


















 下载:
下载:






-
药物和个人护理用品(pharmaceuticals and personal care products, PPCPs)是一类新兴的环境污染物,正在广泛甚至越来越多地用于人类和兽医学。由于在人和动物体内的代谢不完全,部分PPCPs可能随着排泄物排出,最终进入废水处理厂,而传统的水处理工艺难以将该类物质彻底去除,大量残留的污染物可以逃逸到环境并且扩散到水体中,其检出浓度为ng·L−1级至μg·L−1级,且在水体中呈现出伪持久性[1]。PPCPs在环境中存在时对人类和水生物种的健康和安全已构成潜在威胁。其中,普里米酮(primidone, PRM) 被广泛用作抗癫痫药物,是一种典型的难降解PPCP,导致其在废水、地表水以及地下水中普遍检出。由于PRM在水环境中广泛存在且对人体健康具有潜在危害,故需要开发有效的深度处理工艺将其去除。
紫外/氯高级氧化技术因具有反应速率快、二次污染少、可以同时产生非选择性的羟基自由基(·OH)和选择性的活性氯物种(reactive chlorine species, RCS, 如Cl·、 ClO·和Cl2−·等)等优势受到了国内外学者的青睐[2]。当水体中的自由氯(free chlorine, FC)暴露于紫外线下时,除了形成·OH和Cl·(E0=2.47 V)[3]初级自由基外,他们还可以进一步与HOCl或OCl−反应生成次级自由基ClO·和Cl2−·(E0=2.20 V)[4]。与·OH相比,RCS受溶液的影响相对较小,且由于其选择性更强,在有机污染物去除时可优先与含有富电子基团有机化合物如酚类、苯胺类、烯烃和胺类反应[5],对PPCPs的去除表现出强大优势;且FC在UV254 nm处的量子产率和摩尔吸收系数均高于H2O2和过硫酸盐[6-7]。因此,紫外线与氯的结合可以产生协同效应,既能降低氧化剂投加量,又能提高污染物的降解率,被认为是一种极具潜力的高级氧化工艺。
因此,本研究以PRM为目标污染物,研究了紫外/氯体系下PRM的降解效果,系统考察了溶液pH、水体中常见阴离子、出水有机物(EfOM)和Fe3+等反应条件对PRM去除效果的影响,并探讨了PRM在紫外/氯体系中的降解机理和降解路径,以期为PPCPs废水的治理提供参考。
-
1)实验试剂。实验中所用药品及溶剂均为分析纯及以上级别。普里米酮(C12H14N2O2,218.25 Da,东京化学工业有限公司)、次氯酸钠(Alfa Aesar)、甲醇、甲酸、叔丁醇(Sigma试剂)、磷酸二氢钾、三水磷酸氢二钾、氢氧化钠、氯化铁、硝酸钠、碳酸氢钠、异丙醇、硝基苯(NB)、碘化钾、碘酸钾、柠檬酸钠、草酸钠、甲酸钠、乙酸钠、丁二酸(国药集团化学试剂有限公司)、硫代硫酸钠(天津博迪化学试剂公司)、磷酸(四川西陇化工有限公司)、氯化钠(天津福晨化学试剂有限公司)。实验中溶液均由英国Pall公司的Cascada(TM)BIO净水系统制备的超纯水配置。
2) EfOM的提取。实验所用污水取自西安市第三污水处理厂二级出水,水样通过0.45 μm滤膜后储存于4 ℃冰箱备用。将所收集的二级出水用盐酸酸化至pH为2左右后依次通过XAD-8®和XAD-4®树脂吸附富集,用pH=2的甲酸去盐后,用75%的乙腈洗脱吸附有机物,将洗脱液用旋转蒸发仪浓缩后,用冷冻干燥仪去除其中水分获得疏水性有机物 (hydrophobic acids, HPO)和两亲性有机物(transphilic acids, TPI)2种EfOM组分[8]。
-
本实验是在紫外反应箱中进行的,箱体顶部配备了6个8 W的低压汞灯灯管,向下输出光波长254 nm的紫外光谱,反应溶液置于底部中央,其中光源与溶液之间的高度为28 cm,采用碘/碘酸盐化学辐射测定法[9]测得进入反应溶液的平均紫外线通量为1.04 mW·cm−2。反应箱中配有排风扇和磁力搅拌系统,为实验提供稳定均一的反应体系。实验开始前,提前约30 min打开紫外灯,保证反应过程中光源处于稳定状态。作为基础条件,将含有5 μmol·L−1 PRM和70 μmol·L−1 (5 mg·L−1)自由氯的100 mL反应液倒入直径9.5 cm的结晶皿中,置于紫外照射下开始计时。在反应1、2、3、4、5、8、10 min时取1 mL样品置于提前加入淬灭剂Na2S2O3的色谱瓶中(淬灭剂/氧化剂摩尔浓度=2∶1)且于4 h内进行检测。单因素实验则根据预先设计向体系中加入不同浓度的常规阴离子、Fe3+、EfOM等。除特殊说明外,反应溶液pH均为7(5 mmol·L−1磷酸盐缓冲液)。为确保数据准确,所有实验至少重复2次。
-
PRM和NB的浓度采用戴安U3000高效液相色谱仪(HPLC)测定。具体条件如下:分离柱为Pinnacle II C18色谱柱(250 mm×4.6 mm, Pickering公司,英国);流动相为甲醇和1‰磷酸(体积比为50%∶50%),流速为0.8 mL·min−1;柱温为30 ℃;进样体积为20 μL;PRM和NB的检测波长分别为218 nm和270 nm。采用N, N-二乙基对苯二胺分光光度法对体系中余氯浓度进行测定[10]。
自由基的定性分析:通过向反应体系中加入不同浓度的自由基清除剂叔丁醇和异丙醇,根据2种清除剂对不同自由基的反应速率差异,结合PRM的降解速率,得出体系内主要自由基。定量采用NB作为探针,通过式(1)~式(3)计算得出体系中·OH的稳态浓度以及·OH与RCS对PRM在紫外/氯体系中的降解贡献。
式中:k[NB/·OH]为NB与·OH的二级动力学常数,在本研究中为3.9×109 L·(mol·s)−1;k[PRM/·OH] 为PRM与·OH的二级动力学常数,在本研究中为5.71×109 L·(mol·s)−1;c·OH指羟基自由基稳态浓度,mol·L−1;k'NB和k'RPM分别指NB、PRM的准一级动力学常数,min−1;k'·OH、k'RCS、k'UV和k'chlorine分别指·OH、RCS、UV和氯体系降解PRM的一级动力学常数,min−1。
实验中,阴阳离子均采用戴安ICS-1500离子色谱仪进行定量分析。阴离子测定条件:保护柱为Dionex IonPac AG19 (50 mm×4 mm Guard),分析柱为AS19 (250 mm×4 mm, Analytic),抑制器为ASRS-4 mm,以KOH溶液作为流动相,流速设定为1 mL·min−1,进样体积为25 μL。阳离子检测条件:采用Dionex Ion Pac CG12A (50 mm×4 mm Guard)作为保护柱,分析柱为CS12A (250 mm×4 mm, Analytic),使用CSRS-4 mm 阳离子抑制器,DS-5电导检测器。总有机碳(TOC)的测定采用680 ℃催化燃烧氧化-非色散红外吸收法,分析仪器为 TOC-LCPH 测定仪(德国元素Elementar公司)。小分子有机酸的检测使用戴安U3000 HPLC。分离柱为XBridge C18(Waters)色谱柱(250 mm×4.6 mm, 5 μm);流动相为磷酸溶液(pH=2.2),流速0.8 mL·min−1;柱温为30 ℃;进样体积为20 μL;检测波长为210 nm。
通过高分辨率LC-MS和GC-MS鉴定了紫外/氯工艺中PRM降解的芳香性中间产物。采用固相萃取法(solid phase extraction, SPE)对样品进行富集。对于 LC-MS,使用Orbitrap Q-Exactive (Thermoscientific,美国)配备加热电喷雾电离源(检测质量80~1 200 m/z,正离子模式)。在Acquity UPLC BEH C18 柱(1.7 µm×2.1×100 mm, Waters Corporation)上通过Ultra HPLC(Accela 1250, Thermo Scientific)分离化合物,柱温为30 ℃,进样量为10 µL,流动相由超纯水和甲醇组成(均用 0.1% 的甲酸酸化),流速为 0.4 mL·min−1。采用Compound Discover 3.1 软件(Thermo Scientific, 美国)进行质谱解析。对于 GC-MS,采用由GC-7890A 和MS-Accurate-mass QTOF 7200 组成的高分辨率GC-QTOF (安捷伦),在HP5MS (30 m×250 μm×0.25 μm)色谱柱上进行分离(进样量为1 µL)。
-
1)·OH和RCS对PRM降解的贡献。结果表明,单独氯化和单独光照10 min后,
PRM几乎没有降解,而紫外光照与氯的联合作用可以使PRM得到良好的去除(去除率84%),且该反应遵循准一级动力学模型。本实验采用1 μmol·L−1 NB作为探针,在FC为70 μmol·L−1、PRM 为5 μmol·L−1、pH=7时,PRM和NB在紫外/氯体系内的降解情况见图1(a)。图1(b)为PRM降解过程中UV、FC、·OH和RCS的作用贡献。氯化对污染物降解的作用微乎其微,单独光照的贡献较小,而·OH和RCS对PRM降解的贡献分别高达26.2%和70.5%,RCS对PRM一级降解速率的贡献(k'RCS)约为羟基自由基对其贡献(k'·OH)的2.7倍。可见,在中性条件下,紫外/氯体系中RCS对PRM的降解其主导作用,其次为·OH。
2)不同物种在PRM降解体系中的作用。为进一步确认参与PRM在紫外/氯体系降解过程的活性物种,分别在反应中加入不同浓度自由基淬灭剂叔丁醇(TBA)和异丙醇(IPA)。其中,TBA可以快速和·OH、Cl·反应(k[TBA/·OH]=6×108 L·(mol·s)−1、k[TBA/Cl·]=3×108 L·(mol·s)−1),而与Cl2−·反应活性较低(
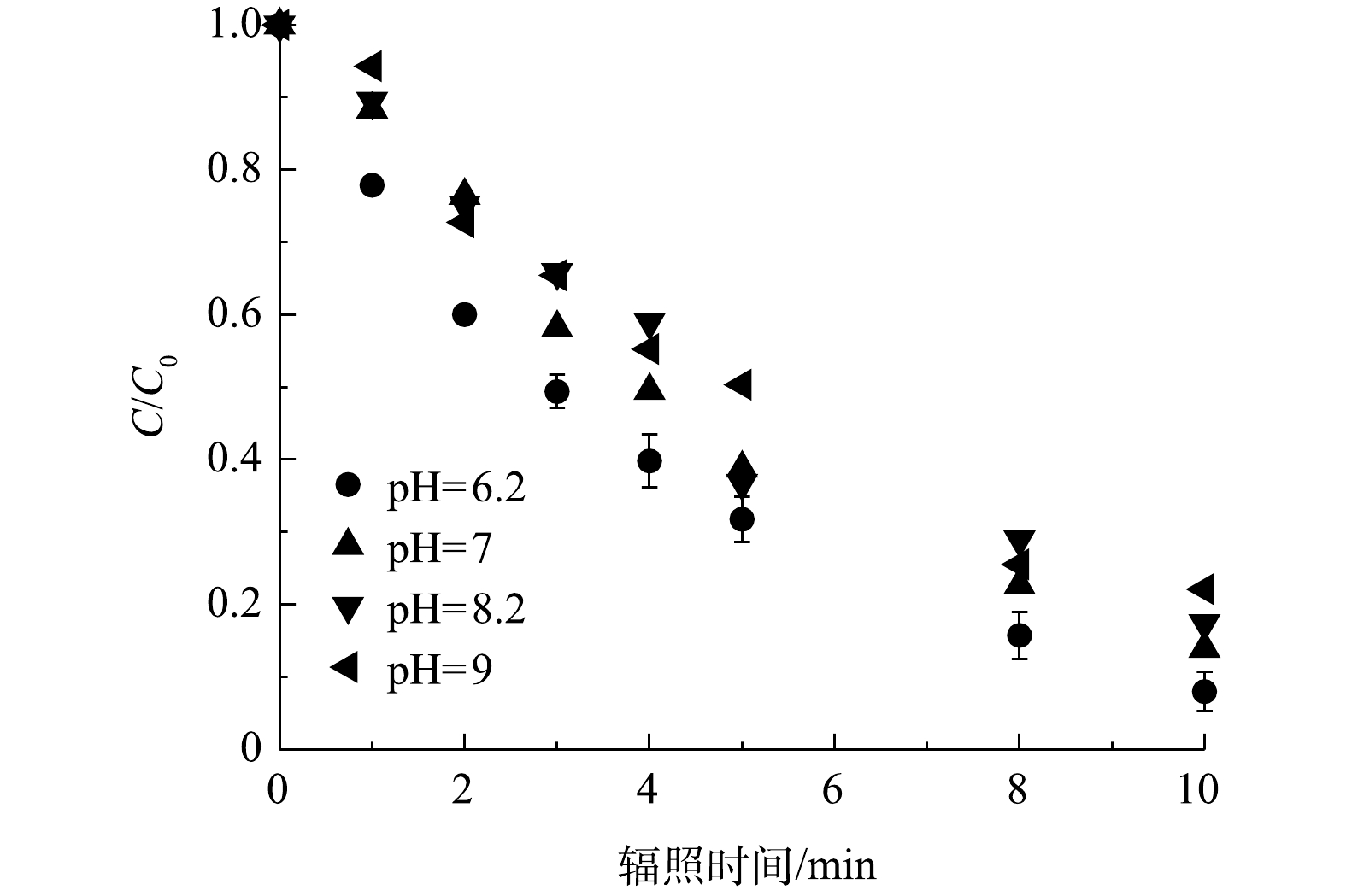
${k_{[{\rm{TBA/Cl}}_2^{\rm{ - }} \cdot ]}} $ =700 L·(mol·s)−1),因此,TBA是一种高效的·OH和Cl·淬灭剂,但其对ClO·的作用仍存在争议[11-13];IPA对·OH、Cl·和Cl2−·均具有清除作用,二级速率常数分别为2.2×109、3.7×109和1.2×105 L·(mol·s)−1,但其与ClO·的反应可忽略不计[14]。同时,pH可以影响体系中的自由基产生和自由基平衡。有研究[4]表明,由于氯的光分解减弱和OCl−的清除作用较高,·OH和Cl·的浓度会随pH的增加而降低,而二次反应产生的ClO·会增多。在pH分别为6.2和8.2的紫外/氯体系中,不同浓度的TBA和IPA对PRM降解影响的结果如图2所示。其中,TBA和IPA在 pH为6.2时对PRM的降解产生影响类似:1 mmol·L−1自由基淬灭剂的引入使PRM的降解率减少了约48%,且淬灭剂浓度的升高未对反应产生影响。在pH为8.2的条件下,PRM的降解同样受到抑制,且TBA和IPA的作用效果与pH为6.2时类似,但抑制作用较小。以上结果表明,Cl2−·对PRM的降解无作用,且TBA与ClO·的反应可忽略不计,此外,ClO·在PRM降解中起重要作用。
-
研究了不同pH下PRM在紫外/氯体系中的降解效果,结果如图3所示。随着溶液pH由6.2增加到9,计算显示PRM的降解速率k'obs由0.24 min−1下降到0.16 min−1,即PRM在酸性条件下的去除高率于碱性条件,在pH 为6.2时污染物去除效果最佳。pH对污染物降解的影响途径主要是影响FC的形态进而影响自由基物种的生成转化速率(pKa(chlorine)=7.5)[15-16]。在碱性条件下,FC(HClO/ClO−)主要以ClO−的形式存在,而HClO/ClO−被认为是紫外/氯体系中最主要的自由基清除剂,其清除效果随污染物的浓度以及与·OH和Cl·反应的速率常数而变化,如与HClO相比,ClO−和·OH、Cl·的反应活性更高(式(4)~式(7))[17-18],导致碱性条件下污染物的降解受到抑制。因此,随着pH的增加,·OH和Cl·的浓度降低,而ClO·几乎是恒定的。其次,HClO(1.45 L·(mol·cm)−1)的量子产率高于ClO−(0.97 L·(mol·cm)−1)[15]。最后,缓冲溶液中的H2PO4−和HPO42−与·OH反应速率(
$k_{[{\rm{H}}_2{\rm{PO}}^-_4/ \cdot{\rm{OH}}]}$ =2×104 L·(mol·s)−1、$k_{[{\rm{H}}{\rm{PO}}^{2-}_4/ \cdot{\rm{OH}}]} $ =1.5×105 L·(mol·s)−1)的变化也会对污染物的降解产生一定影响[11,19]。 -
1)常规阴离子与出水有机物对PRM在紫外/氯体系中降解的影响。实验中考察了Cl−、
${\rm{HCO}}_3^{-} $ 、${\rm{NO}}_3^{-} $ 和EfOM对紫外/氯体系降解PRM的反应动力学影响。在PRM和FC分别为5 μmol·L−1和70 μmol·L−1、溶液pH为7时,反应10 min后,不同阴离子浓度对PRM的降解影响如图4(a)所示。由图4(a)可知,向体系中引入Cl−时,PRM的降解速率基本保持不变,不受Cl−的显著影响,SUN等[6]也观察到了类似的结果。根据前人的研究[20],Cl−可通过式(8)和式(9)对分别·OH和Cl·产生清除作用,由正逆反应速率可知,Cl−的添加可导致Cl2−·显著增加,而Cl·略有下降。此外,虽然HOCl−·能与H+通过式(10)反应生成Cl·,在中性pH条件下该反应的二级速率常数(k=2×103 L·(mol·s)−1)与解离生成·OH的速率常数(k=6.1×109 L·(mol·s)−1)相比可以忽略不计,因此,溶液中·OH的浓度也没有变化。由图4(b)可知,
${\rm{HCO}}_3^{-} $ 对PRM降解过程产生抑制作用,当${\rm{HCO}}_3^{-} $ 浓度由0 mmol·L−1增加到100 mmol·L−1,PRM的降解速率k'obs由0.186 0 min−1下降到0.11 min−1。${\rm{HCO}}_3^{-} $ 对反应的抑制作用可归因于其与污染物对·OH和Cl·的竞争作用(式(11)~式(13))[4],导致溶液中·OH和Cl·的稳态浓度下降,生成了额外的$ {\rm{CO}}^{-}_3 \cdot$ [21],但CO3−·与PRM几乎不反应[22]。在太阳光的照射下,硝酸盐的光解作用使其成为天然水体中自由基的主要来源。已有研究证明,光解
${\rm{NO}}_3^{-} $ 对新兴污染物的降解具有重要作用[23]。硝酸盐的引入可以极大地促进溶液中·OH的生成(式(14)~式(17))[24],同时其光解可以产生活性氮物种(reactive nitrogen species, RNS)如NO3·(2.3~2.5 V)、NO2·(1.03 V)、NO·(0.39 V)、O(3P)和ONOO−(式(18)~式(20))[25]。有研究[26]表明,含N量为5 mg·L−1的硝酸盐水溶液在光解作用下产生的·OH浓度与同等条件下添加10 mg·L−1 H2O2效果相当。此外,RNS对富电子基团化合物具有良好的反应选择性,可以通过电子转移、氢提取或自由基加成的方式将污染物分解[27-28]。例如,NO2·选择性地氧化苯胺、酚基、吩噻嗪、硫醇和抗坏血酸,ONOO−能以相对较高的速率选择性地与酚类基团反应[29]。本研究中,${\rm{NO}}_3^{-} $ 的存在加速了PRM的降解(图4(c)),且随着${\rm{NO}}_3^{-} $ 浓度的增加,氧化反应的准一级动力学速率常数也随之升高。与空白组相比,当体系中${\rm{NO}}_3^{-} $ 增加到100 mmol·L−1时,PRM的降解速率k'obs增加了2.5倍。这说明${\rm{NO}}_3^{-} $ 的光解可以促进PRM的降解。溶解性有机物是一种常见的水基质组分,EfOM是其中一种,对于本体系降解污染物具有重要影响:其特定的官能团可以捕获·OH和RCS等自由基,导致污染物降解速率下降;其发色团可以削弱紫外线照射强度,造成FC光解速率的损失,但其吸收紫外线也可产生·OH等高度反应性的活性自由基,又有助于污染物转化[22,30-31]。它们的活性则因不同区域水体组分的不同和官能团的不同而不同。本研究选取西安市第三污水处理厂二级出水提取的HPO、TPI及两者的混合物HPO+TPI(有机碳含量比为1∶1)共3类EfOM,考察其对紫外/氯体系降解PRM的影响,结果见图4(d)。3种EfOM均显著抑制了PRM在紫外/氯体系中的降解。加入有机碳含量为5 mg ·L−1的HPO后,PRM的降解率由84.4%下降为63.2%,加入相同浓度TPI后PRM的降解率下降为73.5%。在相同条件下,HPO对污染物降解抑制作用最强,这与EfOM的组成、性质及化学结构相关。与TPI相比,HPO芳香性较强,且相对分子质量更大,更易于被活性自由基攻击。据报道,EfOM对紫外光传输的影响也取决于其分子结构,在紫外光范围内,不饱和键的含量与EfOM的吸光度呈正相关,254 nm处的特定吸光度(SUVA)常被用来探测特定组分或DOM的一部分[32],实验前测定到HPO和TPI的SUVA值分别为3.1 L·(cm∙mg)−1和1.96 L·(cm∙mg)−1,较高的SUVA导致HPO在降解转化过程中紫外吸收性更强;此外,较强的疏水性和非极性是HPO的关键指标,而TPI被认为是两亲甚至亲水化合物。基于以上特征,·OH和RCS会更容易受到HPO的影响,从而导致紫外/氯体系中自由基的耗损,造成HPO对PRM的降解有较大的抑制作用。
2) Fe3+的影响。铁(Fe)是地球储量丰富的过渡金属之一,也是水和废水处理过程中适用的元素。在紫外线的照射下,Fe3+(主要以Fe(OH)2+和[Fe(H2O)6]3+形式存在),其可以通过一系列反应产生·OH(式(21)和式(22))[33],作为类Fenton反应显著提高了有机污染物的降解效果,并促进Fe2+的再生;原位生成的Fe2+可以被进一步氧化(式(23)和式(24)),使Fe3+与Fe2+发生高效循环[33];当FC存在时,体系中还可在Fe2+的作用下同时产生·OH和Cl·(式(25)和式(26)),表现出污染控制协同作用。因此,实验中考察了在pH为3的条件下,紫外光诱导的FC(70 μmol·L−1)和Fe3+协同降解PRM的效果(图5(a)),并以1 μmol·L−1 NB为探针定量分析了不同浓度Fe3+存在时体系中生成的各活性物种对5 μmol·L−1 PRM降解的贡献(图5(b)),检测反应前后溶液pH变化误差为±0.2。Fe3+的引入极大地促进了目标污染物的降解,且随着Fe3+的增加污染物的降解率随之增大。当Fe3+的浓度由0 μmol·L−1增加到50 μmol·L−1时,反应1 min后PRM的降解率由25.6%增加到52.3%。结合自由基鉴定结果,可以得出,与基础条件相比,50 μmol·L−1 Fe3+的增加使PRM的准一级动力学常数k'obs变为原来的3.44倍,不同浓度Fe3+存在时PRM降解过程中起主要作用的活性物种为·OH。在PRM的降解体系中,k'·OH和k'RCS分别由0.23 min−1和0.01 min−1增加到0.69 min−1和0.15 min−1。由此可见,Fe3+的引入极大地促进了体系中·OH和RCS的生成。本实验中采用1 μmol·L−1 NB作为·OH探针,发现与对照组相比,50 μmol·L−1 Fe3+的加入使体系内·OH的浓度增加了2.95倍。这主要归因于Fe3+和溶液中生成的Fe2+在紫外光照射下生成了大量的·OH(式(21)、式(22)、式(25));而k'RCS的升高则是由于Cl·浓度的增加(式(26))。上述结果表明,Fe3+的引入可以通过直接光解和与FC协作的双重作用,提高有机污染物的去除效果。
-
1) PRM的降解和小分子有机酸的生成。为系统地阐明PRM在紫外/氯体系中的降解路径,本研究中结合反应过程中污染物的矿化和FC的消耗情况,考察了体系内小分子有机酸、
${\rm{NO}}_3^{-} $ 、${\rm{NO}}_2^{-} $ 和${\rm{NH}}_4^{+} $ 的生成及浓度变化。由图6可知,在污染物和氧化剂初始浓度分别为50 μmol·L−1和200 μmol·L−1、pH为7时,在辐照30 min后溶液中FC消耗完毕,污染物的浓度趋于稳定,而TOC的去除率保持上升。紫外/氯体系中PRM的降解速率k'obs为0.14 min−1,TOC去除率为6.8%。图7为反应1 h后体系中各种小分子有机酸的生成情况,检测出在PRM降解过程中生成了丁二酸、马来酸、乙酸、甲酸和柠檬酸5种。体系中小分子质量的有机酸如甲酸的生成量较高,而丁二酸、柠檬酸的浓度低且增长缓慢,马来酸几乎不能积累。这是因为:随着反应的进行,小分子质量有机酸通常是由于较长链的羧酸如马来酸在苯环开口上的不断氧化所致[34],也表明PRM降解过程中的中间体如丁二酸可以进一步被自由基攻击生成乙酸、甲酸等。其次,辐照10 min后体系中甲酸的生成速度骤降,30 min后积累量有所下降,乙酸几乎没有积累,该现象可能是乙酸、甲酸进一步被攻击从而分解为CO2和H2O所致。辐照45 min后,甲酸趋向于平稳,丁二酸、柠檬酸增速变慢,乙酸、马来酸的消耗变少开始累积,则可能是此时体系内氧化剂剂量和自由基浓度较低产生的结果。
同时,为探究PRM降解过程中N元素的转化规律,检测了体系中
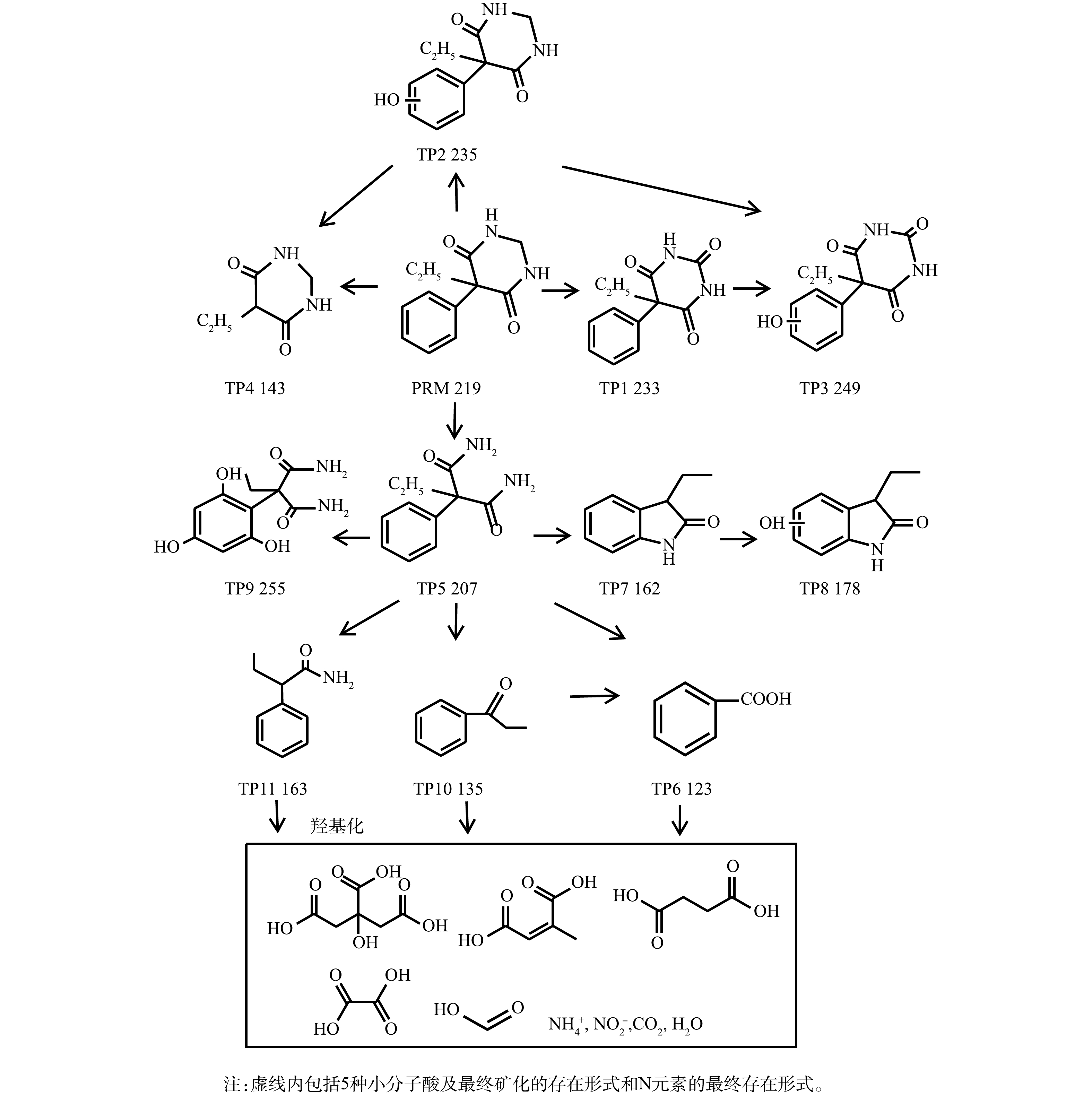
${\rm{NO}}_3^{-} $ 、${\rm{NO}}_2^{-} $ 和${\rm{NH}}_4^{+} $ 的生成情况,结果见图8。可以看出,PRM的降解可以生${\rm{NO}}_2^{-} $ 和${\rm{NH}}_4^{+} $ 。根据以上结果可推测,在·OH的作用下PRM结构中的2个N元素之间发生羟基化,之后可去甲基反应生成—NH2,而后—NH2进一步被—OH取代发生脱氨反应,导致最终N元素以${\rm{NH}}_4^{+} $ 形式去除,而NH4+在强氧化性条件下可转化为${\rm{NO}}_2^{-} $ ,使体系中有${\rm{NO}}_2^{-} $ 检出,因此,溶液中${\rm{NH}}_4^{+} $ 浓度可能出现下降趋势。本研究的实验条件下,PRM降解完成后体系中N元素主要以${\rm{NH}}_4^{+} $ 形式存在,若持续反应或增加氧化剂剂量可能使${\rm{NH}}_4^{+} $ 发生进一步转化。据报道,${\rm{NH}}_4^{+} $ 存在时还可被氧化生成${\rm{NO}}_3^{-} $ [34],但本研究中未检测到。2)污染物降解路径分析。通过高分辨率LC-MS和GC-MS对PRM和FC浓度分别为50 μmol·L−1和200 μmol·L−1、反应pH为7、PRM降解率为93.1%时体系中形成的芳香性中间产物进行了检测,共检测到10种中间体(transformation product, TP),GC-MS分析结果表明,TP8-10对应的信号值在图谱中存在,同时得到新的物种TP11,质荷比(m/z)为163。
根据所得图谱并结合前人相关研究结果推测,本实验得到的PRM在紫外/氯体系中间产物可能结构式与总体转化路径见图9。首先,芳香环和嘧啶环是PRM结构中2个更容易受到·OH亲电攻击的活化位点,这导致PRM的基本衰变途径为芳香环的羟基化,而苯巴比妥(phenobarbital,PBB)作为PRM的活性代谢物同时也是氧化过程中形成的初始中间体(m/z 233, TP1),同时PRM-OH(m/z 235, TP2)也是由于PRM芳香环上发生羟基化而生成,·OH可从PRM-OH的羟基中提取氢原子,形成共振稳定的苯氧基自由基,苯氧基自由基与O2−·/HO2·反应,生成有机过氧化物。PRM-OH通过H2O消除作用形成了相应的醌类化合物TP3 (m/z 249),该类物质在前人研究结果中也有检出[35];同时,·OH也可直接攻击PBB的芳香环从而形成TP3,这在非那西丁的紫外/氯氧化过程有类似的报道[20]。随着反应的进行,体系中产生了一种新的中间体5-Ethy1-1, 3-diazinane-4, 6-dione(EDD, m/z 143, TP4),这是由于PRM和PRM-OH中芳香环与嘧啶环之间的C—C键发生断裂所致。在PRM降解过程中发现的另一个主要中间体是苯乙胺(PEMA, m/z 207, TP5)。其原因是,在自由基的强烈作用下PRM和PBB中的嘧啶环发生断裂,且与PBB一样,PEMA也被证明是PRM的重要代谢物,在紫外/氯高级氧化体系中下可发生进一步氧化[36]。该氧化反应的机理为:首先,PEMA苯环上的支链可轻易转化为羧基,从而在芳香环上形成羧酸基团,其最终被鉴定为产物苯甲酸(BA,m/z 123,TP6);其次,LIU等[37]研究了PEMA的氧化途径,提出伯胺可通过PEMA上的分子内环封闭形成m/z为205的五环化合物,之后发生羟基化产生新的物质(m/z 206),该物质失去一个羧基后经过水解,形成中间体TP7 (m/z 162),最终通过形成羟基加合物形成TP8 (m/z 178),由于·OH对PEMA芳香环的攻击,还可以产生m/z为223和255(TP9)的物质,然而本研究只检测到TP9;第3种途径是产生m/z为208的羟基化中间体,再通过失去羧基形成新的化合物(m/z 164),但本研究在后期只检测到了TP11(m/z 164),而并没有检测到m/z 208,这是因为·OH与PEMA的氧化反应活性极高,很快中间体m/z 208就会转化为TP11。本研究在PRM的氧化过程中检测到了化合物TP10(m/z 135),该物质的产生可能是由于PEMA上的支链被·OH直接氧化,并且随着反应的继续,会进一步转化为BA。
在接下来的反应中,以上中间体可被进一步氧化生成小分子有机酸。据前人报道,PEMA苯环上的支链被氧化形成BA后,可进一步转化为富马酸(C4H4O4)和马来酸等小分子羧酸[35],而在基于O3的高级氧化过程中PRM可被转化为甲酸、乙酸和丁二酸[36]。本研究检测到PRM降解过程中生成了丁二酸、马来酸、柠檬酸、乙酸和甲酸,且溶液中产生的丁二酸可能被高浓度的活性自由基攻击生成乙酸、甲酸等小分子量羧酸,之后被矿化为CO2和H2O。同时,PRM结构中的N元素最终以
${\rm{NO}}_2^{-} $ 和${\rm{NH}}_4^{+} $ 形式存在。 -
1)紫外/氯体系对PRM具有较强的去除能力,且反应遵循一级反应动力学模型。在基础条件下PRM的去除率可达84%。污染物的降解主要是由于溶液中的FC在紫外光照射下生成了具有强氧化性的·OH和RCS。ClO·在PRM降解中起主导作用,其次为·OH,而Cl2−·没有参与污染物的转化过程。
2)酸性条件有利于污染物的降解,因随着pH升高,体系中自由基清除作用逐渐增强,同时FC的量子产率降低;PRM降解的速率常数k'obs几乎不受Cl−的影响,
${\rm{NO}}_3^{-} $ 的引入有效促进·OH的生成进进而促进PRM的降解,${\rm{HCO}}_3^{-} $ 可抑制PRM的降解;在pH为 3条件下引入Fe3+形成的类Fenton体系对污染物降解具有明显的促进作用,且随着Fe3+浓度的提高,k'obs显著提升,这归因于体系中·OH浓度的提升;2种出水有机物(HPO、TPI)通过清除体系中活性自由基对PRM降解产生抑制效果,且由于HPO具有更强的芳香性,对降解抑制效应更强。3) PRM在紫外/氯体系中主要降解机制为连续发生的羟基化过程。PRM首先通过多种途径转化为苯甲酸等物质,同时母体和中间体还可被氧化为丁二酸、马来酸、乙酸和甲酸,直至矿化;与此同时,研究发现PRM结构中氮元素最终以
${\rm{NO}}_2^{-} $ 和${\rm{NH}}_4^{+} $ 的形式存在,且在各种活性自由基的作用下${\rm{NH}}_4^{+} $ 不断转化为${\rm{NO}}_2^{-} $ 。
紫外/氯降解普里米酮的效能和机理
Degradation efficiency and mechanism of primidone by UV/chorine process
-
摘要: 选用普里米酮(primidone, PRM)为目标污染物,研究了紫外/氯高级氧化工艺对PRM的降解效能及反应机理,分别考察了pH、水体常见阴离子(Cl−、
${{\rm{HCO}}_3^{-} }$ 和${{\rm{NO}}_3^{-} }$ )和出水有机物(effluent organic matter, EfOM)对PRM降解效果的影响,并研究了PRM在类Fenton体系中的削减情况;同时,根据溶液总有机碳(TOC)的矿化、芳香性中间产物和小分子有机酸的生成,阐明了在紫外/氯体系中PRM的降解机理。结果表明,当PRM初始浓度为5 μmol·L−1、自由氯浓度为70 μmol·L−1、溶液pH为7时,反应10 min后,PRM的去除率为84%,ClO·对PRM的削减起主导作用,其次为·OH,而Cl2−·对PRM无氧化作用。当pH为6.2时污染物降解效果最佳。在一定范围内,Cl−几乎不影响PRM的降解,${{\rm{HCO}}_3^{-} }$ 因捕获自由基表现为抑制作用,${{\rm{NO}}_3^{-} }$ 因光解产生更多·OH进而加快PRM的分解。当Fe3+浓度为50 μmol·L−1时,PRM降解速率达到最大值0.84 min−1。2种EfOM的引入对PRM降解产生抑制作用,且憎水性EfOM的抑制作用更加显著。TOC矿化实验和降解路径分析结果表明,紫外/氯体系对PRM有一定的矿化作用且PRM首先通过连续的羟基化作用转化为苯甲酸等物质,同时母体化合物和中间体还可继续被氧化为小分子有机酸。PRM中的氮元素最终以${{\rm{NO}}_2^{-} }$ 和${{\rm{NH}}_4^{+} }$ 的形式存在,且在各种活性自由基的作用下${{\rm{NH}}_4^{+} }$ 可转化为${{\rm{NO}}_2^{-} }$ 。Abstract: Primidone (PRM) was selected as target contaminant in this study, and the degradation efficiency and reaction mechanism of PRM in UV/chlorine advanced oxidation process were investigated. The effects of solution pH, common anions (Cl−,${\rm{HCO}}_3^{-} $ and${\rm{NO}}_2^{-} $ ) and effluent organic matter (EfOM) on the degradation of PRM were studied, respectively. PRM decay in Fenton-like system was also studied. Meanwhile, the degradation mechanism of PRM in UV/chlorine system was identified based on TOC mineralization, the formation of aromatic intermediate products and small molecular acid. The results showed that when the initial concentrations of PRM and free chlorine were 5 μmol·L−1 and 70 μmol·L−1, respectively, and solution pH was 7, PRM removal rate was 84% in 10 min. ClO· played a leading role in PRM degradation, followed by ·OH, while Cl2−· did not participate in the conversion process. When solution pH was 6.2, the best degradation effect of pollutant occurred. The PRM degradation was almost unaffected by Cl− within a certain range, and it was inhibited by${\rm{HCO}}_3^{-} $ due to the radical scavenger, while the introduction of${\rm{NO}}_3^{-} $ greatly promoted PRM degradation because of the formation of additional •OH vis photolysis of${\rm{NO}}_2^{-} $ . When the Fe3+ concentration was 50 μmol·L−1, the PRM degradation rate reached the maximum value of 0.84 min−1. The introduction of two kinds of EfOM could inhibit the PRM degradation, and the hydrophobic EfOM played a more significant role. TOC mineralization experiment and degradation path analysis showed that UV/chlorine AOP had a certain mineralization effect on PRM, which first converted to benzoic acid and other substances through continuous hydroxylation, and the precursor and intermediates could continue to be oxidized into small molecular organic acids. It was found that the nitrogen elements in PRM molecular eventually existed in the form of${\rm{NO}}_2^{-} $ and${\rm{NH}}_4^{+} $ , and${\rm{NH}}_4^{+} $ could transform into${\rm{NO}}_2^{-} $ under the attack of various active radicals. -

-
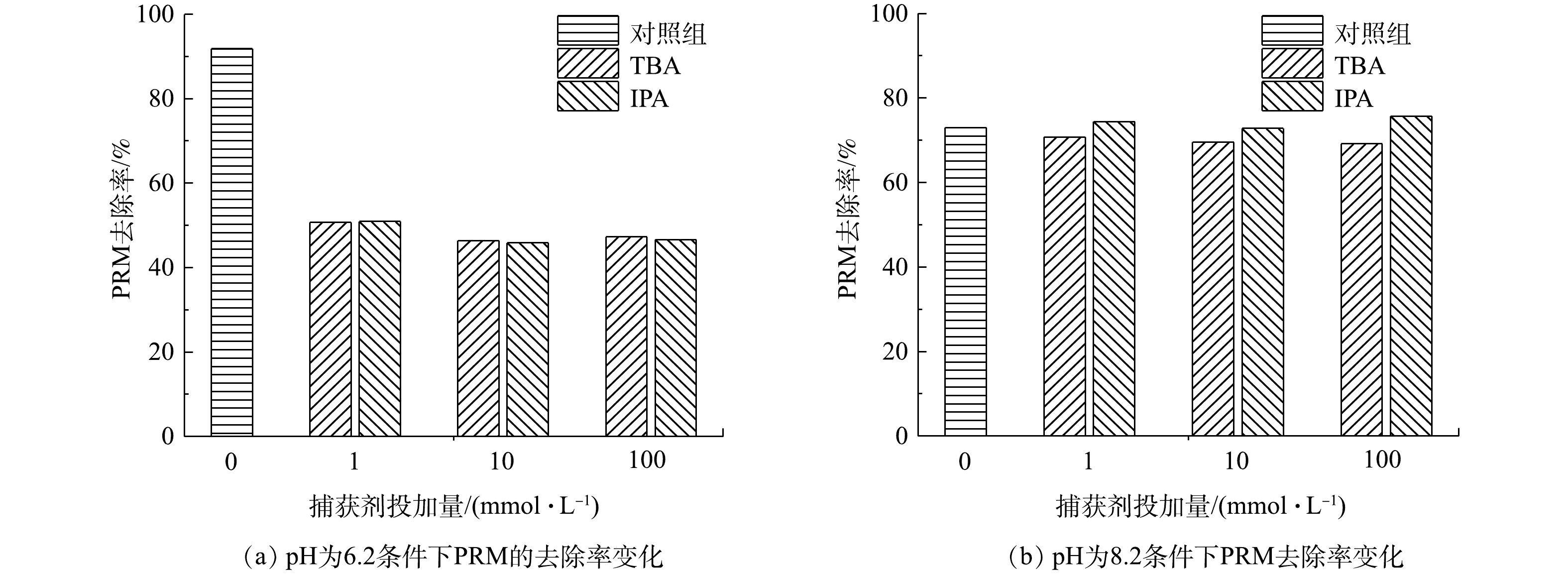
图 2 TBA和IPA在pH=6.2和pH=8.2条件下对紫外/氯体系降解PRM的影响
Figure 2. Effect of TBA and IPA on the degradation of PRM at pH 6.2 and 8.2
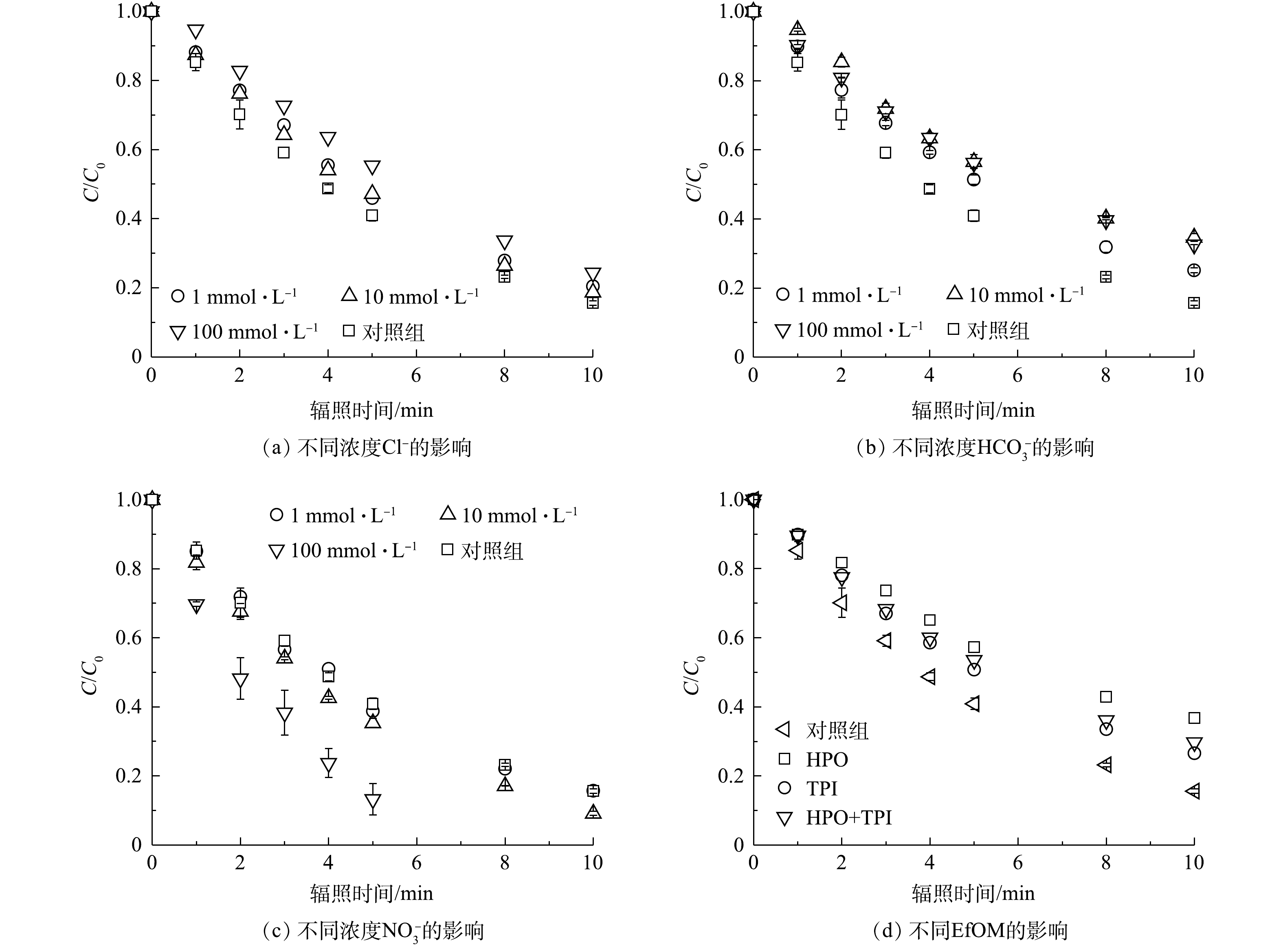
图 4 Cl−、HCO3−、NO3−和 EfOM对PRM降解的影响
Figure 4. Effect of Cl−, HCO3−, NO3− and EfOM on the degradation of PRM.
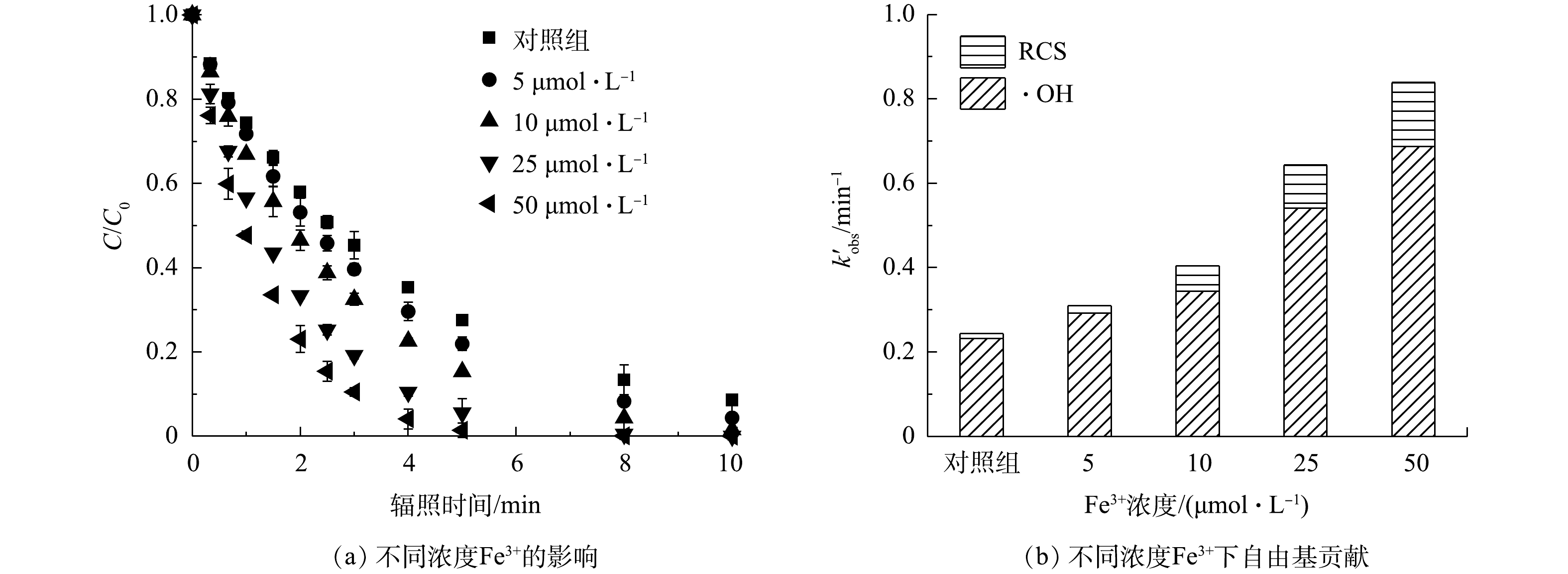
图 5 不同浓度Fe3+对紫外/氯体系降解PRM的影响及自由基贡献
Figure 5. Effect of different concentrations of Fe3+ on PRM degradation by the UV/chlorine and the corresponding contribution of radicals
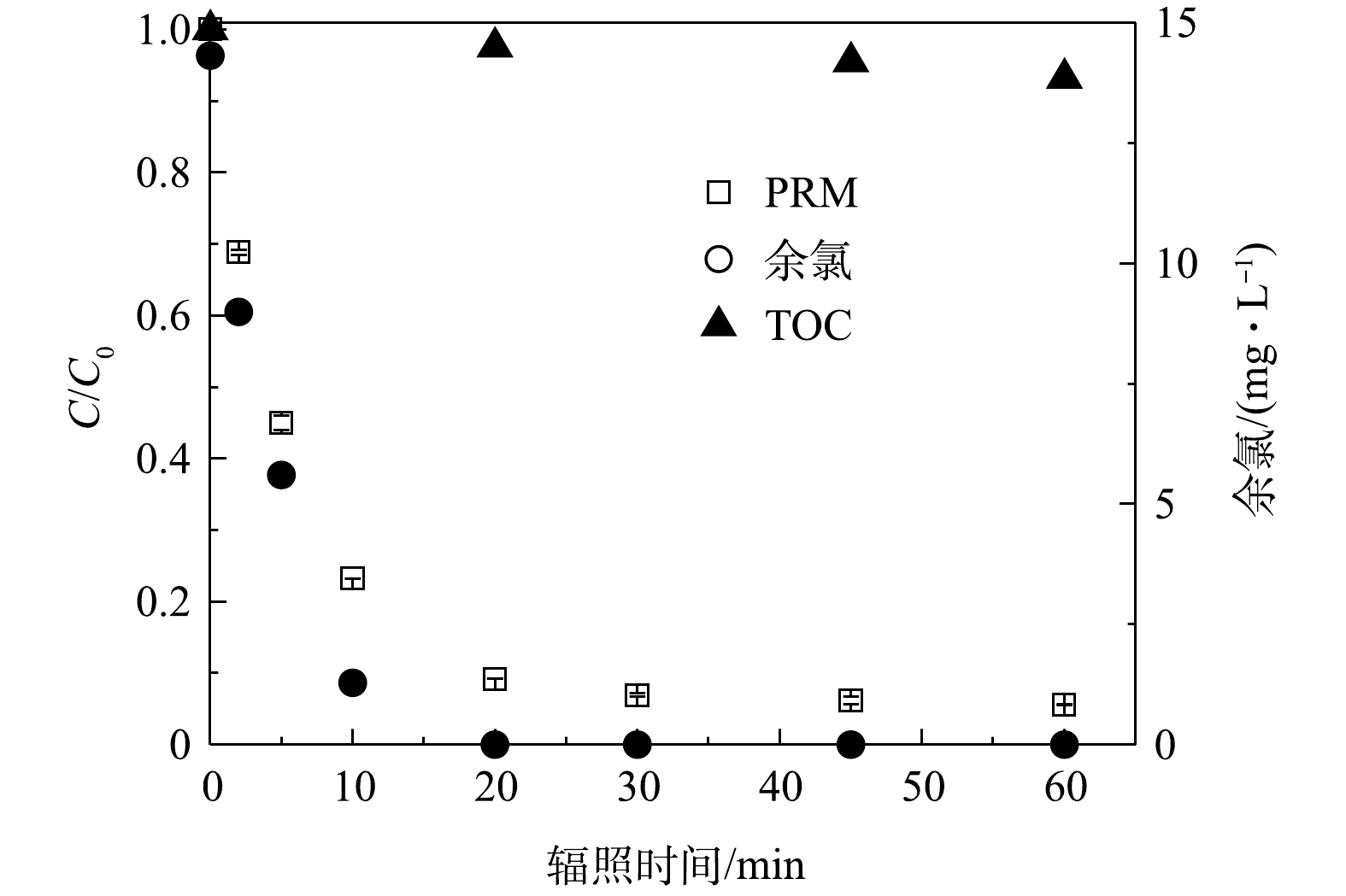
图 6 PRM在紫外/氯体系中的降解、矿化和FC的消耗
Figure 6. PRM degradation, variation of TOC, and consumption of free chlorine in the UV/chlorine process
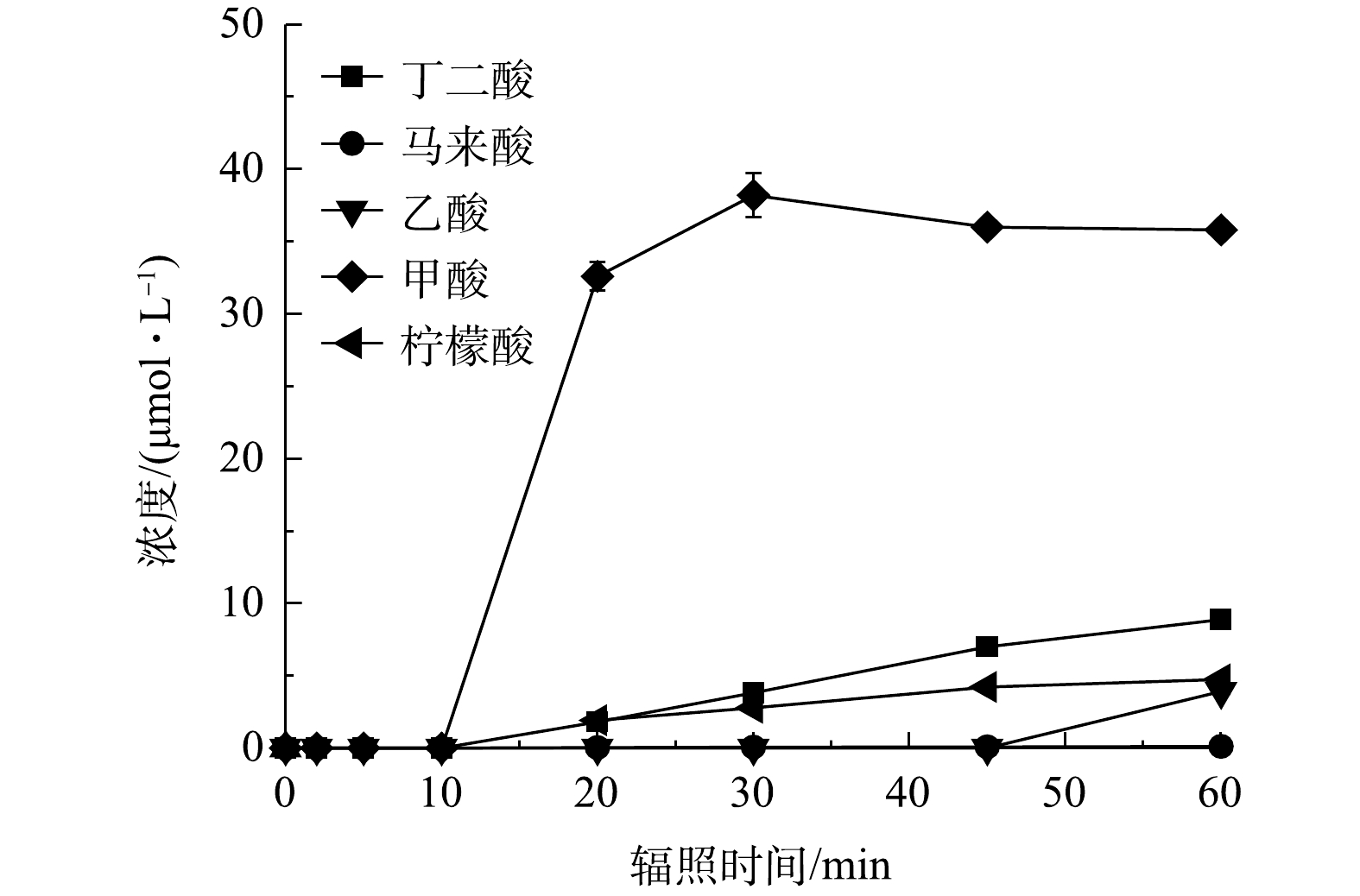
图 7 PRM在紫外/氯体系降解过程中小分子有机酸的浓度变化
Figure 7. Variation in the concentrations of low molecular weight organic acids during PRM degradation in the UV/chlorine process
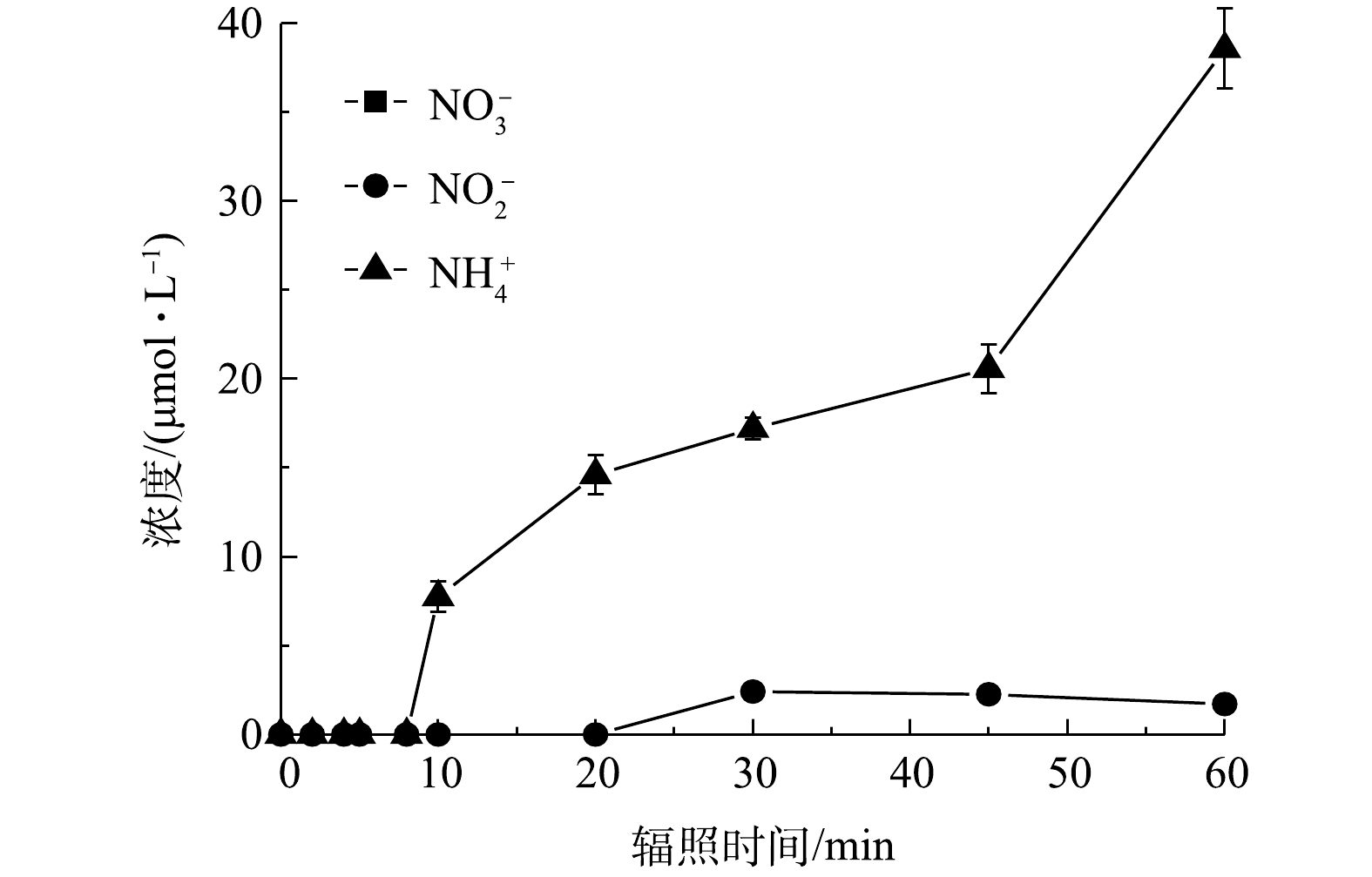
图 8 PRM在紫外/氯体系降解过程中
${{\bf{NO}}_3^{-} }$ 、${{\bf{NO}}_2^{-} }$ 和${{\bf{NH}}_4^{+} }$ 的浓度变化Figure 8. Variation of the concentrations of
${{\rm{NO}}_3^{-} }$ ,${{\rm{NO}}_2^{-} }$ and${{\rm{NH}}_4^{+} }$ during PRM degradation in the UV/chlorine process -
[1] 秀措, 王尘辰, 吕永. 潮汕地区入海河流及水生生物中PPCPs分布特征及风险评估[J]. 环境科学, 2020, 41(10): 4514-4524. [2] 高泽晨, 张天阳, 黄飘怡. 应用紫外/氯组合工艺去除微污染原水中氨氮的特性研究[J]. 环境科学学报, 2019, 39(10): 3427-3433. [3] BEITZ T, BECHMANN W, MITZNER R. Investigations of reactions of selected azaarenes with radicals in water chlorine and bromine radicals[J]. Journal of Physical Chemistry A, 1998, 102(34): 6766-6771. doi: 10.1021/jp980655a [4] FANG J Y, FU Y, SHANG C. The roles of reactive species in micropollutant degradation in the UV/free chlorine system[J]. Environmental Science & Technology, 2014, 48(3): 1859-1868. [5] ZHANG X, HE J, XIAO S, et al. Elimination kinetics and detoxification mechanisms of microcystin-LR during UV/chlorine process[J]. Chemosphere, 2019, 214: 702-709. doi: 10.1016/j.chemosphere.2018.09.162 [6] SUN P, LEE W N, ZHANG R, et al. Degradation of deet and caffeine under UV/chlorine and simulated sunlight/chlorine conditions[J]. Environmental Science & Technology, 2016, 50(24): 13265-13273. [7] 骆靖宇, 李学艳, 李青松. 紫外活化过硫酸钠去除水体中的三氯卡班[J]. 中国环境科学, 2017, 37(9): 3324-3331. doi: 10.3969/j.issn.1000-6923.2017.09.015 [8] DREWES J E, CROUE J P. New approaches for structural characterization of organic matter in drinking water and wastewater effluents[J]. Water Supply, 2002, 2(2): 1-10. doi: 10.2166/ws.2002.0039 [9] BOLTON J R, STEFAN M I, SHAW P S, et al. Determination of the quantum yields of the potassium ferrioxalate and potassium iodide-iodate actinometers and a method for the calibration of radiometer detectors[J]. Journal of Photochemistry and Photobiology A:Chemistry, 2011, 222(1): 166-169. doi: 10.1016/j.jphotochem.2011.05.017 [10] CHENG S, ZHANG X, YANG X, et al. The multiple role of bromide ion in ppcps degradation under UV/chlorine treatment[J]. Environmental Science & Technology, 2018, 52(4): 1806-1816. [11] BUXTON G V, GREENSTOCK C L, HELMAN W P. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O−) in squeous solution[J]. Journal of Physical and Chemical Reference Data, 1988, 17(2): 513-886. doi: 10.1063/1.555805 [12] MERTENS R, VONSONNTAG C. Photolysis(λ=254 nm) of tetrachloroethene in aqueous solution[J]. Photochemistry Photobiol Science, 1995, 85(1/2): 1-9. [13] TETON S. MELLOUKI A, LEBRAS G, et al. Rate constants for reactions of oh radicals with a seires asymmetrical ethers and tert-butyl alcohol[J]. International Chemistry, 1996, 28(4): 291-297. [14] PARKER K M, MITCH W A. Halogen radicals contribute to photooxidation in coastal and estuarine waters[J]. Parkar and Mitch, 2016, 113(21): 5868-5873. [15] WATTS M J, LINDEN K G. Chlorine photolysis and subsequent oh radical production during UV treatment of chlorinated water[J]. Water Research, 2007, 41(13): 2871-2878. doi: 10.1016/j.watres.2007.03.032 [16] 高睿, 杨潇, 冯天宇. 紫外/氯工艺中磺胺类药物的转化机理和毒性评价[J]. 环境化学, 2021, 40(5): 1319-1329. [17] DAUGHTON C G, TERNES T A. Pharmaceuticals and personal care products in the environment: Agents of subtle change?[J]. Enviromental Health Perspectives, 1999, 107(6): 907-938. [18] LIU X, LIANG C, LIU X. Occurrence and human health risk assessment of pharmaceuticals and personal care products in real agricultural systems with long-term reclaimed wastewater irrigation in Beijing, China[J]. Ecotoxicology and Environmental Safety, 2020, 190(1): 1-11. [19] KONG X, JIANG J, MA J, et al. Degradation of atrazine by UV/chlorine: Efficiency, influencing factors, and products[J]. Water Research, 2016, 90(1): 15-23. [20] ZHU Y, WU M, GAO N, et al. Degradation of phenacetin by the UV/chlorine advanced oxidation process: Kinetics, pathways, and toxicity evaluation[J]. Chemical Engineering Journal, 2018, 335(1): 520-529. [21] 罗从伟, 军 马, 进 江. UV/H2O2降解2, 4, 6/三氯苯甲醚动力学及产物研究[J]. 中国环境科学, 2017, 37(5): 1831-1837. doi: 10.3969/j.issn.1000-6923.2017.05.028 [22] WANG Y, COUET M, GUTIERREZ L, et al. Impact of dom source and character on the degradation of primidone by UV/chlorine: Reaction kinetics and disinfection by-product formation[J]. Water Research, 2020, 172: 115463. doi: 10.1016/j.watres.2019.115463 [23] BU L, ZHU N, LI C, et al. Susceptibility of atrazine photo-degradation in the presence of nitrate: Impact of wavelengths and significant role of reactive nitrogen species[J]. Journal of Hazardous Materials, 2020, 388: 121760. [24] RICHARD G. ZEPP J H, HEINZ B. Nitrate-induced photooxidation of trace organic chemicals in water[J]. Environmental Science & Technology, 1987, 21: 443-450. [25] WU Y T, BU L, DUAN X, et al. Mini review on the roles of nitrate/nitrite in advanced oxidation processes: Radicals transformation and products formation[J]. Journal of Cleaner Production, 2020, 273: 123065. doi: 10.1016/j.jclepro.2020.123065 [26] XU L, SUN Y, GAN L, et al. Utilization of photochemical circulation between NO3− and NO2− in water to degrade photoinert dimethyl phthalate: Influence of organic media and mechanism study[J]. Applied Catalysis B:Environmental, 2019, 259: 117958. doi: 10.1016/j.apcatb.2019.117958 [27] NETA P, HUIE R E, ROSS A B. Rate constants for reactions of inorganic radicals in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 1988, 17(3): 1027-1284. doi: 10.1063/1.555808 [28] LI A, ZHANG Z, LI P, et al. Nitrogen dioxide radicals mediated mineralization of perfluorooctanoic acid in aqueous nitrate solution with UV irradiation[J]. Chemosphere, 2017, 188: 367-374. doi: 10.1016/j.chemosphere.2017.08.170 [29] HUANG Y, KONG M, WESTERMAN D, et al. Effects of HCO3- on degradation of toxic contaminants of emerging concern by UV/NO3-[J]. Environmental Science & Technology, 2018, 52(21): 12697-12707. [30] 王雪凝, 张炳亮, 潘丙才. 市政污水二级出水中溶解性有机质在紫外/氯处理过程中的转化特性[J]. 环境科学, 2021, 42(8): 1-18. [31] 赵刘柱, 敏吴, 朱延平. 紫外/氯降解非那西丁影响因素及机理研究[J]. 水处理技术, 2019, 45(3): 69-73. [32] SUN B, WANG Y, XIANG Y, et al. Influence of pre-ozonation of dom on micropollutant abatement by UV-based advanced oxidation processes[J]. Journal of Hazardous Materials, 2020, 391: 122201. doi: 10.1016/j.jhazmat.2020.122201 [33] YUAN Y, FENG L, XIE N, et al. Rapid photochemical decomposition of perfluorooctanoic acid mediated by a comprehensive effect of nitrogen dioxide radicals and Fe3+/Fe2+ redox cycle[J]. Journal of Hazardous Materials, 2020, 388: 121730. [34] DING X, GUTIERREZ L, CROUE J P, et al. Hydroxyl and sulfate radical-based oxidation of RhB dye in UV/H2O2 and UV/persulfate systems: Kinetics, mechanisms, and comparison[J]. Chemosphere, 2020, 253: 126655. doi: 10.1016/j.chemosphere.2020.126655 [35] ZHENG M, DANIELS K D, PARK M, et al. Attenuation of pharmaceutically active compounds in aqueous solution by UV/CaO2 process: Influencing factors, degradation mechanism and pathways[J]. Water Research, 2019, 164(1): 1-11. [36] FIGUEREDO M A, RODRIGUEZ E M, CHECA M, et al. Ozone-based advanced oxidation processes for primidone removal in water using simulated solar radiation and TiO2 or WO3 as photocatalyst[J]. Molecules, 2019, 24(9): 1728-1745. doi: 10.3390/molecules24091728 [37] LIU Y, YAN S, LIAN L, et al. Assessing the contribution of hydroxylation species in the photochemical transformation of primidone (pharmaceutical)[J]. Science of the Total Environment, 2019, 696: 133826. doi: 10.1016/j.scitotenv.2019.133826 -
 点击查看大图
点击查看大图

计量
- 文章访问数: 5569
- HTML全文浏览数: 5569
- PDF下载数: 59
- 施引文献: 0






































