





 下载:
下载:






-
氯代烃作为一种重要的有机溶剂和产品中间体,广泛地应用于机械、电子、皮革、干洗、化工等行业。由于氯代烃生产、使用、储存、处置不当[1],导致地下水中氯代烃污染的问题普遍存在[2]。我国氯代烃污染场地数量多,据文献统计:东部平原4×105个地下水污染场地中,大部分含有氯代烃污染[3-4]。氯代烃污染物毒性大、难降解,多数具有三致效应(致癌、致畸、致突变)[5],被美国国家环保局列为优先控制污染物[6-7],一旦进入水体可持续数十甚至数百年[3],将严重影响人类健康和生态环境。地下水中氯代烃污染修复难度大、经济成本高[8-9],因此,迫切需要有效、经济的技术来降解地下水中氯代烃。
氯代烃污染地下水的修复多采用原位修复方式进行[8, 10]。原位化学氧化技术(in-situ chemical oxidation,ISCO)是向污染区域原位注入氧化剂,使氯代烃污染物被氧化降解为无毒或毒性较小的物质,该技术适用范围广、降解效率高、修复周期短[11-12]。常见的化学氧化剂包括高锰酸盐、过氧化氢、芬顿试剂、过硫酸盐(指二硫酸盐)和臭氧[13-14],其中过硫酸盐的氧化还原电位接近臭氧,大于高锰酸盐、过氧化氢、芬顿试剂,氧化能力较强。臭氧和过氧化氢在地表留存时间短且臭氧在工程应用时需要钻孔通气,高锰酸盐易与含π键结构的有机物反应生成MnO2沉积物,芬顿试剂与活化剂反应速度快、易发生爆炸,而过硫酸盐在常态下更稳定、易溶于水、持久、易传质能很好地适应ISCO的工艺需求[13]。过硫酸盐氧化的主要有效成分是过硫酸根离子(
S2O82− ),可通过加热、过渡金属离子、紫外光、零价铁、碱等单一或复合方法进行活化[15-17]后产生硫酸根自由基(SO4− ·),活化后标准氧化还原电位有所提高,如式(1)~式(2)所示,且活化后反应速率加快[15]。pH是影响过硫酸盐活化的一个重要因素[18]。碱性条件下过硫酸盐本身及其活化产生的
SO−4 ·、OH·可引发一系列自由基链式反应[19],使其反应活性更高,如式(3)~式(6)所示。同时,碱活化可解决过硫酸盐降解水中污染物后反应体系酸化的问题[20],碱活化产生自由基过程缓和、活化效率高、反应时效长为1~3个月。国外关于碱活化过硫酸盐降解污染物的研究已进入工程应用阶段,自2014年以来国内相关研究进展迅速[21]。氧化药剂投加量是反应过程中的关键因素[22],但受污染物消耗、土壤还原性物质消耗等因素影响,在进行规模实施前应先通过实验确定适用的药剂投加比。药剂投加与有效扩散是ISCO的核心环节。常见药剂投加方式包括水力压裂、注射井、高压旋喷注射、浅层搅拌等[23-24],其中高压旋喷注射具有药剂扩散半径大、药剂与污染含水层混合均匀、施工便捷等优点,在国内已被广泛采用。
过硫酸盐与污染物发生氧化反应的产物为
SO2−4 和H+,反应后体系或环境pH会下降[25-26],同时会在一定环境范围内形成SO2−4 富集地区[27]。土壤中SO2−4 浓度一定程度上的增加可以促进微生物活动,但SO2−4 浓度过高会对土壤及地下水造成二次污染[26-28],且过高的SO2−4 会使地下水的腐蚀性增强而影响修复后地块的后续开发建设,另外,长期饮用含高浓度SO2−4 的水源会引发痢疾等疾病。因此,采用过硫酸盐体系氧化修复施工后,应对环境中残留SO2−4 进行跟踪监测、评价和必要的控制。本文以氯代烃污染场地的地下水为研究对象,通过小试实验验证碱活化过硫酸盐对氯代烃污染物的氧化去除效果,从而确定最佳药剂投加比,为场地后续规模化实施提供依据;为进一步验证该项技术对地下水中氯代烃的修复效果,选取地下水污染区域开展中试规模的工程化应用实验,采用高压旋喷注射工艺进行药剂原位注入,将氧化药剂按最佳投加比注入至污染含水层;实施后对地下水中的目标污染物和
SO2−4 的浓度进行跟踪采样监测,并研究地下水中氯代烃的修复效果,以及残留SO2−4 自然降解周期和规律。通过小试实验、中试规模工程应用、跟踪监测,为相关污染场地过硫酸盐氧化修复工程提供参考。
全文HTML












-
1)实验研究场地。所研究的典型氯代烃污染场地位于我国北方某退役化学试剂厂,场地内地下水主要受氯代烃污染严重,主要污染物为氯乙烯、顺-1,2-二氯乙烯、反-1,2-二氯乙烯、三氯乙烯等,污染地下水分布于3~7 m深度内的潜水层。在研究场地内有代表性的位置采集2个初始地下水样品,样品编号分别为GW1、GW2,结果如表1所示。
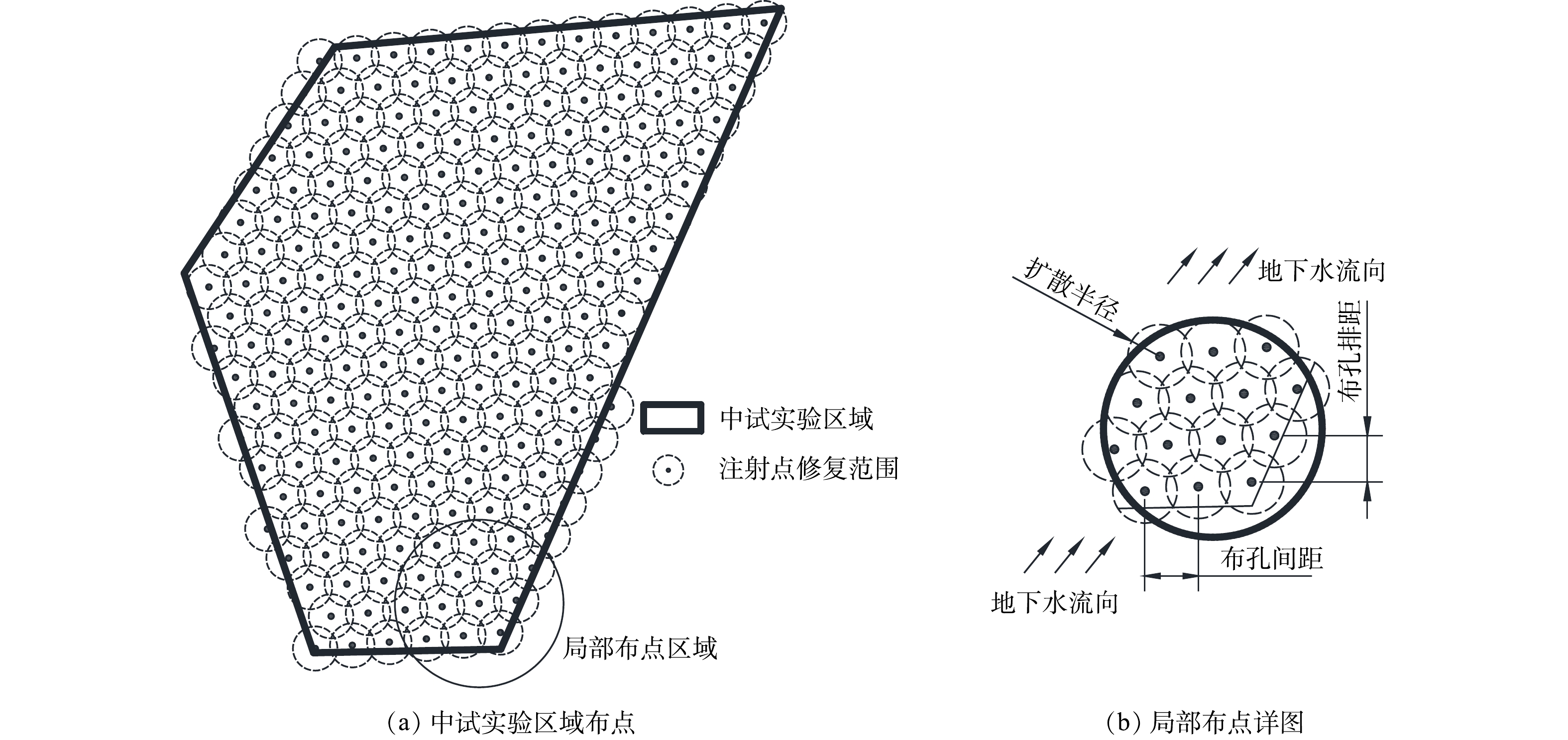
2)中试规模工程应用区域及注药点布设。选取研究场地内地下水受氯代烃污染的典型区域进行中试实验。所选区域的主要污染物为氯乙烯、顺-1,2-二氯乙烯、反-1,2-二氯乙烯、三氯乙烯等,实验前对该区域内的地下水采样调查初始浓度,中试实验区域污染数据见表2。该区域修复面积为1 557.37 m2,地下水修复深度为3~7 m,总计工程量(以含水层土方量计)为6 229.48 m3。实验区域注药点位布设采用三角形布点法[29]进行布设,以保证注射点修复范围全面覆盖实验区域。实验区域及注药点布设如图1所示,共计布置210个注药点。
-
1)小试实验材料。氧化剂选用的是一种基于高纯度过硫酸盐的强氧化剂Klozur药剂[30],该药剂在主导的
SO−4 ·和OH·的作用下,利用强氧化性破坏目标有机污染物,使有机污染物完全矿化或转化为低毒、易自然降解的物质。本实验选用NaOH(分析纯,片状)作为碱活化剂。研究[13]表明,当反应系统pH>10时过硫酸盐氧化性能高,一般调节过硫酸盐氧化体系pH接近11,NaOH是一种良好的碱性活化试剂。2)中试规模工程应用实验材料。碱活化过硫酸盐氧化技术的中试规模工程应用中所采用的修复药剂及设备如下:修复药剂主要包括过硫酸盐氧化剂和碱活化剂,其中过硫酸盐氧化剂使用过硫酸钠(优等品,过硫酸钠质量比≥90.0)、碱活化剂使用工业级液体氢氧化钠(氢氧化钠含量≥32%,液碱),溶剂使用自来水。修复设备主要包括溶配药系统[31]和高压旋喷钻机系统[29]。
-
1)小试实验及采样分析方法。将Klozur药剂和NaOH分别配置成25%(质量百分比)的储备液备用。氧化剂与活化剂投加量质量比为5∶2。量取200 mL实验样品于棕色螺口反应瓶中,分别按投加比(氧化剂体积与样品体积的比值)0.2%、0.5%、1.0%、3.0%添加氧化剂,再添加活化剂,同时做空白对照,实验分组编号如表3所示。药剂添加后混匀,用NaOH溶液调节pH为11,盖紧瓶盖混匀静置。定期调节并维持反应溶液pH为11,所有实验组均在反应28 d后取样检测,且计算分析污染物的去除率。
实验室检测方法参考《土壤和沉积物挥发性有机物的测定 吹扫捕集/气相色谱-质谱法》(HJ 605-2011)[32]标准,通过与待测目标物标准质谱图谱和保留时间比较进行定性、内标法进行定量。污染物去除率根据式(7)进行计算。
式中:
η 为去除率;C0为污染物初始浓度,μg·L−1;C为氧化处理28 d后的污染物浓度,μg·L−1。2)中试规模工程应用实验及采样分析方法。根据中试实验区域污染物的初始污染数据,同时结合实验室研究的最佳药剂投加比结论,确定单孔注药量。单孔注药量根据式(8)计算。
式中:V为单孔注药体积,m3;m为实验区域含水层修复工程质量,kg;n为实验区域注射点数量,个;w为最佳药剂投加比;ρ为药剂密度,kg·m−3。
本研究的中试规模工程应用中,首先利用溶配药系统进行药剂配制,药剂搅拌混匀后通过注药泵送至高压旋喷钻机;根据每个注药点位置,调整钻机就位,采用三重管法[29]进行药剂注射,开启注浆泵同时喷射药剂、液流、压缩空气,将固定量的氧化药剂注入至污染含水层。
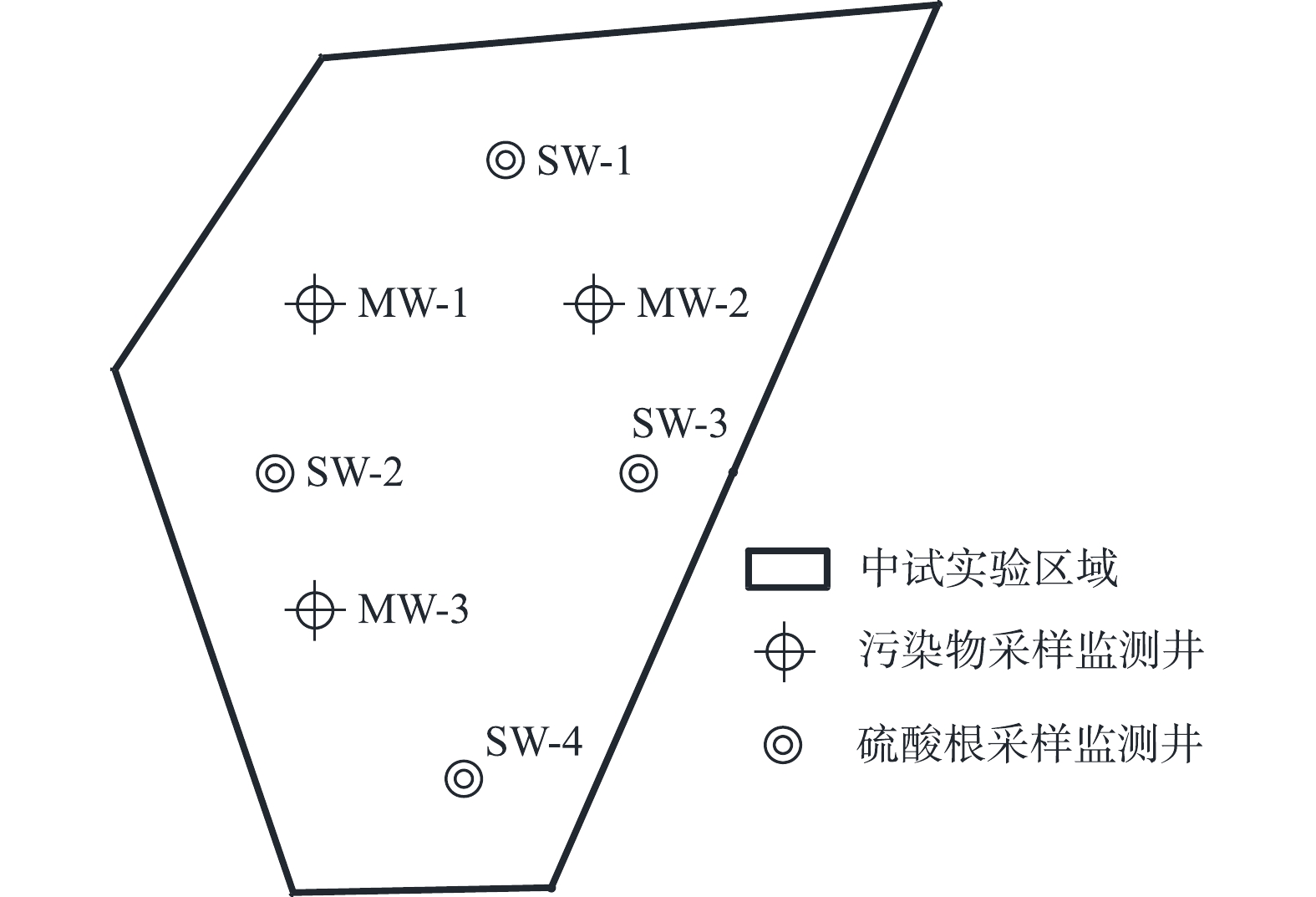
中试修复施工完成30 d后,对区域内地下水进行监测,污染物采样监测井MW-1、MW-2、MW-3和
SO2−4 采样监测井SW-1、SW-2、SW-3、SW-4布点如图2所示,每月采样1次,共采集8个批次。检测指标为地下水中目标污染物(氯乙烯、反-1,2-二氯乙烯、顺-1,2-二氯乙烯、三氯乙烯以及SO2−4 )。根据《污染地块地下水修复和风险管控技术导则》(HJ 25.6-2019)[33]中相关要求,对中试规模工程应用修复达标进行初判。
1.1. 研究场地及实验区域
1.2. 实验材料

1.3. 实验及分析方法



-
实验中选取GW1、GW2水样进行污染物浓度变化及去除率研究。由表1中数据可以看出:GW2整体污染程度比GW1严重,分别研究碱活化过硫酸盐对2种样品中各污染物的去除效果(GW1中三氯乙烯未超标故不作分析),污染物浓度变化及污染物去除率变化如表4、表5所示。表中各实验组投加比信息详见表1。
1)污染物初始浓度对污染物去除率的影响。GW1中氯乙烯、反-1,2-二氯乙烯、顺-1,2-二氯乙烯初始浓度分别为3 310、294.4、5 740 μg·L−1;GW2中氯乙烯、反-1,2-二氯乙烯、顺-1,2-二氯乙烯、三氯乙烯初始浓度分别为6 090、3 550、18 800、4 750 μg·L−1;显然GW2中对应各污染物浓度更高,如表2所示。通过比较2种样品在相同氧化药剂投加比条件下同1种污染物浓度的变化情况,研究污染物初始浓度对污染物去除率的影响。如表4和表5所示,GW1、GW2中各污染物在碱活化过硫酸盐的作用下浓度均呈下降趋势,但降幅有明显差别。在药剂投加比为0.2%时,GW1-2中氯乙烯、反-1,2-二氯乙烯、顺-1,2-二氯乙烯的去除率分别为95.11%、22.55%、55.34%,然而GW2-2中对应污染物的去除率分别为56.16%、15.49%、13.76%,三氯乙烯的去除率为46.95%,可见,在此投加比下GW1中各污染物的去除率均高于GW2;在药剂投加比为0.5%时,GW1-3中氯乙烯、反-1,2-二氯乙烯、顺-1,2-二氯乙烯的去除率分别为95.17%、62.98%、90.93%,而GW2-3中对应污染物的去除率分别为69.29%、25.35%、14.68%,三氯乙烯的去除率为50.32%,同样地,该投加比下GW1中各污染物的去除率也都高于GW2;在药剂投加比为1%时,GW1-4中氯乙烯、反-1,2-二氯乙烯、顺-1,2-二氯乙烯的去除率分别为99.79%、96.84%、99.36%,而GW2-4中对应污染物的去除率分别为74.22%、57.75%、51.38%,三氯乙烯的去除率为74.32%,该投加比下GW1中各污染物的去除率同样都高于GW2。综上,当药剂投加比为0.2%、0.5%、1%时呈现出相同的规律,即:在同一药剂投加比下,污染物初始浓度越高,去除率越低。但随着氧化药剂投加比的增加,2种样品中同一种污染物的去除率差别缩小。直到氧化药剂投加比为3%时,GW1-5、GW2-5中各污染物去除率无明显差异,均达到99%以上。
顾小钢等[34]对热活化过硫酸盐处理地下水中氯代烃的研究结果表明,随着初始浓度的增加,三氯乙烯的去除率逐渐降低,这与本研究的结果一致。因此,在后续的实际工程应用时,根据污染物的初始浓度确定经济、有效的氧化剂最佳投加比。此外,由表4和表5可看出,在未投加氧化药剂时,GW1-1和GW2-1的氯乙烯浓度大幅降低。陈梦舫等[10]的研究表明,氯乙烯的非生物水解作用或脱卤化氢半衰期>10 a、厌氧生物降解半衰期>60 d,本实验的反应时间仅为28 d,据此推测氯乙烯的衰减并非主要由物化与生物降解造成。氯乙烯为沸点-13.9℃的挥发性有机物,可能该污染物在实验条件下存在一定程度的挥发衰减。
2)氧化药剂投加比对污染物去除率的影响及最佳氧化药剂投加比。通过对比分析GW1和GW2在不同药剂投加下各污染物的浓度变化及去除率,以研究氧化药剂投加比对污染物去除的影响规律并确定最佳药剂投加比。如表4和表5所示,GW1在药剂投加比为0.2%、0.5%、1%时,氯乙烯的去除率分别为95.11%、95.17%、99.79%,反-1,2-二氯乙烯的去除率分别为22.55%、62.98%、96.84%,顺-1,2-二氯乙烯的去除率分别为55.34%、90.93%、99.36%;GW2在药剂投加比为0.2%、0.5%、1%时,氯乙烯的去除率分别为56.16%、69.29%、74.22%,反-1,2-二氯乙烯的去除率分别为15.49%、25.35%、57.75%,顺-1,2-二氯乙烯的去除率分别为13.76%、14.68%、51.38%;三氯乙烯的去除率分别为46.95%、50.32%、74.32%。
以上结果表明,反应28 d后,在不同药剂投加比下,GW1和GW2中的污染物去除规律一致,即随着氧化剂投加比提高,GW1和GW2中氯代烃污染物浓度逐渐降低、相应的去除率逐渐提高。由此可见,药剂投加比对污染物去除效果影响显著,氧化剂投加比与污染物的去除率成正比。这可能是由于在相同的反应时间内,氧化剂浓度越高,碱活化产生的自由基越多,从而增加了自由基氧化氯代烃的反应量。这与顾小钢等[34]的研究结论相符。由此可见,氧化药剂投加比是影响污染物去除率最直接、关键的因素,这也与GU等[22]的研究结果相符。
考虑避免药剂过度添加和经济成本,当污染物去除率达到99%以上或不再增加时,基本可认为氧化药剂投加量对于该污染物的去除达到饱和,无须再投加氧化药剂,此时为最佳氧化药剂投加比。对GW1和GW2样品中不同污染物的最佳投加比分析如下:GW1的反-1,2-二氯乙烯去除率,GW2的氯乙烯去除率、反-1,2-二氯乙烯去除率、顺-1,2-二氯乙烯去除率、三氯乙烯的去除率,随氧化药剂投加量变化的规律基本一致,变化线性明显,均在氧化药剂投加比为3%时达到去除率99%以上;GW1的氯乙烯、顺-1,2-二氯乙烯去除率在药剂投加比为1%时达到去除率99%以上,氧化剂投加量继续增加对污染物去除率影响不显著。
在多种氯代烃污染物复合的污染介质中,各污染物对氧化药剂需求规律不同,一定范围内污染物去除率与氧化药剂投加比正相关。综合上述结果可知:处理本场地内与GW1同等反-1,2-二氯乙烯污染程度、GW2同等氯乙烯去除率、反-1,2-二氯乙烯去除率、顺-1,2-二氯乙烯去除率、三氯乙烯污染程度的地下水时,建议最佳氧化剂投加比为3%;处理GW1同等氯乙烯、顺-1,2-二氯乙烯污染程度的地下水时,建议最佳氧化剂投加比为1%。
综上所述,建议在后续工程应用中的氧化剂投加比为1%~3%,具体投加比可根据污染物初始浓度确定。由表3可见,本次中试实验区域的氯乙烯、反-1,2-二氯乙烯、顺-1,2-二氯乙烯、三氯乙烯的超标倍数为分别10.77、3.46、65.57、10.56倍,污染程度与小试实验中GW1相当,因此,建议中试氧化中氧化剂投加比为1%。
-
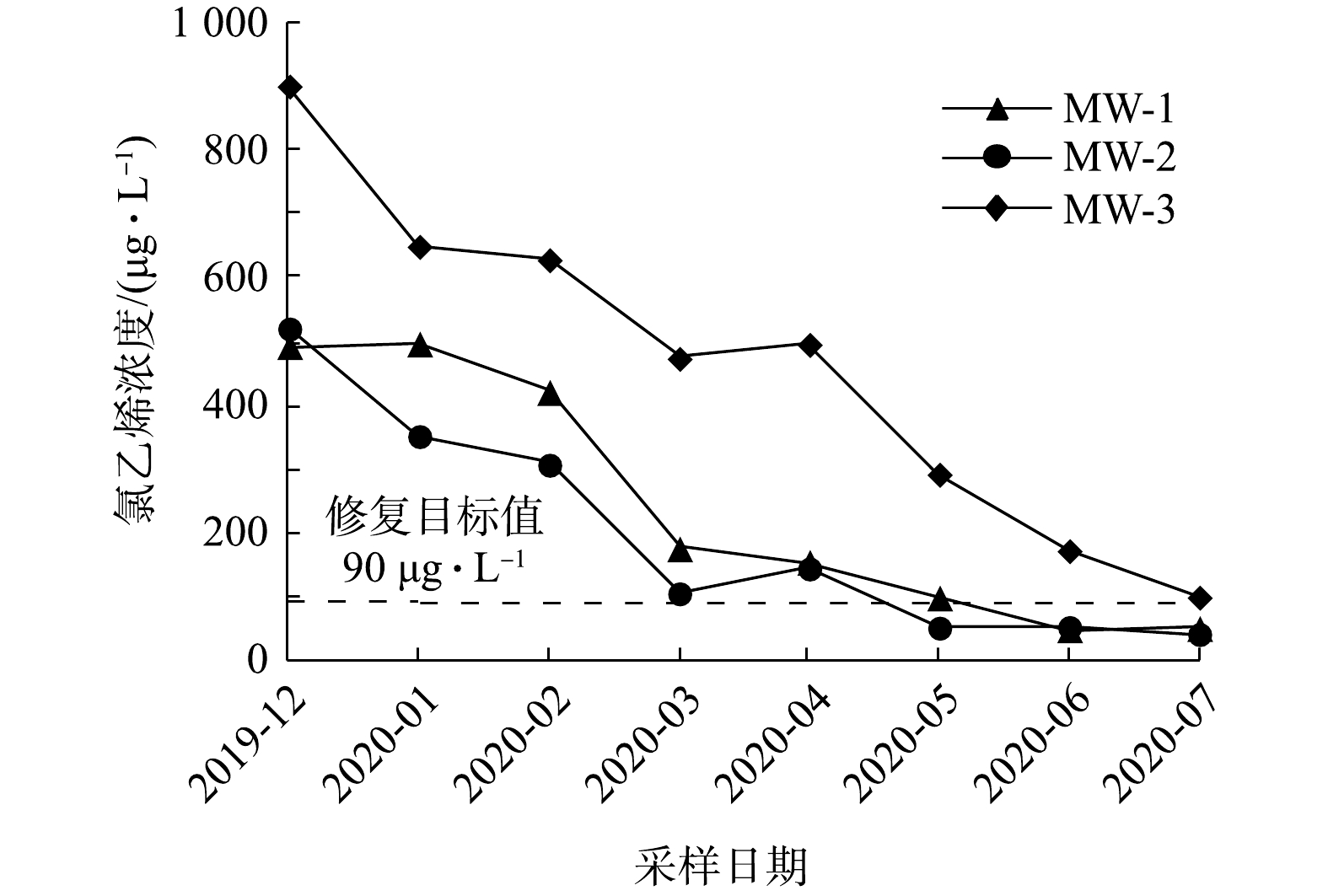
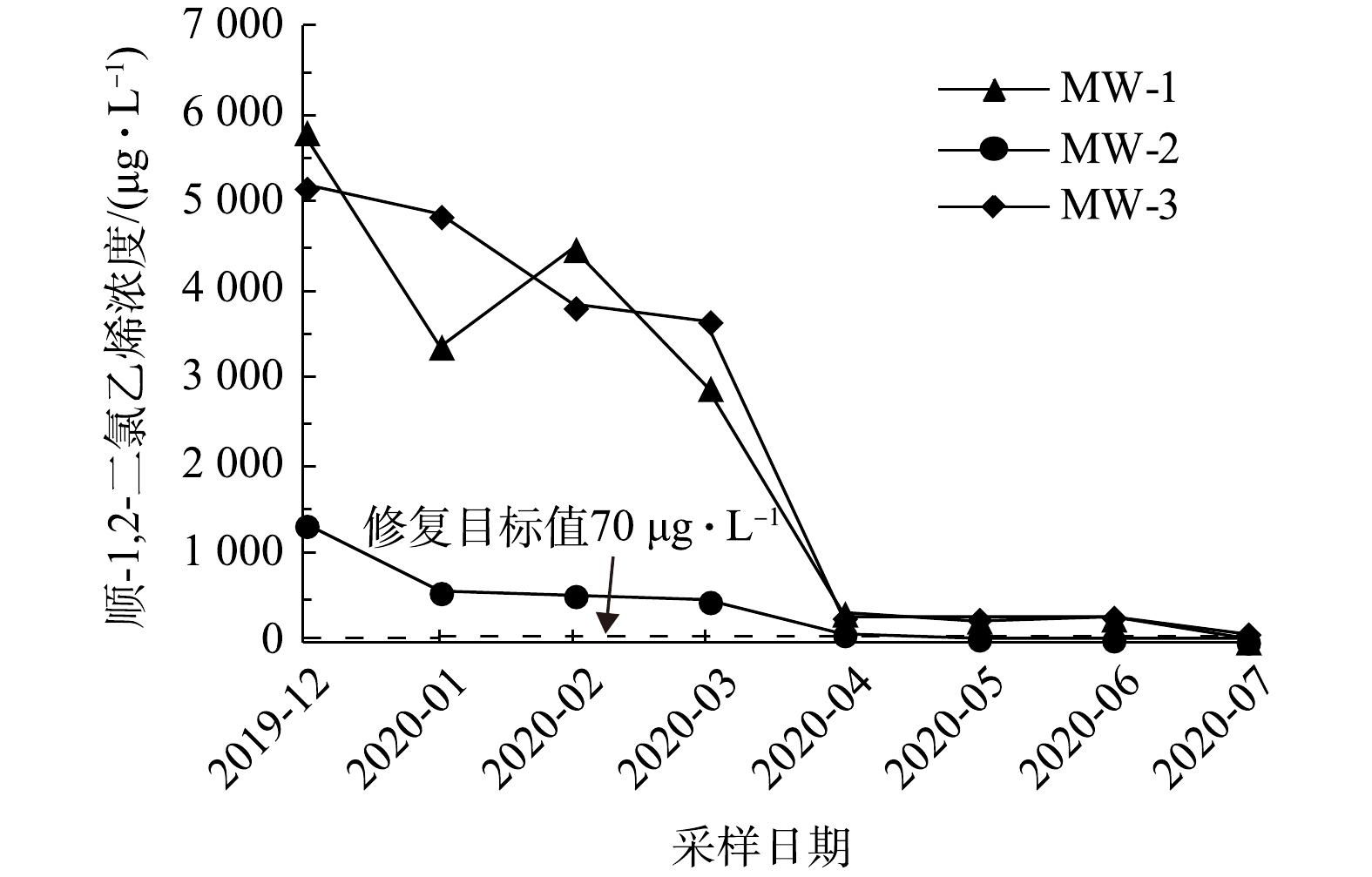
1)实验区地下水中目标污染物浓度的变化。如图3所示,实验区内3个采样点地下水中氯乙烯浓度随修复反应时间的延长而逐渐降低。在第6次采样时,MW-2位置氯乙烯浓度为52 μg·L−1,低于修复目标值;在第7次采样时,MW-1位置氯乙烯浓度为48 μg·L−1,低于修复目标值;在第8次采样时,MW-3位置氯乙烯浓度为100 μg·L−1,已接近修复目标值。
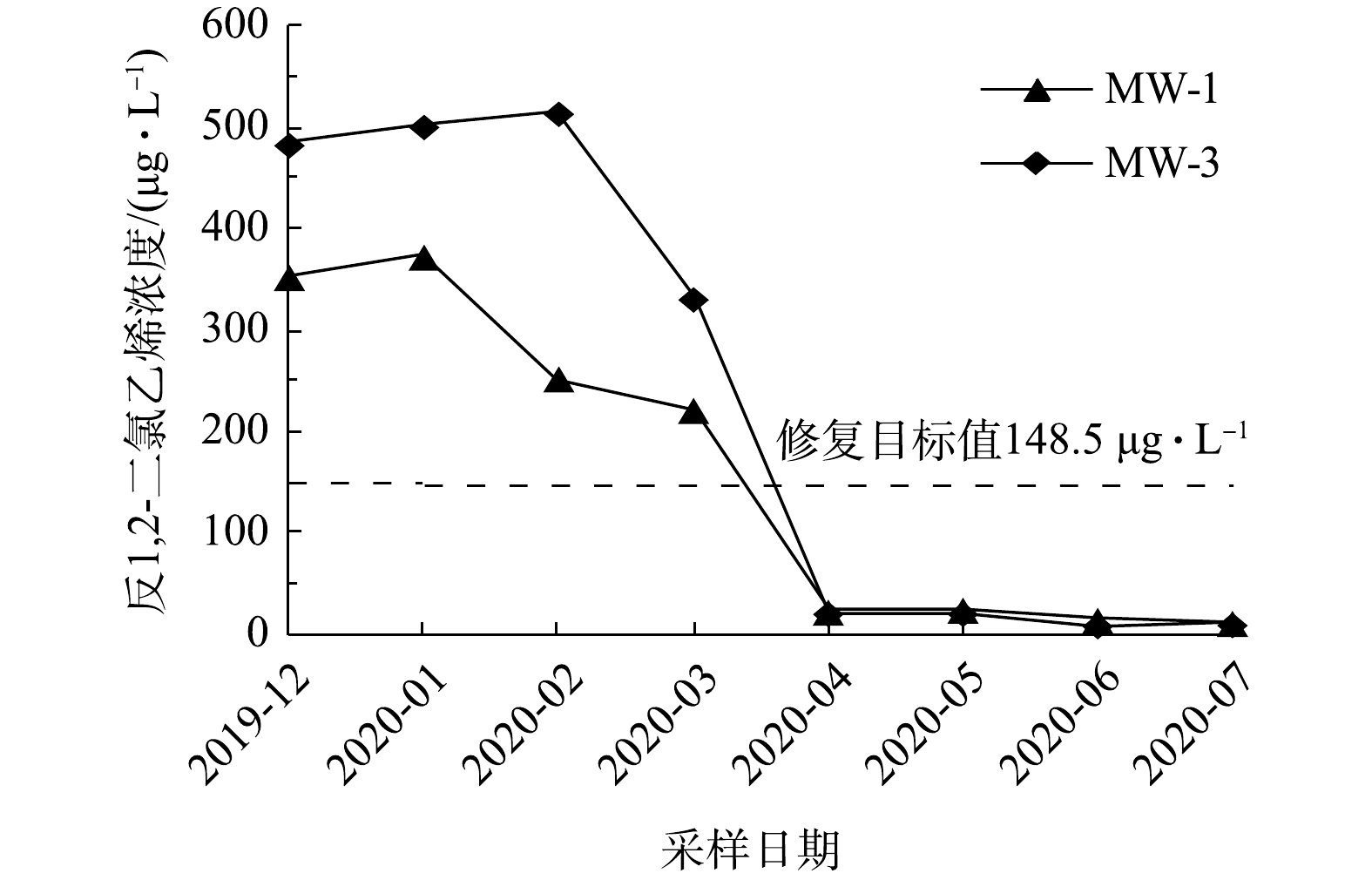
如图4所示,实验区内2个采样点地下水中反-1,2-二氯乙烯浓度(MW-2不超标,在图中无显示)随修复反应时间的延长而逐渐降低。在第5次采样时MW-1和MW-3位置的反-1,2-二氯乙烯浓度分别为23 μg·L−1和21 μg·L−1,均低于修复目标值。
如图5所示,实验区内3个采样点地下水中顺-1,2-二氯乙烯浓度随修复反应时间的延长而逐渐降低。在第6次采样时,MW-2位置顺-1,2-二氯乙烯浓度为58.9 μg·L−1,低于修复目标值;在第8次采样时,MW-1位置顺-1,2-二氯乙烯浓度为28 μg·L−1,低于修复目标值;MW-3位置反-1,2-二氯乙烯浓度为119 μg·L−1,接近修复目标值。
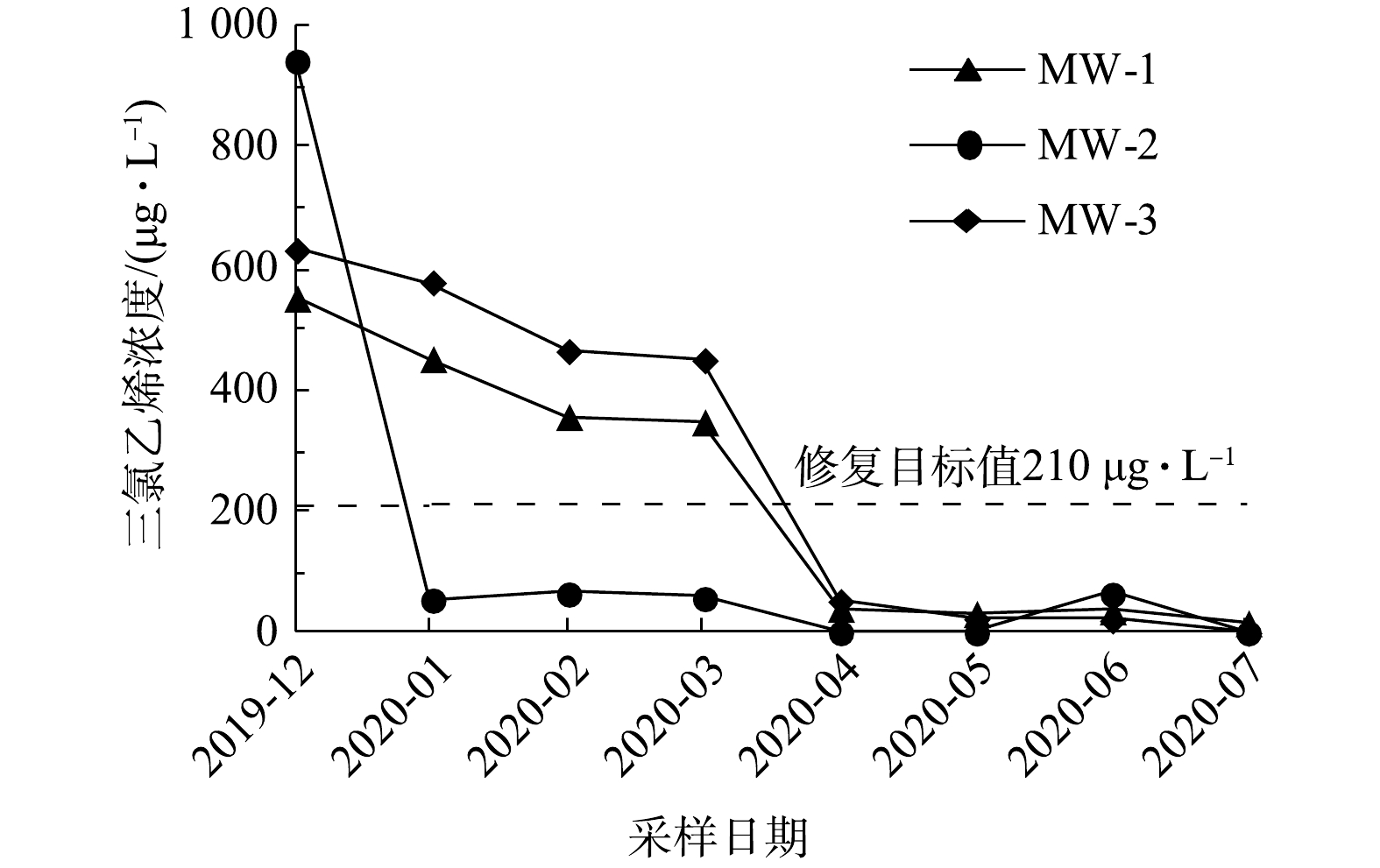
如图6所示,实验区内3个采样点地下水中顺-1,2-二氯乙烯浓度随修复反应时间的延长而逐渐降低。在第2次采样时,MW-2位置三氯乙烯浓度为56.2 μg·L−1,低于修复目标值;在第5次采样时,MW-1和MW-3位置三氯乙烯浓度分别为41.4 μg·L−1和54.4 μg·L−1,均低于修复目标值。
综上所述,在修复施工8个月后,MW-1、MW-2位置4种目标污染物,即氯乙烯、反-1,2-二氯乙烯、顺-1,2-二氯乙烯、三氯乙烯和MW-3位置的顺-1,2-二氯乙烯、三氯乙烯浓度均已低于修复目标值。但MW-3的氯乙烯和反-1,2-二氯乙烯在第8次采样时浓度未达到但已接近修复目标值。这可能是由于该位点局部氯乙烯、反-1,2-二氯乙烯污染较重。为确保修复效果、满足工程工期要求,实际工程应用时针对MW-3位置地下水污染物初始浓度偏高的污染区域,可适当调高氧化剂投加比或进行二次补充注射。
2)实验区地下水中
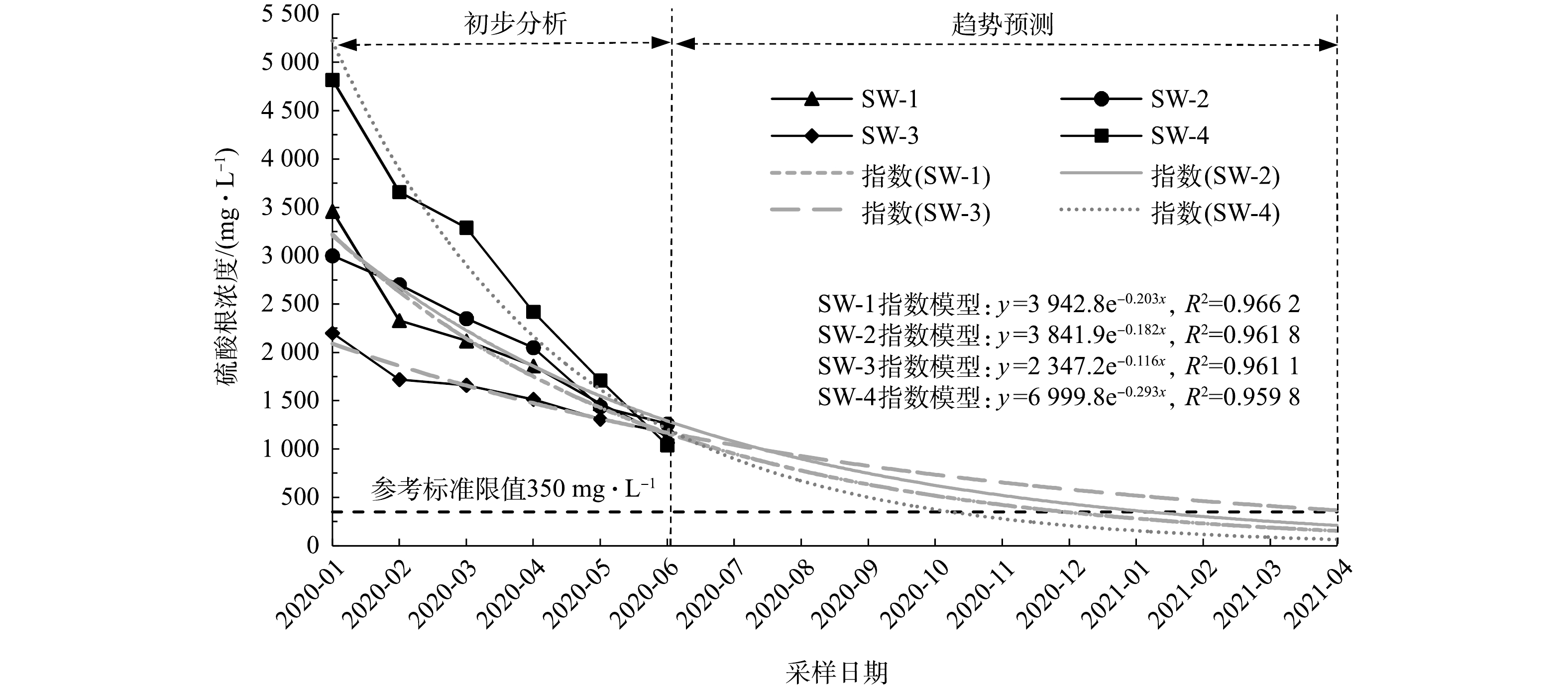
SO2−4 含量变化。实验区修复施工结束后,对实验区内地下水中SO2−4 浓度进行长期监测,实验区内地下水监测井编号为SW-1、SW-2、SW-3、SW-4,监测井位置详见图2。长期监测自注药施工完成、硫酸盐氧化剂与地下水中污染物反应稳定后开始,同时对地下水中的SO2−4 残留浓度进行监测,将所获得的SO2−4 浓度监测数据作为变量,建立SO2−4 浓度随时间变化的估算模型如图7中的 SW-1指数模型、SW-2指数模型、SW-3指数模型、SW-4指数模型,并对模型进行精度验证和分析。SW-1、SW-2、SW-3、SW-4监测井数据估算模型的可决系数(coefficient of determination,R2)分别为0.966 2、0.961 8、0.961 1、0.959 8,均通过回归方程的显著性检验,拟合效果好、预测精度高。如图7所示,注药修复完成后随着时间的延长,实验区内地下水中残留
SO2−4 浓度总体呈持续下降趋势。由于SO2−4 本身存在可作为电子受体被硫酸盐还原菌类微生物利用[35],也可与土壤中本底钙离子、钡离子等形成稳定沉淀[27, 35]等多种自然降解途径。注药完成3个月后,SW-1、SW-2、SW-3、SW-4样品中SO2−4 浓度分别为3 460、3 000、2 200、4 820 mg·L−1,实验区内各监测点位地下水中SO2−4 浓度不同。这可能是由于施工后初期各个点位SO2−4 随药剂注射迁移扩散的距离差和时间差或者自然降解程度不同导致。注药完成8个月后,SW-1、SW-2、SW-3、SW-4样品中SO2−4 浓度分别为1 130、1 260、1 170、1 040 mg·L−1,整个实验区内地下水中残留SO2−4 浓度趋近于同一浓度值。这可能是由于实验区域内的SO2−4 整体分布已趋于均质化。参考《地下水环境质量标准》(GB 14848-2017)[36]中Ⅳ类标准,
SO2−4 标准限制为350 mg·L−1,对注药完成8个月后实验区地下水中残留SO2−4 浓度的趋势线进行预测分析。结果表明:SW-4在注药完成11个月、SW-1和SW-2在注药完成12个月、SW-3在注药完成16个月后的SO2−4 浓度均达到参考标准。整体实验区域考虑最不利因素,预测在注药完成16个月后地下水残留SO2−4 浓度达到参考标准。
2.1. 污染物去除率的影响因素及氧化药剂投加比的优化
2.2.
中试规模工程应用区污染物及残留SO2−4 浓度的变化


















-
1)碱活化过硫酸盐可有效降解所研究典型氯代烃污染场地地下水中的目标污染物,氯代烃去除率随着氯代烃初始浓度升高而降低,碱活化过硫酸盐药剂投加比越大氯代烃去除率越高,建议后续工程应用时氧化药剂投加比范围取1%~3%、中试规模应用时取1%。
2)中试规模应用施工8个月后,实验区的地下水氯代烃污染物基本降低至修复目标值以下,局部浓度偏高区域后续可补充二次注射。中试设计的氧化药剂投加比、药剂注射方式、注射点位布设等参数合理,可为后续大规模修复工程设计和实施提供参考。
3)过硫酸盐氧化后环境中残留
SO2−4 浓度随着时间推移逐渐降低,工程实施区域内的地下水中残留SO2−4 浓度预计在施工第16个月达到《地下水环境质量标准》(GB 14848-2017)中Ⅳ类标准。


