


-
采用静电式空气净化器净化室外新风或室内循环空气,是改善室内空气品质的主要方法之一。一般来说,随着静电式空气净化器的电场强度增大,净化效率会提高,但臭氧产生量亦会增大[1-2]。臭氧是一种具有特殊气味和强氧化性的气体。人体长期暴露于高浓度臭氧会引起呼吸系统疾病,故应采取措施,防止其进入室内环境。臭氧控制措施主要包括两类:优化电极配置以抑制臭氧释放,借助热分解、吸附或催化之类后处理技术分解臭氧。在优化电极配置仍无法抑制臭氧产生的情况下,后者成为研究热点。热分解(含热催化分解)会带来明显的空气温升,吸附法则存在活性炭烧蚀等不利影响,故室温催化是最具实际应用前景的技术。
分解臭氧的催化剂主要活性组分包括贵金属(Ag、Au、Pd、Pt等)和过渡金属氧化物(Mn、Cu、Fe、Co、Ni等)[3]。过渡金属催化剂中,锰氧化物因其低毒性、高活性及优良的可调结构和物理化学性能,已被用作臭氧分解的活性组分[4-5]。锰氧化物存在多种价态和晶型结构。其中,α-MnO2因其具有开放的2×2孔道结构、较大的比表面积、较低的Mn平均氧化态及丰富的表面吸附氧物种,其催化分解臭氧的活性优于其他晶型结构MnO2 [6-7]。同时,α-MnO2中存在能有效捕获臭氧分子的氧空位及丰富的Mn3+/Mn4+氧化还原对,可通过替换孔道结构中部分阳离子或掺杂改性来提高α-MnO2的室温臭氧分解性能[8-9]。但在相对较高湿度下,锰氧化物的催化活性会明显降低。金属Ag价格较低,而且Ag和氧化锰间相互作用可产生更多晶格缺陷和氧空位,从而提高了锰氧化物的还原能力和氧的迁移率[10-11]。因此,采用Ag掺杂不仅可提高催化剂的抗水性,还可改善催化剂的催化活性。本课题组前期研究表明,MnO2或Ag/MnO2催化剂具有优异的室温催化分解臭氧性能[12-14]。
净化室外新风或室内循环空气的静电式空气净化器收尘区极板间距通常仅数毫米,且极板面积大。这意味着待净化空气流经电场空间时,会与极板接触充分,因此,极板涂覆臭氧分解催化材料,有望实现臭氧的原位分解。基于此,本研究采用铝合金板(常用作空气净化器的极板)表面涂覆MnO2或Ag/MnO2催化剂的方法,系统考察涂覆浆料构成(粘结剂的类型和含量、催化剂活性组分)、反应时间和环境条件(臭氧浓度和空气湿度)等因素对催化分解臭氧性能的影响,为优化静电式空气净化器性能以同时实现除尘提效、臭氧控制提供参考。
-
MnO2或Ag/MnO2的制备参照文献[3]。催化剂晶体结构采用D8 Advance型X射线衍射仪(XRD, 德国 Bruker公司)测定;比表面积和孔结构采用ASAP 2020型全自动比表面积及微孔物理吸附仪(BET,美国 Micromeritics公司)测定;表面形貌采用ZEISS Sigma 500 扫描电子显微镜(SEM,德国 Bruker公司)检测。
-
1)表面粗化处理。实验采用表面光滑的铝合金板,其尺寸为100 mm ×55 mm×1 mm。为改善基体与涂层的结合力,需先对铝合金板进行化学粗化处理:先将铝合金板浸泡于40 ℃的混合溶液(NaOH质量浓度为30 g∙L−1,Na2CO3质量浓度为25 g∙L−1 )中约1.5 min,以去除表面的油脂和氧化膜;然后用去离子水冲洗去除多余的碱液;将铝合金板浸泡于混合酸液(将1.5 mol∙L−1 HCl和0.08 mol∙L−1 H2C2O4等体积混合得到)中刻蚀2 h,以便形成粗糙表面,然后用去离子水冲洗除去表面酸液;最后将其置于105 ℃鼓风干燥箱中烘干备用。
2)涂层浆料配制。分别采用复配比为5/7的羧甲基纤维/丁苯橡胶(carboxymethyl cellulose/styrene rubber,CMC/SBR)和海藻酸钠(sodium alginate,SA)作粘结剂配制涂层浆料。浆料配制方法:取一定量的粘结剂溶于去离子水中,少量多次添加MnO2或Ag/MnO2催化剂粉末,并用顶置式电子搅拌器以2 000 r·min−1转速搅拌20 min,得到混合均匀的催化剂浆料。
3)催化剂涂覆。将表面粗化的铝合金板固定在提拉涂膜机夹具上,以10 cm∙min−1的速度浸入催化剂浆料中,浸泡0.5 min后以相同速度提出铝合金板试片。将试片置于105 ℃鼓风干燥箱中干燥1 h初步去除水分,随后以2 ℃∙min−1的速率升温至300 ℃,并在此温度下焙烧20 min,最终获得涂覆催化剂的试片。
-
通过震荡法脱落涂层,以涂层脱落率
ϕ 直观反映催化剂涂层与金属载体的结合性能。将涂层试片放入50 mL烧杯中,下压固定至涡旋振荡仪上,在3 200 r·min−1频率下震荡碰撞1 min。准确称量脱落后的催化剂涂层试片质量,记做m脱落后 。催化剂涂层脱落率计算式为式(1)。式中:
m涂覆前 和m涂覆后 分别代表涂覆前后试片的质量,g。 -
催化剂分解臭氧性能在自行搭建的测试系统中进行(图1)。测试系统由方形石英管、轴流风机、气流均布板、石英试片插槽组成。其中,石英方管的外径尺寸为68 mm×68 mm、壁厚为3 mm、长度为1 000 mm,在距进出口各150 mm处设置1个直径为20 mm的检测口用于检测管道内气体的臭氧浓度、风速和湿度等。风量由微型调频风扇调变。试片插槽是尺寸为60 mm×30 mm×10 mm。试片插槽间距为分别为3、4和5 mm,槽宽1.1 mm、槽深4 mm,将涂覆好的试片嵌入插槽中可组成臭氧分解催化剂试片组进行实验。试片与气流方向平行放置,对应插槽间距为3、4 和5 mm的试片数量分别为18、14和11。模拟气体由臭氧和室内空气组成,臭氧由体积分数5%的 O2/N2合成气经臭氧发生器产生。气体湿度采用美国 Cole-Parmer仪器有限公司生产的7116-CP湿度计检测。
臭氧分解率计算公式如式(2)所示。
式中:
C入口 和C出口 分别为测试系统入口和出口处臭氧质量浓度,mg∙m−3。 -
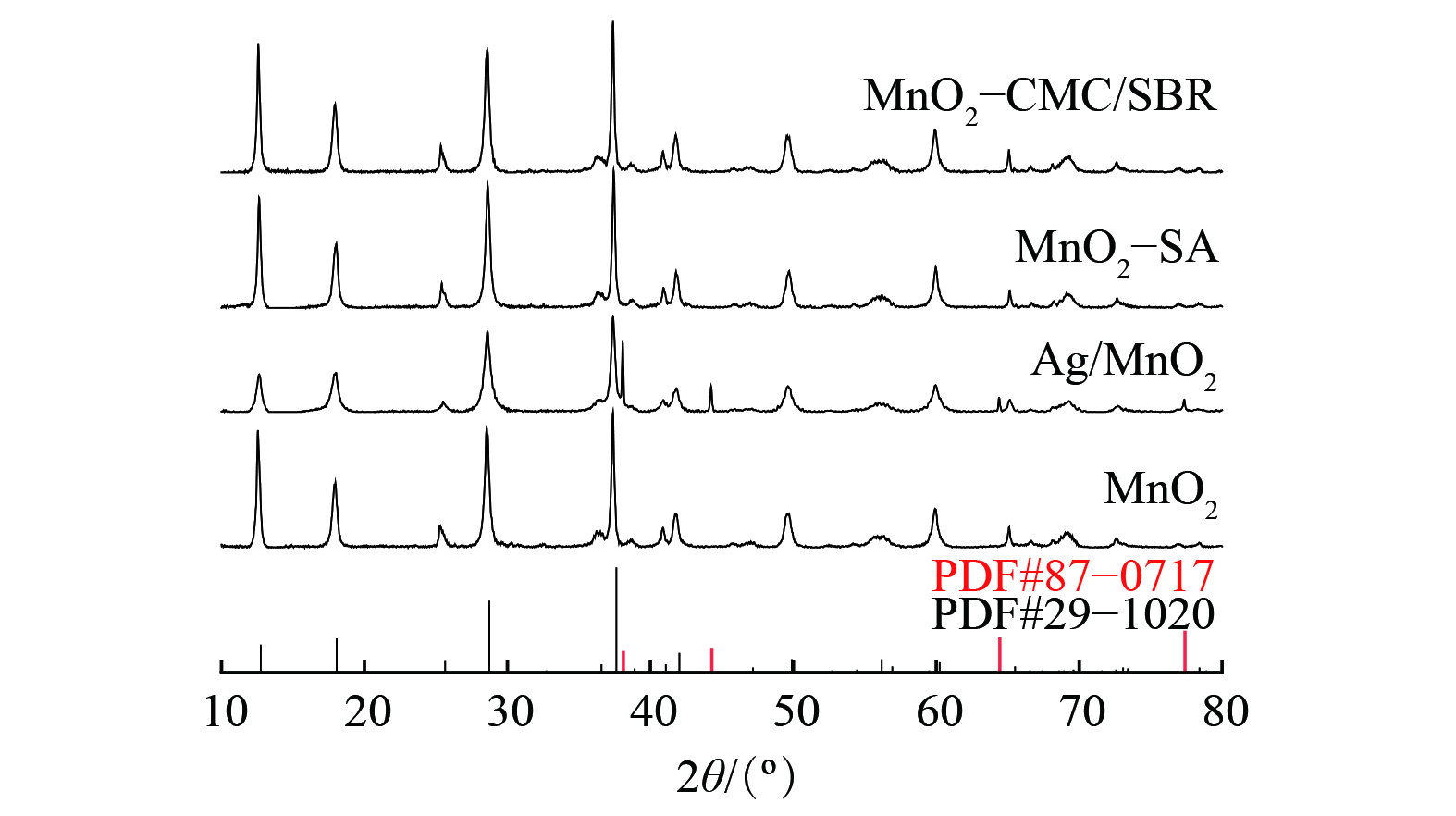
1) XRD、BET表征和脱落率。图2为MnO2和涂层的X射线衍射仪分析结果。MnO2和涂层样品的X射线衍射峰基本相同,皆在2θ角分别为12.7°、18.1°、28.7°、37.6°和60.2°处出现特征峰,与四方晶系锰钾矿MnO2的标准谱图(JCPDS 29-1020)相吻合。这表明添加CMC/SBR复配粘结剂和SA粘结剂均未改变或破坏催化剂的晶体结构。Ag/MnO2除含有锰钾矿MnO2的特征峰(JCPDS 29-1020)外,在2θ角分别为38.1°、44.3°、64.4°、77.4°处存在立方相Ag的特征峰(JCPDS 87-0717)。这表明在催化剂掺杂Ag的制备过程中,部分Ag未进入到MnO2隧道中,而在MnO2表面形成Ag纳米颗粒。
表1为MnO2和涂层的BET,以及脱落率测试结果。添加粘结剂会导致催化剂比表面积减小,且粘结剂含量越高,比表面积越小。平均孔径随粘结剂含量增加而增加,这是由于粘结剂对催化剂微孔产生了堵塞。同时,粘结团聚又在涂层中产生了新孔隙。当粘结剂含量相同时,添加SA粘结剂比添加CMC/SBR复配粘结剂的比表面积更大,这是由于SA热分解温度(220~280℃)低于SBR分解温度(>400℃),经300℃焙烧后SA大部分热分解,MnO2会更充分地暴露[15-16]。
催化剂涂层的脱落率随粘结剂含量的增加而减小。这是由于粘结剂含量越高,对MnO2的粘结强度越大。当粘结剂含量≥1.0%时,MnO2-CMC/SBR和MnO2-SA催化剂的脱落率均小于5%。而当粘结剂含量相同时,催化剂MnO2-CMC/SBR的脱落率小于MnO2-SA,这也同样说明了经300 ℃焙烧后MnO2-SA催化剂中SA粘结剂相比CMC/SBR粘结剂分解得更彻底,故粘结能力下降。
2) SEM表征。利用扫描电子显微镜分别对催化剂及涂层样品进行观察(图3)。图3(a)和(b)表明,MnO2和Ag/MnO2粉末的微观形貌相似,均呈现海胆球状的团簇结构,颗粒直径为1~50 μm。所有团簇体颗粒均为具有规则的长直纳米棒紧密交织团聚而成,纳米棒的长度为200~500 nm,平均直径约20 nm。
图3(c)和(d)表明,添加CMC/SBR复配粘结剂后MnO2纳米棒变短,长直边界线消失,表面出现少量SBR球状突起。纳米棒的平均直径增至40~60 nm后,催化剂出现一定程度团聚。这说明粘结剂对MnO2产生粘结和包覆,且随粘结剂含量的增加SBR球状突起增多,包覆作用增强。图3(e)和(f)表明,添加SA时涂层催化剂纳米棒变短,平均直径增至40~60 nm,无明显突起,相比添加CMC/SBR时纳米棒具有更清晰的边界。这说明催化剂受到SA包覆作用相对更弱些,与BET结果相对应。
-
1) 粘结剂类型及含量的影响。图4为粘结剂类型及含量对臭氧分解性能的影响。图4(a)表明,当CMC/SBR粘结剂质量分数由0.5%增至1.5%时,臭氧分解率由37.9%降至21.7%。类似地,图4(b)表明,当SA粘结剂质量分数由0.5%增至1.5%时,臭氧分解率从53.0%降至39.1%。随着粘结剂含量的增加,催化剂对臭氧的分解性能降低。这可能归因于添加粘结剂对MnO2产生一定包覆和粘结作用,使其比表面积减小,活性组分MnO2不能充分暴露,从而减弱了催化剂与臭氧的接触。由于300 ℃焙烧时,添加的SA相对CMC/SBR热分解得更充分,从而导致添加SA后催化剂比表面积高于相同含量的CMC/SBR,且活性组分MnO2暴露更充分,因此,MnO2-SA比MnO2-CMC/SBR具有更高的臭氧分解率。MnO2-SA0.5%涂层的臭氧分解率为50.2%,而MnO2-SA1.0%涂层的臭氧分解率为46.8%,两者相差不大,而SA含量为0.5%和1.0%时涂层脱落率分别为13.16%和4.15%。因此,综合考虑催化剂涂层结合性能和臭氧分解性能,选取质量分数为1.0%的SA粘结剂进行后续实验。
2) 催化剂活性组分的影响。图5为Ag修饰前后MnO2催化剂的臭氧分解性能。催化剂Ag/MnO2-SA1.0%反应进行2 h后,臭氧分解率为64.4%,而MnO2- SA1.0%的臭氧分解率仅42.1%。Ag修饰可使MnO2催化分解臭氧效率提高约50%。氧化锰表面存在的氧空位是臭氧吸附和分解的主要活性中心。Ag掺杂MnO2后,一方面进入到MnO2隧道中的Ag不仅可更均匀分布在MnO2晶体中,还能显著降低Mn—O配位数,在晶体结构保持完整的同时促进催化剂生成更多的氧空位[9];另一方面,MnO2表面存在的Ag纳米颗粒对臭氧更易形成化学吸附,同时Ag纳米颗粒还与MnO2间形成协同效应,以提高催化剂表面的氧空位浓度,进而提高系统的催化活性[17]。
-
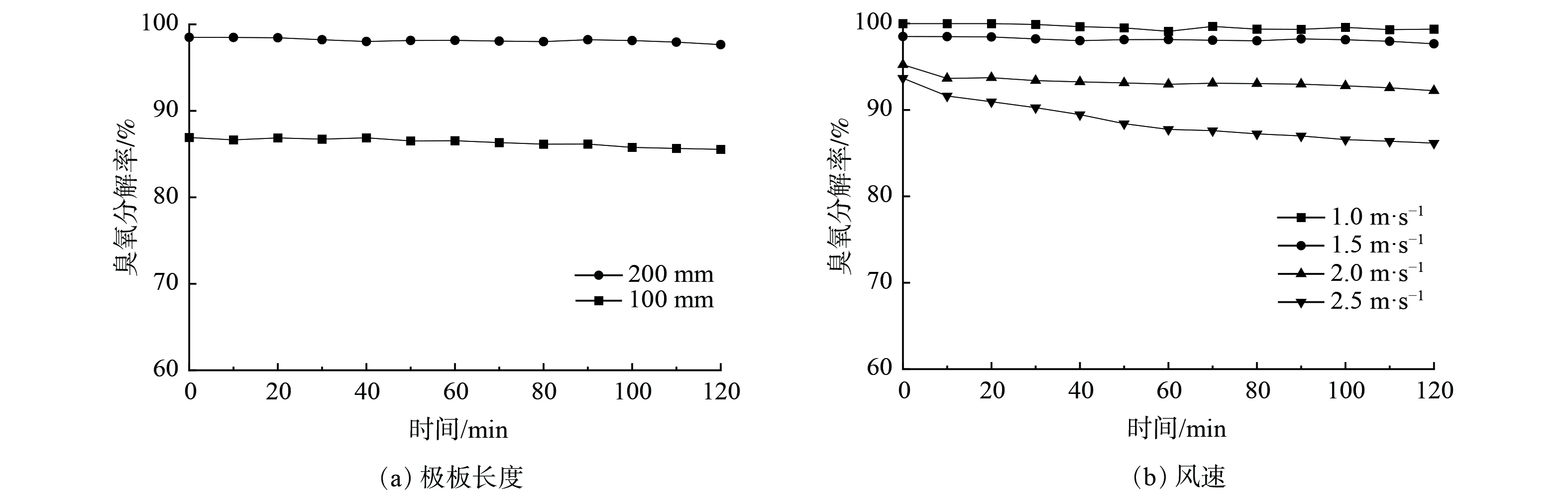
在相同板间距条件下,可通过改变极板长度及气流通过极板速度来调整臭氧分解的反应时间,结果如图6所示。图6(a)表明,极板长度由100 mm增至200 mm,对应反应时间由0.066 s增至0.133 s时,臭氧分解率由85.5%增至98%。图6(b)表明,气流风速从1.0 m·s−1增至2.5 m·s−1,对应的反应时间由0.200 s减至0.080 s时,臭氧分解率从100%逐渐降至86%,且反应0.1 s以上时,臭氧分解率均达到90%以上。
-
1)初始臭氧浓度的影响。图7为初始臭氧质量浓度分别为2.1、10.7和21.4 mg∙m−3时的臭氧分解率,其对应分解率分别为97.6%、92.5%和86.1%,即分解率随着臭氧质量浓度提高而降低。这是由于当臭氧质量浓度增大时,单位体积催化剂所接触的臭氧分子增多,使部分臭氧分子未能参与反应而使分解率下降。进一步地,还考察了初始臭氧质量浓度为2.1 mg∙m−3的臭氧分解稳定性。在24 h内,Ag/MnO2-SA1.0 %催化剂的臭氧分解效率维持在97%以上,以上结果说明该催化剂稳定性良好。
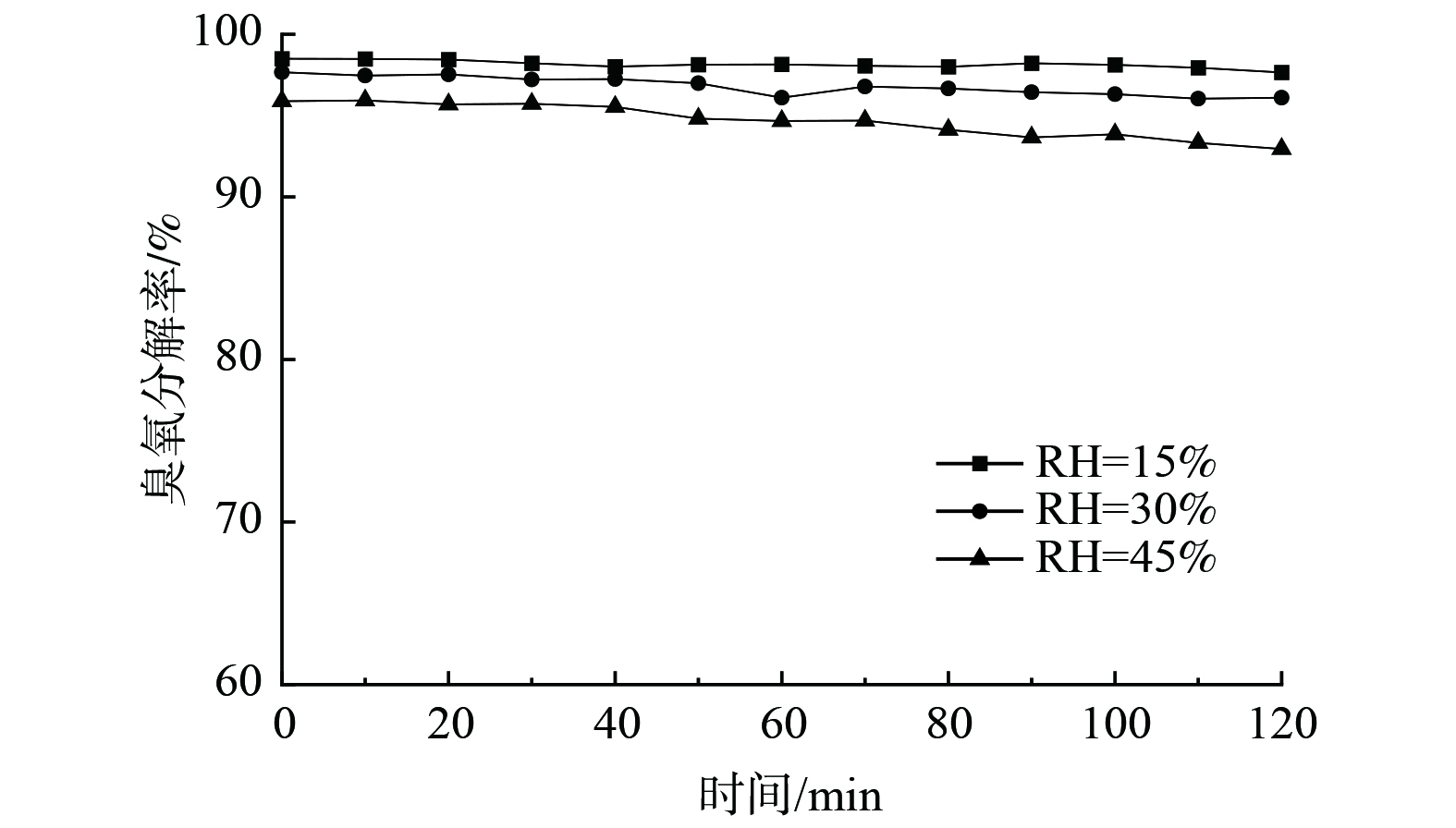
2)空气湿度对臭氧分解性能的影响。空气湿度是影响催化剂催化分解臭氧性能的重要因素。在空气相对湿度分别为15%、30%和45%时,臭氧分解率分别为97.6%、96.1%和92.9%(图8)。催化剂的臭氧分解性能随相对湿度的增加有所下降,这是由于水分子和臭氧在催化剂表面形成了竞争吸附[18]。然而,即使湿度达到45%,仍能保持90%以上的分解率。这是由于掺杂的Ag可显著降低Ag/MnO2表面活性位与水分子间的亲和作用,从而提高Ag/MnO2催化剂的抗水性[9]。
-
1)极板表面涂覆臭氧分解催化剂可实现臭氧原位分解。入口臭氧质量浓度为2.1 mg·m−3、相对湿度为15%、反应时间大于0.1 s时,臭氧分解效率可达90%以上。
2)Ag修饰可使MnO2催化分解臭氧效率提高约50%,但催化剂浆液粘结剂的存在会导致催化分解臭氧效率一定程度下降,需根据催化剂的结合能力和臭氧分解性能合理选择粘结剂及其添加量。
3)在相同反应条件下,提高风速和臭氧浓度,臭氧分解效率下降,可通过增加极板的板长或降低净化器运行风速以获得较大的停留时间,从而使得臭氧高效分解。Ag掺杂可明显改善MnO2的抗水性。当湿度从15%增至45%时,臭氧分解效率从98%降至约92%,分解率仅降低6%。
铝合金极板表面涂覆Mn催化剂及其对臭氧的分解
Ozone decomposition over Mn catalyst coated on aluminum alloy plate
-
摘要: 静电式空气净化器工作时会产生臭氧,因此,可通过在极板表面涂覆臭氧分解催化剂以实现对臭氧的原位分解。研究了6061型铝合金极板表面涂覆Mn基催化剂时,活性组分、粘结剂、反应时间和环境条件等因素对催化分解臭氧性能的影响。结果表明:在实际应用相适应的条件下,臭氧可实现原位高效分解;催化剂构成是影响臭氧分解的关键因素,Ag修饰可使MnO2催化分解臭氧效率提高约50%,但粘结剂的存在会导致催化分解臭氧效率下降;臭氧分解效率随入口臭氧浓度升高或空气湿度增大略有下降。本研究结果可为静电式空气净化器同时实现除尘提效与臭氧控制提供参考。Abstract: Ozone can be generated during the operation of electrostatic air purifier. The in-situ catalytic decompo-sition of ozone is possible by coating catalyst on the electrostatic precipitation plate. In this work, the effects of active composition and binding agent, reaction time and environmental conditions on catalytic decomposition efficiency of ozone were investigated by coating Mn catalysts on the 6061 aluminum alloy plate. The results show that ozone can be effectively decomposed under the conditions matching to actual application. The catalyst compositions are the key factor affecting ozone decomposition performance. The decomposition efficiency increases by 50% or so when the Mn catalyst was modified by dopping Ag. The presence of binding agent makes ozone decomposition efficiency decrease. The ozone decomposition efficiency slightly decreases with the increase of inlet ozone concentration and relative humidity.
-
Key words:
- air purifier /
- electrostatic /
- plate /
- ozone /
- catalytic decomposition
-
城市垃圾填埋处理过程中会产生大量垃圾渗滤液,其成分复杂且含有较多的难降解污染物,同时随着填埋时间的延长,其有机物的性质也会随之改变,很难获取准确信息,给渗滤液的处理带来了困难。溶解性有机物(dissolved organic matter,DOM)是渗滤液的主要成分之一,占总有机碳(total organic carbon,TOC)的80%以上[1]。现有研究[2-3]表明,垃圾渗滤液中的DOM主要包含腐殖酸、富里酸和亲水性有机物这3种成分,DOM中溶解态有机碳(dissolved organic carbon,DOC)通常在800~20 000 mg∙L−1,其中腐殖质在成熟渗滤液中的比例可超过70%[4]。随着填埋时间的延长,DOM的组成也会发生显著变化,通常表现为腐殖质类有机物含量有所增加[5-6]。
为获取渗滤液更多的有机物信息,通常使用光谱法、树脂分离或凝胶色谱分离法结合TOC或光谱进行分析。HUO等[7]采用XAD-8和阳离子交换树脂分离DOM,对各种组分的特性进行了比较,发现随着填埋时间增加,腐殖酸的比例可增加到45%左右,因此,腐殖质类物质通常会成为表征的首要目标物。基于有机物中的多种不同荧光基团[8-9],通过三维荧光可将类腐殖质分子分为类富里酸和类腐殖酸2大类有机物,同时还可分辨类蛋白有机物[10-14]。AFTAB等[13]采用三维荧光分析渗滤液有机物,发现有机物的主要成分是类腐殖质有机物,约占62%,其余多为类蛋白有机物。席北斗等[14]采用三维荧光和紫外可见光谱研究了不同年限垃圾渗滤液中DOM的组成变化,发现年轻垃圾渗滤液中类蛋白有机物是DOM的主要部分,随着填埋时间增加,类腐殖质有机物逐渐增多。在现有色谱分析基础上,有研究者将液相色谱分离法(size-exclusion chromatography,LC)和不同检测器结合,对有机物进行表征,如可将LC与二维荧光检测器[13]、有机碳检测器[15]等组合成为实时检测系统。此系统可将DOM分成不同组分,并可对组分的表征参数进行实时分析,如此可获得同一组分不同表征方式之间的差异,进而获取更多的有机物信息。
渗滤液中有机物通常难以生物降解,因此,氧化预处理工艺对渗滤液有机物去除具有重要意义。臭氧氧化可改变渗滤液中难降解有机物的分子结构,将大分子疏水有机物分解为亲水性小分子,主要包括醇类、有机酸或其他高生物利用度的化合物,改善生物降解性。因此,将臭氧氧化与有机物组分或结构联合分析,对渗滤液的处理具有重要意义。本研究采集不同龄渗滤液,利用液相色谱-有机碳-有机氮-紫外吸收(size-exclusion chromatography-organic carbon detection-organic nitrogen detection-ultraviolet visible detection,LC-OCD-OND-UVD),并结合树脂分离、三维荧光和紫外可见光谱对渗滤液有机物进行了表征和分析,研究了不同年限垃圾渗滤液中DOM的有机物组成特征及臭氧氧化对有机物组分的影响,研究结果可为垃圾渗滤液中的处理提供参考。
1. 材料与方法
1.1 实验水样
渗滤液取自亚热带地区某固废填埋场,1期工程运行25 a后封场,目前已封场约9 a。2期工程于2012年投入使用,目前仍在运行中。1期和2期垃圾渗滤液分别以渗滤液1和渗滤液2表示。2种渗滤液的主要水质特征见表1。
表 1 垃圾渗滤液主要水质指标Table 1. Characteristics of the landfill leachates渗滤液 COD/(mg∙L−1) TOC/(mg∙L−1) UV254/cm−1 B/C 色度 SUVA/(L∙(mg∙m)−1) SS/(mg∙L−1) pH 电导率/(mS∙cm−1) 渗滤液1 1 030 258 5.55 0.29 1 000 2.26 97 8.44 13.51 渗滤液2 6 925 2 211 29.5 0.24 8 000 1.57 167 8.26 48.11 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 水样臭氧氧化
利用自制反应器制备臭氧水,取500 mL一定浓度的臭氧水,置于烧杯中,投加少量垃圾渗滤液原液,使O3/TOC(w:w)分别为3和6。用磁力搅拌器搅拌(300 r·min−1)使其混合均匀,在反应1 min和30 min时分别取水样加入0.5 mL 1 mol·L−1的硫代硫酸钠,消除残余臭氧待测。实验过程设置对照组并在1 min和30 min利用靛蓝法测定臭氧浓度。
1.3 有机物树脂分离
垃圾渗滤液原液经1 μm滤膜(GF/B,Whatman)过滤,调整pH后通过XAD-8和XAD-4树脂柱将有机物分离为亲水性有机物(hydrophilic,HPI)、憎水性中性有机物(hydrophobic neutral,HPO-N)、弱憎水性中性有机物(transphilic neutral,TPI-N)、憎水性酸(hydrophobic acid,HPO-A)和弱憎水性酸(transphilic acid,TPO-A)[16]。组分的TOC浓度采用燃烧氧化-非分散红外吸收法(TOC-L CPH/CPN,岛津)测定。
1.4 LC-OCD-OND-UVD分析
将适宜DOC浓度的渗滤液,用LC-OCD-OND-UVD分析(Model 9,DOC-Labor),3个检测器可实时对有机物进行检测和表征。其中OCD是对有机碳的响应,UVD是对254 nm处的紫外吸收组分(芳香和不饱和结构)的响应,OND是对有机氮(与DOM结合的有机氮)和无机氮(硝酸盐和氨氮)的响应。在线纯化流动相由色谱泵输送至色谱柱,色谱柱用聚丙烯酸酯基聚合物填充,柱前过0.45 μm过滤器。检测条件如下:进样量1 000 μL,流速为2.0 mL·min−1,保留时间70 min,有机物分成5种组分:生物聚合物(biopolymer,BP),腐殖物质(humic substance,HS),腐殖质分解而成的低分子质量类腐殖质物质(building block,BB),低分子中性有机物和酸性有机物(low molecular weight neutral/acid,LMWN/A),有机物组分可通过内置软件ChromCALC进行定性和定量[15]。
1.5 光谱测定
吸收光谱和UV254:紫外可见分光光度计(UV2600,岛津);三维荧光光谱(Lumina,Thermo Scientific):样品DOC浓度稀释至合适范围,激发和发射狭缝宽度为5 nm,激发波长为200~450 nm,发射波长为250~550 nm,扫描速度为1 200 nm·min−1,PMT为700 V,样品荧光光谱减去超纯水的荧光光谱以去除拉曼散射的影响。扫描结果利用EFC软件[17]进行分析。
1.6 其他指标
BOD采用呼吸压差法(BOD TrakⅡ,哈希)测定;COD采用快速消解分光光度法(DR1010+DRB200,哈希)测定;pH采用电极法(FE28,梅特勒)测定;电导率采用电极法(FE38,梅特勒)测定;色度采用比色法(HI96727,哈纳)测定;利用聚苯乙烯磺酸钠(分子质量分别为210 Da、4.3 kDa、6.8 kDa、10 kDa、17 kDa,Polymer Standards Service GmbH,Germany)和水杨酸(138 Da,阿拉丁)作为标准品,进LC-OCD,以OCD作为检测器,保留时间与分子质量呈线性关系。
2. 结果与讨论
2.1 不同龄渗滤液的LC-OCD-OND-UVD特征
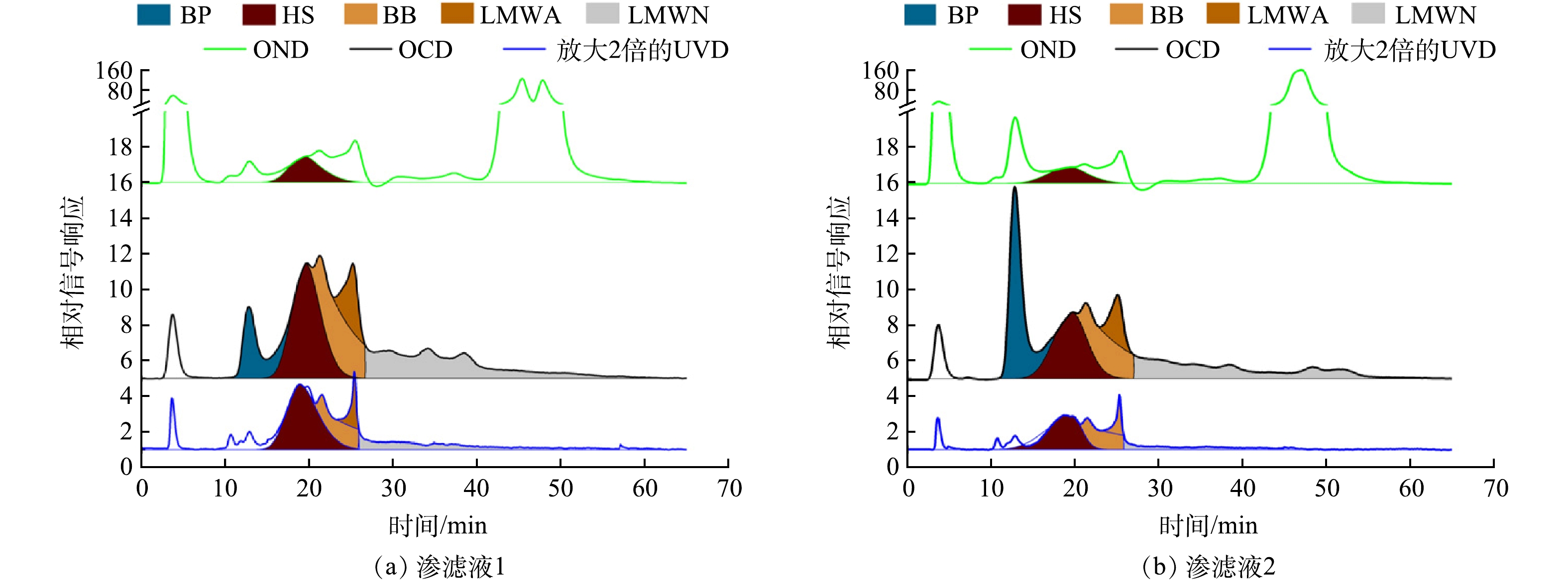
图1为2种垃圾渗滤液中组分对应的LC-OCD-OND-UVD色谱图。在10、20、30、40、50和60 min时的表观相对分子质量分别是19 329、4 327、969、217、49和11。由OCD图谱可知,年轻渗滤液中的BP组分显著高于老龄渗滤液(图2),结合图1的UVD和OND图谱可见,年轻垃圾渗滤液的BP组分表现出较高有机氮含量和较低的UV254吸收特征,说明此组分含氮且双键或共轭结构较少,这可能是二期填埋场接纳新的生活垃圾,大量新的BP类有机物尚未被降解所致。
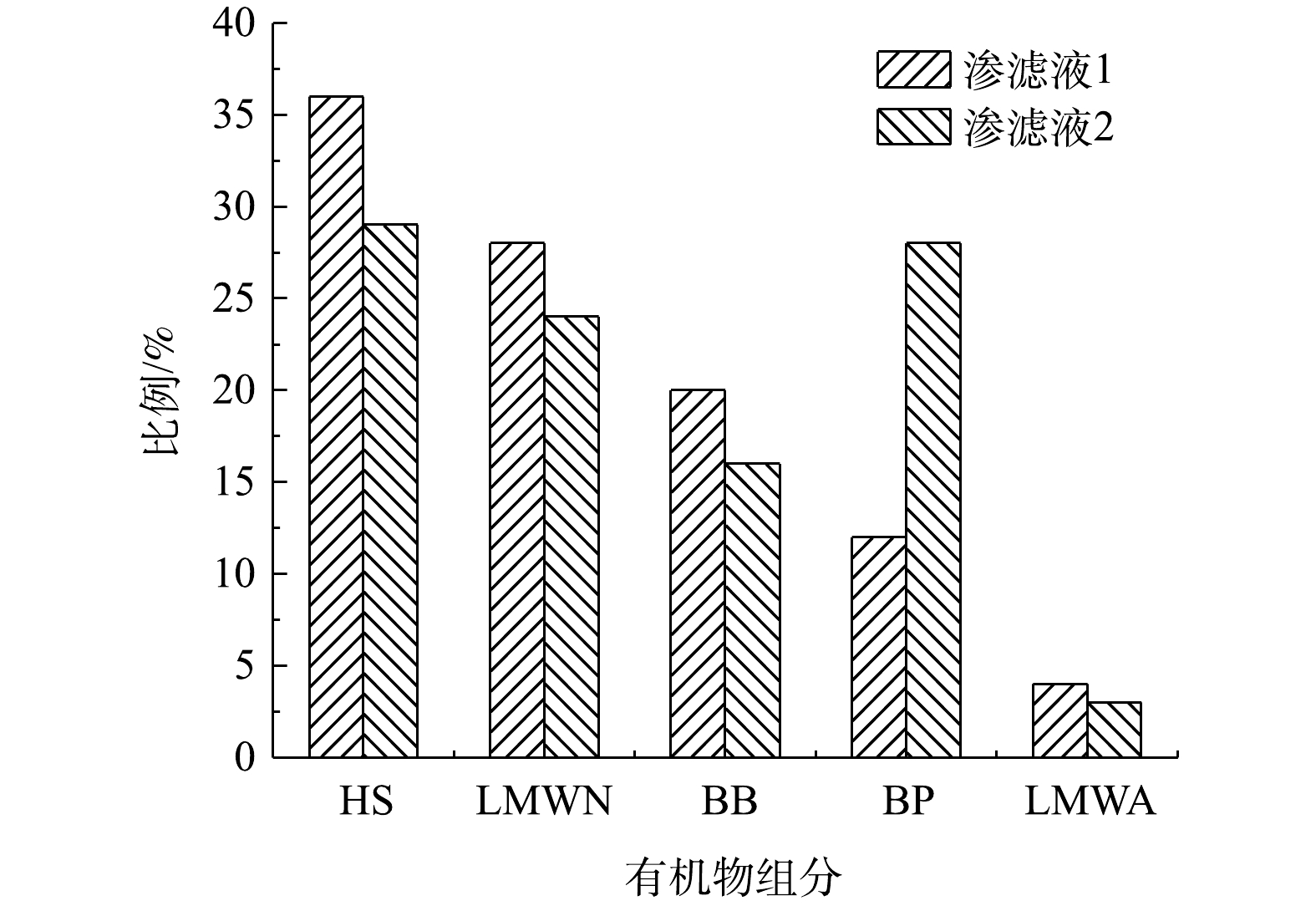 图 2 垃圾渗滤液中基于有机碳的不同有机物组分比例Figure 2. Proportions of different components in the DOM based on organic carbon in landfill leachate
图 2 垃圾渗滤液中基于有机碳的不同有机物组分比例Figure 2. Proportions of different components in the DOM based on organic carbon in landfill leachate老龄渗滤液BP组分含量较年轻渗滤液低,但对应的UV吸收却相差不大,说明经过一定年限填埋后,BP组分中易降解有机物逐渐分解,但其中有UV吸收的类有机物(含芳香和不饱和结构)无法降解。由OND图谱可见,老龄渗滤液BP组分的含氮量降低,但其他组分均出现增加,特别是腐殖质类的HS、BB和LMWA组分的含氮量明显增加。说明BP组分分解后生成较低分子质量的腐殖质类物质[18],甚至是分子质量更小的亲水性有机物。因此,根据氮元素的色谱响应和腐殖质的生成途径,可推测BP组分易于分解,但很可能大部分物质并未脱离垃圾填埋系统,只是转化成分子质量较低的其他物质,特别是腐殖质类有机物。另外,根据UVD和OND在保留时间为11 min时的信号峰,可以推测渗滤液中可能存在一类含氮物质,其分子质量略大于BP组分,有较强的UV吸收,但因其含碳量较低,OCD检测器对其没有很好的响应。
除BP组分外,2种垃圾渗滤液均具有较高的腐殖化程度(图2)。1期和2期渗滤液腐殖质类有机物的比例分别为60%和48%,组分比例大小顺序均是HS>LMWN>BB>LMWA。随着填埋年限的延长,渗滤液中腐殖质类有机物比例逐渐提高,这与大部分文献报道的结果相似[19]。
由图2可见,2种渗滤液大部分有机物组分比例相似,说明填埋后,BP组分会快速分解,形成了相对稳定的低分子中性有机物和腐殖质类有机物。LMWN可能是渗滤液中有机物组分的最终形态,可用生物处理工艺去除,这也是2种渗滤液B/C可达到0.3左右(表1)的主要原因。另外,由图1可知,分子质量不同的3种腐殖质类有机物组分,可能遵循HS-BB-LMWA的分解步骤。且值得注意的是,排在分解链条最后,DOC占比为仅3%~4%(图2)的LMWA,这可能是腐殖质类大分子有机物的降解产物,显示出极强的UV吸收和含氮特征。这类有机物可能由含氮杂环有机物、含氮且有芳香或不饱和结构的物质组成,难以生物降解。因此,垃圾渗滤液的生物处理过程中应重点关注此类有机物,这可能是生物处理取得突破的理论依据。
2.2 不同龄渗滤液的三维荧光光谱特征
渗滤液的三维荧光光谱如图3所示。2种垃圾渗滤液均出现了较强的代表可见光区类富里酸有机物的P1峰(Ex 320 nm/Em 410 nm)和代表紫外区类腐殖酸有机物的P2峰(Ex 250 nm/Em 460 nm)[10,13,20],显示出有机物较强的类腐殖质属性[21-23]。渗滤液1的峰荧光强度显著高于渗滤液2,说明其有机物的腐殖质属性更强,包含更多的双键和芳构化结构,这与图2中一期渗滤液腐殖质类有机物的比例更高的结果相一致。
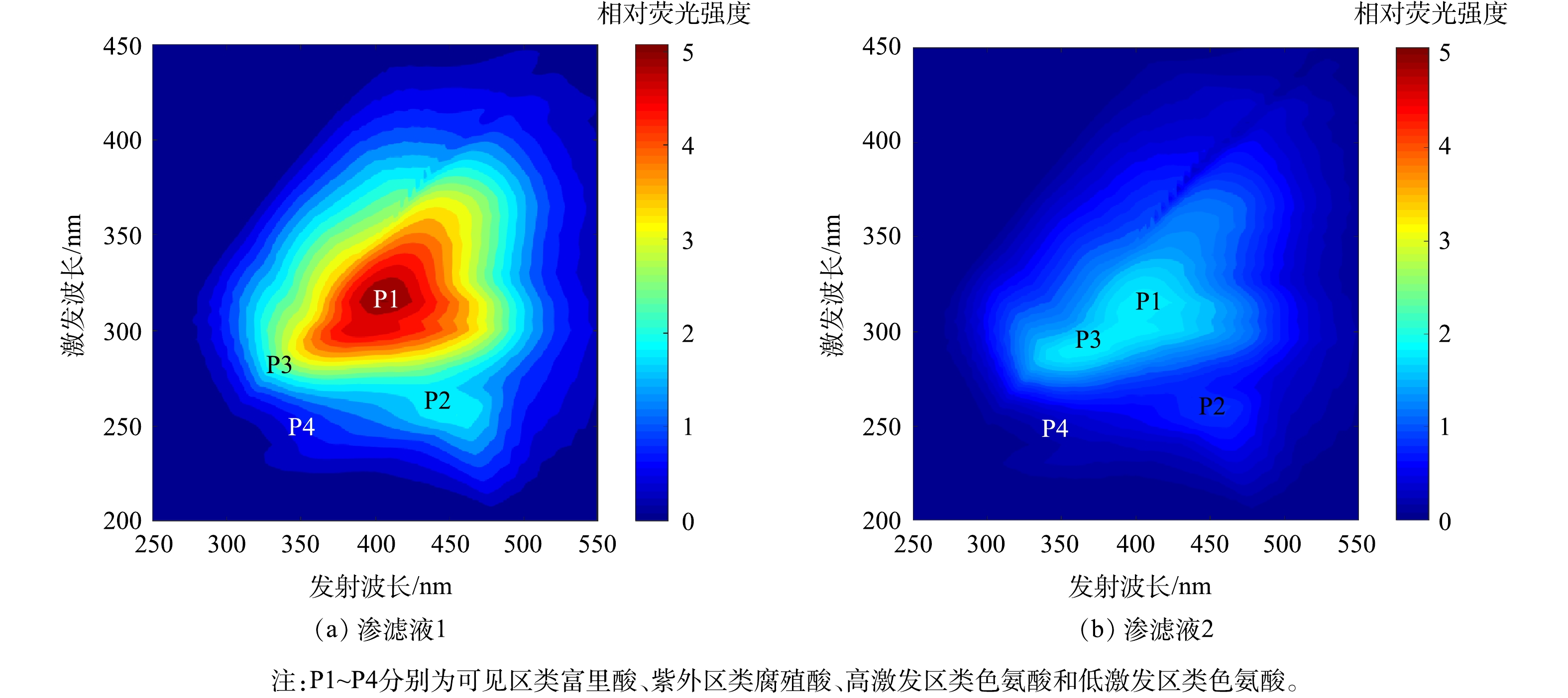 图 3 垃圾渗滤液三维荧光图谱Figure 3. Three-dimensional fluorescence spectra of DOM in landfill leachate
图 3 垃圾渗滤液三维荧光图谱Figure 3. Three-dimensional fluorescence spectra of DOM in landfill leachate渗滤液中的另外2个峰是代表类色氨酸有机物的P3(Ex 280 nm/Em 340 nm)和P4(Ex 230 nm/Em 340 nm),这类物质的荧光响应主要与DOM中含芳环氨基酸结构的类蛋白有机物有关[13,24-25]。年轻渗滤液中易生物降解的类蛋白有机物含量较高,所以渗滤液2的P3和P4特征更明显。此类物质通常具有较高的分子质量,这与渗滤液2中含有大量的BP组分有关(图2)。P3和P4峰的强度在一期渗滤液中的减弱说明微生物优先分解了部分易生物降解的有机物,但尚未显著降低有机物总量,出现了年轻渗滤液的B/C低于成熟渗滤液的现象(表1)。值得注意的是,LC分析中占比较高的LMWN组分(图2)在荧光光谱中应属于易生物氧化降解的小分子有机物区[26],但此区却无明荧光峰。另外,P1周围还有不同程度的延伸峰,说明存在其他性质相近的有机物,这与LC无法将有机物完全分离的结果是一致的(图1)。这表明渗滤液有机物的复杂性,意味着有必要采用多种方法进行相互印证分析,才能获取准确完整的信息。
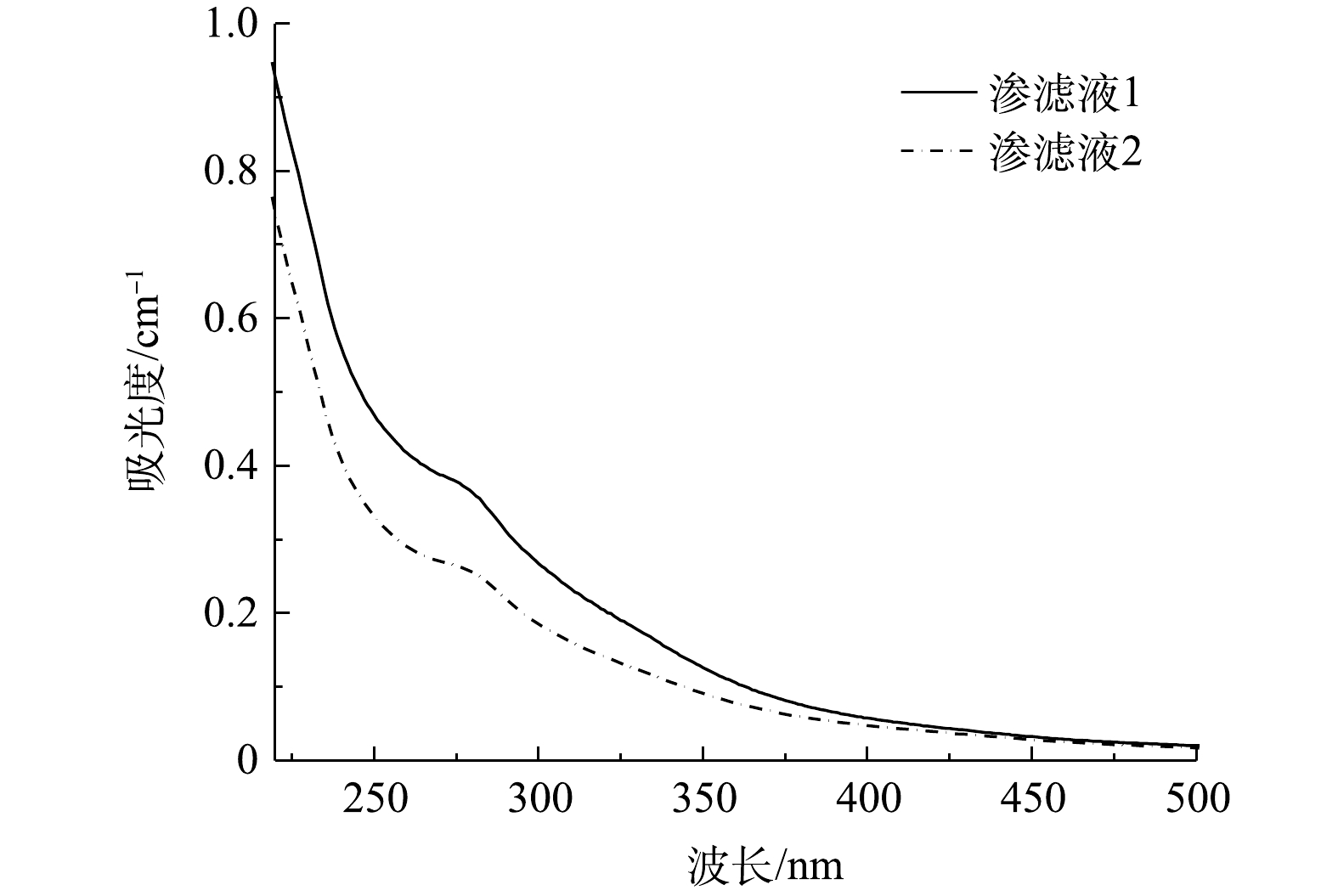
2.3 不同龄渗滤液的紫外可见吸收光谱特征
垃圾渗滤液的紫外可见光谱如图4所示。2种垃圾渗滤液无明显的特征峰,吸光度随着波长的增加呈指数降低,仅在250~300 nm处有微弱的吸收。垃圾渗滤液中DOM成分复杂,但有机物在紫外区较高的吸光度表明渗滤液中腐殖酸类有机物含量较高。而且,随着填埋时间延长,难降解有机物降解生成芳香族有机物;同时,腐殖化进程也生成了更多的腐殖质类有机物,导致有机物的紫外吸收强度更高。1期渗滤液填埋年限更长,因此,有机物的腐殖化程度更高,这与2.1中一期渗滤液中腐殖质类有机物含量更高的结果相吻合。这些难生物降解的有机物在稳定的填埋场渗滤液中逐渐富集,给渗滤液的处理带来了难度。
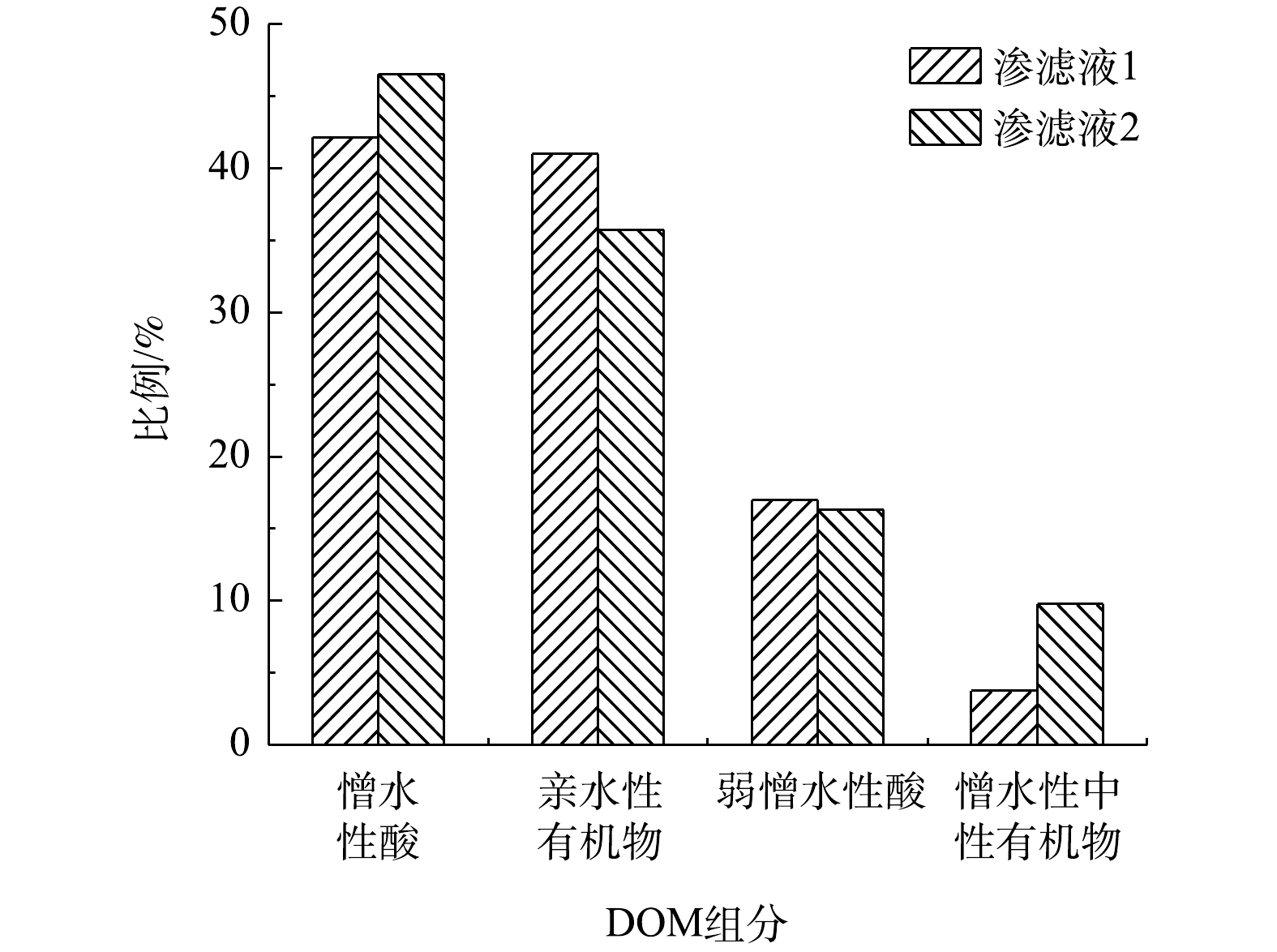
2.4 不同龄渗滤液的有机物树脂分离特征
图5为渗滤液中不同组分所占的比例。2种渗滤液的有机物均表现出中晚期填埋场渗滤液较强的类腐殖质特征,憎水性有机物分别占45.91%和56.3%。这与2个填埋场的运行年限均较长有关,虽然2期填埋场仍在接收填埋垃圾,但早期填埋的垃圾产生的渗滤液含有大量腐殖质和芳香类有机物,使其体现出中晚期填埋场渗滤液的特征。另外,较高比例的亲水性有机物,也是中晚期填埋场的有机物特征之一,此部分有机物主要包括小分子脂肪酸、醛类、糖类化合物等[27-28]。LC和树脂分离均能展示这一有机物特征,且表现出相同的变化规律。但经树脂分离的有机物并未检出LC分析中出现的BP组分。而且,树脂分离的憎水性酸中主要的有机物为腐殖质[27-28],其与LC分离的HS、BB和LMWA性质相似,但比例变化规律相反,这可能与树脂分离方法的环节较多、存在不确定性有关。
2.5 渗滤液中不同有机物组分的三维荧光光谱特征
图6为垃圾渗滤液DOM各组分的三维荧光图谱。1期渗滤液中的憎水性酸组分的三维荧光光谱图类似于渗滤液原液(图3(a)),憎水性类富里酸有机物(P1)和类色氨酸有机物(P3、P4)是憎水性酸的主要成分。通常认为憎水性有机物难以生物降解,但易生物降解的蛋白质类有机物(P3、P4)却保留在憎水性有机物中。结合LC结果可知,与蛋白质类有机物相对应的是BP组分,其对应分子质量接近20 kDa(图1),如此大的分子质量说明有机物具有较长的碳链骨架,导致其水溶性降低,容易被树脂分离到憎水性酸组分中。在2种渗滤液的憎水性酸中,P1和P3相反的强度特征恰好说明不同填埋时期易降解和难降解有机物之间此消彼长的变化规律。
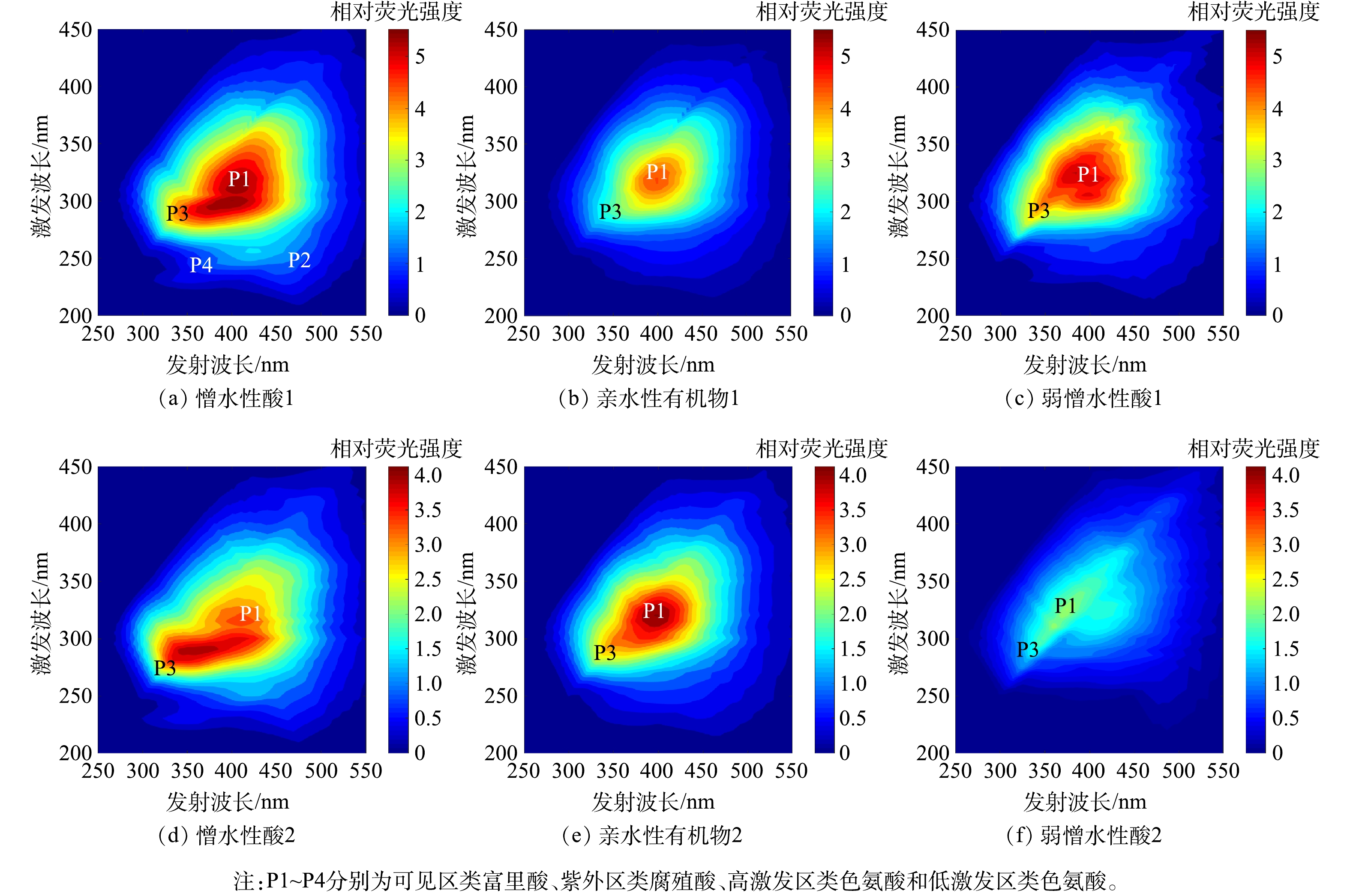 图 6 垃圾渗滤液各DOM组分的三维荧光图谱Figure 6. Three-dimensional fluorescence spectra of DOM components in landfill leachate comprises
图 6 垃圾渗滤液各DOM组分的三维荧光图谱Figure 6. Three-dimensional fluorescence spectra of DOM components in landfill leachate comprises亲水性有机物仅表现出较强的富里酸P1峰,但LC中的极低UVD响应值表明亲水性有机物基本不含类富里酸。其可能的原因是,树脂分离无法将绝大部分类富里酸有机物富集于憎水性酸,这是树脂分离法的固有弊端。亲水性有机物的占比较高(图5),但无显著的荧光峰出现,说明树脂分离的亲水性有机物荧光和UV响应很弱,可能包含醇、醛、酸和小分子脂肪酸等化合物。
2种渗滤液均分离出15%左右的弱憎水性酸,此类物质主要指一些相对分子质量较低的易降解有机物或溶解性微生物代谢产物,因此,1期渗滤液的荧光峰强度更高,这与LC分析中各组分的变化规律相似。
2.6 臭氧氧化对有机物特性的影响
1) TOC、UV254和电导率的变化。图7为臭氧氧化后有机物的变化,a为O3/TOC(w∶w)=3,b为O3/TOC(w∶w)=6时的结果。反应30 min后,TOC、UV254及电导率均降低。臭氧与总有机碳比例由3增大到6时,1期垃圾渗滤液和2期垃圾渗滤液的TOC去除率分别由1%以下提高到10.24%和3.55%。在此静态实验条件下,一方面臭氧存在自分解,会降低参与反应的臭氧量;另一方面渗滤液组分复杂,可能存在其他消耗臭氧的物质。这使得与有机物反应的臭氧量较少,虽然臭氧有较高的氧化还原电位,但要实现有机碳的彻底矿化也相当困难,此结果与大部分研究中TOC去除率不高一致。这也意味着仅靠臭氧矿化有机物会产生相当高的处理成本。
 图 7 臭氧对垃圾渗滤液中的有机物的影响Figure 7. Effect of ozonation on the organics in landfill leachate
图 7 臭氧对垃圾渗滤液中的有机物的影响Figure 7. Effect of ozonation on the organics in landfill leachate氧化对UV254的去除与图8中UV的整体吸收显著降低相吻合,说明臭氧可分解垃圾渗滤液中具有不饱和结构的有机物,但仅仅是改变了其结构[29]。有趣的是,氧化后电导率的变化规律与UV254的变化规律十分相似。电导率取决于溶液的离子组成和离子含量[30],其降低通常意味着带电粒子的减少。一方面,可能因渗滤液呈碱性,溶液中某些低价态金属离子氧化后可能形成高价态的不溶/难溶态金属氢氧化物,如Fe2+可形成Fe(OH)3导致溶液电导率降低。另一方面,臭氧氧化后有机物结构发生变化,金属离子与有机物的螯合也可能随之改变,并使电导率发生变化,但本研究中尚未得出直接的实验证据。
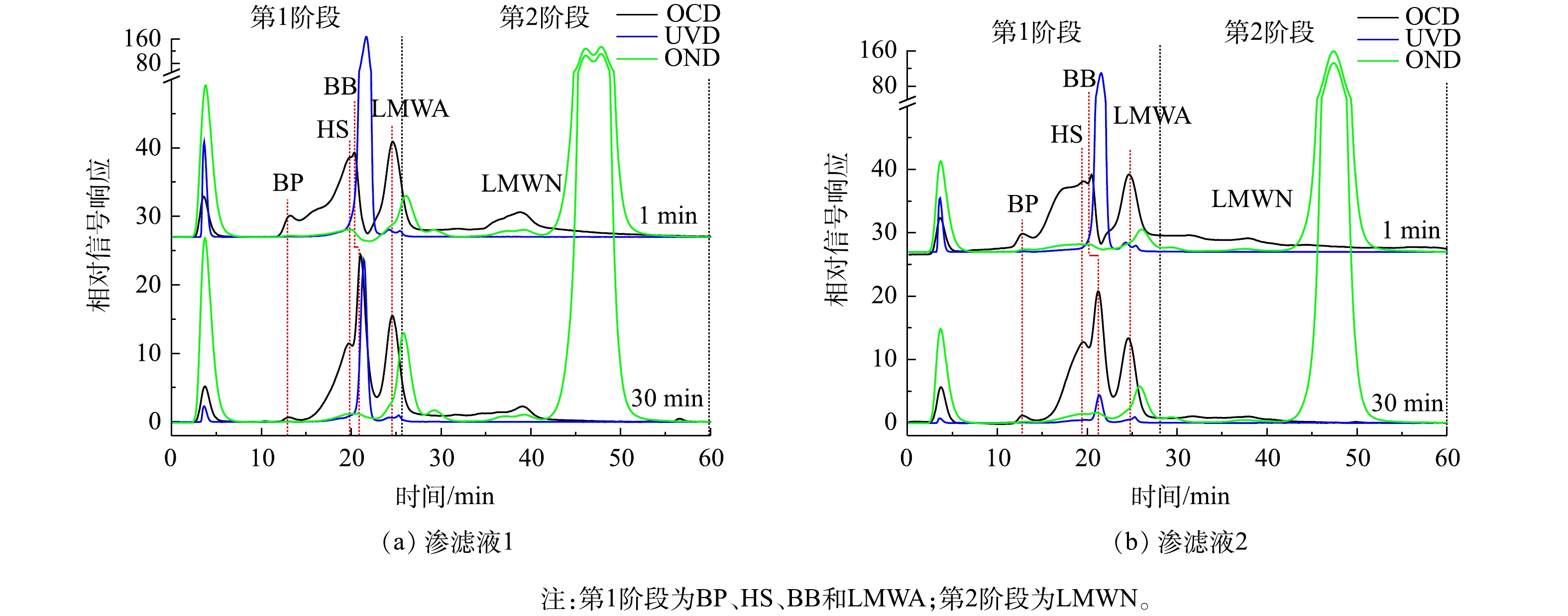
2)臭氧对有机物组分的影响。O3/TOC(w∶w)=6时反应前后水样的LC-OCD-OND-UVD结果如图8所示。由于与臭氧氧化能力相似的的芬顿氧化可以彻底矿化部分LMWN[13,31],所以在30 min后,2种渗滤液中DOC仅有略微降低。氧化后虽然DOC变化不明显,但所有组分的UV254值显著下降,与图7中TOC和UV254变化规律一致,说明较低的臭氧投加量下,臭氧主要改变了有机物结构。
与图1相比,氧化1 min即可使BP组分的OCD响应显著降低,30 min后基本消失,说明臭氧可迅速分解BP组分。随着氧化时间延长,BB峰值显著升高且停留时间略微延长(30 min,图8),这说明易降解的BP和难降解的少量HS最终被氧化为分子质量更小的类腐殖质有机物,这与AFTAB等[13]在氧化渗滤液时发现生成新氧化产物的结果一致。
UV吸收的变化表明有机物在短时间内接触臭氧时,产生了具有较高的UV吸收特性的有机物,但其机理尚不清楚。由图1和图8对比可见,氧化后LMWA的UVD响应降低,说明杂环有机物中的不饱和结构遭到破坏,但碳元素仍旧存在。氧化后BP和LMWA组分的OND响应峰也消失,但在LMWA峰后出现了新的OND响应峰。一方面,说明LMWA仅仅是有机物结构遭到破坏,大部分有机物的分子质量并未降低,因此,其OCD响应基本无变化。OND响应的延后意味着在结构破坏过程中,生成了分子质量更小且含氮的LMWA和LMWN。另一方面,也可能是BP组分的分解直接生成了小分子含氮有机物。
3. 结论
1)憎水性腐殖质类(60%)和亲水性小分子有机物(28%)是中晚期垃圾填埋场渗滤液中有机物的主要组分,低填埋龄渗滤液含有较高(28%)的BP组分。老龄渗滤液有机物具有较高的N/C比,难以生物降解。
2)三维荧光光谱分析结果表明,2种渗滤液均在可见光区类富里酸和紫外区类腐殖酸区出现较强荧光峰,且随着填埋时间的增加,有机物腐殖化程度更高,导致在此区域的荧光强度会更高。
3)较低投加量实验条件下(O3/TOC=3和6),TOC的去除率降低约4%~10%,臭氧可矿化部分LMWN组分并使分子质量小于BP组分的有机物含量有所增加,这提高了可生物降解性。
-
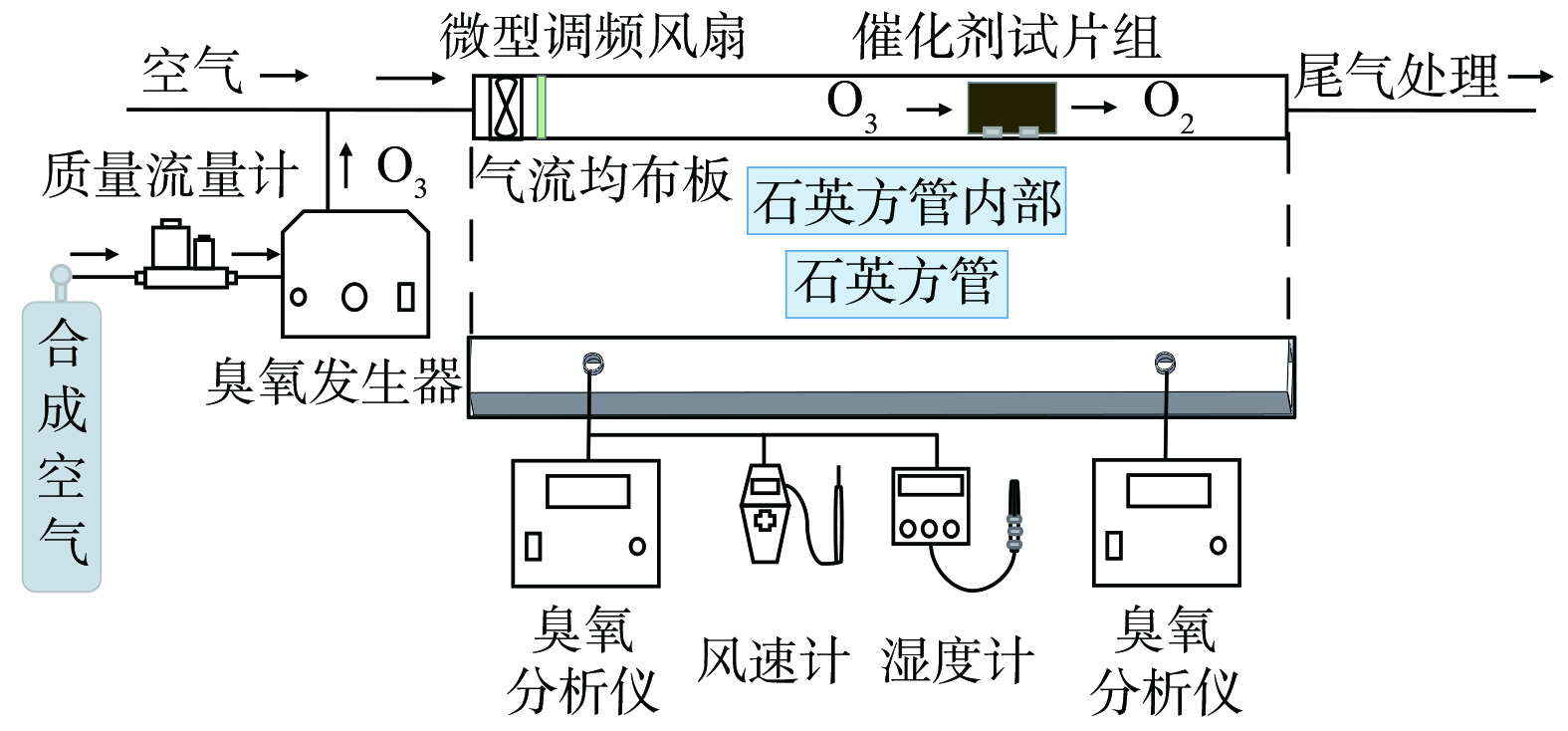
图 1 臭氧催化分解性能测试系统装置示意图
Figure 1. Schematic diagram of ozone catalytic decomposition performance test system
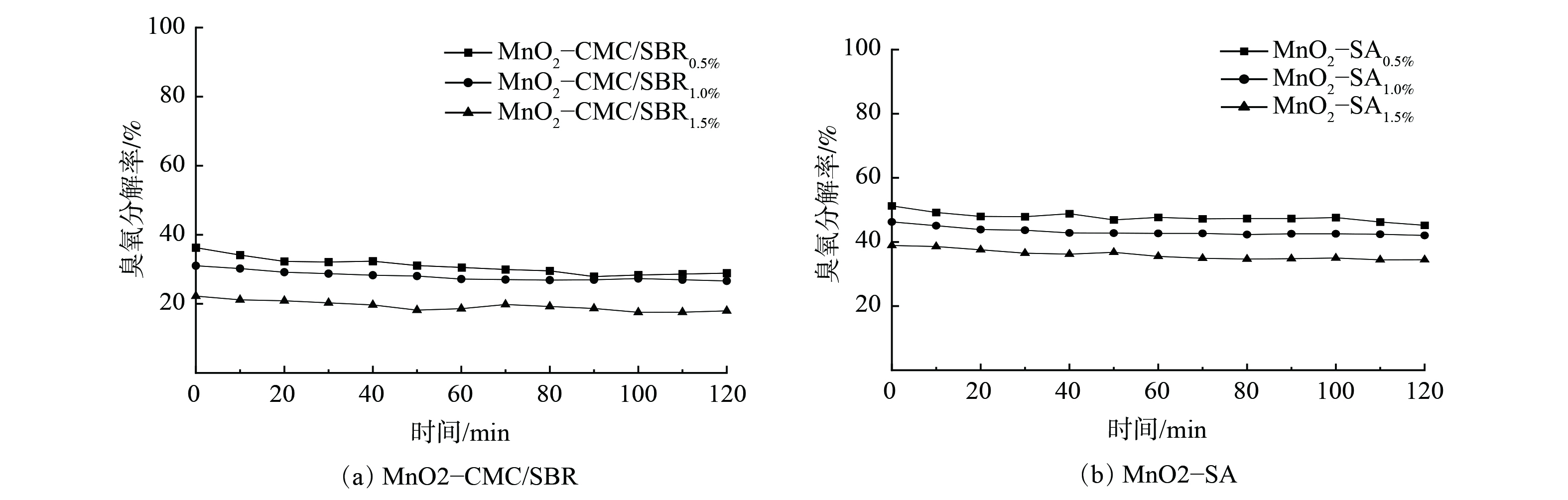
图 4 不同粘结剂及含量对臭氧分解性能的影响
Figure 4. Effect of different binder and content on ozone decomposition
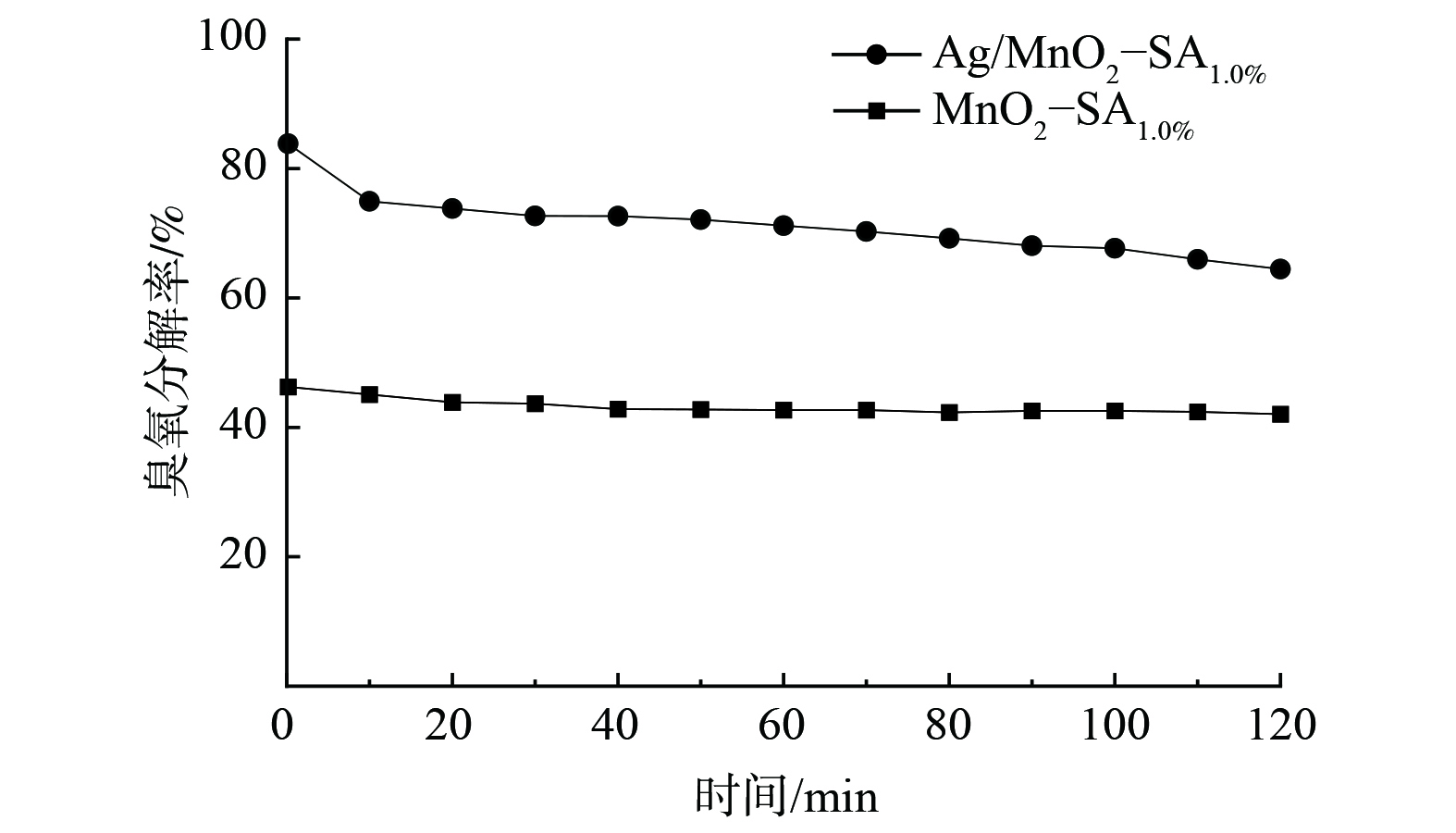
图 5 不同催化剂活性组分对臭氧分解性能的影响
Figure 5. Effect of different active components on ozone decomposition
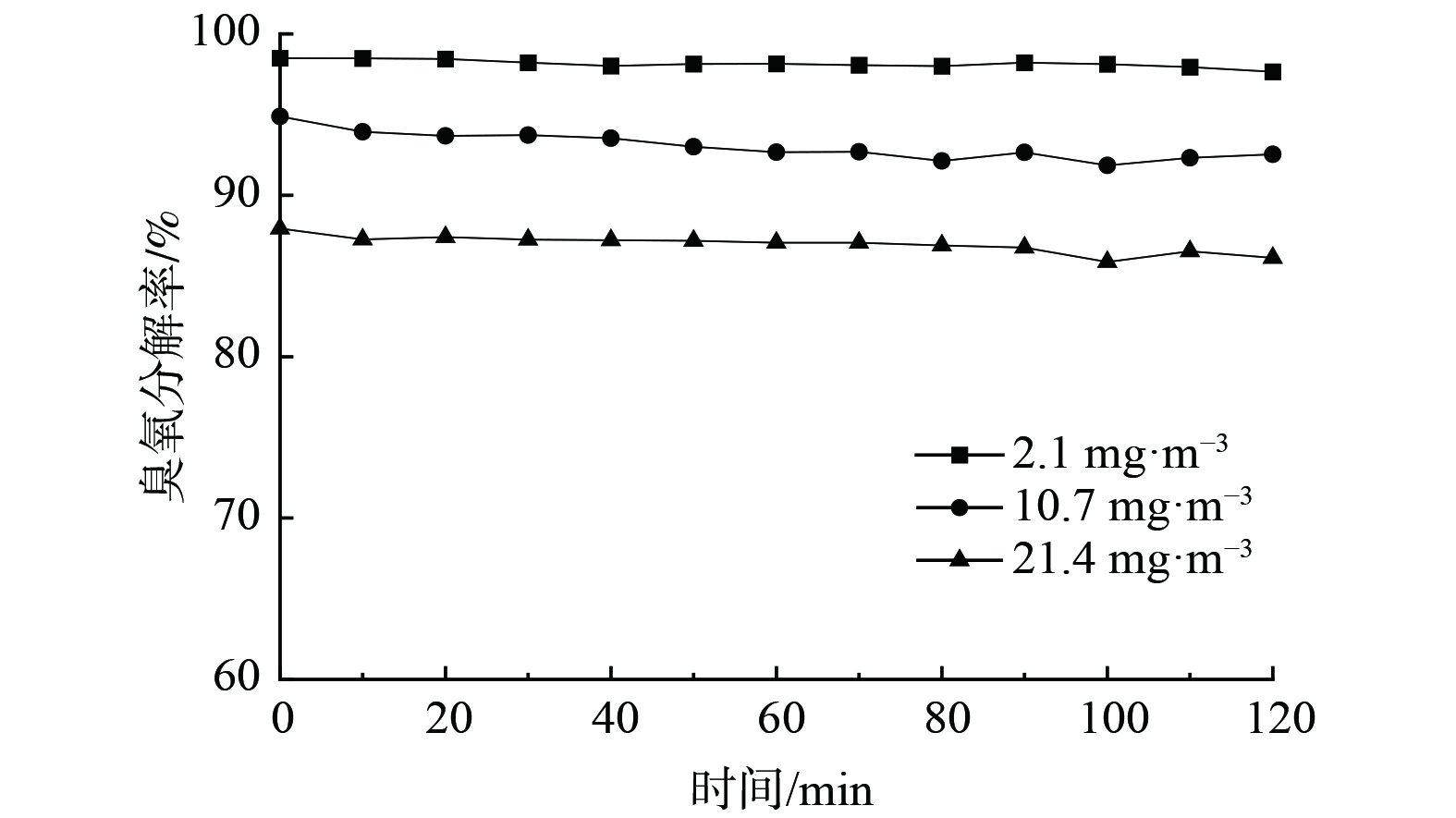
图 7 不同臭氧质量浓度对臭氧分解性能的影响
Figure 7. Effect of different ozone concentration on ozone decomposition
表 1 催化剂的BET和脱落率测试结果
Table 1. BET and the rate of bond failure test results of catalyst
催化剂种类 BET/(m2∙g−1) 孔径/nm 孔体积/(cm3∙g−1) 脱落率/% MnO2 21.3 23.0 0.10 - MnO2-CMC/SBR0.5% 17.1 17.8 0.09 7.18 MnO2- CMC/SBR1.0% 14.3 21.4 0.09 2.15 MnO2- CMC/SBR1.5% 10.1 54.2 0.07 0.84 MnO2- SA0.5% 20.3 21.4 0.09 13.16 MnO2- SA1.0% 16.1 33.5 0.08 4.15 MnO2- SA1.5% 10.7 53.6 0.07 1.25
下载: 导出CSV
-
[1] 李帮俊, 范泽云, 张溢, 等. 高压静电催化耦合净化空气中的臭氧控制及其室内浓度预测模型[J]. 环境工程学报, 2017, 11(7): 4117-4124. [2] 易忠芹, 王宇, 田小兵, 等. 室内环境臭氧污染与净化技术研究进展[J]. 科技导报, 2014, 32(33): 75-77. [3] LI X, MA J Z, HE H. Recent advances in catalytic decomposition of ozone[J]. Journal of Environmental Sciences, 2020, 94: 14-31. doi: 10.1016/j.jes.2020.03.058 [4] DHANDAPANI B, OYAMA S T. Gas phase ozone decomposition catalysts[J]. Applied Catalysis B:Environmental, 1997, 11(2): 129-166. doi: 10.1016/S0926-3373(96)00044-6 [5] JIA J B, ZHANG P Y, CHEN L. Catalytic decomposition of gaseous ozone over manganese dioxides with different crystal structures[J]. Applied Catalysis B-Environmental, 2016, 189: 210-218. doi: 10.1016/j.apcatb.2016.02.055 [6] WANG F, DAI H X, DENG J G, et al. Manganese oxides with rod-, wire-, tube-, and flower-like morphologies: Highly effective catalysts for the removal of toluene[J]. Environment Science & Technology, 2012, 46(7): 4034-4041. [7] ZHANG J H, LI Y B, WANG L, et al. Catalytic oxidation of formaldehyde over manganese oxides with different crystal structures[J]. Catalysis Science & Technology, 2015, 5(4): 2305-2313. [8] ZHU G, ZHU J, LI W, et al. Tuning the K+ concentration in the tunnels of α-MnO2 to increase the content of oxygen vacancy for ozone elimination[J]. Environmental Science & Technology, 2018, 52(15): 8684-8692. [9] DENG H, KANG S Y, MA J Z, et al. Role of structural defects in MnOx promoted by Ag doping in the catalytic combustion of volatile organic compounds and ambient decomposition of O3[J]. Environmental Science & Technology, 2019, 53(18): 10871-10879. [10] QU Z P, BU Y B, QIN Y, WANGY, et al. The improved reactivity of manganese catalysts by Ag in catalytic oxidation of toluene[J]. Applied Catalysis B-Environmental, 2013, 132-133: 353-362. doi: 10.1016/j.apcatb.2012.12.008 [11] GAC W. The influence of silver on the structural, redox and catalytic properties of the cryptomelane-type manganese oxides in the low-temperature CO oxidation reaction[J]. Applied Catalysis B-Environmental, 2007, 75: 107-117. doi: 10.1016/j.apcatb.2007.04.002 [12] HONG W, ZHU T L, SUN Y, et al. Enhancing oxygen vacancies by introducing Na+ into OMS-2 tunnels to promote catalytic ozone decomposition[J]. Environmental Science & Technology, 2019, 53(22): 13332-13343. [13] HONG W, SHAO M P, ZHU T L, et al. To promote ozone catalytic decomposition by fabricating manganese vacancies in ε-MnO2 catalyst via selective dissolution of Mn-Li Precursors[J]. Applied Catalysis B-Environmental, 2020, 274: 13. [14] HONG W, MA J Z, ZHU T L, et al. To enhance water resistance for catalytic ozone decomposition by fabricating H2O adsorption-site in OMS-2 tunnels [J]. Applied Catalysis B-Environmental, 2021, 297: 120466. [15] 席国喜, 田圣军, 成庆堂, 等. 海藻酸钠的热分解研究[J]. 化学世界, 2000(5): 254-258. [16] 钟浩文, 杨启荣, 姚尔人, 等. 丁苯橡胶热解过程的分子动力学研究[J]. 青岛大学学报(工程技术版), 2019, 34(2): 95-99. [17] LI X T, MA J Z, HE H. Tuning the chemical state of silver on Ag-Mn catalysts to enhance the ozone decomposition performance[J]. Environmental Science & Technology, 2020, 54(18): 11566-11575. [18] LIU Y, ZHANG P Y. Removing surface hydroxyl groups of Ce-modified MnO2 to significantly improve its stability for gaseous ozone decomposition[J]. Journal of Physical Chemistry C, 2017, 121(42): 23488-23497. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 3834
- HTML全文浏览数: 3834
- PDF下载数: 73
- 施引文献: 0






