






-
近年来,膜技术已广泛应用于海水淡化、给水处理、污水深度处理等领域,而膜污染仍然是制约膜技术进一步发展的重大挑战之一。膜表面结垢使渗透通量降低[1],导致了更高的能量需求和运行成本[2-3],须不断进行清洗,进而减少膜的使用寿命。膜污染是由膜孔隙及膜面上微生物、胶体、溶质和细胞碎片的不良沉积和积聚引起的[4-5],尽管通过膜改性等防污性能的改善可以一定程度上减缓膜污染,但膜渗透性的增强效果有限[6-7]。
微生物胞外聚合物(extracellular polymeric substances,EPS)被认为是造成膜生物反应器膜污染的关键物质[8]。EPS组成非常复杂,主要由大分子有机物组成,如多聚糖类、蛋白类和核酸等,其表面具有大量的活性官能团和疏水区域,具有极强的吸附和络合能力。在膜污染过程中,EPS会与其他胶体物质在膜表面积累形成致密的胶体层[9-10],因此,EPS的结构和特性会显著影响膜污染的程度。最初研究人员[8-9]通过提取实际EPS来研究EPS的主要组分对膜污染的影响,但由于提取EPS耗时长、效率低且不同提取方法结果不统一,因此,改用模拟EPS溶液(蛋白质、多糖、腐殖酸或其组合)来开展膜污染机理等方面的研究工作。实际EPS和模拟EPS溶液的差异随着研究的深入逐渐开始得到关注,目前公开的报道较少。JIANG等[11]研究了EPS与海藻酸钠在胶体性质上的差异,但是实际EPS与二元甚至多元模拟溶液之间的差异尚不清楚,因此,探究实际EPS和模拟EPS溶液之间的区别具有重要意义。
有研究[12-14]表明,二价阳离子与EPS密切相关,阳离子会桥联EPS中带负电荷的官能团,从而增加EPS和悬浮颗粒间的初始吸附,有助于聚集和稳定生物聚合物,导致胶体和聚合物尺寸增加,其中最常见的阳离子为Ca2+。已有研究[15]表明,Ca2+的存在可显著增加从芽孢杆菌、假单胞菌、沙雷氏菌和耶尔森氏菌等菌种中提取的EPS的絮凝能力,表明Ca2+的存在会对絮凝作用产生影响,从而影响膜的过滤效果。目前,针对二价阳离子,特别是Ca2+存在下实际EPS和模拟EPS的研究较为广泛,但关于对这两者在Ca2+存在下的差异研究却鲜有报道。本研究采用牛血清白蛋白(bovine serum albumin,BSA)和海藻酸钠(sodium alginate,SA)的组合作为模拟溶液,与实际提取的EPS在Ca2+存在的条件下进行比较,采用多种测试分析手段比较两者的粒径分布、官能团、流变特性以及膜污染行为,以期深入认识实际EPS与模拟EPS的差异,旨在探究模拟EPS能否真正替代实际EPS,从而为膜生物反应器污染物质的模拟研究提供理论依据,为膜污染过程中胞外聚合物及其机理研究提供参考。
-
实验所用颗粒污泥取自某污水处理厂曝气池,采用阳离子交换树脂法(CER)提取实际胞外聚合物EPS。将30 mL污泥在4 ℃以6 000 r·min−1离心15 min,将沉淀的污泥重新悬浮在30 mL含有2 mmol·L−1 Na3PO4、4 mmol·L−1 NaH2PO4、9 mmol·L−1 NaCl和1 mmol·L−1 KCl的缓冲溶液(pH=7.0)中[16],加入阳离子交换树脂(Dowex® Na+,Sigma-Aldrich),在4 ℃下以200 r·min−1搅拌1 h。然后通过重力分离取出CER,最后在4 ℃下以10 000 r·min−1离心15 min,除去剩余的污泥和CER,得到上清液经0.45 μm滤头过滤后即为EPS,以备实验使用。
蛋白质测定采用修正后的Lowry法,以牛血清白蛋白作为标准;多糖测定采用苯酚硫酸法,以无水葡萄糖作为标准[17],将蛋白和多糖的总量作为EPS的总量,所有的指标重复测定吸光度。在本研究中,实际提取的胞外聚合物蛋白含量为(116±2.28) mg·g−1,多糖含量为(16±1.86) mg·g−1,将胞外聚合物的实际提取液记为EPS。
-
实验采用牛血清白蛋白(BSA)模拟蛋白,海藻酸钠(SA)模拟多糖,配置与实际提取的EPS组分含量相同的模拟溶液用作实验料液,记为BSA+SA。所有相关试剂全部购自上海阿拉丁试剂,为分析纯级,实验用水均为MilliQ超纯水。
-
在相同的操作条件(0.1 μm PVDF,T=25 ℃,pH=7.0,TMP=0.2 MPa)下,对提取的实际EPS溶液和模拟溶液BSA+SA进行恒压过滤并测定其膜通量随时间的衰减曲线。实验采用孔径为0.1 μm的平板PVDF膜(Millipore,美国),实验前,将膜在超纯水中浸泡6~10 h,以去除表面的保护物质。恒压过滤装置主要由40 L的氮气瓶、减压阀、缓冲瓶、50 mL的Millipore超滤杯(8050,Amicon,美国)、电子天平(JA21002,上海越平科学仪器有限公司)和计算机组成。操作压力由高压氮气钢瓶提供,减压阀用于控制恒压操作;缓冲瓶用于防止压力过大;进料液从超滤杯上端进入,滤液过膜后流入天平上的烧杯中;天平与计算机超级终端连接,实时记录滤液质量变化,再通过折算为体积,最后根据式(1)计算膜通量。
式中:J为膜通量,L·(m2·h)−1;ΔV为过滤滤液体积差值,cm3;A为过滤面积,cm2;Δt为天平计重时间差,s。
恒压过滤过程一般可以用4种模型进行描述:完全堵塞模型、滤饼层过滤模型、标准堵塞模型和中间堵塞模型[18]。4种过滤模型可归纳为简单的数学形式,如式(2)所示。
式中:dt/dV为收集单位滤液所需要的时间,s·cm−3;k为常数;n=0、1、1.5、2,分别对应滤饼层过滤模型、中间堵塞模型、标准堵塞模型和完全堵塞模型。膜过滤堵塞模型的分析使用Origin软件拟合获得。
-
使用Zetasizer Nano ZS(英国马尔文公司)测量粒径及其分布。样品在测量前用孔径为0.22 μm的微滤膜进行过滤;采用Nicolet 6700(美国赛默飞公司)傅里叶红外光谱仪对组成进行分析,测试前须将样品进行真空冷冻干燥,然后将样品与溴化钾混合研磨后进行压片,对样品进行扫描,扫描分辨率为4 cm−1,测定波数为 400~4 000 cm−1;采用S-3400N型扫描电子显微镜(日本日立公司)表征膜的表面形貌,测试前将样品固定在样品台上,为了提高样品的导电性能,采用喷金处理后将样品放入扫描电镜中观察形貌;流变行为在Haake Mar Ⅲ型流变仪(美国赛默飞公司)上测定,流变仪通过水浴控制温度(25±1) ℃,测试使用套筒,进行动态应力扫描和频率扫描以研究流变特性。
-
图1和表1分别为EPS和BSA+SA加入Ca2+前后的粒径分布图和平均粒径值。从图1和表1可知,添加Ca2+前EPS粒径主要集中在90~200 nm,平均粒径为(146±7) nm;BSA+SA粒径分布为30~200 nm,平均粒径为(131±16) nm,比EPS的粒径分布范围稍宽,两者粒径接近,单峰尺寸分布基本重叠。
添加Ca2+后,EPS-Ca2+粒径分布为300~900 nm,平均粒径增大至(498±21) nm;而BSA+SA-Ca2+则主要集中在200~800 nm,平均粒径为(384±11) nm,两者粒径范围差异更明显,且EPS-Ca2+的平均粒径大于BSA+SA-Ca2+的平均粒径。加入Ca2+后,EPS的平均粒径从(146±7) nm增加到(498±21) nm,而BSA+SA的平均粒径仅增加了253 nm,这可能是由于模拟溶液选用成分较为简单的牛血清白蛋白和海藻酸钠作为蛋白和多糖的替代物,而从实际颗粒污泥中提取的EPS蛋白和多糖组分则较为复杂。
当Ca2+添加到溶液后,溶液的平均粒径增大,粒径分布范围变宽,这是由于Ca2+会与海藻酸钠的碳链结构相互作用,形成有高度组织性的结构体并在邻近的海藻酸钠分子间产生架桥作用,最后形成一个密实的凝胶网状结构体[19],因此,当Ca2+存在时,溶液中的多糖会与Ca2+协同形成稳定胶体,导致平均粒径明显增大;同时二价阳离子Ca2+易与溶液中带负电的官能团结合,从而减少电荷斥力,使高聚物分子间易于聚合,使得溶液分子粒径明显增大。已有研究[20-21]发现,溶液加入Ca2+后,高分子趋于卷缩成团,使得溶液颗粒相互积聚,粒径增大,这与本研究结果一致。
-
图2为BSA+SA、EPS及其加入Ca2+后形成的水胶体红外谱。如图2所示,红外光谱中均出现了蛋白质、多糖等多聚物分子内部O—H伸缩振动吸收峰(3 300~3 500 cm−1)、多糖分子内C—H伸缩振动吸收峰(2 900~3 000 cm−1)、羧酸基团中的C=O的伸缩振动吸收峰(1 400 cm−1),而位于1 650、1 540、1 250 cm−1附近振动峰为典型的酰胺谱带(酰胺Ⅰ、Ⅱ和Ⅲ),1 000~1 100 cm−1的特征吸收峰则对应糖苷键C—O—C的拉伸振动。本研究获得的特征官能团的吸收峰与其他研究[22-23]无明显差异,表明EPS的官能团组成与大多数研究相似,其组成物质主要为蛋白质和多糖。
由于EPS和BSA+SA中的羧基、羟基、糖苷键等可与Ca2+发生配位作用形成蛋壳模型[24],因此,红外谱图中特征官能团的位置会发生细微变化。BSA+SA中C=O伸缩振动吸收峰位于1 400 cm−1,加入Ca2+后向右偏移至1 440 cm−1,波数的偏移量远远大于4 cm−1的分辨率,与SARTORI等[25]报道的研究结果一致。而原本位于3 298 cm−1的O—H吸收峰向右偏移至3 394 cm−1,可能是由于Ca2+具有很强的通过O—H基团与多糖结合的能力,导致Ca2+与羟基发生配位作用[26];此外,也有研究[27]表明,Ca2+与溶液中的Na+发生离子交换也可导致吸收峰偏移。对于实际提取的EPS,1 406 cm−1为C=O的伸缩振动吸收峰,加入Ca2+后,该峰向高波数位移17 cm−1,这与EPS中羧基和Ca2+的桥联作用有关[28]。不同的是,原本位于1 512 cm−1的N—H吸收峰向高波数偏移至1 522 cm−1,表明EPS中酰胺的确与Ca2+发生了相互作用。通过红外谱图可知,EPS和BSA+SA主要的官能团种类相似,但添加Ca2+后,红外光谱图在表征官能团特征峰区域时差别较大,产生红外偏移的官能团不同,说明与Ca2+发生作用的官能团种类不同。
-
图3(a)是EPS-Ca2+和BSA+SA-Ca2+水胶体黏弹性模量随应变扫描曲线。由图3(a)可见,BSA+SA-Ca2+的弹性模量G'、黏性模量G''均比EPS-Ca2+的模量高且相差2个数量级,表明BSA+SA-Ca2+水胶体具有较强的水胶体结构。当应变在0.01%~1%变化时,G'和G''的曲线平稳,为线性黏弹区。在线性黏弹区,2种水胶体的G'>G'',说明水胶体的弹性占主导地位,“固体”性质相对明显。随着应变的逐渐增大,G'与G''逐渐减小,且G' 降幅显著高于G'' 降幅,G' 和G'' 相对大小仍为G' >G'',但两者差值逐渐变小,直至达到临界点,表明水胶体结构内部发生了实质性变化。EPS-Ca2+的临界应变约为50%,而BSA+SA-Ca2+的临界应变仅为25%,这是由于BSA+SA-Ca2+水胶体系结构较稳定,随着应变增大,胶体结构被破坏的程度较轻。交点过后,G'和G''持续下降,但黏弹性模量值相对大小呈现G'<G'',说明此时已转变为黏性主导。
图3(b)为EPS-Ca2+和BSA+SA-Ca2+黏弹性模量频率扫描曲线。由图3(b)可见,在频率扫描范围内G' >G'',表明样品均能形成水胶体结构。在整个频率范围内,BSA+SA-Ca2+水胶体的模量G'和G''对频率的相关依赖性低。随着频率的增加,模量G'、G''呈现出极小的增幅趋势且G'和G''互相平行,说明频率的大小对水胶体结构基本不产生影响,BSA+SA-Ca2+具有较好的稳定性。但在高频区,随着频率的增加,EPS-Ca2+的G'、G''出现骤减和骤增,表明EPS-Ca2+的结构体系容易被破坏,这与图3(a)的结果一致。
-
对EPS和BSA+SA进行恒压膜过滤实验来观察膜污染的行为差异,采用SEM对比原始PVDF膜、EPS污染后和BSA+SA污染后的膜面形貌,结果如图4所示。由图4(a)可见,原始PVDF膜面疏松多孔,孔与孔之间相互贯通。EPS和BSA+SA污染后的膜面明显截留了污染物质,导致膜孔堵塞,孔隙率降低,如图4(b)和图4(c)所示。EPS基本将膜孔完全覆盖并且在膜表面形成了污染层,而BSA+SA污染后的膜面仅部分膜孔被堵塞,与EPS污染后的膜面相比,BSA+SA对于膜面的污染程度更轻。
图5(a)为EPS和BSA+SA恒压过滤实验中膜通量随过滤时间的变化曲线。由图5(a)可见,BSA+SA在极短的时间内膜通量迅速下降,在过滤开始5 s后,已下降至初始值(650 L·(m2·h)−1)的0.58倍,之后逐渐趋于平稳,在300 s之后,通量稳定在250 L·(m2·h)−1;而EPS在过滤初始阶段膜通量下降较慢,但随后持续下降,直至稳定后,通量为90 L·(m2·h)−1。EPS恒压过滤后通量降低了近86%,而BSA+SA仅为61%,EPS对膜面的污染程度更为严重,与SEM表征的膜表面污染程度结果一致。图5(b)是加入Ca2+后的膜通量随时间的变化曲线。如图5(b)所示,EPS-Ca2+和BSA+SA-Ca2+均呈现出在短时间内快速下降的通量衰减趋势,FT-IR结果证实溶液中的蛋白质类物质含有多种含氧官能团,这些官能团的存在可使得溶液具有与Ca2+发生离子交换和络合作用的能力,导致粒径的增大;当溶液中存在大量的大粒径污染物质时,随着污染物质不断积累在膜表面,导致有效过滤面积减小,使得通量迅速下降。但BSA+SA-Ca2+引起的通量衰减快于EPS-Ca2+所引起的通量衰减,当过滤时间达到100 s时,BSA+SA-Ca2+引起的通量衰减达到80.8%,明显高于EPS-Ca2+的通量衰减47.7%。随后膜通量缓慢下降,可能是已经形成了污染层,压差被加厚和压实的污染层逐渐抵消。当膜通量保持平稳时,EPS-Ca2+的通量衰减达到了87%,而BSA+SA-Ca2+的通量衰减达到90%。换言之,BSA+SA-Ca2+的膜污染潜能要高于EPS-Ca2+的膜污染潜能,这主要是因为BSA+SA-Ca2+水胶体系结构较稳定,不易被破坏,造成膜污染现象较严重。
为深入研究两者在膜污染中的差别,利用HERMIA[18]所提出的过滤模型进行拟合,恒压过滤过程的模型拟合及可决系数R2结果见图6。如图6所示,EPS-Ca2+对中间堵塞模型、标准堵塞模型和滤饼层堵塞模型的拟合度较好;而BSA+SA-Ca2+则对滤饼层堵塞模型和中间堵塞模型呈现出较好的拟合趋势性,标准堵塞模型和完全堵塞模型拟合度都较差。EPS-Ca2+对中间堵塞模型的拟合度最好,说明有大颗粒参与堵塞膜孔,导致有效过滤面积下降,造成膜污染[29];而BSA+SA-Ca2+最符合滤饼层过滤模型,这是由于膜孔内部逐渐堵满了颗粒,后续颗粒随着过滤的进行逐渐沉积到膜表面,膜表面形成凝胶层并不断压实,阻力不断上升,从而导致较严重的膜污染。同时,单一的污染模型并不能有效准确地描述膜污染的整个过程,因为膜孔堵塞、孔径减小和污染层的形成等均存在于整个恒压过滤过程中,但从膜通量曲线及模型拟合图可以明显观察到,在Ca2+存在的条件下,实际EPS和模拟EPS的堵塞模型存在显著差异。
-
1) EPS-Ca2+的平均粒径大于BSA+SA-Ca2+的平均粒径,加入Ca2+后,絮凝作用使得粒径都有所增大。
2)在官能团特征峰区域,与Ca2+发生作用的官能团不同导致相应的特征吸收峰发生偏移。
3) BSA+SA-Ca2+水胶体系结构较EPS-Ca2+更稳定,导致了在膜污染行为中,BSA+SA-Ca2+絮体不易被破坏,造成更严重的膜污染现象。
4) EPS-Ca2+对中间堵塞模型、标准堵塞模型和滤饼层堵塞模型呈现出较好的拟合度;而BSA+SA-Ca2+则对滤饼层堵塞模型和中间堵塞模型拟合度较好。
5)采用牛血清白蛋白(BSA)和海藻酸钠(SA)来模拟实际EPS组分存在不确定性,未来还需要对EPS中多糖和蛋白的替代物进行进一步研究。
钙离子存在下胞外聚合物及其模拟溶液在膜污染中的差异
Differences of extracellular polymeric substances (EPS) and model solution with regard to membrane fouling in the presence of Ca2+
-
摘要: 针对膜生物反应器膜污染的问题,为探究模拟物质替代实际物质的可行性,以胞外聚合物(EPS)及其模拟溶液(BSA+SA)为研究对象,对比了钙离子(Ca2+)存在下胞外聚合物及其模拟溶液在膜污染中的差异,考察了EPS-Ca2+和BSA+SA-Ca2+在粒径分布、官能团、流变特性和膜污染行为等方面的差异。结果表明:Ca2+使得溶液粒径变大,且EPS-Ca2+的平均粒径大于BSA+SA-Ca2+的平均粒径;不同官能团与Ca2+发生作用导致红外光谱中特征峰发生偏移;BSA+SA-Ca2+水胶体系结构更稳定,随着应变的增大,胶体结构被破坏的程度较轻;在微滤过程中,相同操作条件下,BSA+SA-Ca2+的膜污染现象更为严重。进一步采用Hermia过滤模型拟合发现,EPS-Ca2+和BSA+SA-Ca2+分别对中间堵塞模型和滤饼层过滤模型的拟合程度最高。上述结果可为膜污染过程中胞外聚合物及其膜污染机理研究提供参考。Abstract: The scientific interests in membrane fouling of membrane bioreactors have increased drastically in recent years. In order to investigate the feasibility of simulating substances to replace actual substances, the present study compared the different performance of extracellular polymeric substances (EPS) and model EPS solution (BSA+SA) in the presence of Ca2+ on the membrane fouling. The differences in particle size distribution, functional groups, rheological properties and membrane fouling behaviors of EPS-Ca2+ and BSA+SA-Ca2+ were investigated systematically. The results showed that Ca2+ enlarged the particle size, and the average particle size of EPS-Ca2+ was larger than the that of BSA+SA-Ca2+. Owing to the interactions between different functional groups and Ca2+, the shift of the characteristic peaks in FT-IR spectrum occurred. By comparison of the rheological properties with EPS-Ca2+ hydrocolloid, the BSA+SA-Ca2+ hydrocolloid exhibited a more stable structure which presented a less destruction as the strain increased. In the microfiltration process, the membrane fouling of BSA+SA-Ca2+ was more serious under the same operating conditions. Through the data fitting with Hermia’s filtration model, it was found that EPS-Ca2+ and BSA+SA-Ca2+ obeyed the intermediate blocking model and the cake formation model, respectively. The results would provide basic data for the research of EPS and its membrane fouling mechanism.
-
邻苯二甲酸酯(PAEs)作为增塑剂广泛用于制造和加工塑料产品。PAEs的生产始于20世纪20年代,其用量一直在增长[1-3]。PAEs在制造、使用和处置过程中可以直接或间接地释放到环境中。PAEs普遍存在于大气气溶胶[4-5]、污水和废水处理的污泥[6]、河流和海水/沉积物[7]、饮用水[8]、生物群和空气[9]中。据报道,人类对 PAEs的摄入量可高达70 μg·(kg·d)−1;PAEs对生态系统功能和公共健康存在潜在影响,因而PAEs已引起广泛的关注[10]。短链酯(如邻苯二甲酸二甲酯(DMP))是不同环境介质中最常见的PAEs,作为一种内分泌干扰化学物质,其可能导致人类白细胞的染色体损伤,干扰动物和人类的生殖系统和正常生长发育[11]。因此,研发一种可用于从水域和废水中去除这种污染物的处理技术十分必要。
去除DMP的方法包括催化臭氧法[12],γ-辐射/ H2O2工艺[13],紫外光催化法[14],生物降解处理法[15]等。在众多处理方法中,光催化技术已被证明能够有效处理各种水体污染物。高铁酸钾作为一种高效的电子受体可有效抑制e−/h+重组,其还原产物为Fe3+或Fe(OH)3, 具有絮凝、吸附、共沉淀等功能,是一种绿色氧化剂,但目前关于高铁酸盐和光催化组合降解有机水污染物的研究相对较少。
本研究利用高铁酸钾与TiO2光催化的协同氧化效应降解水中DMP,建立了DMP降解方法,并探究了不同参数对DMP降解效果的影响以及反应过程中在催化剂表面产生Fe—O—(有机)络合物对降解效果的影响。
1. 材料与方法
1.1 试剂与仪器
邻苯二甲酸二甲酯(DMP)、二氧化钛(TiO2)、高铁酸钾(K2FeO4)、磷酸氢二钠(Na2HPO4)、四硼酸钠(Na2B4O7·10H2O)、亚硫酸钠(Na2SO3)均为分析纯;甲醇(CH3OH)为色谱纯。
紫外-可见分光光度计(TU-1810,北京普析通用仪器有限责任公司);紫外灯(8 W,荷兰皇家飞利浦公司);pH计(UB-10,美国Denver Instrument公司);高效液相色谱仪(UltiMate3000,美国Dionex公司);X射线衍射仪(Ultima IV,日本Rigaku公司);磁力搅拌器(78-1,常州国华电器有限公司)。
1.2 实验装置
实验采用自制光催化反应器,它是由石英装置、高压汞灯、磁力搅拌器组成的开放式反应器,实验装置见图1。紫外灯(8 W,365 nm)作为光源,石英反应器放置于磁力搅拌器上距光源8 cm处,反应开始时,开启磁力搅拌器,溶液在搅拌器搅拌下混合均匀。在室温下进行实验,整套实验装置外加紫外线防护装置。
1.3 实验方法
用0.005 mol·L−1 Na2HPO4和0.001 mol·L−1 Na2B4O7·10H2O调节DMP溶液pH为9,目的在于维持高铁酸盐的水溶液稳定性和消除铁酸盐分析中Fe3+的干扰。在光催化实验中,向DMP水溶液中加入一定量的TiO2催化剂;光反应之前,先进行30 min暗反应,以达到吸附/解吸平衡。在反应期间,以不同的时间间隔取样,加入亚硫酸钠[16]终止反应,然后通过0.22 μm 有机滤膜,除去固体颗粒,所有实验均在室温下进行。
1.4 分析方法
采用高效液相色谱仪(HPLC)测量DMP浓度。色谱条件:尖峰II C18柱(5 μm粒度,250 mm×4.6 mm),柱温为30 ℃,使用甲醇/水(70∶30)的流动相,流速为1 mL·min−1,进样量为10 μL,UV检测器波长230 nm。采用紫外-可见分光光度法在505 nm波长下测量高铁酸盐浓度;采用X射线衍射(XRD)仪分析TiO2催化剂反应前后的晶体结构。
2. 结果与讨论
不同因素对DMP降解率的影响见图2。
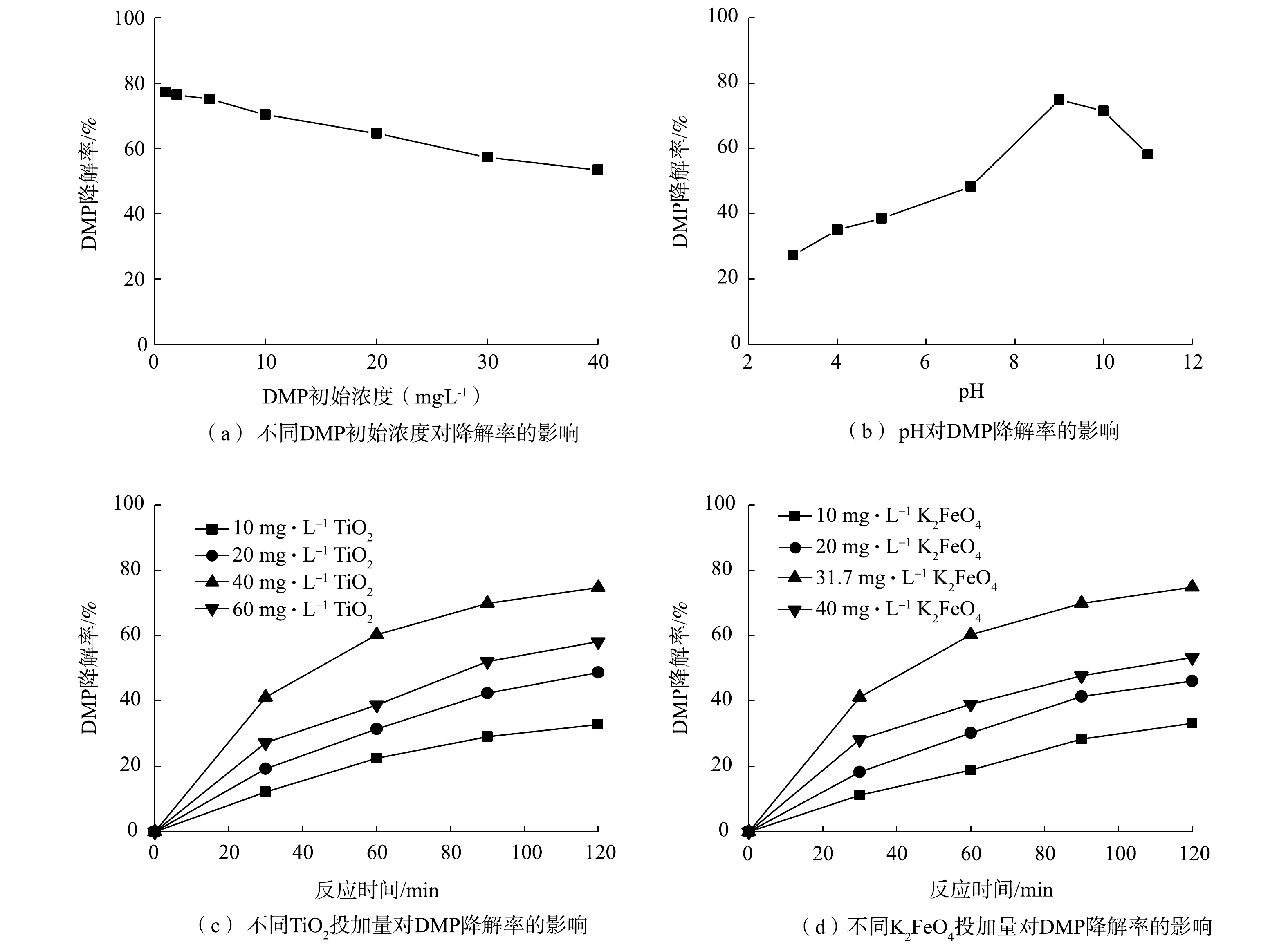 图 2 不同因素对DMP降解率的影响Figure 2. Effect of different factors on the degradation rate of DMP
图 2 不同因素对DMP降解率的影响Figure 2. Effect of different factors on the degradation rate of DMP2.1 DMP初始浓度对DMP降解率的影响
DMP初始浓度对光降解率的影响如图2(a)所示。可以看出,随着DMP初始浓度的增加,DMP的光降解率逐渐降低。水溶液中TiO2的量是恒定的,故催化剂的活性位点数也是恒定的,较高的DMP初始浓度导致分子竞争催化剂的活性位点,使降解速率降低。DMP的初始浓度越高,导致越多的DMP分子被吸附在催化剂的表面上,从而抑制TiO2表面产生光生空穴和光生电子。因此,DMP的最佳初始浓度确定为5 mg·L−1,并在此条件下进行后续的研究。
2.2 溶液pH对DMP降解率的影响
Fe(Ⅵ)在整个pH范围内都是强氧化剂,从式(1)和式(2)中看出,Fe(Ⅵ)在酸性和碱性溶液中的还原电位分别为2.2 V和0.7 V。从图2(b)中可以看出,随着pH的增加,DMP降解率也随之增加,但当pH为9时,降解率随pH的增加开始降低。其原因可能是:在酸性条件下,K2FeO4分解生成的OH−与水溶液中H+发生中和反应,使OH−浓度降低[17];另外,H+使得 K2FeO4在水溶液中不稳定,高铁酸钾的分解速度非常快,还未来得及与DMP反应,自身就已经分解,导致DMP降解率降低。反之,在碱性条件下,反应体系中OH−浓度增加,高铁酸钾自分解速度较缓慢,稳定性增强,DMP降解率较高。综合考虑,在酸性条件下,高铁酸盐稳定性很差,随着pH的升高,高铁酸盐稳定性逐渐加强(碱性>中性>酸性),K2FeO4在pH为9~10的水溶液中稳定性最好[18],故本研究的最佳pH确定为9。
FeO2−4+8H++3e−→Fe3++4H2OE0=2.2V (1) FeO2−4+4H2O+3e−→Fe(OH)3+5OH−E0=0.7V (2) 2.3 TiO2催化剂量对DMP降解率的影响
从图2(c)中看出,TiO2投加量较少时,溶液中催化剂的活性位点较少,导致DMP降解率不高;TiO2投加量逐渐增加,催化剂的活性位点也增加,降解率也随之提高。TiO2投加量为40 mg·L−1时,DMP降解率最高可达到75%;当TiO2投加量超过40 mg·L−1后,进入溶液中可被吸收的光子全部被催化剂吸收,导致催化剂表面产生的活性基团数目保持恒定;继续加大TiO2,投加量不但会造成光散射,而且溶液浑浊度增加,阻碍了紫外光的有效照射,光的吸收效率和反应活性都会下降[19]。
2.4 高铁酸钾浓度对DMP降解率的影响
高铁酸钾投加量对DMP降解效果的影响也较大。由图2(d)中可以看出, 随着高铁酸钾投加量的增加, DMP的降解率先升高后降低。当高铁酸钾投加量达到31.7 mg·L−1时,DMP的降解率最高可达75%。由于高铁酸钾自身会有一定分解, 并且随着高铁酸钾投加量的增大, 高铁酸钾自分解随之加快,可能影响降解效果。若投加过量的铁,会直接影响光照强度,从而降低光催化效率[20]。因此,确定最佳高铁酸钾投加量为31.7 mg·L−1。
2.5 不同体系对DMP降解率的影响
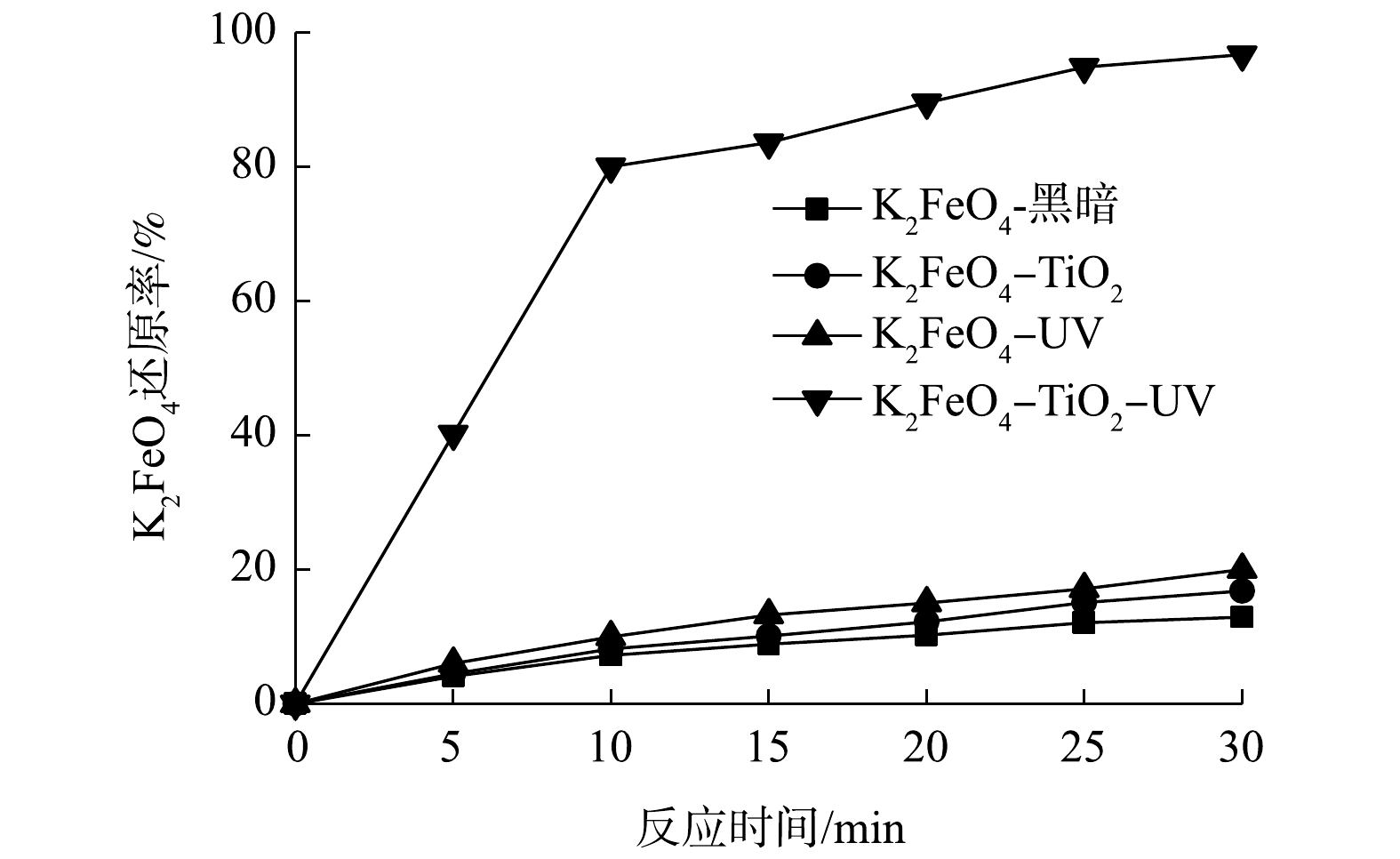
在光催化体系中,存在某些金属离子或氧化剂可以作为电子受体来防止e−和h+电子的快速复合,从而增强目标化合物的光催化降解。从图3中可以看出,高铁酸盐对DMP没有显著的降解效果,DMP降解率只有5%左右,表明高铁酸盐对有机基质的氧化有特定的选择性。在紫外光照和高铁酸盐存在的情况下,DMP的降解效果也较差,表明在没有TiO2的情况下,高铁酸盐和单纯的紫外光照没有明显的协同作用。TiO2光催化氧化降解DMP的效果也不理想,120 min后,DMP只降低15%。然而,与之形成鲜明对比的是,用Fe(VI)-TiO2-UV体系降解DMP的效果是显著的,120 min时,DMP降解率约75%,这表明光催化与高铁酸盐的组合可对DMP的降解产生明显的协同效应。由于高铁酸盐比其他电子受体(如高锰酸盐或过氧化物)具有更高的氧化能力,并且可能还原为高度活性的Fe(Ⅴ),因此,高铁酸盐可以通过电子清除从而起到增强光氧化和有机氧化的作用。
2.6 二氧化钛-高铁酸盐反应体系中的高铁酸盐还原
由图4中可以看出,Fe(Ⅵ)在黑暗中减少地非常缓慢,在单独的UV照射下或者在具有TiO2悬浮液但没有UV照射的情况下仅略快。然而,相比之下,在紫外光照下,二氧化钛悬浮液存在的情况下发生了快速的Fe(Ⅵ)还原。很显然,Fe(Ⅵ)的快速还原是由于它从TiO2催化剂上清除了激发的导带电子。Fe(Ⅵ)还原对催化剂的电子清除大大减少了导带电子(e−)和价带空穴(h+)的复合,从而提高了TiO2光催化反应过程中的量子效率。
2.7 TiO2催化剂的失活与再生
1)TiO2催化剂的失活。使用不同的TiO2催化剂光降解DMP的效果见图5。对采用Fe(Ⅵ)-TiO2-UV工艺降解DMP后的TiO2进行回收利用。由图5可知,回收后的TiO2经水洗后,降解DMP的效果很差;但经1% HCl洗涤后的TiO2在120 min后可降解70%的DMP,类似于新二氧化钛的降解效果。上述结果表明,在对DMP的降解反应过程中,催化剂表面上会形成Fe—O—(有机)络合物,其能够抑制DMP的降解,故可用1% HCl溶液洗涤失活的TiO2催化剂将其再活化。本研究对反复使用后的催化剂的催化降解性能进行了分析。在Fe(Ⅵ)浓度为 31.7 mg·L−1、TiO2为 40 mg·L−1、pH 为 9、反应时间为 120 min的条件下,对5 mg·L−1 的 DMP 进行催化氧化。反应后过滤回收催化剂用1% HCl清洗烘干,在相同反应条件下,对 DMP 进行降解,如此反复使用4次。实验结果表明,第 1 次使用催化剂时,DMP降解率达到了70%;第 2、3、4次重复使用,其降解率依次降低,分别为63.22%、58. 92%、53.33%。这说明催化剂的降解稳定性较好。
 图 5 不同条件下的TiO2对DMP降解率的影响Figure 5. Effect of different TiO2 on the degradation rate of DMP
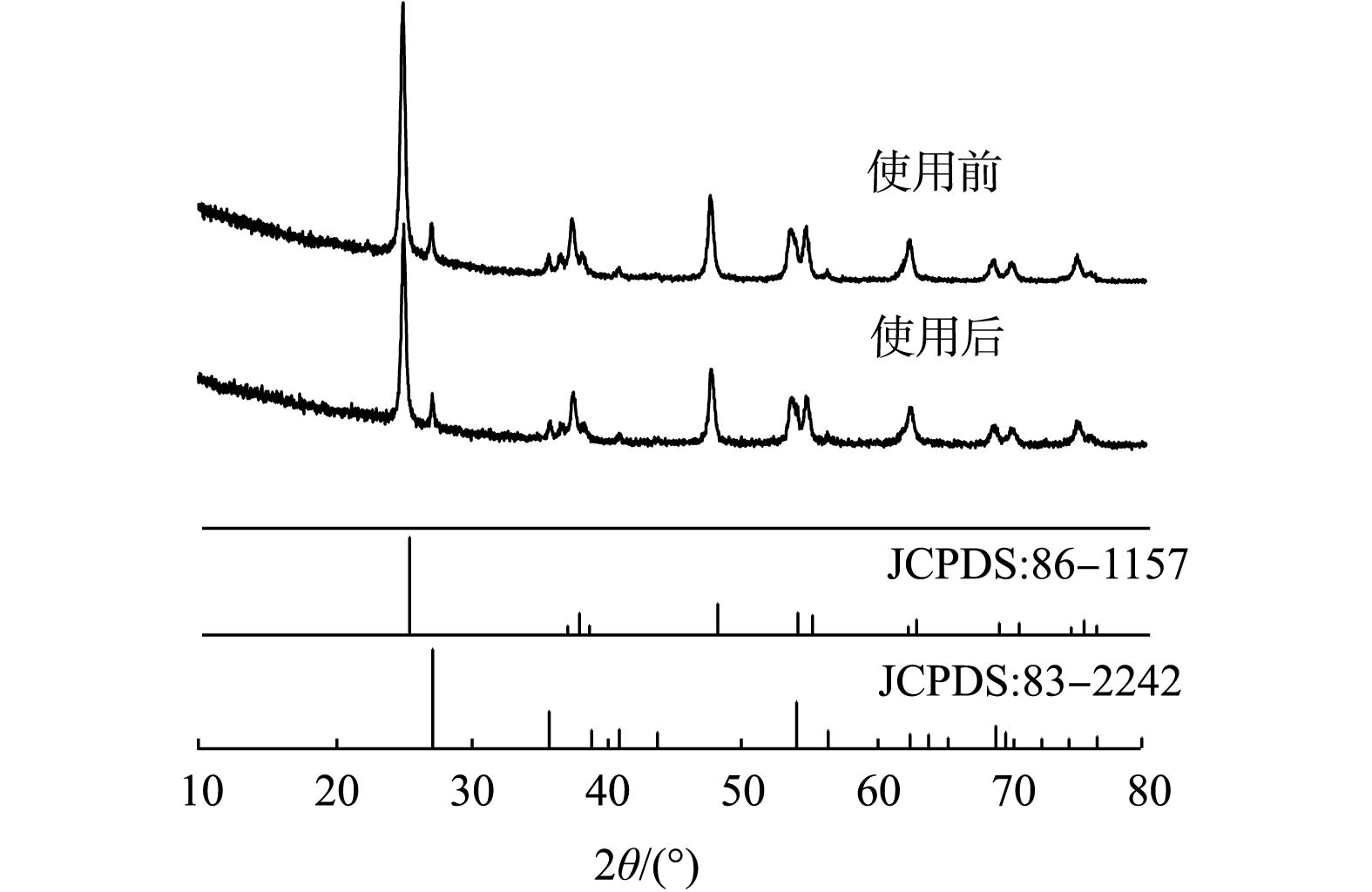
图 5 不同条件下的TiO2对DMP降解率的影响Figure 5. Effect of different TiO2 on the degradation rate of DMP2)未使用和使用过的二氧化钛催化剂的比较。图6是使用前、后TiO2的XRD图谱。由图6可知,XRD特征峰发生在2θ=25.32°、37.86°、48.06°、53.97°、55.08°、62.75°、68.87°、70.33°、75.14°和82.75°,分别对应于锐钛矿TiO2的(101)、(004)、(200)、(105)、(211)、(204)、(116)、(220)、(215)和(224)晶面(JCPDS,No.86-1157);在27.46°,36.08°,41.24°,56.67°这4个特征峰处,分别对应于金红石TiO2的(110)、(101)、(111)和(220)晶面(JCPDS,No.83-2242)[21]。上述结果表明, TiO2催化剂由锐钛矿和金红石的混合相组成。使用前、后TiO2的衍射峰证实了在反应后TiO2催化剂的晶体结构没有发生显著变化。
2.8 Fe(Ⅵ)-TiO2-UV光催化氧化降解实际生产废水中的DMP
Fe(Ⅵ)-TiO2-UV工艺降解实际含DMP废水与浓度为0.32 mg·L−1的DMP模拟水样的结果见图7。模拟水样DMP配置浓度0.32 mg·L−1是基于实际生产废水中DMP的检出浓度,实验中调节水样pH=7,TiO2投加量为40 mg·L−1,K2FeO4投加量为31.7 mg·L−1。结果表明,DMP实际废水和模拟水样在Fe(Ⅵ)-TiO2-UV体系中的降解率分别为67%和78.2%,说明Fe(Ⅵ)-TiO2-UV工艺对实际废水中的DMP降解效果良好,具有实用意义。
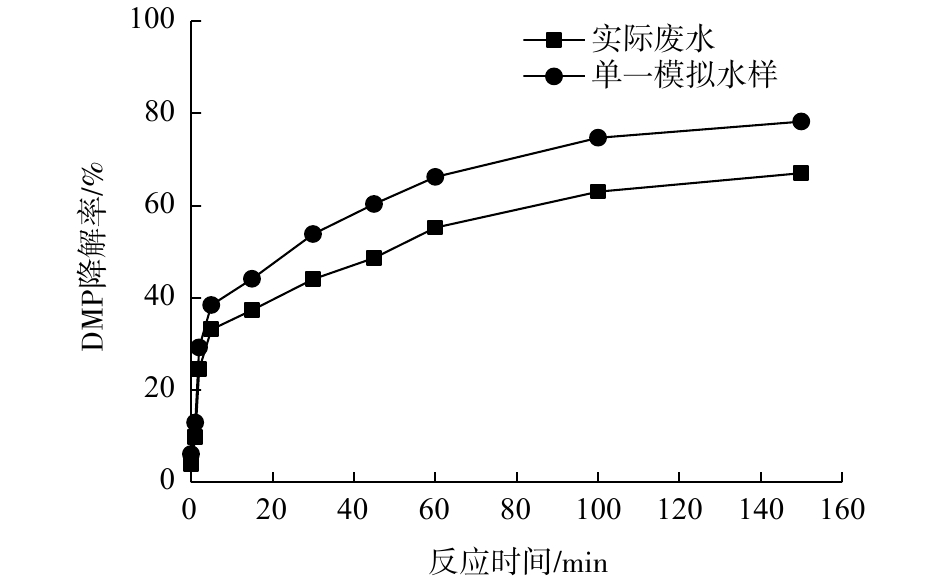 图 7 实际水样与模拟水样中DMP的降解率Figure 7. Degradation rate of DMP in real water samples and simulated water samples
图 7 实际水样与模拟水样中DMP的降解率Figure 7. Degradation rate of DMP in real water samples and simulated water samples2.9 矿化度的分析和降解机制的讨论
由图8可以看出,当反应时间在120 min内,DMP 降解率和矿化率均随反应时间的增加而增大。已知在 120 min 时, DMP 的降解率可达75%,DMP的矿化率仅达到 33. 1% 左右。继续延长反应时间,DMP 降解率和矿化率都趋于平缓。这表明反应体系中紫外光激发 TiO2催化剂所产生的·OH 在反应过程中的协同作用显著,大部分的 DMP 已被转换为较难矿化的有机中间产物。
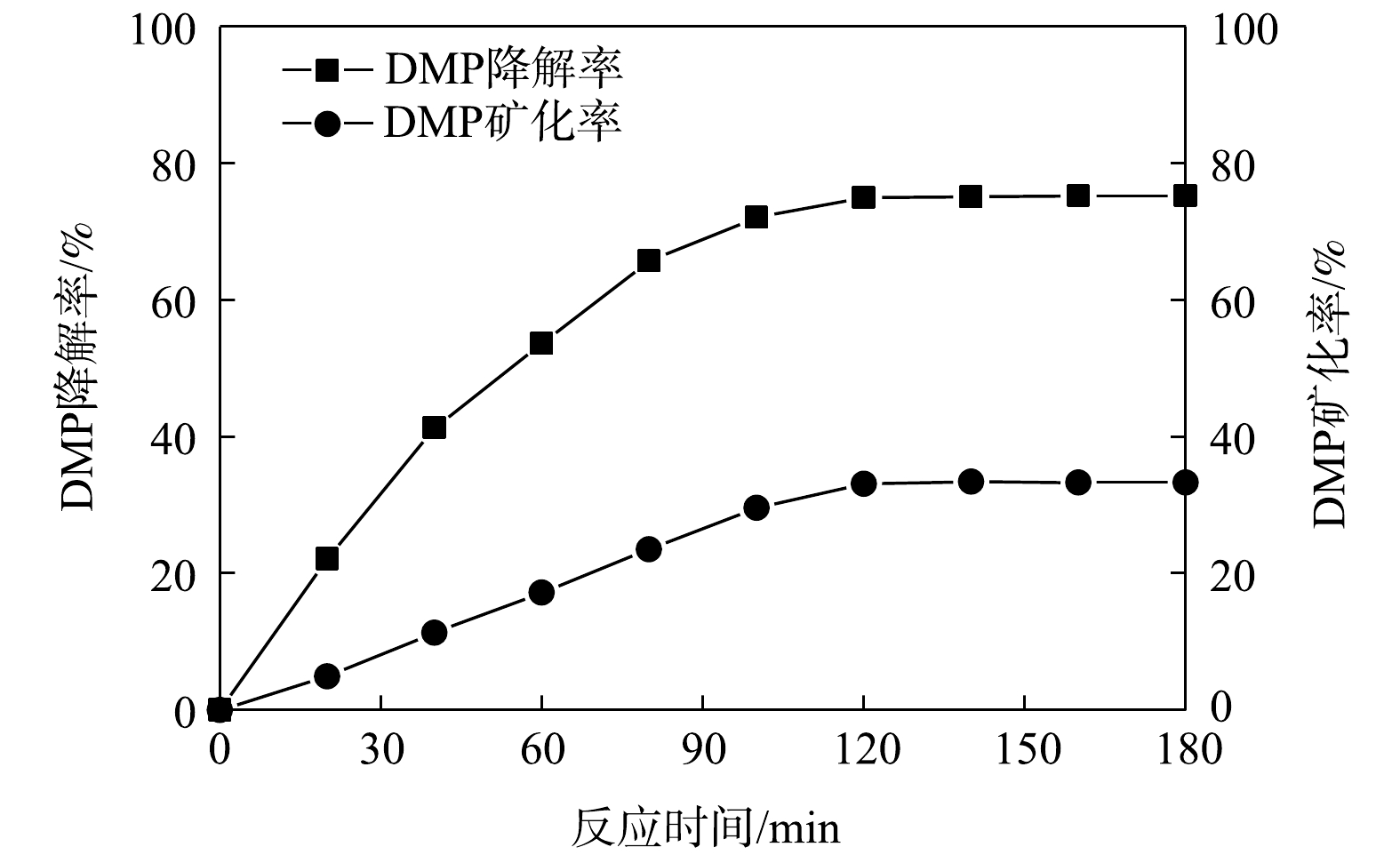 图 8 DMP的光催化降解率和矿化率Figure 8. Photocatalytic degradation rate andmineralization rate of DMP
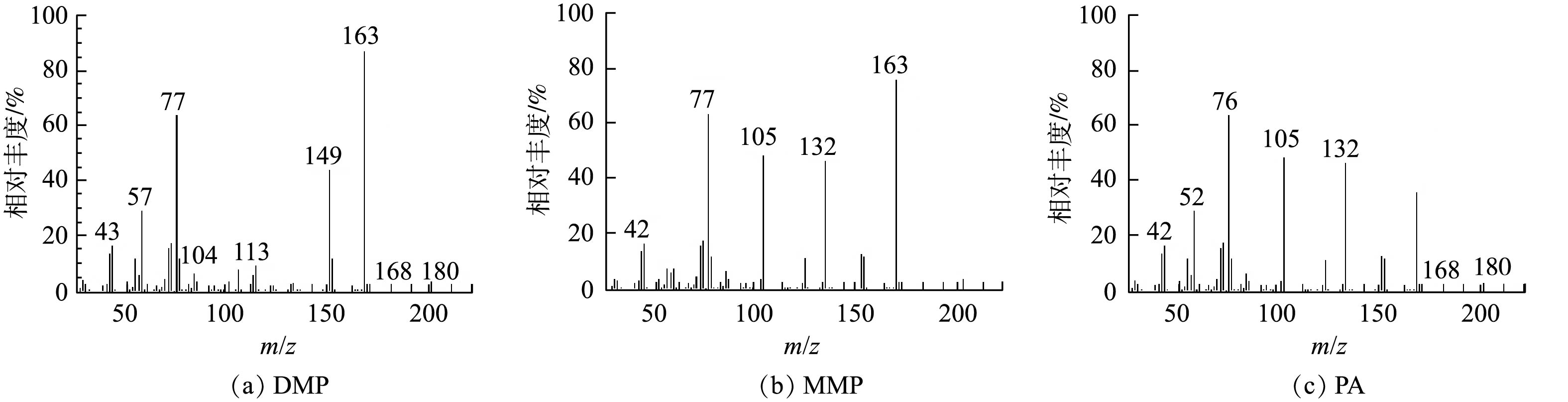
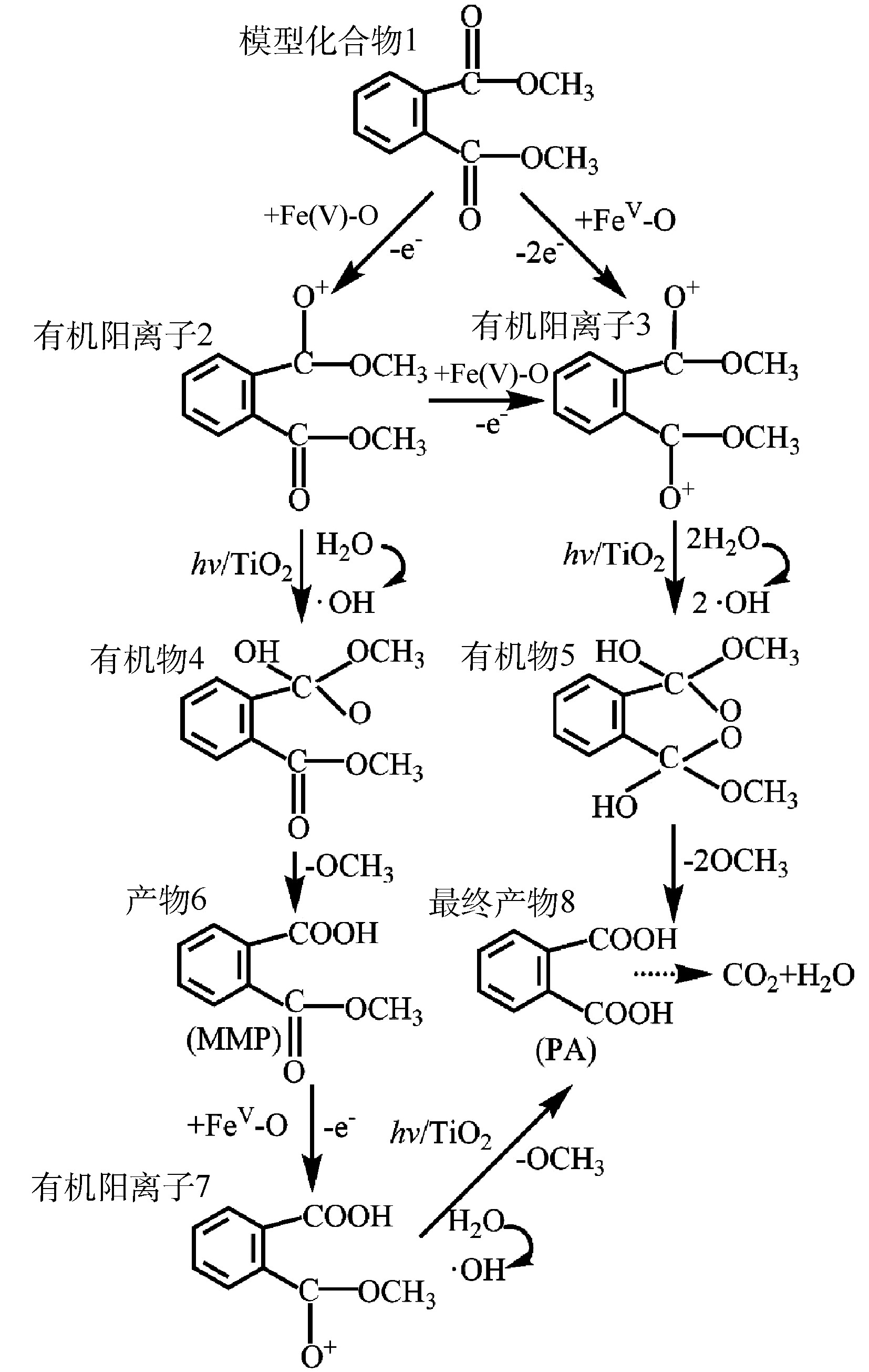
图 8 DMP的光催化降解率和矿化率Figure 8. Photocatalytic degradation rate andmineralization rate of DMPTiO2光催化生成的·OH如何氧化DMP是一个复杂问题,至今未有定论[22]。有研究[23]认为,·OH是从侧链进攻,最终把 DMP 氧化为邻苯二甲酸等。如图9所示,本研究使用GC-MS方法检测了DMP光催化降解中间产物DMP、邻苯二甲酸单甲酯(MMP)和邻苯二甲酸(PA),并在此基础上,推测了Fe(VI)-TiO2-UV体系中可能存在的DMP光催化降解途径(图10)。由图9可知,DMP降解的主要中间产物为PA,其由DMP的电子转移反应和与·OH的反应产生。一旦Fe(Ⅴ)反应失去1个电子或2个电子时,模型化合物1可以分别形成有机阳离子2和3,有机阳离子2可被·OH氧化生成有机物4,有机阳离子3可被·OH氧化生成有机物5,然后再进一步发生反应。有机物4失去一个甲氧基后将生成产物6(MMP),其在Fe(Ⅴ)氧化之后,进一步失去电子生成有机阳离子7,有机阳离子7加入一个羟基,然后失去一个甲氧基,生成最终产物8(PA);或者,有机物5在失去2个甲氧基后,直接生成最终产物8(PA)。最后,在紫外光和·OH的作用下,DMP 及其苯环中间产物发生开环反应,生成小分子有机酸,最后进一步矿化为 CO2和H2O。
3. 结论
1) Fe(Ⅵ)-TiO2-UV工艺对DMP的降解效果优于单独的高铁酸钾和单独的二氧化钛光催化,说明高铁酸钾与TiO2光催化之间存在协同效应。最佳降解条件是DMP初始浓度为5 mg·L−1、pH=9、高铁酸钾和TiO2投加量分别为31.7 mg·L−1和40 mg·L−1,DMP降解效果达到最优。
2) DMP降解过程产生Fe—O—(有机)络合物,造成光催化活性降低,导致DMP降解受到抑制,失活的TiO2催化剂可以用1% HCl溶液再活化。
3)采用Fe(Ⅵ)-TiO2-UV工艺降解实际废水和模拟水样,DMP降解率分别为67%和78.2%,表明K2FeO4协同TiO2光催化降解实际废水中的DMP效果良好,具有实用意义。
4)利用 GC/MS 检测分析,推测Fe(Ⅵ)-TiO2-UV体系光催化降解DMP中生成的自由基首先攻击DMP的侧链,生成中间产物MMP和最终产物PA,然后PA继续分解为小分子有机酸,最后矿化为 CO2和H2O。
-

图 1 EPS、BSA+SA添加Ca2+前后的粒径分布(Ca2+浓度为10 mmol·L−1)
Figure 1. Particle size distribution of EPS and BSA+SA in the absence and presence of Ca2+ (Ca2+ concentration is 10 mmol·L−1)
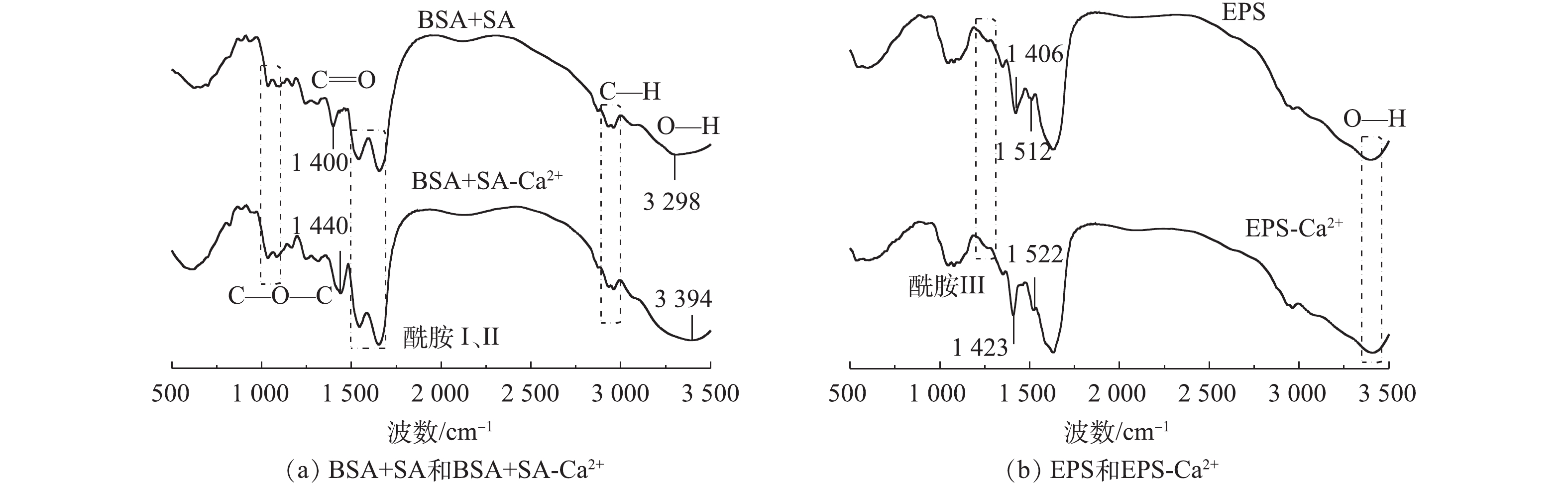
图 2 加入Ca2+前后EPS与BSA+SA的红外光谱图
Figure 2. FT-IR spectra of BSA+SA and EPS in the absence and presence of Ca2+

图 3 EPS-Ca2+和BSA+SA-Ca2+黏弹性模量曲线
Figure 3. Viscoelastic modulus curves of EPS-Ca2+ and BSA+SA-Ca2+

图 6 EPS-Ca2+和BSA+SA-Ca2+恒压过滤过程的模型拟合
Figure 6. Models fitting of constant-pressure filtration behaviors of EPS-Ca2+ and BSA+SA-Ca2+
表 1 EPS和BSA+SA添加Ca2+前后粒径分布
Table 1. Particle size distribution of EPS and BSA+SA in the absence and presence of Ca2+
溶液 粒径/nm 粒径分布/nm EPS 146±7 90~200 BSA+SA 131±16 30~200 EPS-Ca2+ 498±21 300~900 BSA+SA-Ca2+ 384±11 200~800  下载: 导出CSV
下载: 导出CSV
-
[1] WERNER C, KATURI K, ANANDA R H, et al. Graphene-coated hollow fiber membrane as the cathode in anaerobic electrochemical membrane bioreactors: Effect of configuration and applied voltage on performance and membrane fouling[J]. Environmental Science & Technology, 2016, 50(8): 4439-4447. [2] CHENG D, NGO H H, GUO W, et al. Anaerobic membrane bioreactors for antibiotic wastewater treatment: Performance and membrane fouling issues[J]. Bioresource Technology, 2018, 267: 714-724. doi: 10.1016/j.biortech.2018.07.133 [3] SMITH A L, STADLER L B, LOVE N G, et al. Perspectives on anaerobic membrane bioreactor treatment of domestic wastewater: A critical review[J]. Bioresource Technology, 2012, 122: 149-159. doi: 10.1016/j.biortech.2012.04.055 [4] MENG S J, FAN W H, LI X M, et al. Intermolecular interactions of polysaccharides in membrane fouling during microfiltration[J]. Water Research, 2018, 143: 38-46. doi: 10.1016/j.watres.2018.06.027 [5] LIN H J, PENG W, ZHANG M J, et al. A review on anaerobic membrane bioreactors: Applications, membrane fouling and future perspectives[J]. Desalination, 2013, 314: 169-188. doi: 10.1016/j.desal.2013.01.019 [6] ZHAO C Q, ZHANG G Q, XU X C, et al. Rapidly self-assembled polydopamine coating membranes with polyhexamethylene guanidine: Formation, characterization and antifouling evaluation[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2017, 512: 41-50. [7] KIM H C, CHOI B G, NOH J, et al. Electrospun nanofibrous PVDF-PMMA MF membrane in laboratory and pilot-scale study treating wastewater from Seoul Zoo[J]. Desalination, 2014, 346: 107-114. doi: 10.1016/j.desal.2014.05.005 [8] JEONG Y, KIM Y, JIN Y, et al. Comparison of filtration and treatment performance between polymeric and ceramic membranes in anaerobic membrane bioreactor treatment of domestic wastewater[J]. Separation & Purification Technology, 2018, 199: 182-188. [9] GAO W J, QU X, LEUNG K T, et al. Influence of temperature and temperature shock on sludge properties, cake layer structure, and membrane fouling in a submerged anaerobic membrane bioreactor[J]. Journal of Membrane Science, 2012, 421: 131-144. [10] CHEN J R, ZHANG M J, LI F Q, et al. Membrane fouling in a membrane bioreactor: High filtration resistance of gel layer and its underlying mechanism[J]. Water Research, 2016, 102: 82-89. doi: 10.1016/j.watres.2016.06.028 [11] JIANG J K, MU Y, YU H Q. Differences in the colloid properties of sodium alginate and polysaccharides in extracellular polymeric substances with regard to membrane fouling[J]. Journal of Colloid and Interface Science, 2019, 535: 318-324. doi: 10.1016/j.jcis.2018.10.002 [12] MORE T T, YAN S, HOANG N V, et al. Bacterial polymer production using pre-treated sludge as raw material and its flocculation and dewatering potential[J]. Bioresource Technology, 2012, 121: 425-431. doi: 10.1016/j.biortech.2012.06.075 [13] KIM I S, JANG N. The effect of calcium on the membrane biofouling in the membrane bioreactor (MBR)[J]. Water Research, 2006, 40(14): 2756-2764. doi: 10.1016/j.watres.2006.03.036 [14] CAO D Q, SONG X, FANG X M, et al. Membrane filtration-based recovery of extracellular polymer substances from excess sludge and analysis of their heavy metal ion adsorption properties[J]. Chemical Engineering Journal, 2018, 354: 866-874. doi: 10.1016/j.cej.2018.08.121 [15] BALA S S, YAN S, TYAGI R D, et al. Extracellular polymeric substances (EPS) producing bacterial strains of municipal wastewater sludge: Isolation, molecular identification, EPS characterization and performance for sludge settling and dewatering[J]. Water Research, 2010, 44(7): 2253-2266. doi: 10.1016/j.watres.2009.12.046 [16] FROLUND B, PALMGREN R, KEIDING K, et al. Extraction of extracellular polymers from activated sludge using a cation exchange resin[J]. Water Research, 1996, 30(8): 1749-1758. doi: 10.1016/0043-1354(95)00323-1 [17] FROLUND B, GRIEBE T, NIELSEN P H. Enzymatic activity in the activated-sludge floc matrix[J]. Applied Microbiology & Biotechnology, 1995, 43(4): 755-761. [18] HERMIA J. Constant pressure blocking filtration law application to powder-law non-newtonian fluid[J]. Transinstchemeng, 1982, 60(3): 183-187. [19] LEE S, ELIMELECH M. Relating organic fouling of reverse osmosis membranes to intermolecular adhesion forces[J]. Environmental Science & Technology, 2006, 40(3): 980-987. [20] RENBI B, LEOW H F. Microfiltration of activated sludge wastewater: The effect of system operation parameters[J]. Separation & Purification Technology, 2002, 29(2): 189-198. [21] 李方, 吴亮, 杨波, 等. 盐类对模拟胞外聚合物(EPS)膜污染的影响[J]. 环境工程学报, 2014, 8(3): 839-844. [22] 程战利, 武照远, 郭安, 等. A/O的SBR系统中不同方法提取EPS的效果分析[J]. 环境科学与技术, 2015, 38(12Q): 149-153. [23] YANG F L, SHI B Q, MENG F G, et al. Membrane fouling behavior during filtration of sludge supernatant[J]. Environmental Progress & Sustainable Energy, 2010, 26(1): 86-93. [24] GRANT G T, MORRIS E R, REES D A, et al. Biological interactions between polysaccharides and divalent cations: The egg-box model[J]. FEBS Letters, 1973, 32(1): 195-198. doi: 10.1016/0014-5793(73)80770-7 [25] SARTORI C, FINCH D S, RALPH B, et al. Determination of the cation content of alginate thin films by FT-IR spectroscopy[J]. Polymer, 1997, 38(1): 43-51. doi: 10.1016/S0032-3861(96)00458-2 [26] SAJJAD M, KIM K S. Studies on the interactions of Ca2+ and Mg2+ with EPS and their role in determining the physicochemical characteristics of granular sludges in SBR system[J]. Process Biochemistry, 2015, 50(6): 966-972. doi: 10.1016/j.procbio.2015.02.020 [27] 张亚琼, 俞怡晨, 陆轶业, 等. 钙离子存在下海藻酸盐稀溶液的粘度性质[J]. 应用化学, 2006, 23(11): 1259-1263. doi: 10.3969/j.issn.1000-0518.2006.11.016 [28] SOBECK D C, HIGGINS M J. Examination of three theories for mechanisms of cation-induced bioflocculation[J]. Water Research, 2002, 36(3): 527-538. doi: 10.1016/S0043-1354(01)00254-8 [29] 余智勇, 文湘华. 厌氧膜生物反应器中亲疏水性有机物的膜污染特征[J]. 中国环境科学, 2018, 38(7): 2471-2476. doi: 10.3969/j.issn.1000-6923.2018.07.010 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5242
- HTML全文浏览数: 5242
- PDF下载数: 77
- 施引文献: 0
