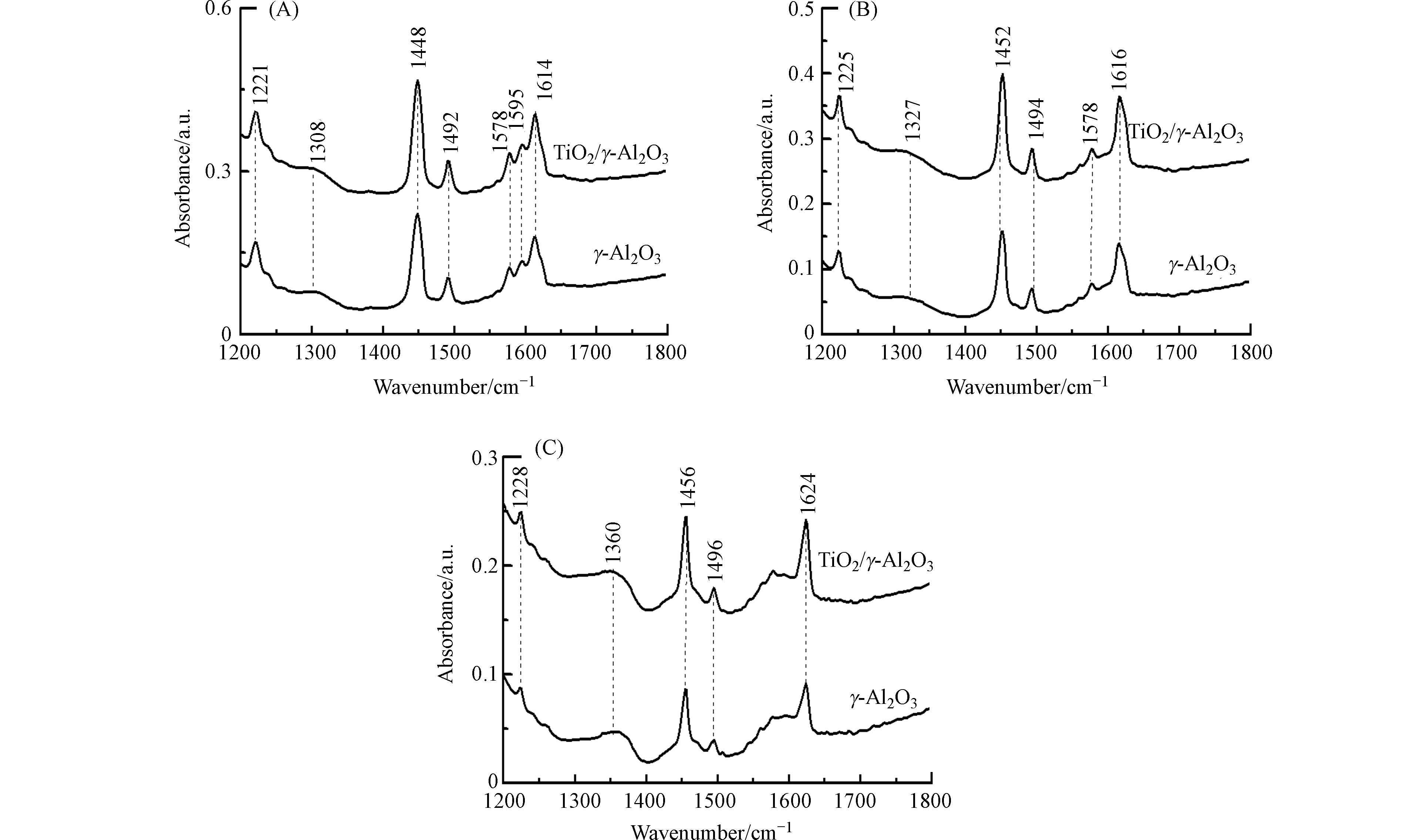
图 4 γ-Al2O3和TiO2/γ-Al2O3在(A)20 ℃,(B)150 ℃,(C)350 ℃脱附的吡啶红外光谱
Figure 4. Pyridine -FTIR spectra of γ-Al2O3 and TiO2/γ-Al2O3 after degassing at 20 ℃(A), 150 ℃(B), 350 ℃(C)
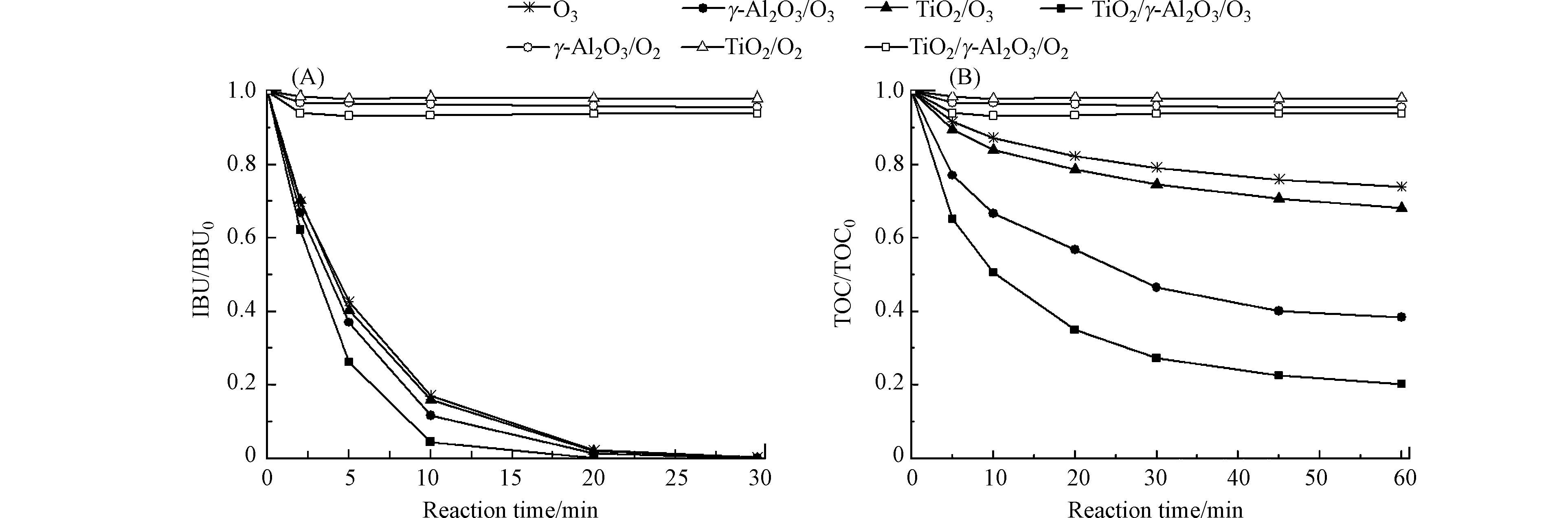
图 5 不同工艺对布洛芬(A)和TOC(B)的去除率
Figure 5. Removal rate of ibuprofen (A) and TOC (B) by different processes
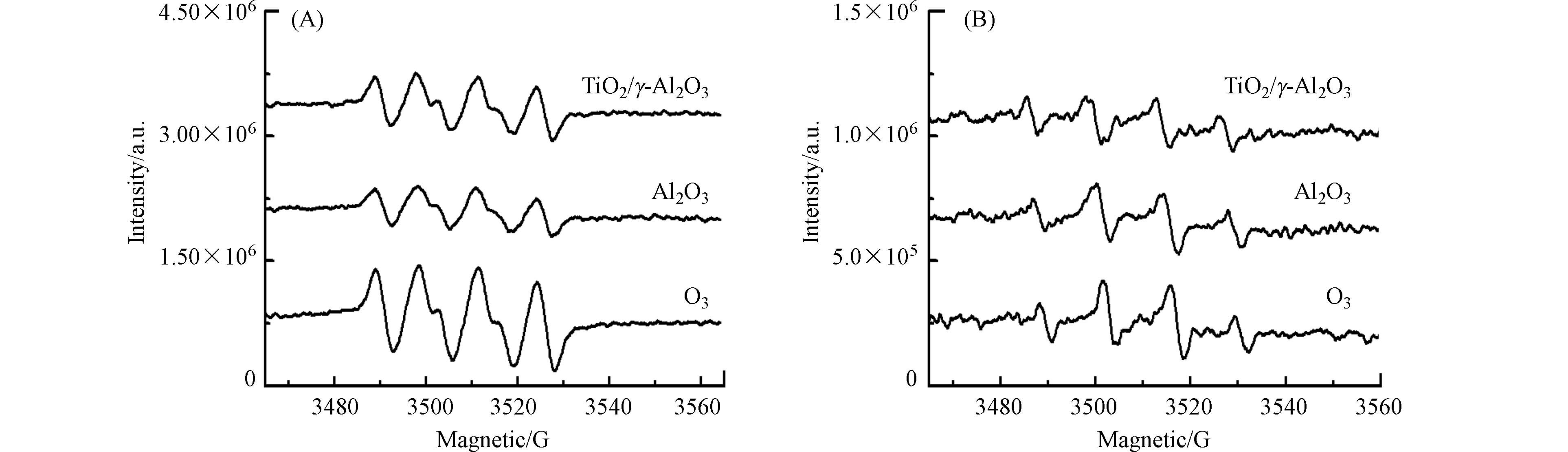
图 6 以BMPO为捕获剂的EPR波谱.BMPO-HO2•/O2•-(A),BMPO-•OH(B)
Figure 6. BMPO spin-trapping EPR spectra recorded in methanol dispersion for BMPO-HO2•/O2•- (A) and aqueous dispersion for BMPO-•OH (B) with ozone
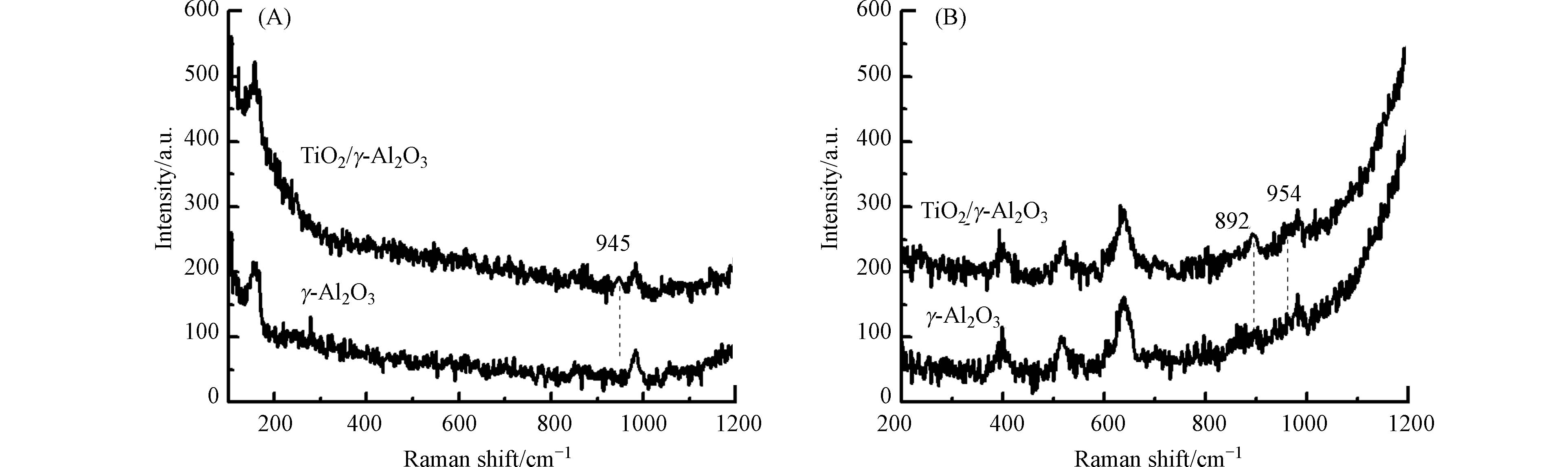
图 7 γ-Al2O3和TiO2/γ-Al2O3催化剂悬浆的拉曼光谱
Figure 7. Raman spectra of γ-Al2O3 and TiO2/γ-Al2O3 aqueous dispersions without (A) and with (B) ozone
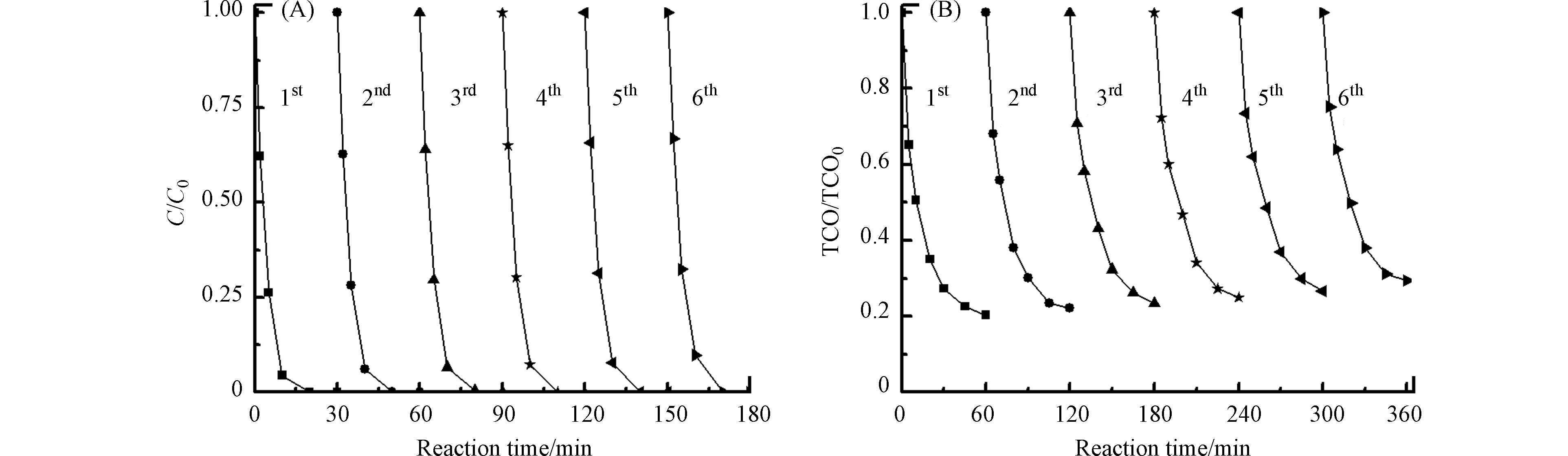
图 8 TiO2/γ-Al2O3催化臭氧氧化布洛芬(A)和TOC(B)的活性与稳定性
Figure 8. Activity and stability of TiO2/γ-Al2O3 catalyst for ozonation of ibuprofen (A) and TOC (B)
表 1 催化剂的比表面、孔径、孔容和孔壁
Table 1. Specific surface area, pore diameter, pore volume, and pore wall of catalysts
催化剂 Catalyst 比表面积/ (m2·g−1) S BET 孔径/ nm Pore diameter 孔容/(cm3·g−1) Pore volume γ-Al2O3 252.81 0.50 6.13 TiO2/γ-Al2O3 221.97 0.47 5.86
下载: 导出CSV
表 2 吡啶-红外光谱测定的不同催化剂的Lewis酸量
Table 2. Amount of Lewis acid sites of different catalysts determined by Pyridine-FTIR
催化剂 Catalyst 总酸量/(μmol·g−1) Total acid 中强酸量/(μmol·g−1) Medium acid 强酸量/(μmol·g−1) Strong acid γ-Al2O3 540.1 197.4 61.1 TiO2/γ-Al2O3 653.8 222.9 85.4
下载: 导出CSV
-
[1] WEBB S, TERNES T, GIBERT M, et al. Indirect human exposure to pharmaceuticals via drinking water [J]. Toxicology Letters, 2003, 142(3): 157-167. doi: 10.1016/S0378-4274(03)00071-7 [2] NICA A V, OLARU E A, BRADU C, et al. Catalytic Ozonation of Ibuprofen in Aqueous Media over Polyaniline–Derived Nitrogen Containing Carbon Nanostructures [J]. Nanomaterials, 2022, 12(19): 3468. doi: 10.3390/nano12193468 [3] VELDHOEN N, SKIRROW R C, BROWN L L Y, et al. Effects of acute exposure to the non-steroidal anti-inflammatory drug ibuprofen on the developing North American Bullfrog (Rana catesbeiana) tadpole [J]. Environmental Science & Technology, 2014, 48(17): 10439-10447. [4] LEGUBE B, KARPEL VEL LEITNER N. Catalytic ozonation: A promising advanced oxidation technology for water treatment [J]. Catalysis Today, 1999, 53(1): 61-72. doi: 10.1016/S0920-5861(99)00103-0 [5] ISSAKA E, AMU-DARKO J N O, YAKUBU S, et al. Advanced catalytic ozonation for degradation of pharmaceutical pollutants―A review [J]. Chemosphere, 2022, 289: 133208. doi: 10.1016/j.chemosphere.2021.133208 [6] NAWROCKI J, KASPRZYK-HORDERN B. The efficiency and mechanisms of catalytic ozonation [J]. Applied Catalysis B:Environmental, 2010, 99(1/2): 27-42. [7] WANG Y, DUAN X, XIE Y, et al. Nanocarbon-Based Catalytic Ozonation for Aqueous Oxidation: Engineering Defects for Active Sites and Tunable Reaction Pathways [J]. ACS Catalysis, 2020, 10(22): 13383-13414. doi: 10.1021/acscatal.0c04232 [8] NAWROCKI J. Catalytic ozonation in water: Controversies and questions. discussion paper [J]. Applied Catalysis B:Environmental, 2013, 142/143: 465-471. doi: 10.1016/j.apcatb.2013.05.061 [9] WANG Y J, YU G. Challenges and pitfalls in the investigation of the catalytic ozonation mechanism: A critical review [J]. Journal of Hazardous Materials, 2022, 436: 129157. doi: 10.1016/j.jhazmat.2022.129157 [10] XU B J, XIAO T C, YAN Z F, et al. Synthesis of mesoporous alumina with highly thermal stability using glucose template in aqueous system [J]. Microporous and Mesoporous Materials, 2006, 91(1/2/3): 293-295. [11] EMEIS C A. Determination of integrated molar extinction coefficients for infrared absorption bands of pyridine adsorbed on solid acid catalysts [J]. Journal of Catalysis, 1993, 141(2): 347-354. doi: 10.1006/jcat.1993.1145 [12] BADER H, HOIGNÉ J. Determination of ozone in water by the indigo method [J]. Water Research, 1981, 15(4): 449-456. doi: 10.1016/0043-1354(81)90054-3 [13] OSTOVAR S, MOUSSAVI G, MOHAMMADI S, et al. Developing a novel Ti-doped γAl2O3 xerogel with high photocatalytic chemical and microbial removal performance: Characterization and mechanistic insights [J]. Chemical Engineering Journal, 2023, 464: 142545. doi: 10.1016/j.cej.2023.142545 [14] ANDONOVA S M, ŞENTÜRK G S, KAYHAN E, et al. Nature of the Ti−Ba Interactions on the BaO/TiO2/Al2O3 NOx Storage System [J]. The Journal of Physical Chemistry C, 2009, 113(25): 11014-11026. doi: 10.1021/jp9005026 [15] MORISHIGE K, KITTAKA S, KATSURAGI S, et al. Thermal desorption and infrared studies of ammonia, amines and pyridines chemisorbed on chromic oxide [J]. Journal of the Chemical Society, Faraday Transactions 1:Physical Chemistry in Condensed Phases, 1982, 78(10): 2947-2957. doi: 10.1039/f19827802947 [16] BING J, HU C, NIE Y, et al. Mechanism of Catalytic Ozonation in Fe2O3/Al2O3@SBA-15 Aqueous Suspension for Destruction of Ibuprofen [J]. Environmental Science & Technology, 2015, 49(3): 1690-1697. [17] YU G, WANG Y, CAO H, et al. Reactive oxygen species and catalytic active sites in heterogeneous catalytic ozonation for water purification [J]. Environmental Science & Technology, 2020, 54(10): 5931-5946. [18] RADHAKRISHNAN R, OYAMA S T. Ozone decomposition over Manganese oxide supported on ZrO2 and TiO2: A kinetic study using in situ laser Raman spectroscopy [J]. Journal of Catalysis, 2001, 199(2): 282-290. doi: 10.1006/jcat.2001.3167 [19] AND R R, OYAMA S, CHEN J G, et al. Electron transfer effects in ozone decomposition on supported Manganese oxide [J]. Journal of Physical Chemistry B, 2001, 105: 4245-4253. doi: 10.1021/jp003246z [20] ZHANG T, LI W W, CROUÉ J P. Catalytic ozonation of oxalate with a cerium supported palladium oxide: An efficient degradation not relying on hydroxyl radical oxidation [J]. Environmental Science & Technology, 2011, 45(21): 9339-9346. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 1445
- HTML全文浏览数: 1445
- PDF下载数: 46
- 施引文献: 0
