

 下载:
下载:







-
海洋飞沫气溶胶(SSA)是指直接从海洋表面向大气喷射的悬浮颗粒[1-2]. 当其进入海洋边界层(marine boundary layer,MBL)后不仅可以通过吸收和散射太阳辐射而直接影响全球辐射平衡,也可以作为云凝结核(cloud condensation nuclei, CCN)和冰核(ice nuclei, IN)间接影响全球气候[3-6]. 据统计,海洋每年向大气输送的SSA可达100—300亿t[7],且远高于沙尘气溶胶[8]. 因此SSA被普遍认为是影响全球气候系统的重要调节因子. 研究发现SSA排放量的增加有助于全球气候的冷却,进而能缓解或抵消由温室气体所引发的全球变暖[9-10]. 此外,SSA在海-气界面水分、盐分、有机物和微生物的传输过程中也发挥着至关重要的作用[11]. 随着全球经济迅速发展,人为活动引起的海洋环境问题日益突出,海洋中的持久性有机污染物[12]、微塑料[13]、过渡金属[14]、藻毒素[15]、细菌和病毒[16]等均可在SSA上富集并远距离传输. 这不仅会影响SSA的化学组成,还会对大气环境和人类健康产生重大影响. 如何准确评估SSA在全球环境和气候变化中的作用成为了当前大气-海洋交叉学科亟需解决的关键科学问题.
自1953年Woodcock等将风速确定为SSA产生的主要驱动力之后[17],一系列将SSA产生与风速相联系的研究接踵而至[18-19]. 虽然风速是目前已知控制SSA产生的关键,但SSA的产生也会受到其它环境因素(海表温度、海水盐度、表面活性剂和浮游微生物活动等)的影响[20]. 由于对这些环境因素的潜在影响机制认识尚不完全,围绕SSA产生过程和相关影响因素的不确定性使其成为一个持续活跃的研究领域. 外场观测虽然可以对SSA进行原位测量,但是存在一定的时空局限性,且难以明确地分离和量化单个环境因素的影响. 针对上述的研究“短板”,近年来科研工作者在实验室中开展了大量模拟工作,并在SSA产生技术方面取得了重大进展. 同时,实验室模拟研究可实现在受控条件下深入探究不同环境因素对SSA产生过程的影响及作用机制,这对SSA的产生机制研究和全球气候效应预测都至关重要. 因此,本文系统总结了近十年来该领域模拟研究的相关进展,重点介绍SSA的产生、粒径分布、化学组成、物质富集以及影响SSA产生的关键环境因素.
-
风驱动产生的SSA粒径大小可跨越几个数量级,一般按其产生机制分为三类:膜滴、射滴和裂滴(图1).
当风速大于5 m·s−1时,波浪破碎使上表层海水夹带大量气泡. 随后气泡上浮至海洋表面,其露出水面的气泡膜不断变薄直至破裂. 气泡破裂过程中气泡膜通过液条断裂[21-23]或快速抖动[24]而产生数百个膜滴. 而后,周围海水将回填气泡破裂后留下的空腔而激起若干垂直喷射的射滴[25]. 当风速大于10 m·s−1时,风可以直接吹裂波峰而产生几十微米至几毫米的裂滴[26],其大气停留时间从几秒到几分钟不等. 由于粒径较大的裂滴大气停留时间短,因此当前研究一般忽略其在大气化学和气候效应中的贡献[2]. 一般而言,气泡破裂产生的SSA粒径范围在0.01—25 μm之间[1],其中膜滴粒径较小,主要集中在亚微米(粒径<1 μm)范围内[24];而射滴粒径较大,贡献了大部分的超微米(粒径>1 μm)颗粒[20]. 膜滴和射滴的产生数量与气泡尺寸大小密切相关. 研究发现气泡尺寸越大产生的膜滴越多. 例如,气泡直径从2 mm增加到6 mm,膜滴数量可从100个增加到1000个,增加一个数量级[27]. 相反,射滴数量随着气泡尺寸的增加而减少. 例如,直径在100—300 μm内的气泡可以产生5—6个射滴,而直径为3 mm的气泡只能产生1个射滴[28]. 此外,膜滴粒径受气泡膜厚度影响. 研究表明气泡膜厚度会随着气泡直径增加而增加,进而容易产生更大的膜滴[23, 29];而射滴粒径约为其破裂气泡直径的1/40[20]. 总之,气泡的破裂过程及尺寸大小共同控制着膜滴与射滴的产生,最终将影响SSA的粒径分布和化学组成[1, 30-32].
-
迄今为止,有关SSA的研究已经做了大量的外场观测工作,但仍然存在两个重要问题亟需解决:(1)原位SSA的产生容易受到多种环境因素耦合的影响,且无法识别各因素在SSA产生过程中的具体作用机制;(2)海洋气溶胶来源复杂,如何区分SSA对海洋气溶胶的贡献依然是个难题. 在这些问题的驱使下,一系列模拟SSA产生的装置应运而生,很好地弥补了外场观测的研究短板. 目前常用的模拟装置有加压雾化器、鼓泡式发生器、跌落式发生器、“Sea Sweep”发生器和波浪破碎式发生器等(图2),具体介绍如下.
(1)加压雾化器:利用伯努利原理将压缩空气直接作用于封闭容器内的海水,高压海水经喷出雾化后形成SSA. 雾化器凭其结构简单、生产效率高等优点在很多陆源气溶胶的实验室模拟中得到广泛应用. 再次强调SSA的产生与气泡的产生与破裂密切相关,但在一些吸湿性增长[33]、N2O5吸收[34]和CCN活性[35]的研究中仍延用雾化发生器来产生SSA. 实验研究表明,该装置产生的SSA粒径分布只有两个模态(约35 nm和约100 nm),与其它模拟方法和外场观测结果不一致[36]. 因此加压雾化器不再适合于SSA的研究.
(2)鼓泡式发生器:将洁净空气通入浸没在水下的烧结玻璃过滤器来产生气泡羽流以模拟膜滴和射滴的形成. 产生的SSA在吹扫空气作用下被载入分析检测设备. 当前设计的鼓泡式发生器形式多样[37-50],有小至容积为500 mL的五颈圆底烧瓶[41],也有大至高为2.1 m的空心圆柱体[37]. 鼓泡式发生器主要通过鼓气流量[51]、多孔过滤器孔径[52]及其水下深度[46]来调节SSA的生成. 例如,研究发现细孔径过滤器产生的SSA质量浓度和数量浓度分别是粗孔径过滤器的57倍和194倍[40]. 虽然鼓泡法一直被用于模拟SSA的产生,但是近年来有越来越多的学者发现它并不是一种理想的模拟产生方法. 这主要是因为其产生的气泡大小更多地取决于所使用过滤器的孔径大小,并不会重现波浪破碎时的气泡尺寸分布,无法实现SSA的仿真模拟.
(3)跌落式发生器:利用水流撞击水面,在水下一定深度内产生气泡羽流,气泡上浮至水面破裂后产生SSA. 按照水流入水的方式可为射流式[36, 43, 53-56]和飞瀑式[31, 57]. 射流式发生器利用喷嘴产生的线状入射水柱撞击水面来产生气泡羽流. 其射流速度和射流高度会改变水下气泡羽流的状态,进而影响SSA的数浓度和中心粒径[53-54]. 飞瀑式发生器以Stokes等[31]在2013年设计的MART(marine aerosol reference tank, MART)为代表. 与射流式发生器的区别在于喷嘴被狭缝替代而形成瀑布状的水帘. 跌落式发生器中的气泡羽流是由水柱/水帘夹带的空气所产生,其空气夹带速率受到冲击速度、流体特性、喷嘴/狭缝设计和下跌高度等因素的影响[54, 58]. 总之,跌落式发生器可以很好地模拟波浪破碎后的气泡尺寸分布[31, 58],而且该装置结构简单、操作便捷,因此在很多模拟研究中都获得了广泛应用.
(4)“Sea Sweep”发生器:由一个0.91 m×0.61 m×0.91 m(长×宽×高)的不锈钢框架构成,如图2(d)所示. 顶部高出不锈钢框架0.3 m,4个锥面由不锈钢板封闭. 该装置由左右两侧的浮筒来维持箱体漂浮状态(图中未展示). 前后两侧保持开放均与海面保持1 cm的距离,并利用1 m·s−1垂直向下的空气“幕”来阻止外界颗粒物对模拟体系的干扰. 在海面以下0.75 m处放置有两个多孔过滤器以产生气泡羽流,随后在锥顶采样口处收集新生的SSA. 同时,“Sea Sweep”发生器随船以0.2 m·s−1的速度前进,以确保海水的实时更新[59]. “Sea Sweep”发生器是外场观测与实验室模拟生成有机结合的典型代表,能在原位产生SSA的同时最大限度的减少外界因素的干扰.
(5)波浪破碎式发生器:加州大学斯克里普斯海洋研究所开发的波浪通道是一个33 m×0.5 m×0.8 m(长×宽×高)的狭长水道,其内部可容纳超104 L的海水. 位于通道中部有一个与底面呈16°的斜坡. 通道一侧在桨叶推动下产生波浪,波浪途经斜坡而发生破碎[60],由此产生的气泡羽流可达液面下0.15 m[57]. 相比前几种发生装置,波浪通道所产生的波浪、气泡尺寸和SSA粒径分布可以更大程度的贴近真实环境[21, 30]. 该通道已被用于研究海洋-大气复杂系统的围隔实验,包括了生物活动对新生SSA颗粒的影响、挥发性有机物(VOCs)的产生以及光化学老化对SSA的影响等研究[60]. 虽然波浪通道是研究SSA的理想装置,但是其巨大的体积容量和高昂的运行成本限制了其广泛应用.
总之,由于生成方法和设计思路的不同,SSA发生器在结构上存在着较大的差异. 但因目前缺少更多的公认参数,只有加压雾化器能够从原理上被排除于SSA的实验室生成,而其余方法均有应用. 不同方法产生的SSA可能在理化性质上存在较大差异,导致一些不同甚至是矛盾的实验结果出现. 因此,基于海浪产生原理开发一种可操作性强、仿真度高的标准化SSA生成方法是当下相关领域仍需解决的技术难题之一.
-
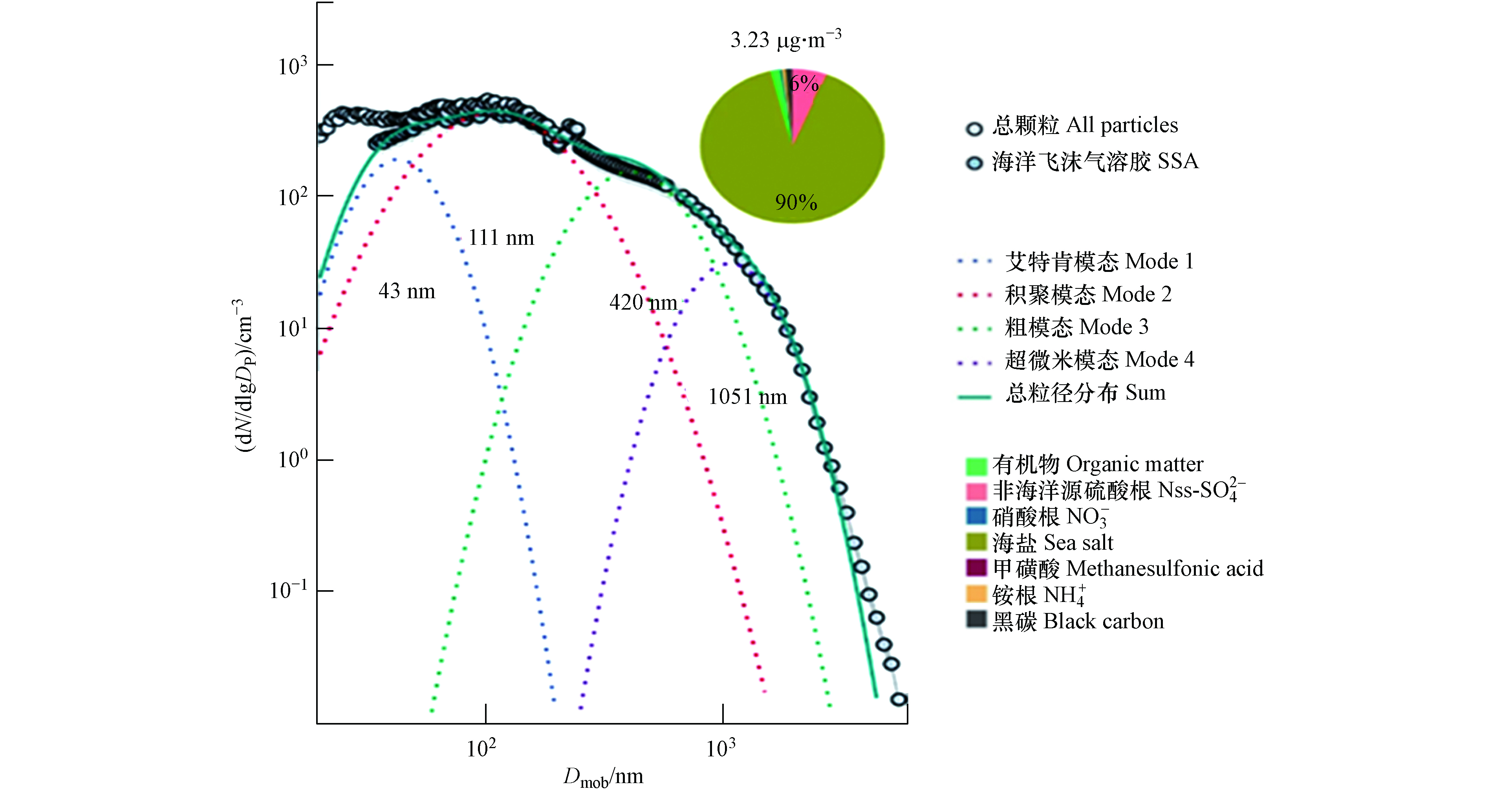
SSA在MBL中常以液滴的形式存在,其直径取决于自身含水量和周围环境相对湿度. 在MBL中环境的相对湿度一般被认为是80%. 从海面刚产生的SSA进入MBL后,会快速与相对湿度为80%的环境到达平衡,此时SSA直径会缩小为初始直径的二分之一;而当相对湿度低于50%后,水分不断蒸干而引发SSA从液滴向固体颗粒的相态转变,其直径将缩小为初始直径的四分之一[1]. 外场观测和实验室研究均表明,对于粒径小于10 μm的颗粒,所得到的SSA粒径分布呈现为多峰分布(图3):(1)在亚微米级分为以25—85 nm为中心的艾特肯模态(Mode 1)、以100—250 nm为中心的积聚模态(Mode 2)和大于250 nm的粗模态(Mode 3);(2)在超微米区域以1—3 μm为中心的超微米模态(Mode 4)[7, 36, 43, 59, 61]. SSA的这种多模态分布表明其产生过程将涉及多种机制,而传统的膜滴和射滴产生机制似乎仍不足以解释[5]. 在不同粒径尺寸的SSA中,亚微米颗粒对SSA数浓度的贡献占绝对优势[7],是大洋上空CCN的主要来源[59]. 与超微米颗粒相比,亚微米颗粒具有更大的比表面积和更复杂的化学组成,所以亚微米颗粒对大气非均相氧化反应和成核过程的影响尤为重要[62]. 随着海洋气溶胶在全球气候效应中的作用日益凸显,亚微米颗粒的相关研究已成为当前该领域的关注热点.
SSA的化学组分极为复杂,与海洋表面的化学组成密切相关. 海洋含有多种多样的无机物和有机物,在上浮气泡的顶托作用下易向海洋表面富集,进而发生从液相向颗粒相的转移. 由此产生的SSA将是无机物与有机物的混合物[63]. 其中无机组分源于海盐,包括Na+、K+、Mg2+、Ca2+和Cl-等,但又与海水中的比例不完全相同. 在一般情况下,从海洋转移到SSA的过程中仅有Na+浓度保持恒定,其它的无机离子则存在一定的富集和损耗[37, 64-66]. 在SSA中有机组分种类繁多,包括脂肪族、糖类、蛋白质、氨基酸和生物组分等[67-71],其对SSA的质量贡献从4%到80%不等[42, 59, 72-73]. 外部混合和内部混合从两个角度描述了SSA的混合状态. 外部混合是指SSA由多种不同化学性质的气溶胶颗粒混合而成. Prather等[30]根据物理性状和化学特性定义了四种主要的颗粒类型,包括海盐(sea salt,SS)、含有机碳海盐(sea salt with organic carbon,SS-OC)、有机物(organic carbon,OC)和生物(biological,Bio). 内部混合是指SSA的单颗粒内部是由不同化学成分组成的混合物. 例如,在SS颗粒中Na+和Cl-的贡献十分显著,而海水中的其他无机离子贡献较小;在SS-OC颗粒中虽仍以NaCl为主,但有机物的比例明显提高;OC颗粒内部则由有机碳主导;在Bio颗粒上则富集了尺寸足够小的细菌或病毒 [14, 57, 74]. 不同类型颗粒的相对丰度与粒径之间存在明显的依赖性[25, 75]. SS颗粒和Bio颗粒主要集中在超微米范围内;而SS-OC和OC颗粒则主要集中在亚微米区域,且随着颗粒物粒径减小,其有机占比不断提高[25, 30, 37]. 扫描电子显微镜结果显示(图4),SS颗粒呈现出明亮且规则的晶体状、OC颗粒一般为深色圆形颗粒、SS-OC颗粒则呈现有机组分包裹无机内核的核壳结构[76]. 有机壳层不仅会改变SSA界面的物质传输,也会改变SSA颗粒的理化性质(如吸湿性[77]、表面张力[78-79]、光散射能力[80]和CCN活化能力[77-78, 81]),进而影响海洋环境乃至全球气候.
-
研究SSA的化学组成对于理解其对大气化学和气候变化的影响至关重要,而不同物质从海水转移到SSA时常表现出富集或损耗,导致无法仅依靠海水的化学组成来预测SSA的化学组成. 通常用富集因子EFc(i)来量化某种物质在SSA上的富集程度[53, 64-65, 79],其被定义为:
其中,
c(i)SSA 是SSA中i 物质的浓度,c(Na+)SSA 是SSA中钠离子的浓度,c(i)sea 是海水中i 物质的浓度,c(Na+)SSA 是海水中钠离子的浓度. EF>1代表SSA中的i 物质富集,EF=1表示既不富集也不抑制,而EF<1则表示抑制. -
近年来,有越来越多的研究报道了无机离子在SSA颗粒中的富集. Kenee等在北大西洋海水的26个单独鼓泡实验中发现,有24个结果显示了Ca2+的EF中位数为1.2[37]. Cochran等在人工海水中加入羧酸盐后观察到Ca2+的EF可高达5,且EF随着颗粒物粒径的减小而增加[82]. Ca2+作为最容易在SSA上富集的无机离子[65, 83-84],因其易与有机物络合密切相关. 若SSA颗粒中的Ca2+不是以有机络合物的形式存在,那么它可能与碳酸盐或碳酸氢盐结合,这将对SSA的pH及其与pH相关的化学过程产生影响[37, 65]. 另一些研究发现,Mg2+与阴离子表面活性剂的优先结合也会促进Mg2+在SSA上的富集[85-86]. 此外,还有研究发现极地海冰形成过程中会发生Na2SO4·10H2O等盐类沉淀,抑制Na+从海水向SSA的转移,因而促进SSA中Mg2+/Na+和K+/Na+比值增加[87]. 这种无机离子在SSA上的富集随海冰季节性变化,将有助于加强对极地冰芯形成的理解. 虽然已有研究证实无机组分会影响SSA颗粒的吸湿性[65, 88-89],但对无机组分的转移机制以及其对SSA理化特性的影响仍需要更深入的研究.
-
有机物从海水转移到SSA的过程具有选择性. 首先,这种选择性会直观地体现在SSA的粒径上. 在糖类富集实验中,观察到细SSA颗粒(<2.5 μm)上的葡萄糖富集程度是粗SSA颗粒(2.5—10 μm)的3倍[64],亚微米SSA中的糖类富集程度比超微米SSA高出1个数量级[67]. 在氨基酸富集实验中,观察到氨基酸在亚微米颗粒上的富集能力比超微米SSA高10—20倍[68]. 这种粒径上的选择性可能是由SSA的产生机制引起的. 气泡在海洋微表层上破裂,由此产生的粒径较小的膜滴将富集大量海洋微表层中的疏水性有机物;而粒径较大的射滴主要来自于气泡的内部气-液界面,主要富含海水中的无机盐和少量的溶解有机碳与颗粒有机碳[47]. 其次,这种选择性体现在有机物的结构性质上. 具有极性酸侧链的游离氨基酸[68]、短链脂肪酸[90-91]和具有表面活性的糖类[92]会优先富集到SSA上. 此外,二价阳离子也会对有机物的选择性富集产生重要影响. 例如,Ca2+可以桥接并稳定海藻酸盐的聚合物链,进而增加海藻酸盐在SSA上的富集[92]. 阴离子可溶性糖可通过二价阳离子的桥接作用共吸附到不溶性表面活性剂棕榈酸微层上,从而增强可溶性糖类在SSA颗粒中的富集[93-94].
-
除无机离子和有机物外,尺寸足够小的细菌和病毒也可以在SSA中富集. 当它们进入海洋大气后,不仅可以在保持传染性前提下进行长距离输送[15, 95],还可以充当CCN和诱导冰成核来影响气候[32, 96-97]. 细菌和病毒在海-气交换过程中发生的相对富集可以通过计算每个基因组的气溶胶化因子(aerosolization factor, AF)来确定,A、B和S分别指在气溶胶、海水和海洋微表层(SSML)中某一基因组在总基因组中的占比[16],公式如下:
Jennifer等[16]的研究表明,大多数同纲或同目的细菌和病毒在SSA上的富集存在分类群相似性,而部分具有选择性基因组的细菌和病毒可能表现出分类群的特异性,例如,疏水性外膜有助于增强细菌和病毒的富集能力. 相比于病毒,细菌更容易富集到SSA上且不易受环境条件的影响.
-
风对海洋表面施压会导致波浪的形成与破裂. 随着风速的增加,SSA产量将非线性增加. Staniec等的研究表明当风速从15 m·s−1增加到32 m·s−1时,海洋飞沫液滴的体积通量从58 mm3· s−1·m−2 增加到3300 mm3· s−1·m-2[98],增幅可达两个数量级. Andreas等的函数模型结果显示,风速增加1倍会引起SSA产量至少增加50倍[99]. 截至现在,源函数是描述SSA产量的最常见方法. 它被定义为在单位时间和面积内,某一粒径范围的SSA通过参考平面(通常为高于海洋表面10 m处的平面)的数量、体积或质量[100]. 目前大多源函数的一般表达式为[101]:
式中,W(U10)是白浪(因夹带大量气泡而具有高反照率的波浪)覆盖率[102],它通常被认为仅依赖于10 m处的风速(U10). FN作为一个形状函数,给出了某粒径(Dp)增量下对应的SSA数量、体积或质量. 比较不同的源函数计算结果发现,SSA年产量存在1—2个数量级的差异[2]. 造成这种差异的原因至少有两个:其一,风驱动SSA形成的动力学机制复杂(如:大气湍流运动、波浪破碎动力学、空气夹带动力学和气泡破裂动力学等[103-104]);其二,仅考虑风速而忽略其它因素的扰动. 虽然从大尺度范围来看风速是决定SSA产生的关键因子,但是在局部过程中其它环境因素对SSA产生的影响也是不容忽视的[103].
-
温度是影响SSA生成的重要因素之一. 温度通过改变海水的理化性质,进而影响气泡羽流、破裂和SSA的产生[36, 50, 105-106]. 整理汇总相关研究进展发现SSA的产生对温度的依赖性结论存在较大争议. Hultin等[107]、Salter等[58]、Zábori等[49-50]和Christiansen等[54]利用射流式发生装置所获得的研究结果表明SSA的数浓度与水温存在一种非线性关系. 当温度从0 ℃增加到约10 ℃时,SSA数浓度显著减少;而温度继续增加到约25 ℃时,SSA数浓度略有增加或几乎不变. 相反,利用MART/miniMART发生装置的模拟结果显示SSA数浓度随着温度的升高而线性增加[108]. 此外,一些鼓泡式发生器的实验结果显示SSA的中心粒径随着温度升高而减小,小粒径分布模态(<300 nm)的SSA数浓度随着温度的升高而降低,而大粒径分布模态(>350 nm)则表现出相反的变化趋势[43, 54, 109]. 目前对这一争议结论的普遍解释是由不同SSA发生装置引发的不同空气夹带率(单位时间内被夹带到海水中的空气体积)所造成的. 鼓泡式发生器的空气夹带率由额定的鼓气流量控制,不受温度影响;射流式发生器的空气夹带率则与水温呈负线性相关[58];而MART的空气夹带率随着温度升高而升高[110]. 此外,一些研究还发现温度会对气泡的羽流特征和尺寸分布等产生影响. 例如,Thorpe等指出水温升高会降低气体溶解度和水体粘度,导致气泡数量和上升速率增加,但这种增加在一定程度上被分子扩散速率的增加而缓解[111]. Nielsere等认为水温变化会显著影响表面气泡的寿命,从而影响SSA的产生[41]. Grythe等[101]统筹分析大量的全球观测数据发现海表温度的升高会引起SSA数浓度增加,这与Liu等[112]在太平洋与大西洋上空的飞机航测结果一致. 但是外场观测无法排除海温与风速之间的强相关性. 综合来看,目前海表温度对SSA生成的影响机制的解释还未达成统一共识.
-
海水盐度作为海洋重要的环境参数之一会对SSA的产生带来一定影响. 纵观全球海洋,大部分海域表层海水盐度在33‰—37‰的范围内变化[51],但在少数海域中盐度可能高于(红海盐度约40 ‰)或低于(波罗的海盐度约8 ‰)该范围. Tyree等[51]和Lv等[40]分别对不同盐度的人工海水进行了鼓泡实验,均得出了SSA数浓度随着盐度增加而增加的结论. Park等采集河流入海区域不同盐度梯度下射流产生的SSA并对其颗粒进行了表征,结果发现随着盐度从0 ‰增加到34 ‰,SSA数浓度增加8—17倍,且中心粒径也随之变大[113]. 研究认为高盐水体容易产生更多更小的气泡,从而产生更多数量的SSA[107, 114]. 但从全球范围来讲,除个别海域外,海洋微小的盐度变化对SSA全球排放通量的影响有限.
-
已有研究发现海洋表层含有各种各样的表面活性物质,包括生物产生的脂肪酸类[69]和微囊藻毒素[115]等,人为排放的全氟烷基酸类[116]和二棕榈酰磷脂酰胆碱(医用气溶胶疗法中的赋形剂)[117]等. 这类物质特有的亲水和疏水基团会使其在海洋表面和海水气泡表面定向排列而显著降低海水的表面张力. 研究发现表面活性物质的种类、浓度和聚合程度等均会对SSA的生成产生不同程度的影响. 例如,丙二酸、果糖和丙二酸钠在一定浓度范围内都会促进SSA的生成,但它们对SSA粒径分布、混合状态和形貌特征的影响不尽相同[40]. 当油酸浓度从0.1 mg·L−1增加至1 mg·L−1时,SSA数浓度明显增加,而继续添加至10 mg·L−1时,数浓度几乎不再发生变化[51]. 乙二醇和不同分子量的聚乙二醇会不同程度地抑制SSA的生成,但会增加颗粒的几何直径. 值得注意的是,随着它们浓度的增加会削弱这种抑制作用,高浓度的聚乙二醇甚至会促进SSA的生成[53]. 此外,表面活性物质还会影响水面气泡的寿命和气泡膜的脆性. Sellegri等的研究发现加入过量的十二烷基硫酸钠会导致水面出现一层泡沫层延长气泡的破裂时间,极大限度抑制了SSA的产生[43]. 虽然表面张力在一定程度上可以解释表面活性物质对SSA生成的影响,但究竟是促进还是抑制SSA的产生在很大程度上是未知的.
-
浮游生物作为海洋有机物的主要生产者,在SSA的形成过程中起到关键作用. 高生产力或发生藻华的水体常伴有高的浮游生物量. 叶绿素-a是衡量生物量大小的常用指标. 模拟研究常采用叶绿素-a浓度来预测SSA上有机物的富集[118-120],但二者相关性的结果存在差异. 一些研究认为SSA中有机物占比与叶绿素-a浓度相关[121-123],而另一些研究认为两者之间没有相关性[79, 124-125]. 浮游植物作为初级生产者能够产生大量有机物,包括溶解有机碳和颗粒有机碳. 不同有机物在SSA上表现出的选择性富集,进而导致并不是所有海水中的有机物均能及时体现在SSA上. 在MART的模拟水华实验中,Santander等推断水华期间海洋有机物向SSA中转移的过程可能是由胞外聚合物主导[126]. 浮游植物的生长导致海水中的溶解有机碳浓度升高,促进SSA颗粒上有机物的富集[127]. Wang等的水华实验表明亚微米和超微米SSA中有机物的富集种类不同,且富集程度取决于有机物源(生物产生)-汇(细菌消耗)过程的动态平衡[32]. 此外,研究发现SSA中腐殖质(HULIS)荧光信号相比于海水而言发生了20 nm的红移,这说明浮游植物虽然不是HULIS从海洋转移到大气的直接驱动因素,但会间接诱导HULIS化学成分的变化[126]. 有趣的是最新的航测结果表明海洋上空SSA数浓度存在昼夜循环规律,这与浮游植物的昼夜活动规律一致,进一步佐证了浮游生物对SSA的产生过程具有重要的调节作用[52, 128-129]. 尽管浮游植物的大量繁殖会导致海洋生物群落结构和生物量的显著变化,但在水华期间SSA的化学组成与浮游生物活动并未建立起明确的内在联系[130].
-
SSA的相关研究是当前全球气候变化研究的重要前沿领域,已成为国际上层海洋-低层大气研究计划(SOLAS)的核心内容之一. 海洋具有自身特有的生物地球化学循环系统,并与陆地-大气多圈层相互作用,进而导致SSA的形成机制、传输条件和理化特性与陆源气溶胶相比具有显著性差异. 尽管近些年已在该领域开展了大量的研究,尤其是在SSA的实验室模拟方面,但仍有诸多具有挑战性的科学问题有待回答与解决:
(1)研究方法:SSA的产生受到气泡尺寸、羽流特征和气泡破裂过程等参数的共同控制,能否准确还原这些参数是模拟真实SSA产生的关键. 由于当前不同SSA发生器的设计思路和装置结构存在显著差异,导致目前无法对SSA的理化性质得到更多的共识. 因此,研发并普及一种标准化的SSA发生器仍是当前和未来实验室模拟SSA研究亟需解决的难题. 突破这一挑战才能实现对参数的精细调控,从而对SSA的粒径分布、化学组成、表面形貌等理化性质进行表征和定量,最终揭示多模态分布下SSA的具体产生机制.
(2)环境行为:SSA的化学组成与形貌特征决定其大气化学命运. 因此亟需开展以SSA为媒介研究物质在海-气界面迁移与转化的全链条综合研究. 聚焦海洋有机物在SSA的产生、迁移与转化行为对多种环境因素耦合影响的响应机制,识别影响该过程的关键调控因子. 另外,如何结合对现有数据的分析,同时开展多海域的大规模调查和模拟,完善SSA的排放源函数关系是今后在SSA环境行为研究方面需要着重思考的问题. 未来还需要着重考虑海洋浮游生物对SSA产生与转化的影响,科学评估浮游生物在SSA形成中所扮演的“角色”.
(3)气候效应:SSA作为CCN引起云辐射强迫的变化是气候评估中最大的不确定因素. SSA的迁移与转移以及对其“汇”过程的定量研究仍然不足,导致当前SSA的研究无法准确实现其环境效应评估. 解决这一研究“瓶颈”的关键在于要量化环境因素变化对海盐气溶胶理化性质的影响,同时精确定量不同环境体系下SSA对CCN贡献的绝对值. 这将有助于全面且系统地认识SSA的形成与转化及其气候效应,并大大提升其对全球气候模型预测的准确性.
海洋飞沫气溶胶的实验模拟
Laboratory simulation of sea spray aerosol
-
摘要: 作为大气气溶胶的重要组成,海洋飞沫气溶胶(sea spray aerosol,SSA)在全球气候变化中发挥着重要作用. SSA的产生过程同时受到海洋和大气环境的共同作用,其理化性质和全球通量存在显著的区域性差异,进而导致目前SSA对全球环境和气候变化的贡献评估存在较大不确定性. 为了认识SSA的形成过程以及不同环境要素对其产生的影响机制,目前已经展开了大量的研究工作,特别是在实验室受控条件下模拟SSA产生方面获得了丰富的研究成果. 本文聚焦SSA实验室模拟研究,重点围绕SSA的产生、化学组成、物质转移以及其对不同环境因素响应方面的研究进展进行了系统总结,并展望了该领域未来的重点研究方向.Abstract: As an important component of atmospheric aerosol, sea spray aerosol (SSA) plays an important role in global climate change. The formation of SSA is affected by both marine and atmospheric environmental factors, leading to significant regional differences in its physical and chemical properties and global flux. This results in great uncertainties in the current assessment of the contribution of SSA to global environment and climate change. In order to understand the formation process of SSA and the impact mechanism of different environmental factors on it, several studies, especially in the simulation of SSA formation under controlled laboratory conditions, have been conducted. This review explores the laboratory simulation research of SSA, with focus on SSA formation related research progress, chemical composition, substance transfer and its response to different environmental factors. We make a systematic summary, and put forward further prospects for future key research directions.
-
海洋飞沫气溶胶(SSA)是指直接从海洋表面向大气喷射的悬浮颗粒[1-2]. 当其进入海洋边界层(marine boundary layer,MBL)后不仅可以通过吸收和散射太阳辐射而直接影响全球辐射平衡,也可以作为云凝结核(cloud condensation nuclei, CCN)和冰核(ice nuclei, IN)间接影响全球气候[3-6]. 据统计,海洋每年向大气输送的SSA可达100—300亿t[7],且远高于沙尘气溶胶[8]. 因此SSA被普遍认为是影响全球气候系统的重要调节因子. 研究发现SSA排放量的增加有助于全球气候的冷却,进而能缓解或抵消由温室气体所引发的全球变暖[9-10]. 此外,SSA在海-气界面水分、盐分、有机物和微生物的传输过程中也发挥着至关重要的作用[11]. 随着全球经济迅速发展,人为活动引起的海洋环境问题日益突出,海洋中的持久性有机污染物[12]、微塑料[13]、过渡金属[14]、藻毒素[15]、细菌和病毒[16]等均可在SSA上富集并远距离传输. 这不仅会影响SSA的化学组成,还会对大气环境和人类健康产生重大影响. 如何准确评估SSA在全球环境和气候变化中的作用成为了当前大气-海洋交叉学科亟需解决的关键科学问题.
自1953年Woodcock等将风速确定为SSA产生的主要驱动力之后[17],一系列将SSA产生与风速相联系的研究接踵而至[18-19]. 虽然风速是目前已知控制SSA产生的关键,但SSA的产生也会受到其它环境因素(海表温度、海水盐度、表面活性剂和浮游微生物活动等)的影响[20]. 由于对这些环境因素的潜在影响机制认识尚不完全,围绕SSA产生过程和相关影响因素的不确定性使其成为一个持续活跃的研究领域. 外场观测虽然可以对SSA进行原位测量,但是存在一定的时空局限性,且难以明确地分离和量化单个环境因素的影响. 针对上述的研究“短板”,近年来科研工作者在实验室中开展了大量模拟工作,并在SSA产生技术方面取得了重大进展. 同时,实验室模拟研究可实现在受控条件下深入探究不同环境因素对SSA产生过程的影响及作用机制,这对SSA的产生机制研究和全球气候效应预测都至关重要. 因此,本文系统总结了近十年来该领域模拟研究的相关进展,重点介绍SSA的产生、粒径分布、化学组成、物质富集以及影响SSA产生的关键环境因素.
1. SSA产生(SSA Formation)
1.1 SSA自然产生
风驱动产生的SSA粒径大小可跨越几个数量级,一般按其产生机制分为三类:膜滴、射滴和裂滴(图1).
当风速大于5 m·s−1时,波浪破碎使上表层海水夹带大量气泡. 随后气泡上浮至海洋表面,其露出水面的气泡膜不断变薄直至破裂. 气泡破裂过程中气泡膜通过液条断裂[21-23]或快速抖动[24]而产生数百个膜滴. 而后,周围海水将回填气泡破裂后留下的空腔而激起若干垂直喷射的射滴[25]. 当风速大于10 m·s−1时,风可以直接吹裂波峰而产生几十微米至几毫米的裂滴[26],其大气停留时间从几秒到几分钟不等. 由于粒径较大的裂滴大气停留时间短,因此当前研究一般忽略其在大气化学和气候效应中的贡献[2]. 一般而言,气泡破裂产生的SSA粒径范围在0.01—25 μm之间[1],其中膜滴粒径较小,主要集中在亚微米(粒径<1 μm)范围内[24];而射滴粒径较大,贡献了大部分的超微米(粒径>1 μm)颗粒[20]. 膜滴和射滴的产生数量与气泡尺寸大小密切相关. 研究发现气泡尺寸越大产生的膜滴越多. 例如,气泡直径从2 mm增加到6 mm,膜滴数量可从100个增加到1000个,增加一个数量级[27]. 相反,射滴数量随着气泡尺寸的增加而减少. 例如,直径在100—300 μm内的气泡可以产生5—6个射滴,而直径为3 mm的气泡只能产生1个射滴[28]. 此外,膜滴粒径受气泡膜厚度影响. 研究表明气泡膜厚度会随着气泡直径增加而增加,进而容易产生更大的膜滴[23, 29];而射滴粒径约为其破裂气泡直径的1/40[20]. 总之,气泡的破裂过程及尺寸大小共同控制着膜滴与射滴的产生,最终将影响SSA的粒径分布和化学组成[1, 30-32].
1.2 SSA模拟产生
迄今为止,有关SSA的研究已经做了大量的外场观测工作,但仍然存在两个重要问题亟需解决:(1)原位SSA的产生容易受到多种环境因素耦合的影响,且无法识别各因素在SSA产生过程中的具体作用机制;(2)海洋气溶胶来源复杂,如何区分SSA对海洋气溶胶的贡献依然是个难题. 在这些问题的驱使下,一系列模拟SSA产生的装置应运而生,很好地弥补了外场观测的研究短板. 目前常用的模拟装置有加压雾化器、鼓泡式发生器、跌落式发生器、“Sea Sweep”发生器和波浪破碎式发生器等(图2),具体介绍如下.
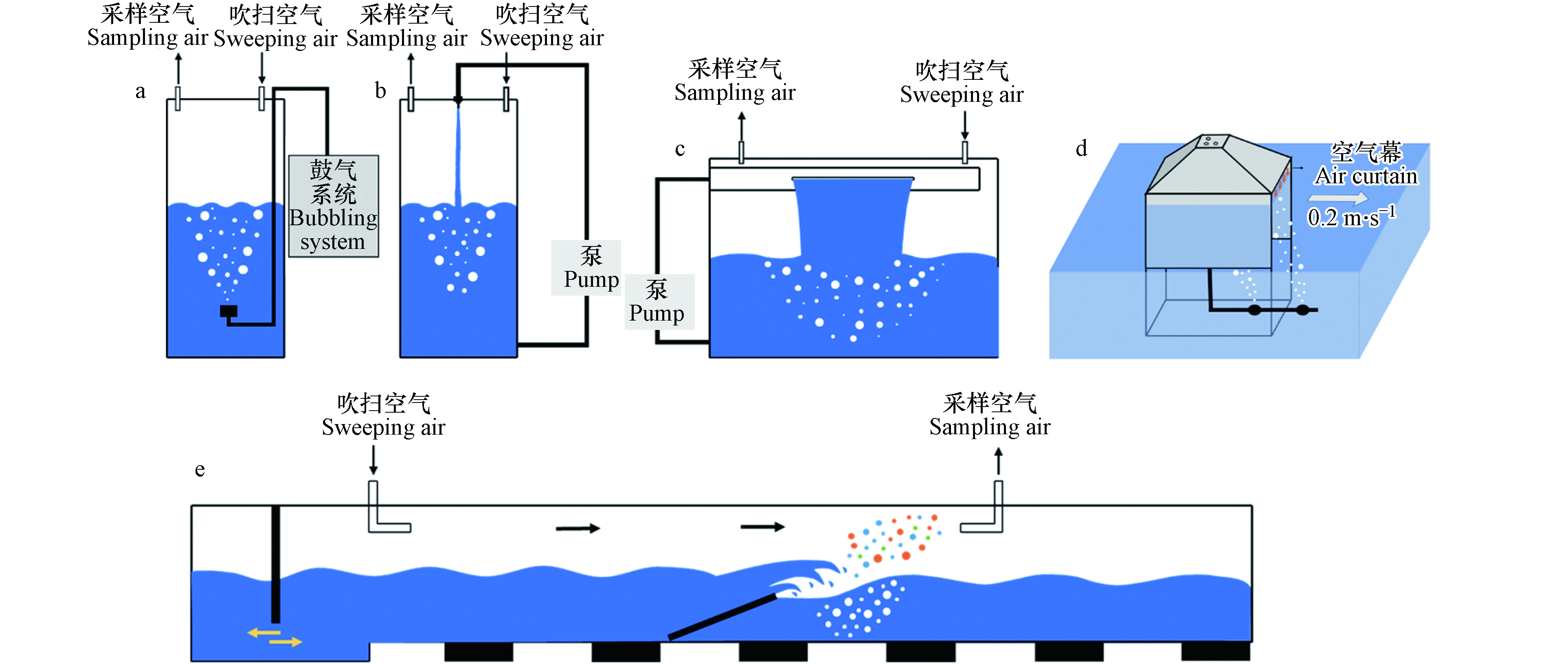 图 2 典型的SSA模拟发生装置Figure 2. Typical SSA simulation generators(a.鼓泡式;b.射流式;c.飞瀑式;d. “Sea Sweep”;e. 波浪破碎式)(a. Bubbling;b. Plunging jet;c. Plunging waterfull;d. “Sea Sweep”;e. Breaking waves)
图 2 典型的SSA模拟发生装置Figure 2. Typical SSA simulation generators(a.鼓泡式;b.射流式;c.飞瀑式;d. “Sea Sweep”;e. 波浪破碎式)(a. Bubbling;b. Plunging jet;c. Plunging waterfull;d. “Sea Sweep”;e. Breaking waves)(1)加压雾化器:利用伯努利原理将压缩空气直接作用于封闭容器内的海水,高压海水经喷出雾化后形成SSA. 雾化器凭其结构简单、生产效率高等优点在很多陆源气溶胶的实验室模拟中得到广泛应用. 再次强调SSA的产生与气泡的产生与破裂密切相关,但在一些吸湿性增长[33]、N2O5吸收[34]和CCN活性[35]的研究中仍延用雾化发生器来产生SSA. 实验研究表明,该装置产生的SSA粒径分布只有两个模态(约35 nm和约100 nm),与其它模拟方法和外场观测结果不一致[36]. 因此加压雾化器不再适合于SSA的研究.
(2)鼓泡式发生器:将洁净空气通入浸没在水下的烧结玻璃过滤器来产生气泡羽流以模拟膜滴和射滴的形成. 产生的SSA在吹扫空气作用下被载入分析检测设备. 当前设计的鼓泡式发生器形式多样[37-50],有小至容积为500 mL的五颈圆底烧瓶[41],也有大至高为2.1 m的空心圆柱体[37]. 鼓泡式发生器主要通过鼓气流量[51]、多孔过滤器孔径[52]及其水下深度[46]来调节SSA的生成. 例如,研究发现细孔径过滤器产生的SSA质量浓度和数量浓度分别是粗孔径过滤器的57倍和194倍[40]. 虽然鼓泡法一直被用于模拟SSA的产生,但是近年来有越来越多的学者发现它并不是一种理想的模拟产生方法. 这主要是因为其产生的气泡大小更多地取决于所使用过滤器的孔径大小,并不会重现波浪破碎时的气泡尺寸分布,无法实现SSA的仿真模拟.
(3)跌落式发生器:利用水流撞击水面,在水下一定深度内产生气泡羽流,气泡上浮至水面破裂后产生SSA. 按照水流入水的方式可为射流式[36, 43, 53-56]和飞瀑式[31, 57]. 射流式发生器利用喷嘴产生的线状入射水柱撞击水面来产生气泡羽流. 其射流速度和射流高度会改变水下气泡羽流的状态,进而影响SSA的数浓度和中心粒径[53-54]. 飞瀑式发生器以Stokes等[31]在2013年设计的MART(marine aerosol reference tank, MART)为代表. 与射流式发生器的区别在于喷嘴被狭缝替代而形成瀑布状的水帘. 跌落式发生器中的气泡羽流是由水柱/水帘夹带的空气所产生,其空气夹带速率受到冲击速度、流体特性、喷嘴/狭缝设计和下跌高度等因素的影响[54, 58]. 总之,跌落式发生器可以很好地模拟波浪破碎后的气泡尺寸分布[31, 58],而且该装置结构简单、操作便捷,因此在很多模拟研究中都获得了广泛应用.
(4)“Sea Sweep”发生器:由一个0.91 m×0.61 m×0.91 m(长×宽×高)的不锈钢框架构成,如图2(d)所示. 顶部高出不锈钢框架0.3 m,4个锥面由不锈钢板封闭. 该装置由左右两侧的浮筒来维持箱体漂浮状态(图中未展示). 前后两侧保持开放均与海面保持1 cm的距离,并利用1 m·s−1垂直向下的空气“幕”来阻止外界颗粒物对模拟体系的干扰. 在海面以下0.75 m处放置有两个多孔过滤器以产生气泡羽流,随后在锥顶采样口处收集新生的SSA. 同时,“Sea Sweep”发生器随船以0.2 m·s−1的速度前进,以确保海水的实时更新[59]. “Sea Sweep”发生器是外场观测与实验室模拟生成有机结合的典型代表,能在原位产生SSA的同时最大限度的减少外界因素的干扰.
(5)波浪破碎式发生器:加州大学斯克里普斯海洋研究所开发的波浪通道是一个33 m×0.5 m×0.8 m(长×宽×高)的狭长水道,其内部可容纳超104 L的海水. 位于通道中部有一个与底面呈16°的斜坡. 通道一侧在桨叶推动下产生波浪,波浪途经斜坡而发生破碎[60],由此产生的气泡羽流可达液面下0.15 m[57]. 相比前几种发生装置,波浪通道所产生的波浪、气泡尺寸和SSA粒径分布可以更大程度的贴近真实环境[21, 30]. 该通道已被用于研究海洋-大气复杂系统的围隔实验,包括了生物活动对新生SSA颗粒的影响、挥发性有机物(VOCs)的产生以及光化学老化对SSA的影响等研究[60]. 虽然波浪通道是研究SSA的理想装置,但是其巨大的体积容量和高昂的运行成本限制了其广泛应用.
总之,由于生成方法和设计思路的不同,SSA发生器在结构上存在着较大的差异. 但因目前缺少更多的公认参数,只有加压雾化器能够从原理上被排除于SSA的实验室生成,而其余方法均有应用. 不同方法产生的SSA可能在理化性质上存在较大差异,导致一些不同甚至是矛盾的实验结果出现. 因此,基于海浪产生原理开发一种可操作性强、仿真度高的标准化SSA生成方法是当下相关领域仍需解决的技术难题之一.
2. SSA的粒径分布与化学组成(Particle size distribution and chemical composition of SSA)
SSA在MBL中常以液滴的形式存在,其直径取决于自身含水量和周围环境相对湿度. 在MBL中环境的相对湿度一般被认为是80%. 从海面刚产生的SSA进入MBL后,会快速与相对湿度为80%的环境到达平衡,此时SSA直径会缩小为初始直径的二分之一;而当相对湿度低于50%后,水分不断蒸干而引发SSA从液滴向固体颗粒的相态转变,其直径将缩小为初始直径的四分之一[1]. 外场观测和实验室研究均表明,对于粒径小于10 μm的颗粒,所得到的SSA粒径分布呈现为多峰分布(图3):(1)在亚微米级分为以25—85 nm为中心的艾特肯模态(Mode 1)、以100—250 nm为中心的积聚模态(Mode 2)和大于250 nm的粗模态(Mode 3);(2)在超微米区域以1—3 μm为中心的超微米模态(Mode 4)[7, 36, 43, 59, 61]. SSA的这种多模态分布表明其产生过程将涉及多种机制,而传统的膜滴和射滴产生机制似乎仍不足以解释[5]. 在不同粒径尺寸的SSA中,亚微米颗粒对SSA数浓度的贡献占绝对优势[7],是大洋上空CCN的主要来源[59]. 与超微米颗粒相比,亚微米颗粒具有更大的比表面积和更复杂的化学组成,所以亚微米颗粒对大气非均相氧化反应和成核过程的影响尤为重要[62]. 随着海洋气溶胶在全球气候效应中的作用日益凸显,亚微米颗粒的相关研究已成为当前该领域的关注热点.
SSA的化学组分极为复杂,与海洋表面的化学组成密切相关. 海洋含有多种多样的无机物和有机物,在上浮气泡的顶托作用下易向海洋表面富集,进而发生从液相向颗粒相的转移. 由此产生的SSA将是无机物与有机物的混合物[63]. 其中无机组分源于海盐,包括Na+、K+、Mg2+、Ca2+和Cl-等,但又与海水中的比例不完全相同. 在一般情况下,从海洋转移到SSA的过程中仅有Na+浓度保持恒定,其它的无机离子则存在一定的富集和损耗[37, 64-66]. 在SSA中有机组分种类繁多,包括脂肪族、糖类、蛋白质、氨基酸和生物组分等[67-71],其对SSA的质量贡献从4%到80%不等[42, 59, 72-73]. 外部混合和内部混合从两个角度描述了SSA的混合状态. 外部混合是指SSA由多种不同化学性质的气溶胶颗粒混合而成. Prather等[30]根据物理性状和化学特性定义了四种主要的颗粒类型,包括海盐(sea salt,SS)、含有机碳海盐(sea salt with organic carbon,SS-OC)、有机物(organic carbon,OC)和生物(biological,Bio). 内部混合是指SSA的单颗粒内部是由不同化学成分组成的混合物. 例如,在SS颗粒中Na+和Cl-的贡献十分显著,而海水中的其他无机离子贡献较小;在SS-OC颗粒中虽仍以NaCl为主,但有机物的比例明显提高;OC颗粒内部则由有机碳主导;在Bio颗粒上则富集了尺寸足够小的细菌或病毒 [14, 57, 74]. 不同类型颗粒的相对丰度与粒径之间存在明显的依赖性[25, 75]. SS颗粒和Bio颗粒主要集中在超微米范围内;而SS-OC和OC颗粒则主要集中在亚微米区域,且随着颗粒物粒径减小,其有机占比不断提高[25, 30, 37]. 扫描电子显微镜结果显示(图4),SS颗粒呈现出明亮且规则的晶体状、OC颗粒一般为深色圆形颗粒、SS-OC颗粒则呈现有机组分包裹无机内核的核壳结构[76]. 有机壳层不仅会改变SSA界面的物质传输,也会改变SSA颗粒的理化性质(如吸湿性[77]、表面张力[78-79]、光散射能力[80]和CCN活化能力[77-78, 81]),进而影响海洋环境乃至全球气候.

3. SSA中的物质转移(Substance transfer in SSA)
研究SSA的化学组成对于理解其对大气化学和气候变化的影响至关重要,而不同物质从海水转移到SSA时常表现出富集或损耗,导致无法仅依靠海水的化学组成来预测SSA的化学组成. 通常用富集因子EFc(i)来量化某种物质在SSA上的富集程度[53, 64-65, 79],其被定义为:
stringUtils.convertMath(!{formula.content}) (1) 其中,
c(i)SSA i c(Na+)SSA c(i)sea i c(Na+)SSA i 3.1 无机离子
近年来,有越来越多的研究报道了无机离子在SSA颗粒中的富集. Kenee等在北大西洋海水的26个单独鼓泡实验中发现,有24个结果显示了Ca2+的EF中位数为1.2[37]. Cochran等在人工海水中加入羧酸盐后观察到Ca2+的EF可高达5,且EF随着颗粒物粒径的减小而增加[82]. Ca2+作为最容易在SSA上富集的无机离子[65, 83-84],因其易与有机物络合密切相关. 若SSA颗粒中的Ca2+不是以有机络合物的形式存在,那么它可能与碳酸盐或碳酸氢盐结合,这将对SSA的pH及其与pH相关的化学过程产生影响[37, 65]. 另一些研究发现,Mg2+与阴离子表面活性剂的优先结合也会促进Mg2+在SSA上的富集[85-86]. 此外,还有研究发现极地海冰形成过程中会发生Na2SO4·10H2O等盐类沉淀,抑制Na+从海水向SSA的转移,因而促进SSA中Mg2+/Na+和K+/Na+比值增加[87]. 这种无机离子在SSA上的富集随海冰季节性变化,将有助于加强对极地冰芯形成的理解. 虽然已有研究证实无机组分会影响SSA颗粒的吸湿性[65, 88-89],但对无机组分的转移机制以及其对SSA理化特性的影响仍需要更深入的研究.
3.2 有机物
有机物从海水转移到SSA的过程具有选择性. 首先,这种选择性会直观地体现在SSA的粒径上. 在糖类富集实验中,观察到细SSA颗粒(<2.5 μm)上的葡萄糖富集程度是粗SSA颗粒(2.5—10 μm)的3倍[64],亚微米SSA中的糖类富集程度比超微米SSA高出1个数量级[67]. 在氨基酸富集实验中,观察到氨基酸在亚微米颗粒上的富集能力比超微米SSA高10—20倍[68]. 这种粒径上的选择性可能是由SSA的产生机制引起的. 气泡在海洋微表层上破裂,由此产生的粒径较小的膜滴将富集大量海洋微表层中的疏水性有机物;而粒径较大的射滴主要来自于气泡的内部气-液界面,主要富含海水中的无机盐和少量的溶解有机碳与颗粒有机碳[47]. 其次,这种选择性体现在有机物的结构性质上. 具有极性酸侧链的游离氨基酸[68]、短链脂肪酸[90-91]和具有表面活性的糖类[92]会优先富集到SSA上. 此外,二价阳离子也会对有机物的选择性富集产生重要影响. 例如,Ca2+可以桥接并稳定海藻酸盐的聚合物链,进而增加海藻酸盐在SSA上的富集[92]. 阴离子可溶性糖可通过二价阳离子的桥接作用共吸附到不溶性表面活性剂棕榈酸微层上,从而增强可溶性糖类在SSA颗粒中的富集[93-94].
3.3 微生物
除无机离子和有机物外,尺寸足够小的细菌和病毒也可以在SSA中富集. 当它们进入海洋大气后,不仅可以在保持传染性前提下进行长距离输送[15, 95],还可以充当CCN和诱导冰成核来影响气候[32, 96-97]. 细菌和病毒在海-气交换过程中发生的相对富集可以通过计算每个基因组的气溶胶化因子(aerosolization factor, AF)来确定,A、B和S分别指在气溶胶、海水和海洋微表层(SSML)中某一基因组在总基因组中的占比[16],公式如下:
stringUtils.convertMath(!{formula.content}) (2) Jennifer等[16]的研究表明,大多数同纲或同目的细菌和病毒在SSA上的富集存在分类群相似性,而部分具有选择性基因组的细菌和病毒可能表现出分类群的特异性,例如,疏水性外膜有助于增强细菌和病毒的富集能力. 相比于病毒,细菌更容易富集到SSA上且不易受环境条件的影响.
4. 影响SSA生成的重要因素(Important factors affecting SSA formation)
4.1 海面风速
风对海洋表面施压会导致波浪的形成与破裂. 随着风速的增加,SSA产量将非线性增加. Staniec等的研究表明当风速从15 m·s−1增加到32 m·s−1时,海洋飞沫液滴的体积通量从58 mm3· s−1·m−2 增加到3300 mm3· s−1·m-2[98],增幅可达两个数量级. Andreas等的函数模型结果显示,风速增加1倍会引起SSA产量至少增加50倍[99]. 截至现在,源函数是描述SSA产量的最常见方法. 它被定义为在单位时间和面积内,某一粒径范围的SSA通过参考平面(通常为高于海洋表面10 m处的平面)的数量、体积或质量[100]. 目前大多源函数的一般表达式为[101]:
stringUtils.convertMath(!{formula.content}) (3) 式中,W(U10)是白浪(因夹带大量气泡而具有高反照率的波浪)覆盖率[102],它通常被认为仅依赖于10 m处的风速(U10). FN作为一个形状函数,给出了某粒径(Dp)增量下对应的SSA数量、体积或质量. 比较不同的源函数计算结果发现,SSA年产量存在1—2个数量级的差异[2]. 造成这种差异的原因至少有两个:其一,风驱动SSA形成的动力学机制复杂(如:大气湍流运动、波浪破碎动力学、空气夹带动力学和气泡破裂动力学等[103-104]);其二,仅考虑风速而忽略其它因素的扰动. 虽然从大尺度范围来看风速是决定SSA产生的关键因子,但是在局部过程中其它环境因素对SSA产生的影响也是不容忽视的[103].
4.2 海表温度
温度是影响SSA生成的重要因素之一. 温度通过改变海水的理化性质,进而影响气泡羽流、破裂和SSA的产生[36, 50, 105-106]. 整理汇总相关研究进展发现SSA的产生对温度的依赖性结论存在较大争议. Hultin等[107]、Salter等[58]、Zábori等[49-50]和Christiansen等[54]利用射流式发生装置所获得的研究结果表明SSA的数浓度与水温存在一种非线性关系. 当温度从0 ℃增加到约10 ℃时,SSA数浓度显著减少;而温度继续增加到约25 ℃时,SSA数浓度略有增加或几乎不变. 相反,利用MART/miniMART发生装置的模拟结果显示SSA数浓度随着温度的升高而线性增加[108]. 此外,一些鼓泡式发生器的实验结果显示SSA的中心粒径随着温度升高而减小,小粒径分布模态(<300 nm)的SSA数浓度随着温度的升高而降低,而大粒径分布模态(>350 nm)则表现出相反的变化趋势[43, 54, 109]. 目前对这一争议结论的普遍解释是由不同SSA发生装置引发的不同空气夹带率(单位时间内被夹带到海水中的空气体积)所造成的. 鼓泡式发生器的空气夹带率由额定的鼓气流量控制,不受温度影响;射流式发生器的空气夹带率则与水温呈负线性相关[58];而MART的空气夹带率随着温度升高而升高[110]. 此外,一些研究还发现温度会对气泡的羽流特征和尺寸分布等产生影响. 例如,Thorpe等指出水温升高会降低气体溶解度和水体粘度,导致气泡数量和上升速率增加,但这种增加在一定程度上被分子扩散速率的增加而缓解[111]. Nielsere等认为水温变化会显著影响表面气泡的寿命,从而影响SSA的产生[41]. Grythe等[101]统筹分析大量的全球观测数据发现海表温度的升高会引起SSA数浓度增加,这与Liu等[112]在太平洋与大西洋上空的飞机航测结果一致. 但是外场观测无法排除海温与风速之间的强相关性. 综合来看,目前海表温度对SSA生成的影响机制的解释还未达成统一共识.
4.3 海水盐度
海水盐度作为海洋重要的环境参数之一会对SSA的产生带来一定影响. 纵观全球海洋,大部分海域表层海水盐度在33‰—37‰的范围内变化[51],但在少数海域中盐度可能高于(红海盐度约40 ‰)或低于(波罗的海盐度约8 ‰)该范围. Tyree等[51]和Lv等[40]分别对不同盐度的人工海水进行了鼓泡实验,均得出了SSA数浓度随着盐度增加而增加的结论. Park等采集河流入海区域不同盐度梯度下射流产生的SSA并对其颗粒进行了表征,结果发现随着盐度从0 ‰增加到34 ‰,SSA数浓度增加8—17倍,且中心粒径也随之变大[113]. 研究认为高盐水体容易产生更多更小的气泡,从而产生更多数量的SSA[107, 114]. 但从全球范围来讲,除个别海域外,海洋微小的盐度变化对SSA全球排放通量的影响有限.
4.4 表面活性物质
已有研究发现海洋表层含有各种各样的表面活性物质,包括生物产生的脂肪酸类[69]和微囊藻毒素[115]等,人为排放的全氟烷基酸类[116]和二棕榈酰磷脂酰胆碱(医用气溶胶疗法中的赋形剂)[117]等. 这类物质特有的亲水和疏水基团会使其在海洋表面和海水气泡表面定向排列而显著降低海水的表面张力. 研究发现表面活性物质的种类、浓度和聚合程度等均会对SSA的生成产生不同程度的影响. 例如,丙二酸、果糖和丙二酸钠在一定浓度范围内都会促进SSA的生成,但它们对SSA粒径分布、混合状态和形貌特征的影响不尽相同[40]. 当油酸浓度从0.1 mg·L−1增加至1 mg·L−1时,SSA数浓度明显增加,而继续添加至10 mg·L−1时,数浓度几乎不再发生变化[51]. 乙二醇和不同分子量的聚乙二醇会不同程度地抑制SSA的生成,但会增加颗粒的几何直径. 值得注意的是,随着它们浓度的增加会削弱这种抑制作用,高浓度的聚乙二醇甚至会促进SSA的生成[53]. 此外,表面活性物质还会影响水面气泡的寿命和气泡膜的脆性. Sellegri等的研究发现加入过量的十二烷基硫酸钠会导致水面出现一层泡沫层延长气泡的破裂时间,极大限度抑制了SSA的产生[43]. 虽然表面张力在一定程度上可以解释表面活性物质对SSA生成的影响,但究竟是促进还是抑制SSA的产生在很大程度上是未知的.
4.5 浮游生物
浮游生物作为海洋有机物的主要生产者,在SSA的形成过程中起到关键作用. 高生产力或发生藻华的水体常伴有高的浮游生物量. 叶绿素-a是衡量生物量大小的常用指标. 模拟研究常采用叶绿素-a浓度来预测SSA上有机物的富集[118-120],但二者相关性的结果存在差异. 一些研究认为SSA中有机物占比与叶绿素-a浓度相关[121-123],而另一些研究认为两者之间没有相关性[79, 124-125]. 浮游植物作为初级生产者能够产生大量有机物,包括溶解有机碳和颗粒有机碳. 不同有机物在SSA上表现出的选择性富集,进而导致并不是所有海水中的有机物均能及时体现在SSA上. 在MART的模拟水华实验中,Santander等推断水华期间海洋有机物向SSA中转移的过程可能是由胞外聚合物主导[126]. 浮游植物的生长导致海水中的溶解有机碳浓度升高,促进SSA颗粒上有机物的富集[127]. Wang等的水华实验表明亚微米和超微米SSA中有机物的富集种类不同,且富集程度取决于有机物源(生物产生)-汇(细菌消耗)过程的动态平衡[32]. 此外,研究发现SSA中腐殖质(HULIS)荧光信号相比于海水而言发生了20 nm的红移,这说明浮游植物虽然不是HULIS从海洋转移到大气的直接驱动因素,但会间接诱导HULIS化学成分的变化[126]. 有趣的是最新的航测结果表明海洋上空SSA数浓度存在昼夜循环规律,这与浮游植物的昼夜活动规律一致,进一步佐证了浮游生物对SSA的产生过程具有重要的调节作用[52, 128-129]. 尽管浮游植物的大量繁殖会导致海洋生物群落结构和生物量的显著变化,但在水华期间SSA的化学组成与浮游生物活动并未建立起明确的内在联系[130].
5. 结论与展望(Conclusion and prospective)
SSA的相关研究是当前全球气候变化研究的重要前沿领域,已成为国际上层海洋-低层大气研究计划(SOLAS)的核心内容之一. 海洋具有自身特有的生物地球化学循环系统,并与陆地-大气多圈层相互作用,进而导致SSA的形成机制、传输条件和理化特性与陆源气溶胶相比具有显著性差异. 尽管近些年已在该领域开展了大量的研究,尤其是在SSA的实验室模拟方面,但仍有诸多具有挑战性的科学问题有待回答与解决:
(1)研究方法:SSA的产生受到气泡尺寸、羽流特征和气泡破裂过程等参数的共同控制,能否准确还原这些参数是模拟真实SSA产生的关键. 由于当前不同SSA发生器的设计思路和装置结构存在显著差异,导致目前无法对SSA的理化性质得到更多的共识. 因此,研发并普及一种标准化的SSA发生器仍是当前和未来实验室模拟SSA研究亟需解决的难题. 突破这一挑战才能实现对参数的精细调控,从而对SSA的粒径分布、化学组成、表面形貌等理化性质进行表征和定量,最终揭示多模态分布下SSA的具体产生机制.
(2)环境行为:SSA的化学组成与形貌特征决定其大气化学命运. 因此亟需开展以SSA为媒介研究物质在海-气界面迁移与转化的全链条综合研究. 聚焦海洋有机物在SSA的产生、迁移与转化行为对多种环境因素耦合影响的响应机制,识别影响该过程的关键调控因子. 另外,如何结合对现有数据的分析,同时开展多海域的大规模调查和模拟,完善SSA的排放源函数关系是今后在SSA环境行为研究方面需要着重思考的问题. 未来还需要着重考虑海洋浮游生物对SSA产生与转化的影响,科学评估浮游生物在SSA形成中所扮演的“角色”.
(3)气候效应:SSA作为CCN引起云辐射强迫的变化是气候评估中最大的不确定因素. SSA的迁移与转移以及对其“汇”过程的定量研究仍然不足,导致当前SSA的研究无法准确实现其环境效应评估. 解决这一研究“瓶颈”的关键在于要量化环境因素变化对海盐气溶胶理化性质的影响,同时精确定量不同环境体系下SSA对CCN贡献的绝对值. 这将有助于全面且系统地认识SSA的形成与转化及其气候效应,并大大提升其对全球气候模型预测的准确性.
-
[1] VERON F, Ocean spray[J]. Annual Review of Fluid Mechanics, 2015, 47: 507-538. [2] de LEEUW G, ANDREAS E L, ANGUELOVA M D, et al. Production flux of sea spray aerosol [J]. Reviews of Geophysics, 2011, 49(2): RG2001. [3] BROOKS S D, THORNTON D C O. Marine aerosols and clouds [J]. Annual Review of Marine Science, 2018, 10: 289-313. doi: 10.1146/annurev-marine-121916-063148 [4] HENDRICKSON B N, BROOKS S D, THORNTON D C O, et al. Role of sea surface microlayer properties in cloud formation [J]. Frontiers in Marine Science, 2021, 7: 596225. doi: 10.3389/fmars.2020.596225 [5] XU W, OVADNEVAITE J, FOSSUM K N, et al. Sea spray as an obscured source for marine cloud nuclei [J]. Nature Geoscience, 2022, 15(4): 282-286. doi: 10.1038/s41561-022-00917-2 [6] FOSSUM K N, OVADNEVAITE J, CEBURNIS D, et al. Summertime primary and secondary contributions to Southern Ocean cloud condensation nuclei [J]. Scientific Reports, 2018, 8: 13844. doi: 10.1038/s41598-018-32047-4 [7] QUINN P K, COFFMAN D J, JOHNSON J E, et al. Small fraction of marine cloud condensation nuclei made up of sea spray aerosol [J]. Nature Geoscience, 2017, 10(9): 674-679. doi: 10.1038/ngeo3003 [8] BEER C G, HENDRICKS J, RIGHI M, et al. Modelling mineral dust emissions and atmospheric dispersion with MADE3 in EMAC v2.54 [J]. Geoscientific Model Development, 2020, 13(9): 4287-4303. doi: 10.5194/gmd-13-4287-2020 [9] CONNOLLY P J, MCFIGGANS G B, WOOD R, et al. Factors determining the most efficient spray distribution for marine cloud brightening [J]. Philosophical Transactions:Mathematical, Physical and Engineering Sciences, 2014, 372(2031): 20140056. [10] LATHAM J, SMITH M H. Effect on global warming of wind-dependent aerosol generation at the ocean surface [J]. Nature, 1990, 347(6291): 372-373. doi: 10.1038/347372a0 [11] SCHIFFER J M, MAEL L E, PRATHER K A, et al. Sea spray aerosol: Where marine biology meets atmospheric chemistry [J]. ACS Central Science, 2018, 4(12): 1617-1623. doi: 10.1021/acscentsci.8b00674 [12] JOHANSSON J H, SALTER M E, NAVARRO J C A, et al. Global transport of perfluoroalkyl acids via sea spray aerosol [J]. Environmental Science:Processes & Impacts, 2019, 21(4): 635-649. [13] YANG S Y, ZHANG T, GAN Y Q, et al. Constraining microplastic particle emission flux from the ocean [J]. Environmental Science & Technology Letters, 2022, 9(6): 513-519. [14] GUASCO T L, CUADRA-RODRIGUEZ L A, PEDLER B E, et al. Transition metal associations with primary biological particles in sea spray aerosol generated in a wave channel [J]. Environmental Science & Technology, 2014, 48(2): 1324-1333. [15] SEM K, JANG M, PIERCE R, et al. Characterization of atmospheric processes of brevetoxins in sea spray aerosols from red tide events [J]. Environmental Science & Technology, 2022, 56(3): 1811-1819. [16] MICHAUD J M, THOMPSON L R, KAUL D, et al. Taxon-specific aerosolization of bacteria and viruses in an experimental ocean-atmosphere mesocosm [J]. Nature Communications, 2018, 9: 2017. doi: 10.1038/s41467-018-04409-z [17] WOODCOCK A H, KIENTZLER C F, ARONS A B, et al. Giant condensation nuclei from bursting bubbles [J]. Nature, 1953, 172(4390): 1144-1145. doi: 10.1038/1721144a0 [18] MONAHAN E C, O'MUIRCHEARTAIGH I G. Whitecaps and the passive remote sensing of the ocean surface [J]. International Journal of Remote Sensing, 1986, 7(5): 627-642. doi: 10.1080/01431168608954716 [19] NILSSON E D, RANNIK, SWIETLICKI E, et al. Turbulent aerosol fluxes over the Arctic Ocean: 2. Wind-driven sources from the sea [J]. Journal of Geophysical Research:Atmospheres, 2001, 106(D23): 32139-32154. doi: 10.1029/2000JD900747 [20] LEWIS R, SCHWARTZ E. Sea Salt Aerosol Production: Mechanisms, Methods, Measurements and Models—A Critical Review[M]. Washington, D. C. : American Geophysical Union, 2004. [21] DEANE G B, STOKES M D. Scale dependence of bubble creation mechanisms in breaking waves [J]. Nature, 2002, 418(6900): 839-844. doi: 10.1038/nature00967 [22] WANG Y, BOUROUIBA L. Non-isolated drop impact on surfaces [J]. Journal of Fluid Mechanics, 2018, 835: 24-44. doi: 10.1017/jfm.2017.755 [23] LHUISSIER H, VILLERMAUX E. Bursting bubble aerosols [J]. Journal of Fluid Mechanics, 2012, 696: 5-44. doi: 10.1017/jfm.2011.418 [24] JIANG X H, ROTILY L, VILLERMAUX E, et al. Submicron drops from flapping bursting bubbles [J]. Proceedings of the National Academy of Sciences of the United States of America, 2022, 119(1): e2112924119. doi: 10.1073/pnas.2112924119 [25] QUINN P K, COLLINS D B, GRASSIAN V H, et al. Chemistry and related properties of freshly emitted sea spray aerosol [J]. Chemical Reviews, 2015, 115(10): 4383-4399. doi: 10.1021/cr500713g [26] 吕辰. 海洋飞沫气溶胶参与下的烯烃醚臭氧化反应[D]. 济南: 山东大学, 2020. LV C. Ozonolysis of vinyl ethers with the participation of sea spray aerosol[D]. Jinan: Shandong University, 2020(in Chinese).
[27] BLANCHARD D C. The electrification of the atmosphere by particles from bubbles in the sea [J]. Progress in Oceanography, 1963, 1: 73-202. doi: 10.1016/0079-6611(63)90004-1 [28] CIPRIANO R J, BLANCHARD D C. Bubble and aerosol spectra produced by a laboratory ‘breaking wave’ [J]. Journal of Geophysical Research Atmospheres, 1981, 86(C9): 8085. doi: 10.1029/JC086iC09p08085 [29] KE W R, KUO Y M, LIN C W, et al. Characterization of aerosol emissions from single bubble bursting [J]. Journal of Aerosol Science, 2017, 109: 1-12. doi: 10.1016/j.jaerosci.2017.03.006 [30] PRATHER K A, BERTRAM T H, GRASSIAN V H, et al. Bringing the ocean into the laboratory to probe the chemical complexity of sea spray aerosol [J]. Proceedings of the National Academy of Sciences of the United States of America, 2013, 110(19): 7550-7555. doi: 10.1073/pnas.1300262110 [31] STOKES M D, DEANE G B, PRATHER K, et al. A Marine Aerosol Reference Tank system as a breaking wave analogue for the production of foam and sea-spray aerosols [J]. Atmospheric Measurement Techniques, 2013, 6(4): 1085-1094. doi: 10.5194/amt-6-1085-2013 [32] WANG X F, SULTANA C M, TRUEBLOOD J, et al. Microbial control of sea spray aerosol composition: A tale of two blooms [J]. ACS Central Science, 2015, 1(3): 124-131. doi: 10.1021/acscentsci.5b00148 [33] BRABAN C F, ADAMS J W, RODRIGUEZ D, et al. Heterogeneous reactions of HOI, ICl and IBr on sea salt and sea salt proxies [J]. Physical Chemistry Chemical Physics:PCCP, 2007, 9(24): 3136-3148. doi: 10.1039/b700829e [34] MCNEILL V F, PATTERSON J, WOLFE G M, et al. The effect of varying levels of surfactant on the reactive uptake of N2O5 to aqueous aerosol [J]. Atmospheric Chemistry and Physics, 2006, 6(6): 1635-1644. doi: 10.5194/acp-6-1635-2006 [35] MOORE R H, INGALL E D, SOROOSHIAN A, et al. Molar mass, surface tension, and droplet growth kinetics of marine organics from measurements of CCN activity [J]. Geophysical Research Letters, 2008, 35(7): L07801. [36] FUENTES E, COE H, GREEN D, et al. Laboratory-generated primary marine aerosol via bubble-bursting and atomization [J]. Atmospheric Measurement Techniques, 2010, 3(1): 141-162. doi: 10.5194/amt-3-141-2010 [37] KEENE W C, MARING H, MABEN J R, et al. Chemical and physical characteristics of nascent aerosols produced by bursting bubbles at a model air-sea interface [J]. Journal of Geophysical Research Atmospheres, 2007, 112(D21): D21202. doi: 10.1029/2007JD008464 [38] WISE M E, FRENEY E J, TYREE C A, et al. Hygroscopic behavior and liquid-layer composition of aerosol particles generated from natural and artificial seawater [J]. Journal of Geophysical Research Atmospheres, 2009, 114(D3): D03201. [39] PARK J Y, LIM S, PARK K. Mixing state of submicrometer sea spray particles enriched by insoluble species in bubble-bursting experiments [J]. Journal of Atmospheric and Oceanic Technology, 2014, 31(1): 93-104. doi: 10.1175/JTECH-D-13-00086.1 [40] LV C, TSONA N T, DU L. Sea spray aerosol formation: Results on the role of different parameters and organic concentrations from bubble bursting experiments [J]. Chemosphere, 2020, 252: 126456. doi: 10.1016/j.chemosphere.2020.126456 [41] NIELSEN L S, BILDE M. Exploring controlling factors for sea spray aerosol production: Temperature, inorganic ions and organic surfactants [J]. Tellus B:Chemical and Physical Meteorology, 2020, 72(1): 1801305. doi: 10.1080/16000889.2020.1801305 [42] MODINI R L, HARRIS B, RISTOVSKI Z D. The organic fraction of bubble-generated, accumulation mode Sea Spray Aerosol (SSA) [J]. Atmospheric Chemistry and Physics, 2010, 10(6): 2867-2877. doi: 10.5194/acp-10-2867-2010 [43] SELLEGRI K, O'DOWD C D, YOON Y J, et al. Surfactants and submicron sea spray generation [J]. Journal of Geophysical Research Atmospheres, 2006, 111(D22): D22215. doi: 10.1029/2005JD006658 [44] MOORE M J K, FURUTANI H, ROBERTS G C, et al. Effect of organic compounds on cloud condensation nuclei (CCN) activity of sea spray aerosol produced by bubble bursting [J]. Atmospheric Environment, 2011, 45(39): 7462-7469. doi: 10.1016/j.atmosenv.2011.04.034 [45] GASTON C J, FURUTANI H, GUAZZOTTI S A, et al. Unique ocean-derived particles serve as a proxy for changes in ocean chemistry [J]. Journal of Geophysical Research Atmospheres, 2011, 116(D18): D18310. doi: 10.1029/2010JD015289 [46] KING S M, BUTCHER A C, ROSENOERN T, et al. Investigating primary marine aerosol properties: CCN activity of sea salt and mixed inorganic-organic particles [J]. Environmental Science & Technology, 2012, 46(19): 10405-10412. [47] WANG X F, DEANE G B, MOORE K A, et al. The role of jet and film drops in controlling the mixing state of submicron sea spray aerosol particles [J]. Proceedings of the National Academy of Sciences of the United States of America, 2017, 114(27): 6978-6983. doi: 10.1073/pnas.1702420114 [48] RASMUSSEN B B, NGUYEN Q T, KRISTENSEN K, et al. What controls volatility of sea spray aerosol?Results from laboratory studies using artificial and real seawater samples [J]. Journal of Aerosol Science, 2017, 107: 134-141. doi: 10.1016/j.jaerosci.2017.02.002 [49] ZÁBORI J, KREJCI R, STRÖM J, et al. Comparison between summertime and wintertime Arctic Ocean primary marine aerosol properties [J]. Atmospheric Chemistry and Physics, 2013, 13(9): 4783-4799. doi: 10.5194/acp-13-4783-2013 [50] ZÁBORI J, KREJCI R, EKMAN A M L, et al. Wintertime Arctic Ocean Sea water properties and primary marine aerosol concentrations [J]. Atmospheric Chemistry and Physics, 2012, 12(21): 10405-10421. doi: 10.5194/acp-12-10405-2012 [51] TYREE C A, HELLION V M, ALEXANDROVA O A, et al. Foam droplets generated from natural and artificial seawaters [J]. Journal of Geophysical Research Atmospheres, 2007, 112(D12): D12204. doi: 10.1029/2006JD007729 [52] KEENE W C, LONG M S, REID J S, et al. Factors that modulate properties of primary marine aerosol generated from ambient seawater on ships at sea [J]. Journal of Geophysical Research:Atmospheres, 2017, 122(21): 11961-11990. doi: 10.1002/2017JD026872 [53] LIU L R, DU L, XU L, et al. Molecular size of surfactants affects their degree of enrichment in the sea spray aerosol formation [J]. Environmental Research, 2022, 206: 112555. doi: 10.1016/j.envres.2021.112555 [54] CHRISTIANSEN S, SALTER M E, GOROKHOVA E, et al. Sea spray aerosol formation: Laboratory results on the role of air entrainment, water temperature, and phytoplankton biomass [J]. Environmental Science & Technology, 2019, 53(22): 13107-13116. [55] HULTIN K A H, NILSSON E D, KREJCI R, et al. In situ laboratory sea spray production during the Marine Aerosol Production 2006 cruise on the northeastern Atlantic Ocean [J]. Journal of Geophysical Research Atmospheres, 2010, 115(D6): D06201. [56] FACCHINI M C, RINALDI M, DECESARI S, et al. Primary submicron marine aerosol dominated by insoluble organic colloids and aggregates [J]. Geophysical Research Letters, 2008, 35(17): L17814. doi: 10.1029/2008GL034210 [57] COLLINS D B, ZHAO D F, RUPPEL M J, et al. Direct aerosol chemical composition measurements to evaluate the physicochemical differences between controlled sea spray aerosol generation schemes [J]. Atmospheric Measurement Techniques, 2014, 7(11): 3667-3683. doi: 10.5194/amt-7-3667-2014 [58] SALTER M E, NILSSON E D, BUTCHER A, et al. On the seawater temperature dependence of the sea spray aerosol generated by a continuous plunging jet [J]. Journal of Geophysical Research:Atmospheres, 2014, 119(14): 9052-9072. doi: 10.1002/2013JD021376 [59] BATES T S, QUINN P K, FROSSARD A A, et al. Measurements of ocean derived aerosol off the coast of California [J]. Journal of Geophysical Research:Atmospheres, 2012, 117(D21): D00V15. [60] SAUER J S, MAYER K J, LEE C, et al. The Sea Spray Chemistry and Particle Evolution study (SeaSCAPE): Overview and experimental methods [J]. Environmental Science. Processes & Impacts, 2022, 24(2): 290-315. [61] MODINI R L, FROSSARD A A, AHLM L, et al. Primary marine aerosol-cloud interactions off the coast of California [J]. Journal of Geophysical Research:Atmospheres, 2015, 120(9): 4282-4303. doi: 10.1002/2014JD022963 [62] DONALDSON D J, VAIDA V. The influence of organic films at the air-aqueous boundary on atmospheric processes [J]. Chemical Reviews, 2006, 106(4): 1445-1461. doi: 10.1021/cr040367c [63] BIGG E K, LECK C. The composition of fragments of bubbles bursting at the ocean surface [J]. Journal of Geophysical Research Atmospheres, 2008, 113(D11): D11209. doi: 10.1029/2007JD009078 [64] JAYARATHNE T, SULTANA C M, LEE C, et al. Enrichment of saccharides and divalent cations in sea spray aerosol during two phytoplankton blooms [J]. Environmental Science & Technology, 2016, 50(21): 11511-11520. [65] SALTER M E, HAMACHER-BARTH E, LECK C, et al. Calcium enrichment in sea spray aerosol particles [J]. Geophysical Research Letters, 2016, 43(15): 8277-8285. doi: 10.1002/2016GL070275 [66] SCHWIER A N, SELLEGRI K, MAS S, et al. Primary marine aerosol physical flux and chemical composition during a nutrient enrichment experiment in mesocosms in the Mediterranean Sea [J]. Atmospheric Chemistry and Physics, 2017, 17(23): 14645-14660. doi: 10.5194/acp-17-14645-2017 [67] HASENECZ E S, JAYARATHNE T, PENDERGRAFT M A, et al. Marine bacteria affect saccharide enrichment in sea spray aerosol during a phytoplankton bloom [J]. ACS Earth and Space Chemistry, 2020, 4(9): 1638-1649. doi: 10.1021/acsearthspacechem.0c00167 [68] TRIESCH N, van PINXTEREN M, SALTER M, et al. Sea spray aerosol chamber study on selective transfer and enrichment of free and combined amino acids [J]. ACS Earth and Space Chemistry, 2021, 5(6): 1564-1574. doi: 10.1021/acsearthspacechem.1c00080 [69] COCHRAN R E, LASKINA O, JAYARATHNE T, et al. Analysis of organic anionic surfactants in fine and coarse fractions of freshly emitted sea spray aerosol [J]. Environmental Science & Technology, 2016, 50(5): 2477-2486. [70] FU P Q, KAWAMURA K, USUKURA K, et al. Dicarboxylic acids, ketocarboxylic acids and glyoxal in the marine aerosols collected during a round-the-world cruise [J]. Marine Chemistry, 2013, 148: 22-32. doi: 10.1016/j.marchem.2012.11.002 [71] HAWKINS L N, RUSSELL L M. Polysaccharides, proteins, and phytoplankton fragments: Four chemically distinct types of marine primary organic aerosol classified by single particle spectromicroscopy [J]. Advances in Meteorology, 2010, 2010: 1-14. [72] FUENTES E, COE H, GREEN D, et al. On the impacts of phytoplankton-derived organic matter on the properties of the primary marine aerosol–Part 2: Composition, hygroscopicity and cloud condensation activity [J]. Atmospheric Chemistry and Physics, 2011, 11(6): 2585-2602. doi: 10.5194/acp-11-2585-2011 [73] SCHWIER A N, ROSE C, ASMI E, et al. Primary marine aerosol emissions from the Mediterranean Sea during pre-bloom and oligotrophic conditions: Correlations to seawater chlorophyll a from a mesocosm study [J]. Atmospheric Chemistry and Physics, 2015, 15(14): 7961-7976. doi: 10.5194/acp-15-7961-2015 [74] PRATT K A, DEMOTT P J, FRENCH J R, et al. In situ detection of biological particles in cloud ice-crystals [J]. Nature Geoscience, 2009, 2(6): 398-401. doi: 10.1038/ngeo521 [75] KALUARACHCHI C P, OR V W, LAN Y, et al. Size-dependent morphology, composition, phase state and water uptake of nascent submicrometer sea spray aerosols during a phytoplankton bloom [J]. ACS Earth and Space Chemistry, 2022, 6(1): 116-130. doi: 10.1021/acsearthspacechem.1c00306 [76] LEE H D, WIGLEY S, LEE C, et al. Physicochemical mixing state of sea spray aerosols: Morphologies exhibit size dependence [J]. ACS Earth and Space Chemistry, 2020, 4(9): 1604-1611. doi: 10.1021/acsearthspacechem.0c00153 [77] KOEHLER K A, KREIDENWEIS S M, DEMOTT P J, et al. Water activity and activation diameters from hygroscopicity data - Part II: Application to organic species [J]. Atmospheric Chemistry and Physics, 2006, 6(3): 795-809. doi: 10.5194/acp-6-795-2006 [78] LEE H D, MORRIS H S, LASKINA O, et al. Organic enrichment, physical phase state, and surface tension depression of nascent core–shell sea spray aerosols during two phytoplankton blooms [J]. ACS Earth and Space Chemistry, 2020, 4(4): 650-660. doi: 10.1021/acsearthspacechem.0c00032 [79] CRAVIGAN L T, MALLET M D, VAATTOVAARA P, et al. Sea spray aerosol organic enrichment, water uptake and surface tension effects [J]. Atmospheric Chemistry and Physics, 2020, 20(13): 7955-7977. doi: 10.5194/acp-20-7955-2020 [80] LI J, CARLSON B E, YUNG Y L, et al. Scattering and absorbing aerosols in the climate system [J]. Nature Reviews Earth & Environment, 2022, 3(6): 363-379. [81] KANAKIDOU M, SEINFELD J H, PANDIS S N, et al. Organic aerosol and global climate modelling: A review [J]. Atmospheric Chemistry and Physics, 2005, 5(4): 1053-1123. doi: 10.5194/acp-5-1053-2005 [82] COCHRAN R E, JAYARATHNE T, STONE E A, et al. Selectivity across the interface: A test of surface activity in the composition of organic-enriched aerosols from bubble bursting [J]. The Journal of Physical Chemistry Letters, 2016, 7(9): 1692-1696. doi: 10.1021/acs.jpclett.6b00489 [83] MUKHERJEE P, REINFELDER J R, GAO Y. Enrichment of calcium in sea spray aerosol in the Arctic summer atmosphere [J]. Marine Chemistry, 2020, 227: 103898. doi: 10.1016/j.marchem.2020.103898 [84] LECK C, SVENSSON E. Importance of aerosol composition and mixing state for cloud droplet activation over the Arctic pack ice in summer [J]. Atmospheric Chemistry and Physics, 2015, 15(5): 2545-2568. doi: 10.5194/acp-15-2545-2015 [85] GAO Q, LECK C, RAUSCHENBERG C, et al. On the chemical dynamics of extracellular polysaccharides in the high Arctic surface microlayer [J]. Ocean Science, 2012, 8(4): 401-418. doi: 10.5194/os-8-401-2012 [86] CASILLAS-ITUARTE N N, CALLAHAN K M, TANG C Y, et al. Surface organization of aqueous MgCl2 and application to atmospheric marine aerosol chemistry [J]. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(15): 6616-6621. doi: 10.1073/pnas.0912322107 [87] HARA K, OSADA K, YABUKI M, et al. Seasonal variation of fractionated sea-salt particles on the Antarctic coast [J]. Geophysical Research Letters, 2012, 39(18): L18801. [88] WELLER R, WAGENBACH D. Year-round chemical aerosol records in continental Antarctica obtained by automatic samplings [J]. Tellus B:Chemical and Physical Meteorology, 2007, 59(4): 755-765. doi: 10.1111/j.1600-0889.2007.00293.x [89] PARK K, KIM J S, MILLER A L. A study on effects of size and structure on hygroscopicity of nanoparticles using a tandem differential mobility analyzer and TEM [J]. Journal of Nanoparticle Research, 2009, 11(1): 175-183. doi: 10.1007/s11051-008-9462-4 [90] CARTER-FENK K, ALLEN H. Collapse mechanisms of nascent and aged sea spray aerosol proxy films [J]. Atmosphere, 2018, 9(12): 503. doi: 10.3390/atmos9120503 [91] SONG Y R, LI J L, TSONA N T, et al. Enrichment of short-chain organic acids transferred to submicron sea spray aerosols[J]. The Science of the Total Environment, 2022, 851(Pt 1): 158122. [92] HASENECZ E S, KALUARACHCHI C P, LEE H D, et al. Saccharide transfer to sea spray aerosol enhanced by surface activity, calcium, and protein interactions [J]. ACS Earth and Space Chemistry, 2019, 3(11): 2539-2548. doi: 10.1021/acsearthspacechem.9b00197 [93] CARTER-FENK K A, DOMMER A C, FIAMINGO M E, et al. Calcium bridging drives polysaccharide co-adsorption to a proxy sea surface microlayer [J]. Physical Chemistry Chemical Physics:PCCP, 2021, 23(30): 16401-16416. doi: 10.1039/D1CP01407B [94] SCHILL S, BURROWS S, HASENECZ E, et al. The impact of divalent cations on the enrichment of soluble saccharides in primary sea spray aerosol [J]. Atmosphere, 2018, 9(12): 476. doi: 10.3390/atmos9120476 [95] KIRKPATRICK B, PIERCE R, CHENG Y S, et al. Inland transport of aerosolized Florida red tide toxins [J]. Harmful Algae, 2010, 9(2): 186-189. doi: 10.1016/j.hal.2009.09.003 [96] MCCLUSKEY C S, HILL T C J, MALFATTI F, et al. A dynamic link between ice nucleating particles released in nascent sea spray aerosol and oceanic biological activity during two mesocosm experiments [J]. Journal of the Atmospheric Sciences, 2017, 74(1): 151-166. doi: 10.1175/JAS-D-16-0087.1 [97] ICKES L, PORTER G C E, WAGNER R, et al. The ice-nucleating activity of Arctic Sea surface microlayer samples and marine algal cultures [J]. Atmospheric Chemistry and Physics, 2020, 20(18): 11089-11117. doi: 10.5194/acp-20-11089-2020 [98] STANIEC A, VLAHOS P, MONAHAN E C. The role of sea spray in atmosphere–ocean gas exchange [J]. Nature Geoscience, 2021, 14(8): 593-598. doi: 10.1038/s41561-021-00796-z [99] ANDREAS E L. A new sea spray generation function for wind speeds up to 32 m s–1 [J]. Journal of Physical Oceanography, 1998, 28(11): 2175-2184. doi: 10.1175/1520-0485(1998)028<2175:ANSSGF>2.0.CO;2 [100] 赵栋梁. 海洋飞沫及其对海—气相互作用影响的研究进展 [J]. 地球科学进展, 2012, 27(6): 624-632. ZHAO D L. Progress in sea spray and its effects on air-sea interaction [J]. Advances in Earth Science, 2012, 27(6): 624-632(in Chinese).
[101] GRYTHE H, STRÖM J, KREJCI R, et al. A review of sea-spray aerosol source functions using a large global set of sea salt aerosol concentration measurements [J]. Atmospheric Chemistry and Physics, 2014, 14(3): 1277-1297. doi: 10.5194/acp-14-1277-2014 [102] MONAHAN E C, MUIRCHEARTAIGH I. Optimal power-law description of oceanic whitecap coverage dependence on wind speed [J]. Journal of Physical Oceanography, 1980, 10(12): 2094-2099. doi: 10.1175/1520-0485(1980)010<2094:OPLDOO>2.0.CO;2 [103] DEIKE L. Mass transfer at the ocean–atmosphere interface: The role of wave breaking, droplets, and bubbles [J]. Annual Review of Fluid Mechanics, 2022, 54: 191-194. doi: 10.1146/annurev-fluid-030121-014132 [104] DEIKE L, GHABACHE E, LIGER-BELAIR G, et al. Dynamics of jets produced by bursting bubbles [J]. Physical Review Fluids, 2018, 3: 013603. doi: 10.1103/PhysRevFluids.3.013603 [105] ANGUELOVA M D, WEBSTER F, Whitecap coverage from satellite measurements: A first step toward modeling the variability of oceanic whitecaps[J]. Journal of Geophysical Research Atmospheres, 2006, 111(C3): C03017. [106] SALTER M E, ZIEGER P, ACOSTA NAVARRO J C, et al. An empirically derived inorganic sea spray source function incorporating sea surface temperature [J]. Atmospheric Chemistry and Physics, 2015, 15(19): 11047-11066. doi: 10.5194/acp-15-11047-2015 [107] HULTIN K A H, KREJCI R, PINHASSI J, et al. Aerosol and bacterial emissions from Baltic Seawater [J]. Atmospheric Research, 2011, 99(1): 1-14. doi: 10.1016/j.atmosres.2010.08.018 [108] FORESTIERI S D, MOORE K A, MARTINEZ BORRERO R, et al. Temperature and composition dependence of sea spray aerosol production [J]. Geophysical Research Letters, 2018, 45(14): 7218-7225. doi: 10.1029/2018GL078193 [109] MÅRTENSSON E M, NILSSON E D, de LEEUW G, et al. Laboratory simulations and parameterization of the primary marine aerosol production [J]. Journal of Geophysical Research:Atmospheres, 2003, 108(D9): 4297. [110] CALLAGHAN A H, STOKES M D, DEANE G B. The effect of water temperature on air entrainment, bubble plumes, and surface foam in a laboratory breaking-wave analog [J]. Journal of Geophysical Research:Oceans, 2014, 119(11): 7463-7482. doi: 10.1002/2014JC010351 [111] THORPE S A, BOWYER P, WOOLF D K. Some factors affecting the size distributions of oceanic bubbles [J]. Journal of Physical Oceanography, 1992, 22(4): 382-389. doi: 10.1175/1520-0485(1992)022<0382:SFATSD>2.0.CO;2 [112] LIU S, LIU C C, FROYD K D, et al. Sea spray aerosol concentration modulated by sea surface temperature [J]. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(9): e2020583118. doi: 10.1073/pnas.2020583118 [113] PARK J, JANG J, YOON Y J, et al. When river water meets seawater: Insights into primary marine aerosol production[J]. The Science of the Total Environment, 2022, 807(Pt 2): 150866. [114] MAY N W, AXSON J L, WATSON A, et al. Lake spray aerosol generation: A method for producing representative particles from freshwater wave breaking [J]. Atmospheric Measurement Techniques, 2016, 9(9): 4311-4325. doi: 10.5194/amt-9-4311-2016 [115] XU M L, TSONA N T, LI J L, et al. Atmospheric chemical processes of microcystin-LR at the interface of sea spray aerosol [J]. Chemosphere, 2022, 294: 133726. doi: 10.1016/j.chemosphere.2022.133726 [116] CASAS G, MARTÍNEZ-VARELA A, ROSCALES J L, et al. Enrichment of perfluoroalkyl substances in the sea-surface microlayer and sea-spray aerosols in the Southern Ocean [J]. Environmental Pollution (Barking, Essex:1987), 2020, 267: 115512. doi: 10.1016/j.envpol.2020.115512 [117] van ACKER E, de RIJCKE M, LIU Z X, et al. Sea spray aerosols contain the major component of human lung surfactant [J]. Environmental Science & Technology, 2021, 55(23): 15989-16000. [118] PARTANEN A I, DUNNE E M, BERGMAN T, et al. Global modelling of direct and indirect effects of sea spray aerosol using a source function encapsulating wave state [J]. Atmospheric Chemistry and Physics, 2014, 14(21): 11731-11752. doi: 10.5194/acp-14-11731-2014 [119] VIGNATI E, FACCHINI M C, RINALDI M, et al. Global scale emission and distribution of sea-spray aerosol: Sea-salt and organic enrichment [J]. Atmospheric Environment, 2010, 44(5): 670-677. doi: 10.1016/j.atmosenv.2009.11.013 [120] SPRACKLEN D V, ARNOLD S R, SCIARE J, et al. Globally significant oceanic source of organic carbon aerosol [J]. Geophysical Research Letters, 2008, 35(12): L12811. [121] RINALDI M, DECESARI S, FINESSI E, et al. Primary and secondary organic marine aerosol and oceanic biological activity: Recent results and new perspectives for future studies [J]. Advances in Meteorology, 2010, 2010: 310682. [122] O'DOWD C D, FACCHINI M C, CAVALLI F, et al. Biogenically driven organic contribution to marine aerosol [J]. Nature, 2004, 431(7009): 676-680. doi: 10.1038/nature02959 [123] PARK J, KIM D, LEE K, et al. Effect of phytoplankton biomass in seawater on chemical properties of sea spray aerosols [J]. Marine Pollution Bulletin, 2016, 110(1): 231-237. doi: 10.1016/j.marpolbul.2016.06.058 [124] RINALDI M, FUZZI S, DECESARI S, et al. Is chlorophyll-a the best surrogate for organic matter enrichment in submicron primary marine aerosol? [J]. Journal of Geophysical Research:Atmospheres, 2013, 118(10): 4964-4973. doi: 10.1002/jgrd.50417 [125] FRENEY E, SELLEGRI K, NICOSIA A, et al. Mediterranean nascent sea spray organic aerosol and relationships with seawater biogeochemistry [J]. Atmospheric Chemistry and Physics, 2021, 21(13): 10625-10641. doi: 10.5194/acp-21-10625-2021 [126] SANTANDER M V, SCHIFFER J M, LEE C, et al. Factors controlling the transfer of biogenic organic species from seawater to sea spray aerosol [J]. Scientific Reports, 2022, 12: 3580. doi: 10.1038/s41598-022-07335-9 [127] LEE C, SULTANA C M, COLLINS D B, et al. Advancing model systems for fundamental laboratory studies of sea spray aerosol using the microbial loop [J]. The Journal of Physical Chemistry A, 2015, 119(33): 8860-8870. doi: 10.1021/acs.jpca.5b03488 [128] LONG M S, KEENE W C, KIEBER D J, et al. Light-enhanced primary marine aerosol production from biologically productive seawater [J]. Geophysical Research Letters, 2014, 41(7): 2661-2670. doi: 10.1002/2014GL059436 [129] FLORES J M, BOURDIN G, KOSTINSKI A B, et al. Diel cycle of sea spray aerosol concentration [J]. Nature Communications, 2021, 12: 5476. doi: 10.1038/s41467-021-25579-3 [130] QUINN P K, BATES T S. The case against climate regulation via oceanic phytoplankton sulphur emissions [J]. Nature, 2011, 480(7375): 51-56. doi: 10.1038/nature10580 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 6386
- HTML全文浏览数: 6386
- PDF下载数: 199
- 施引文献: 0







