




 下载:
下载:








-
砷来源广泛,包括火山喷发、岩石风化等自然来源以及采矿、冶金等人为来源[1-2]。在全球范围内,土壤中砷的平均质量分数为1.8 mg·kg−1,而我国土壤中砷平均质量分数达到9.2 mg·kg−1,超过世界水平的5倍[3]。我国云南、贵州、四川等西南地区的土壤中砷背景值远超全国土壤背景值[4]。土壤中的砷通过食物链进入人体后,可引发色素沉着、慢性肺病、心血管疾病和神经系统紊乱等健康问题[5]。因此,对砷污染土壤的修复十分迫切。
电动修复是常用的一种砷污染土壤修复方法,其利用电渗析、电迁移等电动效应使砷酸根和亚砷酸根定向迁移,从而降低土壤中砷的总量[6-7]。但常规电动修复技术对砷的修复效果有限,KARACA等[8]对沉积物中的砷进行电动修复时发现,运行18 d后砷几乎没有被去除。电极逼近法为电动修复的一种,其在电动过程中每隔一段时间将电极向某一方向移动一定距离,以此来影响土壤pH、氧化还原电位 (Eh) 等环境因子,而砷的溶解性和迁移性与环境因子密切相关。YAO等[9]发现,相比于固定电极法 (FE-EK) ,阴极逼近法 (AC-EK) 通过提高阴极区域pH可将砷的去除率提高4倍。付博等[10]发现,当pH<4时,随着pH的降低,粗砂和细砂中砷的溶出量不断增加。周一敏等[11]发现,当Eh较低时,五价砷[As(V)]会转化为移动性更高的三价砷[As(III)],另外还能驱动土壤中砷的释放。由此可见,电动逼近技术对提高砷污染土壤修复效果具有很大潜力。
目前,常采用向土壤中加入化学药剂[2,7]、增设渗透反应墙[6,12]等方式提高砷去除率,但基于电极逼近技术对砷污染土壤进行修复的研究尚很缺乏。基于此,本研究采用不同的电极逼近方式对砷污染土壤进行修复。研究不同逼近方式对总砷[As(T)]的分布以及As(III)、As(V)迁移转化的影响,探究捕集室土壤中砷赋存形态的转化,以期为砷污染场地修复提供技术参考和理论依据。
-
供试土壤采自辽宁大连某污染场地,经风干并研磨后过20目标准筛备用。供试土壤基本理化性质为:pH为7.17,Eh为273.5 mV,电导率为2 012.5 μS·cm−1,Al、Fe、Ca质量分数分别为40.13、44.17、102.55 g·kg−1,As(T)、As(III)、As(V)质量分数分别为355.08、120.32、234.76 mg·kg−1。其中,As(T)质量分数超过《土壤环境质量 建设用地土壤污染风险管控标准 (试行) 》 (GB 36600-2018) [13]第一类用地筛选值 (20 mg·kg−1) 的17倍。
-
如图1(a)所示,实验装置主体由土壤室和捕集室组成,捕集室置于实验装置中部,可自由取出,2侧为土壤室。取样点位置如图1(b)所示,从阳极到阴极划分为阳极区 (S1~S3) 、捕集室 (S4) 、阴极区 (S5~S7) 3部分,S1~S7每个区域设置3个取样点,将3个取样点的土壤混合后作为该区域的代表性土壤。
-
实验共设置4个电动处理组,分别为FE-EK、AC-EK、阳极逼近处理组 (AA-EK) 、两极逼近处理组 (AAC-EK) 。其中,FE-EK处理组不移动电极;AC-EK处理组的阴极电极每隔10 d向阳极方向移动4 cm,共移动2次;AA-EK处理组的阳极电极每隔10 d向阴极方向移动4 cm,共移动2次;AAC-EK处理组的阳极电极和阴极电极每隔10 d相向各移动4 cm,共移动2次。各电动处理组土壤室内均填装1 600 g污染土壤,捕集室内填装400 g混有质量分数为20% Fe2O3的污染土壤。另取400 g混有Fe2O3的污染土壤,不通电,作为电动处理组的对照组 (CK) 。
实验以不锈钢电极为电极,电压恒定为24 V,处理时间30 d。实验过程中每隔4~5 d采用重量法补充去离子水,保持土壤含水率为30%。取样间隔为10 d,移动电极后的无电场区域不再继续取样。
-
本研究中总能耗和单位修复能耗的计算方法见式(1)和式(2)[14]。
式中:E为总能耗,kWh;U为实验电压,V;I为电流,A;t为修复时间,h。
式中:E0为单位修复能耗,kWh·mg−1;c0和c30为第0 天和第30天时捕集室中As(T)质量分数,mg·kg−1;m为捕集室中土壤质量,kg。
-
电流使用电流监控装置监测并记录。pH和电导率使用pH计 (PHS-3C型,上海仪电科学仪器股份有限公司) 和电导率仪 (CON700,美国Eutech公司) 测定[6]。Eh参考《土壤 氧化还原电位的测定 电位法》 (HJ 746—2015) [15],使用便携式ORP测定仪 (TR-901型,上海仪电科学仪器股份有限公司) 测定。As(T)质量分数利用HNO3-HF-HClO4对土壤进行分步消解[16],并用电感耦合等离子体质谱仪 (ICAPRQ,美国Thermo Fisher Scientific公司) 测定。As(III)质量分数参考ZHENG等[17]以及张静等[18]的提取方法,并用原子荧光光谱仪 (AFS-9700A,北京海光仪器有限公司) 测定[19]。As(V)质量分数为As(T)与As(III)的差值。砷赋存形态参考XU等[7]的方法依次提取可交换态砷 (Ex-As) 、铝结合态砷 (Al-As) 、铁结合态砷 (Fe-As) 以及钙结合态砷 (Ca-As) ,并用ICP-OES (Avio 220 Max,美国PerkinElmer公司) 测定。残渣态砷 (Res-As) 测定方法同As(T)。
-
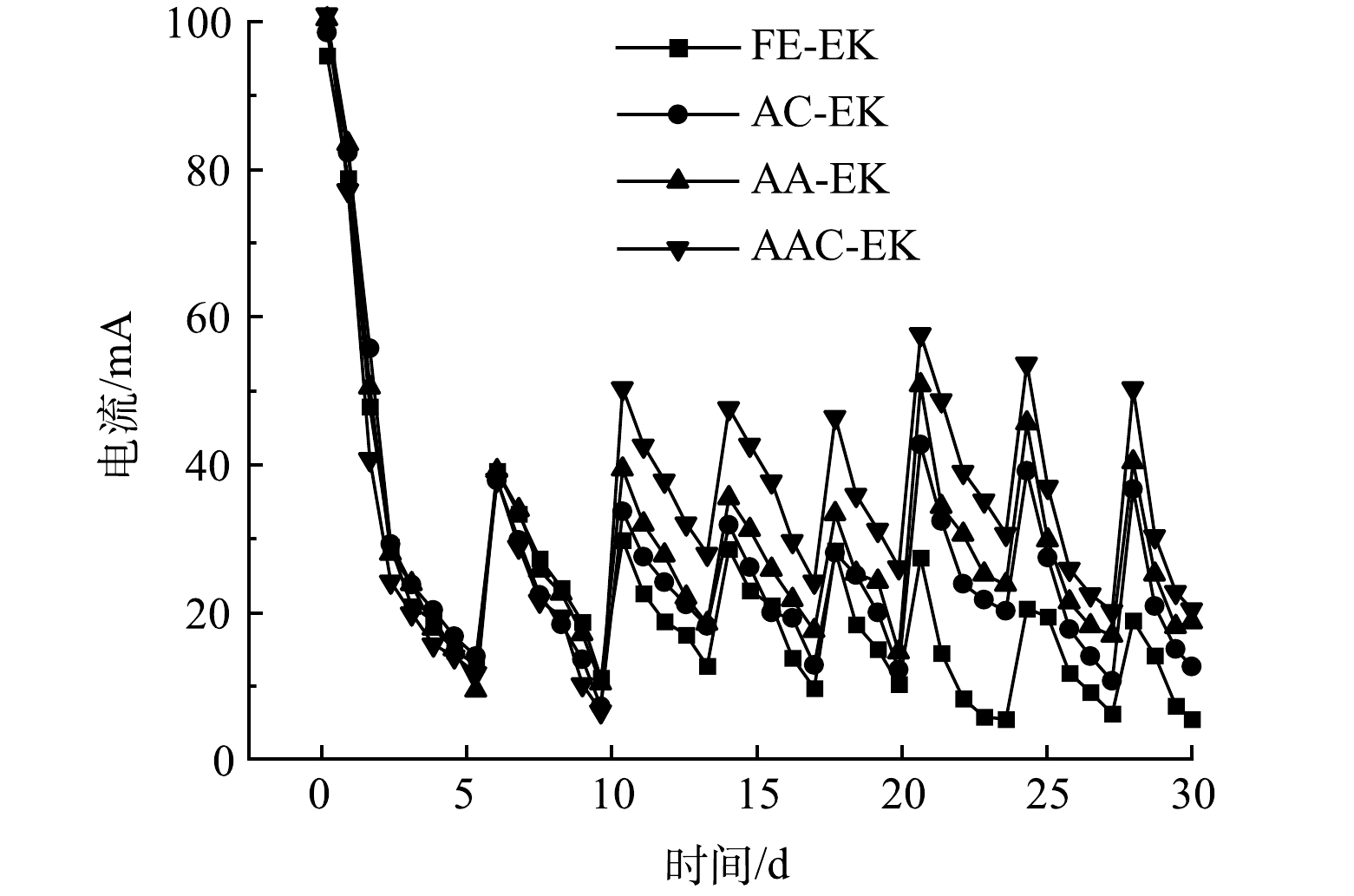
如图2所示,各处理组在移动电极前的电流值相似,表明各处理组间的平行性较好。通电后电流在短时间内即达到最大值,约为100 mA;随后电流值迅速下降,至第5 d时仅为9.42~14.04 mA;第5 d补水后电流值又迅速上升。这是因为,电动初始时土壤中含有大量可移动离子;而后电解水产生的H+和OH−被不断中和,孔隙水中的离子强度降低[9];补水后土壤中的可移动离子数量又有所增加 [20-21]。电导率常用来表示土壤孔隙液中溶解离子的数量[22]。各处理组的电导率变化如图3所示,表现为两极高、中间低。这归因于阴离子和阳离子不断迁往两极[6],降低了中间区域可溶性离子数量。各处理组电导率在S1、S4、S5区域存在显著差异 (p<0.05) 。
运行10 d后,电极逼近处理组的电流值高于固定电极处理组,以第20 d为例,FE-EK、AC-EK、AA-EK以及AAC-EK的电流值分别为27.36、42.64、50.74、57.68 mA。这主要是因为,随着电极的移动,土壤有效长度缩短,提高了系统电流[9]。因AAC-EK的两极间距最短,所以AAC-EK的电流值又高于AC-EK和AA-EK。AC-EK的电流值低于AA-EK主要是因为AC-EK能提高阴极区pH,容易生成氢氧化物、碳酸盐等不溶性和非导电物质,降低系统电流[21]。
-
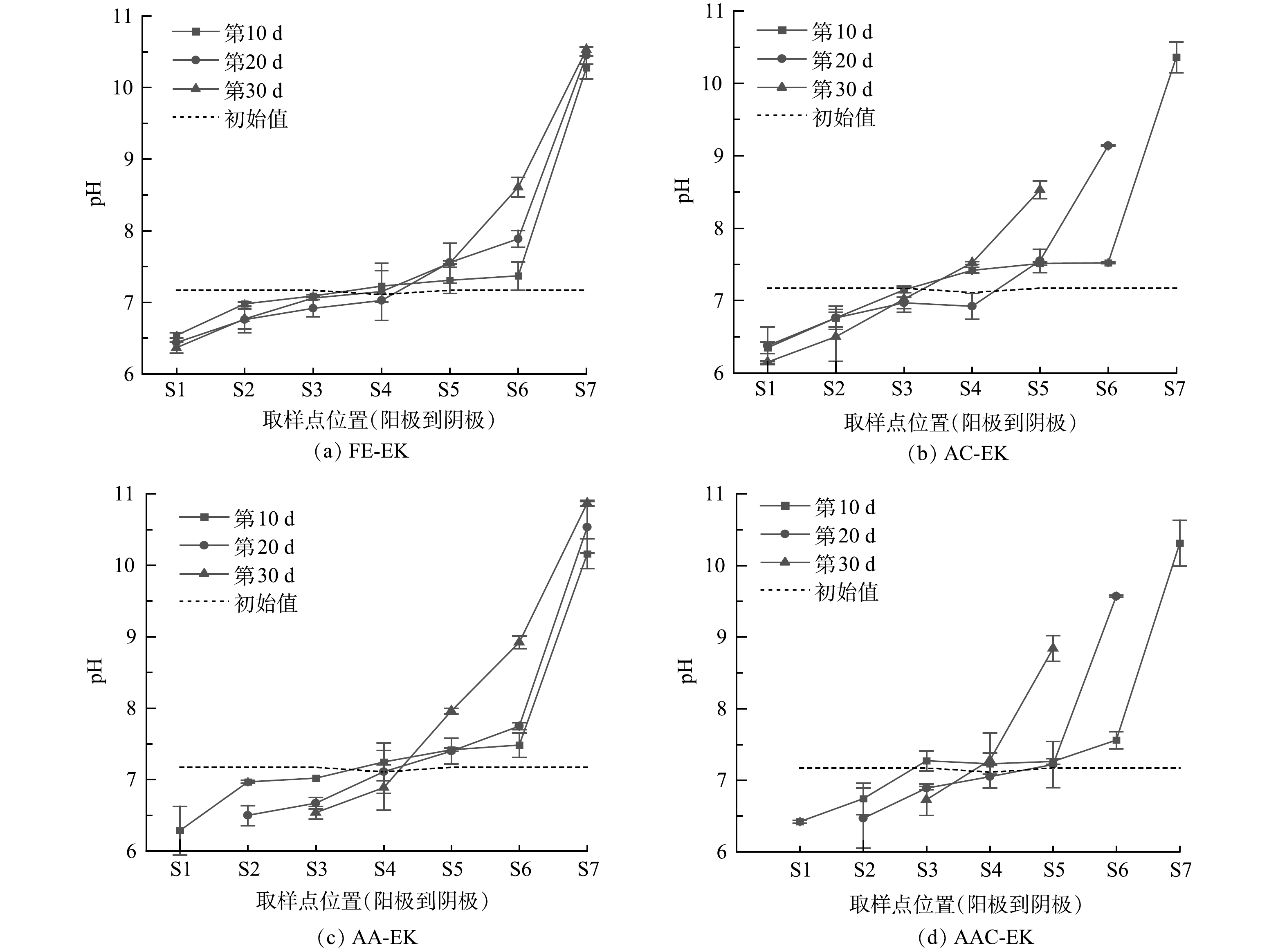
土壤室土壤初始pH为7.17,捕集室土壤初始pH为7.11。如图4所示,土壤pH从阳极至阴极呈逐渐增大趋势,且阴极区变化幅度高于阳极区。这是因为,在外加电场作用下,阳极和阴极因发生水解反应分别生成H+和OH−[9]。AA-EK能够促进阳极区pH降低,例如其S2区域在10~20 d降低0.47,高于FE-EK下降幅度,但AA-EK并未能阻止阴极区的pH升高,这可能是由于土壤的酸缓冲性能较高,向阴极移动的H+在到达阴极区前就被消耗殆尽。反之,AC-EK的阴极电极不断向阳极靠近,使其阴极区pH随时间的推移逐渐升高。由于AAC-EK电流值最高,导致其S2~S6区域的pH变化幅度一般高于AC-EK、AA-EK或FE-EK。
-
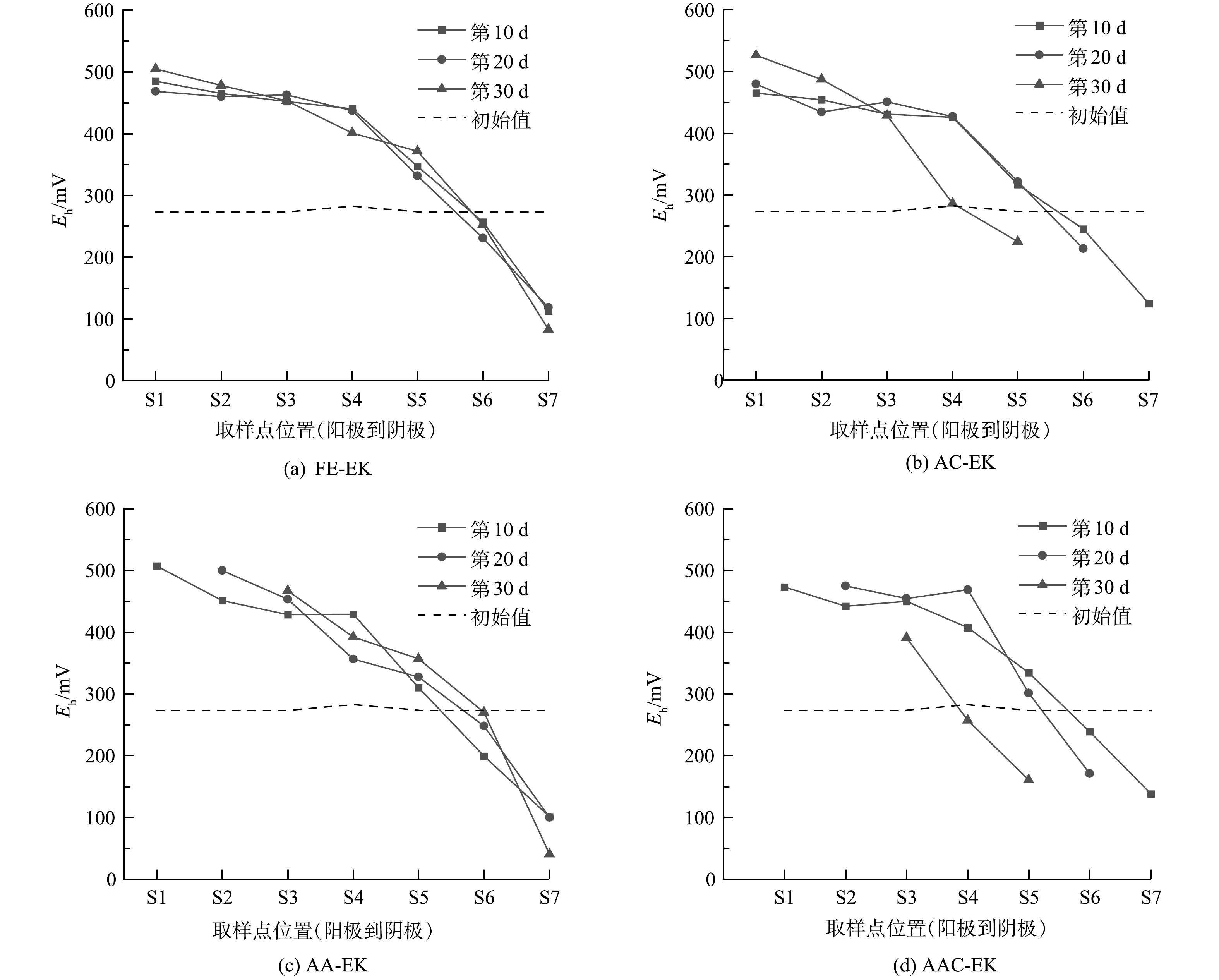
如图5所示,土壤室土壤初始Eh为273.5 mV,捕集室土壤初始Eh为282.5 mV。电动结束后,土壤Eh表现为从阳极到阴极逐渐降低的分布趋势。其中,S1~S5区域的Eh一般高于初始值,S6~S7区域的Eh低于初始值。阳极Eh的升高主要源于水电解反应产生的氧气及活性自由基;而阴极Eh的降低主要源于水解反应产生氢气,使阴极土壤处于还原气氛。与FE-EK相比,阳极电极的移动促进阳极区Eh升高,而阴极电极的移动促进阴极区Eh降低。以AC-EK为例,其第30 d时S5区域的Eh比FE-EK低147 mV,与SHEN等[23]的研究结果一致。
-
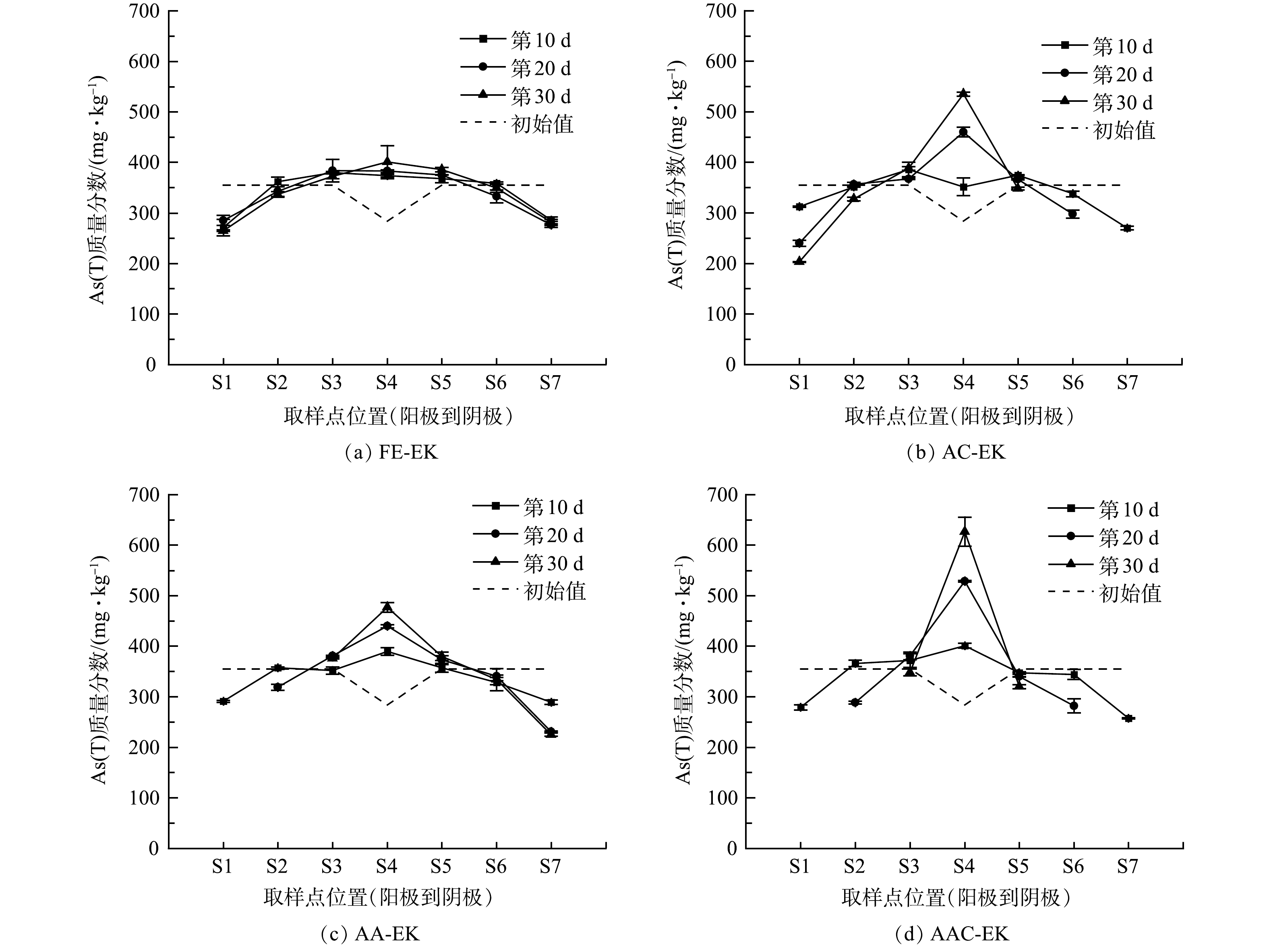
如图6所示,土壤室土壤初始As(T)质量分数为355.08 mg·kg−1,捕集室土壤初始As(T)质量分数为283.97 mg·kg−1。修复过程中,土壤中As(T)从两极区域向中间区域聚集,并最终呈现两极低、中间高的分布趋势。As(T)分布的变化是因为,As(T)在电场作用下同时受到电迁移和电渗析作用,一方面,带负电荷的H2AsO4−、HAsO42−、H2AsO3−等随电迁移迁往阳极;另一方面,溶解于土壤孔隙水中的砷随电渗流迁往阴极[24],导致两极及其附近区域As(T)质量分数降低。由于土壤中对砷吸附能力较强的铝、铁、钙等元素较多,可与砷形成不可移动的沉淀,导致砷移动性显著降低;此外,捕集室中Fe2O3对砷具有很强的吸附能力,迁移至此的砷难以继续向两极迁移,使得捕集室中As(T)质量分数不断升高。运行30 d后,AC-EK、AA-EK以及AAC-EK捕集室中As(T)质量分数与初始值相比显著升高 (p<0.05) ,S1、S7区域As(T)质量分数显著性降低 (p<0.05),以AAC-EK处理组As(T)质量分数显著性降低点位最多 (S1、S2、S5、S6、S7) ,而FE-EK捕集室中As(T)质量分数与初始值相比无显著性差异 (p>0.05) ,仅S1区域As(T)显著性降低 (p<0.05) ,这表明电极逼近对As(T)的迁移具有显著促进作用。
运行30 d后,FE-EK的As(T)整体迁移率最低 (15.38%) ,AAC-EK的As(T)整体迁移率最高 (31.50%) ,AC-EK与AA-EK居于2者之间 (27.25%、21.65%) 。AC-EK之所以能促进砷的迁移主要因为以下几个方面:首先,电极间距的缩短增大了系统电流,加速了砷的迁移;其次,阴极电极的移动增大了阴极区土壤pH,使土壤对带负电荷的砷酸根和亚砷酸根吸附能力减弱[2],且OH−能置换出以含氧阴离子形式存在的砷[25];此外,阴极电极的移动还降低了土壤Eh,使Fe(III)向Fe(II)转化,Fe(OH)3等铁系物因此发生溶解[26],砷因失去吸附相被释放到土壤溶液中,有利于砷的迁移。AA-EK因电极间距的缩短增大了系统电流,同样能促进As(T)的迁移;但因为其阳极区域pH不断降低,增强了土壤对砷的吸附,导致促进效果不明显。虽然AAC-EK阳极区pH也较低,但它的电流值最高,且其阴极区pH最高,Eh最低,有利于砷的解吸,所以AAC-EK对As(T)的迁移效果优于AC-EK和AA-EK。
如图7所示,FE-EK、AC-EK、AA-EK、AAC-EK的总电能耗依次为373.46、449.59、496.46、572.64 kWh,单位修复能耗依次为7.98、4.47、6.44、4.18 kWh·mg−1。可见,总电能耗最高的AAC-EK的单位修复能耗最低。这是因为,当电压一定时,单位修复能耗除了与电流强度有关还与污染物迁移量有关,AAC-EK捕集室中的As(T)的增加量为FE-EK的2.93倍。
-
初始土壤中,As(V)为无机砷的主要形式,约为As(III)的1.95倍。电动处理30 d后As(V)的分布如图8(a)所示。As(V)表现为中间高、两极低的分布趋势,FE-EK、AC-EK、AA-EK、AAC-EK捕集室中As(V)质量分数依次升高60.62%、120.61%、93.99%、162.86%。这是因为,阴极带负电荷的As(V)不断移向阳极,在迁移过程中,pH逐渐降低,As(V)迁移能力随之下降;且中间区域的Fe2O3对As(V)有强亲和力,导致As(V)移动至捕集室后难以继续移动,并最终停滞在捕集室;另外,由于电渗析流会带动部分溶解于土壤间隙液中As(V)向阴极迁移,导致阳极区的As(V)也有不同程度的降低。各处理组间As(V)分布差异主要集中在阴极区,AC-EK和AAC-EK能够升高阴极区pH,进而提高砷的移动性,所以这2个处理组阴极区的As(V)质量分数低于AA-EK和FE-EK;又因为AA-EK电流较大,所以其阴极区的As(V)质量分数又低于FE-EK。
As(III)的分布如图8(b)所示。As(III)与As(V)分布趋势一致,为中间高、两极低。这是因为,阳极区土壤pH<9.2,As(III)以不带电的分子形式 (H3AsO3) 存在,主要受电渗析作用迁往阴极[27];在阴极区,越接近阴极土壤pH越高,As(III)又以分子形式向含氧酸根形式转化,带负电荷的亚砷酸根 (H2AsO3−、HAsO32−、AsO33−) 逐渐增多,并随电迁移迁往阳极,导致S6、S7区域的As(III)质量分数低于S5区域。对比来看,各处理组阳极区As(III)质量分数从低到高依次为AAC-EK、AA-EK、AC-EK、FE-EK。处理组间的差异可能与电流强度有关,当电流较大时电渗析作用较强,更多的As(III)受电渗析作用迁移向阴极,所以电流越大阳极区的As(III)残留量越低,同时使得捕集室中As(III)质量分数越高。
由于土壤Eh普遍升高,导致部分As(III)转化为As(V)。运行30 d后,FE-EK、AC-EK、AA-EK、AAC-EK各点位As(III)平均质量分数较初始值分别降低9.78%、7.81%、13.65%、4.09%。与此同时,As(V)质量分数随之升高。有研究指出,As(III)的毒性高于As(V)[3],因此,经电动修复土壤中砷的毒性被降低。比较而言,AA-EK因能提高阳极区Eh,所以对As(III)的削减量最高;AAC-EK虽然也能提高阳极区Eh,但其阴极区Eh明显降低,所以对As(III)的总体削减效果较差。
-
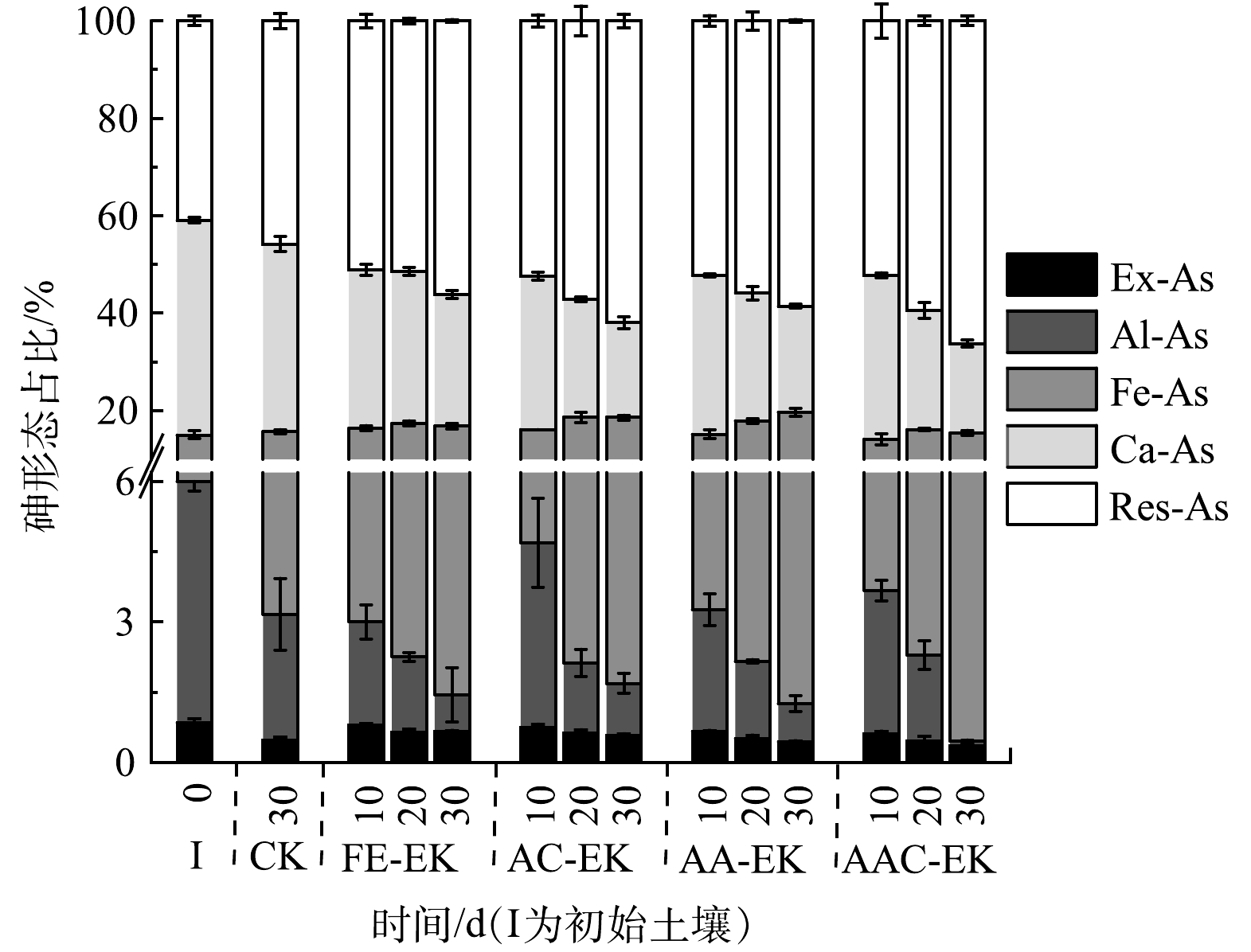
各处理组捕集室中砷的形态分布如图9所示。初始土壤中各形态砷占比从低到高依次为Ex-As (0.84%)、Al-As (5.16%)、Fe-As (9.05%)、Res-As (40.88%)、Ca-As (44.07%)。砷在电场的作用下不断向捕集室中迁移,并在Fe2O3的作用下发生赋存形态的明显转化,表现为Ex-As、Al-As、Ca-As占比下降,Fe-As和Res-As占比上升。对比各处理组砷赋存形态占比可知,FE-EK处理组的Ex-As最终占比最高,AA-EK、AAC-EK处理组的Ex-As最终占比较低,分别为0.44%和0.36%;FE-EK处理组的Res-As最终占比最低,AAC-EK处理组Res-As占比最终最高,达64.98%,为CK的1.42倍。
Ex-As占比的降低是因为Fe2O3的加入为砷提供了更多的吸附位点,使Ex-As转化为Fe-As。由于AA-EK、AAC-EK的电流较大,且阳极区pH相对较低,电极腐蚀后产生的Fe2+/Fe3+在随电迁移迁往阴极的过程中因pH逐渐增大而被沉淀于捕集室中,进一步加强了对Ex-As的吸附,导致其Ex-As占比较低。Al-As占比的降低也可能是受Fe2O3的影响。胡丽琼等[28]研究发现,当向砷污染水稻土中加入Fe2O3的量达到0.5 mg·kg−1时,Al-As已降至检测限以下。Res-As占比的升高一方面是由于Al-As、Ca-As向Res-As转化;另一方面,砷被铁吸附后形成Fe-As双核或单基配体化合物,或通过发生化学反应使沉淀于铁氧化物表面的砷酸盐形成砷酸铁沉淀,进而生成Res-As [29-31]。不同赋存形态砷的生物有效性从大到小依次为Ex-As>Ca-As>Al-As>Fe-As>Res-As[29]。可见,经电动修复后,砷的生物有效性大幅度降低。因AAC-EK处理组Ex-As占比最低,Res-As占比最高,所以处理效果最好。
-
1) 相比于固定电极,3种电极逼近方式通过影响环境因子 (pH、Eh) 以及系统电流,对As(T)的迁移具有促进作用,以AAC-EK的As(T)整体迁移率最高 (31.50%) ,且单位修复能耗最低。
2) 砷的价态转化受Eh影响,电动修复后,各处理组As(III)平均质量分数较初始值有所降低,As(V)平均质量分数较初始值有所升高。
3) 电动联合Fe2O3施用可使砷的形态从Ex-As、Al-As、Ca-As向Fe-As、Res-As转化,降低捕集室中的砷的生物有效性,以AAC-EK的稳定化效果最佳。由此可见,AAC-EK在修复砷污染土壤方面具备很大潜力。
电动修复过程中电极逼近对土壤砷迁移与形态转化的影响
Effect of the approaching electrode on the soil arsenic migration and speciation transformation during electrokinetic remediation
-
摘要: 以高浓度砷污染土壤为修复对象,探究电极逼近法耦合捕集室对砷污染土壤的修复效果。实验设置4个处理,分别为固定电极 (FE-EK) 、阴极逼近 (AC-EK) 、阳极逼近 (AA-EK) 和两极逼近 (AAC-EK) 。结果表明,AC-EK、AA-EK以及AAC-EK对总砷[As(T)]的迁移具有促进作用,表现为捕集室中As(T)质量分数与初始值相比显著升高 (p<0.05) ,而FE-EK捕集室中As(T)质量分数与初始值相比无显著性差异 (p>0.05) ,As(T)整体迁移率以AAC-EK最高 (31.50%) ,FE-EK最低 (15.38%) 。由于土壤整体氧化还原电位升高,使得FE-EK、AC-EK、AA-EK、AAC-EK处理组三价砷平均质量分数较初始值分别降低9.78%、7.81%、13.65%、4.09%。砷的生物有效性在Fe2O3和电动效应的联合作用下不断降低,表现为可交换态砷、铝结合态砷、钙结合态砷向铁结合态砷、残渣态砷转化。本研究结果表明,AAC-EK促进As(T)迁移的效果最好,可交换态砷占比最低,且单位修复能耗最低,具有良好的砷污染土壤修复潜力。Abstract: The remediation effect of the approaching electrode technique coupled with a capture chamber was evaluated using soil contaminated with a high concentration of arsenic. Four treatments were set up in the experiment: fixed-electrode electrokinetic (FE-EK), approaching cathode electrokinetic (AC-EK), approaching anode electrokinetic (AA-EK), and approaching anode and cathode electrokinetic (AAC-EK) techniques. The results demonstrated that AC-EK, AA-EK, and AAC-EK techniques promoted the migration of total arsenic [As(T)]: the mass fraction of As(T) in their capture chambers was significantly higher than the initial value(p<0.05), while that in the capture chamber of FE-EK did not differ significantly from the initial value(p>0.05). The overall migration rates of As(T) in AAC-EK was the highest (31.50%), and that in FE-EK was the lowest (15.38%). The average mass fraction of trivalent arsenic in FE-EK, AC-EK, AA-EK, and AAC-EK techniques decreased by 9.78%, 7.81%, 13.65%, and 4.09%, respectively, compared with the initial value owing to an increase in the overall soil redox potential. Under the combined influence of Fe2O3 and electric-influence, the bioavailability of arsenic was continuously reduced, as evidenced by the conversion of exchangeable arsenic, aluminum-bound arsenic, and calcium-bound arsenic to iron-bound arsenic and residual arsenic. This study showed that AAC-EK was associated with the highest As(T) migration, lowest proportion of exchangeable arsenic, and lowest energy consumption per unit of remediation. Thus, the AAC-EK technique had good potential for remediating arsenic-contaminated soil.
-
为实现“碳中和”目标,低碳脱氮新技术的开发及其工程化应用成为污水处理领域的研究热点。污水生物脱氮的主要途径为传统的硝化反硝化反应,但其工程应用仍存在能耗高且难实现低碳化的问题。厌氧氨氧化(anaerobic ammonium oxidation,anammox)可在厌氧条件下,以氨氮为电子供体、亚硝氮为电子受体,实现氨氮和亚硝氮的同步脱除并生成氮气。与传统硝化反硝化工艺相比,基于厌氧氨氧化的新工艺可有效降低曝气成本、减少污泥产量及有机碳源需求量,是未来污水生物脱氮的重要发展方向。为更好地宣传该领域最新成果,《环境工程学报》编辑部邀请李玉友、刘思彤、陈荣3位教授组织了“厌氧氨氧化生物脱氮理论与技术发展”专题。本文梳理和总结厌氧氨氧化的发现过程、理论研究、工艺发展、工程应用及其新动态,并作为该专题的代序言。
1. 厌氧氨氧化生物反应的发现及对菌种的初步探索
1.1 厌氧氨氧化生物反应的发现
1977年,奥地利理论化学家BRODA Engelbert运用热力学理论推断并提出存在一种生物反应,可利用氨氮和亚硝氮生成氮气(式1)[1]。然而,传统学者认为:由于氨氮具有化学惰性,要氧化氨氮需要氧气和混合功能的加氧酶[2],故厌氧氨氧化是不可行的[2]。
NH+4+NO–2→N2+H2O,ΔG=−358kJ⋅mol−1 (1) 20世纪90年代,荷兰Gist Brocades公司的MULDER Arnold发现该公司的反硝化中试流化床装置中存在一个有趣的现象:在厌氧条件下,当污水中含有硝氮时,反应器中氨氮明显减少并伴有氮气的产出。该现象被命名为厌氧氨氧化(anaerobic ammonium oxidation)[3]。MULDER Arnold将此现象介绍给了荷兰代尔夫特理工大学的KUENEN Gijs教授。1995年,KUENEN Gijs的学生VAN DE GRAAF Astrid通过向基质中加入N15标记
NH+4 NO−2 1.2 对厌氧氨氧化菌的初步探索
KUENEN Gijs团队从1997年开始对anammox展开系统性的基础研究。首先,STROUS Marc利用序批式反应器将厌氧氨氧化菌纯化至70%以上,确定了anammox菌隶属于浮霉菌门(Planctomycete),将实验株命名为Candidatus Brocadia anammoxidans,并提出了厌氧氨氧化反应的化学计量式(式(2))[5-6]。由式(2)可看出,anammox菌的增殖系数非常低,消耗每摩尔
NH+4 NH+4+1.32NO–2+0.066HCO–3+0.13H+→1.02N2+0.26NO–3+2.03H2O+0.066CH2O0.5N0.15 (2) 随后,KUENEN Gijs团队与澳大利亚昆士兰大学FUERST John A团队合作,基于电子显微镜对厌氧氨氧化细胞结构的观察,发现其独特的隔膜结构,并将其命名为厌氧氨氧化体(Anammoxosome)[9]。STROUS Marc和JETTEN Mike S M对神秘的厌氧氨氧化菌进行了宏基因分析,确认了潜在的中间代谢产物羟氨(NH2OH)和肼(N2H4)及潜在的代谢路径[10]。基于宏基因分析结果,JETTEN Mike S M的学生DE ALMEIDA Naomi M进一步确定了关键的厌氧氨氧化酶在不同细胞结构中的位置,并提出厌氧氨氧化体是厌氧氨氧化生物反应的关键细胞结构[11]。
2. 厌氧氨氧化菌的培养和富集
现有厌氧氨氧化菌的培养和富集技术有5种:1) KUENEN Gijs和JETTEN Mike S M团队利用序批式反应器富集絮状anammox污泥[5-6];2)荷兰代尔夫特理工大学VAN LOOSDRECHT Mark培育厌氧氨氧化菌时形成了厌氧氨氧化颗粒污泥[12];3)美国马里兰大学生物技术研究所SCHREIER Harold J团队利用移动床反应器形成厌氧氨氧化菌附着在移动床反应器(moving bed biofilm reactor,MBBR)载体上[13];4)日本熊本大学的古川憲治团队使用固定载体富集厌氧氨氧化菌[14];5)VAN LOOSDRECHT Mark团队通过膜生物反应器培养菌种时形成了厌氧氨氧化游离细胞悬浮液,并修正了厌氧氨氧化反应的化学计量式(即式(3))[15]。2010年,浙江大学郑平教授团队的唐崇俭(现就职于中南大学)利用升流式厌氧污泥床反应器(up-flow anaerobic sludge bed/blanket,UASB)富集出高污泥浓度厌氧氨氧化颗粒污泥,最大容积氮去除速率达到74.3~76.7 kg·(m3·d)−1[16]。郑平教授的另一个学生金仁村(现就职于杭州师范大学)对多种anammox抑制物(包括重金属、抗生素等)的作用进行了机理研究,为厌氧氨氧化反应应用于光伏行业、制药行业以及农牧业废水的处理提供了参考[17]。
NH+4+1.146NO–2+0.071HCO–3+0.057H+→0.986N2+0.161NO–3+2.002H2O+0.071CH1.72O0.31N0.2(厌氧氨氧化菌) (3) 式中:CH1.72O0.31N0.2为厌氧氨氧化菌生物质的化学式。
图1概述了厌氧氨氧化生物反应的发现、探索,以及厌氧氨氧化菌的富集技术研究,为该研究的初步阶段。
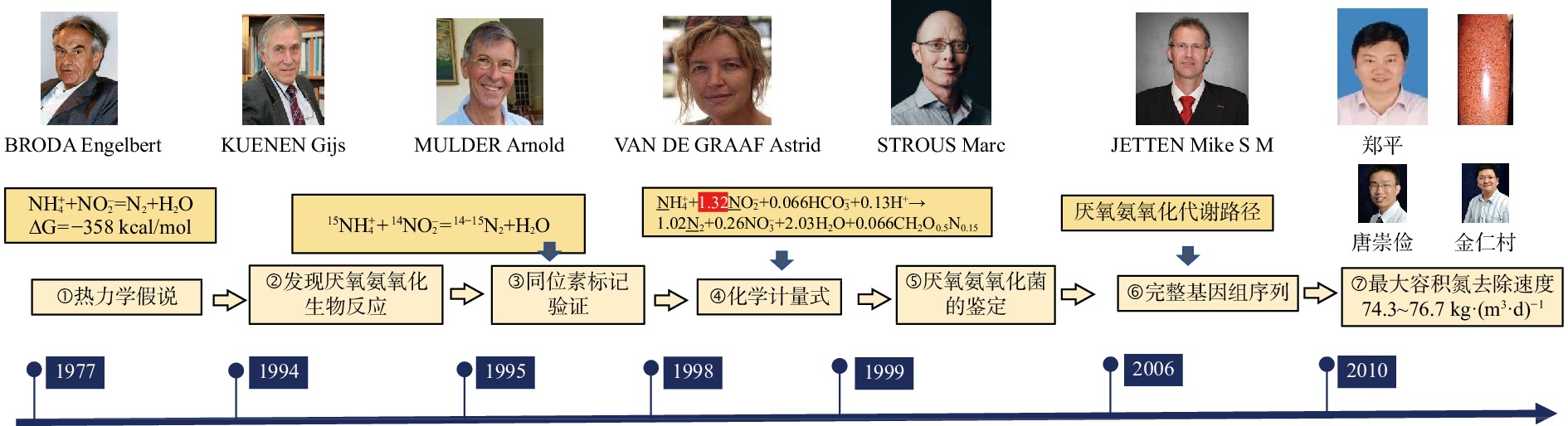 图 1 厌氧氨氧化生物反应发现与探索进程Figure 1. Discovery and exploration of biological reaction of anammox
图 1 厌氧氨氧化生物反应发现与探索进程Figure 1. Discovery and exploration of biological reaction of anammox3. 厌氧氨氧化技术的早期工程应用
1)一段式工艺的演进。1998年,比利时根特大学的VERSTRAETE Willy团队采用序批式反应器率先提出氧限制型自养硝化反硝化工艺(oxygen-limited autotrophic nitrification-denitrification,OLAND) [18]。2001年,JETTEN Mike S M团队以氧气为限制条件控制部分亚硝化过程,利用序批式反应器提出了基于亚硝酸盐的完全自养脱氮CANON(Completely Autotrophic Nitrogen removal Over Nitrite)的一段式工艺。该工艺的反应式为式(4)[19]。
NH3+0.85O2→0.44N2+0.11NO–3+1.43H2O+0.14H+ (4) 2005年,古川憲治团队使用固定床生物膜培养富集厌氧氨氧化菌,并提出一段式部分亚硝化-厌氧氨氧化脱氮工艺(single-stage nitrogen removal using anammox and partial nitritation,SNAP)[20]。由于厌氧氨氧化反应会产生硝氮,其比例达进水总氮的7.5%~11.2%,故需要进一步去除。2008年,大连理工大学杨凤林团队结合反硝化工艺提出了同步部分亚硝化、厌氧氨氧化和反硝化工艺(simultaneous partial nitrification, anammox and denitrification,SNAD),以去除剩余的硝氮[21]。
2)针对高浓度含氮污水的改进工艺。1997—1998年,德国的80 m3的全程自养脱氮(Deammonification,DEMON)工艺[22]和瑞士的33 m3生物转盘反应器在处理高浓度氨氮的垃圾渗滤液时均发生厌氧氨氧化反应[23]。2001—2006年,VAN LOOSDRECHT Mark及其同事与帕克公司合作,用两年时间在荷兰鹿特丹市启动了第1个两段式SHARON®-Anammox®脱氮示范工程,用以处理污泥消化回流液[12,24]。该工艺的前段SHARON(Single reactor system for High-rate Ammonium Removal Over Nitrite)是采用1 800 m3完全混合式的部分亚硝化工艺(partial nitrification,PN),而PN出水进入70 m3内循环(internal circulation,IC)厌氧反应器,并通过厌氧氨氧化颗粒污泥转化为氮气[12,25]。2004年,WETT Bernhard和HELL Martin在奥地利Strass污水处理厂采用500 m3序批式反应器用两年半时间成功启动一段式厌氧氨氧化悬浮污泥脱氮工艺(DEMON®)(反应式为式(5))[26]。该工艺基于硝化反应产H+原理,通过检测pH调节曝气实现部分亚硝化,并采用旋流分离器和微筛选择器截留污泥,以控制污泥停留时间、减少厌氧氨氧化菌流失[27]。
NH3+0.804O2+0.071HCO–3→0.436N2+0.009C5H7O2N(氨氧化菌)+0.028CH2O0.5N0.15(厌氧氨氧化菌)+0.111NO–3+1.46H2O+1.038H+ (5) 2010年,为进一步去除厌氧氨氧化反应产生的硝氮,台湾省交通大学林志高团队在台北市成功启动同步部分亚硝化、厌氧氨氧化和反硝化(SNAD)工艺(384 m3),用以处理垃圾渗滤液[28]。2014年,美国俄克拉荷马市蓝色平原污水处理厂建立全球最大的侧流全程自养脱氮工艺(DEMON®)。该污水处理厂的氮处理能力为11 800 kg·d−1,并且节省了60%的能耗、减少了90%的污泥[29]。
截止2014年,全球范围内采用厌氧氨氧化工艺建成的商用污水处理厂已超过100座,但这些污水处理厂主要用于处理高氨氮浓度的污水[30]。
3)针对低浓度含氮污水(市政污水)的改进工艺。2016年,新加坡公用事业局的曹业始及其同事在樟宜再生水厂的市政污水处理单元(处理量为20×104t·d−1)中实现了部分厌氧氨氧化工艺(处理贡献率>30%)[31]。该工艺较传统脱氮工艺降低了10%~30%能耗,减少了10%~40%池容[31]。2019年,北京工业大学彭永臻院士团队利用移动床生物膜反应器(MBBR)改造西安第四污水厂(处理量为25×104t·d−1),实现了部分厌氧氨氧化工艺(处理贡献率>16%)[32]。2021年,日本东北大学李玉友教授团队在市政污水处理厂的厌氧膜生物反应器出水后实现了中试规模一段式流动载体型的部分亚硝化-厌氧氨氧化工艺(处理贡献率>89%),并开发了中试规模一段式部分亚硝化-厌氧氨氧化-羟基磷酸钙型的脱氮磷回收工艺(partial nitritation/anammox-hydroxyapatite,PNA-HAP)[33–35]。
4. Anammox工艺开发新进展
1)利用甲烷型反硝化耦合厌氧氨氧化工艺。2013年,澳大利亚昆士兰大学袁志国团队提出利用甲烷型反硝化耦合厌氧氨氧化的工艺[36]。该工艺中,厌氧甲烷氧化反硝化(denitrifying anaerobic methane oxidation,DAMO)菌可利用厌氧氨氧化反应产出的硝氮及发酵液中的溶解性甲烷,进而生成亚硝氮或者氮气(反应见式(6)和(7))。之后,亚硝氮还可继续被厌氧氨氧化菌利用。该工艺有3个优势:提高脱氮效率;去除厌氧出水中溶解性甲烷,防止温室气体甲烷释放至大气中;无需额外补充有机物就能去除硝氮。
NO–3+0.25CH4→0.25NO–2+0.5H2O+0.25CO2 (6) NO–2+0.375CH4+H+→0.5N2+1.25H2O+0.375CO2 (7) 2)部分反硝化耦合厌氧氨氧化工艺。2013年,北京工业大学彭永臻院士团队提出了部分反硝化(
NO−3 NO−2 NO−3 NO−2 NO–3+有机物→NO–2+CO2 (8) 3)同步脱氮回收磷型颗粒污泥工艺。2018年,李玉友团队利用膨胀颗粒污泥床(expanded granular sludge bed,EGSB),在厌氧氨氧化颗粒污泥的基础上耦合化学结晶法,实现了磷的回收(反应式为式(9))。该工艺中形成的新型anammox-HAP颗粒污泥外层为厌氧氨氧化生物膜,可实现脱氮,其内核为碱式磷酸钙结晶[39]。这种颗粒污泥的沉降速度可达到300 m·h−1,使得系统污泥流失率大大减小[40]。该工艺已发展出一段式和两段式2种类型,在温度为7~35 ℃、进水总氮浓度为200~1 500 mg·L−1条件下,已长期运行并高效地实现了氮的去除和磷的回收[41-42]。
NH+4+1.146NO–2+0.071HCO–3+0.057H++0.285Ca2++0.171PO3−4→0.986N2+0.161NO−3+2.002H2O+0.071CH1.72O0.31N0.2(厌氧氨氧化菌)+0.285Ca10(PO4)6OH2(羟基磷酸钙) (9) 式中:CH1.72O0.31N0.2为厌氧氨氧化菌生物质的化学式。
4)反硝化聚磷酸微生物耦合厌氧氨氧化工艺。2018年,阿里格尔穆斯林大学BASHEE Farrukh团队和北京工业大学彭永臻院士团队先后提出利用反硝化聚磷酸菌回收污水中磷的耦合厌氧氨氧化工艺。该工艺既可减少厌氧氨氧化产出硝氮,又能高效地实现磷的回收[43-44]。利用该工艺处理市政污水时,出水的平均TN为5.0 mg·L−1、平均
NH+4 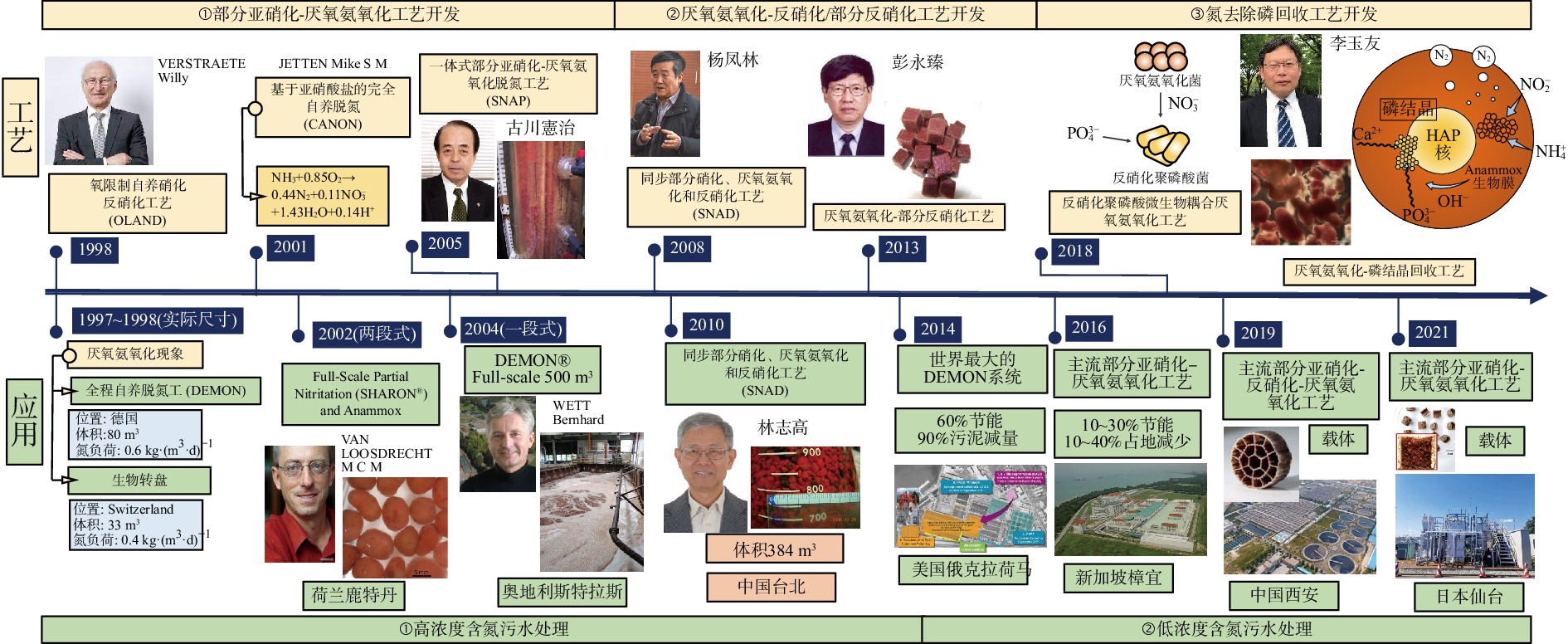 图 2 厌氧氨氧化的工艺开发与应用进展Figure 2. Progress in development and application of anammox process
图 2 厌氧氨氧化的工艺开发与应用进展Figure 2. Progress in development and application of anammox process5. 展望
如图2所示,历经多年的发展,厌氧氨氧化工艺开发已取得诸多新进展新突破,但是,在厌氧氨氧化的实际工程应用中仍存在菌种倍增时间长、亚硝氮来源受限、易发生抑制、运行不稳定等诸多问题。如何高效富集anammox细菌、快速启动反应器,以及如何优化调控
NO−2 NH+4 NO−3 -
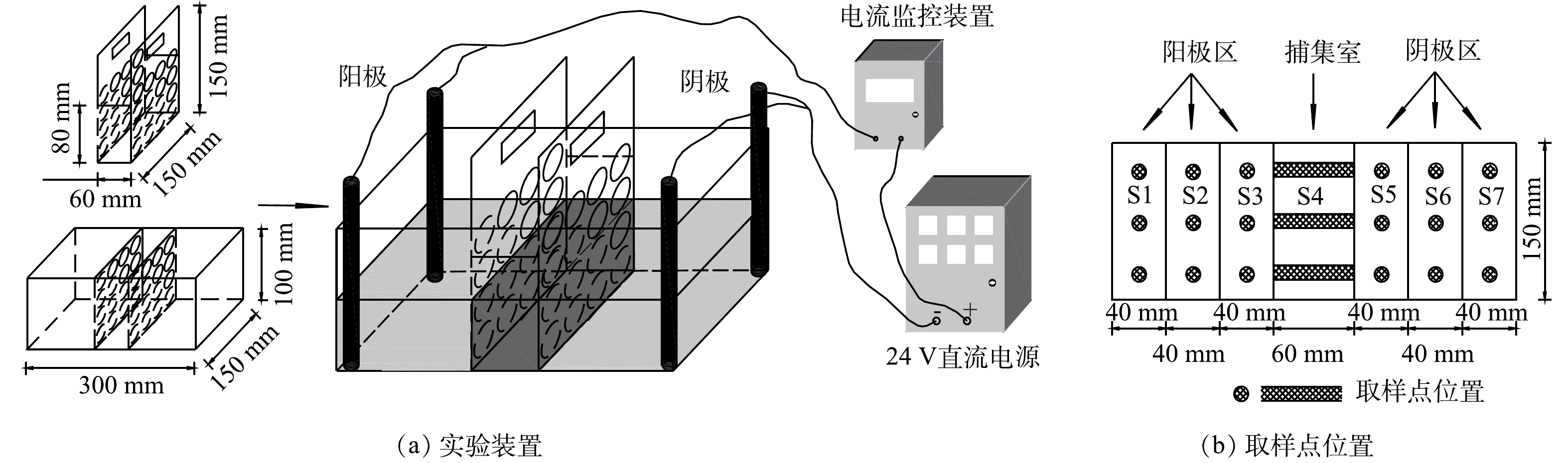
图 1 实验装置及取样点位置
Figure 1. Schematic diagram of experimental setup and sampling positions
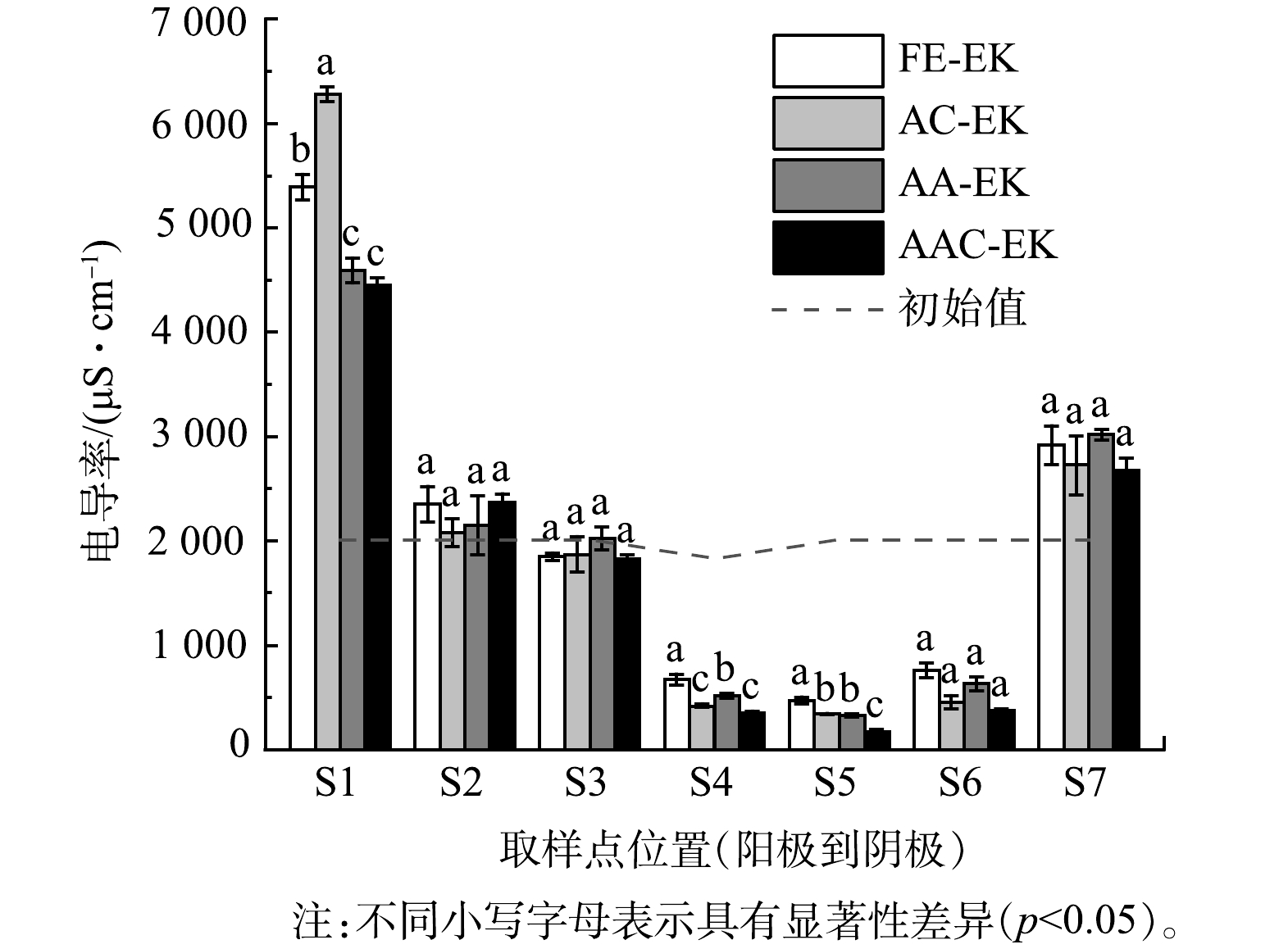
图 3 电动修复后土壤电导率分布
Figure 3. Distribution of conductivity in soil after electrokinetic treatment
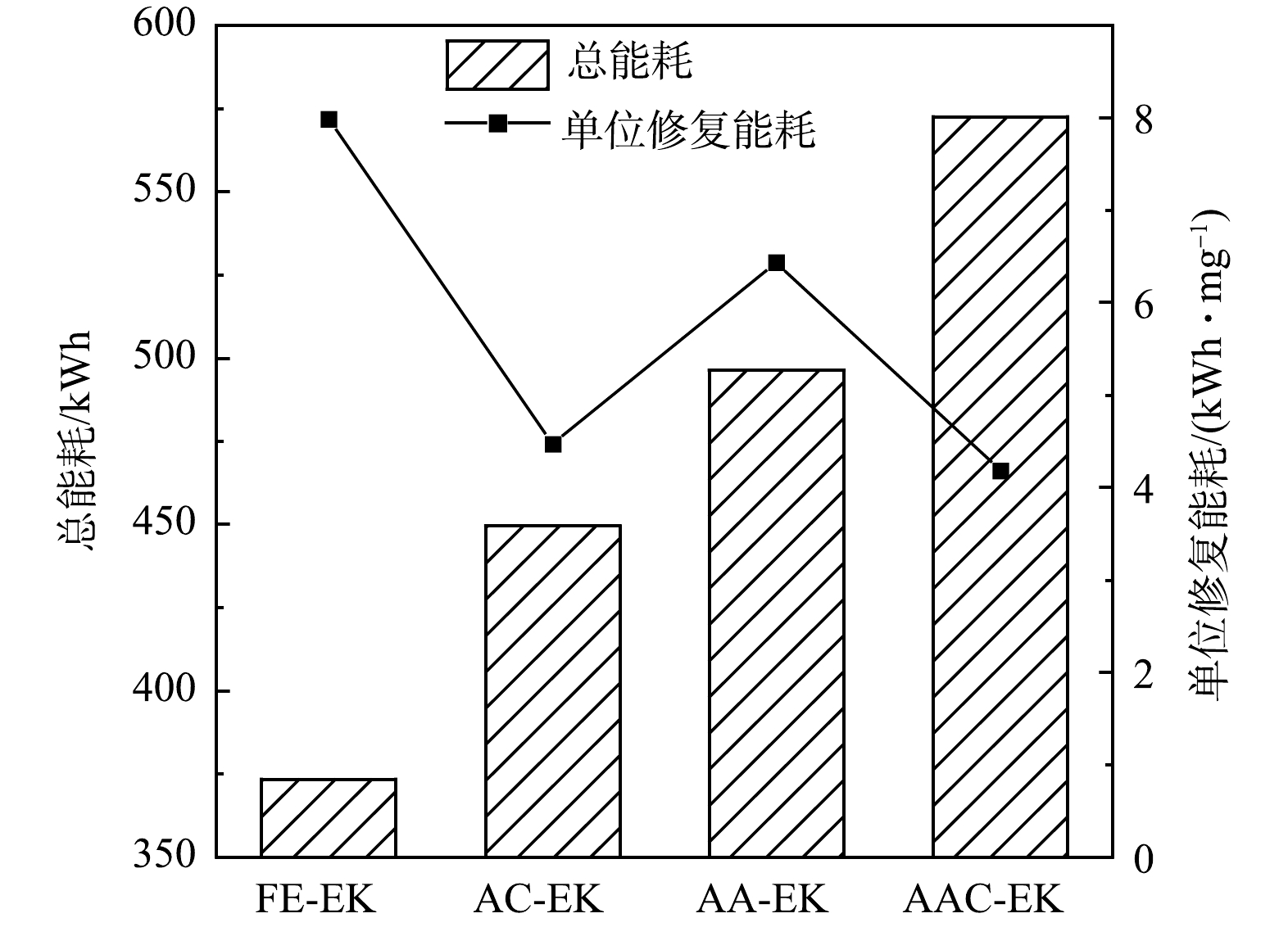
图 7 不同电动处理组总能耗及单位修复能耗
Figure 7. Total energy consumption and energy consumption per unit of remediation of different electrokinetic treatments

图 8 电动结束后土壤中As(V)和As(III)分布
Figure 8. Distributions of As(V) and As(III) in soil after electrokinetic treatment
-
[1] 赵宇, 艾雯妍, 文思颖, 等. 微生物-植物联合修复镉砷污染农田土壤技术与应用[J]. 生态毒理学报, 2022, 17(6): 144-162. [2] LI J P, DING Y, WANG K L, et al. Comparison of humic and fulvic acid on remediation of arsenic contaminated soil by electrokinetic technology[J]. Chemosphere, 2020, 241: 125038. doi: 10.1016/j.chemosphere.2019.125038 [3] 吕紫娟, 王华伟, 吴雅静, 等. 纳米零价铁物相转变对砷污染土壤稳定化效果和潜在毒性的影响[J]. 环境工程, 2022, 40(3): 24-31. [4] 骆永明, 滕应. 我国土壤污染的区域差异与分区治理修复策略[J]. 中国科学院院刊, 2018, 33(2): 145-152. doi: 10.16418/j.issn.1000-3045.2018.02.003 [5] ALKA S, SHAHIR S, IBRAHIM N, et al. Arsenic removal technologies and future trends: A mini review[J]. Journal of Cleaner Production, 2021, 278: 123805. doi: 10.1016/j.jclepro.2020.123805 [6] MA C Z, LI J P, XIA W, et al. Effect of additives on the remediation of arsenic and chromium co-contaminated soil by an electrokinetic-permeable reactive barrier[J]. Environmental Science and Pollution Research, 2022, 29(8): 11966-11975. doi: 10.1007/s11356-021-16357-1 [7] XU Y F, LU Q Q, LI J P, et al. Effect of humus on the remediation of arsenic-contaminated soil by electrokinetic technology[J]. Environmental Technology & Innovation, 2021, 21(14): 101297. [8] KARACA O, CAMESELLE C, BOZCU M. Opportunities of electrokinetics for the remediation of mining sites in Biga peninsula, Turkey[J]. Chemosphere, 2019, 227: 606-613. doi: 10.1016/j.chemosphere.2019.04.059 [9] YAO W K, CAI Z P, SUN S Y, et al. Electrokinetic-enhanced remediation of actual arsenic-contaminated soils with approaching cathode and Fe0 permeable reactive barrier[J]. Journal of Soils and Sediments, 2020, 20(3): 1526-1533. doi: 10.1007/s11368-019-02459-4 [10] 付博, 王刚, 张志彬, 等. pH与Eh对郑州北郊水源地沉积物中砷溶出的影响[J]. 青岛理工大学学报, 2013, 34(4): 99-103. [11] 周一敏, 黄雅媛, 刘凯, 等. 典型铁、锰矿物对稻田土壤砷形态与酶活性的影响[J]. 环境科学, 2022, 43(5): 2732-2740. [12] JI D L, ZHANG J, MENG F S, et al. Species and distribution of arsenic in soil after remediation by electrokinetics coupled with permeable reactive barrier[J]. Water, Air, & Soil Pollution, 2020, 231(12): 567. [13] 中华人民共和国生态环境部. 土壤环境质量建设用地土壤污染风险管控标准(试行): GB 36600—2018[S]. 北京: 中国环境科学出版社, 2018. [14] 尹静玄, 王平, 徐海音, 等. 耐镉细菌联合电动技术修复镉污染土壤的研究[J]. 环境科学学报, 2020, 40(6): 2212-2219. [15] 中华人民共和国生态环境部. 土壤氧化还原电位的测定 电位法: HJ 746—2015[S]. 北京: 中国环境科学出版社, 2015. [16] 刘向磊, 孙文军, 文田耀, 等. 三酸分步消解-电感耦合等离子体质谱法测定土壤详查样品中23种金属元素[J]. 岩矿测试, 2020, 39(5): 793-800. [17] ZHENG J, HINTELMANN H, DIMOCK B, et al. Speciation of arsenic in water, sediment, and plants of the Moira watershed, Canada, using HPLC coupled to high resolution ICP–MS[J]. Analytical and Bioanalytical Chemistry, 2003, 377(1): 14-24. doi: 10.1007/s00216-003-1920-3 [18] 张静, 刘晓端, 江林. 土壤中不同形态砷的分析方法[J]. 岩矿测试, 2008(3): 179-183. doi: 10.3969/j.issn.0254-5357.2008.03.005 [19] 黄中情, 杨常亮, 张璟, 等. 碳酸氢盐对沉积物中砷迁移转化的影响[J]. 环境科学与技术, 2020, 43(11): 69-75. doi: 10.19672/j.cnki.1003-6504.2020.11.009 [20] 孟欣, 李刚, 高鹏, 等. 高羊茅对电动-微生物修复石油污染土壤的影响[J]. 农业环境科学学报, 2020, 39(7): 1532-1539. doi: 10.11654/jaes.2019-1438 [21] BESSAIM M M, KARACA O, MISSOUM H, et al. Effect of imposed electrical gradient on removal of toxic salt contaminants from alkali-saline low permeable soil during electrokinetic remediation[J]. Arabian Journal of Geosciences, 2020, 13(14): 1-12. [22] XU H T, CANG L, SONG Y, et al. Influence of electrode configuration on electrokinetic-enhanced persulfate oxidation remediation of PAH-contaminated soil[J]. Environmental Science and Pollution Research, 2020, 27(35): 44355-44367. doi: 10.1007/s11356-020-10338-6 [23] SHEN Z M, ZHANG J D, QU L Y, et al. A modified EK method with an I−/I2 lixiviant assisted and approaching cathodes to remedy mercury contaminated field soils[J]. Environmental Geology, 2009, 57(6): 1399-1407. doi: 10.1007/s00254-008-1418-6 [24] 周丽玮, 王航, 刘阳生. 转换电极的电动力强化植物修复高浓度砷污染土壤[J]. 环境工程, 2020, 38(10): 228-233. doi: 10.13205/j.hjgc.202010036 [25] SHIN S Y, PARK S M, BAEK K. Electrokinetic removal of As from soil washing residue[J]. Water, Air, & Soil Pollution, 2016, 227(7): 223. [26] 周实际, 杜延军, 倪浩, 等. 压实度对铁盐稳定化砷、锑污染土特性的影响及机制研究[J]. 岩土力学, 2022, 43(2): 432-442. doi: 10.16285/j.rsm.2021.1474 [27] RYU S R, JEON E K, BAEK K. A combination of reducing and chelating agents for electrolyte conditioning in electrokinetic remediation of As-contaminated soil[J]. Journal of the Taiwan Institute of Chemical Engineers, 2017, 70: 252-259. doi: 10.1016/j.jtice.2016.10.058 [28] 胡立琼, 曾敏, 雷鸣, 等. 含铁材料对污染水稻土中砷的稳定化效果[J]. 环境工程学报, 2014, 8(4): 1599-1604. [29] 蒋毅, 刘雅, 辜娇峰, 等. 三元复合调理剂对土壤镉砷赋存形态和糙米镉砷累积的调控效应[J]. 环境科学, 2021, 42(1): 378-385. doi: 10.13227/j.hjkx.202006126 [30] 邓天天, 胡烨, 刘帅霞, 等. Fe2O3@GO聚合物对水中As3+的吸附特性表征[J]. 生态与农村环境学报, 2018, 34(10): 930-938. [31] 蒋成爱, 吴启堂, 陈杖榴. 土壤中砷污染研究进展[J]. 土壤, 2004, 36(3): 264-270. doi: 10.13758/j.cnki.tr.2004.03.007 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2495
- HTML全文浏览数: 2495
- PDF下载数: 127
- 施引文献: 0
