















-
全程自养脱氮(completely autotrophic nitrogen removal over nitrite, CANON)工艺将短程硝化和厌氧氨氧化(anaerobic ammonium oxidation, ANAMMOX)2个过程置于同一反应器内进行[1]。相对于传统生物脱氮技术,CANON工艺具有无需外部碳源、节省曝气量和污泥产率低的显著优势[2]。在CANON工艺中,氨氧化细菌(ammonia oxidizing bacteria, AOB)与ANAMMOX菌起主要作用,AOB以O2为电子受体,将NH4+-N氧化成NO2−-N;ANAMMOX菌以NO2−-N为电子受体,将剩余NH4+-N氧化成N2,并生成少量的NO3−-N[3]。由于ANAMMOX所需要的NO2−-N是由短程硝化提供的,因此,稳定的短程硝化是CANON工艺稳定运行的必要条件。然而,由于亚硝酸盐氧化菌(nitrite oxidizing bacteria, NOB)与AOB生长所需的环境条件较为相似,系统中往往同时存在有AOB与NOB,若NOB过量增殖,NO2−-N被氧化为NO3−-N,则会导致短程硝化被破坏。
有诸多因素会对短程硝化造成影响,如pH、温度、溶解氧(dissolved oxygen, DO)和游离氨(free ammonia, FA)等[4]。其中,DO是控制CANON工艺中短程硝化和ANAMMOX稳定实现的关键参数。曝气过量使得DO质量浓度偏高,则会造成NOB的增殖和对ANAMMOX菌的抑制作用;曝气不足使得DO质量浓度偏低,则会使AOB活性受到抑制,氨氧化速率随之下降,ANAMMOX菌无法获得充足的NO2−-N基质进而导致脱氮效率降低[5-6]。李冬等[7]在序批式反应器(sequencing batch reactor, SBR)内构建了CANON工艺,结果表明,在DO为0.05~0.10 mg∙L−1的反应器中,ANAMMOX菌的活性不受影响,并在DO为0~0.40 mg∙L−1内通过增大曝气量提高总氮去除率;当DO达到0.5 mg∙L−1时,ANAMMOX菌的活性受到抑制,导致CANON工艺被破坏,但却实现了稳定的短程硝化,亚氮积累率达到93.35%。王会芳等[8]在以陶粒作为填料,研究了DO对生物膜CANON反应器的影响时发现,当DO超过1.75 mg∙L−1时,反应器中ΔNO3−-N/ΔTN值为0.239,严重偏离了理论值0.127,表明短程硝化遭到了严重破坏。JOSS等[9]在采用CANON工艺的污水处理厂中发现,由于2 h的过量曝气,DO质量浓度达到1 mg∙L−1以上,造成了对ANAMMOX菌活性的抑制,NO2−-N也开始积累,DO和NO2−-N质量浓度同时升高使得NOB在反应器内大量增殖,CANON反应器出水NO3−-N质量浓度从低于15 mg∙L−1升高到200 mg∙L−1以上,短程硝化和ANAMMOX均被破坏。
上述研究主要针对的是DO质量浓度对CANON工艺中短程硝化和ANAMMOX运行稳定性的破坏,但就CANON反应器中短程硝化运行失稳后调控恢复的相关报道较少。其中短程硝化过程的破坏主要表现为系统ΔNO3−-N/ΔTN值与理论值0.127的偏离,即系统中NOB开始发挥作用,ΔNO3−-N增加的同时,ΔTN也会由于NOB与ANAMMOX菌对NO2--N底物的竞争而减少,导致ΔNO3−-N/ΔTN值偏大。且由于采用的反应器形式不同以及污泥特性等方面的差异,研究者们对于CANON工艺中最佳DO质量浓度以及DO对CANON工艺中功能菌活性的影响还并未形成统一认识。本研究以CANON工艺中遭破坏的短程硝化为主要研究对象,通过限氧的方式对NOB活性进行抑制,从而恢复短程硝化,同步考察了NOB与ANAMMOX菌活性的变化,探究了DO质量浓度对CANON工艺中短程硝化恢复过程的影响。
-
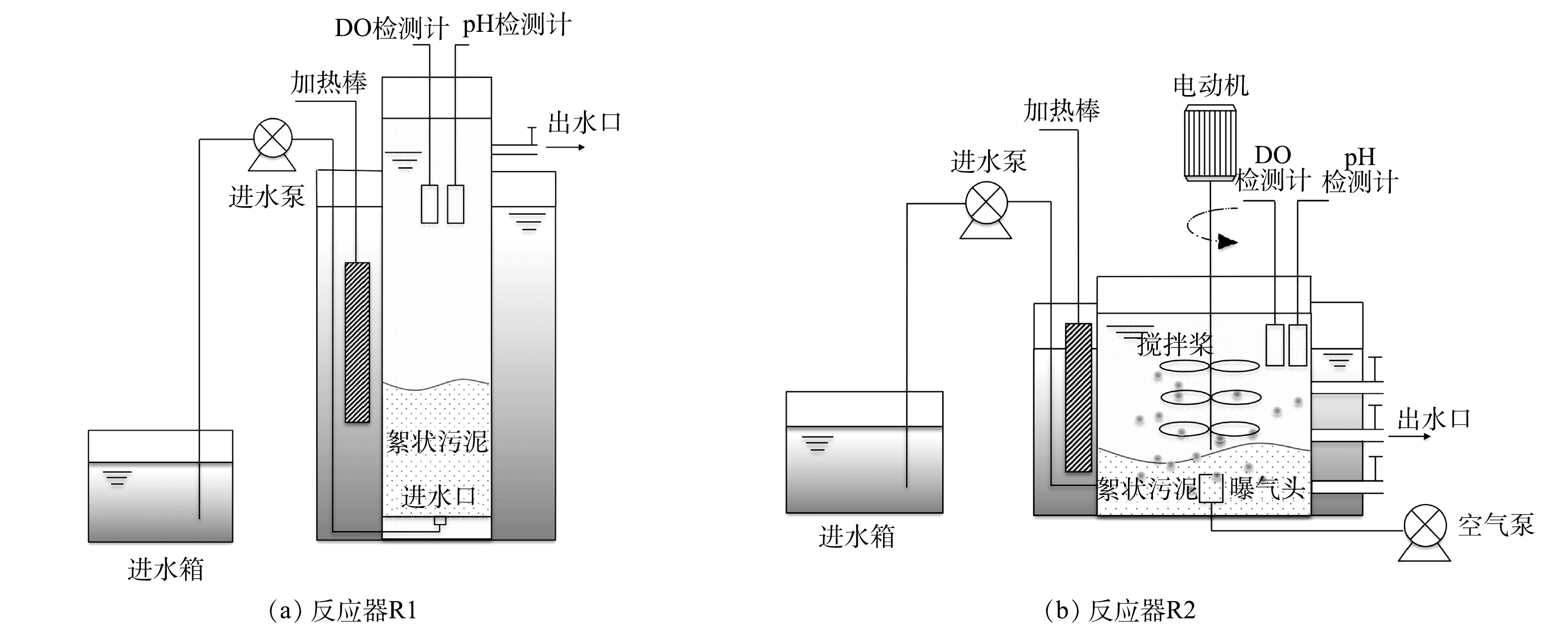
实验的不同阶段在不同的反应器中进行。其中,在连续流反应器R1中进行抑制期短程硝化恢复实验,而后,为保证曝气阶段的活性污泥混合反应完全,并避免污泥流失,改用序批式反应器R2进行过渡期和好氧期的短程硝化恢复实验。
连续流反应器R1由圆柱状有机玻璃制成,其内径为10 cm,高度为1.2 m,有效容积为8 L。反应器采用底部进水,上部出水的形式;R2反应器由圆柱状有机玻璃制成,其内径和高度分别为20 cm和27 cm,有效容积为5 L。反应器内部设有曝气及搅拌装置,外部通过水浴放置加热棒维持温度恒定。反应器每日运行6个周期,每周期总时长为4 h,其中进水10 min,反应200 min,沉淀20 min,出水10 min,由定时器自动控制各个装置的开关运行。排水比为50%。装置如图1所示。
-
各反应器进水均为无机人工配水,采用自来水进行配制,并采用NH4Cl、NaNO2或二者以一定比例混合作为主要氮源,添加适量的NaHCO3、NaH2PO4以及1 mL·L−1微量元素浓缩液Ⅰ和Ⅱ[10]。
反应器R1、R2中污泥均接种自课题组现有中试MBBR中短程硝化遭破坏的CANON工艺污泥。中试MBBR运行期间的ΔNO3−-N/ΔTN平均值为0.274,该值反映的是CANON工艺中的短程硝化效果[11],显著大于理论值,表明短程硝化性能较差。
-
取课题组现有中试MBBR当中的絮状污泥至反应器R1中,1~73 d内按照1:1.32的比例在进水中添加NH4+-N与NO2−-N强化ANAMMOX菌活性,初始进水流量设定为0.22 L∙h−1,即水力停留时间(hydraulic retention time, HRT)约为36 h,在实验初期采用较长的HRT有利于ANAMMOX菌的增长和持留[12]。同时采取密封措施,减少空气中的氧气进入,营造ANAMMOX菌的适宜生长环境,并对NOB的活性进行抑制。第74天时将反应器R1中污泥转移至反应器R2中,该过程中未对反应器中的污泥进行淘洗,尽量保持了污泥的原有形态和数量,避免了对实验的影响,并在此阶段逐步增加曝气量以及减少进水NO2−-N质量浓度以强化AOB的活性,使反应器由ANAMMOX工艺逐步转化为CANON工艺。反应器共连续运行130 d,运行工况如表1所示。
分别对连续运行第1天和第80天的污泥进行高通量测序,考察微生物群落结构的变化规律,并在连续运行的第1、28、36、48、58、90以及110天进行批次实验测定ANAMMOX菌以及NOB活性。
-
1) NH4+-N使用纳氏试剂分光光度法检测;NO2−-N使用N-(1-萘基)-乙二铵光度法检测;NO3−-N使用紫外分光光度法检测;pH使用METTLER TOLEDO F2 基础型便携式pH计检测;温度和DO使用METTLER TOLEDO FG4基础型便携式溶氧仪检测。TN= NH4+-N+NO2−-N+NO3−-N [8]。FA质量浓度按式(1)计算。
式中:
CNH+4-N 为混合液中NH4+-N质量浓度,mg∙L−1;T为混合液温度,℃。2)活性测定。以TN比降解速率q作为ANAMMOX活性指标,根据式(2)进行计算。
式中:ΔTN为反应过程中TN质量浓度变化量,mg∙L−1;t为反应时间,h;M为混合液挥发性悬浮固体质量浓度,g∙m−3。
采用NOB的比耗氧速率(specific oxygen uptake rate, SOUR)r表征NOB活性[14],根据式(3)进行计算。
式中:D1为不添加底物时单位实验时间的DO质量浓度差,mg·(L·h)−1;D2为添加NO2−-N底物时单位实验时间的DO质量浓度差,mg·(L·h) -1。
3)微生物分析。委托上海生工生物工程有限公司进行高通量测序,对污泥样品进行宏基因组16S rDNA测序,分析微生物群落结构[15]。
-
1)抑制期。反应器共连续运行130 d。如图2(a)所示,在抑制初期0~3 d,NH4+-N去除率下降,NH4+-N的平均出水质量浓度为124.6 mg∙L−1。由图2(b)可见,NO2−-N去除率由74.3%降低至38.1%,ΔNO2−-N/ΔNH4+-N值大于ANAMMOX特征理论值1.32[8]。这表明此时ANAMMOX作用不明显。由于运行初期的进水流量相对较大,初始进水流量为0.22 L∙h−1,且未对进水中的DO进行吹脱,反应器内DO高于0.6 mg∙L−1,超过了ANAMMOX菌对DO的最适质量浓度0.15~0.30 mg∙L−1 [16]。此外,如图3所示,运行初期FA质量浓度接近10 mg∙L−1。有研究表明,该水平FA质量浓度会对ANAMMOX菌的活性存在一定的抑制作用[17]。如图2(c)和图2(d)所示,0~3 d内的ΔNO3−-N/ΔTN值均高于理论值0.127,出水NO3−-N质量浓度偏高,TN去除率由42.7%下降至24.5%。这表明此时短程硝化效果不理想,反应器内主要进行的是AOB以及NOB利用进水中的DO进行氨氧化以及亚硝酸盐氧化反应。
在第7天将进水流量减半至0.11 L∙h−1,由于AOB和NOB持续消耗进水中的DO进行硝化反应,导致DO质量浓度在第28天下降至0.33 mg∙L−1。在此阶段,由图2(a)和图2(b)可见,NH4+-N和NO2−-N虽均有不同程度的去除,但NH4+-N平均去除率仅为49.0%,NO2−-N去除率则稳定在95.0%以上,ΔNO2−-N/ΔNH4+-N平均值为2.02,远高于ANAMMOX反应计量理论值1.32。这表明ANAMMOX尚未成为反应器内的主要反应。此外,如图2(c)所示,ΔNO3−-N/ΔTN值小于0,远低于ANAMMOX反应计量理论值0.127。由图2(d)可见,TN去除率有所上升。由于本研究采用的是无机配水,排除了反硝化细菌利用外源有机物进行反硝化脱氮的因素。其原因可能为,DO缺乏导致部分好氧菌因底物匮乏死亡,之后转化为可降解有机物,反硝化菌利用其作为碳源进行内源呼吸代谢[18]。
在第30天,将进水流量还原至初始进水流量0.22 L∙h−1。如图2(a)~(d)所示,当反应器内DO质量浓度降低至0.19 mg∙L−1时,NH4+-N和NO2−-N去除率分别升高至98.7%和98.9%,ΔNO3−-N/ΔTN由负值开始升高,TN去除率达到93.1%,此时的ΔNO2−-N/ΔNH4+-N值为1.36,与理论值1.32相比稍有偏高。这表明NH4+-N与NO2−-N基本实现同步去除,ANAMMOX作用得到提升,但反应器内依然存在硝化反硝化作用。
在31~73 d,分别于第35天以及第49天将进水流量提高至初始进水流量的2倍(0.44 L∙h−1)和4倍(0.88 L∙h−1)。如图2(b)和图2(d)可知,ΔNO2−-N/ΔNH4+-N平均值为1.37,TN去除负荷升高至1.08 kg∙(m3∙d)−1。这表明ANAMMOX菌活性得到提升,ANAMMOX占主导作用,但偏高的ΔNO2−-N/ΔNH4+-N值说明系统中依旧存在NOB参与的亚硝酸盐氧化,本阶段较低的DO并不能完全抑制NOB,若要进一步淘汰系统中的NOB,可能需要更严格的限氧条件。同时,由图2(c)可以看出,ΔNO3−-N/ΔTN值继续升高向理论值趋近,但仍低于0.127。这表明仍有死亡微生物作为可生物降解有机物参与反硝化过程,异养菌同NOB对DO的竞争,使得NOB难以利用有限的DO进行亚硝酸盐氧化反应,有利于对短程硝化的进一步恢复。
2)过渡期。在此阶段,进水NH4+-N质量浓度由150 mg∙L−1降低至125 mg∙L−1;进水NO2−-N质量浓度由200 mg∙L−1降低至50 mg∙L−1。因ANAMMOX反应基质NO2−-N减少,第74天时,由图2(a)和图2(d)可见,NH4+-N和TN去除率偏低,分别为42.8%和39.9%。通过过渡期恢复曝气,AOB活性预期可逐步恢复,以保证NO2−-N来源,第91天时,NH4+-N和TN去除率分别上升至82.4%和70.0%。此外,由于ΔNH4+-N和ΔNO2−-N反映的是AOB与ANAMMOX菌共同作用的结果,不宜继续采用ΔNO2−-N/ΔNH4+-N作为衡量指标[11]。由图2(c)可见,过渡期的出水NO3−-N质量浓度增长量较抑制期减小,同时,ΔNO3−-N/ΔTN值由过渡初期时0.207下降至0.105。这说明,由于ANAMMOX菌的活性得到强化,NOB难以同ANAMMOX菌竞争NO2−-N,同时,由于DO限制,AOB较好的溶解氧亲和能力使其在与NOB竞争DO的过程中获得优势,短程硝化效果得到改善。
3)好氧期。在此阶段,进水NH4+-N质量浓度由125 mg∙L−1提高至200 mg∙L−1;进水NO2−-N质量浓度由50 mg∙L−1降低至0。由于NO2−-N质量浓度的进一步限制,在第92天时,由图2(a)和图2(d)可见,NH4+-N和TN去除率较前一阶段急剧下降。随后,NH4+-N和TN去除率逐渐得到提升。根据VAN DER STAR等[19]的研究结果,NOB对NO2−-N的半饱和常数为12~955 μmol∙L−1,而ANAMMOX菌对NO2−-N的半饱和常数为0.2~3 μmol∙L−1,即ANAMMOX菌对NO2−-N的亲和能力更强,从而在NO2−-N质量浓度较低时能够先于NOB将其利用。此外,随着AOB活性在好氧期的增强,氨氧化对系统中DO的消耗,进一步抑制了NOB的活性,并提升了ANAMMOX的脱氮效果。运行第130天,如图2(a)~(d)所示,NH4+-N去除率升高至75.0%,NO2−-N去除率维持较高水平,TN去除负荷达到0.86 kg∙(m3∙d)−1,ΔNO3−-N/ΔTN值为0.136,较理论值(0.127)偏高。此结果说明NOB仍有一定活性,系统中的NOB没有被完全淘汰。
-
ANAMMOX菌活性批次实验进水NH4+-N和NO2−-N质量浓度分别为43 mg∙L−1和57 mg∙L−1;NOB活性批次实验进水NH4+-N和NO2−-N质量浓度分别为0和50 mg∙L−1。如图4所示,ANAMMOX菌在限氧抑制初期活性恢复较缓慢,此时,污泥处于对反应器内DO质量浓度波动式下降的适应阶段。此后,ANAMMOX菌活性迅速升高,由0.11 kg∙(kg∙d)−1(以VSS计)升高至0.34 kg∙(kg∙d)−1,但NOB的SOUR值自抑制期开始始终呈下降趋势,由第1天的1.68 kg∙(kg∙d)−1(以VSS计)降低至第110天的0.57 kg∙(kg∙d)−1,说明抑制效果显著。在本研究中,在污泥以絮状污泥为主且未形成明显颗粒污泥的条件下,ANAMMOX菌活性依然得到了强化,仅在当反应器进入过渡期恢复曝气后,由于好氧条件的限制,ANAMMOX菌活性开始下降,于第110天下降至0.26 kg∙(kg∙d)−1,但与反应初期的0.08 kg∙(kg∙d)−1相比,ANAMMOX菌活性仍有较大提升。若在后续长期运行中形成颗粒污泥,或许可进一步提升ANAMMOX菌的活性。此外,当解除限氧抑制恢复曝气后,NOB活性并未出现回升趋势,但直至反应运行的第110天,NOB活性也并未完全丧失,表明限氧条件虽然能够有效抑制NOB的活性,但不能彻底淘汰NOB,并且在后续长期运行过程中,存在NOB逐渐克服抑制作用进行增殖从而再次使短程硝化遭到破坏的风险。
-
分别取连续运行第1天和第80天反应器中污泥进行高通量测序,抑制期前后系统中的微生物群落结构变化规律如图5所示。短程硝化恢复过程中,检测到的ANAMMOX菌有Candidatus Kuenenia和Candidatus Brocadia。其中,Candidatus Kuenenia为优势菌属,其丰度由7.91%增长至13.12%。有研究表明,Candidatus Brocadia适于在严格厌氧环境下生长,而Candidatus Kuenenia则可以接受混合液中存在少量的溶解氧[16],表明抑制期的低DO质量浓度可促进Candidatus Kuenenia的生长。同时,反应器中存在少量的Candidatus Brocadia,其丰度在抑制前的絮状污泥中为0.87%,经过过渡期的NO2−-N质量浓度波动后,丰度降至0.01%以下。有研究表明,在SBR中对Candidatus Kuenenia和Candidatus Brocadia的选择可能是基于对限制性底物(如NO2−-N)的亲和力,Candidatus Kuenenia对NO2−-N的亲和性能较强[19]。因此,在本实验有限的进水NO2−-N质量浓度条件下,Candidatus Kuenenia占据了优势生长地位。AOB中的优势菌属为Nitrosomonas,由于抑制期的低DO质量浓度,导致AOB的生长受到抑制,相对丰度由27.43%降低至8.67%。NOB的主要菌属为Nitrospira,NOB在抑制期前后的特征较为一致,即在微生物群体中的相对丰度较低,平均低于AOB以及ANAMMOX菌一个数量级。其中,经抑制后的污泥中Nitrospira相对丰度由抑制前的0.01%以下升高至1.03%,即NOB相对丰度经过限氧抑制后反而上升。
此外,还检测到几种反硝化菌属,相对丰度均在抑制期后有所提高,如Thauera、Pseudomonas和Comamonas,均被证明具有利用有机物为碳源进行反硝化的能力[20-22],由于进水中未添加有机物,因此,这进一步印证了抑制期间可能发生的内源反硝化过程。
-
本研究中,Nitrospira为检测出的主要NOB菌属,这与传统污水处理硝化菌种一致[23]。经过抑制期的短程硝化恢复,活性批次实验结果表明NOB活性抑制情况良好,然而,丰度却有所升高。可见,NOB的活性和生物量的变化趋势并不同步。类似的结果也出现在课题组同期进行的SBR间歇曝气恢复的短程硝化实验中,反应器恢复短程硝化后,AOB活性稳定的同时其丰度显著下降,而NOB虽检测不到活性,但其相对丰度呈上升趋势[24]。
普遍认为,AOB的氧半饱和常数小于NOB[25],在低DO条件下,AOB的生长速率几乎不受影响,而 NOB的生长速率显著降低。然而,杨庆等[26]对此的研究表明,低DO系统内AOB和NOB的氧半饱和常数分别为0.281 mg∙L−1和0.280 mg∙L−1,两者非常接近,并且NOB的生长速率远大于AOB。结合分析本实验中AOB和NOB丰度的变化,推测其原因可能为:1)抑制期低DO质量浓度可限制AOB的生长和氨氧化反应速率,生长速率较NOB缓慢,导致AOB相对丰度降低;2)尽管限氧条件抑制了NOB的活性,但抑制期进水NO2−-N质量浓度较高,充足的底物质量浓度为NOB在不利条件下生长提供了可能性,因此NOB相对丰度升高;3)在抑制期间反应器不能做到与空气完全隔绝,难免有氧气进入反应器,且实验进水为DO未经吹脱的自来水人工配制,因此,反应器内DO会从实验进水中得到一定补充,导致NOB仍可利用DO进行增殖;4)本实验进水为无机配水,不含外源有机物,因此,减弱了异养菌同NOB对DO的竞争作用,进一步提升了NOB利用DO进行增殖的空间。
USHIKI等[27]对NOB基因组的分析结果表明:Nitrospira含有的编码细胞色素氧化酶bd基因,能够在低氧条件下被激活,使细菌在限氧环境下也能够完成细胞生长的过程。张亮等[28]认为,当受到抑制时,NOB或可通过提高丰度等方式克服抑制作用。可见,NOB仍有部分活性规律尚未明确,这些特性均给CANON工艺中针对NOB的抑制带来了挑战。
值得一提的是,新加坡樟宜再生水厂地处热带,污水温度在(30
± 2)℃,适合ANAMMOX菌的增殖并有利于实现短程硝化,但其在由测流向主流ANAMMOX工艺转换的过程中,同样存在对NOB抑制的问题[29]。一方面,侧流工艺中的高NH4+-N质量浓度(500~1 500 mg∙L−1)使得FA质量浓度高于抑制NOB的阈值(0.08~0.82 mg∙L−1)[30],而主流工艺中城市污水较低的NH4+-N质量浓度(30~100 mg∙L−1)使得NOB抑制较为困难;另一方面,AOB、NOB及ANAMMOX的活性-温度系数在不同温度下的差异较大[31],主流工艺中季节变换导致的城市污水温度有波动,使得污泥微生物种群和数量发生改变,同样对NOB的抑制以及ANAMMOX菌的持留带来挑战。通过对本研究及樟宜再生水厂在以上两方面运行条件的对比,可以在一定程度上解释本研究中NOB无法被完全抑制的原因:一是本研究中的NH4+-N质量浓度在长期运行过程中处于较低水平,因此,缺乏FA对NOB的持续抑制作用;二是本研究进水温度维持在25℃左右,与ANAMMOX菌的适宜中温条件有一定偏离。此外,针对NOB在系统中的相对丰度变化,孙延芳等[32]关于絮体污泥龄(sludge retention time, SRT)对CANON工艺运行稳定性的影响研究中发现,不淘洗絮体污泥时,系统NOB丰度显著增加,并通过定量PCR技术分析得出,絮体SRT时间越长,ANAMMOX菌的丰度越高。因此,有必要在后续的长期运行中持续关注微生物的丰度变化及系统稳定性,结合控制合理污泥龄的手段来实现功能菌(AOB和ANAMMOX菌)的有效持留和NOB的淘洗。
-
1)经过抑制期的限氧,ANAMMOX活性由0.08 kg∙(kg∙d)−1上升至0.34 kg∙(kg∙d)−1,TN去除率可达88.6%,TN去除负荷最高可达1.08 kg∙(kg∙d)−1,ANAMMOX占主导作用。
2) Candidatus Kuenenia为ANAMMOX菌优势菌属,Nitrospira为NOB的主要菌属,经过抑制期恢复,二者的相对丰度分别由7.91%和低于0.01%增长至13.12%和1.03%。
3)通过短程硝化恢复,ΔNO3−-N/ΔTN值由0.318逐渐恢复至0.136,短程硝化基本恢复,但较理论值0.127稍有偏高,仍存在亚硝酸盐氧化过程。
4)连续运行期间,NOB活性降低显著,但相对丰度有所提高,于微生物群体中仍具有一定的相对丰度,在后续运行中有适应抑制作用并进行增殖的可能,CANON工艺短程硝化仍具有被破坏的风险。
CANON工艺中短程硝化恢复及ANAMMOX强化策略
Short-cut nitrification recovery and ANAMMOX enhancement strategy in CANON process
-
摘要: 为了对CANON工艺中遭破坏的短程硝化进行恢复,并对ANAMMOX菌的活性进行强化,在第1阶段(抑制期)采用连续流反应器,限制DO质量浓度为0.1~0.4 mg∙L−1,利用限氧条件对NOB活性进行抑制,投加NH4+-N和NO2−-N,经过73 d运行,ANAMMOX菌活性由0.08 kg∙(kg∙d)−1(以VSS计)上升至0.34 kg∙(kg∙d)−1;NOB比耗氧速率(SOUR)由1.68 kg∙(kg∙d)−1(以VSS计)降低至0.79 kg∙(kg∙d)−1,活性显著降低,系统TN去除率由42.7%升高至88.6%,NH4+-N和NO2−-N同步去除,ΔNO3−-N/ΔTN值向理论值0.127趋近。第2阶段(过渡期)、第3阶段(好氧期)采用SBR进行,分别将DO维持在0.4~0.7 mg∙L−1和0.7~1.0 mg∙L−1,至第130天,NOB活性降低至0.57 kg∙(kg∙d)−1,TN去除负荷达到0.86 kg∙(m3∙d)−1,ΔNO3−-N/ΔTN值由抑制前的0.318降低至0.136,短程硝化基本恢复。在短程硝化恢复过程中,ANAMMOX菌优势菌属Candidatus Kuenenia的相对丰度由7.91%增长至13.12%,但NOB的主要菌属Nitrospira的相对丰度由低于0.01%增至1.03%,表明在后续长期运行过程中,依然存在短程硝化遭到破坏的风险。Abstract: In order to recover the short-cut nitrification in CANON process and enhance the activity of anaerobic ammonium oxidation (ANAMMOX) bacteria, at the first stage (inhibition period), a continuous flow reactor was used and the dissolved oxygen (DO) concentration was limited to 0.1-0.4 mg∙L−1, so as to inhibit the activity of NOB by oxygen limitation. At the same time, NH4+-N and NO2−-N was added. After 73 days of operation, the activity of ANAMMOX bacteria increased from 0.08 kg∙(kg∙d)−1 (calculated as VSS) to 0.34 kg∙(kg∙d)−1. The specific oxygen uptake rate (SOUR) of nitrite oxidizing bacteria (NOB) decreased from 1.68 kg∙(kg∙d)−1 (calculated as VSS) to 0.79 kg∙(kg∙d)−1, indicating a significant decrease in the activity of NOB. The total nitrogen (TN) removal rate of the system increased from 42.7% to 88.6%, NH4+-N and NO2−-N were removed simultaneously, and the ΔNO3−-N/ΔTN value approached to the theoretical value of 0.127. The second stage (transition period) and the third stage (aerobic period) were carried out by SBR mode, and the DO concentration maintained at 0.4~0.7 mg∙L−1 and 0.7~1.0 mg∙L−1, respectively. On the 130th day, the activity of NOB decreased to 0.57 kg∙(kg∙d)−1, and the TN removal load reached 0.86 kg∙(m3∙d)−1. Meanwhile, the ΔNO3−-N/ΔTN value decreased from 0.318 before the inhibition to 0.136, and the short-cut nitrification was basically recovered. In the process of short-cut nitrification recovery, the relative abundance of the dominant ANAMMOX bacteria, Candidatus Kuenenia, increased from 7.91% to 13.12%, while the relative abundance of the main NOB bacteria, Nitrospira, increased from less than 0.01% to 1.03%, this indicated that a risk of short-cut nitrification destruction will still exist in the following long-term operation.
-
盐酸强力霉素(DC)是一种半合成的四环素类抗生素。大量的四环素直接排泄到环境中,对生态系统和人类健康具有潜在风险。目前,已在水生环境中被普遍检测出此类抗生素[1]。由于其复杂的结构,DC不能通过常规的生物处理工艺被有效地去除。因此,通常采用许多物理和化学处理方法予以去除,例如吸附[2]、基于臭氧的高级氧化过程[3]、光催化[4]等。光催化方法由于其低成本,高效率和环境友好性等特点被广泛应用于处理印染废水[5]、抗生素废水[6]等。类石墨氮化碳(g-C3N4)因其合适的带隙,无毒,稳定性好而被认为是潜在的去除有机污染物的可见光光催化剂。但该催化剂存在对可见光的响应效率较低,光生电子和空穴的重组率较高等缺陷[7]。因此,需要寻找有效的方法去改善g-C3N4的光催化性能。其中,构建基于g-C3N4的异质结复合材料是最有效的方法之一,这可以有效地促进光诱导电荷的分离并加速光催化反应进程[8]。宋思扬等[9]通过化学浴沉淀法制备了Co掺杂的FeOOH与石墨相氮化碳复合材料(Co-FeOOH/g-C3N4),以罗丹明B(RhB)为目标污染物,在最佳反应条件下,Co-FeOOH、g-C3N4和Co-FeOOH/g-C3N4对RhB的去除率分别为23.7%、59.6%和91.5%。CeO2作为一种活性稀土金属氧化物,由于具有Ce4+和Ce3+的化合价变化而引起了广泛关注。Ce4+和Ce3+的氧化还原循环将改善光生电子和空穴对的界面电荷转移和分离速率[10]。据报道,CeO2的CB和VB分别为−0.39 eV和2.50 eV,而g-C3N4的CB和VB分别为−1.13 eV和1.57 eV[11],因此,CeO2和g-C3N4因具有良好匹配的能带结构而可以形成高效的异质结构。HUMAYUN等[12]制备了g-C3N4/CeO2,在可见光下考察了其对2,4-二氯苯酚(2,4-DCP)的降解效果,发现羟基自由基(˙OH)是降解2,4-DCP的主要活性物质。此外,基于密度泛函方法的理论算术,铈具有像Pt一样的能垒,而g-C3N4负载的铈可以产生更多的活性位点[13]。目前的研究中,CeO2/g-C3N4仅作为光催化剂降解污染物[14-16],但由于CeO2可以与H2O2产生类芬顿反应[17],因此,构建新型CeO2/g-C3N4非均相光芬顿体系,有望进一步提高对污染物的降解效率。
基于上述原因,本研究通过水热法制备了CeO2/g-C3N4,并在可见光下采用光催化-芬顿法降解盐酸强力霉素(DC)。使用扫描电子显微镜(SEM)、透射电子显微镜(TEM)、X射线粉末衍射(XRD)、X射线光电子能谱(XPS)、电阻抗能谱(EIS)、电子自旋共振(ESR)和漫反射光谱(UV-Vis)等手段对合成的CeO2/g-C3N4进行了表征;分别考察了初始pH、H2O2浓度、不同的铈掺杂量、催化剂用量和DC浓度对DC降解的影响,优化了反应条件;评价了复合光催化剂的重复使用性和稳定性;探讨了CeO2/g-C3N4光催化-芬顿体系的降解机理。
1. 材料与方法
1.1 实验试剂与仪器
试剂:六水合硝酸铈、糠醇、异丙醇、对苯醌、乙二胺四乙酸二钠、盐酸(均为分析纯,国药集团化学试剂有限公司);30%双氧水(优级纯,国药集团);尿素(分析纯,上海麦克林生化科技有限公司);5,5-二甲基-1-吡咯啉-N-氧化物(DMPO,98%,上海九鼎生物科技有限公司);盐酸强力霉素(纯度96%,上海阿拉丁生化科技有限公司);实验用水为超纯水。
仪器:KSL-1100X控温马弗炉(合肥科晶材料技术有限公司);XPA光化学反应仪(南京胥江机电厂);UV-2600紫外-可见分光光度计(日本岛津公司);Gemini 50扫描电子显微镜(德国卡尔蔡司公司);ESCALAB250Xi型光电子能谱仪(美国赛默飞世尔公司);X-Pert PRO MPD固定靶X射线衍射仪(荷兰帕纳科公司);JES-FA200电子顺磁共振波谱仪(日本捷欧路公司)。
1.2 材料制备
采用水热法[18]用于制备CeO2/g-C3N4复合催化剂。首先称取5.0 g尿素置于坩埚中,以2.5 ℃·min−1的升温速率加热至550 ℃,煅烧2 h,待冷却至室温后,将淡黄色固体取出并研磨得到粉末状的g-C3N4;将0.4 g的g-C3N4溶于50 mL去离子水中,超声波处理30 min;将一定量的Ce(NO3)3·6H2O添加到悬浮液中,搅拌30 min后将其转移到高压釜中,并在160 ℃下加热10 h,再将其冷却到室温,并用去离子水反复洗涤几次,最后在90 ℃下干燥8 h,得到浅黄色材料。对所制备的催化剂命名为X%CeO2/g-C3N4。按照与上述相同的方法,还制备了纯CeO2。
1.3 实验方法
在光催化反应仪(XPA-7)中进行了DC的降解实验。为了使反应溶液温度恒定在(18±2) °C,仪器中的光反应装置与冷却装置连接。垂直可见光源是500 W氙气灯(用滤光片过滤了波长小于420 nm的紫外线)。首先,使用浓度为0.5 mol·L−1的NaOH或H2SO4溶液调节DC溶液的初始pH。之后,将称量的0.025 g CeO2/g-C3N4放入到50 mL DC溶液中,并在黑暗中搅拌30 min达到吸附平衡,转速为500 r·min−1。最后,加入5 mmol·L−1 H2O2并打开灯反应120 min。在此过程中,每20 min取1 mL反应悬浮液样品并使用0.22 μm PES滤头进行过滤。将反应后的溶液进行离心分离,使用纯水洗涤离心3次,进行真空干燥,得到反应后的CeO2/g-C3N4。并再按照上述步骤反复4次,考察其重复性和稳定性。
1.4 分析方法
将获得的样品通过高效液相色谱(HPLC)测量DC浓度。测试DC浓度的最佳流动相是0.01 mol·L−1的乙二酸,甲醇和乙腈的混合溶液(乙二酸∶甲醇∶乙腈=65∶17∶18),检测波长为357 nm,柱温为30 ℃。
2. 结果与讨论
2.1 催化剂表征结果
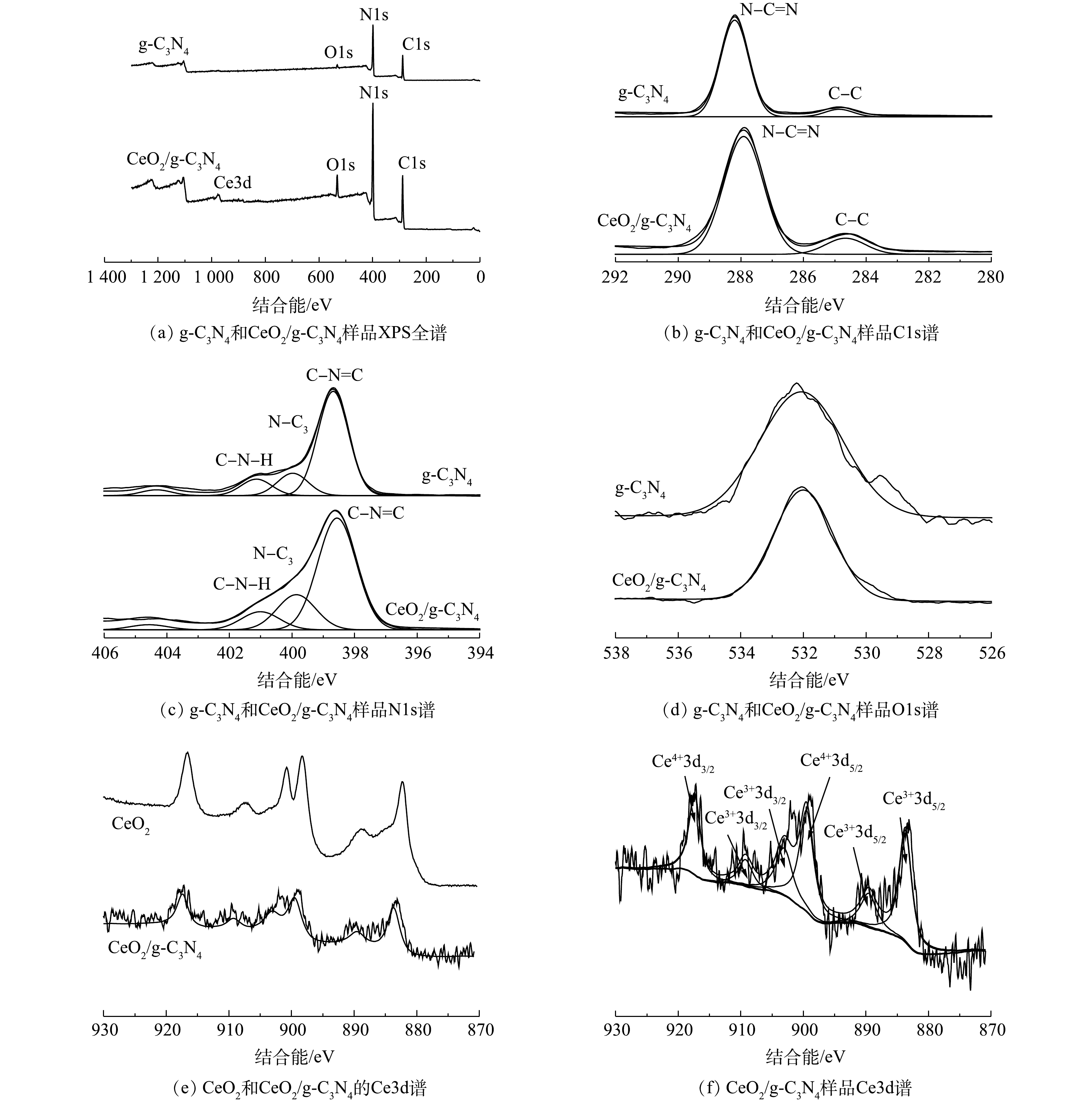
通过粉末XRD分析研究了所制备的纳米复合材料中各相的晶体结构。图1显示了X%CeO2/g-C3N4、CeO2和g-C3N4的XRD谱图。由图1可以看出,g-C3N4的谱图中显示出有2个明显的特征衍射峰,典型的衍射峰位于13.1°和27.6°,可分别归因与g-C3N4(100)和(002)晶面[19]。纯CeO2和X%CeO2/g-C3N4复合材料的XRD光谱显示出立方CeO2的典型XRD图谱(JCPDS 78-0694)[20]。在28.8°、33.3°、47.7°、56.58°和59.34°处CeO2的特征衍射峰对应(111)、(200)、(220)、(311)和(222)平面[21]。从图1中可以看出,在X%CeO2/g-C3N4复合材料中,显示了位于27.6°的特征衍射峰,这表明存在g-C3N4相。
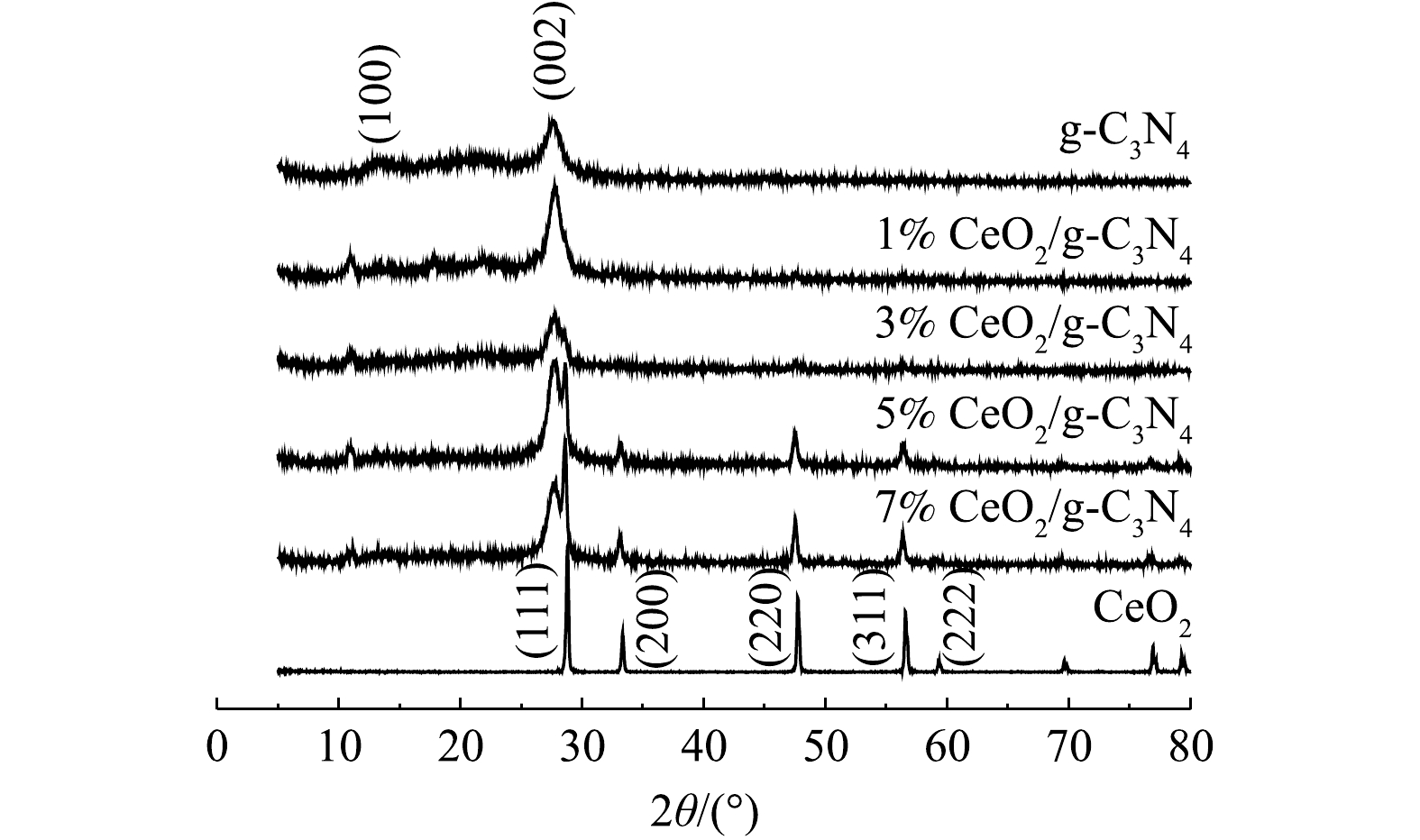 图 1 CeO2/g-C3N4,CeO2和g-C3N4的XRD图Figure 1. XRD patterns of CeO2/g-C3N4, CeO2 and g-C3N4
图 1 CeO2/g-C3N4,CeO2和g-C3N4的XRD图Figure 1. XRD patterns of CeO2/g-C3N4, CeO2 and g-C3N4为了表征CeO2/g-C3N4的形态和微观结构,进行了SEM分析。图2(a)和图2(b)显示了g-C3N4和5%CeO2/g-C3N4的SEM图像。所有的催化剂均含有介孔状结构,在图2(b)中,CeO2颗粒均匀分布在g-C3N4的表面,这表明CeO2已经成功掺杂到g-C3N4中并且不会改变其结构。
图3显示了g-C3N4、CeO2和X%CeO2/g-C3N4的FTIR光谱。由图3中可以看出,对于X%CeO2/g-C3N4复合材料,显示了g-C3N4的典型分子结构,也观察到g-C3N4的所有特征吸收峰,这证实了复合材料中有g-C3N4的骨架。1 247~1 637 cm−1的强吸收带(在1 247 cm−1和1 637 cm−1处具有特征峰)可以归因于CN杂环的典型拉伸振动[22]。3 000~3 600 cm−1处的峰应该是NH的拉伸振动吸收峰[23]。在含Ce催化剂的制备中,CeO2是通常产生的物质,且在500~700 cm−1处观察到Ce—O拉伸振动[24]。上述表征结果表明,在掺杂Ce后,g-C3N4的主要结构并未发生明显变化。此外,由于掺杂含量低和峰重叠影响,因此,看不到Ce相关基团的振动带[25]。
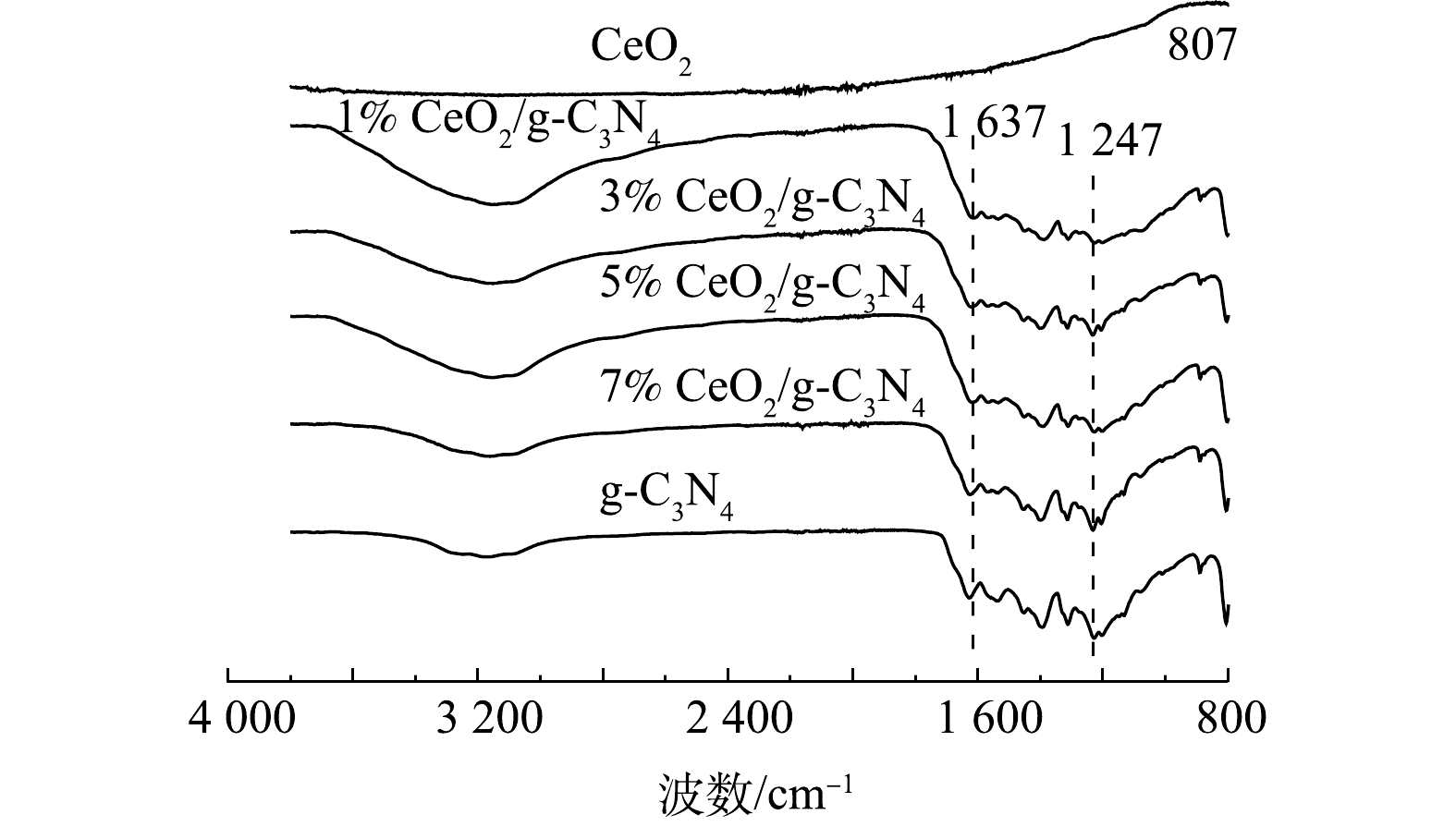 图 3 CeO2/g-C3N4,CeO2和g-C3N4的FTIR图Figure 3. FTIR spectra of CeO2/g-C3N4, CeO2 and g-C3N4
图 3 CeO2/g-C3N4,CeO2和g-C3N4的FTIR图Figure 3. FTIR spectra of CeO2/g-C3N4, CeO2 and g-C3N4图4(a)显示了g-C3N4和CeO2/g-C3N4的所有元素的全光谱。在g-C3N4的XPS光谱中发现了C、N、O的峰,在CeO2/g-C3N4的XPS光谱中发现了C、N、O和Ce的峰,这表明CeO2已成功引入g-C3N4。可以看出,C和N是主要元素。图4(b)显示了g-C3N4和CeO2/g-C3N4的Cls光谱。g-C3N4的C1s谱可分解为2个不同的高斯-洛伦兹峰,中心峰的结合能为284.88 eV和288.21 eV。284.88 eV(19.82%)处的峰可归因于表面无定形碳的C—C配位,288.21 eV(80.18%)处的峰可归因于C—N或C—(N)3[26]。CeO2/g-C3N4的C1s光谱与g-C3N4的相似,中心峰的结合能为283.6 5eV和287.78 eV。在g-C3N4的N1s光谱中(图4(c)),可以观察到3个峰:398.69 eV(76.54%)处的峰可归因于sp2轨道杂化的芳族氮(C—N=C);399.93 eV(10.46%)处的峰是由于sp3轨道杂化N—(C)3引起的; 401.14 eV处的的峰(9.28%)可归因于C—N—H组[27]。在CeO2/g-C3N4的N 1s光谱中,中心峰的结合能为398.04、399.88和401.17 eV。在532.3 eV处的O1s峰与在催化剂表面上的羟基基团或水分子的存在有关[15, 28](图4(d))。图4(e)显示了纯CeO2和所制备的CeO2/g-C3N4的Ce3d光谱,在纯CeO2的Ce3d光谱观察到6个峰,分别位于882.3、888.9、898.3、900.8、907.3和916.7 eV,而在CeO2/g-C3N4的Ce3d光谱也观察到相对应的6个峰。在CeO2/g-C3N4的Ce3d光谱中(图4(f)),结合能峰位于883.6 eV和889.5 eV,说明存在Ce3+3d5/2,结合能峰位于899.4 eV,则说明存在Ce4+3d5/2,而结合能峰位于903.1 eV和909.2 eV则说明Ce3+3d3/2的存在,结合能峰位于917.3 eV则是由于Ce4+3d3/2的存在,证明Ce以Ce(Ⅲ)和Ce(Ⅳ)态的形式存在[11, 29]。CeO2/g-C3N4峰的位置相较于纯CeO2有所偏移,这可能是由于g-C3N4与CeO2之间存在相互作用[15]。
为了研究催化剂的光吸收性能,测量了g-C3N4和不同含量CeO2/g-C3N4的UV-Vis漫反射光谱。从图5(a)中可以看出,随着引入Ce掺杂剂,CeO2/g-C3N4的吸收边缘在大约420~460 nm处出现红移。这可能是因为Ce和g-C3N4之间的共轭和电荷转移。此外,他们的吸收范围更宽更强,从而增强了可见光吸收和光催化性能。掺杂CeO2可以改善催化剂的光吸收性能,不同CeO2含量的催化剂的光吸收性能差别不大,其中5%CeO2/g-C3N4的光吸收性能略好。此外,基于UV-vis DRS数据,通过Kubelk-Munk方法(式(1))计算了CeO2、g-C3N4和CeO2/g-C3N4的带隙值。
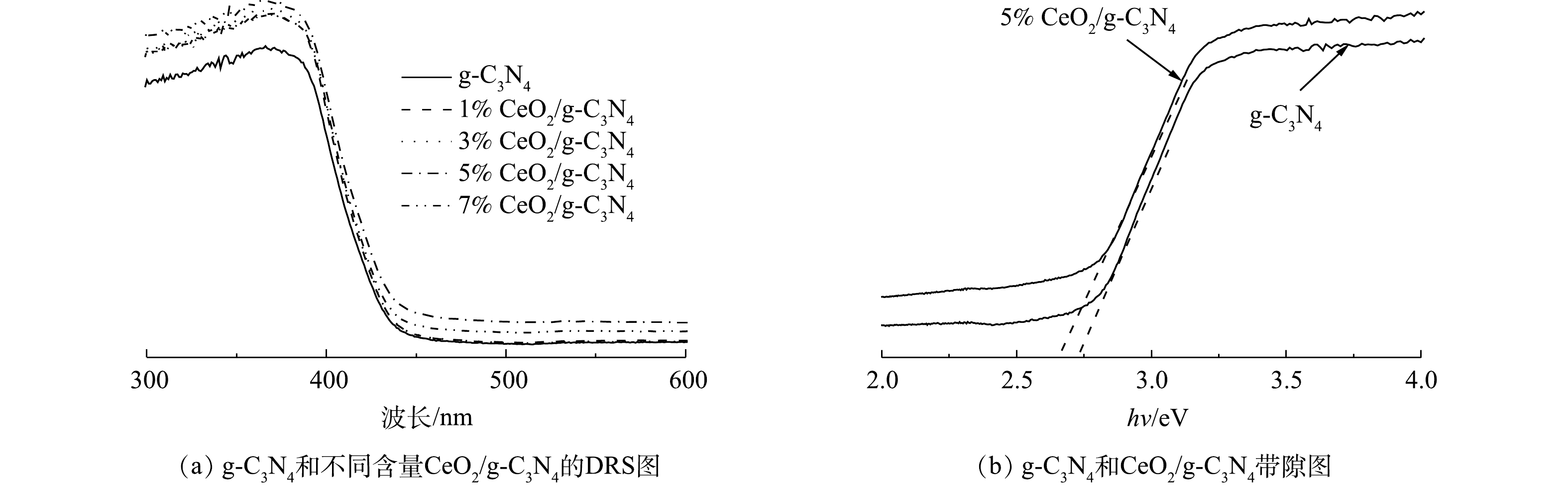 图 5 g-C3N4和CeO2/g-C3N4的DRS和带隙能谱图Figure 5. DRS and band gap spectra of g-C3N4 and CeO2/g-C3N4
图 5 g-C3N4和CeO2/g-C3N4的DRS和带隙能谱图Figure 5. DRS and band gap spectra of g-C3N4 and CeO2/g-C3N4αhv=A(hv−Eg)n/2 (1) 式中:α,hν,Eg和A分别代表吸收系数,光能,光带隙能量和常数。
n取决于半导体中的跃迁特性(直接跃迁n = 1;间接跃迁n = 4)。对于g-C3N4,n=1[30]。根据式(1)计算得出,g-C3N4和5%CeO2/g-C3N4的带隙分别为2.73 eV和2.59 eV(图5(b))。带隙变窄有利于光吸收,这意味着激发电子从价带(VB)跃迁至导带(CB)所需的能量更少,通过掺杂Ce可以增强光的吸收。由此可见,Ce掺杂对g-C3N4的作用可以扩展可见光吸收,最终导致光催化活性的提高。
为了更好地了解CeO2/g-C3N4中的光诱导电流分离行为,对其进行了电化学阻抗谱(EIS)的测量,结果如图6所示。EIS电化学阻抗谱上的电弧反映了电极/电解质界面处电荷转移层的电阻。较小的电弧半径表示较低的电阻和较高的电荷转移效率[31]。由图6(a)可以看出,CeO2/g-C3N4复合光催化剂的电弧半径小于g-C3N4,其中5%CeO2/g-C3N4催化剂的电弧半径最小,这表明,在5%CeO2/g-C3N4复合光催化剂界面处的电子-空穴对的转换和分离更有效。为了进一步评估不同催化剂的电荷分离效率,对其进行了瞬态光电流响应的测量。图6(b)显示了纯g-C3N4和CeO2/g-C3N4的光电流响应。当打开和关闭光源时,CeO2/g-C3N4产生的光电流最高,这表明与纯g-C3N4相比,CeO2/g-C3N4复合光催化剂具有更低的电子-空穴对复合率。
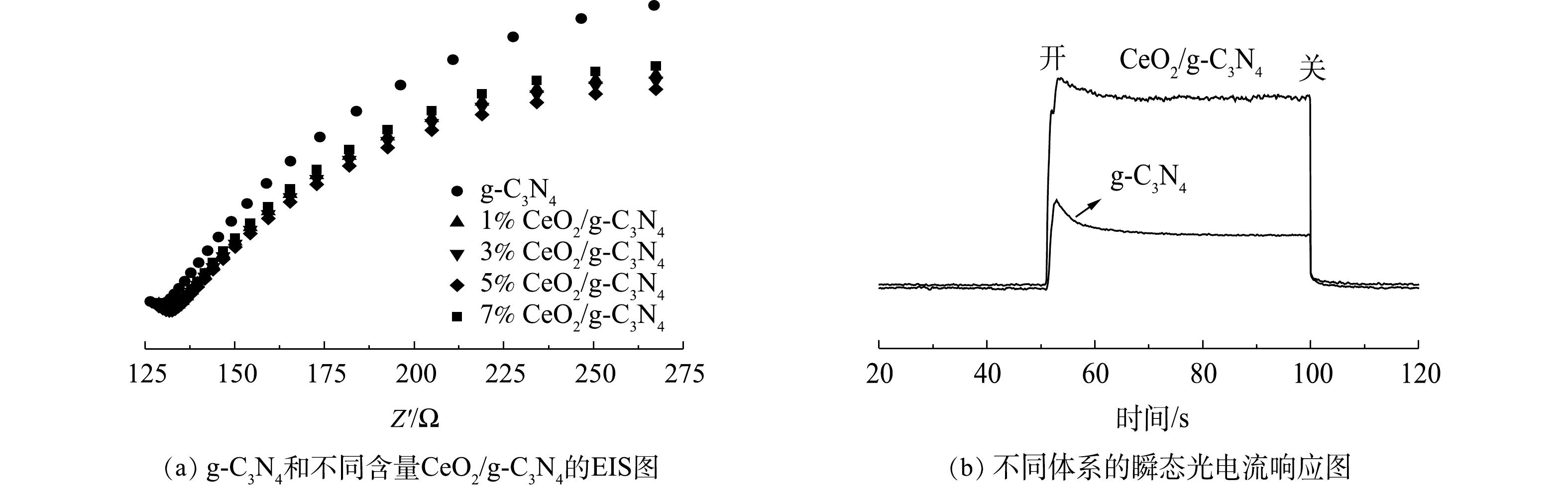 图 6 g-C3N4和CeO2/g-C3N4的EIS和瞬态光电流响应Figure 6. EIS and transient photocurrent response of g-C3N4 and CeO2/g-C3N4
图 6 g-C3N4和CeO2/g-C3N4的EIS和瞬态光电流响应Figure 6. EIS and transient photocurrent response of g-C3N4 and CeO2/g-C3N42.2 CeO2/g-C3N4催化降解盐酸强力霉素的性能
1)不同体系下DC的降解效果。为了探索CeO2/g-C3N4的光催化活性,在催化剂投加量为500 mg·L−1,初始H2O2浓度为5 mmol·L−1,初始pH为2.0,DC浓度为10 mg·L−1的条件下进行实验。图7显示了DC在不同的反应体系中的降解效果,这些体系分别是非均相芬顿体系,光催化体系和光催化-芬顿体系。为排除催化剂的吸附作用对污染物浓度变化的影响,制备的催化剂在光照实验前均进行了30 min避光搅拌。由图7可见,单独的CeO2/g-C3N4在黑暗条件下对DC的吸附去除率只有5.1%。在含有H2O2/Vis系统中,DC的去除率为11.9%,这表明,在没有催化剂的情况下,H2O2在可见光下对DC的氧化能力有限。在g-C3N4和CeO2/g-C3N4的光催化体系中DC的去除率在120 min内分别为38.1%和46.9%。在单独CeO2的和5%CeO2/g-C3N4的非均相芬顿体系中,在120 min内对DC的去除率为27.3%和31.5%。但是,在5%CeO2/g-C3N4的光芬顿体系中,DC的去除率在120 min内可达到99.1%。上述结果表明,5%CeO2/g-C3N4复合催化剂的去除率高于其他催化剂。
2)初始pH对DC降解效率的影响。如图8所示,当初始pH为2.0、3.0和5.0时,DC的去除率分别为97.3%、81.8%和73.5%。当初始pH进一步提高至中性条件时,DC降解则受到抑制。在初始pH为7.0时,DC的去除率降低至62.2%。该结果可能是由于,在酸性条件下,溶液中存在大量H+离子,促进了Ce与H2O2发生芬顿反应产生·OH。另一方面,当溶液pH偏高时,H2O2容易分解为H2O和O2[32]。
3)不同铈掺杂量对DC去除率的影响。如图9所示,当使用CeO2/g-C3N4作为催化剂时,对DC的去除率大大增加。随着铈掺杂量由1%增加到5%,DC去除率由67%提高至97%;而当铈掺杂量增加到7%时,对DC的去除率降低到58%。有研究表明,g-C3N4中金属离子的存在会引起表面缺陷的形成,一方面,可以通过增加掺杂金属的量来改善催化反应性;另一方面,过量的CeO2物种可能充当载流子的复合中心并覆盖表面上的活性位点,从而降低了光催化效率[12,33]。本实验研究结果表明,当铈掺杂量达到5%时表现出最佳的降解效果。
4) CeO2/g-C3N4投加量对DC去除效果的影响。图10显示了在120 min下,不同质量浓度催化剂(0.25、0.5、0.75和1.0 g·L−1)对DC的去除效果。随着催化剂投加量由0.25 g·L−1增加到0.5 g·L−1,DC的去除率在120 min内由70.1%增加到99.3%。这是因为,对于一定浓度的DC溶液,在一定的催化剂用量范围内,催化剂浓度的增加可以增加活性位点,从而提高DC的降解效率。但是,当催化剂质量浓度从0.5 g·L−1增加到1.0 g·L−1时,一方面过量的催化剂会使不透明性增加,光透过率降低,从而会阻碍光和活性位点在催化剂表面的渗透,导致DC去除率下降[34];另一方面,催化剂投加量过大也有可能导致产生自由基过多,造成自我淬灭[35]。
 图 10 CeO2/g-C3N4投加量对DC的降解影响Figure 10. Effects of CeO2/g-C3N4 dosage on DC degradation
图 10 CeO2/g-C3N4投加量对DC的降解影响Figure 10. Effects of CeO2/g-C3N4 dosage on DC degradation5) H2O2浓度对DC去除效果的影响。图11显示了加入不同H2O2浓度对CeO2/g-C3N4复合催化剂降解DC的影响。当H2O2浓度由1 mmol·L−1增加到5 mmol·L−1时,DC去除率在100 min内由75%提高到97.3%。该结果表明,随着H2O2浓度的增加,DC去除率有所增加。但是,当H2O2的浓度进一步增加到7 mmol·L−1时,DC的去除率会降低至86%。这是因为当H2O2浓度低于临界值时,催化反应产生的˙OH自由基的数量随H2O2浓度的加大而增加;相反,当H2O2浓度高于临界值时,生成的˙OH可能被过量的H2O2捕获而形成活性较低的˙HO2自由基[36](式(2)~(4))。大量的˙OH被消耗,˙OH无法与DC有效反应,导致去除率降低和H2O2浪费。
H2O2+⋅OH→⋅HO2+H2O (2) ⋅HO2+⋅OH→H2O+O2 (3) ⋅OH+⋅OH→H2O2 (4) 6) DC初始浓度对其去除效果的影响。图12反映了DC的初始浓度对其降解效率的影响。可以看出,DC的去除率与其初始浓度成反比。当DC的初始浓度为10 mg·L−1时,可以在120 min内完全降解;但是,当DC的初始质量浓度增加到20 mg·L−1和30 mg·L−1时,反应120 min后的DC去除率为70.6%和56.5%。这是因为过量的DC分子将占据催化剂表面部位并阻止其与H2O2接触,从而无法生成足够的羟基自由基来降解DC[37]。
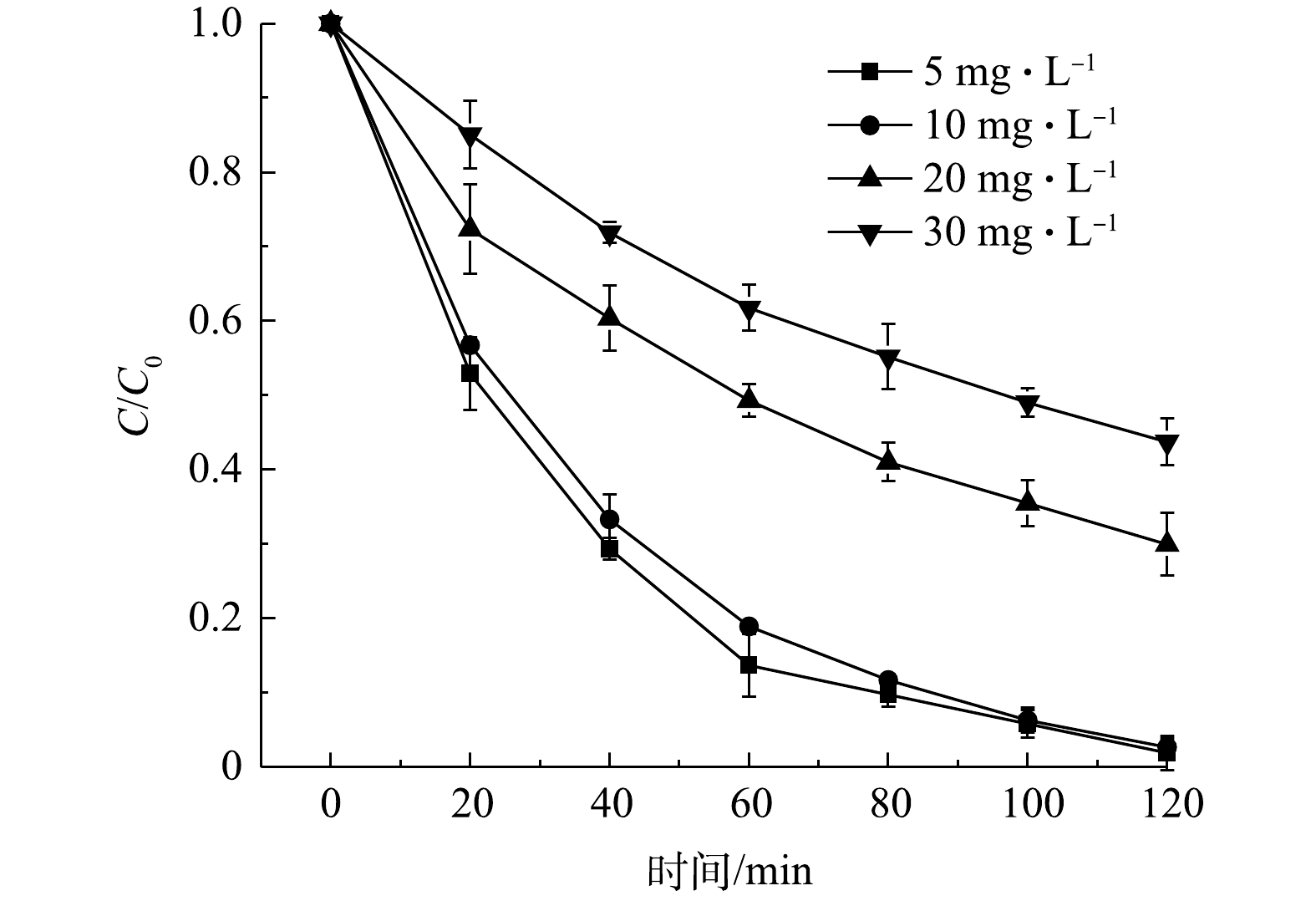 图 12 DC初始浓度对DC的降解影响Figure 12. Effects of different DC concentration on DC degradation
图 12 DC初始浓度对DC的降解影响Figure 12. Effects of different DC concentration on DC degradation2.3 CeO2/g-C3N4催化剂的重复性与稳定性
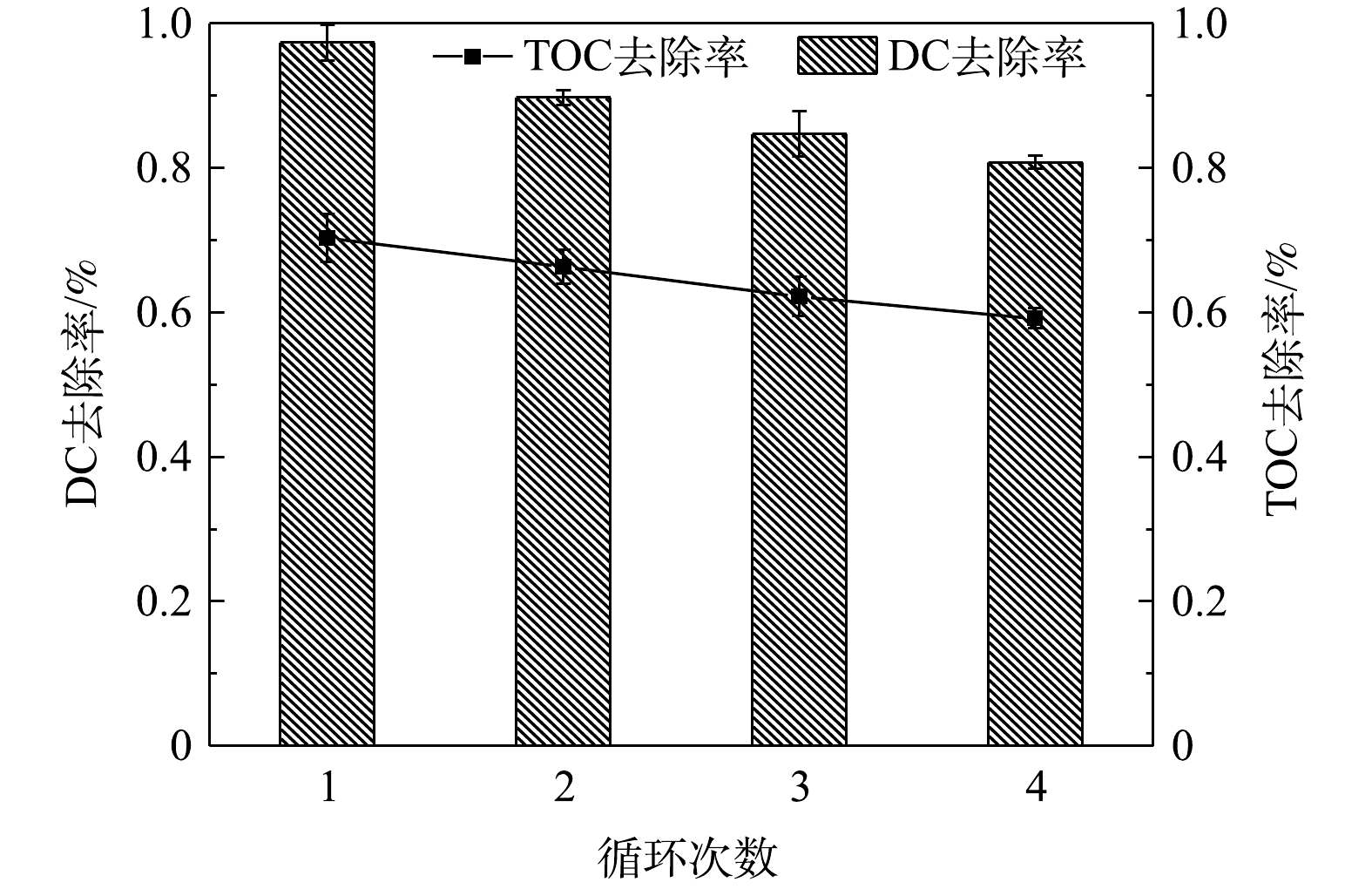
催化剂的重复性与稳定性是技术实际应用中的重要因素。为了评估CeO2/g-C3N4的化学稳定性和重复使用性,在可见光下对光催化剂进行了4次连续的重复实验。在每个反应之后,通过静置分离,然后用纯水反复洗涤并真空干燥,干燥后的样品研磨收集以便用于后续降解实验。实验结果如图13所示。经过4次循环,DC的去除率从97.8%降低到81.5%,仅下降16.3%。TOC的去除率从69.2%降低到58.5%,仅下降为10.7%。此外,在循环过程中光催化剂的量略有减少。上述结果表明,CeO2/g-C3N4具有较高的重复性和稳定性。
2.4 CeO2/g-C3N4催化H2O2降解DC机理
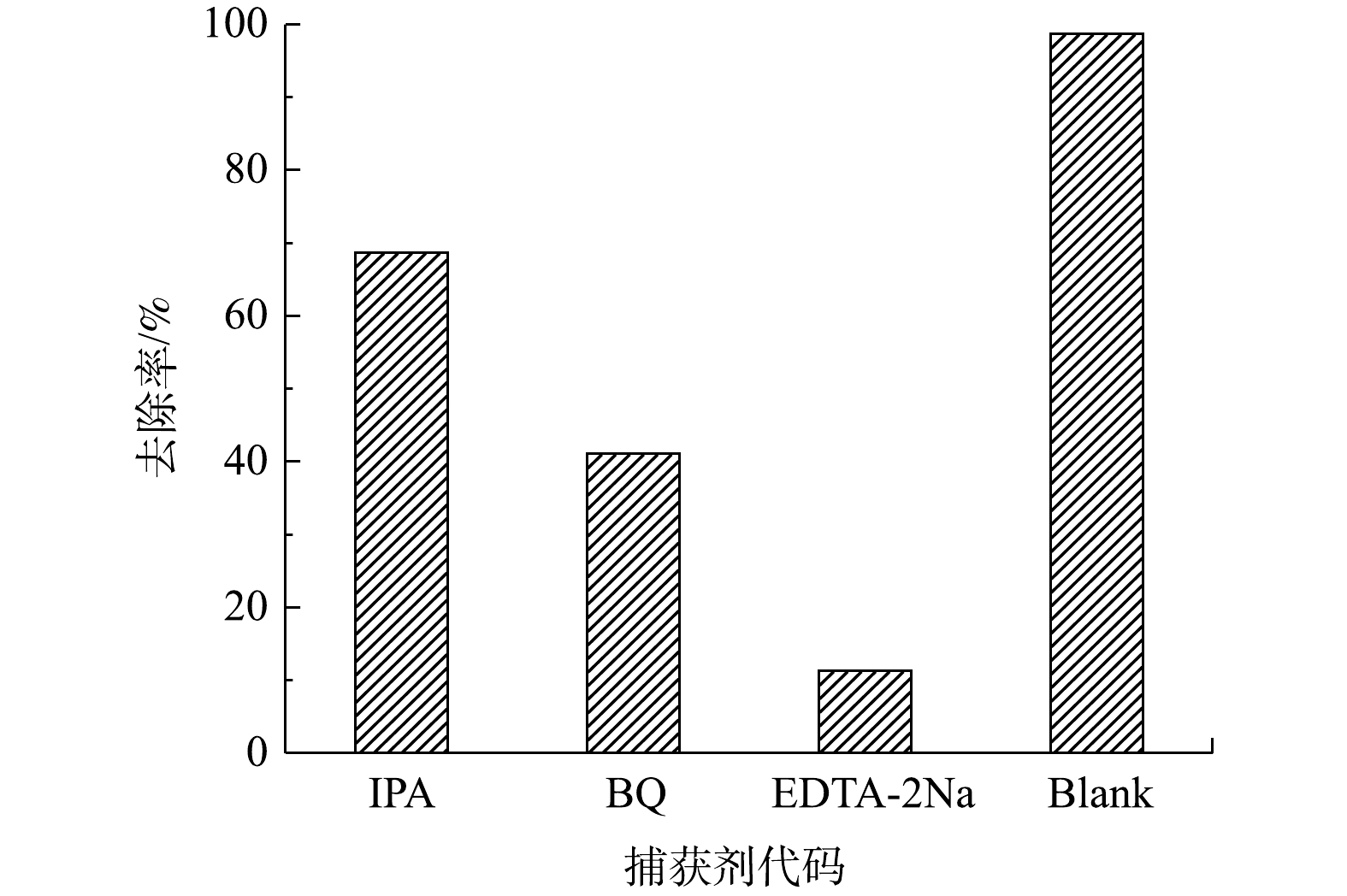
为了确定CeO2/g-C3N4/H2O2体系降解DC反应中主要的自由基种类,进行了自由基淬灭实验。用于实验的淬灭剂为异丙醇(˙OH)、对苯醌(
⋅O−2 ⋅O−2 ⋅O−2 ⋅O−2 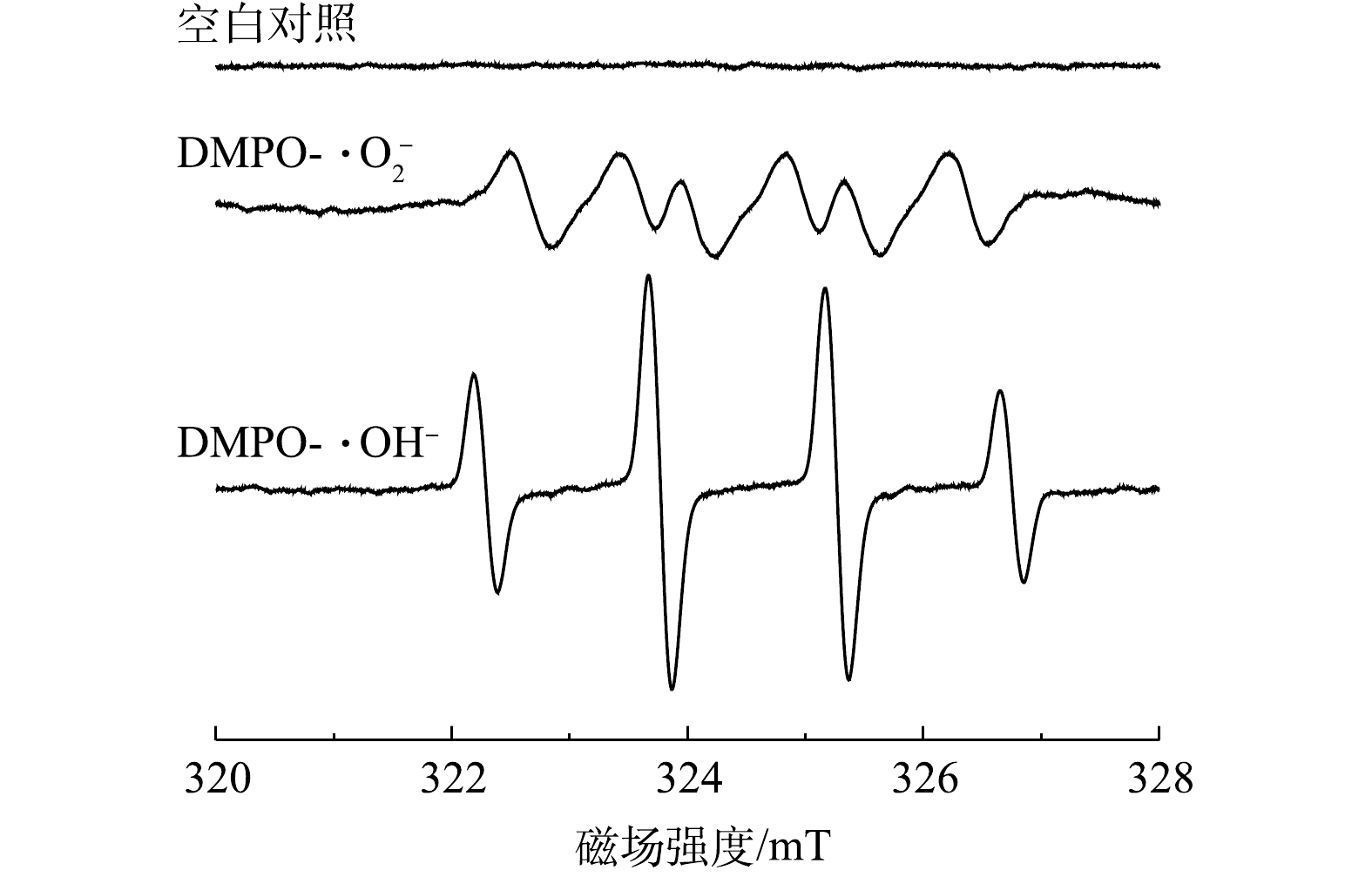 图 15 CeO2/g-C3N4体系中通过DMPO捕获˙OH和
图 15 CeO2/g-C3N4体系中通过DMPO捕获˙OH和⋅O−2 Figure 15. ESR spectra of ˙OH and⋅O−2  图 16 CeO2/g-C3N4反应前后的Ce3d谱图Figure 16. Ce3d spectras before and after the CeO2/g-C3N4 reaction
图 16 CeO2/g-C3N4反应前后的Ce3d谱图Figure 16. Ce3d spectras before and after the CeO2/g-C3N4 reaction综合以上信息可以推测,CeO2/g-C3N4催化H2O2降解DC的催化机理为:首先,g-C3N4在见光照下产生光生电子和空穴h+。g-C3N4和CeO2的CB分别为−1.09 eV和−0.79 eV,VB分别为1.61 eV和2.03 eV[39]。因为g-C3N4和CeO2的CB比E0 (O2/
⋅O−2 ⋅O−2 ⋅O−2 g-C3N4+hv→g-C3N4(h++e−) (5) O2+e−→⋅O−2 (6) Ce4++e−→Ce3+ (7) Ce3++H2O2→Ce4++⋅OH+OH− (8) Ce4++H2O2→Ce3++⋅OOH+H+ (9) h++H2O2→⋅O−2+2h+ (10) ⋅O−2+h++⋅OH+DC→小分子/离子 (11) 3. 结论
1)在pH为2.0、H2O2为5 mmol·L−1、催化剂投加量为0.5 g·L−1的最佳条件下,5%CeO2/g-C3N4可有效去除10 mg·L−1的DC,DC去除率可达到99.1%。
2)在可见光和H2O2同时存在下催化降解DC,CeO2/g-C3N4的光催化活性比纯g-C3N4的光催化活性有明显提高,其中5%CeO2/g-C3N4显示最优的催化活性,反应速率是g-C3N4的2.6倍,分别比单独的光催化体系和非均相芬顿体系的去除率提高了61%和72%。上述结果说明,光催化技术和非均相芬顿技术之间存在协同效应。
3) CeO2/g-C3N4降解DC可能的反应机理为:光催化促进了类芬顿反应中Ce4+和Ce3+的循环,也提高光生电子-空穴分离效率。循环实验结果表明,CeO2/g-C3N4具有很好的重复利用性。
-
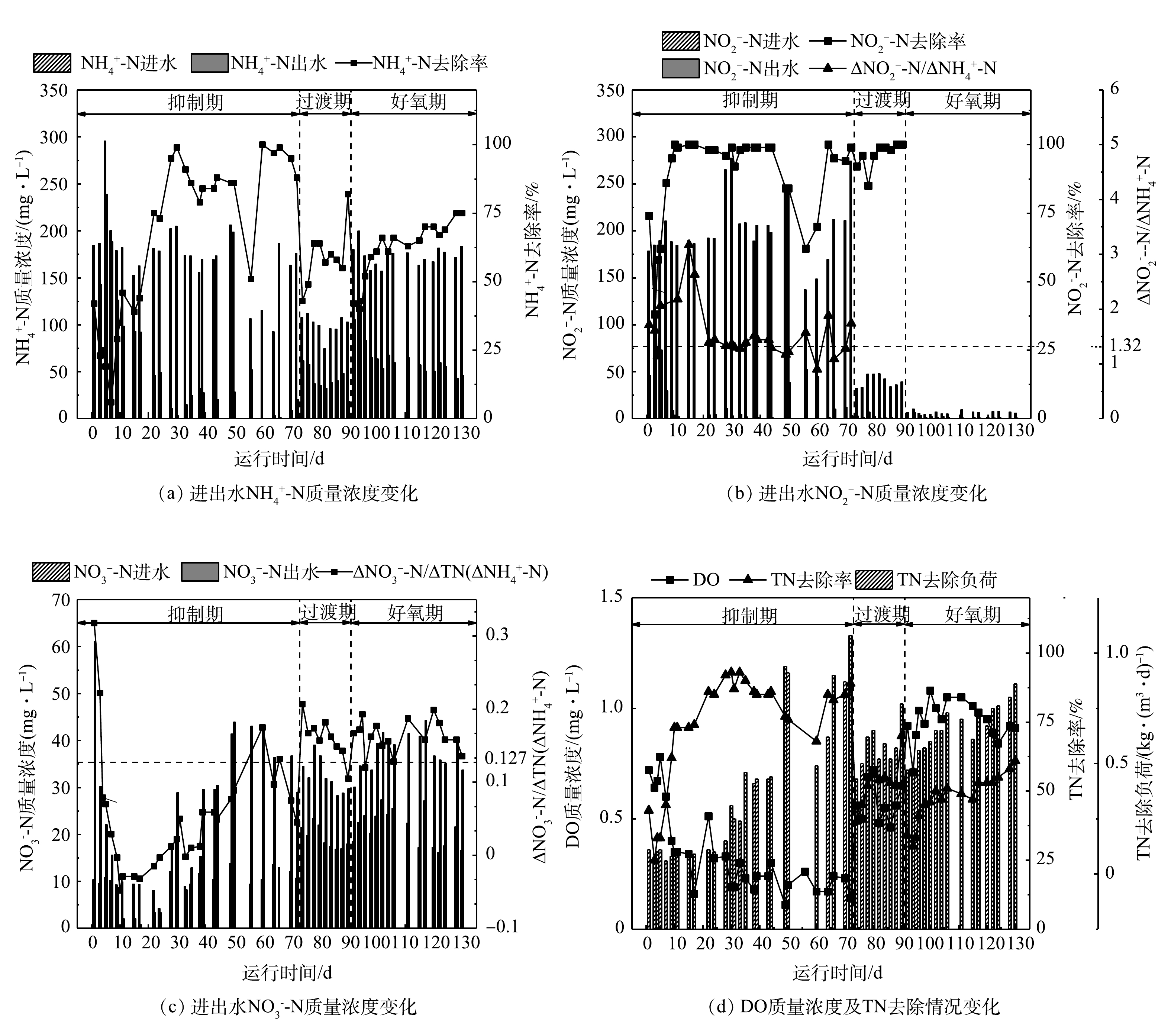
图 2 反应器连续运行期间各氮素质量浓度变化
Figure 2. Variation of nitrogen concentration during continuous operation of the reactor
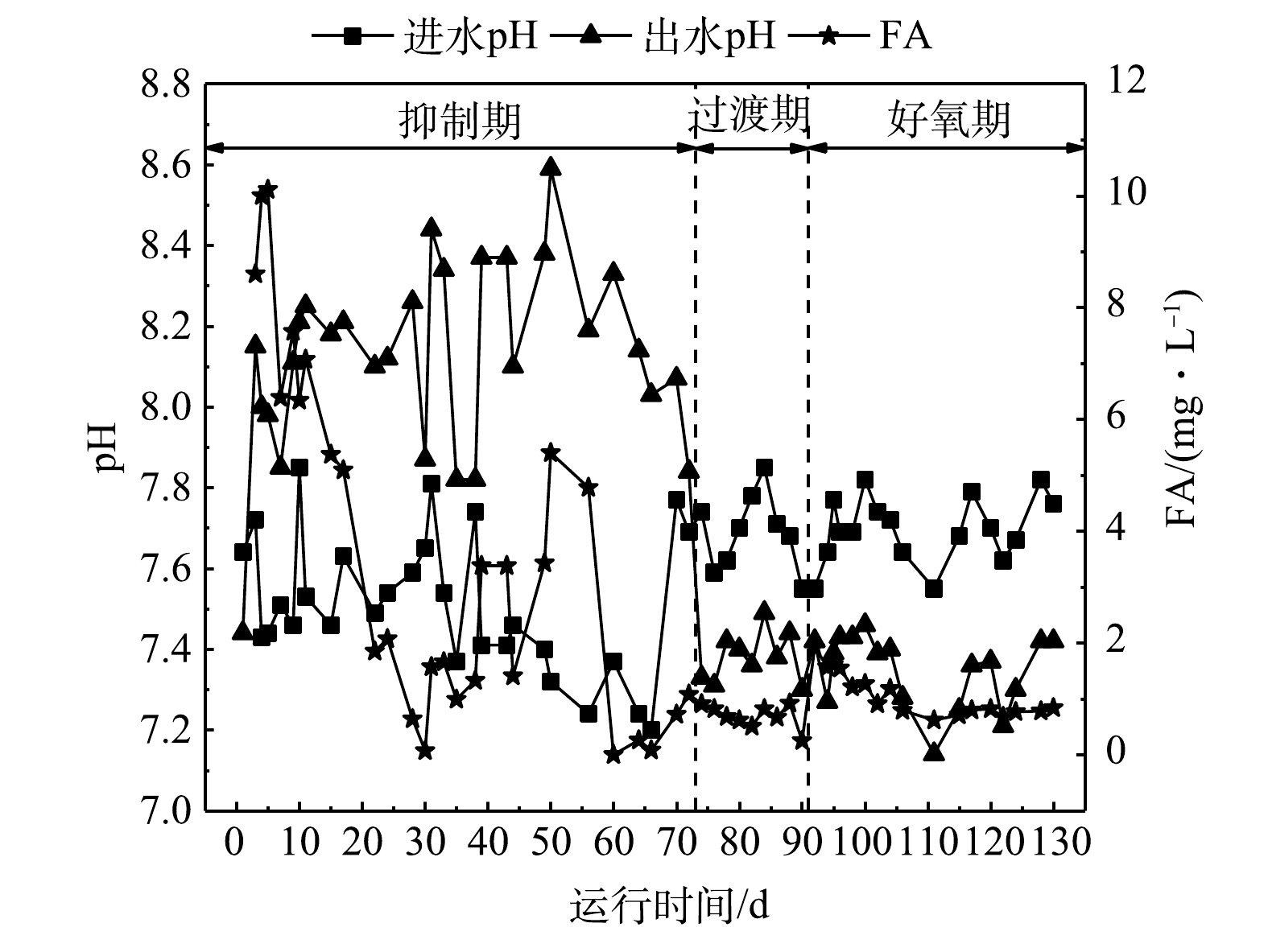
图 3 反应器连续运行期间pH和FA质量浓度变化
Figure 3. Variation of pH and FA concentrations during continuous operation of the reactor

图 4 反应器连续运行过程中ANAMMOX菌及NOB活性变化
Figure 4. Variation of ANAMMOX bacteria and NOB activity during continuous operation of the reactor
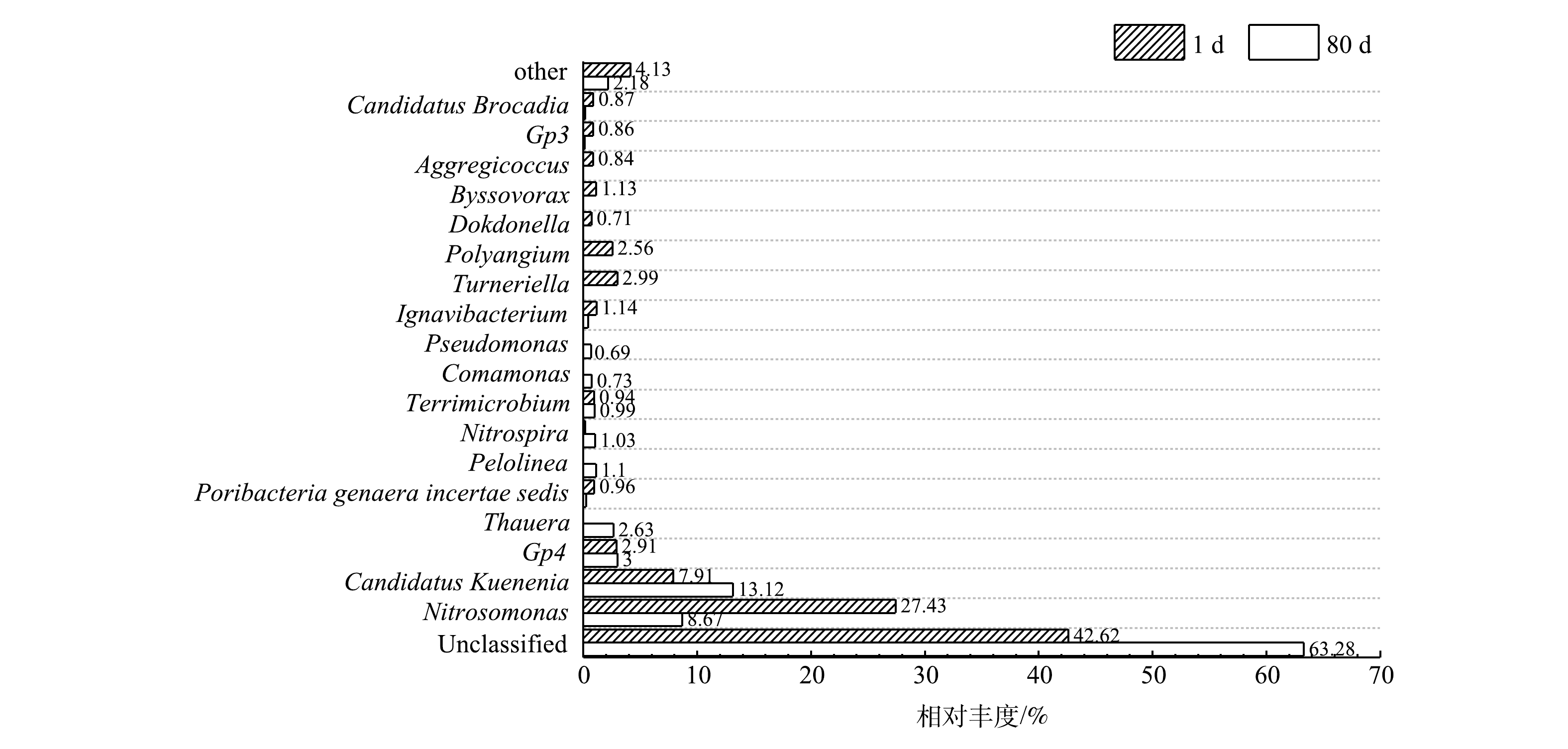
图 5 反应器中主要菌属相对丰度变化
Figure 5. Microbial community structure of sludge on the 80th day of operation
表 1 反应器连续运行工况
Table 1. Continuous operating conditions of oxygen-limited aeration reactor
反应器 阶段 运行时间/d 进水流量/(L∙h−1) NH4+-N/(mg∙L−1) NO2−-N/(mg∙L−1) NO3−-N/(mg∙L−1) PO43-−P/(mg∙L−1) 温度/℃ pH 曝气量/(mL∙min−1) DO/(mg∙L−1) R1 抑制期 1~73 0.22 150 200 0~10 10 20±5 7.3~7.5 0 0.1~0.4 R2 过渡期 74~91 — 125 50 0~10 10 25 7.8~8.0 100 0.4~0.7 好氧期 92~130 — 200 0 0~10 10 25 7.5~8.0 200 0.7~1.0  下载: 导出CSV
下载: 导出CSV
-
[1] 张铃敏, 常青龙, 史勤, 等. CANON工艺短程硝化恢复调控及微生物种群结构变化[J]. 中国环境科学, 2019, 39(6): 2354-2360. doi: 10.3969/j.issn.1000-6923.2019.06.015 [2] ZUO L S, YAO H, LI H Y, et al. Modeling of completely autotrophic nitrogen removal process with salt and glycine betaine addition[J]. Chemosphere, 2020, 264(2): 1-8. [3] STROUS M, KUENEN G J, JETTEN M S M. Key physiology of anaerobic ammonium oxidation[J]. Applied and Environmental Microbiology, 1999, 65(7): 3248-3250. doi: 10.1128/AEM.65.7.3248-3250.1999 [4] 聂铭, 李振轮. 水体中亚硝酸盐积累的生物过程及影响因素研究进展[J]. 生物工程学报, 2020, 36(8): 1493-1503. [5] 张姚, 韩海成, 王伟刚, 等. 溶解氧对CANON颗粒污泥自养脱氮性能的影响[J]. 中国环境科学, 2017, 37(12): 4501-4510. doi: 10.3969/j.issn.1000-6923.2017.12.012 [6] HAN M, VLAEMINCK S E, AL-OMARI A, et al. Uncoupling the solids retention times of flocs and granules in mainstream deammonification: A screen as effective out-selection tool for nitrite oxidizing bacteria[J]. Bioresource Technology, 2016, 221: 195-204. doi: 10.1016/j.biortech.2016.08.115 [7] 李冬, 崔少明, 梁瑜海, 等. 溶解氧对序批式全程自养脱氮工艺运行的影响[J]. 中国环境科学, 2014, 34(5): 1131-1138. [8] 王会芳, 付昆明, 左早荣, 等. 水力停留时间和溶解氧对陶粒CANON反应器的影响[J]. 环境科学. 2015(11): 4161-4167. [9] JOSS A, DERLON N, CYPRIEN C, et al. Combined nitritation-anammox: Advances in understanding process stability[J]. Environmental Science & Technology, 2011, 45(22): 9735. [10] GRAAF A V D, BRUIJN P D, ROBERTSON L A, et al. Autotrophic growth of anaerobic ammonium-oxidizing micro-organisms in a fluidized bed reactor[J]. Microbiology. 1996, 142(8): 2187-2196. [11] 付昆明, 周厚田, 苏雪莹, 等. 生物膜短程硝化系统的恢复及其转化CANON工艺的过程[J]. 环境科学, 2017, 38(4): 1536-1543. [12] 刘成良, 李天煜, 刘可慧, 等. 氨氮废水的厌氧氨氧化生物脱氮研究[J]. 生态环境学报, 2010, 19(9): 2172-2176. doi: 10.3969/j.issn.1674-5906.2010.09.026 [13] ZEKKER I, RIKMANN E, TENNO T, et al. Modification of nitrifying biofilm into nitritating one by combination of increased free ammonia concentrations, lowered HRT and dissolved oxygen concentration[J]. Journal of Environmental Sciences, 2011, 23(7): 1113-1121. doi: 10.1016/S1001-0742(10)60523-2 [14] 赵文钊, 裴浩, 王超, 等. 校园污水脱氮除磷处理系统的运行策略及其对污泥性能的影响[J]. 环境工程学报, 2020, 14(11): 3053-3062. doi: 10.12030/j.cjee.201912128 [15] 李炳荣, 曹特特, 王林, 等. 低氧条件下A2/O工艺对城市污水脱氮处理的中试研究[J]. 中国环境科学, 2019, 39(1): 134-140. doi: 10.3969/j.issn.1000-6923.2019.01.014 [16] JETTEN M S M, NIFTRIK L V, STROUS M, et al. Biochemistry and molecular biology of anammox bacteria[J]. Critical Reviews in Biochemistry and Molecular Biology. 2009, 44(2/3): 65-84. [17] 毛文龙, 杨瑞丽, 王晓君, 等. 由anammox转为CANON工艺的调控策略及微生物响应特性[J]. 环境工程学报, 2021, 15(7): 2488-2501. doi: 10.12030/j.cjee.202103122 [18] 冯平, 周少奇. 常温下厌氧氨氧化生物膜反应器的启动研究[J]. 环境科学与技术, 2010, 33(6): 19-22,34. [19] VAN DER STAR W R L, MICLEA A I, VAN DONGEN U G J M, et al. The membrane bioreactor: a novel tool to grow anammox bacteria as free cells[J]. Biotechnology and Bioengineering, 2008, 101(2): 286-294. doi: 10.1002/bit.21891 [20] LU L, WANG B J, ZHANG Y, et al. Identification and nitrogen removal characteristics of Thauera sp. FDN-01 and application in sequencing batch biofilm reactor[J]. Science of the Total Environment, 2019, 690: 61-69. doi: 10.1016/j.scitotenv.2019.06.453 [21] 陈均利, 彭英湘, 刘锋, 等. 异养硝化-好氧反硝化菌脱氮特性研究进展[J]. 环境科学与技术, 2020, 43(5): 41-48. [22] SU J F, YANG S, HUANG T L, LI M, et al. Enhancement of the denitrification in low C/N condition and its mechanism by a novel isolated Comamonas sp. YSF15[J]. Environmental pollution, 2020, 256(1): 1-7. [23] 付昆明, 付巢, 李慧, 等. 主流厌氧氨氧化工艺的运行优化及其微生物的群落变迁[J]. 环境科学, 2018, 39(12): 5596-5604. [24] 付昆明, 杨宗玥, 廖敏辉, 等. 中试MBBR反应器启动CANON工艺及其短程硝化[J]. 环境科学. 2020, 41(3): 1393-1400. [25] HANAKI K, WANTAWIN C, OHAGKI S. Nitrification at low-levels of dissolved oxygen with and without organic loading in a suspended growth reactor[J]. Water Research, 1990, 24(3): 297-302. doi: 10.1016/0043-1354(90)90004-P [26] 杨庆, 杨玉兵, 杨忠启, 等. 溶解氧对短程硝化稳定性及功能菌群的影响[J]. 中国环境科学, 2018, 38(9): 3328-3334. doi: 10.3969/j.issn.1000-6923.2018.09.016 [27] USHIKI N, JINNO M, FUJITANI H, et al. Nitrite oxidation kinetics of two Nitrospira strains: The quest for competition and ecological niche differentiation[J]. Journal of Bioscience and Bioengineering. 2017, 123(5): 581-589. [28] 张亮, 张树军, 彭永臻. 污水处理中游离氨对硝化作用抑制影响研究[J]. 哈尔滨工业大学学报, 2012, 44(2): 75-79. doi: 10.11918/j.issn.0367-6234.2012.02.016 [29] CAO Y S, VAN LOOSDRECHT MARK C M, DAIGGER G T. Mainstream partial nitritation-anammox in municipal wastewater treatment: Status, bottlenecks, and further studies[J]. Applied Microbiology and Biotechnology, 2017, 101(4): 1365-1383. doi: 10.1007/s00253-016-8058-7 [30] ANTHONISEN A C, LOEHR R C, PRAKASAM T B, et al. Inhibition of nitrification by ammonia and nitrous acid[J]. Journal Water Pollution Control Federation, 1976, 48(5): 835-852. [31] LOTTI T, KLEEREBEZEM R, ABELLEIRA-PEREIRA J M, et al. Faster through training: The anammox case[J]. Water Research, 2015, 81(Sep.15): 261-268. [32] 孙延芳, 韩晓宇, 张树军, 等. 颗粒+絮体污泥CANON工艺的启动与SRT影响研究[J]. 环境科学, 2017, 38(2): 672-678. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 4171
- HTML全文浏览数: 4171
- PDF下载数: 116
- 施引文献: 0


