





















 下载:
下载:

</td><td class="table_top_border" align="center" valign="middle">N<sub>2</sub>O积累峰值/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">N<sub>2</sub>O积累速率/(mg·(g·h)<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">N<sub>2</sub>O降解速率/(mg·(g·h)<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">N<sub>2</sub>O最大转化率/%</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">0</td><td class="table_top_border2" align="center" valign="middle">5.012</td><td class="table_top_border2" align="center" valign="middle">1.280</td><td class="table_top_border2" align="center" valign="middle">—</td><td class="table_top_border2" align="center" valign="middle">5.44</td></tr><tr><td align="center" valign="middle">5</td><td align="center" valign="middle">7.712</td><td align="center" valign="middle">1.969</td><td align="center" valign="middle">—</td><td align="center" valign="middle">7.85</td></tr><tr><td align="center" valign="middle">10</td><td align="center" valign="middle">8.293</td><td align="center" valign="middle">2.352</td><td align="center" valign="middle">2.353</td><td align="center" valign="middle">8.70</td></tr><tr><td align="center" valign="middle">20</td><td align="center" valign="middle">11.235</td><td align="center" valign="middle">4.781</td><td align="center" valign="middle">5.093</td><td align="center" valign="middle">11.28</td></tr><tr><td align="center" valign="middle">40</td><td align="center" valign="middle">15.232</td><td align="center" valign="middle">9.722</td><td align="center" valign="middle">5.249</td><td align="center" valign="middle">15.32</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">100</td><td class="table_bottom_border" align="center" valign="middle">22.123</td><td class="table_bottom_border" align="center" valign="middle">18.828</td><td class="table_bottom_border" align="center" valign="middle">7.914</td><td class="table_bottom_border" align="center" valign="middle">24.05</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula>-N/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">N<sub>2</sub>O积累峰值/(mg·L<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">N<sub>2</sub>O积累速率/(mg·(g·h)<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">N<sub>2</sub>O降解速率/(mg·(g·h)<sup>−1</sup>)</td><td class="table_top_border" align="center" valign="middle">N<sub>2</sub>O最大转化率/%</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">0</td><td class="table_top_border2" align="center" valign="middle">5.012</td><td class="table_top_border2" align="center" valign="middle">1.280</td><td class="table_top_border2" align="center" valign="middle">—</td><td class="table_top_border2" align="center" valign="middle">5.44</td></tr><tr><td align="center" valign="middle">5</td><td align="center" valign="middle">7.712</td><td align="center" valign="middle">1.969</td><td align="center" valign="middle">—</td><td align="center" valign="middle">7.85</td></tr><tr><td align="center" valign="middle">10</td><td align="center" valign="middle">8.293</td><td align="center" valign="middle">2.352</td><td align="center" valign="middle">2.353</td><td align="center" valign="middle">8.70</td></tr><tr><td align="center" valign="middle">20</td><td align="center" valign="middle">11.235</td><td align="center" valign="middle">4.781</td><td align="center" valign="middle">5.093</td><td align="center" valign="middle">11.28</td></tr><tr><td align="center" valign="middle">40</td><td align="center" valign="middle">15.232</td><td align="center" valign="middle">9.722</td><td align="center" valign="middle">5.249</td><td align="center" valign="middle">15.32</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">100</td><td class="table_bottom_border" align="center" valign="middle">22.123</td><td class="table_bottom_border" align="center" valign="middle">18.828</td><td class="table_bottom_border" align="center" valign="middle">7.914</td><td class="table_bottom_border" align="center" valign="middle">24.05</td></tr></tbody>
</table></div></foreignObject></svg>)






















-
脱氮是污水处理流程中一个重要的环节,强化脱氮处理能避免污水排放后形成氮素污染。在传统生物脱氮过程中,包括硝化和反硝化2个过程[1]。其中,反硝化是在缺氧条件下,以有机碳源作为电子供体用于产能和细胞合成,同时在硝酸盐还原酶(nitrate reductase, Nar)、亚硝酸盐还原酶(nitrite reductase, Nir)、一氧化氮还原酶(nitric oxide reductase, Nor)和氧化亚氮还原酶(nitrous oxide reductase, Nos)的参与下,将
NO−3 -N逐步还原为N2[2]。反硝化的中间产物N2O是一种强温室气体[3]。有研究[4]指出,N2O是21世纪排放的主要臭氧消耗物质,其在大气中的浓度每增加一倍,将导致全球升温0.3 ℃[5]。因此,N2O的排放控制日益引起国家政府的重视。N2O作为反硝化过程的中间产物之一,当其还原速率小于生成速率时,即可出现N2O的积累。与其他反硝化酶相比,
NO−2 -N更容易影响Nos酶的活性,从而抑制N2O的还原,导致N2O的积累[6-9]。许多研究[10-13]表明,NO−2 -N是引起反硝化过程中N2O积累和释放的关键因素。为减少污水脱氮过程中N2O的释放,国内外的科研人员围绕生物脱氮过程中N2O的产生机理进行了大量研究[8, 14-17],但控制反应参数以减少N2O排放的效果还不甚良好。本研究以葡萄糖为碳源,在保证初始氮素总量一致的条件下,改变NO−2 -N占氮素总量的比例,监测了反应器内NO−3 -N、NO−2 -N、N2O的浓度以及耗氧有机污染物的浓度(以COD计)等指标的变化,考察了不同初始条件对反硝化过程中脱氮效果的影响,探究了NO−2 -N占比对反硝化的影响,进而分析其对N2O产生的影响。本研究可为控制反硝化过程中N2O的排放提供参考。
全文HTML
-
采用500 mL广口瓶进行批次实验。取实验室原有的SBR反应器中已经驯化适应了以葡萄糖为碳源的反硝化污泥混合液。为确保污泥中不再残留化学物质,将其经沉淀、离心、去除上清液,加入清水后再次进行沉淀、离心、去除上清液,重复3次。将去除上清液后的污泥置于500 mL广口瓶中,加入20 mg·L−1 NH4Cl(以N计)、8 mg·L−1 NaH2PO4(以P计)、10 mg·L−1酵母浸膏,摇晃均匀以配成悬浮液。使用3 mol·L−1 HCl和NaOH稀溶液调节瓶中pH为6.5。将插有2根橡胶管的瓶塞将瓶口密封,随后通过橡胶管向广口瓶中持续通入5 min氮气,以去除混合液中氧气,最后用夹子夹紧2根橡胶管,使批次实验在缺氧条件下运行。将
NO−3 -N、NO−2 -N和耗氧有机污染物(以COD计)按所需浓度分别配成50 mL浓缩液,在反应开始时,立即注射入广口瓶中,并将广口瓶置于磁力搅拌器上进行搅拌,转速为150 r·min−1。同时,设置平行实验,文中实验结果均为取平行实验的平均值。 -
通过磁力搅拌器控制温度为22 ℃。MLSS和MLVSS分别为2.62 g·L−1和2.35 g·L−1。实验在COD/N比为6.5的条件下进行,其中耗氧有机污染物的浓度(以COD计)为650 mg·L−1。改变实验用水中
NO−2 -N占总氮素的比例,同时保证氮素浓度总和为100 mg·L−1,具体指标如表1所示。并将对应的FNA计算结果显示于表1中。实验过程中,每隔10 min取水样测反应器中各项水质指标(NO−3 -N、NO−2 -N、溶解态N2O),持续100 min。每次取样结束后向广口瓶中通入适量氮气以补充瓶内气压。 -
本实验中,各污染物的检测指标均参考《水和废水检测分析方法》[18]进行:
NO−3 -N采用紫外分光光度法;NO−2 -N采用N-(1-萘基)-乙二铵光度法;COD采用重铬酸钾法;气态N2O采用气体收集法;溶解态N2O采用岛津气相色谱仪GC-2014通过顶空平衡法进行测量;pH采用Testr 30型pH计进行测量。FNA浓度按照式(1)进行计算。
式中:CFNA为游离亚硝酸浓度,mg·L−1;
Ct,NO2 为总亚硝酸盐浓度,mg·L−1;T为温度,℃。各反硝化酶的电子消耗速率按照式(2)~式(5)进行计算[19];各反硝化酶之间的电子排布按照式(6)进行计算;N2O最大转化率按照式(7)进行计算。
式中:
rNar,e 、rNir,e 、rNor,e 、rNos,e 分别为Nar、Nir、Nor、Nos的电子消耗速率,mmol·(g·h·L)−1;rNO−3−N 、rNO−2-N 、rNO-N 、rN2O-N 分别为NO−3 -N、NO−2 -N、NO、N2O的消耗量,mg·(g·h)−1。式中:R为电子分布率;rx,e为各反硝化酶的电子消耗速率,mmol·(g·h·L)−1。
式中:
η 为N2O最大转化率;A为N2O最大积累量,mg·L−1;Δρ为各氮素的变化量(以氮计),mg·L−1。
1.1. 实验方法
1.2. 运行条件
1.3. 分析项目及检测方法
-
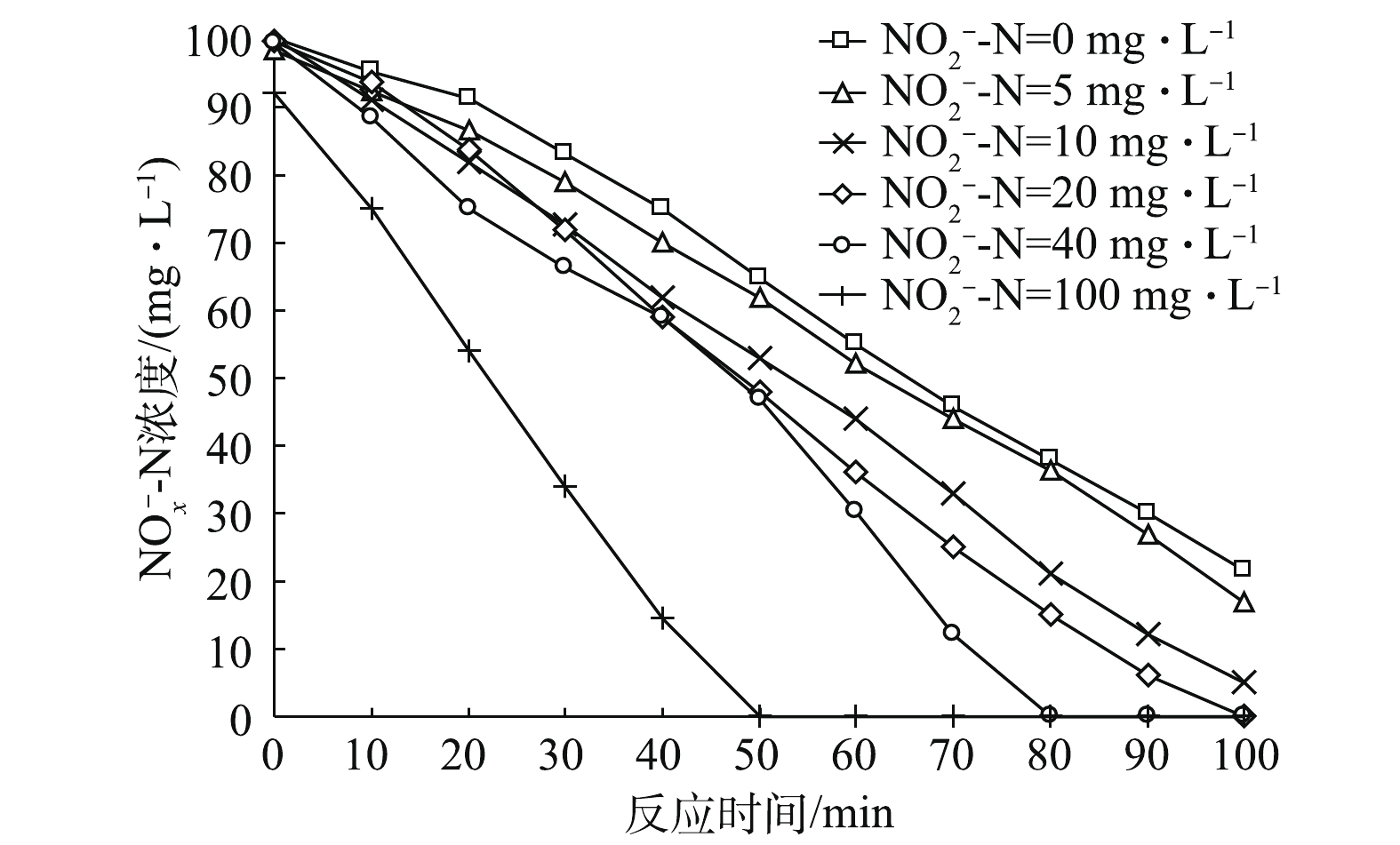
在控制初始
NO−x -N总量一致的情况下,通过改变初始NO−2 -N浓度,使其初始占比不同,反应器内NO−x -N的变化如图1所示。由图1可知,在不同初始NO−2 -N占比条件下,NO−x -N的变化基本呈线性变化趋势。总体而言,随着初始NO−2 -N占比的提高,NO−x -N的降解速率加快,即反硝化脱氮效率升高。当NO−2 -N的初始浓度为0、5和10 mg·L−1时,即NO−2 -N占总氮素比例为0%、5%和10%时,反硝化过程在100 min内尚未结束。而当初始NO−2 -N占20%、40%和100%时,反应均在100 min内结束。当初始NO−2 -N占比为100%时,反硝化速率最快,NO−x -N在50 min时即降至0。初始NO−2 -N占比为0%、10%、20%、40%、100%对应的NO−x -N去除率分别为0.021 4、0.022 1、0.024 3、0.025 4、0.031 7和0.047 0 g·(g·h) −1。若将反硝化路径简化成2步反硝化模型[20-21],反应式如式(8)所示。在反应式(8)中,
NO−3 -N为氮源的反硝化过程,需要经由NO−2 -N,再进行下一步反应。若氮源全部为NO−2 -N,则反硝化过程只需一步反应,故脱氮路程便相较于前者缩短了。因此,初始NO−2 -N的占比越高,反硝化速率越快。王少坡等[22]在内源反硝化脱氮过程中也得出了类似结论。不同初始
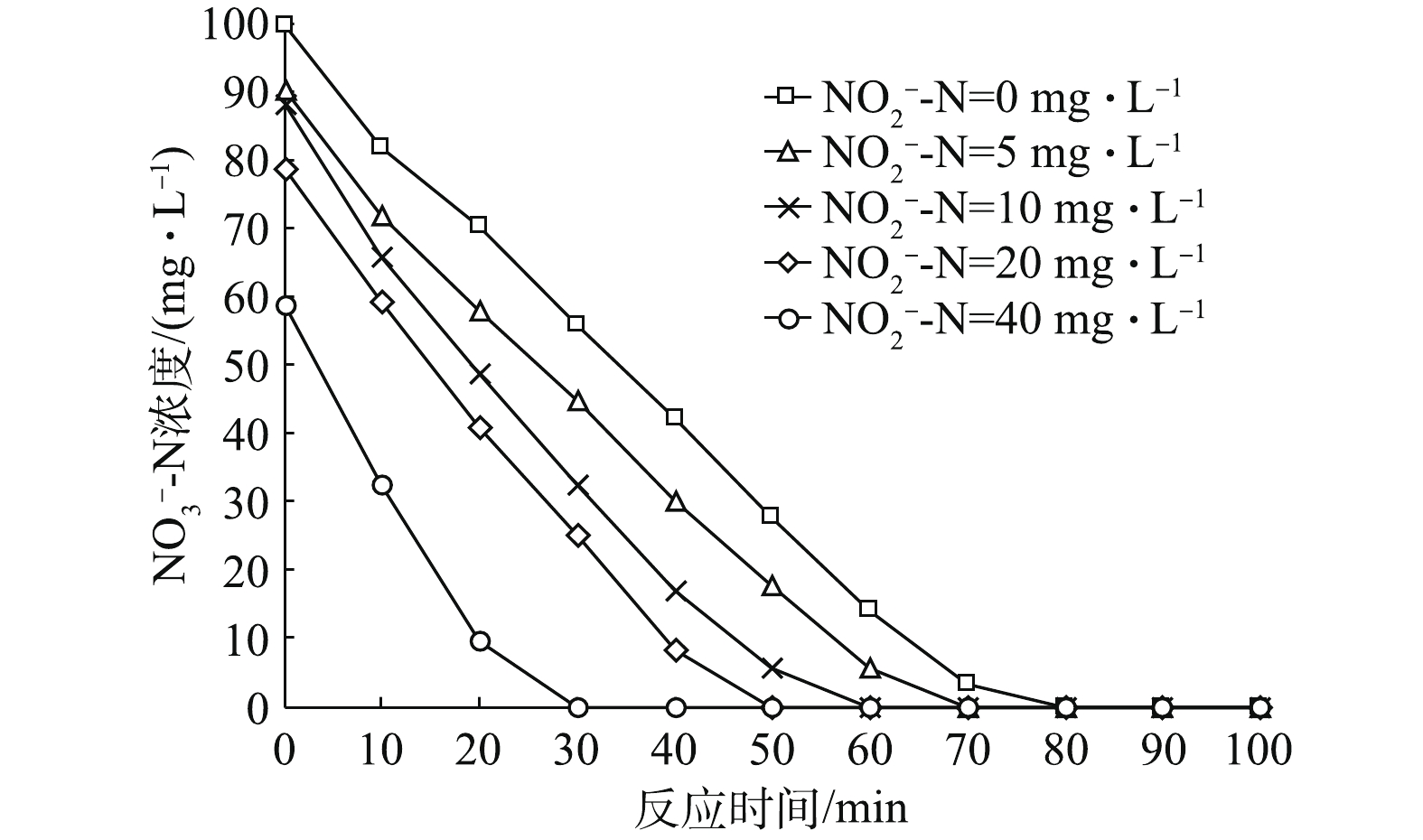
NO−2 -N占比条件下,反应器内NO−3 -N的变化情况如图2所示。其中,各初始NO−2 -N占比条件下(初始NO−2 -N浓度为100 mg·L−1时,系统内主要进行NO−2 -N的反硝化,因此,不讨论此浓度下NO−3 -N浓度的变化情况),NO−3 -N均在80 min内迅速降解完毕,降解速率分别为0.032 5、0.032 9、0.037 5、0.040 2和0.050 1 g·(g·h)−1。由此可见,随着初始NO−2 -N占比的升高,初始NO−3 -N浓度降低,反应过程中NO−3 -N降解速率加快。这主要归为以下2点原因:首先,相对Nir酶而言,Nar酶对电子的亲和力更强,因此,溶液中NO−3 -N浓度优先得到降解;其次,在污泥浓度和耗氧有机污染物的浓度(以COD计)一定的情况下,即供给的电子和接收位点(酶)的数量一定时,电子受体(NO−3 -N)越少,反应速率越快,则所需时间越少。图3为各初始
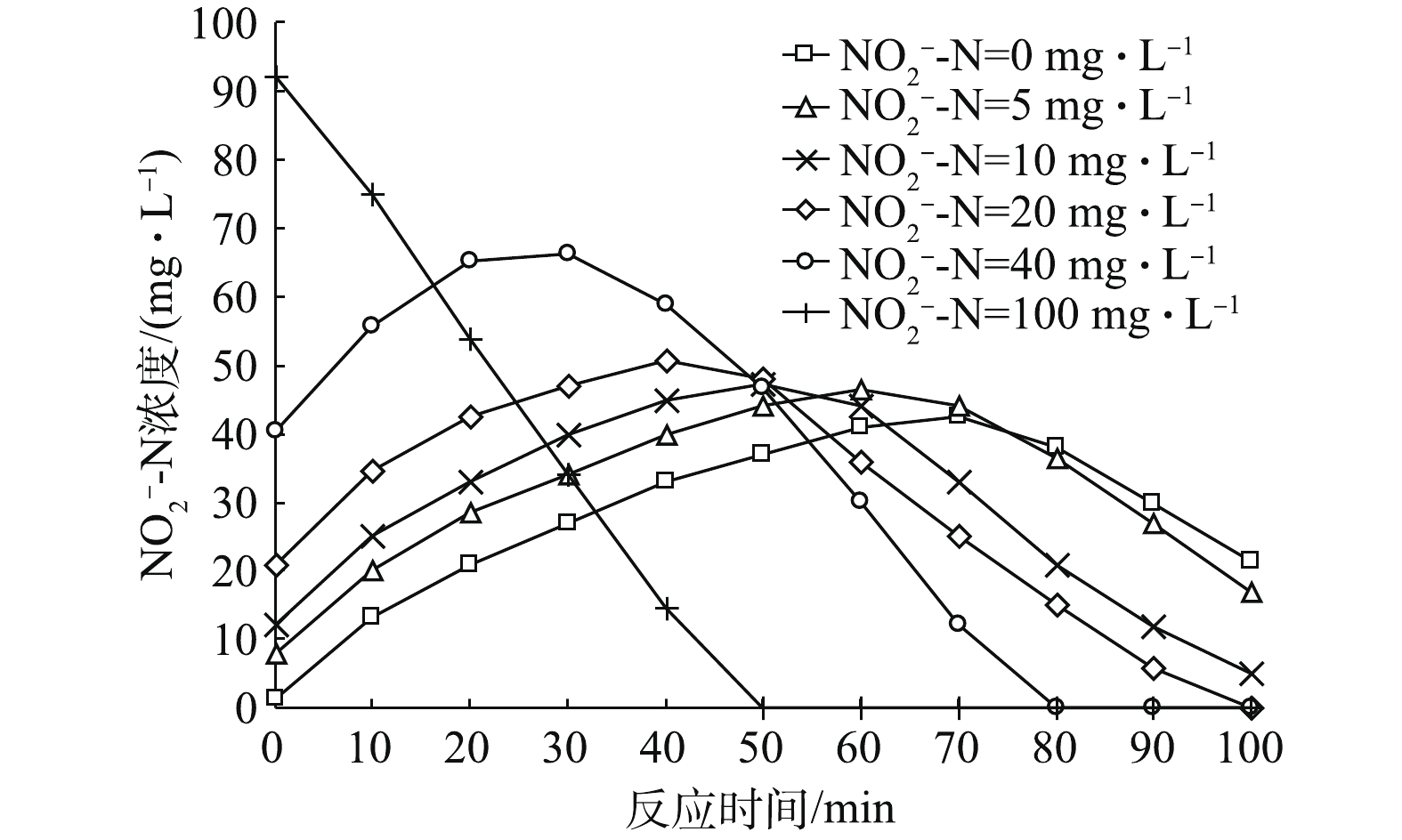
NO−2 -N占比条件下,反应器内NO−2 -N浓度随时间变化规律图。由图3可以看出,NO−2 -N浓度随时间基本呈先增加后减少的趋势(初始NO−2 -N占比为100%时,因为溶液中不含NO−3 -N,故其没有NO−2 -N积累情况出现,NO−2 -N浓度变化趋势即为NO−x -N浓度变化趋势,之前已作分析,因此不再分析)。各初始NO−2 -N浓度条件下,NO−2 -N浓度达到最大积累值的时间分别为70、60、50、40和30 min,其分别对应NO−3 -N浓度变化曲线(图2)中NO−3 -N降至0的时间点,即NO−2 -N浓度在NO−3 -N即将耗尽时达到最大值。另外,NO−2 -N浓度的最高积累值随初始NO−2 -N占比的升高而增加,分别为42.5、46.5、47.2、50.8和66.2 mg·L−1。这是因为:在初始NO−2 -N占比不断增加的同时,NO−2 -N降解又具有滞后性导致其不断积累;同时,由图5(a)可见,随着初始NO−2 -N占比的增加,Nar和Nir酶之间的电子消耗速率落差加剧,NO−3 -N降解速率变快,进而导致NO−2 -N积累速率变快,分别为0.015 8、0.016 3、0.017 9、0.019 1和0.021 9 g·(g·h)−1。因此,初始NO−2 -N浓度高以及积累速率高共同导致高初始浓度条件下NO−2 -N最高积累值大。 -
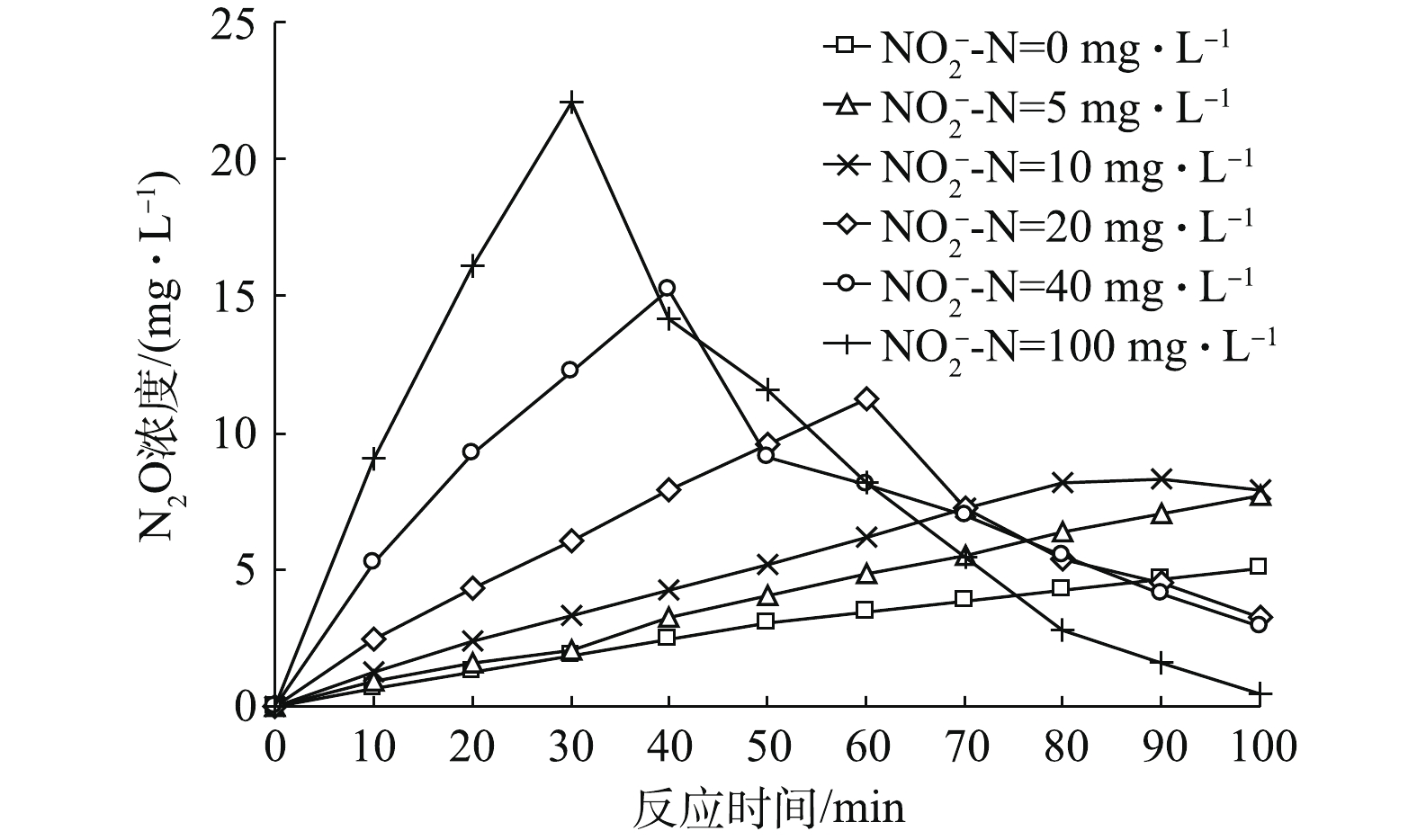
反硝化过程中,N2O在不同初始
NO−2 -N占比条件下随时间变化趋势如图4所示。由图4可知,当初始NO−2 -N占比为0时,N2O呈缓慢上升趋势;当初始NO−2 -N占比为5%时,N2O呈缓慢上升趋势;当初始NO−2 -N浓度为10%时,N2O呈先缓慢上升之后略微下降的趋势;当初始NO−2 -N浓度继续增大后,N2O先迅速升高后显著下降。图4中,初始
NO−2 -N占比为0%和5%时的N2O呈现一直上升的趋势,原因为截至100 min时,NO−3 -N还原尚未结束,所以N2O浓度保持上升趋势。当初始NO−2 -N占比为10%时,N2O的变化呈现先升高后保持稳定并未出现显著下降的趋势,笔者猜测,这可能与剩余COD有关。有研究[23]表明,以葡萄糖为碳源的反硝化过程最佳碳氮比为6~7(COD/NO−3 -N),因而在本研究中,在COD/N=6.5、初始NO−2 -N为0的情况下,反应结束时剩余的COD量已经很少,故只有很少量溶解态N2O浓度得以被降解,大多数仍溶解于水中。N2O的剩余浓度随着初始NO−2 -N占比的升高而减少,这是由NO−3 -N和NO−2 -N作为电子受体时消耗不等量的COD导致的。以葡萄糖为碳源时,NO−3 -N和NO−2 -N完全被降解的代谢方程分别如式(9)和式(10)所示。比较式(9)和式(10)可知,1 mol
NO−3 -N被降解所需的葡萄糖是1 molNO−2 -N所需的1.67倍,因此,在NO−x -N浓度一定的条件下,初始NO−2 -N占比愈高,反硝化消耗的碳源就愈少,那么剩余COD就会相对愈高,当NO−3 -N和NO−2 -N降解完之后,溶解态N2O的降解速率愈快,导致剩余愈少。总体而言,当反应时间足够长且在碳源充足的条件下,N2O的积累应呈先升高后降低的趋势。值得注意的是,随着初始
NO−2 -N占比的升高,反硝化过程中N2O的积累峰值越高,峰值后又被降解的N2O的量也越多,且在截至100 min时,系统中剩余量越少。经计算可知,随着初始NO−2 -N占比的升高,N2O的积累速率变快、N2O积累量的最高值逐渐升高,且N2O的转化率逐渐变大,具体数值如表2所示。其中当初始NO−2 -N占总氮素比例为100%时,N2O最大转化率达到24.05%。通过计算得到各初始
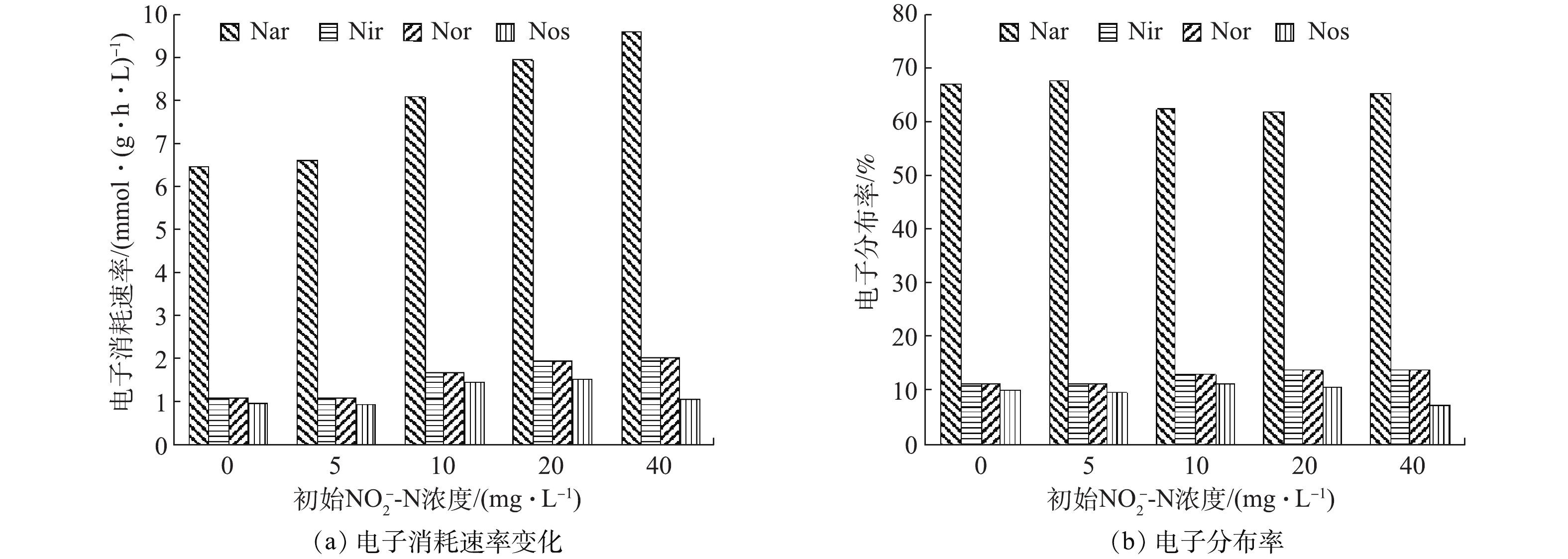
NO−2 -N占比条件(0、5、10、20、40)下的FNA分别为0.001 1~0.032 7、0.006 2~0.035 8、0.009 3~0.036 3、0.016 0~0.039 1、0.031 2~0.050 9,而初始NO−2 -N占比为100%时,初始FNA高达0.070 8。因此,FNA浓度随着初始NO−2 -N浓度的升高而升高,这也许是导致N2O产生量增加的原因。有研究[24-26]表明,FNA浓度的升高可抑制Nos酶的活性,减弱其竞争电子的能力,进而导致不完全反硝化的发生,导致N2O大量产生。各反硝化酶的电子消耗速率和电子分布率如图5所示。由于NO在低于10 ℃以下才会产生积累[27],因此,可认为在本实验条件下,反硝化过程中不会出现NO的积累现象,即Nir酶和Nor酶的降解速率相等。由图5可见,随着初始
NO−2 -N占比的不断增加,Nar、Nir和Nor的电子消耗速率基本呈逐渐加快的趋势,这是因为,初始NO−2 -N占氮源比值越大,则反硝化所需COD越少,那么在COD量和微生物量一定的情况下,电子的有效碰撞概率越高,进而电子传递速率越快。初始NO−2 -N占0%、5%、10%、20%和40%时反硝化酶的电子消耗比例分别为1∶0.169∶0.169∶0.151、1∶0.168∶0.168∶0.143、1∶0.209∶0.209∶0.182、1∶0.222∶0.222∶0.172和1∶0.210∶0.210∶0.111。结合图5(b)可见,随着初始NO−2 -N占比的升高,Nir酶和Nos酶之间的电子分布率落差逐渐增大。也就是说,Nos酶竞争电子能力减弱,这就将导致更多的N2O产生积累,即NO−2 -N与N2O的产生有非常明显的相关性[28]。初始
NO−2 -N占比的升高将导致FNA浓度的升高,同时反硝化酶的电子分布率发生改变,导致大量N2O产生。这说明,NO−2 -N和FNA与N2O之间有一定的相关关系,但无明确的因果关系。究竟是NO−2 -N还是FNA导致N2O产生积累?此问题还需进一步研究。
2.1.
初始NO−2![]()
![]()
-N占比对脱氮效果的影响
2.2.
初始NO−2![]()
![]()
-N占比对N2O产生的影响
-
1)控制氮素浓度总和为100 mg·L−1,随着初始
NO−2 -N浓度的升高,反硝化脱氮效率升高。其中,NO−2 -N浓度随时间呈先增加后减少的趋势;初始NO−2 -N占比越高,NO−3 -N降解速率越快,NO−2 -N积累也越多。2) N2O积累基本呈先上升后下降的趋势。随着初始
NO−2 -N占比的升高,N2O的积累速率变快,最高达18.828 mg·(g·h)−1;N2O积累最高值逐渐增大,最大为22.123 mg·L−1;N2O的转化率逐渐变大至24.05%。3)随着初始
NO−2 -N占比的升高,FNA浓度升高,更多的N2O产生积累。FNA浓度可能是导致N2O大量积累的原因。
