







-
零价镁(zero-valent magnesium, ZVMg)是一种银白色的碱土金属,具有化学性质活泼、低密度 (1.74 g/cm3)、高强度和地壳中高丰度的特点,镁合金已经在航空、育种、自动化和电器工业中有广泛应用[1]。ZVMg的活性较强,在污染物的还原降解方面具有诸多优势[2-3]:ZVMg的氧化还原电位(−2.37 V)是零价铁(ZVI,−0.44 V)的5.4倍,具有更强的还原活性;Mg(OH)2的沉淀平衡常数(Ksp)为7.08×10−12 (25 ℃),显著高于Fe(OH)2 (7.943×10−16)[4],因此ZVMg表面形成的Mg(OH)2氧化膜比ZVI表面形成的Fe(OH)3膜更疏松易溶,有利于ZVMg传递电子;ZVI只适用于厌氧环境,而ZVMg能同时用于有氧和无氧环境[5-6];镁广泛存在于天然环境中,是植物光合作用和人体细胞必需的元素之一,对环境友好。因此,ZVMg作为一种环境友好型高效还原剂,在环境修复领域具有较大应用潜力。
ZVMg应用于污染物去除的研究始于20世纪末期[7],1998~2021年间,Web of Science上可统计的关于ZVMg降解污染物的文章50篇左右,而国内几乎没有相关研究。ZVMg与水之间发生强烈的腐蚀-钝化作用可能是限制其应用的重要因素[8];此外,对于ZVMg进入水环境后的生态影响及较高的使用成本等问题,目前尚缺少详细研究结果以支持ZVMg的环境应用。由于ZVMg具有负电位(<<−1 VSHE),其与水反应将在短时间内生成大量的H2[8],同时pH快速升至10以上,并且Mg2+和OH−生成的Mg(OH)2沉淀层抑制Mg传递电子,导致ZVMg的还原效率降低[9-10],这与ZVI在水中的钝化机制类似。同时,由于ZVMg具有较高的氧化还原电势,很难通过化学还原方法得到纳米级的ZVMg,目前主要集中于微米颗粒的研究[11]。因此,虽然ZVMg在去除环境污染物方面具有较多优势,但目前仍存在较多问题。本文旨在详细介绍ZVMg的制备方法与性质特征,重点评述ZVMg独特的腐蚀特性和近年来ZVMg材料用于处理不同污染物的研究进展,指出现有研究存在问题及未来应用面临的挑战,并展望了ZVMg未来的研究方向,以期为ZVMg在环境修复领域的研究与应用提供关键理论基础。
-
SOLANKI et al[12]提出可以利用NaBH4通过湿化学还原方法得到ZVMg纳米颗粒,但是由于ZVMg自身较强的还原特性,通常认为很难通过湿化学还原方法制备ZVMg。目前多数研究通过机械球磨法获得微纳米尺寸的ZVMg颗粒。该方法利用高速转动的研磨小球与罐体之间的碰撞作用力来改变颗粒表面的微观结构,降低颗粒粒径,增大比表面积,而且机械作用力能破坏金属表面的氧化层,使颗粒表面产生更多缺陷结构,增加活性位点[10, 13-14]。球磨过程中还需要添加过程控制剂,以防止或减弱球磨材料发生冷焊作用[15-16]。导电性能较好的石墨通常被用作球磨镁粉的控制剂[17-19],此外,活性炭(active carbon,AC)是一种较好的控制剂,并且活性炭是良好的多孔碳材料,对疏水性有机污染物具有强烈的吸附性能[19],有利于提高污染物的去除效率。
球磨ZVMg的常见方法:利用Red Devil 5400系列油漆搅拌器,配备传统托盘承载研磨罐体,容器和磨球为不锈钢材质。镁粉(76 g)和石墨(9 g)混合置于研磨罐(内径5.5 cm,高17 cm)内,加研磨球,磨球与粉末的质量比3:1,容器充入氮气或氩气后密封,球磨约30~45 min、球磨转速为670 r/min[10, 18, 20] 。但有学者利用行星式球磨仪(BM40,北京格瑞德曼仪器设备有限公司)制备得到了微米级镁粉颗粒[21]。球磨参数:4.5 g镁粉、0.5 g石墨与100.0 g氧化锆小球混合,置于不锈钢球磨罐(内径7 cm,高10 cm)中密封,设定球磨仪的转速为300 r/min,球磨过程每运行5 min、暂停10 min,共循环9次。
此外,合成ZVMg材料的方法还包括:氢气还原氧化镁(500 ℃以上);碱金属还原镁盐;氢化镁脱氢;在四氢呋喃条件下气化金属镁;超声电化学方法等[6, 22-24]。但这些方法的操作难度较高,实验室条件下较难制备ZVMg的微纳米颗粒,比较而言,机械球磨法制备ZVMg材料更为可行。
-
在Mg表面掺杂其他过渡金属形成具有催化还原作用的双金属,是强化Mg还原效率的常见方法。Mg双金属合成方法与纳米Fe双金属的合成方法类似。一般是将Mg颗粒沉浸在第二相金属盐溶液中,利用Mg自身的强还原性,使催化金属盐在液相中直接被还原并负载到Mg颗粒表面,形成许多具有催化作用的金属“小岛”[11]。这种合成方法具有以下优势:操作过程简单;材料合成重复性好;催化金属均匀负载在Mg颗粒表面;可通过改变溶剂的种类、反应温度或添加不同的稳定剂等方式,合成不同的Mg双金属材料[25]。
-
有研究发现,球磨后ZVMg颗粒的比表面积为3.62 m2/g,是球磨前颗粒比表面积 (0.83 m2/g)的4.36倍[26],比纳米零价铁(nZVI)的比表面积(10~50 m2/g)[27]低一个数量级。但是由于nZVI颗粒自身强烈的磁引力作用,它们进入水溶液后容易形成链状或树枝状的聚集体,导致nZVI颗粒粒径比原来增大几十甚至几百倍[28],而ZVMg颗粒则不存在磁性吸引导致的颗粒团聚问题。
MOGHARBEL et a[19]通过扫描电镜(scanning electron microscopy,SEM)图发现ZVMg和AC球磨后为圆粒状颗粒,见图1(a)和(b)。但魏鹏刚等[21]的研究发现,ZVMg与石墨(C)球磨后由不规则的粒状变为较光滑的片状结构,见图1(c)和(d)。
ZHANG et al[26]利用X-射线衍射(XRD)分析了ZVMg/C表面的晶体结构,发现ZVMg的特征衍射峰包括32.16°、34.40°、36.62°、47.74°、57.26°和63.06°,见图2,这与SICILIANO et al[4]报道的XRD分析结果一致。ZVMg与水溶液反应后,ZVMg的特征衍射峰信号显著减弱,而在2θ 为 18.5°、38.0°、50.8°、58.7°和62.1°处出现了Mg(OH)2的特征衍射峰,并且通过扫描电镜(SEM)谱图,见图3[26],也证明了ZVMg表面形成了花瓣状疏松多孔的Mg(OH)2氧化层结构[29]。
在空气中自然老化6 d后,ZVMg/C的XRD衍射峰与新鲜制备的ZVMg/C几乎没有差异,见图2[26],表明老化时间内空气对ZVMg/C的氧化作用比较微弱。并且通过高分辨率透射电镜(HRTEM)分析并没有发现ZVMg/C颗粒表面形成明显的氧化壳结构,见图4[26],与nZVI的壳-核结构存在明显差异[30],因此,ZVMg在空气中老化速率较慢。
-
当ZVMg颗粒进入水溶液中,将自发与水发生电化学腐蚀反应,生成氢氧化镁[Mg(OH)2] 和氢气 (H2),见式 (1) 。尽管O2/H2O的标准氧化还原电位(+1.23 V)明显高于Mg2+/Mg的标准氧化还原电位 (E0SHE=−2.37 V),但事实证明溶解氧的浓度对ZVMg的溶解程度和反应速率几乎没有影响,因此一般认为溶解氧对ZVMg的氧化作用微乎其微[31-33]。
分解反应式,见式(2 ~ 4):
式中,ZVMg腐蚀产生释放Mg2+,同时水中产生大量OH−,导致pH上升到10.5左右,而Mg(OH)2在pH>10.7将形成饱和沉淀[34]。当Mg与其他过渡金属形成微电解体系,Mg表面的过渡金属作为阴极有助于加速阳极Mg的腐蚀作用;而Mg腐蚀形成的Mg(OH)2中,有一部分用于修复Mg表面的氧化膜。因此水溶液中,Mg表面的腐蚀-氧化作用形成了一套循环调控机制。Mg颗粒表面独特的腐蚀特性具有较大的环境应用潜力,这使得ZVMg可以自然存储在环境中,仍然可以保持其还原活性。LEE et al[31]研究了粒状ZVMg在开放的缓冲溶液中的溶解动力学,结果表明,浓度为10~50 mg/L的ZVMg在200 min内可以完全溶解,主要的氧化剂为水分子而非氧气。
-
ZVMg或Mg的双金属材料已被证明对多种有机和无机污染物具有还原降解功能,并具有较高的去除效率和反应速率,见表1。ZVMg去除有机或重金属污染物的概念模型图,见图5。与ZVI结构类似,ZVMg同样具有核-壳结构,ZVMg单质表面覆盖着一层镁氧化物或氢氧化物氧化层,但氧化层的结构具有分散性和多孔状,有利于内部ZVMg传递电子。理论上,ZVMg与ZVI发生还原作用的机理基本一致,过程主要包括:1)有机污染物,尤其是卤代烷烃或芳香烃污染物主要通过得电子和加氢脱氯作用被还原[10, 35],见式(5);2)重金属阳离子作为电子受体,直接接受ZVMg单质传递的电子,从高价态被还原为低价态,见式(6),如Cr(VI)被ZVMg还原为Cr(III)[2];3)还原后的重金属离子,如Cr(III)可能形成Cr(OH)3沉淀在ZVMg表面或与Mg2+发生共沉淀作用[2];4)一些相对稳定的无机盐离子,如高氯酸盐和硝酸盐也可能被ZVMg还原。如,硝酸盐离子能直接被ZVMg还原为氨根离子[36];5)而某些不易被还原的金属离子也有可能单纯被ZVMg吸附。此外,在ZVMg单质的还原过程中,同时伴随着ZVMg与水的腐蚀反应产生过量H2,尽管H2通常被认为在没有催化的条件下很难具有还原作用,但有研究发现Mg-水反应过程中,H2的逐渐生成会增加阳极ZVMg的电位,这是一个较为特殊的现象[31]。因此有研究者提出ZVMg可能首先生成了Mg+,并且与水分子反应首先产生一个活性中间体:水合氢电子(eaq−),它是生成H2的前体物,同时也是一种良好的还原剂,能够与污染物发生电子传递作用[31]。
-
在已有研究报道中,有机污染物被ZVMg或Mg的双金属降解研究多数在酸性有机溶剂体系中开展,以避免Mg与水分子的剧烈反应导致Mg利用效率下降的问题,有机溶剂主要包括乙醇[45]、1:1乙醇/乳酸乙酯[46]和丙酮[47]等。如,美国中佛罗里达大学YESTREBSKY课题组[10, 46]以冰醋酸 (1% v/v) 作为活化剂,利用球磨后的ZVMg/C分别在1:1乙醇/乳酸乙酯 (v/v) 和无水乙醇溶液体系中降解六氯苯和五氯酚。
与单独ZVMg材料相比,Mg双金属材料能显著提高污染物的降解效率[12]。常见过渡金属包括Pd[11, 18, 45-48]、Ni[41]、Cu[41]和Ag[14]等。Mg的双金属材料已经被证明能有效去除各种高毒性的氯代有机物,如滴滴涕[49]、六氯苯[10]、多氯联苯[5, 11, 42]和氯酚类[12-13, 46]等物质。关于Mg的双金属降解氯代有机污染物的作用机理,加氢脱氯作用普遍被认为是高氯代有机污染物 (氯苯类、多氯联苯) 逐渐被转化为低毒性、易生物降解的低氯代有机物的主要过程[11, 42, 48]。活性氢原子 (H·) 被认为是铁基或镁基双金属体系在活化催化条件下产生的主要还原物质。CWIERTNY et al[50]甚至提出吸附在金属催化剂表面H·的量决定了其还原能力。PATEL et al[38]发现在水溶液中,单独ZVMg难以降解五氯酚,但利用ZVMg在五氯酚溶液中原位还原Ag+生成Mg/Ag (Ag=3.1w%) 双金属后,可以降解约35%的五氯酚;加入醋酸 (6.25%) 后,ZVMg可去除80%以上的五氯酚,而Mg/Ag双金属可以在1 h内降解90%以上的五氯酚。此外,PATEL et al[51]也发现原位还原合成的Mg/Pd双金属比非原位合成的材料对五氯酚降解更有效。而且,单独ZVMg和Mg/Pd双金属对五氯酚的降解效果优于Fe/Pd双金属和单纯ZVI[13]。MORALES[52]研究证实,Mg/Pb双金属在室温和常压条件下可将苯酚氢化成环己醇和环己酮。Mg/Pd也被用于有机氯农药和二噁英的去除研究,结果表明,滴滴涕、滴滴滴和滴滴伊可以被Mg/Pd完全还原脱氯至烃类化合物,且脱氯过程中无有毒中间体生成[49, 53]。THANGADURAI et al[47]利用Mg/Pd实现了对硫丹的完全脱硫和脱氯,最终产物为碳氢化合物。BEGUM et al[54]也证实了Mg/Zn体系可将硫丹和林丹这2种内分泌干扰物完全脱氯成碳氢化合物。
-
虽然ZVMg被证明在有机溶剂体系中能有效去除目标污染物,但是添加有机溶剂容易导致环境次生污染、增加环境修复成本等问题,对环境和经济并不友好,在有机体系中ZVMg很难原位应用于场地地下水的修复技术中。在有限的水溶液体系研究中,ZVMg仍然可以实现对污染物的降解目的,但可能存在反应速率下降、溶液pH显著上升导致Mg钝化等问题。ZHANG et al[26]探索了ZVMg降解地下水典型污染物三氯乙烯 (TCE) 的应用潜力和内在作用机制。结果表明,在未调节pH的水溶液体系中,10 g/L的球磨ZVMg/C能够将初始浓度为38 μmol/L的TCE在24 h降解91%,与其在添加冰醋酸(1 vol.%)的有机溶剂体系中的降解率相当。TCE通过加氢脱氯途径被降解生成甲烷 (62.51%)、正己烷 (11.86%) 和乙烷 (7.40%) 等烯烃和炔烃产物去除,见图6(a)。在添加或未添加冰醋酸 (1 vol.%) 的水溶液中,TCE降解速率常数 (KSA) 分别为1.42×10−1和9.31×10−2 L/(m2·h)。在TCE水溶液中,ZVMg的利用率约为60%,见图6(b)[26],与对照实验(未添加TCE的背景溶液)中ZVMg的利用率相当,表明ZVMg/C在2种体系中具有相似的供电子能力。此外,研究表明,ZVMg与水反应后,由于pH短时间(~10 min)内上升到11左右,导致Mg2+会迅速生成Mg(OH)2沉淀。在未调节溶液pH条件下,投加量为10 g/L的ZVMg反应后溶液中Mg2+的浓度约为1.05 mg/L,远低于白云质石灰岩含水层中Mg2+的背景浓度(22~43 mg/L)[55],同时也远低于我国《地下水质量标准:GB/T 14848—2017》I类水的总硬度指标(≤150 mg/L)[56]。其次,通过老化实验发现,当ZVMg/C在空气中自然暴露2、4和6 d,TCE的KSA分别降至2.52×10−2、2.07×10−2 和1.90×10−2 L/(m2·h),这主要是因为空气氧化使得ZVMg的含量由最初的85.2%下降了5.3%~7.3%,研究结果初步证明了ZVMg/C对工业污染场地地下水氯代烃污染去除的应用潜力。
-
Cr(VI) 作为最常见的无机污染物之一,可以被Fe2+、硫化零价铁、ZVI等还原剂转化为毒性较低、迁移性较差的Cr(III),但在中性、碱性或溶解氧存在条件下,Cr(VI) 的还原效率较低,而LEE et al[31]则发现ZVMg能在中性缓冲溶液中将Cr(VI) 完全还原去除,研究还发现,当质量比ZVMg:Cr(VI) <100时,ZVMg会被水分子快速消耗,并且Cr(VI)被还原生成的Cr(III) 会大量沉淀在ZVMg表面,因而降低了ZVMg的还原速率,但是提高ZVMg的投加量将显著提高还原效率。LEE et al[31]提出,Cr(VI) 并非被ZVMg直接还原,而是被ZVMg-水反应生成的活性中间物质 (eaq− 和/或H·) 还原:1)Mg与水反应生成活性中间物质I:Mg + H2O → Mg2+ + I;2)Cr(VI) 与活性中间物质I反应生成Cr(III):Cr(VI) + I → Cr(III);3)同时活性中间物质I与另一种活性物质S反应可以生成氢气:I + S → H2。
ZVMg也能高效处理水中硝酸盐,MIRABI et al[36]发现用Mg/AC可以完全去除水中硝酸盐,而且在相同实验条件下,Mg/AC体系对硝酸盐的去除效率显著优于Fe/AC。ILERI et al[9]通过超声活化ZVMg还原水中硝酸盐,发现单独超声或单独ZVMg均不能有效降低硝酸盐的浓度,但两者联合可将50 mg/L的NO3-N在60 min内降解90%以上,降解产物主要是亚硝酸盐和氮气,并且可以通过增加超声功率或ZVMg剂量提高亚硝酸盐转化成氮气的比例。
-
溶液中背景电解质对ZVMg腐蚀或溶解的影响主要分为抑制和促进作用[57]。在溶液pH为10~11条件下,Mg在不同电解质溶液中的腐蚀速率为NaCl > Na2SO4 > MgSO4 > NaIO4;并且Mg在Na2SO4溶液中的腐蚀速率要比在MgSO4溶液中低2个数量级,这可能是由于Na2SO4能促进ZVMg表面形成Mg(OH)2氧化膜的缘故[31]。
AGARWAL et al[39]利用Mg/Pd降解2-氯联苯时也考察了不同阴离子对降解速率的影响,见图7。在Mg/Pd投加量为4 g/L,2-氯联苯初始浓度为4 mg/L,阴离子浓度为50 mmol/L条件下,研究发现醋酸根 (CH3COO−)、氯离子 (Cl−)、碳酸氢根 (HCO3−)和磷酸一氢根 (HPO32−)均可作为质子供体增强Mg/Pd的腐蚀性;而乙二胺四乙酸 (EDTA)、硫酸根 (SO42−) 和磷酸二氢根 (H2PO3−) 分别与2-氯联苯溶液共存时,则表现为先促进后抑制的作用。这是由于EDTA作为螯合剂容易与Mg2+结合,因而最初能降低Mg(OH)2膜的形成,增强ZVMg的腐蚀性,但是EDTA可能迫使过多ZVMg进入溶液,反而降低了Mg对2-氯联苯的降解速率。SO42−是一种弱腐蚀剂,一般被认为能够加速ZVMg的腐蚀,但该研究中,SO42−可能导致ZVMg表面形成了较厚的氧化膜,正如BARIL et al[33]发现将Mg浸入0.1 mol/L的Na2SO4溶液3.5 h,ZVMg表面形成了较厚的膜,但这层孔隙性较好的膜没有完全抑制ZVMg的腐蚀,因而说明SO42−最初促进了ZVMg腐蚀,但随着膜增厚而逐渐降低腐蚀速率。NO3−的存在也会降低2-氯联苯的脱氯速率,因为NO3−可以与污染物竞争电子,被还原为NH4+。
魏鹏刚等[21]也考察了不同阴离子 (Cl−、HCO3−、SO42−和NO3−) 对ZVMg/C降解TCE的影响,见图8。在高浓度 (50 mmol/L) 或低浓度 (50 mmol/L) 共存阴离子溶液中,ZVMg/C降解TCE的反应速率在前2 h内均受到不同程度的抑制作用,但2 h后又呈现出不同的影响趋势,可分为促进、抑制和无影响3种作用,且同种离子在不同浓度下对TCE降解的影响程度也有差异。具体来说,Cl−具有先抑制后促进作用;而SO42−对ZVMg的腐蚀影响较小;HCO3−和NO3−则具有明显的抑制作用。但是也有研究发现共存Cl−对Mg/Pd降解五氯酚速率有明显的抑制作用[14]。因此,与阴离子对ZVI降解污染物的影响类似,不同阴离子对金属还原剂的腐蚀影响并不完全一致,这可能取决于离子-金属-污染物三者的共同作用。
-
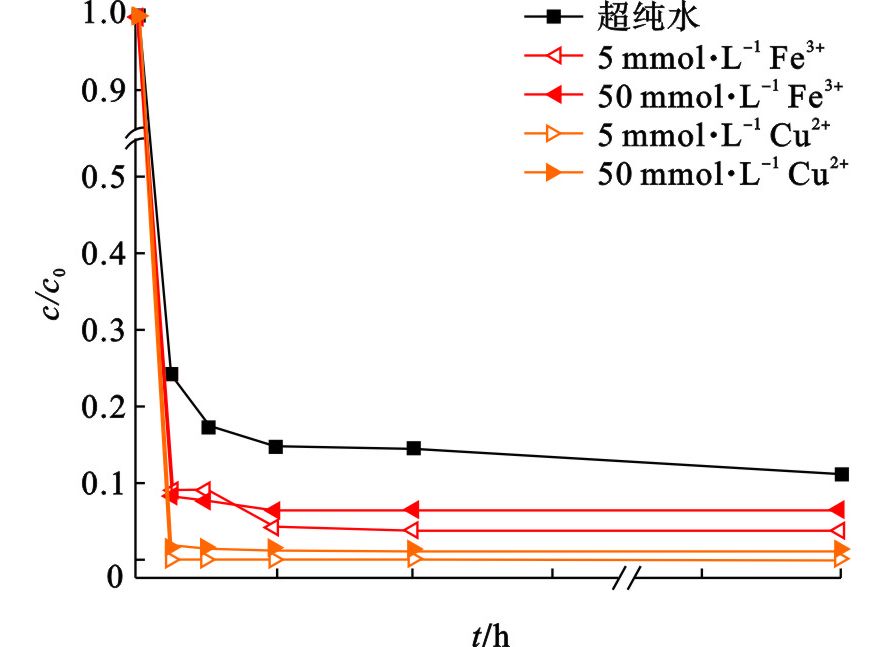
魏鹏刚等[21]研究了水溶液中不同浓度的Fe3+和Cu2+与TCE共存时对其降解的影响,发现两者均可以提高ZVMg的还原活性。相比于对照实验,TCE的降解率从原来的89%分别提升至94%~96%和99%~100%,见图9,说明共存金属阳离子对ZVMg起到了催化活化作用,提高了ZVMg的利用率[11, 48];而且共存Cu2+能够使TCE在15 min内被完全降解,比共存Fe3+的反应速率更快,这主要是由于Mg/Cu的电势差 (2.71 V) 高于Mg/Fe (1.93 V)[3],对Mg的催化效果更显著。并且,双金属形成有利于降低整个反应体系的pH (<10.5),这将抑制Mg(OH)2的沉淀,进而减缓了ZVMg表面的钝化作用。因此,溶液中共存的过渡金属阳离子能优先被ZVMg原位还原并对Mg起到化学催化作用,显著提高ZVMg的还原活性。
-
ZVMg材料虽然在环境修复领域具有较大的应用潜力,对多种难降解污染物具有较高的还原去除效率,但仍有许多关键科学问题亟待解决。
(1)ZVMg与水的腐蚀-钝化作用显著影响ZVMg与污染物的相互作用,但该过程对ZVMg降解污染物的长期作用机制目前尚不十分清楚。
(2)ZVMg与环境持久性难降解污染物的相互作用机理及关键影响因素有待进一步研究,尤其是ZVMg的强还原作用对地下水中高浓度、高异质性和高复合有机污染物的去除效果尚不可知,对相关作用机制的认知还处于空白。
(3)在特定环境条件下,水化学成分与ZVMg的相互作用及其对污染物降解的构-效关系研究目前还很缺乏,关于ZVMg在水环境中的老化作用和对污染物的长效机制及其对生态环境的长期影响尚未见报道。
(4)地下水是一个复杂的环境体系,涉及地下水、地层结构、地球化学和水化学等多种特征的综合作用,因此地下水污染修复不仅仅是修复材料在水溶液中对污染物的处理或降解。ZVMg材料在降解污染物的处理工艺、批量生产、与修复技术和设备上的匹配和工程应用方面还需要大量的基础研究和实践探索。
-
ZVMg作为一种绿色高效的还原材料,相比于ZVI,具有反应活性更高,表面不易钝化、在空气中更稳定和制备工艺简单等特点,已经被证明对多种难降解污染物具有很好的去除效果,在环境污染修复领域中具有重要的应用潜力,但ZVMg在水溶液中的化学稳定性、使用安全性和长效机制还有待于进一步研究,未来实际应用的可能性还需要大量验证工作。ZVMg材料的研发、改性和降解污染物机理研究和工程化应用,以及材料对生态环境的影响研究将成为未来研究的重点。ZVMg材料的研发应用,或将成为ZVI修复材料的替代产品,为地下水污染的绿色可持续修复提供先进理论基础和关键技术储备。
零价镁材料在环境修复领域的研究进展
Research progresses of zero-valent magnesium application in environmental remediation
-
摘要: 零价镁(ZVMg)金属化学活性强,在空气中易稳定保存,是一种环境友好的高效还原型修复材料,对多种有机污染物和重金属具有很好的去除效果,具有较大的环境修复应用潜力。文章综述了近20年来 ZVMg对环境污染物去除的研究进展,重点阐述了ZVMg材料的还原特性、制备方法及其对污染物的去除效率和反应机理,指出了目前ZVMg材料应用于环境修复所面临的问题与挑战,并展望了未来ZVMg在污染场地治理领域的研究方向和应用前景。Abstract: Zero-valent magnesium (ZVMg) is an environment-friendly and efficient reductant with a high reactivity and a good stability in the atomsphere. It has an excellent performance on the removal of numerous organic and heavy metal pollutants, and a great application potential in the environmental remediation. This paper reviews on the research progresses of ZVMg to remove environmental pollutants in the past 20 years, focusing on ZVMg’s reducing characteristics and preparation methods, pollutants removal efficiency and reaction mechanisms. Accordingly, the present difficulties and challenges on the application of ZVMg in the field of the environmental remediation are highlighted. Future research areas and application prospects for the utilization of ZVMg-based materials in the environmental remediation are also prospected.
-
在水处理领域,基于硫酸根自由基(SO4·−)的高级氧化技术是近年来的一个研究热点. 与芬顿技术相比,SO4·−的氧化还原电位为2.5—3.1 V,高于羟基自由基(·OH,2.7 V),半衰期远比·OH长,pH的适应范围宽[1]. 固态过硫酸盐(PS)比液态过氧化氢(H2O2)要利于运输和存储,因此在处理水中污染物时具有更高的能效和实际操作优势. 过硫酸盐自身的氧化能力较弱,通常需要将其活化产生氧化性更强的活性物质(如SO4·−等自由基)来发挥氧化作用.
在活化过硫酸盐的方法中,碳基材料因其较大的比表面积、sp2和sp3杂化的碳骨架结构、良好的导电性能以及非金属特性而被广泛用于研究[2,3]. 目前,石墨烯、碳纳米管、活性炭、介孔炭和生物炭是备受关注的几种材料. 与其他碳材料相比,生物炭具有制备和原料来源上的"减碳"和成本优势,是一种环保的吸附剂和催化剂. 然而,与石墨化的碳纳米管和石墨烯相比,直接焙烧得到的生物炭主要由非晶碳组成,其催化活化过硫酸盐的能力有限,通常需要适当改性以提高其催化性能. Hou等将生物炭与适量氧化石墨烯复合,成功将材料活化过硫酸盐降解苯酚的去除率从原始生物炭的20%提高到100%[2]. 另一类活化过硫酸盐的优秀催化剂是过渡金属(如锰、铁、钴)及其氧化物[4]. 然而,这些金属及其氧化物容易团聚,活性位点难以充分利用. 将它们负载到生物炭上不仅可以提高活性金属及其氧化物的分散性和稳定性,还可以发挥生物炭自身的吸附能力,增强材料活化过硫酸盐的能力[5].
孔道结构和比表面积是影响碳材料活化过硫酸盐的另一个重要因素. 由于受到生物质原料的限制,生物炭的孔容和比表面积通常较低. 为了增加生物炭的孔道结构,可以采用CO2、氧气和水蒸气等氧化碳壁产生孔道,也可以通过化学活化剂(如氯化锌、磷酸、强碱等)活化新增孔容[6]. 近年来,高锰酸钾开始用于活化碳材料[7]. Hu等[8]利用高锰酸钾溶液对生物炭进行浸渍,显著提高了生物炭的比表面积. 此外,作为高锰酸钾的分解产物,锰氧化物活化过硫酸盐的性能优异,在自然界中的丰度高,对环境友好且生物毒性较低. 因此,利用高锰酸钾改性生物炭有望提高生物炭在高级氧化技术中的潜力.
垂序商陆是一种入侵植物,对本地物种和生态系统危害较大[9]. 此外,垂序商陆生物量大,生长快,适应性强,分布广[10]. 将垂序商陆转化成生物炭用于处理水环境中的污染物,对减污降碳意义较大. 本研究选择垂序商陆作为生物炭的原料,经高锰酸钾溶液处理,不同温度下焙烧制得锰改性生物炭,以水中常见污染物苯酚作为目标污染物,探讨该碳材料活化PDS降解苯酚的性能.
1. 材料与方法(Materials and methods)
1.1 实验试剂与材料
使用的生物质为垂序商陆茎秆部分,采自南京市浦口区龙王山脚;苯酚、过二硫酸钾(PDS)、甲醇(MeOH)、叔丁醇(TBA)、L-组氨酸(L-His)以及硫酸钠购于阿拉丁试剂有限公司;乙醇(EtOH)、氢氧化钠、盐酸、硫酸、氯化钠、硝酸钠、高锰酸钾以及二氧化锰购于国药集团化学试剂有限公司. 实验过程中所用试剂均为分析纯.
1.2 材料的制备和表征
将洁净并风干的垂序商陆茎秆截成小段,超声清洗并烘干后,使用磨碎机研磨成粉. 粉碎后的生物质在氮气气氛下于700 ℃或850 ℃焙烧,保持2 h,自然冷却至室温,得到的生物炭分别标记为700BC和850BC,用乙醇和超纯水洗涤数次,烘干,研磨后备用.
用0.12 mol·L−1的KMnO4溶液浸泡研磨成粉的生物质24 h,过滤. 为了使KMnO4充分负载至生物质上,再用0.06 mol·L−1的KMnO4溶液二次浸泡24 h,过滤后水洗数次并真空干燥,在氮气氛围下碳化,碳化条件和后续洗涤步骤同上,制得的改性生物炭分别标记为Mn-700BC和Mn-850BC.
材料的形貌特征和物相分别通过扫描电镜(SEM,日本日立SU1510)和X射线衍射(XRD,日本岛津,XRD-6100)描述;材料表面的元素价态通过X射线光电子能谱(XPS,美国赛默飞Nexsa)进行分析;材料的表面官能团和碳结构分别利用傅立叶红外光谱(FT-IR,美国尼高力iS5)和拉曼光谱仪(美国赛默飞,DXR2)测试;材料的孔道信息在氮气吸附-脱附仪(美国麦克,ASAP2020)上采集.
1.3 苯酚降解实验
将材料置于锥形瓶中,加入10 mL超纯水,在25 ℃下用磁力搅拌器搅拌30 min,使材料被水充分润湿后,加入40 mL一定浓度的苯酚溶液,使苯酚初始浓度为0.16 mmol·L−1,并调节初始pH=9. 待30 min达到表观吸附平衡后,加入适量PDS溶液,使PDS浓度为2 mmol·L−1. 在不同时间间隔里取样,过滤,定量滤液迅速与定量甲醇混合以抑制滤液中活性物质对苯酚的降解,混匀后用分光光度法在270 nm处测定苯酚浓度.
在评价材料活化降解苯酚的性能实验中,考察了苯酚初始浓度、PDS浓度、pH以及共存无机阴离子的影响. 此外,利用EtOH、TBA和L-His分别作为SO4·−和·OH、·OH、1O2的捕获剂,探讨这些活性组分在苯酚降解中的作用.
2. 结果与讨论(Results and discussion)
2.1 材料的表征
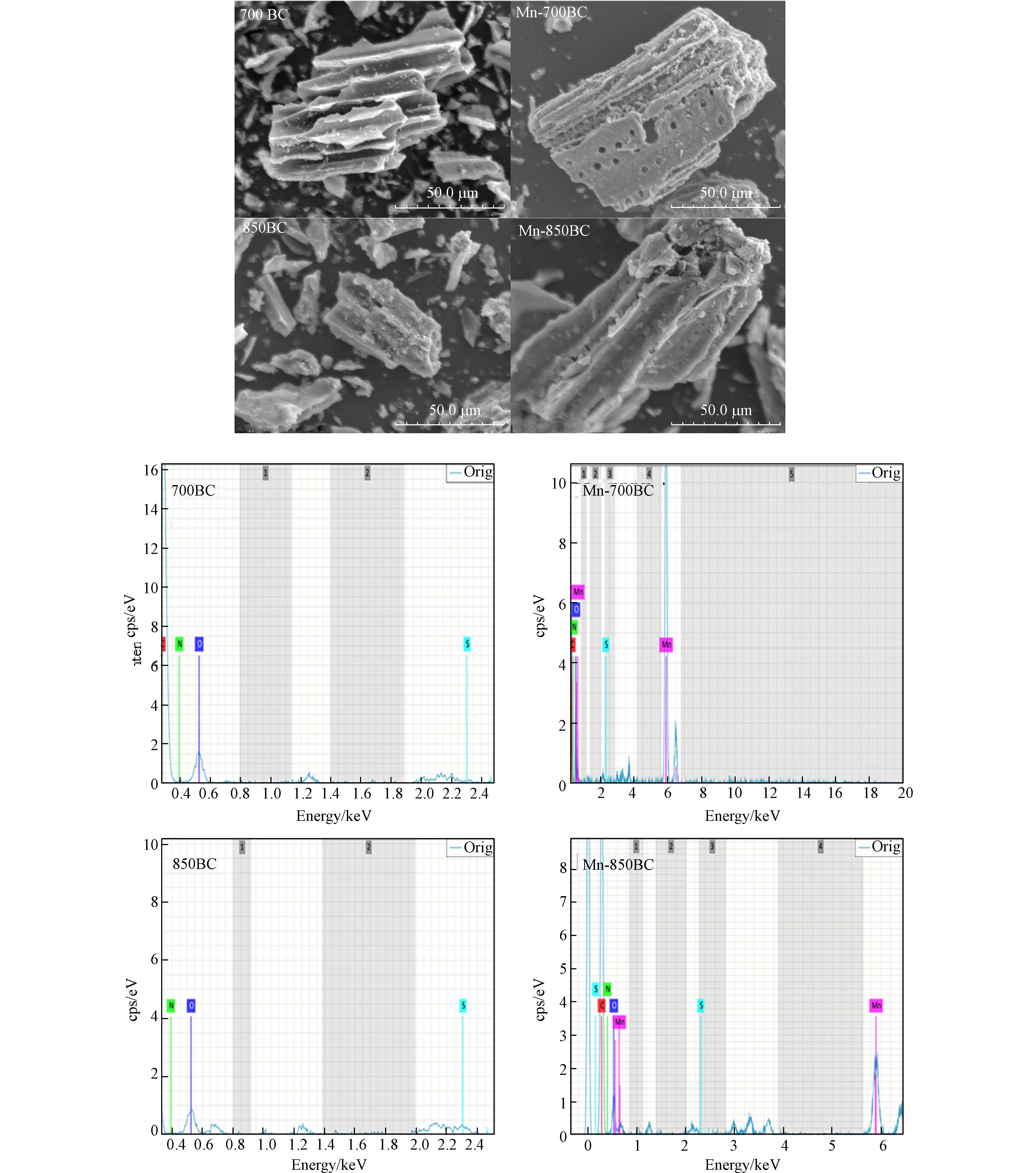
图1展示了700BC、Mn-700BC、850BC和Mn-850BC的SEM照片. 生物炭的外观形状大多不规则,Mn-700BC和Mn-850BC的形状相对于700BC和850BC没有发生明显变化,但表面出现了一些新的孔洞,这是因为在高温碳化时,碳壁在高锰酸钾活化下发生了二次裂解. EDS能谱(图1)分析证实Mn-700BC和Mn-850BC中存在锰的掺杂.
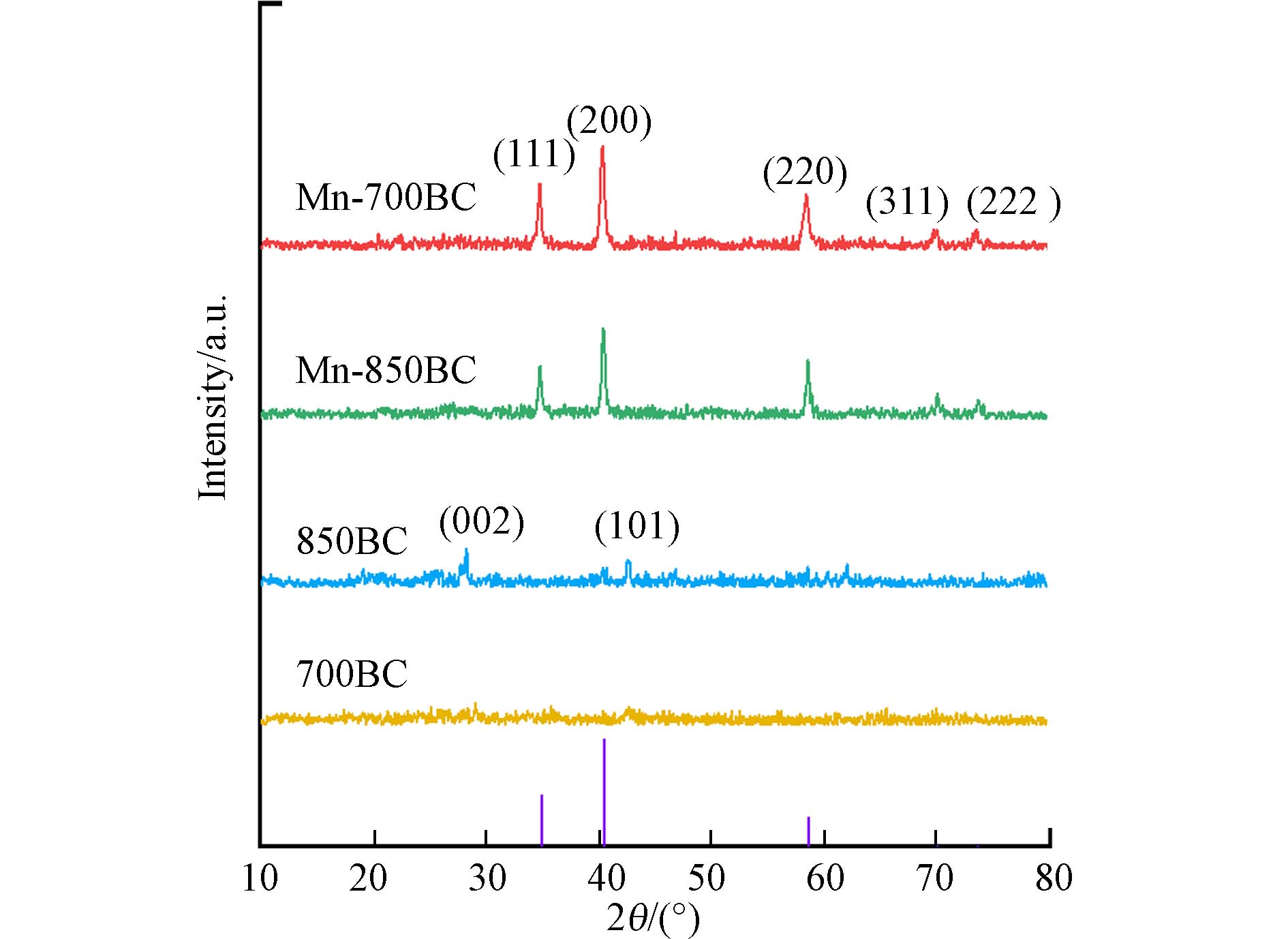
图2是700BC、Mn-700BC、850BC和Mn-850BC的XRD图谱. 从图2可以看出,原始生物炭没有出现金属元素锰等的衍射峰. 但是,Mn-700BC经过锰改性后,材料在34.91°、40.55°、58.72°、70.18°和73.79 °处均出现了衍射峰,对应于方锰矿MnO(JCPDSNo.07-0230). 由文献可知,二氧化锰会以无定形的形式存在于生物炭载体上[11]. 结合高锰酸钾的高温分解反应,推测改性生物炭上可能存在无定形二氧化锰. 对照700BC和850BC的图谱,发现850BC在2θ在26°和44°处各有1个衍射峰,是典型石墨晶面,说明850BC材料的石墨化程度高于700BC[10].
XPS结果(见图3)可证实上述锰氧化物的存在. 改性的两个材料均出现了Mn 2p能谱峰,642 eV附近的能谱峰揭示锰的氧化态为Mn2+/3+,642 eV和654 eV附近的能谱差值约为11.6 eV,说明材料中的锰具有Mn3+和Mn4+的混合价态,但以Mn4+居多[7,12]. C 1s在结合能为284.8 eV出现的能谱峰对应于C=C键[13]. O 1s能谱图中,结合能为532 eV附近和530 eV附近分别对应于缺陷氧羟基(C—OH)和晶格氧(Mn—O)的能谱峰[14 − 15].
 图 3 材料的XPS图谱 (a)全扫(b)Mn 2p (c) C 1s和(d)O 1sFigure 3. The XPS profiles of (a) XPS spectrum (b) Mn 2p (c) C 1s (d) O 1s.
图 3 材料的XPS图谱 (a)全扫(b)Mn 2p (c) C 1s和(d)O 1sFigure 3. The XPS profiles of (a) XPS spectrum (b) Mn 2p (c) C 1s (d) O 1s.从图4(a)的FT-IR光谱中可以发现,4种生物炭在1112 cm−1、1627 cm−1和3417 cm−1处均出现明显的吸收峰,对应于—C—O、羧基/酯基/醛基的C=O和—OH的伸缩振动峰[16 − 18]. 700BC和Mn-700BC在1410 cm−1处的—CH的弯曲振动峰比850BC和Mn-850BC明显,体现了碳化温度对官能团的影响,即低的碳化温度有利于保留材料表面的官能团[19].
 图 4 改性前后生物炭的(a)FT-IR光谱和(b)拉曼光谱Figure 4. (a) FT-IR spectra and (b) Raman spectra of biochar before and after modification
图 4 改性前后生物炭的(a)FT-IR光谱和(b)拉曼光谱Figure 4. (a) FT-IR spectra and (b) Raman spectra of biochar before and after modification4个材料的拉曼光谱(图4(b))均出现了D带和G带. 在1320 cm−1处的D带表示碳材料的缺陷和无序性,1580 cm−1处的G带反映了材料的石墨化程度. 相对于700BC,850BC的ID/IG值较大,石墨化结构的无序性增加,材料缺陷位点相对较多,即碳化温度的提高增加了材料的缺陷度[20],这和牛粪消化产物、苹果树修剪的废弃物所制备的生物炭结果相类似[21 − 22]. 此外,锰氧化物的掺入,也提高了材料的缺陷度.
氮气吸附-脱附实验可以反映材料经改性后孔道的结构变化. 图5(a)是4种材料的氮气吸附-脱附等温线. 在压力很低时,4种材料的氮气吸附量急剧上升,说明它们均含有一定量的微孔孔道;随着压力的升高,氮气吸附量增长缓慢,在中等压力下,因毛细凝聚作用出现的滞后环反映出材料中还有中孔孔道,且改性材料的滞后环更明显,说明锰改性增加了材料的中孔比例,这和孔分布结果一致(图5(b)). 从表1可知,850 ℃下焙烧的生物炭,其孔容高于700 ℃的材料,说明提高温度有利于增加生物炭的孔道结构;同样,高锰酸钾和碳壁在高温下的反应也可在一定程度上增加材料的孔道,使Mn-850BC的比表面积高于850BC. 然而,Mn-700BC的比表面积略低于700BC的比表面积,这可能是锰氧化物颗粒堵塞了部分微孔孔道.
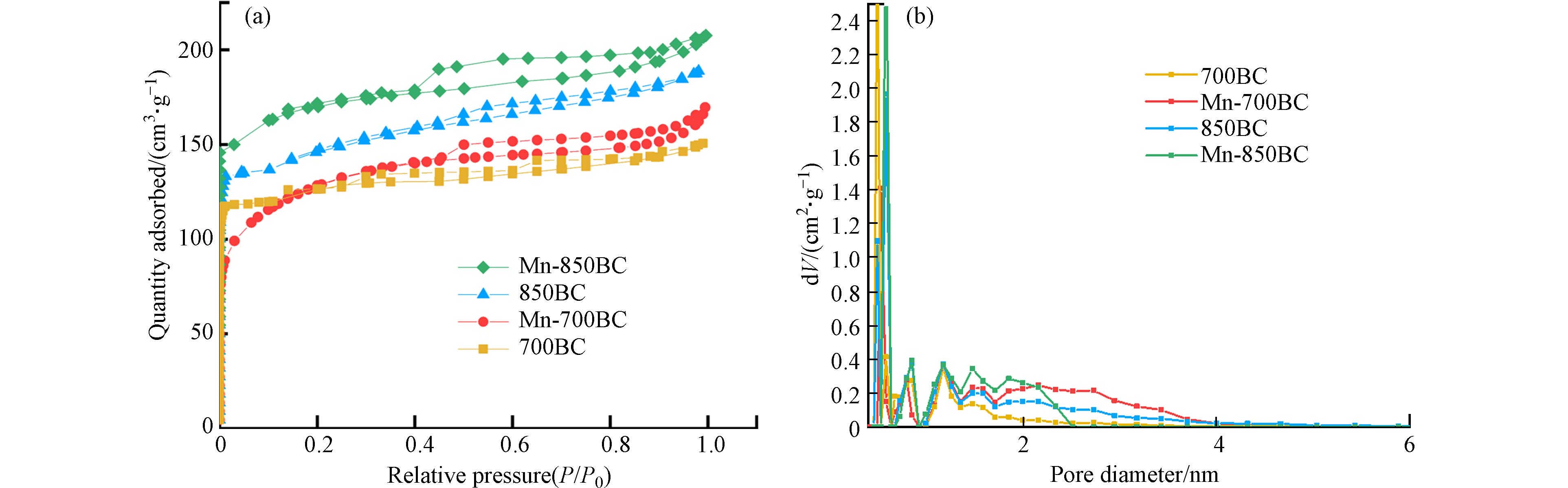 图 5 (a)氮气吸附-脱附等温曲线和(b)孔径分布图(DFT方法)Figure 5. (a) Nitrogen adsorption-desorption isothermal curves and (b) Pore size distributions表 1 材料的孔结构参数Table 1. Pore structural parameters of the tested materials
图 5 (a)氮气吸附-脱附等温曲线和(b)孔径分布图(DFT方法)Figure 5. (a) Nitrogen adsorption-desorption isothermal curves and (b) Pore size distributions表 1 材料的孔结构参数Table 1. Pore structural parameters of the tested materials比表面积a/(m2·g−1) Specific surface area 总孔容b/(cm3·g−1)Total pore volume 微孔孔容c/(cm3·g−1) Mesopore volume 中孔孔容d/(cm3·g−1)Mesopore volume 700BC 423 0.22 0.18 0.04 Mn-700BC 412 0.25 0.13 0.12 850BC 512 0.29 0.21 0.08 Mn-850BC 570 0.33 0.22 0.11 a:Brunauer-Emmet-Teller(BET)方法计算 b:取相对压力为0.97处 c:t-plot方法 d:总孔容-微孔 | Show Table DownLoad:
CSV
DownLoad:
CSV
2.2 材料活化PDS降解苯酚
2.2.1 不同材料活化PDS比较
在评估材料的催化活性之前,首先考察了这些碳材料对苯酚的吸附情况,具体结果见图6(a). 在30 min内,苯酚在4种材料上基本能够达到吸附平衡,其中Mn-850BC、Mn-700BC、850BC和700BC对苯酚的去除率分别为38%、33%、73%和20%. 图6(b)展示了材料活化PDS降解苯酚的实验结果. 结果表明,锰改性生物炭降解苯酚的效果远优于商业MnO2,显示了这种锰改性方法具有一定的应用前景. 在材料吸附苯酚达到表观平衡后,加入PDS溶液,700BC/PDS几乎不能降解苯酚,850BC/PDS对苯酚的降解率约为5%,表明850BC石墨化的碳结构对活化PDS可能有一定贡献;而锰改性的两种材料Mn-700BC和Mn-850BC在吸附平衡后仍可降解约60%左右的苯酚,说明负载的锰氧化物是降解苯酚的重要成分,且主要通过活化PDS产生活性物质降解苯酚,它与苯酚之间直接的氧化还原不是主要降解路径. 此外,研究结果显示,β-MnO2等金属氧化物的添加大大加速了矿物、黏土等活化过硫酸盐产生SO4·−和·OH的速率,使苯的降解速率提高了两个数量级[23]. 除了可以产生SO4·−和·OH等活性物种,锰氧化物在活化过硫酸盐时,还可以在材料表面形成亚稳中间体,通过与过硫酸盐进一步反应生成1O2,以非自由基路径氧化污染物[24-25] .
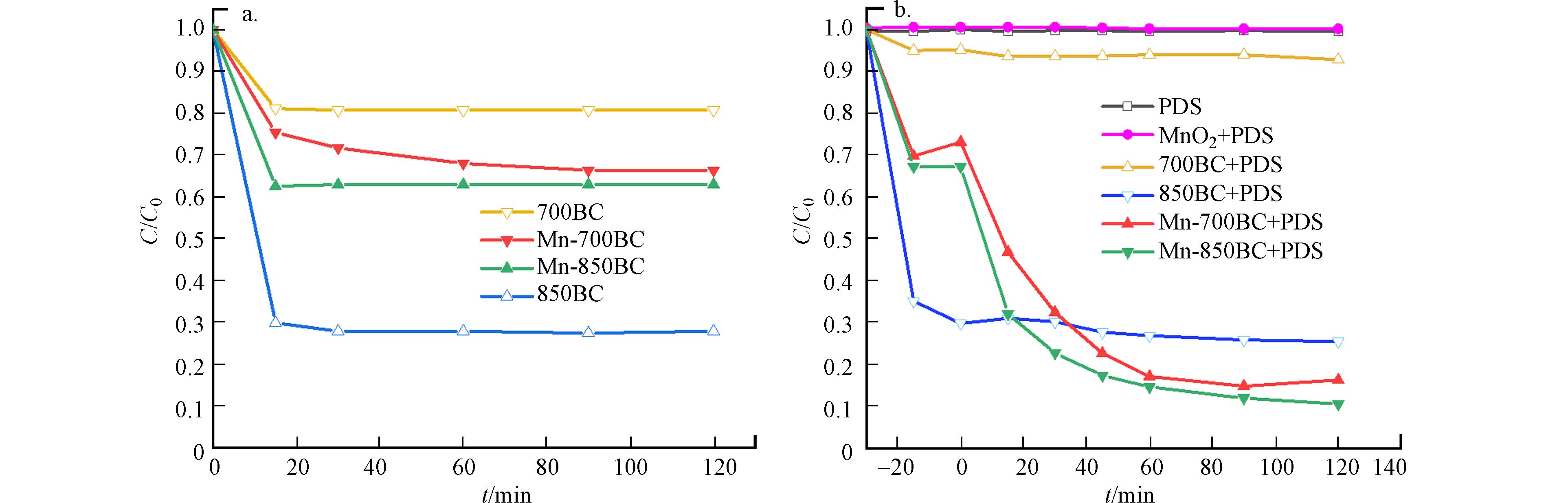 图 6 (a)不同材料对苯酚的吸附(b)不同材料活化过硫酸盐降解苯酚Figure 6. (a) Adsorption of phenol by different materials (b) Activation of persulfate for phenol degradation by different materials
图 6 (a)不同材料对苯酚的吸附(b)不同材料活化过硫酸盐降解苯酚Figure 6. (a) Adsorption of phenol by different materials (b) Activation of persulfate for phenol degradation by different materials2.2.2 PDS和苯酚初始浓度的影响
非均相催化反应中,反应组分浓度对降解速率的影响至关重要. 图7显示了在不同苯酚初始浓度及PDS浓度下锰改性碳材料活化降解苯酚的结果. 随着PDS浓度的增加,苯酚的降解速率逐渐增加,但浓度增加到5 mmol·L−1时,降解速率没有明显变化. 提高溶液中PDS含量可以加快苯酚的降解,但催化材料的活性位点数量有限,不能有效活化过多的PDS. 此外,过量的S2O82-会与逐渐增多的SO4·−反应,削弱对苯酚的降解[26]. 在苯酚初始浓度为0.16—0.53 mmol·L−1范围内,随着浓度的提高,苯酚的去除趋于缓慢. 用准一级动力学方程拟合,结果见表2. 拟合的相关系数R2均在0.94以上,说明苯酚的降解过程可以用准一级动力学方程描述. 浓度越高,速率常数k1值越小,反应速率越慢. 一般而言,非均相催化反应过程包括反应组分的扩散和吸附、表面反应、产物的脱附和扩散这些步骤. 在这些过程中,推测组分的内扩散和吸附过程对降解速率影响较大. 随着苯酚浓度的增加,前期占据于材料孔口附近的苯酚分子会阻碍后续分子进入孔道深处,扩散路径增加,使这些分子到达内表面位点的时间延长.
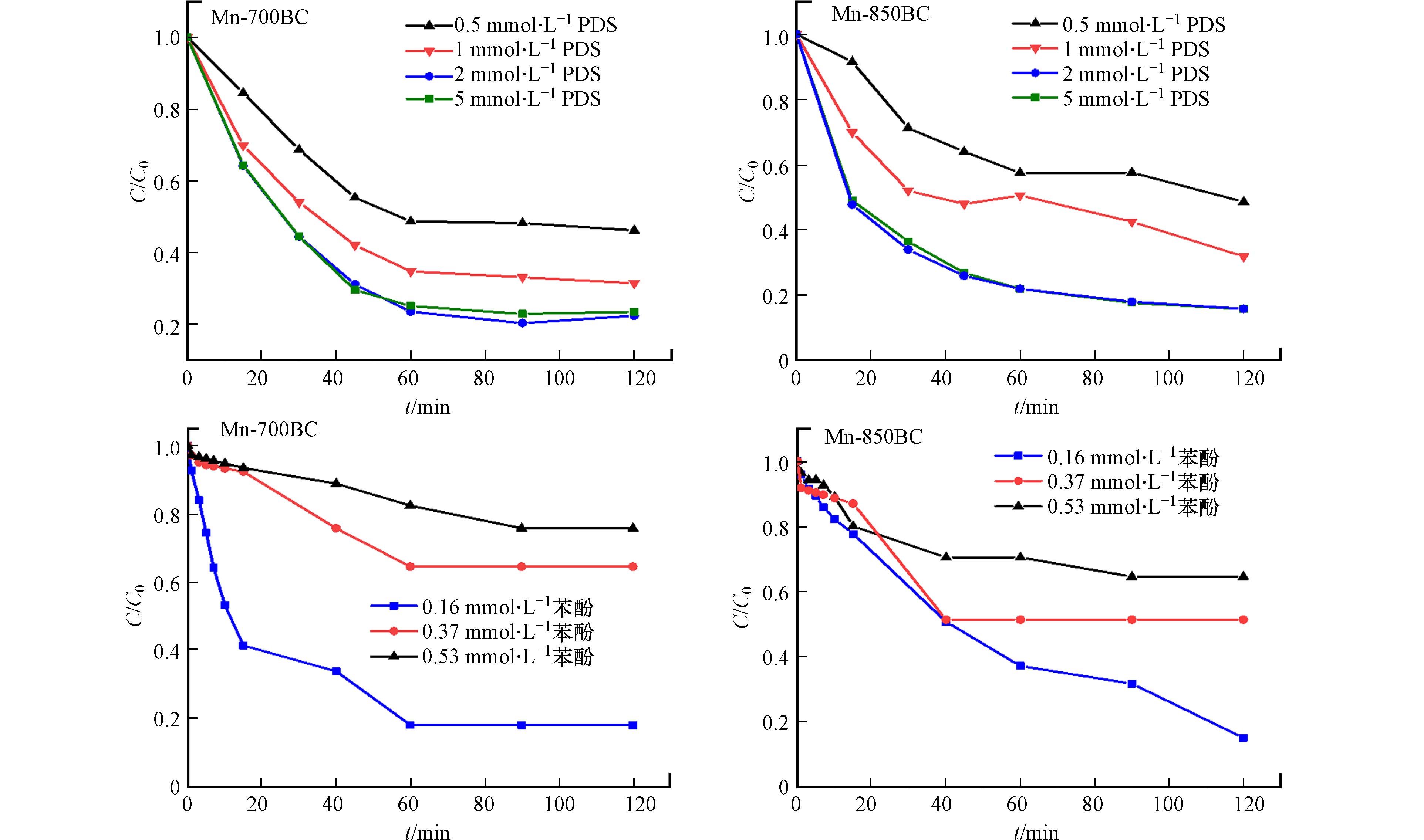 图 7 PDS与苯酚初始浓度对材料活化过硫酸盐降解苯酚的影响Figure 7. Effect of PDS initial concentrations and phenol initial concentrations on activation of persulfate for phenol degradation
图 7 PDS与苯酚初始浓度对材料活化过硫酸盐降解苯酚的影响Figure 7. Effect of PDS initial concentrations and phenol initial concentrations on activation of persulfate for phenol degradation为了阐明上述推测,采用Langmuir-Hinshelwood模型来描述反应组分的吸附过程对催化反应速率的影响[27,28]. 该模型的简易方程为式(1, 2):
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) 其中,r0为反应初活性,选择15 min以内的数据进行计算以避免降解中间产物或产物引起的竞争吸附. θs为苯酚或PDS在材料表面的覆盖率,k为反应速率常数,b为材料对苯酚或PDS的吸附平衡常数.
表 2 材料活化PDS降解苯酚的一级动力学拟合结果Table 2. Fitting results of phenol degradation using the first order kinetic model材料Materials 苯酚浓度/(mmol·L−1)Phenol concentration 表观速率常数k1 /(min−1)Apparent rate constant 相关系数R2Correlation coefficient Mn-700BC 0.16 0.0271 0.94 0.37 0.0070 0.98 0.53 0.0029 0.99 Mn-850BC 0.16 0.0188 0.97 0.37 0.0108 0.98 0.53 0.0089 0.97 | Show TableDownLoad:
CSV
Langmuir-Hinshelwood模型认为,催化反应速率与吸附的反应组分浓度成比例关系. 由图8(a)可知,两个材料的反应初活性均随苯酚浓度的增加而增加,Mn-700BC和Mn-850BC的初活性分别由79.11 mg·g−1·h−1和67.15 mg· g·h−1增加到132.94 mg ·g−1·h−1和470.91 mg ·g−1·h−1. 相类似地,在实验范围内,提高PDS浓度同样使反应初活性增强. 将1/r0与1/C苯酚作图可知,两者具有良好线性关系(R2>0.95),说明锰改性碳材料活化PDS降解苯酚能够用Langmuir-Hinshelwood模型描述,吸附于材料表面的苯酚浓度是该催化反应的控制步骤. 同样地,1/r0与1/CPDS之间也存在较好的线性关系,吸附的PDS浓度在苯酚降解中也起重要作用.
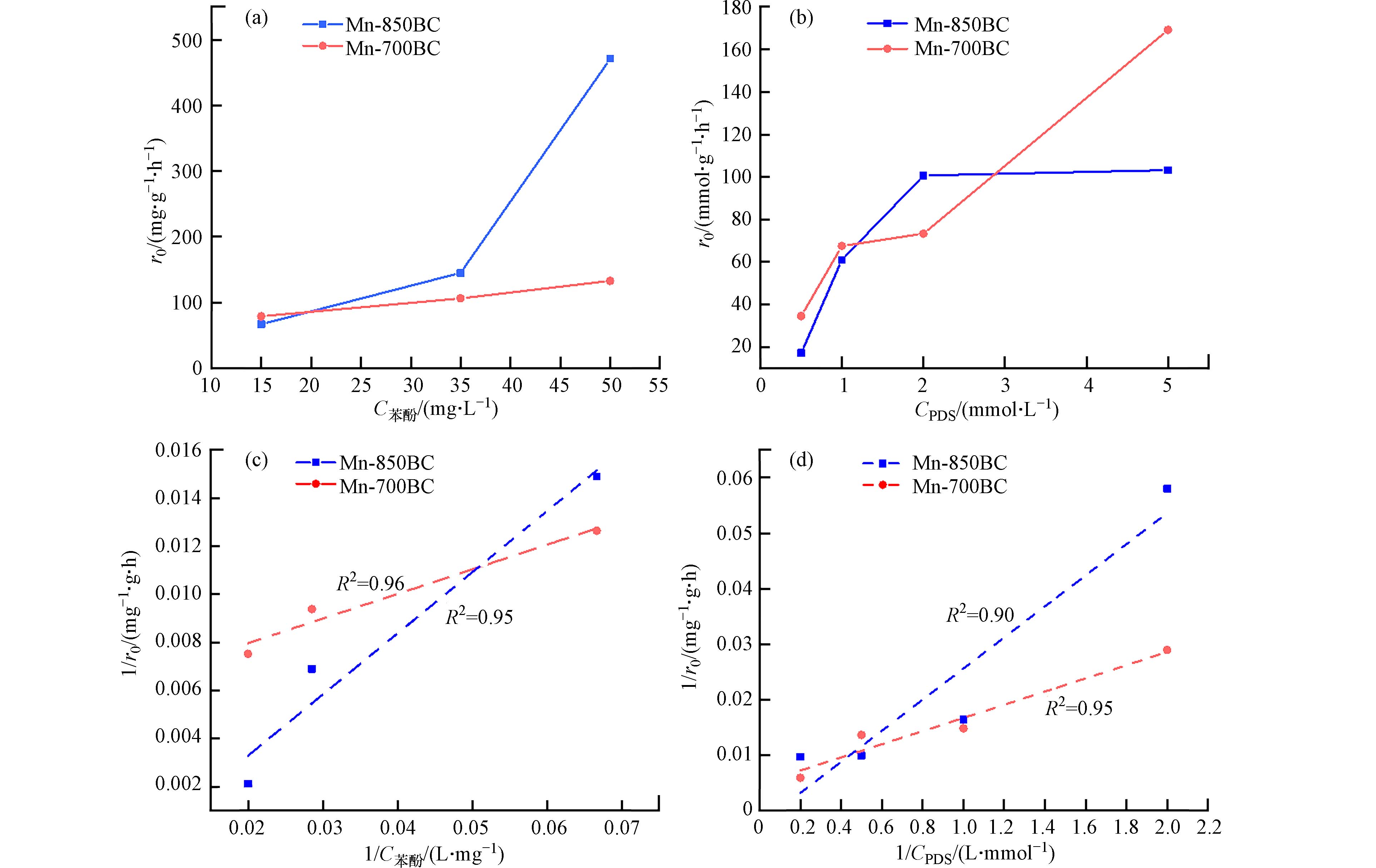 图 8 (a)、(b)苯酚及PDS初始浓度对反应初活性的影响以及(c)、(d)Langmuir-Hinshelwood拟合曲线Figure 8. (a) (b)Effect of phenol or PDS concentrations on initial reactivities and (c) (d) Langmuir-Hinshelwood fitting curves
图 8 (a)、(b)苯酚及PDS初始浓度对反应初活性的影响以及(c)、(d)Langmuir-Hinshelwood拟合曲线Figure 8. (a) (b)Effect of phenol or PDS concentrations on initial reactivities and (c) (d) Langmuir-Hinshelwood fitting curves2.2.3 pH和共存阴离子的影响
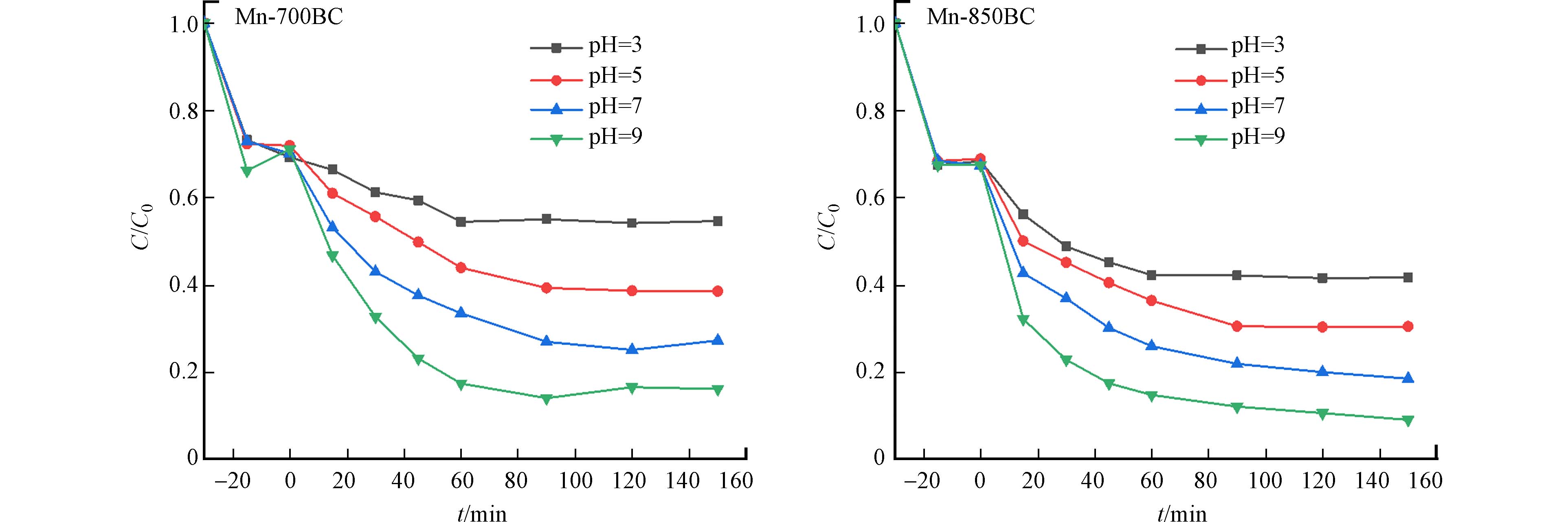
溶液pH可以改变材料表面的电荷特性以及污染物和活化剂PDS的化学特性,影响PDS与材料、苯酚与材料的相互作用,从而对活化和降解产生影响. 根据图9,在接近于实际水体的偏中性环境下,苯酚的去除率在80%左右,体现出这两种改性材料活化PDS降解苯酚的pH适应性. 在实验pH范围内,提高溶液pH有利于增强材料的活化效果. 这和生物炭负载γ-MnO2 复合材料活化过一硫酸盐降解对氯苯酚的结果相似[29]. 在酸性环境下,H+容易和S2O8−中的O—O基团形成氢键,不利于PDS与材料的接触,影响自由基的产生[30];其次,pH的提高能够增加过硫酸盐生成SO4·−和·OH的速率,Guan等通过表观摩尔吸收系数实验和半定量法测定两种自由基等方式验证了上述推测[31]. 同时,增加溶液中OH−的浓度,有利于材料表面羟基的形成,增加了电子转移的活性位点 [32].
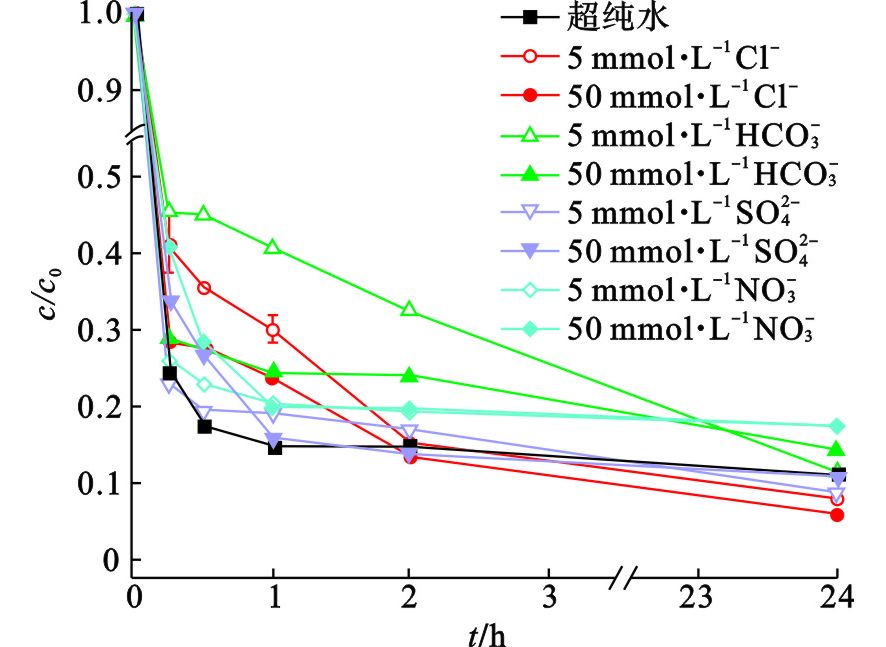
天然水体中含有大量阴离子,如Cl−、SO42-、NO3−等,存在的这些阴离子对催化过程的影响不可避免. 本文研究了水体中主要阴离子与苯酚共存时的去除效果,结果见图10.
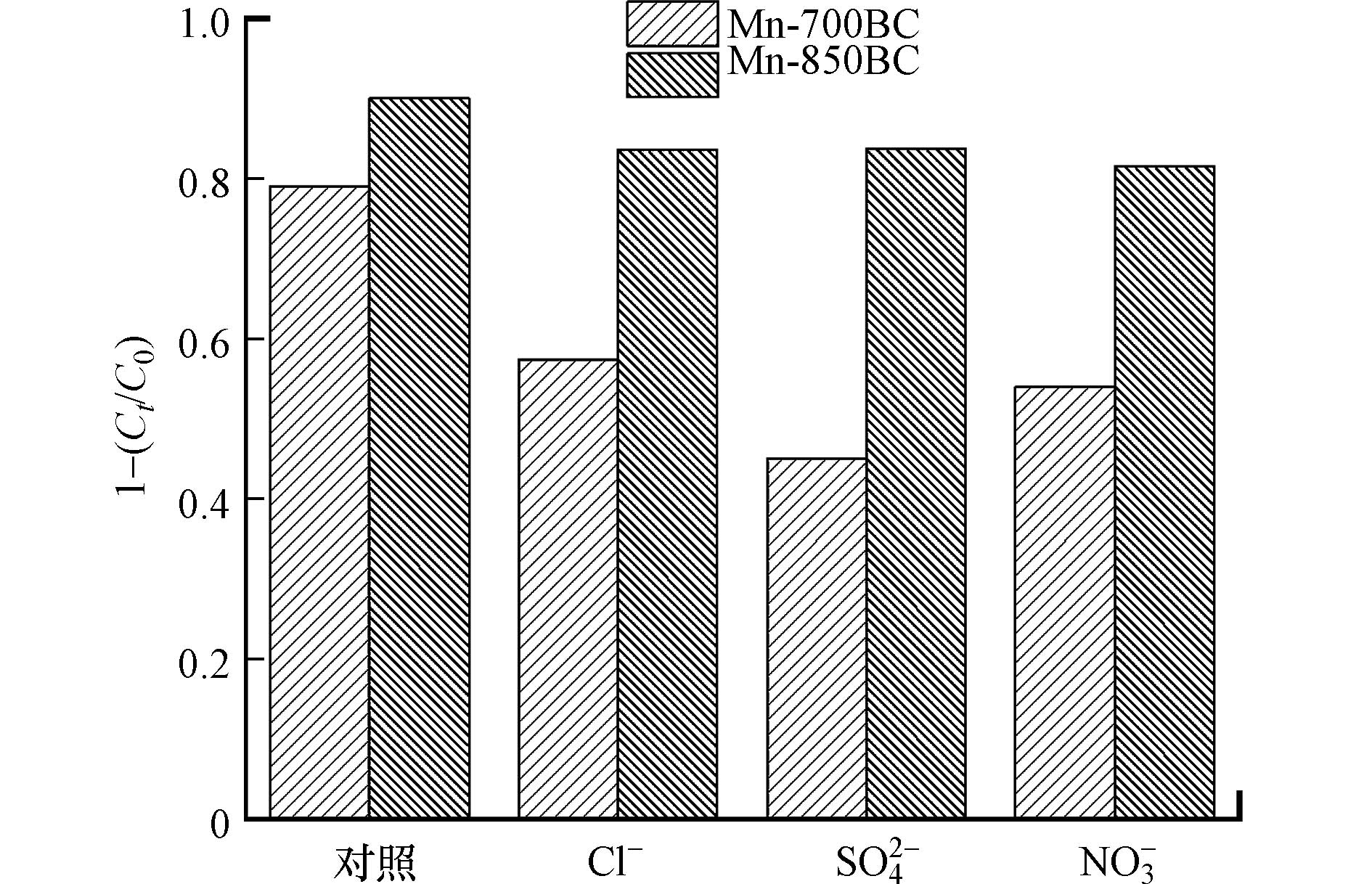 图 10 无机阴离子Cl−、SO42-、NO3−对去除苯酚的影响Figure 10. Effect of inorganic anions, Cl−, SO42-, and NO3−, on the removal of phenol
图 10 无机阴离子Cl−、SO42-、NO3−对去除苯酚的影响Figure 10. Effect of inorganic anions, Cl−, SO42-, and NO3−, on the removal of phenol当Cl−、SO42-和NO3−浓度均为20 mmol·L−1时,对苯酚的降解均显示出一定的抑制作用,SO42-的抑制程度最明显. 例如,对于Mn-700BC,分别向溶液中加入Cl−、NO3−和SO42-时,与不含这些阴离子的对照样相比,苯酚的降解效率分别减少了27%、30%和39%. 由前述结果可知,吸附于材料表面的过硫酸根离子浓度是影响活化的重要因素,那么,共存的阴离子会与过硫酸根离子竞争材料表面的活性位点,影响PDS的活化[12]. SO42-的化合价比Cl−和NO3−高,与材料表面Mnx+的亲和力更大. 另一方面,这些阴离子可通过反应或非反应方式降低体系中活性物种的氧化能力. Cl−和NO3−会与苯酚竞争SO4·−和·OH,生成Cl·和Cl2−·、 ·NO3 和 ·NO2 [33],这些自由基的氧化能力比SO4·−和·OH低,从而降低了苯酚的降解效率;SO42-的加入,使体系中SO42-的浓度过高,降低了SO4·−/SO42-的氧化还原电位,在一定程度上阻碍了PDS的活化. 值得注意的是,Mn-700BC受抑制的程度高于Mn-850BC. 这可能跟材料的特性差异导致PDS活化路径不完全一致有关. 众多文献结果显示,阴离子主要影响以自由基氧化为主的活化降解过程,对电子转移介导的非自由基氧化路径影响较小[34 − 36]. 从红外光谱可以看到,Mn-700BC的含氧官能团比Mn-850BC丰富,有利于自由基的产生,但不利于PDS吸附于材料表面形成亚稳态复合体以氧化污染物,非自由基氧化作用较弱[19]. 除此以外,Mn-850BC的缺陷比Mn-700BC多,石墨化程度亦较高,这些都有利于提高基于电子转移的非自由基氧化的作用[21,37].
2.3 活性氧的判定
为了阐明该体系中的主要活性氧物质,我们探讨了活性氧捕获剂存在时苯酚的降解情况,具体结果如图11所示. 加入TBA后,Mn-700BC和Mn-850BC对苯酚的去除率分别从原来的84%和90%减少至14%和30%,这表明催化剂活化PDS产生了·OH,而·OH在氧化苯酚时发挥着重要作用. 乙醇作为·OH和SO4·−的淬灭剂,更大程度上抑制了苯酚的降解,意味着SO4·−和·OH同样参与了苯酚的降解过程. 在添加L-His后, Mn-700BC和Mn-850BC对苯酚的去除率降低至24%和55%,这表明体系中有1O2参与的非自由基途径存在. 添加淬灭剂后,Mn-700BC活化PDS降解苯酚的受抑制程度均高于Mn-850BC. 这可能是由于Mn-700BC的孔道和比表面积较低,活性位点相对较少,淬灭剂竞争活性物种更严重. 另外,由于C=O等官能团的差异,Mn-700BC活化PDS时自由基路径占比更大,体现出更强的受抑制程度.
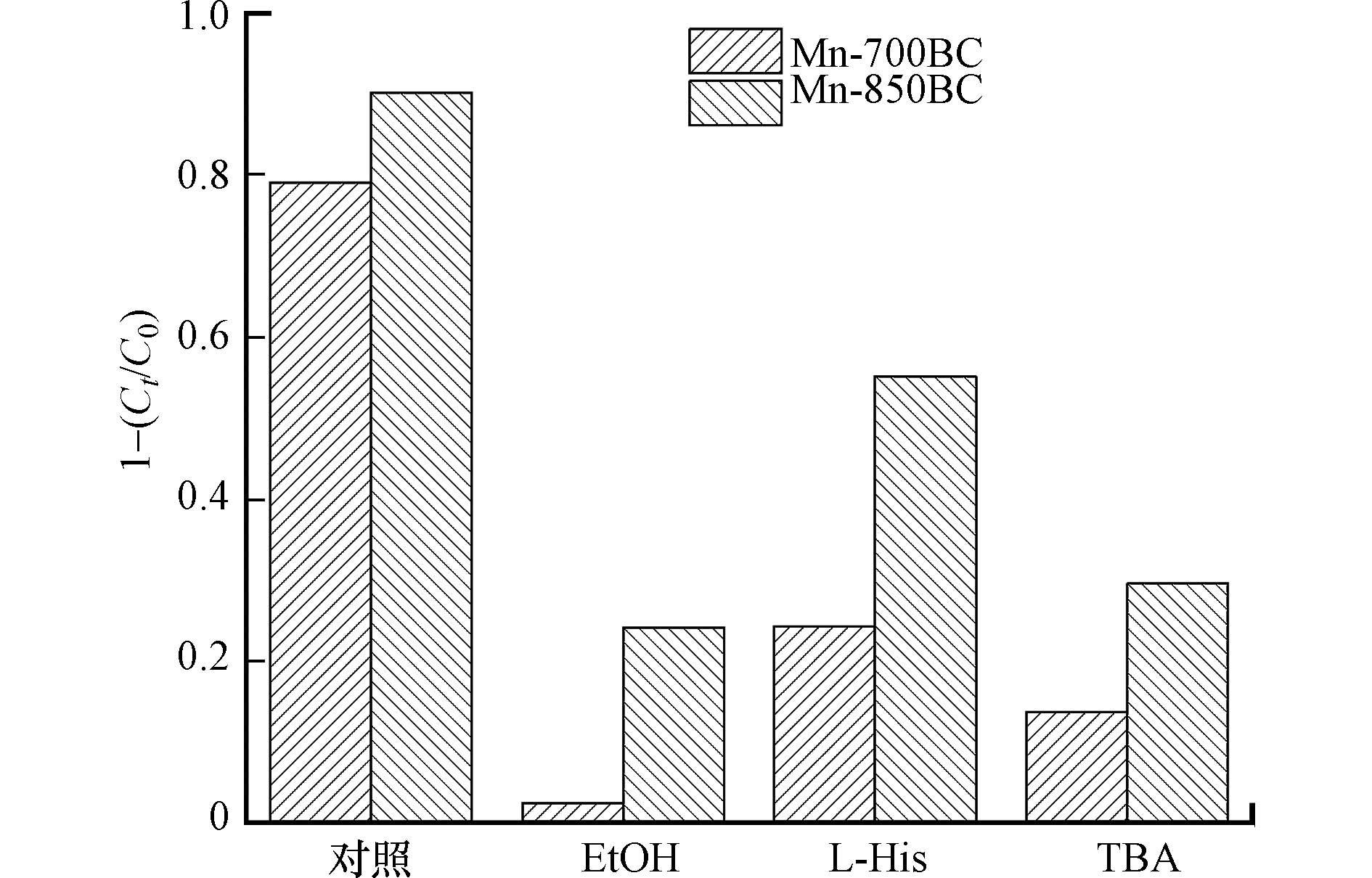 图 11 捕获剂EtOH、L-His和TBA对去除苯酚的影响Figure 11. Effect of quenching agents EtOH, L-His and TBA on the removal of phenol
图 11 捕获剂EtOH、L-His和TBA对去除苯酚的影响Figure 11. Effect of quenching agents EtOH, L-His and TBA on the removal of phenolEPR分析进一步揭示了活化过程中产生的自由基. 采用甲基吡啶N-氧化物(DMPO)作为SO4·−和·OH自由基自旋捕获剂,所得波谱如图12所示. 在700BC、850BC、Mn-700BC和Mn-850BC中,观察到强度比例为1:2:2:1的DMPO-OH加成物峰,以及6个较弱的DMPO-SO4·−加成物峰,表明这些材料在活化PDS时产生了·OH和SO4·−[38]. 在其他锰氧化物与炭的复合材料活化过硫酸盐时,也常鉴定出·OH和SO4·−[39]. 这些加成物信号峰的强度大小顺序为Mn-850BC > Mn-700BC > 850BC > 700BC,反映出活化PDS产生的这些自由基的含量差异,这与之前的降解结果一致.
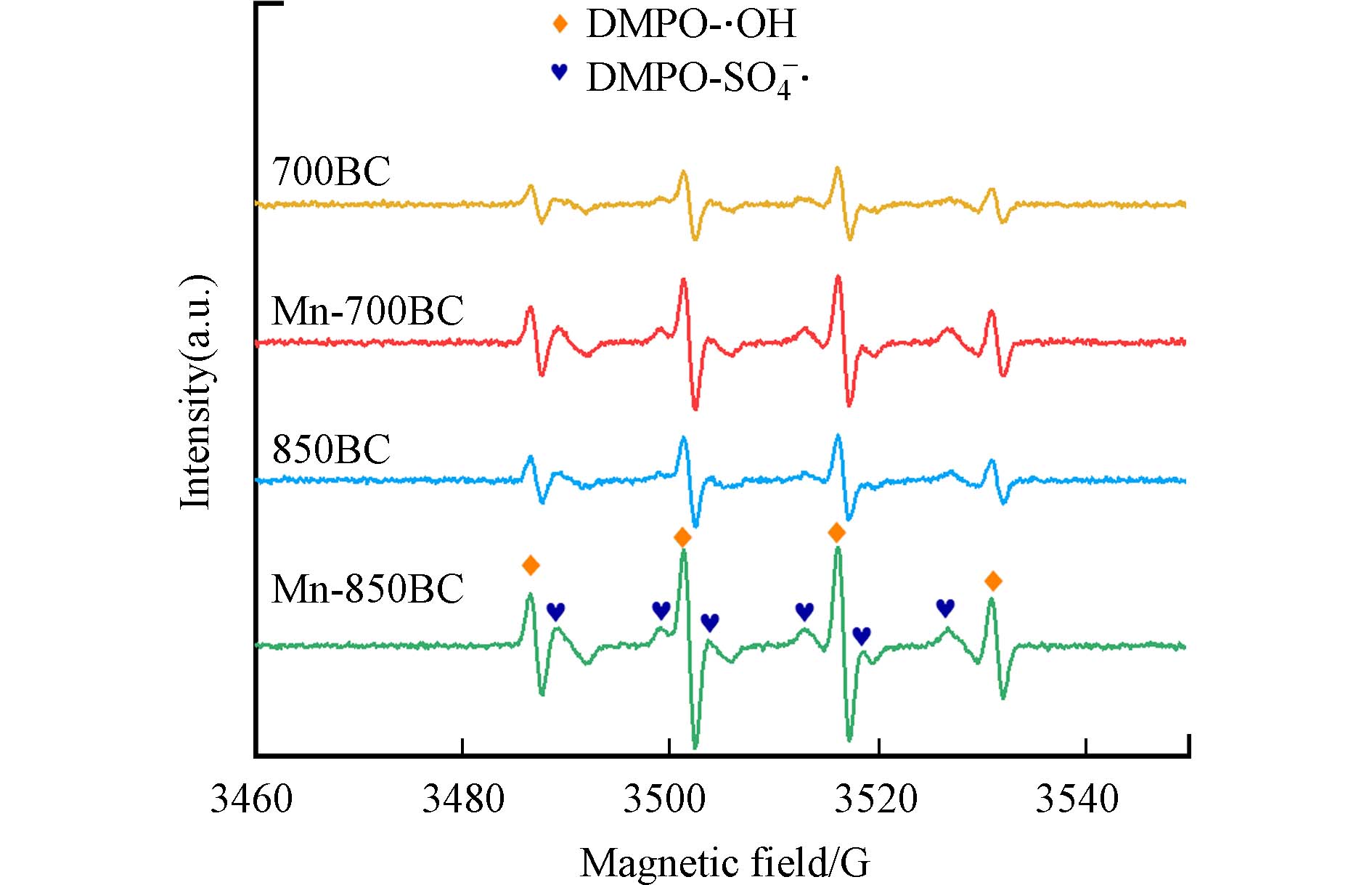 图 12 不同材料的电子顺磁共振波谱图Figure 12. Electron paramagnetic resonance spectra of different materials
图 12 不同材料的电子顺磁共振波谱图Figure 12. Electron paramagnetic resonance spectra of different materials3. 结论(Conclusions)
1)将垂序商陆茎末浸泡于高锰酸钾溶液再高温焙烧,成功将锰氧化物负载到生物炭上,且这种改性方式增加了生物炭的孔道结构. XRD和XPS结果显示,Mn以Mn(Ⅱ)、Mn(Ⅲ)和Mn(Ⅳ)混合价存在;随着焙烧温度的升高,材料的石墨化程度和缺陷增加,但表面含氧官能团含量降低.
2)锰改性生物炭活化PDS降解苯酚的效果显著优于未改性生物炭和商业二氧化锰,材料表面锰氧化物起主要活化作用;苯酚和PDS的初始浓度对改性材料活化降解苯酚的活性影响可用 Langmuir- Hinshelwood模型描述,说明其降解速率受PDS和苯酚的吸附控制;pH在3—9范围内时,提高溶液pH可提高苯酚的降解效率;溶液中共存阴离子Cl−、NO3−、SO42-均可在一定程度上抑制材料活化PDS降解苯酚的效果.
3)自由基捕获实验和EPR结果显示,在锰改性生物炭/过硫酸盐体系中,·OH和SO4·−等自由基路径是氧化苯酚的主要方式,同时还伴有1O2参与的非自由基路径.
-
表 1 ZVMg或Mg的双金属材料还原去除污染物研究总结
材料 制备方法 投加量/g·L−1 污染物 初始浓度/mg·L−1 反应溶液 反应速率常数/min−1 (除非特别标注) 去除率/% 参考文献 商业镁粉 - 2 硝酸盐 50 水溶液 0.35 91 [37] 商业镁粉 - 0.65 硝酸盐 90 水溶液 - 80 [36] Mg/Ag 湿式化学沉淀法 5 五氯苯酚 10 丙酮、乙醇 30 a > 85 [14, 38] Mg/Pd 湿式化学沉淀法 25 2-氯联苯 4 丙酮 0.33 > 90 [11] Mg/Pd 湿式化学沉淀法 10, 12 多氯联苯 3 乙醇 - > 90 [11] Mg/Pd 湿式化学沉淀法 4、6 2-氯联苯 4 乙醇、丙酮 - > 99 [39] ZVMg/Ag 湿式化学沉淀法 0.5 4-氯苯酚 10 - - 99 [12] Mg/Zn 湿式化学沉淀法 2.5 对硝基苯酚 19.8 - 0.066 9 - [40] Mg/Cu、Mg/Ni、Mg/Zn 湿式化学沉淀法 5 2,4-二硝基苯甲醚 250 水溶液 - 35~100 [41] Mg/Pd 机械球磨 25 多氯联苯 5 甲醇 0.002 26~0.007 16 a > 90 [5] Mg/Pd 机械球磨 25 多氯联苯 20 甲醇 1.72×10−4 a 80 [42] ZVMg 机械球磨 50 多氯联苯 1 乙醇 - > 94 [43] Mg/Pd 机械球磨 25 多氯联苯 10 甲醇 0.001 76 a - [44] ZVMg/C 机械球磨 25~50 多环芳烃 44.9~250 乙醇/乙酸乙酯 0.000 128~0.004 3 66~97 [18, 20, 45] ZVMg/C 机械球磨 50 五氯苯酚 20 乙醇 0.038 3~0.237 37~99 [18, 20, 45] ZVMg/C 机械球磨 50 六氯苯 20 乙醇/乳酸乙酯 0.222 99 [10] ZVMg, ZVMg/AC 机械球磨 50 八氯二苯并呋喃、2,8-二氯二苯并呋喃 20 乙醇 0.000 269~0.251 9 > 99 [19] 2-丁氧基乙醇 0.000 503~0.338 ZVMg 超声活化 5 硝酸盐 50 水溶液 - 90 [9] 注: a 表示反应速率常数的单位为L·(min·g)−1;- 表示文献里未说明。
下载: 导出CSV
-
[1] AYYILDIZ O, ACAR E, ILERI B. Sonocatalytic reduction of hexavalent chromium by metallic magnesium particles[J]. Water, Air, and Soil Pollution, 2016, 227(10): 1 − 9. [2] LEE G, PARK J, HARVEY O R. Reduction of Chromium(VI) mediated by zero-valent magnesium under neutral pH conditions[J]. Water Research, 2013, 47(3): 1136 − 1146. doi: 10.1016/j.watres.2012.11.028 [3] AGARWAL S, AL-ABED S R, DIONYSIOU D D. Chapter 25 - magnesium-based corrosion nano-cells for reductive transformation of contaminants[M]// Nanotechnology Applications for Clean Water (Second Edition). Oxford: William Andrew Publishing, 2014: 395-403. [4] SICILIANO A, CURCIO G M, LIMONTI C. Chemical denitrification with Mg0 particles in column systems[J]. Sustainability (Basel, Switzerland), 2020, 12(7): 2984. [5] DEVOR R, CARVALHO-KNIGHTON K, AITKEN B, et al. Dechlorination comparison of mono-substituted PCBs with Mg/Pd in different solvent systems[J]. Chemosphere, 2008, 73(6): 896 − 900. doi: 10.1016/j.chemosphere.2008.07.006 [6] IMAMURA H, NOBUNAGA T, KAWAHIGASHI M, et al. Preparation and hydriding behavior of magnesium metal clusters formed in low-temperature cocondensation: application of magnesium for hydrogen storage[J]. Inorganic Chemistry, 1984, 23(16): 2509 − 2511. doi: 10.1021/ic00184a027 [7] DOYLE J G, MILES T, PARKER E, et al. Quantification of total polychlorinated biphenyl by dechlorination to biphenyl by Pd/Fe and Pd/Mg bimetallic particles[J]. Microchemical Journal, 1998, 60(3): 290 − 295. doi: 10.1006/mchj.1998.1668 [8] THOMAS S, MEDHEKAR N V, FRANKEL G S, et al. Corrosion mechanism and hydrogen evolution on Mg[J]. Current Opinion in Solid State and Materials Science, 2015, 19(2): 85 − 94. doi: 10.1016/j.cossms.2014.09.005 [9] ILERI B, AYYILDIZ O, APAYDIN O. Ultrasound-assisted activation of zero-valent magnesium for nitrate denitrification: Identification of reaction by-products and pathways[J]. Journal of Hazardous Materials, 2015, 292: 1 − 8. doi: 10.1016/j.jhazmat.2015.03.004 [10] GARBOU A M, LIU M, ZOU S, et al. Degradation kinetics of hexachlorobenzene over zero-valent magnesium/graphite in protic solvent system and modeling of degradation pathways using density functional theory[J]. Chemosphere, 2019, 222: 195 − 204. doi: 10.1016/j.chemosphere.2019.01.134 [11] AGARWAL S, AL-ABED S R, DIONYSIOU D D. Enhanced corrosion-based Pd/Mg bimetallic systems for dechlorination of PCBs[J]. Environmental Science & Technology, 2007, 41(10): 3722 − 3727. [12] SOLANKI J N, MURTHY Z V P. Reduction of 4-chlorophenol by Mg and Mg–Ag bimetallic nanocatalysts[J]. Industrial & Engineering Chemistry Research, 2011, 50(24): 14211 − 14216. [13] MORALES J, HUTCHESON R, NORADOUN C, et al. Hydrogenation of phenol by the Pd/Mg and Pd/Fe bimetallic systems under mild reaction conditions[J]. Industrial & Engineering Chemistry Research, 2002, 41(13): 3071 − 3074. [14] PATEL U D, SURESH S. Effects of solvent, pH, salts and resin fatty acids on the dechlorination of pentachlorophenol using magnesium–silver and magnesium–palladium bimetallic systems[J]. Journal of Hazardous Materials, 2008, 156(1-3): 308 − 316. doi: 10.1016/j.jhazmat.2007.12.021 [15] 戴乐阳, 陈清林, 林少芬, 等. 高能球磨中促进粉体细化的主要因素研究[J]. 材料导报, 2009, 23(22): 59 − 61. doi: 10.3321/j.issn:1005-023X.2009.22.018 [16] 李学问. 机械球磨制备超细晶Mg-3Al-Zn合金及其组织性能的研究[D]. 哈尔滨: 哈尔滨工业大学, 2010. [17] SONG J, KIM H, KIM H, et al. Refinement behavior of coarse magnesium powder by high energy ball milling (HEBM)[J]. Journal of Korean Powder Metallurgy Institute, 2010, 17(4): 302 − 311. doi: 10.4150/KPMI.2010.17.4.302 [18] ELIE M R, WILLIAMSON R E, CLAUSEN C A, et al. Application of a magnesium/co-solvent system for the degradation of polycyclic aromatic hydrocarbons and their oxygenated derivatives in a spiked soil[J]. Chemosphere, 2014, 117: 793 − 800. doi: 10.1016/j.chemosphere.2014.10.042 [19] MOGHARBEL A T, YESTREBSKY C L. Dechlorination comparison of octachlorodibenzofuran over ball-milled zero-valent magnesium with and without activated carbon in different solvent systems[J]. Journal of Environmental Chemical Engineering, 2019, 7(2): 102950. doi: 10.1016/j.jece.2019.102950 [20] ELIE M R, CLAUSEN C A, YESTREBSKY C L. Reductive degradation of oxygenated polycyclic aromatic hydrocarbons using an activated magnesium/co-solvent system[J]. Chemosphere, 2013, 91(9): 1273 − 1280. doi: 10.1016/j.chemosphere.2013.02.031 [21] 魏鹏刚, 韩璐, 赵迎新, 等. 球磨零价镁/石墨(ZVMg/C)降解水中三氯乙烯[J]. 环境化学, 2022, 41(01): 276 − 287. [22] RIEKE R D. Preparation of highly reactive metal powders and their use in organic and organometallic synthesis[J]. Accounts of Chemical Research, 2002, 10(8): 301 − 306. [23] BARTMANN E, BOGDANOVI B, JANKE N, et al. Active magnesium from catalytically prepared magnesium hydride or from magnesium anthracene and its uses in the synthesis[J]. Chemische Berichte, 1990, 123(7): 1517 − 1528. doi: 10.1002/cber.19901230712 [24] HAAS I, GEDANKEN A. Synthesis of metallic magnesium nanoparticles by sonoelectrochemistry[J]. Chemical Communications, 2008, 15: 1795 − 1797. [25] TOSHIMA N, YONEZAWA T. Bimetallic nanoparticles–novel materials for chemical and physical applications[J]. New Journal of Chemistry, 1998, 22(11): 1179 − 1201. doi: 10.1039/a805753b [26] ZHANG W Y, WEI P G, CHEN M, et al. Trichloroethylene dechlorination rates, pathways, and efficiencies of ZVMg/C in aqueous solution[J]. Journal of Hazardous Materials, 2021, 417: 125993. doi: 10.1016/j.jhazmat.2021.125993 [27] SUN Y P, LI X Q, CAO J S, et al. Characterization of zero-valent iron nanoparticles[J]. Advances in Colloid and Interface Science, 2006, 120(1-3): 47 − 56. doi: 10.1016/j.cis.2006.03.001 [28] TIRAFERRI A, CHEN K L, SETHI R, et al. Reduced aggregation and sedimentation of zero-valent iron nanoparticles in the presence of guar gum[J]. Journal of Colloid and Interface Science, 2008, 324(1-2): 71 − 79. doi: 10.1016/j.jcis.2008.04.064 [29] LIU M H, WANG Y H, CHEN L T, et al. Mg(OH)2 supported nanoscale zero valent iron enhancing the removal of Pb(II) from aqueous aolution, Acs[J]. ACS APPL Mater Inter, 2015, 7: 7961 − 7969. doi: 10.1021/am509184e [30] LING L, HUANG X Y, LI M R, et al. Mapping the reactions in a single zero-valent iron nanoparticle[J]. Environmental Science & Technology, 2017, 51(24): 14293 − 14300. [31] LEE G, PARK J. Reaction of zero-valent magnesium with water: Potential applications in environmental remediation[J]. Geochimica et Cosmochimica Acta, 2013, 102: 162 − 174. doi: 10.1016/j.gca.2012.10.031 [32] BELDJOUDI T, FIAUD C, ROBBIOLA L. Influence of homogenization and artificial aging heat treatments on corrosion behavior of Mg-Al alloys[J]. Corrosion, 1993, 49(9): 738 − 745. doi: 10.5006/1.3316126 [33] BARIL G, PÉBÈRE N. The corrosion of pure magnesium in aerated and deaerated sodium sulphate solutions[J]. Corrosion Science, 2001, 43(3): 471 − 484. doi: 10.1016/S0010-938X(00)00095-0 [34] SONG G, ATRENS A. Understanding magnesium corrosion–a framework for improved alloy performance[J]. Advanced Engineering Materials, 2003, 5(12): 837 − 858. doi: 10.1002/adem.200310405 [35] REN Y, KANG S, ZHU J. Mechanochemical degradation of hexachlorobenzene using Mg/Al2O3 as additive[J]. Journal of Material Cycles and Waste Management, 2015, 17(4): 607 − 615. doi: 10.1007/s10163-015-0398-3 [36] MIRABI M, GHADERI E, RASOULI SADABAD H. Nitrate reduction using hybrid system consisting of zero valent magnesium powder/activated carbon (Mg0/AC) from water[J]. Process Safety and Environmental Protection, 2017, 111: 627 − 634. doi: 10.1016/j.psep.2017.08.035 [37] KUMAR M, CHAKRABORTY S. Chemical denitrification of water by zero-valent magnesium powder[J]. Journal of Hazardous Materials, 2006, 135(1-3): 112 − 121. doi: 10.1016/j.jhazmat.2005.11.031 [38] PATEL U, SURESH S. Dechlorination of chlorophenols by magnesium–silver bimetallic system[J]. Journal of Colloid and Interface Science, 2006, 299(1): 249 − 259. doi: 10.1016/j.jcis.2006.01.047 [39] AGARWAL S, AL-ABED S R, DIONYSIOU D D. Impact of organic solvents and common anions on 2-chlorobiphenyl dechlorination kinetics with Pd/Mg[J]. Applied Catalysis B:Environmental, 2009, 92(1-2): 17 − 22. doi: 10.1016/j.apcatb.2009.07.029 [40] KHAN S R, BATOOL M, JAMIL S, et al. Synthesis and characterization of Mg–Zn bimetallic nanoparticles: selective hydrogenation of p-nitrophenol, degradation of reactive carbon black 5 and fuel additive[J]. Journal of Inorganic and Organometallic Polymers and Materials, 2020, 30(2): 438 − 450. doi: 10.1007/s10904-019-01202-3 [41] HADNAGY E, MAI A, SMOLINSKI B, et al. Characterization of Mg-based bimetal treatment of insensitive munition 2, 4-dinitroanisole[J]. Environmental Science and Pollution Research International, 2018, 25(24): 24403 − 24416. doi: 10.1007/s11356-018-2493-1 [42] DEVOR R, CARVALHO-KNIGHTON K, AITKEN B, et al. Mechanism of the degradation of individual PCB congeners using mechanically alloyed Mg/Pd in methanol[J]. Chemosphere, 2009, 76(6): 761 − 766. doi: 10.1016/j.chemosphere.2009.05.007 [43] MALONEY P, DEVOR R, NOVAES-CARD S, et al. Dechlorination of polychlorinated biphenyls using magnesium and acidified alcohols[J]. Journal of Hazardous Materials, 2011, 187(1-3): 235 − 240. doi: 10.1016/j.jhazmat.2011.01.006 [44] COUTTS J L, DEVOR R W, AITKEN B, et al. The use of mechanical alloying for the preparation of palladized magnesium bimetallic particles for the remediation of PCBs[J]. Journal of Hazardous Materials, 2011, 192(3): 1380 − 1387. doi: 10.1016/j.jhazmat.2011.06.052 [45] ELIE M R, CLAUSEN C A, GEIGER C L. Reduction of benzo[a]pyrene with acid-activated magnesium metal in ethanol: A possible application for environmental remediation[J]. Journal of Hazardous Materials, 2012, 203-204: 77 − 85. doi: 10.1016/j.jhazmat.2011.11.089 [46] GARBOU A M, CLAUSEN C A, YESTREBSKY C L. Comparative study for the removal and destruction of pentachlorophenol using activated magnesium treatment systems[J]. Chemosphere, 2017, 166: 267 − 274. doi: 10.1016/j.chemosphere.2016.09.139 [47] THANGADURAI P, SURESH S. Reductive transformation of endosulfan in aqueous phase using magnesium–palladium bimetallic systems: A comparative study[J]. Journal of Hazardous Materials, 2013, 246-247: 245 − 256. doi: 10.1016/j.jhazmat.2012.12.031 [48] AGARWAL S, AL-ABED S R, DIONYSIOU D D, et al. Reactivity of substituted chlorines and ensuing dechlorination pathways of select PCB congeners with Pd/Mg bimetallics[J]. Environmental Science & Technology, 2009, 43(3): 915 − 921. [49] ENGELMANN M D, DOYLE J G, CHENG I F. The complete dechlorination of DDT by magnesium/palladium bimetallic particles[J]. Chemosphere, 2001, 43(2): 195 − 198. doi: 10.1016/S0045-6535(00)00163-6 [50] CWIERTNY D M, BRANSFIELD S J, ROBERTS A L. Influence of the oxidizing species on the reactivity of iron-based bimetallic reductants[J]. Environmental Science & Technology, 2007, 41(10): 3734 − 3740. [51] PATEL U, SURESH S. Dechlorination of chlorophenols using magnesium–palladium bimetallic system[J]. Journal of Hazardous Materials, 2007, 147(1-2): 431 − 438. doi: 10.1016/j.jhazmat.2007.01.029 [52] MORALES J. Dechlorination of chlorinated phenols by catalyzed and uncatalyzed Fe(0) and Mg(0) particles[J]. Journal of Hazardous Materials, 2002, 90(1): 97 − 108. doi: 10.1016/S0304-3894(01)00336-3 [53] GAUTAM S K, SURESH S. Dechlorination of DDT, DDD and DDE in soil (slurry) phase using magnesium/palladium system[J]. Journal of Colloid and Interface Science, 2006, 304(1): 144 − 151. doi: 10.1016/j.jcis.2006.08.052 [54] BEGUM A, GAUTAM S K. Dechlorination of endocrine disrupting chemicals using Mg0/ZnCl2 bimetallic system[J]. Water Research, 2011, 45(7): 2383 − 2391. doi: 10.1016/j.watres.2011.01.017 [55] USGS. Study and interpretation of the chemical characteristics of natural water[M]. Washington D C, USA: United States Geological Survey Water-Supply, 1992, 2254. [56] 中华人民共和国国家质量监督检验检疫总局, 中国国家标准化管理委员会. 地下水质量标准: GB/T 14848—2007[S]. 北京: 中国标准出版社, 2017. [57] GULBRANDSEN E. Anodic behaviour of Mg in HCO3−/CO32− buffer solutions. Quasi-steady measurements[J]. Electrochimica Acta, 1992, 37(8): 1403 − 1412. doi: 10.1016/0013-4686(92)87014-Q -

 点击查看大图
点击查看大图

计量
- 文章访问数: 3249
- HTML全文浏览数: 3249
- PDF下载数: 36
- 施引文献: 0
