






-
生物炭价格低廉、来源广泛,作为一种良好吸附剂,其在有机污染物去除方面表现出良好的应用前景,已经引起了广泛的关注[1]。土壤中添加少量的生物炭即可对农药的吸附性能提高40~2 500倍[2],因此,有学者[3-4]针对生物炭吸附性能进行了深入研究。然而,近期有研究[5]发现,生物炭热解制备过程中会形成相对稳定、寿命较长的环境持久性自由基(environmental persistent free radicals, EPFRs),可能与有机污染物存在反应,导致有机污染物的降解。
通过检测发现热解(200~700 ℃)生物炭表面存在大量EPFRs。有研究表明,EPFRs在与有机污染物相互作用的同时,还能够活化水分子[6]或者溶解氧[7],产生一些小分子自由基,且这些物质能够与有机污染物发生氧化反应,进而将其降解。此外,在紫外光照下,液相中氧气会被生物炭EPFRs激发产生单线态氧,诱导产生活性氧成分(ROS),主要包含羟基自由基( · OH)和过氧阴离子自由基(
O⋅−2 )等[8-9]。这些ROS是液相中的小分子自由基,具有寿命短、反应活性强的特点[10-11],故能促进有机污染物的降解[12]。在水稻秸秆制备的生物炭对RhB的吸附实验中,同样观察到明显的降解现象[13]。但低温制备(自由基信号强度弱)生物炭降解程度略高于中温制备(自由基信号强)的生物炭,这与之前研究中有机污染物降解程度与生物炭EPFRs信号强度呈正比的结果[5]不一致。因此,生物炭降解可能须考虑更多因素。低温制备的生物炭在制备过程中有机质燃烧不充分,导致在液相环境中会溶出较多的溶解性有机质(dissolved organic matter, DOM)。DOM成分主要包括溶解性炭黑等物质,这些物质在光照条件下会产生光化学反应过程。目前也有研究[14]指出,DOM对有机污染物降解的贡献。因此,在生物炭-RhB体系中,RhB降解程度与自由基信号强度不完全相关,可能还须进一步考虑DOM及其与EPFRs的相互作用对有机污染物降解的影响。
本研究对不同温度条件下制备生物炭颗粒(其EPFRs强度和DOM含量均不同)对RhB吸附和降解进行了定量分析,以区分固相上的吸附与降解,并通过光照与暗反应区分EPFRs和DOM在生物炭降解RhB中的贡献,进一步了解生物炭-有机污染物之间的相互作用机制,以期为生物炭处理有机污染物的实际应用提供参考。
-
乙腈(C2H3N, 99.9%)、甲醇(CH3OH, 99.9%)、高纯氮气(N2, 99.999%)、高纯氧气(O2, 99.999%)。高效液相色谱仪(1100HPLC,美国安捷伦公司);比表面积分析仪(JW-BK132F,微精高博);总有机碳分析仪(TOC Select,德国元素分析系统公司Vario);有机元素分析(Variomicrocube,德国元素分析系统公司);电子顺磁共振波谱仪(A300-6/1EPR,德国布鲁克拜厄斯宾);傅里叶红外光谱仪(Tensor 27,德国布鲁克拜厄斯宾);紫外中光灯UV MID LIGHT(Labino AB MPXL PS135 UV)。
-
将水稻秸秆置于马弗炉中,先通氮气30 min(气体流速保持在50 cm3·min−1),排净空气,并在不同温度(标记为R-0、R-200、R-500、R-1000)热解过程中始终保持氮气的通入。恒温热解4 h,自然冷却至室温后,取出生物炭。
-
采用比表面积分析仪测定不同温度制备的生物炭比表面积;采用总有机碳分析仪测定不同温度生物炭上清液中TOC含量;采用有机元素分析测定不同温度生物炭C、N、O、H、S元素的含量;采用EPR测定不同温度生物炭的自由基信号强度;采用FT-IR测定生物炭颗粒官能团种类;采用HPLC测定实验过程中上清液RhB浓度。
-
生物炭光降解RhB实验:分别称取40 mg上述不同温度下的生物炭,按照固液比1∶500配置溶液,每组实验设置2个平行样;将一组样品置于暗反应中144 h,另一组样品先置于暗反应中72 h,再置于紫外光照下继续反应72 h;用高效液相色谱仪测定上清液中RhB的浓度,同时设置RhB溶液空白对照,排除紫外光对RhB降解产生的干扰。
待反应结束后,离心移除上清液,向生物炭固体颗粒中加入20 mL乙腈,将剩余固体用乙腈萃取2次。通过计算,得出不同温度制备的生物炭对RhB的降解量。
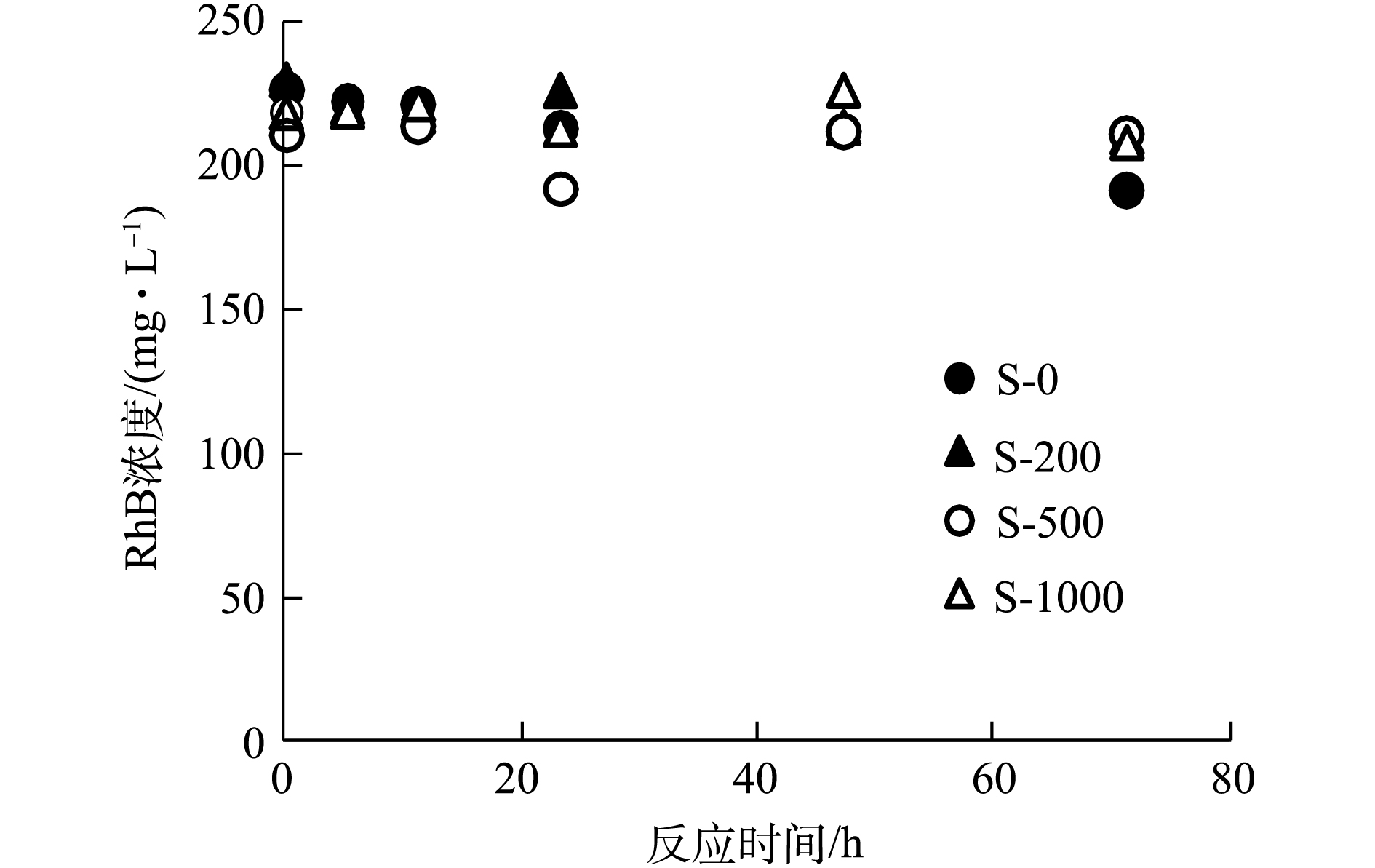
不同温度制备生物炭DOM降解RhB实验:称取10 g不同温度制备的生物炭放入洗瓶中,分别加入500 mL高纯水,放入摇床振荡12 h(25 ℃、60 r·min−1)后取出,静置24 h,取上清液过0.45 μm滤膜,用抽滤机进行抽滤,收集滤液,按照生物炭制备温度标记为S-0、S-200、S-500、S-1000;分别加入S-0、S-200、S-500、S-1000,使RhB溶液浓度定量为200 mg·L−1,配置2组平行,并将2组样品分别置于暗、光反应中72 h。
羟基自由基( · OH)淬灭实验:称取3组40 mg上述温度生物炭,加入摩尔浓度分别为0、3.7、11.1 mol·L−1的叔丁醇(tert-butanol,TBA),淬灭液相中的 · OH。然后按照上述固液比分别加入40 mg·L−1的RhB溶液,放入摇床中反应72 h(25 ℃、60 r·min−1)后,离心取上清液,用HPLC测定,剩余固体用乙腈萃取2次,同时设置RhB溶液加入TBA作为空白对照,排除TBA对RhB降解的干扰。
超氧阴离子自由基(
O⋅−2 )对照实验:称取2组40 mg R-0、R-200、R-500、R-1000,分别加入80 mg·L−1 RhB溶液,在加入生物炭进行反应之前,将配置好的RhB溶液一组通入N2,另一组通入O2,通气时间为30 min;按反应时间2、4、6、12、24、36、72 h取样,测定上清液中RhB的浓度,剩余固体用乙腈萃取2次;同时设置RhB溶液空白对照。 -
从表1中可以看出,生物炭的比表面积随着温度的升高不断增大[15],从19.5 m2·g−1上升到136.8 m2·g−1,这说明温度是影响生物炭比表面积的关键因素。C元素的百分含量不断上升,从39.1%升高到52.3%,H/C原子个数比随着温度的增高而减少,从1.62下降到0.52。当热解温度从200 ℃增加到1 000 ℃,生物炭浸泡后,上清液中的总有机碳(total organic carbon, TOC)含量从370.0 mg·L−1降低到14.6 mg·L−1,这说明热解温度越高,水分和有机组分更容易挥发,导致有机碳逐渐缩合,进而使得上清液TOC含量随温度升高随之减少现象。其中,R-200的TOC含量高于R-0,这可能是由于有机质的不充分燃烧,到最后其在液体中更容易释放。而高温制备生物炭TOC含量迅速下降,因为有机质在热解过程中被逐渐分解,因此,上清液中TOC含量降低。
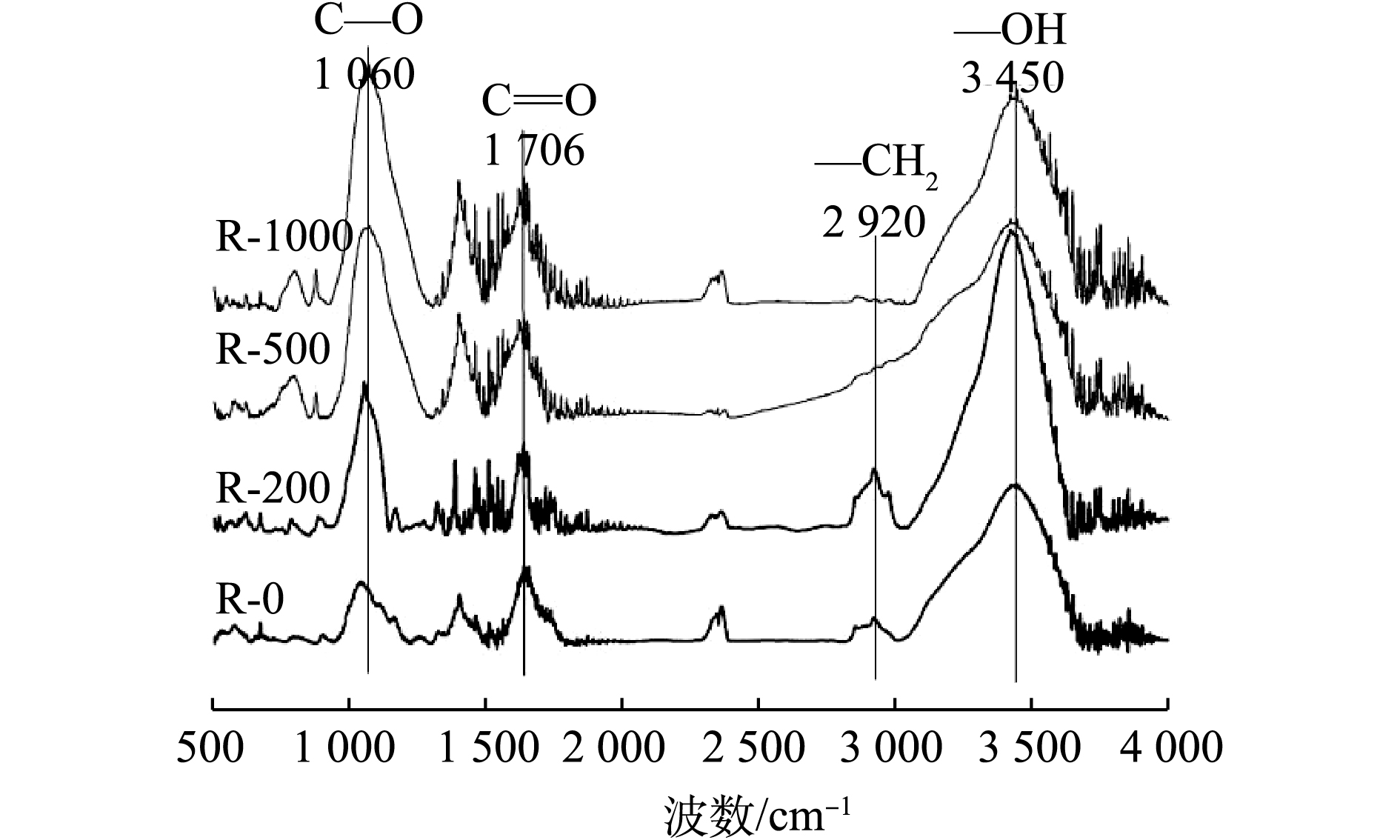
生物炭表面含有丰富的官能团,其类型可以通过傅里叶红外光谱(FT-IR)来进行测定。图1是不同温度下所制备生物炭的红外光谱。由图1可以看到,随着制备温度的升高,生物炭表面官能团发生了改变。低温生物炭在3 450 cm−1处有吸收峰,普遍认为此处应为—OH官能团,当温度超过200 ℃时,此处的吸收峰逐渐减弱[16]。在波数2 920 cm−1处,普遍认为是脂肪烃和烷烃的—CH2基团的伸缩振动产生的,该振动在R-200中增强,而在R-500、R-1000中发生明显的减弱,这说明温度升高到200 ℃时,大量烷烃基团生成,随着温度的进一步增高,又逐渐被热解[13]。1 706 cm−1处普遍认为是羧基中C=O键伸缩振动产生,在4种生物炭样品中均存在[17]。而随着生物炭热解温度的增高,1 060 cm−1处的酯基上C—O伸缩振动增强,此时C、O元素主要以芳香族结合态存在。
-
从生物炭的性质上看,经过高温裂解后的生物炭孔隙增加,比表面积增大[18](表1),故从理论上讲,增加热解温度会导致生物炭对RhB的吸附能力增强,但在生物炭与RhB的反应动力学中没有观察到这样的现象(图2)。本研究从固体颗粒上萃取RhB,区分吸附与降解的贡献(图3)。由图2可知,在暗反应72 h后,后续加光照条件,对RhB的去除几乎没有影响。而图3显示,紫外光照普遍增大了RhB的降解,尤其是R-200在光照以后,RhB的降解程度显著提高。相对而言,光照条件对R-0、R-500和R-1000体系的降解能力影响不大。值得注意的是,在未添加生物炭的实验组中,RhB溶液在避光和紫外光照中的降解现象均不显著。因此,观察到的RhB降解与生物炭的引入密切相关。
EPFRs的类型通常以g因子值来区别。当g因子值大于2.004 0时,EPFRs是以氧为中心自由基;g因子值小于2.003 0时,EPFRs是以碳为中心自由基;而当g因子值为2.003 0~2.004 0时,EPFRs为以氧为中心和以碳为中心的复合自由基体系。从不同制备温度的生物炭的电子顺磁共振(EPR)图谱(图4)来看,200 ℃生物炭g因子值大约为2.004 08,故表明是典型的以氧为中心自由基,500 ℃生物炭g因子值大约为2.003 23。因此,自由基类型主要为以氧为中心和以碳为中心的自由基存在的复合自由基体系[18]。原始生物质的EPR信号相对微弱,随着热解温度的升高,EPR信号可在500 ℃时达到最大,烧制温度的继续升高,导致有机质组分的大量损失,EPR信号急剧减弱,因此,在1 000 ℃烧制温度时,生物炭检测到的EPR信号几乎为0。本研究中EPR信号的变化与RhB的降解程度没有显示较好的关联性。虽然R-500的EPR信号比R-200高出2个数量级,然而R-500生物炭对RhB的降解量却低于R-200,因此,除了EPFRs的影响外,RhB的降解可能须考虑更多的影响因素。
-
DOM是生物炭中未充分燃烧的可溶性有机组分。有研究[19]表明,DOM在光照条件下会诱导产生单线态氧等氧化性物质。因此,我们猜测,RhB在生物炭吸附体系中的降解可能包括2个过程:一个是如我们前期研究中指出的,生物炭上的EPFRs与RhB接触反应,发生了在固相颗粒上的降解[5];另一个过程可能与DOM的反应活性有关。比如,紫外光照射后,DOM受光照激发,与生物炭颗粒EPFRs诱导水分子或者溶解氧产生ROS( · OH和
O⋅−2 )[7, 20],发生光化学反应,促使RhB在液相的降解。而R-500和R-1000由于炭化程度较高,DOM的释放量较少,导致RhB在R-500和R-1000系统中的低降解,而R-0中EPFRs信号强度极弱,故无法诱导产生ROS,产生降解。值得提出的是,为明确DOM在RhB降解中的作用,本研究将不同温度生物炭中的DOM与生物炭颗粒分离出来,在紫外光照条件下进行DOM对RhB的降解实验,结果如图5所示。分离后DOM在光照条件下对RhB没有明显的降解效果。 -
· OH是ROS的一个重要组成,为识别其在RhB降解中的贡献,本研究进行了 · OH淬灭的对照实验。实验对不同温度制备的生物炭添加了不同量的叔丁醇(tert-butanol,TBA),与未添加TBA进行对比,其对RhB的降解量如图6所示。随着TBA添加量从3.7 mol·L−1增加到11.1 mol·L−1,RhB的降解没有被明显地抑制。如果 · OH是RhB降解的主要因素,TBA的引入必然会导致RhB降解率明显降低。实验结果(图6)表明,在生物炭-RhB体系中, · OH可能不是RhB降解的主要原因。
O⋅−2 是ROS中的一个重要组成部分[20]。在生物炭-RhB体系中通入O2,可以促进O⋅−2 的形成。为进一步验证O⋅−2 在RhB降解中的作用,在反应体系中分别通入N2和O2。如图7所示,在2种条件下,生物炭对RhB的吸附作用没有明显差异,但是RhB的降解程度因为通入气体的不同有显著的差别。在R-200体系中,O2条件下,RhB降解效率提高了50%,但是在R-500和R-1000体系中,RhB的降解程度受气体条件影响不明显。因此,在R-0和R-200体系中,O⋅−2 对RhB降解作用可能更加显著,这一结果与DOM浓度高低也有很好的相关性。根据上述实验结果可知,生物炭体系对有机化学物质的降解可能与生物炭颗粒中的EPFRs和溶出的DOM的相互作用有关。低温制备的生物炭中溶出DOM多,在紫外光照下,R-200溶出DOM与EPFRs相互作用,诱导产生ROS结合产生
O⋅−2 (水相中的O2通过得到电子形成O⋅−2 [21]),促进降解RhB,而在R-0体系中,能够检测到的EPFRs信号强度极其微弱。因此,即使上清液中溶出较多的DOM,在紫外光照下也无法产生足量的O⋅−2 以降解RhB。同样,R-500生物炭中DOM含量低,虽然能检测到的EPFRs信号较强,但是能够诱导产生的O⋅−2 数量较少。因此,紫外光照对RhB降解促进并不明显。 -
1)随着热解温度的增高,生物炭比表面积相应增大,但RhB的去除率却未随着比表面积的增加而增大。因此,在生物炭-RhB体系中,不仅存在对RhB的吸附作用,还包括对RhB的降解;但EPFRs的存在并不能完全地解释生物炭-RhB体系中的降解原因。
2)在光照条件下,低温制备的生物炭对RhB降解效果明显,但高温制备的生物炭的降解效果并不明显。这是因为,低温制备的生物炭中DOM含量较高,添加紫外光后,DOM与EPFRs相互作用产生了以
O⋅−2 为主要成分的ROS,促进了RhB的降解,故DOM和EPFRs是光催化降解的2个必要因素,缺一不可。3)目前,对生物炭吸附体系中有机污染物的降解已有初步认识,但仍需要开展更多研究来区分有机污染物在生物炭固体颗粒上和在液相中的降解机制。
生物炭中溶解性有机质光催化降解罗丹明B
Photocatalysis degradation of rhodamine B by dissolved organic matter of biochars
-
摘要: 为了区分生物炭对有机物降解的因素,通过控制光照条件、气体氛围、 · OH淬灭等实验对生物炭降解有机染料罗丹明B(rhodamine-B, RhB)的过程进行了考察;采用元素分析、电子顺磁共振、总有机碳分析仪对生物炭颗粒、持久性自由基(environmental persistent free radicals, EPFRs)及溶解性有机质(dissolved organic matter, DOM)进行了表征测定;研究了不同实验条件下,不同热解温度制备的水稻秸秆生物炭对RhB的吸附和降解效果。结果表明:在200 ℃和500 ℃下所制备的生物炭中检测到明显的EPFRs信号,但其强度与RhB的降解程度不匹配;200 ℃制备的生物炭中DOM含量显著高于其他温度条件下制备的生物炭;在光降解实验中,紫外光能明显促进200 ℃生物炭对RhB降解;气体氛围实验进一步证明紫外光可诱导DOM与生物炭颗粒中EPFRs相互作用形成大量的活性氧组分(主要为
O⋅−2 ),进而促进了其对RhB的降解。Abstract: In order to identify the factors affecting the degradation of organic pollutants on biochar, the process of photocatalytic degradation of biochar-rhodamine B (RhB) system was investigated through controlling illumination conditions, gas atmosphere, and ·OH quenching. The biochar composition, environmental persistent free radicals (EPFRs) and dissolved organic matter (DOM) of biochar were characterized by the elemental analysis (EA), electron paramagnetic resonance (EPR), and total organic carbon analyzer (TOC), respectively. The RhB adsorption and degradation effect on the rice straw biochar prepared at different pyrolysis temperatures were studied under different experimental conditions. The results showed that significant EPFRs signals could be detected in biochars prepared at 200 ℃ and 500 ℃, but their intensities didn’t match the degradation degree of RhB. The DOM content in biochar prepared at 200 ℃ was significantly higher than other types of biochar. In the photodegradation experiment, UV light could significantly promote the RhB degradation by biochar prepared at 200 ℃. The gas atmosphere experiments further confirmed that UV light could induce the interaction between DOM and EPFRs in biochar particles and the formation a large amount of active oxygen components (mainly wereO⋅−2 ), which promoted the RhB degradation.-
Key words:
- biochars /
- rhodamine-B /
- free radical /
- dissolved organic matter /
- photocatalysis
-
抗生素是一种具有抗菌活性的药物[1],可以用于预防和治疗微生物引起的多种疾病[2]。近年来,滥用抗生素带来的生态环境问题已经成为全球性热点关注问题。水环境中残留抗生素的污染分布范围广,具有毒性大、浓度低、难降解、易生物富集等特性[3]。目前降解抗生素的常用方法有物理吸附[4]、化学氧化[5]和生物降解[6]等。其中光催化氧化技术由于有效性、低成本、高稳定性和环境友好性被广泛用于降解抗生素废水[7]。
三氧化钨(WO3)是n型纳米结构半导体,其禁带宽度约为2.6~2.8 eV,制备成本低、绿色环保、具有优异化学稳定性和良好的光催化性能[8-11],因此,被认为是一种有潜力代替TiO2的光催化材料。但由于光生电子和空穴复合率高,其在光催化领域的应用受到了限制,有研究指出构建Z型异质结结构有助于提高WO3的光催化活性[12-15]。
近年来,高分子石墨氮化碳(g-C3N4)被报道为一种新型的无金属光催化剂,其具有2.7 eV的可见光响应窄带隙[16]。g-C3N4制备简单、具有优异的吸附性能和稳定的化学性质,常被用作载体材料。苏跃涵等[17]制备出二维超薄g-C3N4,提高了光催化过程对于恩诺沙星的降解。YU等[18]使用微波加热法制备出金字塔状g-C3N4阵列,其具有较大的比表面积,光生载流子分离效率高,表现出优异的光催化活性,对罗丹明B的脱色率高达99.5%。有研究表明,g-C3N4/WO3异质结材料具有良好的光催化性能[19-20],采用球磨法合成的g-C3N4/WO3具有较高的比表面积,导致光生载流子在可见光下分离和迁移增强,且对罗丹明B的光催化活性明显增强。然而目前将g-C3N4与WO3进行复合并用于降解四环素类抗生素的研究较少,对于g-C3N4/WO3光催化降解抗生素机理的研究较欠缺。
本研究通过原位水热法制备出g-C3N4/WO3复合光催化材料。分析了不同g-C3N4含量的g-C3N4/WO3复合材料的形貌结构和光电性能,并评价了其对土霉素溶液的光催化降解性能和稳定性。最后通过自由基淬灭实验探寻g-C3N4/WO3光催化降解机理。本研究制备的具有高效光生载流子分离、优异氧化还原能力和高吸附能力的Z型异质结光催化剂,对抗生素的去除具有一定的应用价值,可为光催化氧化技术处理抗生素废水提供参考。
1. 材料与方法
1.1 g-C3N4/WO3复合材料的制备
首先采用热缩聚法合成层状g-C3N4。将装有尿素的氧化铝坩埚放入马弗炉中550 ℃下煅烧4 h,自然冷却至室温后收集黄色固体,用蒸馏水和乙醇洗涤3次后在60 ℃下烘干研磨备用。
以二水钨酸钠化合物作为前驱体通过水热反应制备g-C3N4/WO3复合材料。先称取3.30 g Na2WO4·2H2O和一定量的NaCl结构导向剂溶解于40 mL去离子水中,均匀揽拌20 min至原料完全溶解,再加入不同质量(0.3、0.6、1.0、3.0 g)的g-C3N4,搅拌均匀。随后,向溶液中缓慢滴入3 mmol·L−1盐酸溶液同时不断搅拌以调节体系pH至2.0,形成有黄色沉淀的悬浮液。持续揽拌1 h后转移到容量为100 mL的不锈钢高压反应釜中密封并在160 ℃的烘箱中加热24 h。自然冷却至室温后将固体产物进行离心,用蒸馏水和乙醇洗涤3次,最后在60 ℃下烘干(根据g-C3N4不同的添加量,分别将样品命名为WG-0.3、WG-0.6、WG-1.0、WG-3.0,其中W代表WO3,G代表g-C3N4,数字代表复合材料中g-C3N4的添加量分别为0.3、0.6、1.0、3.0 g)。
1.2 WO3/g-C3N4复合材料性能测试
采用PANalytical公司的PW3040/60型X射线衍射仪XRD对复合材料进行晶体结构分析;使用ESCLAB250型X射线光电子能谱仪XPS分析复合材料的表面化学组成,不同元素的XPS谱图以C1s结合能284.8 eV为基准进行校正;采用JEOL公司的JSM-7001F型发射扫描电子显微镜SEM,获取复合材料的微观形貌特征和尺寸。
采用日立公司F-4500型荧光分光光度计测得复合材料的光致发光谱图,观察光催化剂被光激发后电子空穴对的复合状态。复合材料的光电化学性能通过电化学工作站在三电极系统中进行测试,包括瞬态光电流和电化学阻抗谱EIS,用于评估载流子的分离和迁移效率。其中,复合材料作为工作电极,Pt电极作为对电极,饱和甘汞电极作为参比电极,电解质溶液为0.5 mol·L−1硫酸钠溶液。工作电极通过如下方法制备:称取10 mg粉末样品分散在1 mL超纯水溶液中,再加入50 uL Nafion乙醇溶液,超声30 min形成悬浮液,然后在ITO玻璃上滴加150 uL悬浮液,室温下晾干进行光电测试。光电测试所用的光源为北京泊菲莱公司所生产的300 W氙灯(型号PLS-SXE300C)。在EIS测试时,电场变化频率围0.1 Hz~100 kHz,电解质溶液为0.5 mol·L−1硫酸钠溶液。采用安捷伦公司CARY300/PE lambda 750S型光谱仪测得复合材料的紫外-可见漫反射光谱UV-Vi并确定光响应范围及吸收强度。
1.3 WO3/g-C3N4复合材料光催化降解OTC实验
实验采用420 nm滤波片滤除小波长光,300 W氙灯作为光源。每组实验加入50 mg复合材料至含有100 mL 20 mg·L−1 OTC溶液的反应器中。先将混合溶液在黑暗条件下搅拌40 min,使OTC在样品上达到吸附平衡。然后开始光照,每隔20 min取样5 mL,经高速离心除去沉淀,取上清液用于测定HitachiU-3500紫外-可见分光光度计在360 nm处的吸光度值,测定OTC浓度。
实际工程应用中,光催化剂的稳定性至关重要,通过将反应后的复合材料对20 mg·L−1 OTC溶液进行光催化降解的重复实验测试其稳定性。每完成1组循环实验后,将复合材料离心、洗涤、过滤和干燥后用于下1组实验。循环实验次数为3次。
1.4 光催化降解机理实验
在光催化降解OTC的过程添加草酸钠(Na2C2O4,10 mmol·L−1)、异丙醇(IPA,10 mmol·L−1)和超氧化物淬灭剂(TEMPOL,10 mmol·L−1),分别作为空穴(h+)、羟基自由基(·OH)和超氧自由基(·O2−)的淬灭剂。将3种淬灭剂分别加入到含有50 mg WG-0.6的100 mL的OTC溶液中,在黑暗条件下持续搅拌40 min,使OTC在样品上达到吸附平衡。开始光照后,每隔20 min取样5 mL经高速离心除去沉淀,取上清液使用HitachU-3500紫外-可见分光光度计测定在360 nm处的吸光度值。
2. 结果与讨论
2.1 形貌与结构分析
XRD被用于表征复合材料的晶体结构,如图1(a)所示,g-C3N4存在由层面结构堆积而形成的位于27.41的(002)晶面,而块体g-C3N4具有的来自芳香体系的特征晶面间堆积的(100)晶面非常微弱,表明g-C3N4已成功分层剥离。对于具有不同g-C3N4量的g-C3N4/WO3复合材料,观察到所有复合材料的WO3的特征衍射峰,与标准粉末衍射卡(JCPDS)no.35-1001相一致,均为六方相WO3[21]。g-C3N4的(002)衍射峰位置与WO3(101)(200)晶面的衍射峰位置接近,因此,不同g-C3N4含量的g-C3N4/WO3复合材料的主要特征衍射峰与原始WO3的特征衍射峰相近。随着g-C3N4质量含量的增加,WO3的相对衍射峰强度逐渐减弱。此外,当g-C3N4的投加量达到3.0 g时,WO3的衍射图样没有被探测到,表明WO3已经被g-C3N4完全覆盖,同时g-C3N4的(001)晶面的衍射峰也消失。以上结果表明层状g-C3N4与WO3纳米粒子成功地复合。
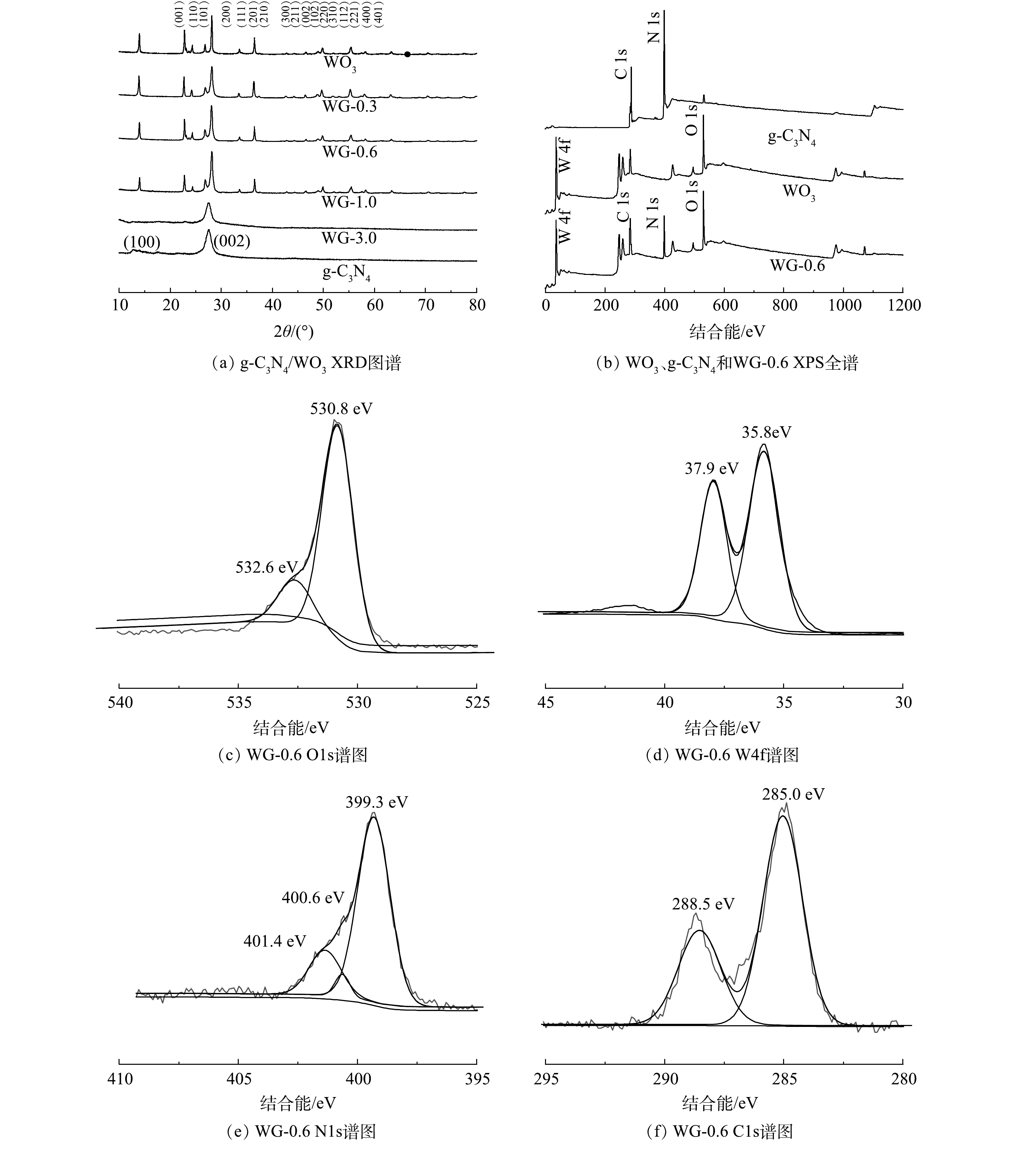 图 1 g-C3N4/WO3复合材料的XRD和XPS图谱Figure 1. XRD pattern and XPS spectra of g-C3N4/WO3 composite
图 1 g-C3N4/WO3复合材料的XRD和XPS图谱Figure 1. XRD pattern and XPS spectra of g-C3N4/WO3 composite为深入了解g-C3N4、WO3和WG-0.6复合材料表面元素的化学状态,进行XPS测试。XPS全谱图显示WG-0.6复合材料中存在氧、钨、氮和碳元素。O1s、W4f、N1s和C1s的高分辨率光谱如图1(b)所示。对于图1(c)中的O1s,该峰可分解成530.8 eV和532.6 eV的2个峰,分别对应于W-O-W和W-O-H的氧结合物种。如图1(d)所示,在W4f图谱中结合能分别位于35.8 eV和37.9 eV的W4f7/2和W4f5/2峰,表明WG-0.6中的W为W6+的特征。在N1s光谱中,在401.4、400.6和399.3 eV处可识别出3个峰,分别对应C-N-H,N-(C)3和sp2杂化氮(C=N-C),从而证实sp2键石墨氮化碳的存在,如图1(e)所示。如图1(f)所示,C1s的高分辨率光谱可分为2个峰值,分别为285.0 eV对应sp2C-C键和288.5 eV对应含氮芳环中sp2键的碳(N-C=N)[22]。
使用扫描电镜SEM对光催化剂的结构及形貌分析。由图2(a)可见,WO3为大量纳米棒团聚形成的均匀三维花状微球,其单体为长约500~800 nm,直径约为30 nm的纳米棒。由图2(b)可见,g-C3N4则是通过g-C3N4纳米片的聚集而构建的,结构纹理清晰,表现出典型的带褶皱层状结构。由图2(c)可见,较小的WO3纳米棒修饰在大颗粒层状g-C3N4上,分散性及形貌特征与花状微球形的WO3明显不同。可以观察到,其表面没有表现出明显的g-C3N4层状结构,这意味着g-C3N4纳米片被WO3纳米棒均匀覆盖。纳米棒覆盖在纳米片上的堆积结构增加了材料比表面积,从而可提高其对OTC的吸附能力,促进光催化剂与污染物的接触,有利于提高对OTC的降解率。
2.2 光电化学性能研究
测试WO3、g-C3N4和g-C3N4/WO3复合材料的光致发光谱图,结果如图3(a)所示,以确定光生载流子分离的效率。g-C3N4的光致发光光谱在450 nm附近有一强发射峰,这可能与g-C3N4的光生电子和空穴的复合有关。g-C3N4/WO3复合材料的光致发光强度明显低于g-C3N4,g-C3N4/WO3复合材料中光生电子和空穴的复合受到极大的抑制,而WG-0.6的光致发光强度最低,表明适当的g-C3N4/WO3复合比例可形成更有效的光生载流子分离路径。
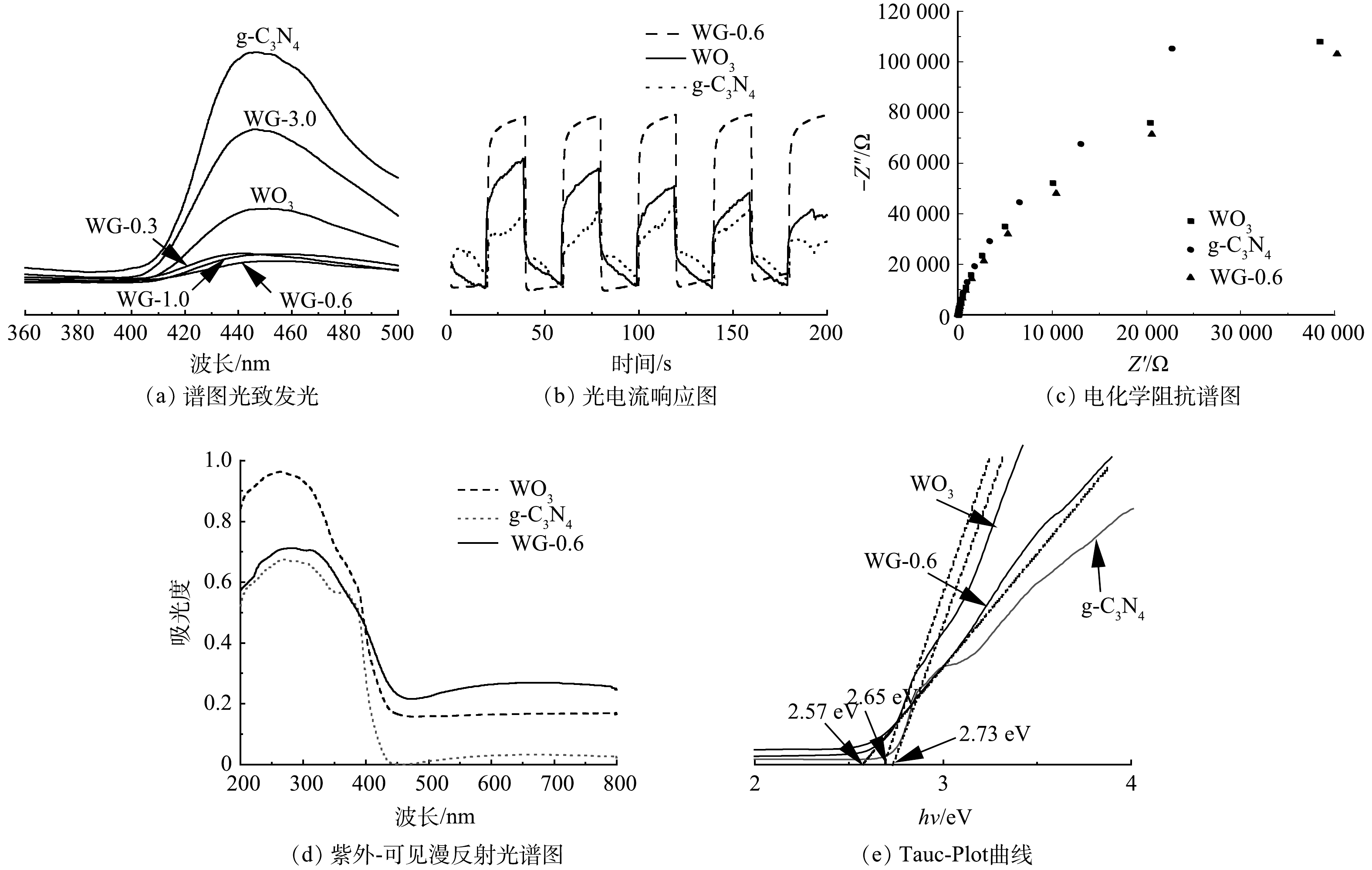 图 3 不同催化剂的光致发光谱图、光电流响应图、电化学阻抗谱图、紫外-可见漫反射光谱图和Tauc-Plot曲线Figure 3. Photoluminescence spectra, photocurrent responses, electrochemical impedance spectroscopy, UV-Vis spectra and Tauc-Plot of different catalysts
图 3 不同催化剂的光致发光谱图、光电流响应图、电化学阻抗谱图、紫外-可见漫反射光谱图和Tauc-Plot曲线Figure 3. Photoluminescence spectra, photocurrent responses, electrochemical impedance spectroscopy, UV-Vis spectra and Tauc-Plot of different catalysts为了进一步研究g-C3N4/WO3复合材料的异质结对光生电子和空穴分离效率的影响,测试了g-C3N4、WO3和WG-0.6可见光照射下的瞬态光响应电流,如图3(b)所示。在4个关灯周期过程中的光电流密度与照射时间关系曲线。当灯关闭时,光电流密度接近于零,当灯打开时,光电流密度迅速增加并稳定在一定值。结果显示,WG-0.6表现出较强的光电流密度,表明其具有较高的光生载流子分离率,有利于提高其对可见光的活性。
g-C3N4、WO3和WG-0.6的电化学阻抗谱(EIS)也验证了光电流测试结果。电化学阻抗测试中Nyquist曲线中半圆的直径与电荷迁移电阻有关,Nyquist曲线半圆直径越小说明电子迁移阻力越小[23]。图3(c)为g-C3N4、WO3和WG-0.6的电化学阻抗谱图。相比其他2种材料,WG-0.6的圆弧半径更小,表明电子转移阻力更低,光生载流子的分离更迅速。
紫外-可见漫反射光谱显示,g-C3N4、WO3和g-C3N4/WO3复合材料均在可见光区有典型的半导体吸收。如图3(d)所示,在450 nm附近观察到WO3的吸收边缘,与其他学者研究一致[24]。g-C3N4在约430 nm处显示出吸收边缘。相比于g-C3N4与WO3,WG-0.6在可见光区域表现出较高的吸收强度,且吸收边带有明显的红移。由此可见,通过在WO3上负载g-C3N4改变了原材料的能带结构,从而可增强其对可见光的吸收响应。
根据Tauc-Plot曲线,如图3(e)所示估算光催化剂的带隙,得到g-C3N4、WO3和WG-0.6的带隙分别为2.73、2.65和2.57 eV。综上所述,g-C3N4和WO3构建的Z型异质结结构有利于促进光生载流子的分离。
2.3 光催化降解土霉素效果
通过光催化降解土霉素实验来研究不同光催化材料的活性。如图4(a)所示,暗反应阶段显示各光催化材料都具有较好的吸附能力。当g-C3N4的添加量为0.6 g时,g-C3N4/WO3复合材料的光催化活性最好。随着g-C3N4添加量从0增加到0.6 g,g-C3N4/WO3的光催化活性增强。当g-C3N4含量超过0.6 g时,g-C3N4/WO3的光催化活性下降。结果表明,g-C3N4的含量对于g-C3N4/WO3复合材料的活性有很大影响,其中g-C3N4的最佳添加量为0.6 g。当g-C3N4的添加量高于0.6 g时,WO3的量不足以在g-C3N4和WO3之间构建有效的异质结来分离和转移光生电子空穴对。当g-C3N4的含量过小时,WO3的含量过高导致复合材料的WO3壳变厚,外层的WO3远离g-C3N4,电荷分离会更低效,光生电子和空穴在移动过程中更容易复合,会降低光催化降解率。
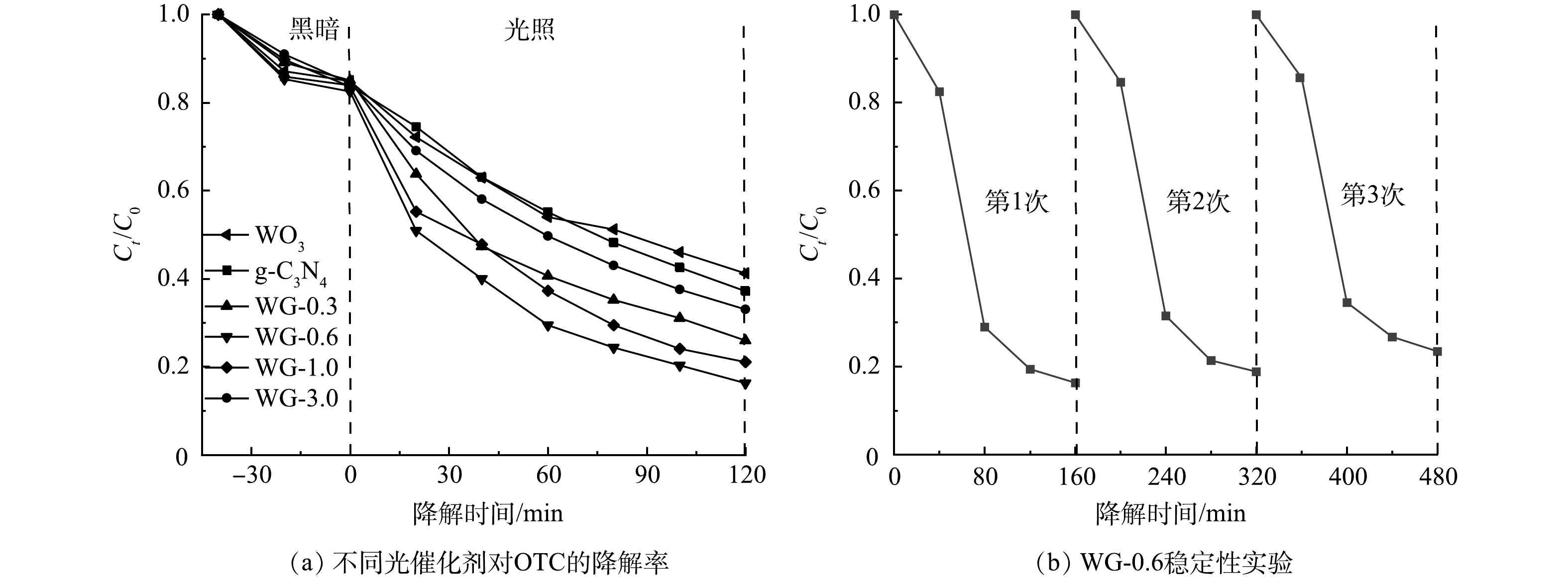 图 4 不同光催化剂对OTC的降解率和WG-0.6稳定性实验Figure 4. The degradation rate of OTC by different photocatalysts and photocatalyst stability experiment of WG-0.6
图 4 不同光催化剂对OTC的降解率和WG-0.6稳定性实验Figure 4. The degradation rate of OTC by different photocatalysts and photocatalyst stability experiment of WG-0.6为评价g-C3N4/WO3复合光催化剂的稳定性和重复使用性,在相同条件下用WG-0.6光催化降解OTC 3次,结果如图4(b)所示。OTC经3次光催化降解循环后,WG-0.6的光催化性能略有下降。3次循环实验后,OTC的光催化降解率在120 min内达到77%,表明g-C3N4/WO3复合材料具有良好的光催化稳定性。活性减弱的主要原因是催化剂在循环过程中有所损失。
2.4 光催化降解机理
为研究WG-0.6光催化氧化过程中的主要活性物种,进行自由基淬灭实验。如图5(a)所示,光照120 min后,空白对照组和加入Na2C2O4(h+淬灭剂)、TEMPOL(·O2−淬灭剂)、IPA(·OH淬灭剂)后对OTC的光降解率分别为86%、30.2%、66.9%、74.7%,说明h+、·O2−和·OH共同参与g-C3N4/WO3光催化降解OTC反应。其中加入Na2C2O4光催化降解率下降高达55.8%,说明h+在g-C3N4/WO3光催化降解OTC中起主要作用。半导体的导带位置EVB和价带位置ECB可根据经验式(1)~(2)计算[25]。
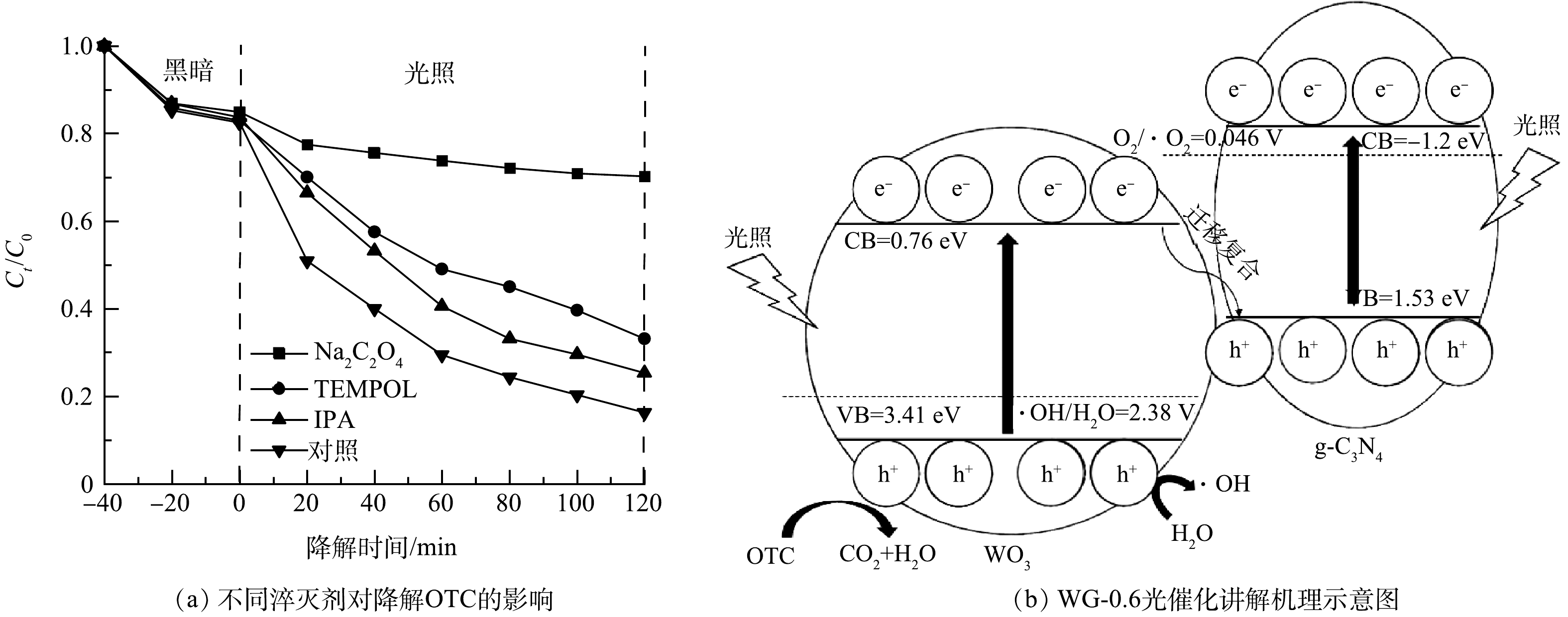 图 5 不同淬灭剂对降解OTC的影响和WG-0.6光催化降解机理示意图Figure 5. Influence of OTC degradation by different radical scavengers and photocatalytic mechanism of WG-0.6
图 5 不同淬灭剂对降解OTC的影响和WG-0.6光催化降解机理示意图Figure 5. Influence of OTC degradation by different radical scavengers and photocatalytic mechanism of WG-0.6EVB=X−Ee+0.5Eg (1) ECB=EVB−Eg (2) 式中:X、Ee和Eg分别表示半导体的绝对电负性、自由能和带隙能量。WO3和g-C3N4的X分别为6.59 eV和4.67 eV。Ee大约为4.5 eV,WO3和g-C3N4的Eg分别为2.65 eV和2.73 eV。因此,WO3和g-C3N4的EVB计算分别为3.41 eV和1.53 eV,ECB计算分别为0.76 eV和−1.2 eV。如图5(b)所示,在可见光照射下,g-C3N4和WO3产生光生电子空穴对。因为g-C3N4的ECB比E0(O2/·O2−)=0.046 V更低,g-C3N4导带上的电子可以将溶解的O2还原成·O2−。因为WO3的EVB比E0(·OH/H2O)=2.38 V更高,WO3价带上的空穴可以与水反应生成·OH。WO3导带上的电子开始迁移并与g-C3N4的价带上的空穴复合。最后,OTC在h+、·O2−和·OH的共同作用下被降解。
3. 结论
1)通过原位一步水热法成功合成了g-C3N4/WO3异质结材料。WO3纳米棒均匀地负载在层状的g-C3N4上,形成g-C3N4/WO3异质结结构,光生电子和空穴复合率降低,光吸收范围和吸收强度增强。
2) WG-0.6在可见光照射120 min后OTC降解率高达86%,并且在循环使用3次后仍具有较好催化活性,表现出较高的稳定性。
3) h+、·O2−和·OH共同参与g-C3N4/WO3光催化降解OTC反应,其中h+起主要作用。
-
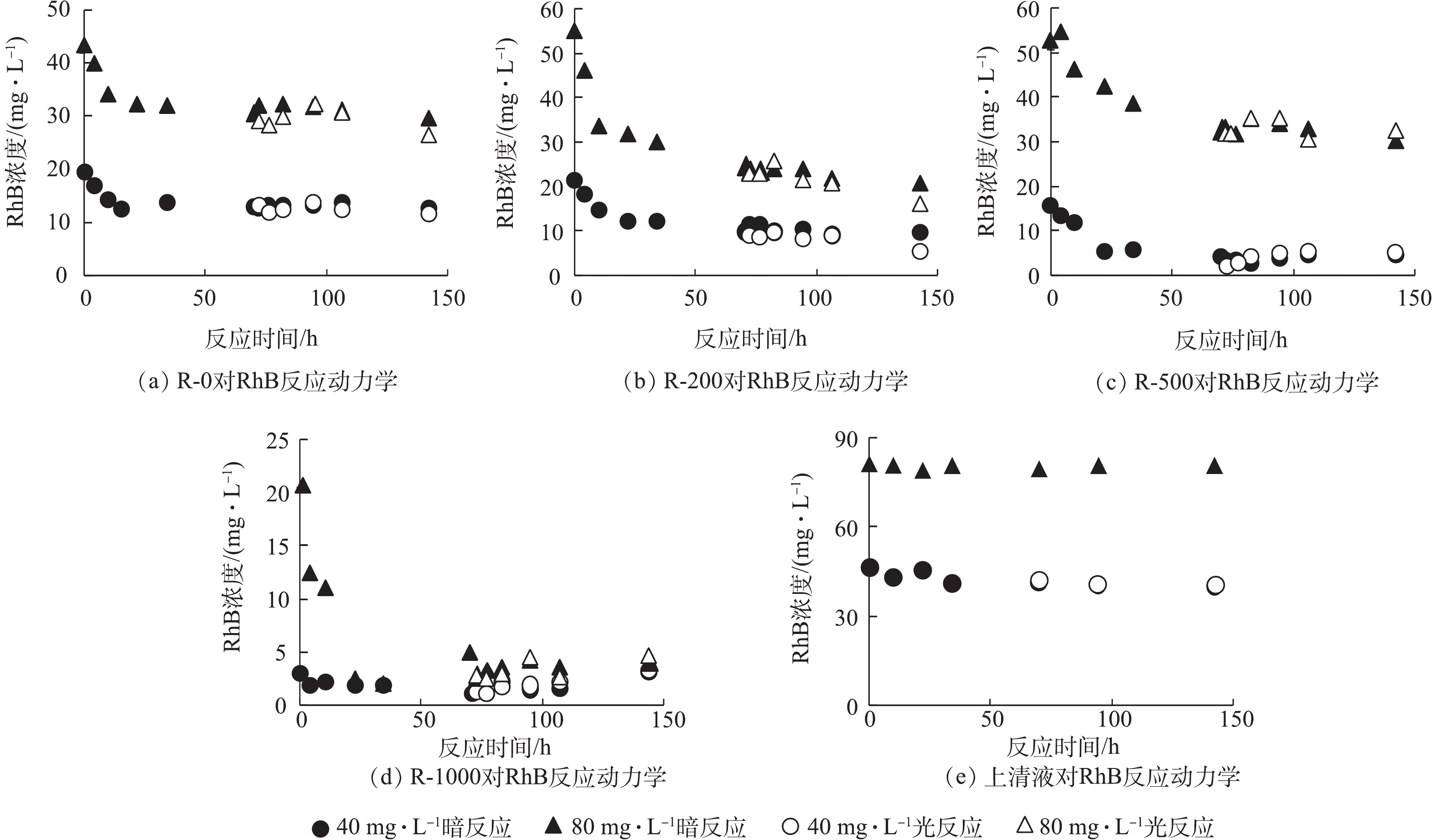
图 2 不同温度制备的生物炭对RhB反应动力学
Figure 2. RhB degradation kinetics on biochar prepared at different temperatures
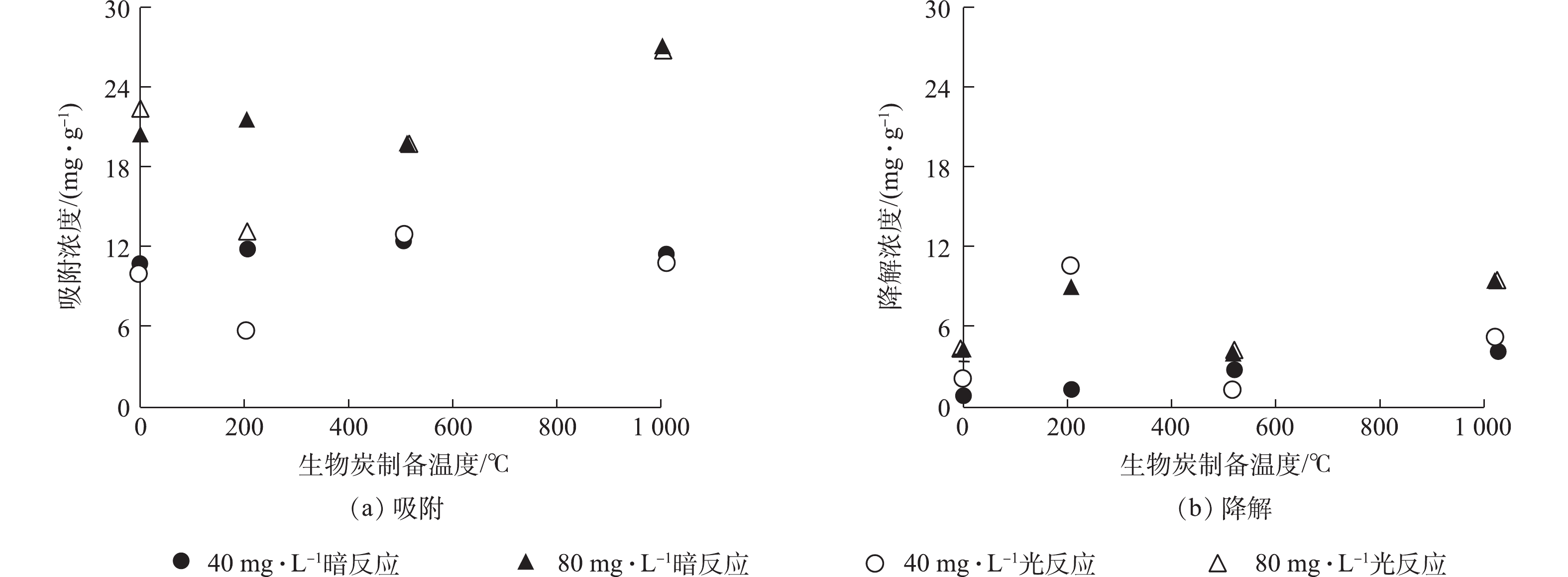
图 3 不同温度下生物炭对RhB的吸附与降解
Figure 3. Adsorption and degradation on biochar prepared at different temperatures
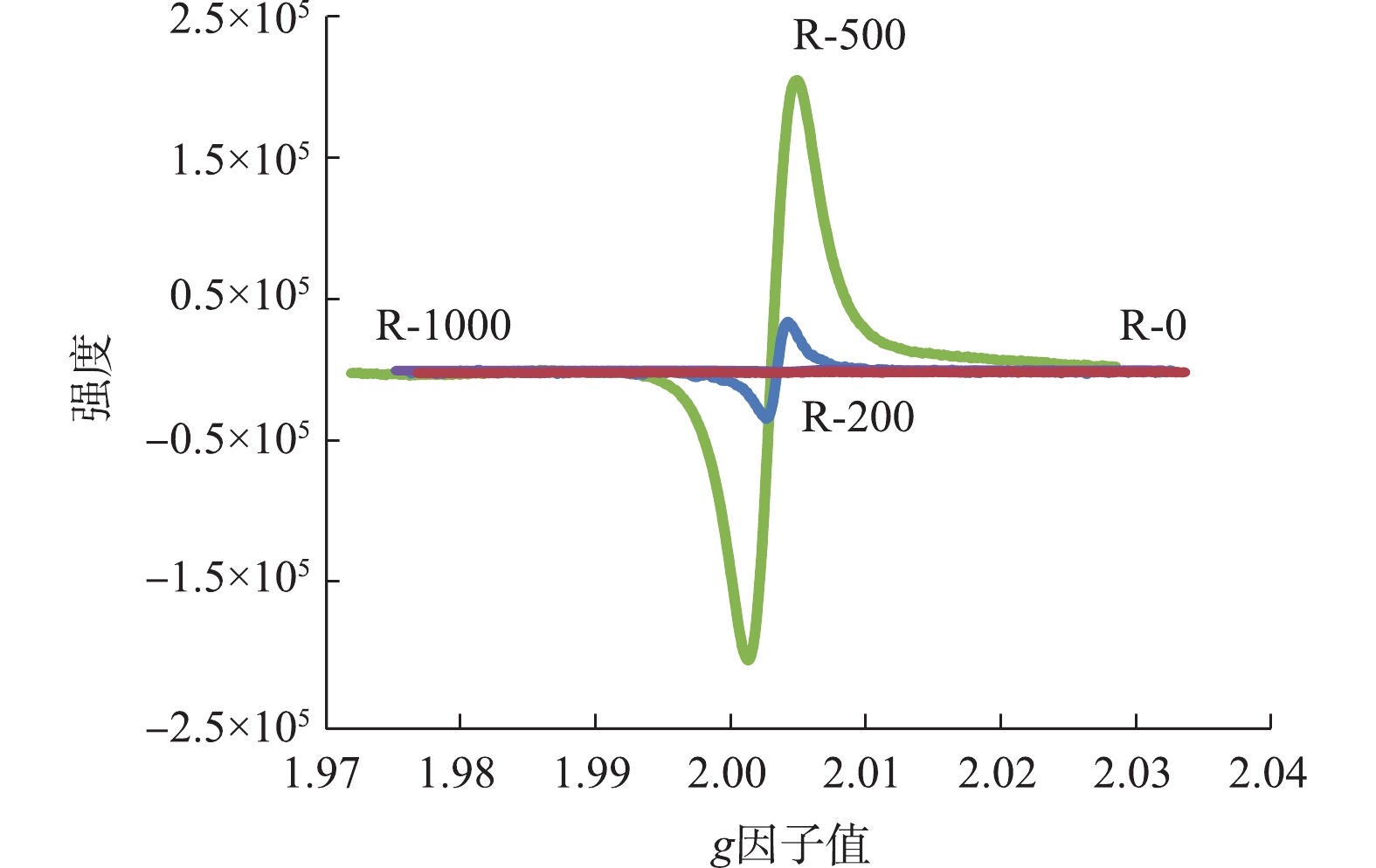
图 4 不同温度制备的生物炭自由基信号
Figure 4. Free radical signal of biochar prepared atdifferent temperatures
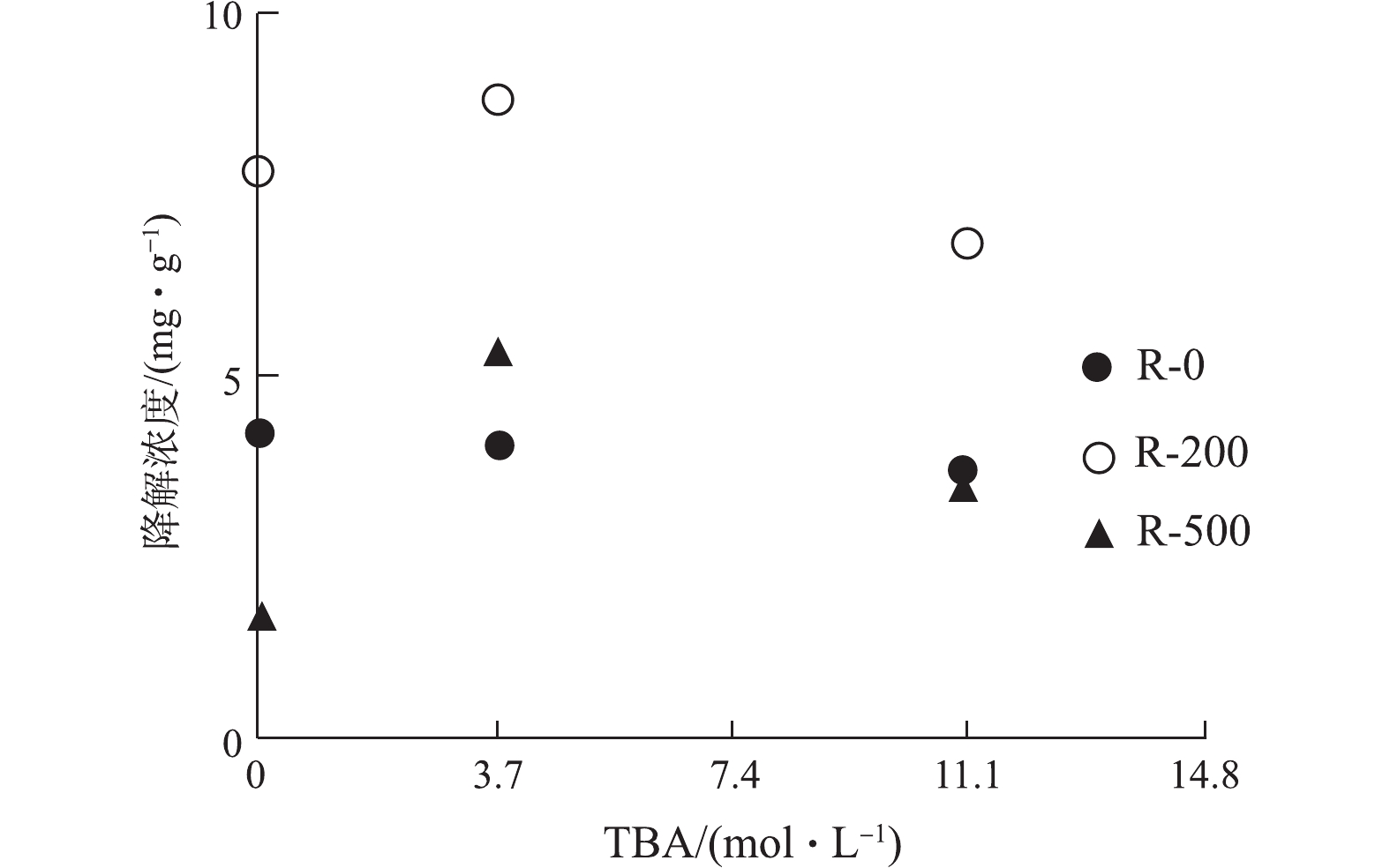
图 6 不同TBA的量对生物炭降解RhB的抑制
Figure 6. RhB degradation inhibition on biochar bydifferent amount of TBA
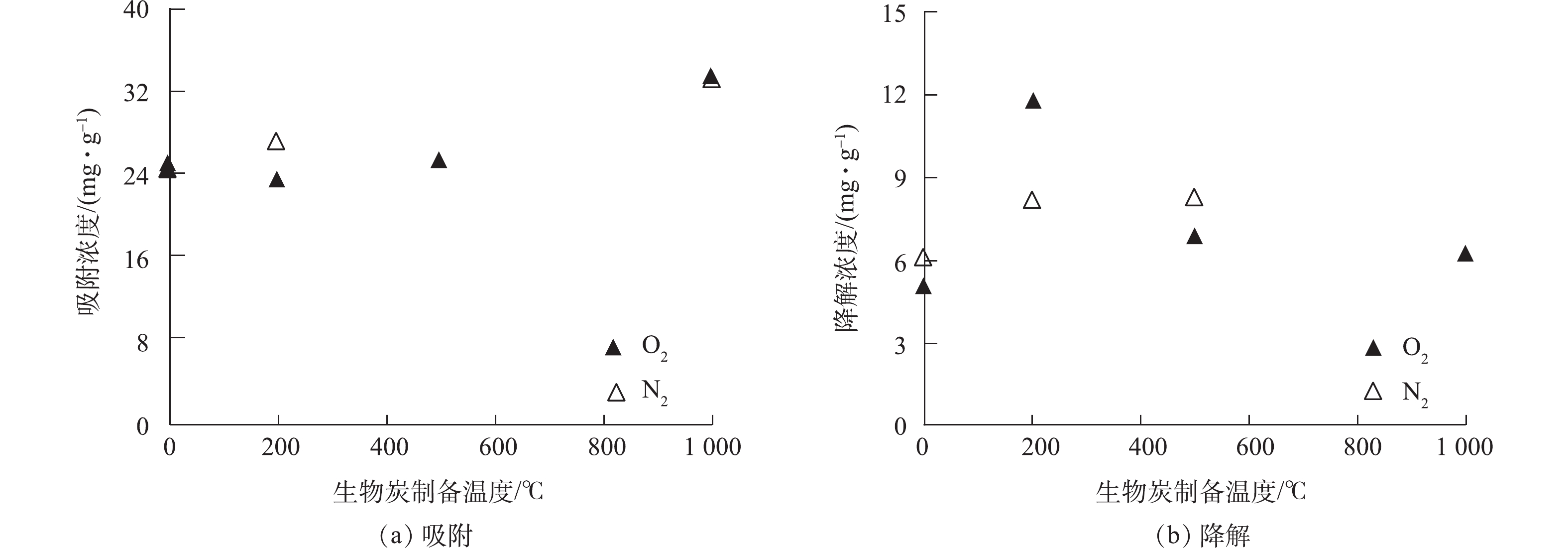
图 7 RhB在生物炭上吸附与降解(受氧气的影响)
Figure 7. Adsorption and degradation of RhB on biochar (influenced by oxygen)
表 1 水稻秸秆生物炭的基本理化性质
Table 1. Physicochemical properties of biochar prepared from rice straw pyrolyzation
生物炭 比表面积/(m2·g−1) 总有机碳/(mg·L−1) 元素含量% 原子比 N C H S O H/C (O+N)/C R-0 19.50 240.32 0.61 39.12 5.27 0.43 39.56 1.62 0.77 R-200 28.81 369.98 0.76 44.45 5.16 0.34 35.39 1.39 0.61 R-500 97.07 29.17 0.85 52.41 2.32 0.50 13.88 0.53 0.21 R-1000 136.77 14.63 0.87 52.35 2.29 0.49 13.92 0.52 0.21  下载: 导出CSV
下载: 导出CSV
-
[1] KOOKANA R S, SARMAH A K, ZWIETEN L V, et al. Biochar application to soil: Agronomic and environmental benefits and unintended consequences[J]. Advances in Agronomy, 2011, 112: 103-143. doi: 10.1016/B978-0-12-385538-1.00003-2 [2] 王宁, 侯艳伟, 彭静静, 等. 生物炭吸附有机污染物的研究进展[J]. 环境化学, 2012, 31(3): 287-295. [3] LI J, LIANG N, JIN X, et al. The role of ash content on bisphenol A sorption to biochars derived from different agricultural wastes[J]. Chemosphere, 2017, 171: 66-73. doi: 10.1016/j.chemosphere.2016.12.041 [4] GHAFFAR A, GHOSH S, LI F, et al. Effect of biochar aging on surface characteristics and adsorption behavior of dialkyl phthalates[J]. Environmental Pollution, 2015, 206: 502-509. doi: 10.1016/j.envpol.2015.08.001 [5] YANG J, PAN B, LI H, et al. Degradation of p-nitrophenol on biochars: Role of persistent free radicals[J]. Environmental Science & Technology, 2016, 50(2): 694-700. [6] FANG G, GAO J, LIU C, et al. Key role of persistent free radicals in hydrogen peroxide activation by biochar: Implications to organic contaminant degradation[J]. Environmental Science & Technology, 2014, 48(3): 1902-1910. [7] FANG G, ZHU C, DIONYIOU D D, et al. Mechanism of hydroxyl radical generation from biochar suspensions: Implications to diethyl phthalate degradation[J]. Bioresource Technology, 2015, 176: 210-217. doi: 10.1016/j.biortech.2014.11.032 [8] VELASCO L F, FONSECA I M, PARRA J B, et al. Photochemical behaviour of activated carbons under UV irradiation[J]. Carbon, 2012, 50(1): 249-258. doi: 10.1016/j.carbon.2011.08.042 [9] VELO-GALA I, LOPEZ-PENALVER J J, SANCHEZ-POLO M, et al. Activated carbon as photocatalyst of reactions in aqueous phase[J]. Applied Catalysis B: Environmental, 2013, 142(10): 694-704. [10] WEN G, WANG S J, MA J, et al. Oxidative degradation of organic pollutants in aqueous solution using zero valent copper under aerobic atmosphere condition[J]. Journal of Hazardous Materials, 2014, 275(2): 193-199. [11] JIANG B, DAI D, YAO Y, et al. The coupling of hemin with persistent free radicals induces a nonradical mechanism for oxidation of pollutants[J]. Chemical Communications, 2016, 52: 9566-9569. doi: 10.1039/C6CC02973F [12] F UH, LIU H, MAO J, et al. Photochemistry of dissolved black carbon released from biochar: Reactive oxygen species generation and phototransformation[J]. Environmental Science & Technology, 2016, 50(3): 1218-1226. [13] 李丹, 金修齐, 王朋, 等. 水稻秸秆生物炭对罗丹明B的吸附与降解[J]. 环境工程学报, 2017, 11(9): 5195-5200. doi: 10.12030/j.cjee.201609022 [14] ZHOU Z, CHEN B, QU X, et al. Dissolved black carbon as an efficient sensitizer in the photochemical transformation of 17β-estradiol in aqueous solution[J]. Environmental Science & Technology, 2018, 52(18): 10391-10399. [15] WARNOCKD D L, JOHANNES, KUYPER T W, et al. Mycorrhizal responses to biochar in soil: Concepts and mechanisms[J]. Plant & Soil, 2007, 300: 9-20. [16] 陈再明, 陈宝梁, 周丹丹. 水稻秸秆生物碳的结构特征及其对有机污染物的吸附性能[J]. 环境科学学报, 2013, 33(1): 9-19. doi: 10.11654/jaes.2013.01.002 [17] CHEN B L, ZHOU D D, ZHOU L Z. Transitional adsorption and partition of nonpolar and polar aromatic contaminants by biochars of pine needles with different pyrolytic temperatures[J]. Environmental Science and Technology, 2008, 42(14): 5137-5143. doi: 10.1021/es8002684 [18] CRUZ A L N D, COOK R L, LOMNICKI S M, et al. Effect of low temperature thermal treatment on soils contaminated with pentachlorophenol and environmentally persistent free radicals[J]. Environmental Science & Technology, 2012, 46(11): 5971-5978. [19] FANG G, LIU C, WANG Y, et al. Photogeneration of reactive oxygen species from biochar suspension for diethyl phthalate degradation[J]. Applied Catalysis B: Environmental, 2017, 214: 34-45. doi: 10.1016/j.apcatb.2017.05.036 [20] CHEN N, HUANG Y, HOU X, et al. Photochemistry of hydrochar: Reactive oxygen species generation and sulfadimidine degradation[J]. Environmental Science & Technology, 2017, 51(19): 11278-11287. [21] CHONGM N, JIN B, CHOW C, et al. Recent developments in photocatalytic water treatment technology: A review[J]. Water Research, 2010, 44(10): 2997-3027. doi: 10.1016/j.watres.2010.02.039 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 6489
- HTML全文浏览数: 6489
- PDF下载数: 117
- 施引文献: 0
