


-
醛酮类羰基化合物是环境中一类重要污染物[1-4],是我国及世界许多国家和地区重点监测的环境污染物之一。环境中醛酮类化合物一次污染源主要来源于工业排放、植被释放、香烟烟雾、烹饪油烟以及化石燃料和植物燃烧[5-9] 等。大气中VOCs的光氧化反应是醛酮类羰基化合物二次排放的主要来源[10]。在水[11-12]、土壤和沉积物[13-14],尤其是大气[15-16]等介质中均有不同程度的检出。
测定醛酮类羰基化合物主要有GC法[17]、GC/MS(/MS)法[7-18],毛细管电泳-紫外法[19]、HPLC法[13-15, 20]以及HPLC/MS法[12, 21]等。空气样品醛酮类化合物中最常用的方法是用涂敷2,4-二硝基苯肼(DNPH)的硅胶管采集,使目标物与DNPH在酸性介质中发生衍生化反应生成醛酮-DNPH腙衍生物,用反相液相色谱法分离,紫外检测器检测。
LC对比GC和GC/MS法的缺点是分辨率低、分析时间长、溶剂用量大,但GC或GC/MS只能分析样品在进样系统不发生分解且能产生较高不受干扰色谱或质谱峰的羰基化合物或羰基化合物衍生物,而大部分高沸点羰基化合物衍生物容易分解,使得GC或GC/MS分析的目标物种类受到很大限制。LC-MS(/MS)对比LC-UV主要优势在于其选择性和灵敏度,但LC法仪器成本低、方法稳定性好也是其不可被替代的优势。Ochs等[22-23] 建立了RRLC-APCI(-)-MS/MS法测定31种羰基化合物,并与RRLC-UV法的线性、相关系数、检测限和灵敏度进行了比较,RRLC-UV法灵敏度更高;RRLC-APCI(-)-MS/MS检测限为0.71—10.3 µg·L−1,略低于RRLC-UV法,但方法定性准确更高。Zurek等[24]对比了APCI和ESI正离子模式下测定羰基化合物与4-二甲氨基-6-(4-甲氧基-1-萘基)-1,3,5-三嗪-2-肼(DMNTH) 的Hantzsch衍生反应产物。随着高分辨质谱技术的发展,化合物定性准确性又有了大幅提高。孟志娟等[25] 将Orbitrap GC-MS用于农产品中70种农药残留的快速筛查分析,陈溪等[26]用UPLC-Q-Orbitrap MS对水中112种药品及个人护理品进行了筛查和定量测定,112 种PPCPs 的定量限可达 0.002—0. 8 µg·L−1,准确性和灵敏度大大提高。目前还未发现使用液相色谱/高分辨质谱分析醛酮类化合物的报道。
本论文深入研究了25种羰基化合物-DNPH衍生物的HPLC-UV、UPLC-ESI-MS/MS和UPLC-ESI-Q- Orbitrap MS的最佳分析条件,建立了各自对应分析方法,并对分析结果的分离度、分辨率、检出限、线性范围和定性准确性等分析特征进行了比较评价。使用3种方法分别对加标模拟样及天津市大气实际样品进行了分析测定,明确3种方法各自特点和适用范围,Orbitrap MS还可对样品中非靶标污染物进行准确识别,并可通过理化性质相似的已知浓度化合物响应因子比较,提供半定量结果,为复杂环境样品污染物精准识别和准确检测提供了有力技术支持。
-
高效液相色谱仪,带二极管阵列检测器(Agilent1260,美国Agilent公司);超高效液相色谱-串联质谱仪,带电喷雾离子源(Agilent 1290/6460,美国Agilent公司);超高效液相色谱-电喷雾离子源-四极杆/静电场轨道阱高分辨质谱仪(Ultimate 3000-Q Exactive Focuse,美国Thermo Fisher公司);N-EVAP111氮吹仪(Organomation公司);Milli-Q超纯水系统(美国Millipore公司);色谱柱1:Agilent ZORBAX Extend-C18,4.6 mm×250 mm,5 µm;色谱柱2 Agilent ZORBAX Eclipse Plus C18,2.1 mm×100 mm,1.8 μm (美国Agilent公司);色谱柱3:Thermo Accucore RP-MS,2.1 mm×100 mm,2.6 µm(美国Thermo公司);Cleanert DNPH-Silica醛酮气体采样管 (200 mg·3mL−1)及臭氧去除管(240 mg·3mL−1)(天津艾杰尔飞诺美公司);0.22 µm尼龙水相针式滤膜(美国Supelco公司)。
25种醛酮-DNPH标准溶液,15 mg·L−1(美国ChemTek公司),分别为甲醛、乙醛、丙烯醛、丙酮、糠醛、丙醛、巴豆醛/丁烯醛、丁酮、甲基丙烯醛、丁醛、苯甲醛、异戊醛、环己酮、戊二醛、戊醛、对,间,邻-甲基苯甲醛、甲基异丁基酮、己醛、2,5-二甲基苯甲醛、庚醛、辛醛、壬醛和癸醛的DNPH衍生物;16种醛酮类标准液1000 mg·L−1(包括甲醛、乙醛、丙烯醛、丙酮、丙醛、丁烯醛、丁酮、丁醛、苯甲醛、异戊醛、戊醛、对,间,邻-甲基苯甲醛、己醛、2,5-二甲基苯甲醛,CHEM SERVICE公司),其余9种醛酮为标准品,均为色谱纯 (DIKMA公司);2,4-二硝基苯肼(DNPH) (99.5%,百灵威公司);甲醇、乙腈(液相色谱纯,MERCK公司);超纯水,经Milli-Q超纯水器纯化得到。
-
分别取9种醛酮类标准品用乙腈配制成质量浓度为1000 mg·L−1的混合储备溶液,与16种醛酮类标准溶液一起,各取100 µL于10 mL容量瓶中,用乙腈定容,配制成10 mg·L−1的25种醛酮类混合标准工作溶液,低于4 ℃冰箱保存。
-
环境空气按照HJ 683—2014和EPA TO-11的规定采集样品,在真空泵的作用下,气体样品经除臭氧小柱去除臭氧后,用 DNPH柱吸附,样品中醛酮类化合物与DNPH发生衍生化反应。采样流量0.5 L·min−1,采气体积100 L。采样管用密封帽将两端管口封闭,放入避光密封袋中,低于4 ℃保存,时间不超过7 d。
-
用乙腈洗脱采样管,洗脱方向与采样时气流方向相反,将洗脱液收集于5 mL容量瓶中,用氮吹仪浓缩至≤0.5 mL(液相色谱法浓缩至≤1.0 mL),若存在未溶解的橘红色颗粒物,用少量乙腈溶解,乙腈含量应≤0.5 mL,用纯水定容至1.0 mL,混匀,经针头过滤器过滤,滤液收集于2 mL棕色样品瓶中,待测。
-
(1)色谱柱1;柱温:40 ℃;流动相:A相为纯水,B相为乙腈;检测波长:360 nm;进样量:10 μL。流速:1.0 mL·min−1。
(2)梯度洗脱程序:0—15 min,60%B; 15—23 min,60%—100%B;23—34 min,100%B;34—34.1 min,100%—60%B;34—36 min,60%B。
-
(1)色谱柱2;柱温:40 ℃;流动相 A:1 mmol·mol−1乙酸铵/水溶液;流动相B:乙腈;流速:0. 3 mL·min−1;进样量:10 µL。
(2)梯度洗脱程序:0—18 min,60%—70%B;18—23 min,70%—90%B;23—26 min,90%B;26—26.1 min,95%—60%B;26.1—30 min,60%B。
(3)质谱条件:离子源:ESI,负离子模式;干燥气流量:10 L·min−1,温度:300 ℃;毛细管电压:4000 V;监测方式:MRM(多反应监测)。优化后得到25种醛酮腙的MRM条件见表1。
-
(1)色谱柱3;柱温:40 ℃;流动相A:1 mmol·mol−1乙酸铵/水溶液;流动相B:乙腈;流速:0.3 mL·min−1;进样量:10 µL。
(2)梯度洗脱:0—5 min, 50% B; 5—15 min, 50%—60% B; 15—20 min, 60%—70% B; 20—24 min, 70%—80% B; 24—25 min, 80%—95% B; 25—28 min, 95% B; 28—28.1 min, 95%—50% B; 28.1—30 min, 50% B。
(3)离子化模式:HESI电离源;扫描模式:负离子模式;毛细管温度: 325 ℃;加热温度: 350 ℃;鞘气: N2,流速 40 arb;辅助气: N2,流速10 arb;喷雾电压:2800 V; 透镜电压:60 V。
(4)扫描模式:全扫描/数据依赖二级(Full MS/dd-MS2)用于筛查、定性和定量,Full MS参数:分辨率 70000,自动增益控制目标(AGC target) 1×106,最大驻留时间(maximum IT) 100 ms,扫描范围 m/z50—600;dd-MS2参数:分辨率17500,AGC target2×105,最大驻留时间60 ms,动态排除8.0 s;NCE/Stepped NCE 25、45、65,Loop count 3,Isolation window 1.0 m/z。
-
对25种浓度为150 µg·L−1羰基化合物-DNPH混合标准溶液用3种分析仪器在优化的质谱条件下分别进行分离测定,通过优化各自的最佳流动相梯度,各自分别考察了柱1:Agilent ZORBAX Extend-C18、柱2:Agilent ZORBAX Eclipse Plus C18和柱3:Thermo Accucore RP-MS的分离效果。25种目标物中含丙烯醛、丙酮、糠醛;环己酮、戊二醛、异戊醛、戊醛;邻,间,对-甲基苯甲醛等3组难分离物质。
UV法用化合物的特征紫外吸收波长以及其保留时间定性,大部分化合物最佳吸收波长均为360 nm,难分离物质需要单标辅助定性,否则容易造成目标峰识别错误,这对化合物分离的要求更高。通过调整流动相梯度,只有柱1能够实现所有化合物识别(对,间-甲基苯甲醛能够在峰顶分离,环己酮和戊二醛分离最差,仅能分辨出是两种物质接近共流出),其他两根色谱柱均完全不能实现这两组难分离物质对的识别,因此UV法选择柱1进行分析测定。
质谱法均为串联超高效液相色谱,色谱柱容量比常压液相色谱小,溶剂效应影响较大,因此待分析物质溶剂中有机相比例不能高于流动相的初始比例,否则容易出现色谱峰变形,使分离变差。柱2和柱3均为超高压液相色谱柱,适用于质谱的分析。通过优化各自最佳的分离条件,两根色谱柱在各自质谱仪上分辨率相差不大,最终MS/MS法选择柱2,Orbitrap MS法以柱3为分析柱。柱2和柱3除对,间-甲基苯甲醛不能实现分离外,其余化合物在30 min之内均可获得良好的分离度和尖锐对称的色谱峰形.
由于MS/MS法是以特征离子对和保留时间定性、特征子离子定量,Orbitrap MS法以保留时间和具有精确质量数的二级特征碎片离子定性、母离子定量,因此对,间-甲基苯甲醛合并定量外,其他保留时间接近化合物均可不受对方干扰,即可准确定性定量。具体色谱分析条件优化结果见1.5节,本研究3根色谱柱25种目标物出峰顺序略有不同,具体见图1—(a),(b),(c)。
-
羰基化合物-DNPH结构使得ESI离子源采用正/负离子模式都有可能,通过负离子模式以甲醇−1 mmol/mol乙酸铵/水溶液为流动相,正离子模式以甲醇−0.1%甲酸/水溶液为流动相,在MS/MS法分别优化最佳子离子及最佳离子响应、最佳framgenger和CE值,Orbitrap MS优化了最佳NCE值条件下,测定各自ESI(+/−)模式下的分离度和响应值,结果发现在相同的分离情况下,负离子模式对大多数目标物具有更高的响应值。图2即为MS/MS法ESI(+/−)总离子流图的比较。Orbitrap MS ESI(+/−)母离子提取质谱图对比发现同样是ESI(−)比ESI(+)具有更高的响应值,更适合分析25种醛酮类羰基化合物。具体质谱分析优化条件结果见1.5节。
-
分别量取一定量的羰基化合物-DNPH标准溶液于1.0 mL棕色进样瓶中,用纯水定容,混匀。配制成质量浓度分别为0.45、0.75、1.50、3.00、4.50、7.50、15.0、30.0、45.0、75.0、150、300、450、750、1500 µg·L−1的标准系列,在各自优化的色谱和质谱条件下,UV以目标物色谱峰面积、MS/MS法以母离子对应的定量子离子峰面积、Orbitrap MS以母离子的提取色谱峰面积为纵坐标,以目标物的质量浓度为横坐标,绘制标准曲线,具体线性范围、相关系数测定结果见表2。由测定结果可见,3组曲线相关系数r均大于 0.990,UV法比MS/MS法线性范围更宽,UV法所有目标物在15—1500 µg·L−1内线性关系良好,而MS/MS法线性最高点仅为300 µg·L−1。Orbitrap MS法测定浓度最低,约为0.45—300 µg·L−1。方法检出限测定按照HJ168—2010规定的方法,采用向空白醛酮采样管中加标的方式(不同检测方法加标量分别为各化合物线性最低点对应的量,按采样量为100 L,定容体积为1.0 mL计算),测定7组模拟样品,按照样品测定方法进行测定,并计算标准偏差,以3.14倍标准偏差计算方法检出限,测定结果见表2。由表2可见,各目标物检出限 UV法略高于MS/MS法,分别在0.12—0.40 µg·m−3和0.08—0.60 µg·m−3之间,Orbitrap MS法最低,在1.1—13 ng·m−3之间,更适用于环境低浓度样品的测定。
-
向3个6组空白醛酮采样管口分别加入25种醛酮标准混合溶液,按照样品采集方法,用高纯氮气代替实际样品,模拟采集环境空气中醛、酮类化合物,连续采样 1 h,制成加标量分别为75 ng的模拟样品,按照样品测定方法进行测定,计算加标回收率平均值和相对标准偏差。具体测定结果见表3。25种目标物的UV法、MS/MS法和Orbitrap MS测得平均回收率范围分别为68.9%—98.8%,67.9%—97.6%和66.5%—107%,相对标准偏差范围分别为4.9%—10%,6.9%—18%和5.6%—11%,精密度和准确度均能够满足测定要求。
-
将所建立的方法应用于来自天津市区某采样点于2020年3月9日(样品1)和3月27日(样品2)采集的环境空气样品,采用相同的前处理方法,制备好的试样分别分成3份,使用不同分析仪器针对25种羰基化合物分别进行了定性和定量测定。
测定结果见根据测定结果可知,2个样品中除戊二醛未检出、样品2苯甲醛未检出外,Orbitrap法24种目标物均有检出,且其保留时间及一级母离子精确质量数测定值与理论值相差均<5×10−6,二级质谱5个碎片与筛查谱库离子碎片一致,测得24种目标物平均浓度在0.007—4.84 µg·m−3之间,其中浓度最高分别为丙酮、甲醛、乙醛、壬醛和己醛,浓度均高于1.00 µg·m−3;浓度最低依次是甲基苯甲醛、甲基丙烯醛、糠醛、丁烯醛和甲基异丁基酮。3种方法UV法和MS/MS法检出的化合物均远少于Orbitrap法,UV法检出最少。3种方法均能检出的目标物相对偏差均小于20%。
-
本研究通过优化流动相梯度、最佳离子化模式和离子对、最佳碰撞能量等色谱质谱条件,建立了分别用HPLC-UV、UPLC-ESI-MS/MS和UPLC-ESI-Q-Orbitrap MS法同时测定空气中25种醛酮类羰基化合物的分析方法并进行了比较。研究结果表明3种检测方法线性相关系数、精密度和准确度均良好,MS方法定性准确性高,检出限较UV法低,而Orbitrap MS法最低,可达ng·m−3水平。UV法适用于检测相对高浓度样品,线性范围最宽;MS/MS法适用于测定中等浓度样品;而Orbitrap MS法适用于环境中低浓度样品检测,同时通过分析碎片离子,根据裂解规律、母离子和子离子精确质量,还可扩展筛查其他可能存在的非靶标羰基化合物。3种方法应用于实际样品测定发现Orbitrap MS法能够准确定性定量测定的化合物种类最多,UV法最少,但3种法均能检出的目标物相对偏差均小于20%。3种方法各有侧重,相互补充,为环境醛酮类羰基化合物准确定性定量提供了完善的技术手段,同时为其他适用于液相色谱和液相色谱质谱测定的化合物方法建立提供了可靠的例证。
液相色谱和液相色谱质谱测定空气中25种醛酮类化合物
Determination of 25 aldehydes and ketones in air by HPLC and UHPLC-MS
-
摘要: 本研究建立了高效液相色谱-紫外检测法(HPLC-UV,UV)、超高效液相色谱-电喷雾离子源-串联四极杆质谱法(UPLC-ESI-MS/MS,MS/MS)和超高效液相色谱-电喷雾离子源-三重四极杆/静电场轨道阱高分辨质谱法(UPLC-ESI-Q-Orbitrap MS,Orbitrap MS)分别同时测定空气中25种醛酮类羰基化合物的分析方法。优化了流动相梯度、碎裂电压和碰撞能等参数。结果发现,3种方法标准曲线相关系数r均大于 0.990,UV法比MS/MS法线性范围更宽,在30—1500 µg·L−1内线性关系良好,而MS法线性最高点仅为300 µg·L−1内,Orbitrap MS法适合检测浓度最低,约为0.45—300 µg·L−1。方法检出限UV法略高于MS/MS法,分别在0.12—0.40 µg·m−3和0.08—0.60 µg·m−3之间,Orbitrap MS法最低,在1.1—13 ng·m−3之间。加标量为75 ng的模拟样品UV法、MS/MS法和Orbitrap MS测得平均回收率范围分别为68.9%—98.8%、67.9%—97.6%和66.5%—107%,相对标准偏差范围分别为4.9%—10%、6.9%—18%和5.6%—11%。3种方法检测实际样品,UV法和MS/MS法检出化合物分别为9种和14种,均远少于Orbitrap MS法的24种。3种方法均能检出的目标物测定结果相对偏差均小于20%,均可用于醛酮类羰基化合物检测,但MS法定性较UV法更加准确,UV法可以测定高浓度样品,MS/MS法适合测定中等浓度样品,而Orbitrap MS不仅适用于测定极低浓度样品,同时还可用于筛查非靶标目标物,为污染物识别和防治提供了更加有力的技术手段。Abstract: Three methods of qualitative and quantitative determination of 25 aldehydes and ketones in air by high performance liquid chromatography with ultraviolet detection (HPLC-UV), ultrahigh performance liquid chromatography with electrospray ion source and tandem quadrupole mass spectrometry (UPLC-ESI-MS/MS), and ultrahigh performance liquid chromatography with electrospray ion source and quadrupole/electrostatic field orbitrap high resolution mass spectrometry (UPLC-ESI-Q-Orbitrap MS) were developed.Some key test conditions like gradient of mobile phase, mode of positive and negative ions, fragmentation voltage, collision energy (CE) and Orbitrap MS screening library were optimized. The results showed that the regression coefficients of the calibration curves (r) were all greater than 0.990. The linear ranges of UV method were wider than those of MS/MS method, with the range of 30—1500 µg·L−1, while the highest linear point of MS method was only 300 µg·L−1. The liner ranges of Orbitrap MS method were about 0.45—300 µg·L−1. The method detection limits (MDL) of UV and MS/MS were between 0.12—0.40 µg·m−3 and 0.08—0.60 µg·m−3 respectively, while the Orbitrap MS was between 1.1—13 ng·m−3, which was the lowest of the three methods. The average 75 ng spiked recoveries for UV, MS/MS and orbitrap MS methods were between 68.9%—98.8%, 67.9%—97.6% and 66.5%—107%, and the relative standard deviations were 4.9%—10%, 6.9%—18% and 5.6%—11% respectively. The compounds numbers of field samples detected by UV method and MS/MS method were 9 and 14 respectively, far less than 24, which detected by Orbitrap MS method. The relative deviations of the test results for the targets detected out by all three methods were less than 20%. However, MS methods were more accurate than UV method. UV method could be used to determine high concentration samples, and MS/MS method was suitable for the determination of medium concentration samples. Furthermore Orbitrap MS method is not only suitable for determining very low concentration samples, but also for screening non-target objects, which provides a more powerful techniques for pollutants identification and control.
-
Key words:
- aldehydes and ketones /
- optimization and comparison /
- high performance liquid chromatography with ultraviolet detection (HPLC-UV) /
- ultrahigh performance liquid chromatography with electrospray ion source and tandem quadrupole mass spectrometry (UPLC-ESI-MS/MS) /
- ultrahigh performance liquid chromatography with electrospray ionization / quadrupole / electrostatic field orbitrap high resolution mass spectrometry (UPLC-ESI-Q-Orbitrap MS)
-
汞具有长距离迁移性和生物富集性,可以在大气中停留数月至1 a,随气流长距离传输,造成远离污染源地区环境汞污染。我国是全球大气汞排放量最大的国家,大约占全球大气汞排放量的30%,所以我国汞控制排放对全球汞减排具有重要意义[1-2]。因此,我国作为全球大气汞排放量最大的国家,汞排放控制也得到全球的广泛关注,面临巨大的履约压力。2021年,WU等[2-3]探究了2021年我国重点行业大气汞排放量为314 t,其中燃煤电厂和工业锅炉排放量分别为36 t和24 t,占总排放量的19.1%。因此,煤炭燃烧是我国汞排放控制的重点行业。
相较于国外燃煤行业专门汞控制技术(活性炭喷射技术),我国燃煤行业普遍采用污控措施协同控制措施降低烟气汞排放浓度。以燃煤电厂为例,目前燃煤电厂的污控措施基本都具有选择性催化还原(selective catalytic reduction,SCR)催化剂、除尘设备、湿法脱硫设备(wet fuel gas desulfurization device,WFGD)。汞在烟气中主要存在3种形态:气态元素汞(Hg0)、气态氧化汞(Hg2+)和颗粒态汞(Hgp)[4-6]。Hg2+和Hgp分别被WFGD和除尘设备控制捕集下来,捕集效率高达80%和99%以上。Hg0不易溶于水和被除尘捕集,所以燃煤行业烟气汞主要以Hg0形式存在。因此,燃煤行业汞减排关键在于通过催化剂将Hg0转化为Hg2+,进而被脱硫设备捕集。
目前,燃煤电厂普遍采用钒钨钛催化剂作为商业脱硝催化剂,但V2O5的高毒性、致癌性等特点限制了钒钨钛催化剂的使用。氧化铈(CeO2)具有良好储氧能力和氧化还原特性,可以作为催化剂V2O5的替代活性组分[7-9]。以往的研究表明,Ce基催化剂表现出良好的NOx还原能力或Hg0氧化能力,但单一的CeO2难以满足NOx还原和Hg0氧化双重催化需求,构建多活性位点多反应区域是满足NOx和Hg0协同控制的重要条件。CeMnTi催化剂具备良好的Hg0氧化性能和NOx低温还原性能,但CeO2高氧化性使SO2转化为Ce(SO4)2或Ce2(SO4)3,造成催化剂失活[10-12]。基于以上研究,开发SCR烟气条件下具备Hg0高效氧化和良好抗硫性能的催化剂尤为必要。
基于以上研究结果与不足,考虑到NH3、SO2和Hg0酸碱性的差异,本研究以CeO2为活性位点、TiO2为载体、WO3和CuO为改性剂,制备多种Ce基催化剂,构建酸碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力,探究了催化剂协同催化脱除和抗硫反应机制,为燃煤行业多污染协同控制和多设备协同控汞提供理论依据。
1. 材料及方法
1.1 催化剂的制备
本研究采用德固赛公司生产的商业TiO2 (P25) 作为催化剂的载体。基于已有研究[13]结果,将一定量的Ce(NO3)3·6H2O和草酸溶解于去离子水,按照Ce:W:Cu:Ti比例添加一定量的(NH4)10H2(W2O7)6和Cu(NO3)2,再添加一定量的P25形成混合溶液,微波处理15 min。将混合溶液搅拌24h,然后再放入110 ℃烘箱中干燥12 h,最后在马弗炉中500 ℃焙烧4h后,自然降温到25 ℃,制备出不同Ce基催化剂。将催化剂研磨压片过筛得到40~60目催化剂样品用于汞氧化实验。所得催化剂包括Ce5Ti(Ce∶Ti=5∶95)、Ce5W9Ti(Ce∶W∶Ti=5∶9∶86)以及Cu5Ce5W9Ti(Cu∶Ce∶W∶Ti=5∶5∶9∶81)。
1.2 催化剂的表征分析
本研究采用X射线衍射(XRD)、X射线光电子能谱(XPS)、H2程序升温还原测试(H2-TPR)、CO2程序升温脱附分析(CO2-TPD)和X射线透射电镜(TEM)等表征手段对反应前后的催化剂进行表征,分析催化剂表面理化性质,探究汞氧化反应机制。XRD 物相分析采用Bruker D8 (德国布鲁克公司),管电流30 mA,管电压40 kV,扫描角度2θ为10°~90°,扫描速度为2 °·min−1。XPS分析采用X射线光电子能谱仪(Thermo ESCALAB 250XI)分析催化剂表面Cu、Ce、W、Ti、O、S、N、Cl和Hg元素的价态。H2-TPR实验采用AutoChem II
2920 型化学吸附仪 (Micrometritics Co.),实验步骤如下:取 50 mg 样品(40~60 目),在纯氧气氛下 400 ℃ 预处理 30 min;降温到50 ℃后用He吹扫15 min;然后通入体积分数为10% H2+90%Ar混合气,待仪器基线平稳后,以10 ℃·min−1速率升温至600 ℃,采用TCD 检测耗氢量。CO2-TPD实验步骤如下:取100 mg 样品 (40~60 目),在氦气氛下经500 ℃预处理30 min;冷却至50 ℃,再通入30 mL·min−1的CO2吸附至饱和,氦气吹扫1h 后开始TPD 实验,以10 ℃·min−1 速率升温至800 ℃,采用TCD 检测。1.3 烟气模拟实验
本研究采用固定床实验装置探究催化剂汞氧化性能。该固定床实验装置包括四部分:模拟烟气配气系统、汞源发生系统、催化反应系统和汞浓度与形态测试系统。催化反应系统通过加热系统精准调控反应温度,温度误差≤2 ℃。模拟烟气配气系统包括高纯氮气(N2)、氧气(O2)、标准混合气(HCl、SO2、NO和NH3等)和80 μg·m−3 Hg0,标准混合气均采用N2作为平衡气。以上标准气体均采用质量流量控制器控制气体添加,混合气体总流量为1L·min−1。空速是催化剂催化氧化性能的重要实验参数,本实验催化剂空速约为100 000 h−1。研究中各种模拟烟气组分见表1。反应前后烟气中Hg0和Hg2+的浓度通过烟气分析仪分析(Thermo Fisher 80i)。汞氧化率通过式(1)计算。
表 1 汞催化氧化实验模拟烟气组分Table 1. The components of simulated flue gas in mercury oxidation experiments序号 组分 温度 元素比例 1 基本烟气组分 100~450 ℃ 6%O2+N2 2 SCR烟气组分 200、300和400 ℃ 6%O2+134 mg·m−3 NO+76 mg·m−3 NH3+N2 3 SCR+HCl烟气组分 200、300和400 ℃ 6%O2+134 mg·m−3 NO+76 mg·m−3 NH3+16 mg·m−3 HCl+N2 4 SCR+SO2烟气组分 200、300和400 ℃ SCR组分+1 429 mg·m−3 SO2 5 SCR+HCl+SO2烟气组分 200、300和400 ℃ SCR组分+16 mg·m−3 HCl+1 429 mg·m−3 SO2 | Show Table DownLoad:
CSV
DownLoad:
CSV
η=CHg2+CHgt×100% (1) 其中:η为汞氧化率,%;CHg2+和CHgt 分别代表反应管出口烟气Hg2+浓度和入口烟气总汞浓度,μg·m−3。所有数据为3组平行实验的平均值,实验数据相对误差不超过10%。
2. 结果与讨论
2.1 不同Ce基催化剂的汞氧化性能
在基本烟气组分条件下,本研究对比了Ce5Ti、Ce5W9Ti和Cu5Ce5W9Ti催化剂的汞氧化性能。如图1所示,Ce作为一种中温活性组分,Ce5Ti和Ce5W9Ti在中温区间(250~400 ℃)表现出最佳的汞氧化性能,Ce5Ti和Ce5W9Ti催化剂汞氧化率分别达到58.4%~69.1%和68.8%~81.3%。Ce5Ti和Ce5W9Ti在低温区间(100~200 ℃)的汞氧化率普遍较低,分别低于53.1%和62.5%,表现出较差的低温活性。CuO掺杂的Cu5Ce5W9Ti表现出最佳的汞氧化性能,在300 ℃下汞氧化率达到最高,为88.8%。在中温区间,Cu5Ce5W9Ti的汞氧化率稳定在86.3%~88.8%。反应温度进一步上升到450 ℃,3种催化剂的汞氧化率均降低,说明温度过高使CeO2的催化氧化活性降低。与Ce5Ti和Ce5W9Ti相比,Cu5Ce5W9Ti在低温区间(100~200 ℃)表现出较高的汞氧化率,为76.3%~85.5%,表明CuO的掺杂可显著提升Ce基催化剂低温氧化活性。
 图 1 Ce基催化剂对汞的氧化性能(基本烟气组分)Figure 1. Mercury oxidation efficiency of various Ce-based catalysts (basic flue gas)
图 1 Ce基催化剂对汞的氧化性能(基本烟气组分)Figure 1. Mercury oxidation efficiency of various Ce-based catalysts (basic flue gas)2.2 SCR烟气组分对Ce基催化剂汞氧化性能影响
本研究对比了3种催化剂在基本烟气组分和SCR烟气组分条件下的汞氧化活性的差异性。如图2所示,在300 ℃下,相较于基本烟气组分,Ce5Ti催化剂在SCR烟气组分下汞氧化率明显降低,由62.7%下降到37.2%。在SCR烟气组分条件下,Ce5W9Ti催化剂汞并没有表现出明显的降低,汞氧化率由73.8%下降到62.4%,表明WO3掺杂明显提高了Ce5W9Ti催化剂对SCR反应条件的适应性。有研究[14-15]表明,SCR烟气组分可降低催化剂的汞氧化活性,这主要归咎于NH3与Hg0对活性位点的竞争吸附作用,高浓度的NH3分子明显降低了Hg0与活性位点接触的可能性。Cu5Ce5W9Ti催化剂SCR烟气组分条件下汞氧化表现与Ce5W9Ti相似,汞氧化率略微降低,由88.8%下降到81.3%,依然表现出较好的SCR烟气组分的适应性。
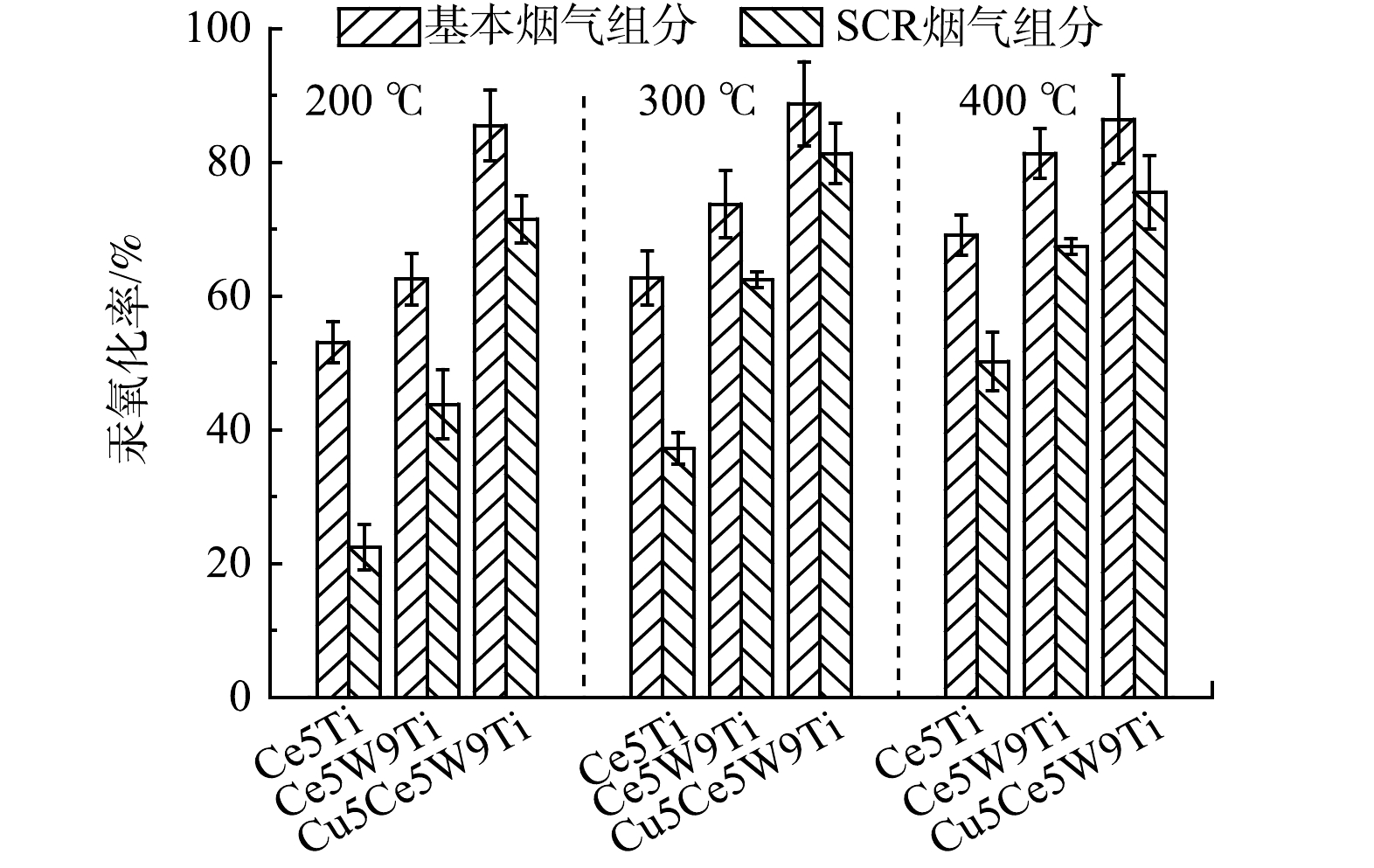 图 2 SCR烟气组分对Ce基催化剂汞氧化性能影响Figure 2. Mercury oxidation efficiency of various Ce-based catalysts at SCR flue gases
图 2 SCR烟气组分对Ce基催化剂汞氧化性能影响Figure 2. Mercury oxidation efficiency of various Ce-based catalysts at SCR flue gases在200 ℃和400 ℃的条件下,SCR烟气组分对催化剂具有相似的的影响。在200 ℃下,SCR烟气组分对Ce5Ti和Ce5W9Ti催化剂汞氧化抑制作用较为明显,而在400 ℃下,SCR烟气组分的抑制作用相对减少。这可能是因为Ce5Ti和Ce5W9Ti催化剂缺失低温活性组分,低温条件下NH3吸附量增加,汞氧化性能较低;而高温条件下NH3吸附量降低,减少了对Hg0抑制吸附作用,提高催化剂汞氧化性能。
2.3 SO2和HCl组分对Ce5W9Ti和 Cu5Ce5W9Ti催化剂汞氧化性能影响
SO2和HCl是燃煤烟气普遍存在的酸性气体,对催化剂汞氧化性能具有重要影响。通过以上实验筛选,本研究对比了Ce5W9Ti和 Cu5Ce5W9Ti催化剂在SCR烟气组分、SCR+SO2烟气组分、SCR+HCl烟气组分以及SCR+SO2+HCl烟气组分中的汞氧化性能变化,分析SO2和HCl对Ce5W9Ti和Cu5Ce5W9Ti催化剂影响。
如图3所示,在SCR烟气组分(300 ℃)下,1 429 mg·m−3 SO2 的添加明显降低了Ce5W9Ti催化剂的汞氧化性能,汞氧化率从62.4%降低到41.4%,表明SO2严重抑制了Ce5W9Ti催化剂汞氧化活性。有研究表明:高浓度SO2极容易吸附在CeO2活性位点,形成稳定的硫酸铈,使Ce基活性位丧失储氧能力[14]。SO2同样降低了Cu5Ce5W9Ti催化剂的汞氧化性能,但汞氧化率下降幅度较小,由81.3%下降到78.8%。该结果表明CuO的掺杂明显提高了催化剂的抗硫性能,可能是CuO的掺杂增加了反应活性位点或是保护了Ce基活性位点,使得Cu5Ce5W9Ti在SO2存在条件下依然具有较高的汞氧化性能。在200 ℃下,SO2对Ce5W9Ti和Cu5Ce5W9Ti的汞氧化性能抑制作用进一步提高。常化振等[16]研究表明,在低温条件下SO2与NH3形成大量的亚硫酸氢铵,覆盖了催化剂表面的活性位点,导致催化剂活性较低。在400 ℃条件下,SO2的抑制作用减弱,可能因为高温抑制了SO2吸附和亚硫酸氢铵的形成[16]。
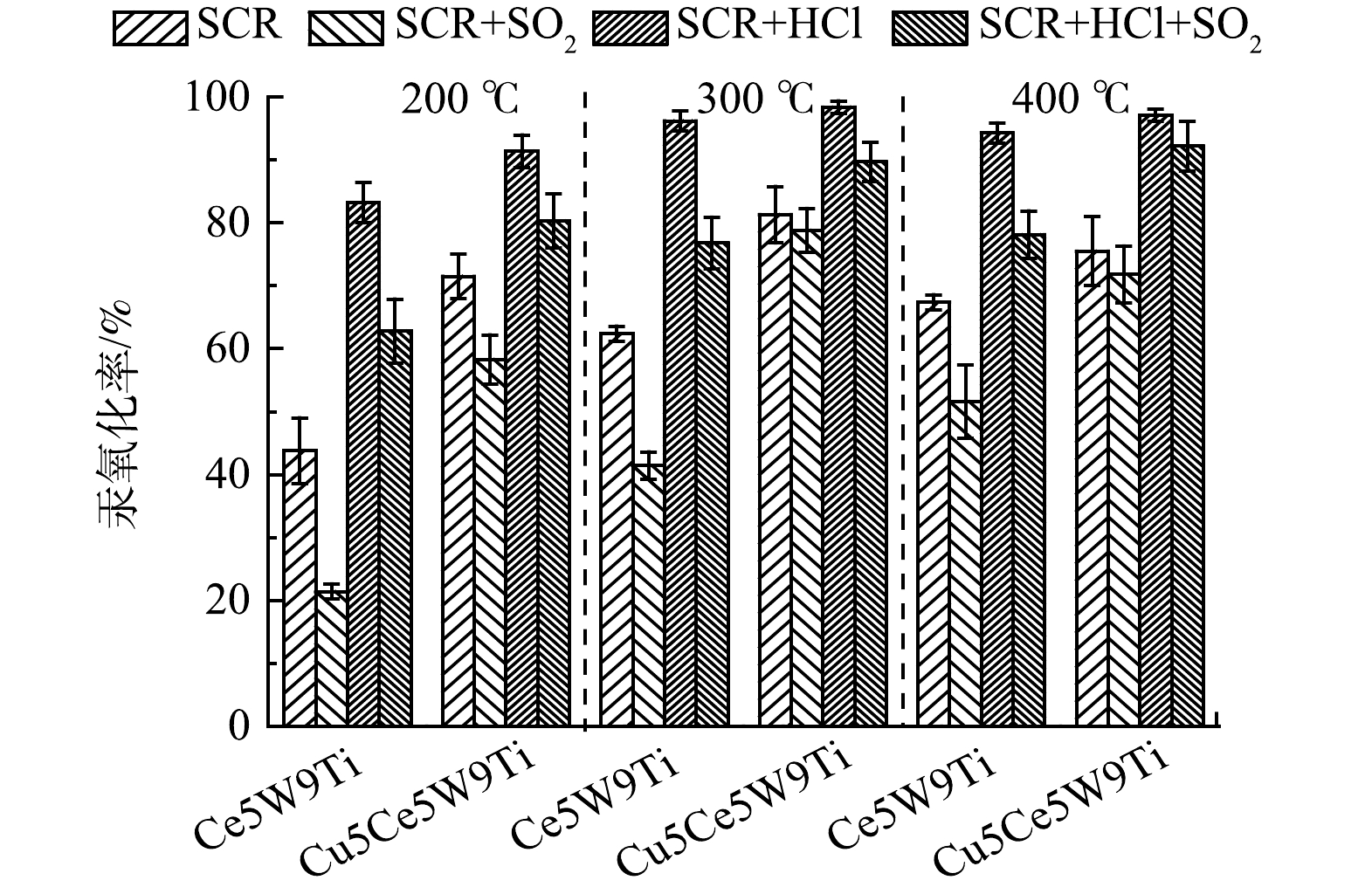 图 3 SO2和HCl对Ce基催化剂汞氧化性能影响Figure 3. Mercury oxidation efficiency of various Ce-based catalysts at the presence of SO2 and HCl
图 3 SO2和HCl对Ce基催化剂汞氧化性能影响Figure 3. Mercury oxidation efficiency of various Ce-based catalysts at the presence of SO2 and HClHCl的添加明显提高了Ce5W9Ti和 Cu5Ce5W9Ti催化剂汞氧化性能,汞氧化率分别提高到96.2%和98.4%。该结果表明HCl的添加可能增加了新的活性位点,促进了汞氧化率。进一步添加1 429 mg·m−3 SO2到模拟烟气,Ce5W9Ti和 Cu5Ce5W9Ti催化剂出现下降,但Ce5W9Ti和 Cu5Ce5W9Ti催化剂在SCR+HCl+SO2烟气组分下汞氧化率(76.8%和89.7%)依然明显高于SCR组分下汞氧化率(62.4%和81.3%)。该结果表明相较于SO2,HCl更具有竞争力,在催化剂表面依然形成新反应活性位,促进了Hg0的吸附和氧化。在200 ℃和400 ℃下,HCl显著提高了Ce5W9Ti和 Cu5Ce5W9Ti的汞氧化率和抗硫性能。
2.4 理化表征
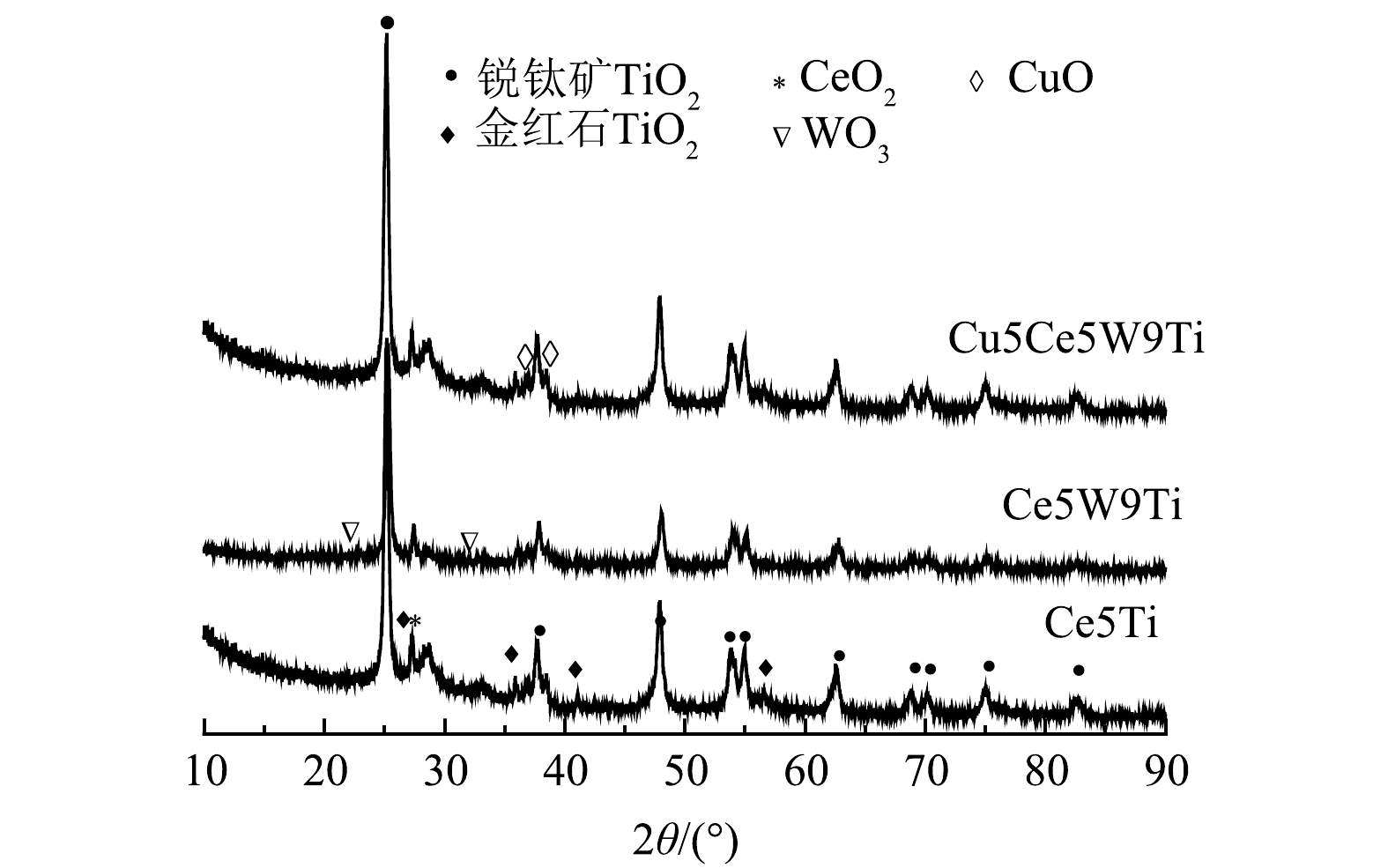
1) XRD分析。采用XRD技术分析了不同Ce基催化剂的晶相(图4)。3种催化剂XRD谱图主要以锐钛矿和红金石2种形态TiO2特征峰为主(JCPDS 21-
1272 ),符合载体P25组分80.0%锐钛矿和20.0%红金石组成特征。3种催化剂在2θ=28.35°位置出现一个微弱的CeO2晶峰,表明Ce在载体表面高度分散,具备较高的催化氧化活性。Ce5W9Ti和 Cu5Ce5W9Ti催化剂都没有出现明显的WO3晶峰,说明元素W在催化剂表面同样高度分散。Cu5Ce5W9Ti催化剂在35.5o和38.6o位置出现了微弱的CuO(111)特征峰(JCPDS 48-1548 ),证明Cu活性组分绝大部分均匀分布在催化剂表面。基于以上结果,Ce、W和Cu元素在载体表面高度均匀分散[13]。2) TEM分析。采用TEM分析了不同Ce基催化剂表面的元素晶体形态,结果见图5。如图5(a)所示,催化剂载体P25主要以立方体形态存在。如图5(b)和图5(c)所示,Ce5Ti和Ce5W9Ti催化剂表面CeO2以(111)晶型存在,晶格间距为0.31 nm,与XRD的分析结果一致。Ce5W9Ti催化剂表面没有发现WO3晶体,但Ce与Ti掺杂形成钛铀矿CeTi2O6(110)[17],晶体间距为0.34 nm,说明W元素的高度分散有利于Ce与Ti原子的螯合掺杂,可形成稳定的氧空位。如图5(d)所示,Cu5Ce5W9Ti催化剂表面出现间距为0.28 nm的CuO(110)晶格条纹和间距为0.31 nm的CeO2(111)晶格条纹,CuO与CeO2界面形成掺杂晶体界面。在界面位置,CuO、CeO晶体中的原子被相互替代,形成晶体错位,晶体条纹边界发生扭曲转移,促进大量Cu+和Ce3+形成,使催化剂表面形成氧空位,从而提高催化剂的储氧和氧化还原能力[18]。
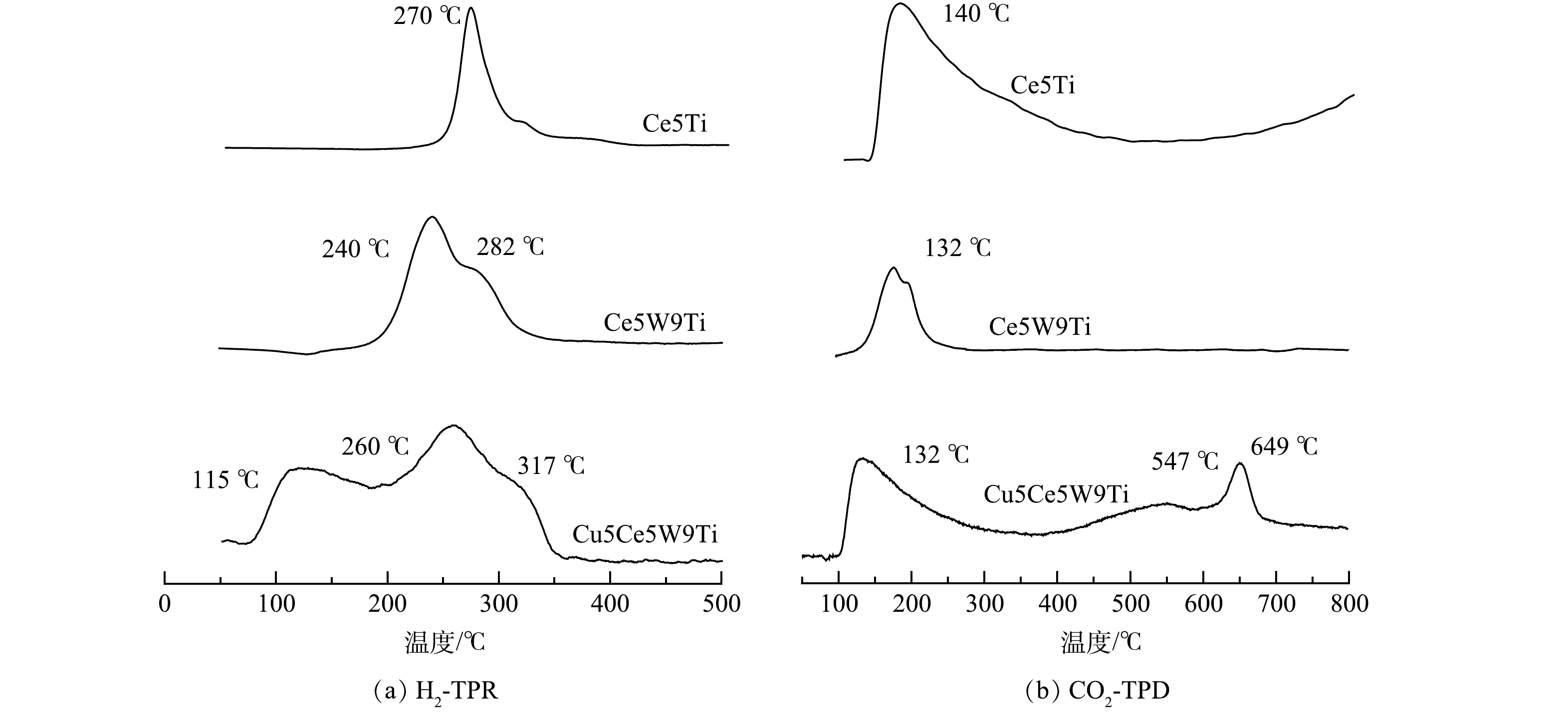
3) TPR和TPD分析。本研究采用H2-TPR和CO2-TPD技术分析了Ce5Ti、Ce5W9Ti和 Cu5Ce5W9Ti催化剂氧化位点和活性位点酸碱性。如图6(a)所示,Ce5Ti催化剂H2-TPR图谱出现一个270 ℃的主峰,代表CeO2形成的氧空位或活性位点。Ce5W9Ti催化剂的H2-TPR图谱中出现240 ℃和282 ℃ 2个主峰,结合TEM分析结果,除了CeO2活性位点,Ce与Ti掺杂形成的钛铀矿CeTi2O6(110)建 立新的活性位点[17]。因此,Ce5Ti、Ce5W9Ti催化剂在中温区间(250~400 ℃)表现出良好的汞氧化性能。如图6(a)所示,Cu5Ce5W9Ti催化剂的H2-TPR图谱中出现115、260和317 ℃ 3个主峰。260 ℃和317 ℃代表中温CeO2活性位点和Ce-Ti活性位点,115 ℃代表低温CuO活性位点,表明CuO的掺杂可提高催化剂在低温区域活性位点的形成。因此,Cu5Ce5W9Ti催化剂在图1表现出较高的低温(100~200 ℃)汞氧化性能。
如图6(b)所示,通过CO2-TPD分析了3种催化剂表面活性位点的酸碱性。有研究[19-20]表明,根据CO2脱附温度可将活性位点酸碱性分为弱碱(100~250 ℃)、中强碱(250~600 ℃)和强碱(600~800 ℃)。在Ce5Ti的CO2-TPD图谱中出现一个脱附主峰(140 ℃),表明CO2主要为弱碱活性位点;Ce5W9Ti的脱附主峰温度进一步降低到132 ℃,表明酸性氧化物WO3在催化剂表面的高度分散可进一步降低催化剂碱性强度。Cu5Ce5W9Ti的CO2-TPD图谱包含3个特征峰,即132、547和649 ℃ [21-22]。在SCR+SO2烟气条件下,强酸性气体SO2优先与碱性活性位(Cu—O—Ce和CuO)反应,从而保护了CeO2活性位依然保持高活性和氧化性,因此,Cu5Ce5W9Ti在SCR+SO2烟气条件下依然表现出良好的汞氧化性能(图3)。
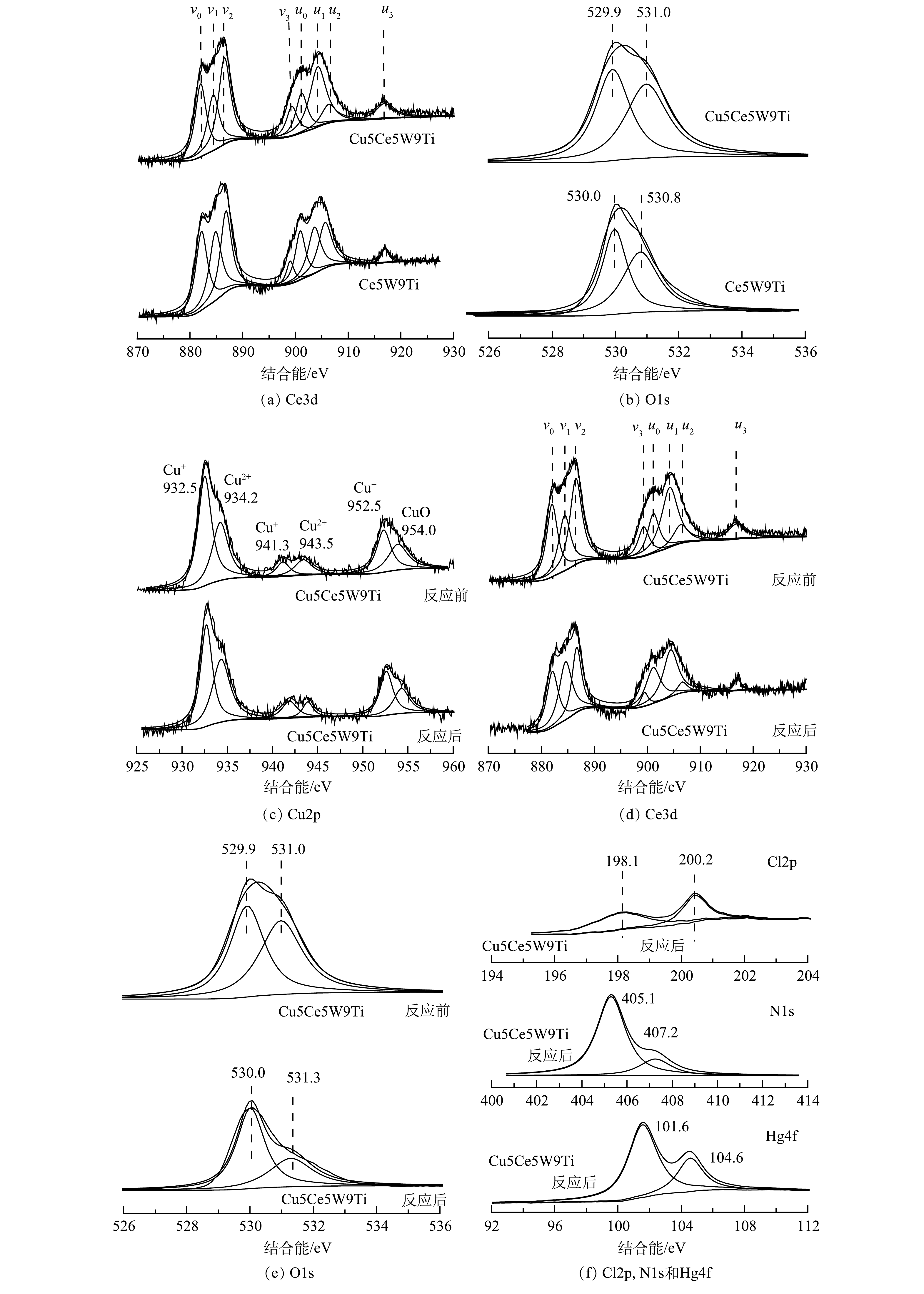
4) XPS分析。本研究采用XPS技术分析催化剂表面各种元素价态,探究催化剂汞氧化机制。如图7(a)所示,Ce元素能谱图包含8个分峰:u0、u1、u2、u3、v0、v1、v2和v3。其中u1和v1代表Ce3+;u0、u2、u3、v0、v2和v3代表Ce4+,Ce3+一般作为氧空位或是吸附氧位点[23]。Ce5W9Ti催化剂Ce3+峰面积和Ce4+峰面积比例(Ce3+/Ce4+)为31.2%,Cu5Ce5W9Ti 的Ce3+/Ce4+面积比例上升到39.2%。该结果与TEM分析结果一致,说明CuO与CeO2掺杂形成晶体错位,形成更多Ce3+和氧空位。
如图7(b)所示,Ce5W9Ti和Cu5Ce5W9Ti催化剂表面O元素2个特征峰:529.9~530.0 eV特征峰代表晶格氧,530.8~531.0 eV特征峰代表活性氧[24-25]。活性氧一般是氧空位吸附的高活性吸附氧,为Hg0氧化的关键活性位点。Ce5W9Ti催化剂活性氧特征峰面积占比为38.7%,Cu5Ce5W9Ti的活性氧特征峰面积占比为45.8%,该结果与Ce3+/Ce4+面积比例上升一致。如图7(c)所示,Cu5Ce5W9Ti催化剂表面在932.5 eV和934.2 eV的2个主要特征峰分别代表Cu+和CuO[26]。Cu5Ce5W9Ti催化剂表面Cu主要以Cu+形式存在,这与Cu、Ce原子掺杂形成氧空位界面保持一致。Cu5Ce5W9Ti催化剂反应前后Cu+/CuO比例由69.4%下降到57.2%,这可能是部分Cu+形成的活性氧与SO2反应形成大量的CuSO4,因此,Cu2+比例明显上升。如图7(d)所示,Cu5Ce5W9Ti 的Ce3+/Ce4+面积比例由反应前的39.2%下降到反应后的35.4%,说明部分Ce3+活性位在反应过程中转化为稳定的Ce4+,降低了催化剂的吸氧释氧能力。如图7(e)所示,Cu5Ce5W9Ti催化剂反应前后表面O元素2个特征峰发生明显变化。Cu5Ce5W9Ti催化剂活性氧特征峰面积占比有反应前的45.8%下降到反应后的35.3%,说明大量活性氧参与了Hg0氧化和NOx还原反应。
XPS表征没有发现反应前的Cu5Ce5W9Ti催化剂表面存在Cl、N和Hg原子。如图7(f)所示,SCR+SO2+HCl条件反应后的Cu5Ce5W9Ti催化剂表面Cl、N和Hg元素的价态。Cu5Ce5W9Ti催化剂表面Cl存在Cl−离子(198.1 eV)和活性Cl*(200.2 eV)2个特征峰[17, 23]。大面积活性Cl*特征峰的出现说明HCl在Cu5Ce5W9Ti催化剂表面吸附并转化为活性Cl*,促进Hg0转化为HgCl2。NO在催化剂表面XPS能谱图出现硝酸根(407.2 eV)和NO2(405.1 eV)2个特征峰。NO2特征峰面积要远大于硝酸根特征峰面积,说明NO在催化剂上主要以更高活性的NO2或是亚硝酸盐官能团形式存在。反应后的Cu5Ce5W9Ti催化剂表面存在Hg元素101.6 eV和104.6 eV两个特征峰,表明吸附在催化剂表明的汞主要以 Hg2+形式存在。
2.5 Ce基催化剂汞氧化机制分析
基于3种Ce基催化剂汞氧化实验和理化性质表征结果,本研究发现单一活性位点的Ce5Ti和Ce5W9Ti催化剂中温区间(250~400 ℃)表现出较高汞催化氧化活性(图1),但在SCR烟气组分下Ce5Ti的汞氧化率明显下降(图2),这归咎于NH3与Hg0竞争Ce基活性位点。WO3的掺杂提高了Ce5W9Ti在SCR烟气组分下的汞氧化率,为62.4%。CO2-TPD分析结果说明WO3的掺杂增加了Ce5W9Ti催化剂酸性(图6),增加了强碱性气体NH3的吸附位点,降低了NH3对Hg0吸附氧化竞争。同时XRD和TEM分析结果表明高度分散的WO3可促进Ce与Ti的耦合掺杂形成钛铀矿CeTi2O6(110),提高氧空位的比例。CuO的掺杂提高了Cu5Ce5W9Ti催化剂在低温区间(100~200 ℃)的汞氧化率(76.3%~85.5%),并且在SCR和SCR+SO2烟气条件下表现出较高的汞氧化率(81.3%和78.8%)。H2-TPR分析结果说明Cu掺杂增加Cu5Ce5W9Ti催化剂低温(115 ℃)活性位点,因此,提高催化剂的低温氧化活性;CO2-TPD分析结果表明W、Cu掺杂使Cu5Ce5W9Ti催化剂具备弱碱、中强碱、强碱3种活性位,可有效地分离酸性气体SO2、碱性气体NH3和Hg0吸附氧化反应区域,从而提高催化剂的抗硫性(图6)。TEM分析说明CuO和CeO2晶体掺杂的界面,发生原子替代和晶体错位,形成更多的Ce3+和Cu+作为氧空位,吸附更多的活性氧作为氧化位点,这一结论也在H2-TPR和XPS分析中得到证实(图6(a)和图7)。HCl和NO可与催化剂表面的活性氧反应,形成新的活性位点Cl*和NO2,促进Hg0氧化。综上所述,单一的活性位点难以满足NOx还原和Hg0协同控制;基于NH3、SO2和Hg0酸碱性的差异,构建强弱碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力,是提高催化剂协同污控性能和抗硫性重要途径。
3. 结论
1)单一活性位点的Ce5Ti和Ce5W9Ti催化剂在中温区间(250~400 ℃)表现出较高的汞氧化率,分别为58.4%~69.1%和68.8%~81.3%,但在低温区间(100~200 ℃)下的汞氧化率较低(低于53.1%和62.5%)。CuO掺杂提高了Cu5Ce5W9Ti催化剂低温活性,提高低温区间(100~200 ℃)汞氧化率至76.3%~85.5%。
2) WO3以不定晶型的形式高度分散于催化剂的表面,可促进Ce与Ti原子的替代和掺杂,形成钛铀矿CeTi2O6(110),提高氧空位的暴露;同时酸性氧化物WO3的掺杂可增加催化剂表面酸性活性位点,促进强碱性气体NH3的吸附,进而降低NH3和Hg0对Ce基氧化位点的竞争吸附,因此,Ce5W9Ti催化剂在SCR烟气组分下依然保持较高的汞氧化性能。
3) CuO、WO3引入在Cu5Ce5W9Ti催化剂表面形成强弱碱性多反应活性区域,分离NH3、SO2和Hg0吸附反应区域,降低NH3、SO2和Hg0的竞争吸附,提高催化剂Hg0氧化能力和抗硫性能,为多污控措施协同控制汞排放提供关键技术理论。
-
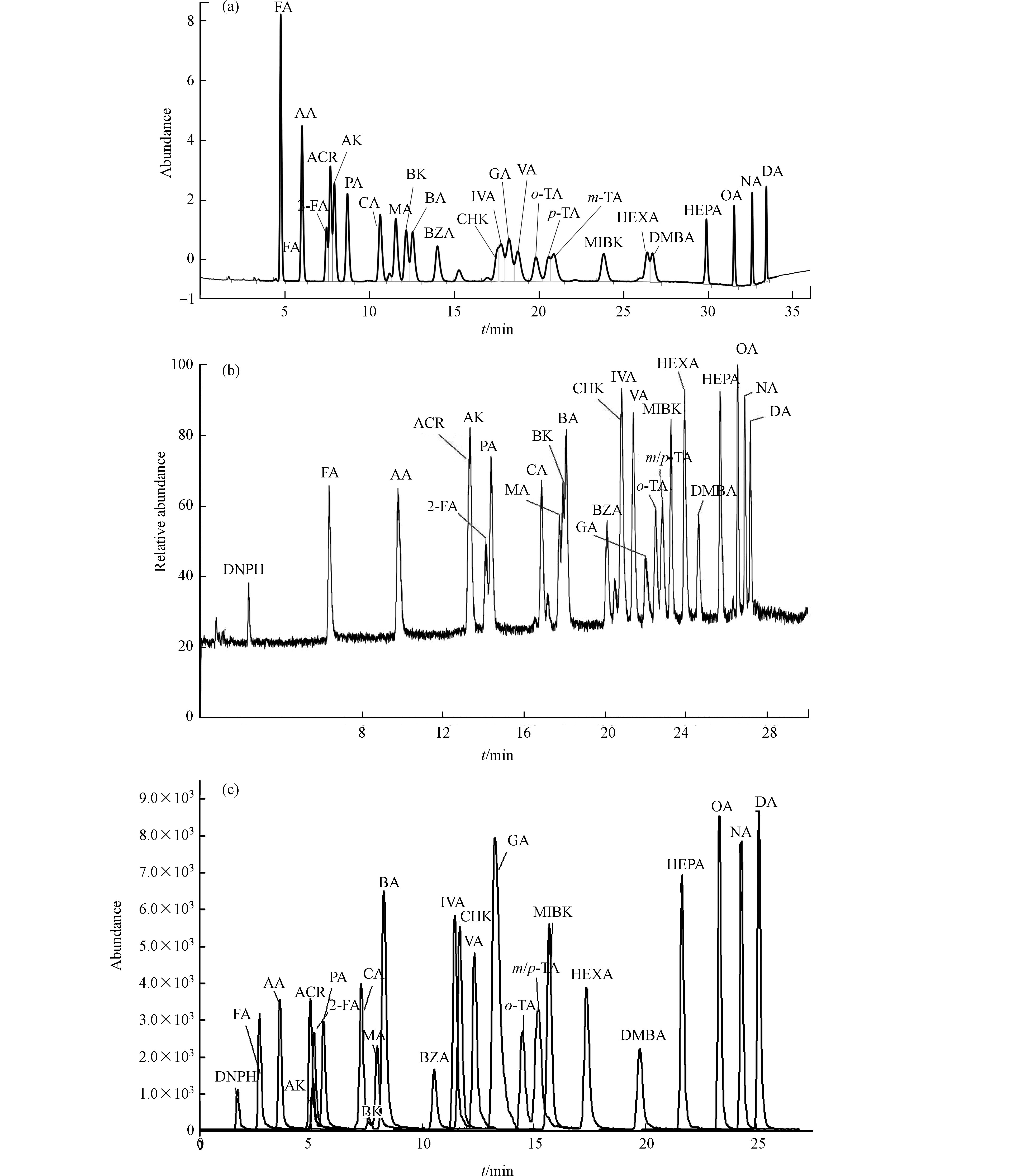
图 1 25种醛酮腙标样UV色谱图/质谱图(a)UV色谱图;(b)Orbitrap MS提取母离子质谱图;(c)MS/MS 提取离子质谱图.
Figure 1. Chromatograms of 25 carbonyl derivatives: (a) by UVD; (b) by Orbitrap MS; and (c) by MS/MS.
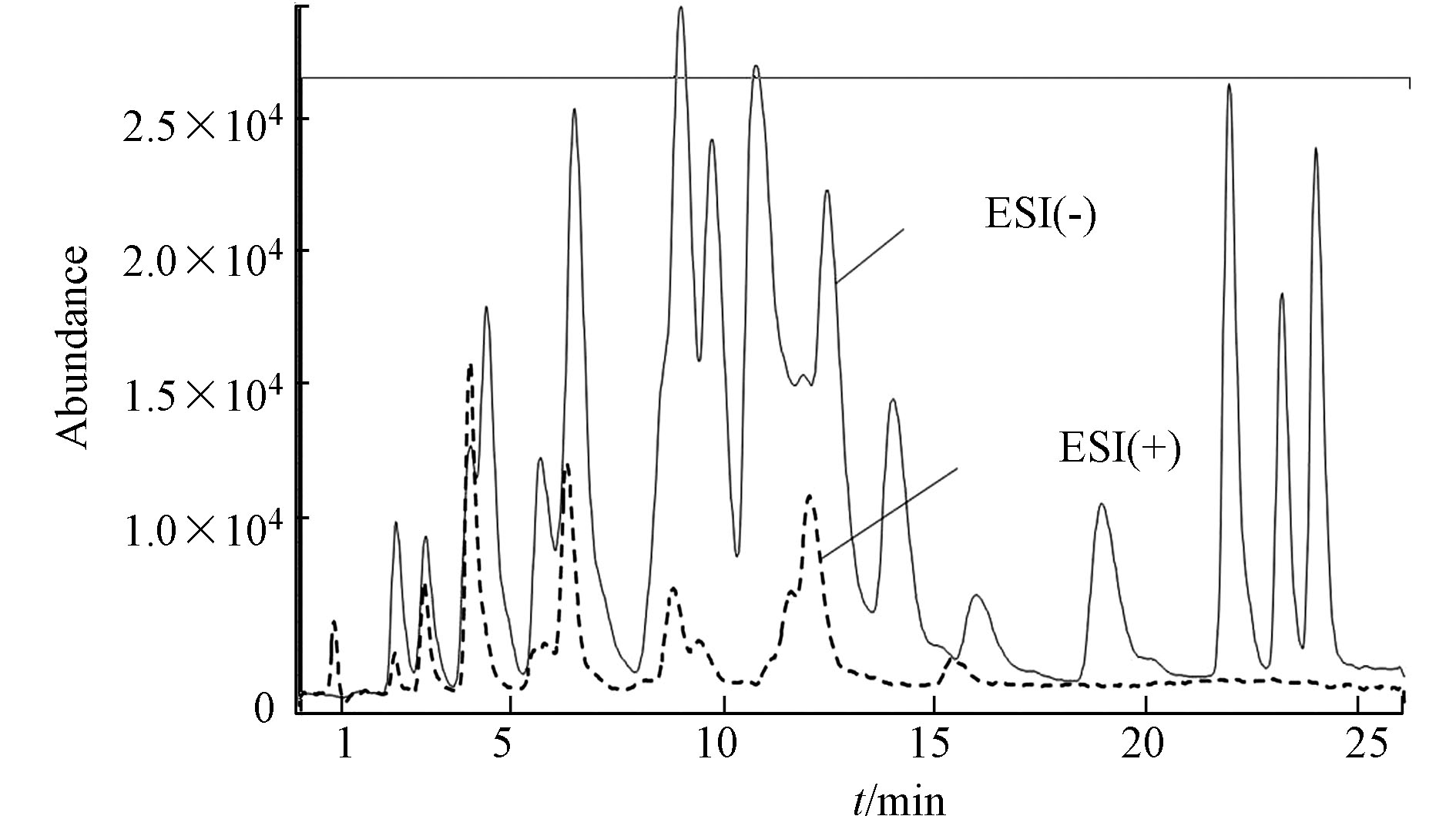
图 2 25种醛酮腙标样ESI(+/−)-MS/MS总离子流比较
Figure 2. TIC abundance comparison of 25 carbonyl hydrazones by ESI(+/−)-MS/MS
表 1 目标化合物的多反应监测条件
Table 1. MRM parameters for target compounds
序号No. 化合物缩写Compound abbreviation 化合物Compound 分子式Molecular formula 母离子Precursor ions(m/z) 子离子Product ions(m/z) 碎裂电压/VFragmenter 碰撞能/eVCE 1 DNPH 2,4-二硝基苯肼2,4-dinitrophenylhydrazine C6H6N4O4 197 137 60 8 167.1 4 2 FA 甲醛-DNPHFormaldehyde-DNPH C7H6N4O4 209 163.1 100 0 151.1 0 3 AA 乙醛-DNPHAcetaldehyde-DNPH C8H8N4O4 223 46.1 100 16 163.1 0 4 ACR 丙烯醛-DNPHAcrolein-DNPH C9H8N4O4 235 158.2 100 4 163.1 28 5 PA 丙醛-DNPHPropionaldehyde-DNPH C9H10N4O4 237.1 46.1 100 20 163.1 16 6 AK 丙酮-DNPHActone-DNPH C9H10N4O4 237.1 46.1 100 20 122.0 0 7 CA 巴豆醛/丁烯醛-DNPHCrotonaldehyde-DNPH C10H10N4O4 249.1 172.1 100 4 46.2 12 8 MA 甲基丙烯醛-DNPHMethacrolein-DNPH C10H10N4O4 249.1 46.2 100 12 172.1 4 9 BK 丁酮-DNPHButanone-DNPH C10H12N4O4 251.1 152.1 100 8 122.0 16 10 BA 丁醛-DNPHButylaldehyde-DNPH C10H12N4O4 251.1 152.1 100 8 122.0 16 11 IVA 异戊醛-DNPHIsovaleraldehyde-DNPH C11H14N4O4 265.1 152.1 100 12 163.1 4 12 VA 戊醛-DNPHValeraldehyde-DNPH C11H14N4O4 265.1 152.1 100 12 163.1 4 13 2-FA 糠醛-DNPH2-Furaldehyde-DNPH C11H8N4O5 275 46.1 100 12 228.0 4 14 CHK 环己酮-DNPHCyclohexanone-DNPH C12H14N4O4 277.1 247.2 100 4 231.1 12 15 HEXA 己醛-DNPHHexaldehyde-DNPH C12H16N4O4 279.1 152.0 100 12 122.0 28 16 MIBK 甲基异丁基酮-DNPHMethyl Isobutyl Ketone-DNPH C12H16N4O4 279.1 152.0 100 12 122.0 28 17 BZA 苯甲醛-DNPHBenzaldehyde-DNPH C13H10N4O4 285.1 46.2 100 20 163.1 20 18 HEPA 庚醛-DNPHHeptaldehyde-DNPH C13H18N4O4 293.1 152.1 100 12 163.2 4 19 o-TA 邻-甲基苯甲醛-DNPHo-Tolualdehyde-DNPH C14H12N4O4 299.1 162.9 100 8 252.1 8 20,21 p,m-TA 对,间-甲基苯甲醛-DNPHp,m-Tolualdehyde-DNPH C14H12N4O4 299.1 162.9 100 8 252.1 8 22 OA 辛醛-DNPHOctanal-DNPH C14H20N4O4 307.1 152.1 100 20 163.0 8 23 DMBA 2,5-二甲基苯甲醛2,5-Dimethylbenzaldehyde-DNPH C15H14N4O4 313.1 181.0 140 20 163.0 20 24 NA 壬醛-DNPHNonanal-DNPH C15H22N4O4 321.1 152.0 140 20 46.1 56 25 DA 癸醛-DNPHDecanal-DNPH C16H24N4O4 335.2 152.1 140 20 163.0 8 26 GA 戊二醛-DNPHGlutaraldehyde-DNPH C17H16N8O8 459.1 182.1 140 12 179.0 8
下载: 导出CSV
表 2 不同方法测定25种醛酮类化合物线性方程、相关系数、方法检出限比较
Table 2. Comparison of linear equations, correlation Coefficients, method detection limits of 25 carbonyl derivatives by different methods
化合物compound HPLC-UV法 HPLC-ESI-MS/MS法 HPLC-ESI-Q-Orbitrap MS法 线性方程Linear equations 线性范围/ (µg·L−1)Linear range 相关系数r 检出限/ (µg·m−3)MDL 线性方程Linear equations 线性范围/(µg·L−1)Linear range 相关系数r 检出限/ (µg·m−3)MDL 线性方程Linear equations 线性范围/(µg·L−1)Linear range 相关系数r 检出限/ (ng·m−3)MDL FA y=0.362x+0.807 3.0—1500 0.999 0.12 y=226x+55.6 15—300 0.999 0.15 y=7.43e6x+2.026e6 0.75—300 0.999 7.5 AA y=0.267x+0.670 7.5—1500 0.999 0.15 y=259x+130 7.5—300 0.997 0.08 y=1.0917x−3.0536 0.75—300 0.999 4.6 2-FA y=0.0981x+0.256 15—1500 0.999 0.25 y=143x+181 7.5—300 0.996 0.08 y=1.06e7x−6.78e6 0.75—300 0.999 6.5 ACR y=0.231x+0.679 15—1500 0.999 0.15 y=213x+80.2 7.5—300 0.994 0.08 y=1.47e6x−1.57e6 0.75—300 0.999 4.5 AK y=0.209x+0.486 15—1500 1.000 0.16 y=121x+176 7.5—300 0.995 0.08 y=6.87e6x−1.16e6 1.50—300 0.999 15 PA y=0.207x+0.524 15—1500 0.999 0.16 y=430x+545 7.5—300 0.997 0.08 y=1.16e7x−5.58e6 0.75—300 0.999 8.5 CA y=0.182x+0.365 15—1500 0.999 0.20 y=342x+396 7.5—300 0.997 0.08 y=1.53e7x−5.54e6 0.75—150 0.999 9.1 MA y=0.184x+0.722 15—1500 0.999 0.20 y=352x+290 7.5—300 0.999 0.08 y=1.51e7x−6.12e5 0.45—300 0.999 5.1 BK y=0.153x+0.338 15—1500 0.999 0.21 y=764x−10.6 7.5—300 0.994 0.08 y=7.04e6x−3.74e6 1.50—300 0.999 15 BA y=0.160x+0.388 15—1500 0.999 0.21 y=889x+109 7.5—300 0.998 0.08 y=1.47e7x−9.78e4 0.75—300 0.999 7.5 BZA y=0.125x+0.255 15—1500 0.999 0.25 y=165x+70.3 15—300 0.998 0.15 y=1.58e7x-4.47e6 0.45—75.0 0.999 4.5 CHK y=0.0911x+0.310 30—1500 0.999 0.28 y=191x+172 30—300 0.998 0.30 y=7.20e6x-2.61e6 0.45—150 0.999 3.8 IVA y=0.113x+1.16 30—1500 0.999 0.30 y=538x+109 7.5—300 0.998 0.08 y=3.14e7x−9.02e6 0.45—300 0.999 4.1 GA y=0.191x+1.10 30—1500 0.999 0.30 y=1939x1549 7.5—300 0.998 0.08 y=1.69e7x−2.16e6 1.50—75.0 0.999 15 VA y=0.119x+1.33 30—1500 0.999 0.35 y=798x+29.3 7.5—300 0.999 0.08 y=1.50e7x+1.90e6 0.45—300 0.999 4.5 o−TA y=0.105x+1.50 30—1500 0.999 0.38 y=325x+276 30—300 0.995 0.30 y=1.63e7x−6.86e6 0.75—75.0 0.999 5.5 p−TA y=0.0879x+0.496 30—1500 0.999 0.38 y=567x+283 60—600 0.997 0.60 y=2.66e7x+4.00e6 1.5—150 0.999 5.5 m−TA y=0.121x−0.0047 30—1500 0.999 0.38 MIBK y=0.120x+0.258 30—1500 0.999 0.38 y=838x+346 7.5—300 0.998 0.08 y=1.09e7x−6.70e6 0.45—300 0.999 4.5 DMBA y=0.102x+0.927 30—1500 0.999 0.40 y=278x+266 30—300 0.998 0.30 y=1.48e7x−4.44e6 0.45—75.0 0.999 3.5 HEXA y=0.0951x+0.241 30—1500 0.999 0.40 y=521x+266 15—300 0.993 0.15 y=1.58e7x+6.15e6 0.45—300 0.999 3.8 HEPA y=0.108x+0.378 15—1500 0.999 0.20 y=509x+111 7.5—300 0.997 0.08 y=1.53e7x+2.39e6 0.45—300 0.999 2.5 OA y=0.0954x+0.192 15—1500 0.999 0.20 y=553x+738 7.5—300 0.996 0.08 y=1.74e7x−4.66e6 0.45—75.0 0.999 1.1 NA y=0.0897x+0.117 15—1500 1.000 0.21 y=638x+116 7.5—300 0.994 0.08 y=1.81e7x−2.58e6 0.45—75.0 0.999 1.1 DA y=0.0840x+0.709 15—1500 0.999 0.21 y=854x+308 7.5—300 0.997 0.08 y=1.82e7x+2.24e6 0.45—75.0 0.999 1.1
下载: 导出CSV
表 3 UV法、MS/MS法和Orbitrap法精密度、准确度和实际样品测定结果比较
Table 3. Comparison of the accuracy, precision and real sample test results by UV, MS/MS and Orbitrap methods respectively
化合物Compound 加标样/% Spiked sample(n=6) 样品1/(µg·m−3) Sample 1 样品2/(µg·m−3) Sample 2 UV MS/MS Orbitrap MS UV MS/MS Orbitrap MS UV MS/MS Orbitrap MS 回收率Recovery RSD 回收率Recovery RSD 回收率Recovery RSD FA 94.7 10.4 90.3 12 97.9 7.1 2.31 2.06 2.25 2.45 2.70 2.56 AA 98.8 7.3 90.7 8.1 107 8.9 2.05 1.78 1.96 1.80 1.76 1.92 2-FA 83.6 7.1 86.1 7.9 88.5 5.6 ND ND 0.052 ND ND 0.017 ACR 75.8 7.5 67.9 6.9 74.1 6.2 ND 0.146 0.136 ND ND 0.049 AK 81.3 7.4 70.3 12 74.5 11 3.27 3.13 3.80 2.78 2.67 2.84 PA 90.9 5.6 89.5 13 94.5 7.1 0.33 0.25 0.300 0.25 0.22 0.229 CA 84.1 5.9 78.7 8.8 81.5 8.9 ND ND 0.10 ND ND 0.013 MA 85.3 6.6 80.2 9.1 83.3 7.8 ND ND 0.033 ND ND 0.016 BK 77.4 9.7 78.6 15 66.5 6.7 0.64 0.702 0.656 0.35 0.41 0.364 BA 68.9 5.5 75.6 8.2 85.3 7.9 0.41 0.468 0.477 0.22 0.26 0.235 BZA 91.1 5.6 92.5 9.6 97.4 9.7 ND ND 0.114 ND ND 0.043 CHK 75.6 8.8 70.7 14 86.7 8.2 ND 0.46 0.048 ND ND 0.040 IVA 80.9 7.4 78.9 17 94.8 9.9 ND 0.12 0.108 ND ND 0.027 GA 71.7 9.8 72.5 18 77.1 11 ND ND ND ND ND ND VA 79.3 5.8 75.1 12 99.4 7.5 ND 0.33 0.274 ND 0.12 0.117 o−TA 79.3 4.9 80.1 11 98.4 8.1 ND ND 0.015 ND ND ND p−TA 92.9 7.6 97.6 12 101 8.7 ND ND 0.009 ND ND ND m−TA 90.5 6.7 ND ND MIBK 82.2 5.4 78.4 8.8 77.7 6.2 ND ND 0.040 ND ND 0.015 DMBA 90.8 7.1 91.2 16 93.2 8.6 ND ND 0.024 ND ND 0.007 HEXA 84.8 7.6 83.4 8.8 79.4 9.2 ND 0.40 0.334 0.50 0.63 0.536 HEPA 86.5 7.3 78.3 11 81.0 7.8 ND 0.16 0.192 0.31 0.29 0.322 OA 93.2 8.4 87.8 9.3 90.9 5.7 0.47 0.54 0.518 0.83 0.78 0.895 NA 90.9 6.7 88.7 10 86.4 9.1 2.29 2.12 2.40 4.45 4.37 4.84 DA 93.4 7.9 92.2 15 90.4 7.7 0.43 0.31 0.336 0.77 0.71 0.820 注:ND., 未检出. ND., not detected.
下载: 导出CSV
-
[1] O’BRIEN P J, SIRAKI A G, SHANGARI N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health [J]. Crit Rev Toxicol, 2005, 35: 609-662. doi: 10.1080/10408440591002183 [2] SKYBOVA M, LENICEK J, RYCHTECKA A N, et al. Determination of volatile organic compounds in the atmosphere and their influence on ozone formation [J]. Fresenius Environmental Bulletin, 2006, 15(12): 1616-1623. [3] ATKINSON R. Gas-phase tropospheric chemistry of organic compounds: A review [J]. Atmos Environ, 1990, 24A: 1-41. [4] GARAYCOECHEA J I, CROSSAN G P, LANGEVIN F, et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells [J]. Nature, 2018, 553: 171-177. doi: 10.1038/nature25154 [5] PANG X, LEE X. Temporal variations of atmospheric carbonyls in urban ambient air and street canyons of a Mountainous city in Southwest China [J]. Atmospheric Environment, 2010, 44: 2098-2106. doi: 10.1016/j.atmosenv.2010.03.006 [6] HUANG J A, FENG Y L, FU J M, et al. A method of detecting carbonyl compounds in tree leaves in China [J]. Environmental Science and Pollution Research, 2010, 17: 1129-1136. doi: 10.1007/s11356-009-0277-3 [7] 崔婷惠, 李恒, 唐石云, 等. 超临界流体色谱与气相色谱-质谱联用测定卷烟主流烟气中醛酮类化合物的研究 [J]. 分析测试学报, 2018, 39(7): 766-771. doi: 10.3969/j.issn.1004-4957.2018.07.002 CUI T H, LI H, TANG S Y, et al. Determination of aldehydes and ketones compounds in mainstream cigarette smoke by supercritical fluid chromatography and gas chromatography-mass spectrometry [J]. Journal of Instrumental Analysis, 2018, 39(7): 766-771(in Chinese). doi: 10.3969/j.issn.1004-4957.2018.07.002
[8] 史纯珍, 姜锡, 姚志良, 等. 烹饪油烟羰基化合物排放特征 [J]. 环境工程学报, 2015, 9(3): 1376-1380. doi: 10.12030/j.cjee.20150364 SHI C Z, JIANG X, YAO Z L, et al. Carbonyl compounds emission characteristics in cooking fumes [J]. Chinese Journal of Environmental Engineering, 2015, 9(3): 1376-1380(in Chinese). doi: 10.12030/j.cjee.20150364
[9] KIM K H, CHOI B, PARK S, et al. Emission characteristics of compression ignition (CI) engine using diesel blended with hydrated butanol [J]. Fuel, 2019, 257(1): 1-9. [10] LI J, CHEN J Y, JI Y M, et al. Solar light induced transformation mechanism of allyl alcohol to monocarbonyl and dicarbonyl compounds on different TiO2: A combined experimental and theoretical investigation [J]. Chemosphere, 2019, 232(9): 287-295. [11] SUKHAREV S, MARIYCHUK R, ONYSKO M, et al. Fast determination of total aldehydes in rainwaters in the presence of interfering compounds [J]. Environmental Chemistry Letters, 2019, 17(3): 1405-1411. doi: 10.1007/s10311-019-00875-z [12] ZWIENER C, FRIMMEL F H. LC-MS analysis in the aquatic environment and in water treatment technology–a critical review [J]. Analytical and Bioanalytical Chemistry, 2004, 378: 862-874. doi: 10.1007/s00216-003-2412-1 [13] 崔连喜, 李利荣, 吴宇峰, 等. 液相色谱法测定土壤和沉积物中15种醛酮类化合物 [J]. 理化检验:化学分册, 2018, 54(4): 484-487. CUI L X, LI L R, WU Y F, et al. LC determination of 15 carbonyl compounds in soil and sediment [J]. Physical Testing and Chemical Analysis part B:Chemical Analysis, 2018, 54(4): 484-487(in Chinese).
[14] 李利荣, 关玉春, 吴宇峰, 等. 2, 4-二硝基苯肼衍生-固相萃取-液相色谱法测定环境固体基质中15种醛酮类羰基化合物的含量 [J]. 理化检验:化学分册, 2018, 54(7): 841-846. LI L R, GUAN Y C, WU Y F, et al. Determination of 15 aldehyde and ketone carbonyl compounds in environmental solid matrix by liquid chromatography with 2, 4-dinitrophenylhydrazine derivatization and solid phase extraction [J]. Physical Testing and Chemical Analysis part B:Chemical Analysis, 2018, 54(7): 841-846(in Chinese).
[15] QIAN X, SHEN H Q, CHEN Z M. Characterizing summer and winter carbonyl compounds in Beijing atmosphere [J]. Atmospheric Environment, 2019, 214: 116845. doi: 10.1016/j.atmosenv.2019.116845 [16] 景盛翱. 上海市典型区域大气羰基化合物水平研究 [J]. 环境污染与防治, 2017, 39(7): 713-716. SHENG J A. Study on the level of ambient carbonyl compounds in typical regions of Shanghai [J]. Environmental Pollution & Control, 2017, 39(7): 713-716(in Chinese).
[17] MANDIC A I, SEDEJ I J, SAKAC M B, et al. Static headspace gas chromatographic method for aldehyde determination in crackers [J]. Food Analytical Methods, 2013, 6(1): 61-68. doi: 10.1007/s12161-012-9415-5 [18] CALEJO I, MOREIRA N, ARAUJO A M, et al. Optimization and validation of a HS-SPME-GC-IT/MS method for analysis of carbonyl volatile compounds as biomarkers in human urine: Application in a pilot study to discriminate individuals with smoking habits [J]. Talanta, 2016, 148: 486-493. doi: 10.1016/j.talanta.2015.09.070 [19] PEREIRA E A, REZENDE M O O, TAVARES M F M. Analysis of low molecular weight aldehydes in air samples by capillary electrophoresis after derivatization with 4-hydrazinobenzoic acid [J]. J Sep Sci, 2004, 27(1-2): 28-32. doi: 10.1002/jssc.200301665 [20] GUO S J, CHEN M, TAN J H. Seasonal and diurnal characteristics of atmospheric carbonyls in Nanning, China [J]. Atmospheric Research, 2016, 169: 46-53. doi: 10.1016/j.atmosres.2015.09.028 [21] TIE C, HU T, JIA Z X, Zhang J L. Derivatization strategy for the comprehensive characterization of endogenous fatty aldehydes using HPLC-multiple reaction monitoring [J]. Anal Chem, 2016, 88: 7762-7768. doi: 10.1021/acs.analchem.6b01756 [22] OCHS S M, FASCIOTTI M, BARRETO R P, et al. Optimization and comparison of HPLC and RRLC conditions for the analysis of carbonyl-DNPH derivatives [J]. Talanta, 2010, 81(1-2): 521-529. doi: 10.1016/j.talanta.2009.12.036 [23] OCHS S M, FASCIOTTI M, NETTO A D P. Analysis of 31hydrazones of carbonyl compounds by RRLC-UV and TTLC-MS(/MS): A comparison of methods [J]. Journal of Spectroscopy, 2015(6b): 1-11. [24] ZUREK G, KARST U. Liquid chromatography-mass spectrometry method for the determination of aldehydes derivatized by the hantzsch reaction [J]. J Chromatogr A, 1999, 864: 191-197. doi: 10.1016/S0021-9673(99)01041-9 [25] 孟志娟, 孙文毅, 赵丽敏, 等. 气相色谱-静电场轨道阱高分辨质谱快速筛查农产品中 70 种农药残留 [J]. 分析化学, 2019, 47(8): 1227-1234. MENG Z J, SUN W Y, ZHAO L M, et al. Rapid screening of 70 kinds of pesticide residues in agricultural products by gas chromatography-electrostatic field orbit trap high resolution mass spectrometry [J]. Chinese Journal of Analytical Chemistry, 2019, 47(8): 1227-1234(in Chinese).
[26] 陈溪, 吴慈, 王龙祥, 等. 基于超高效液相色谱-高分辨质谱技术的水中 112 种药品和个人护理用品的高通量筛查和定量方法 [J]. 色谱, 2019, 36(11): 1147-1157. CHEN X, WU C, WANG L X, et al. High-throughput screening and quantitative analysis method of 112 pharmaceutical and personal care products in water based on ultra-high performance liquid chromatography with high-resolution mass spectrometry [J]. Chinese Journal of Chromatography, 2019, 36(11): 1147-1157(in Chinese).
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 4172
- HTML全文浏览数: 4172
- PDF下载数: 133
- 施引文献: 0
