



-
近年来,随着城市化进程的加速,许多原本位于城区的化工企业被迫从城市中心地带迁出,从而遗留了大量的污染场地[1-3]。其中,有机污染场地中的土壤和地下水污染问题较为突出,由此带来的环境和健康风险严重威胁着人们的生产和生活。因此,对其中污染严重的场地遗址进行修复已刻不容缓[4-6]。
针对有机污染场地的修复,近些年来,国内应用异位热脱附技术较多,且积累了大量的工程经验及成功案例。但是,应用异位热脱附技术修复有机污染场地的过程中尚存在着二次污染难控制、社会影响较大等问题。尤其涉及污染较深的场地,不适合使用异位热脱附技术[7-9]。原位热脱附技术(in-situ thermal desorption,ISTD)是通过对土壤直接或间接加热,促使有机污染物挥发,由固相、液相向气相转化,进而对其进一步收集并去除的技术[9]。ISTD作为修复土壤和地下水中有机污染的一项重要技术,在修复低渗透性土壤、污染深度较深且有机物污染严重的场地中具有很大的优势和应用前景[10-11]。
ISTD按照不同的加热方式可分为蒸汽加热[12-13]、电阻加热[14-15]和热传导加热[16]。电热脱附技术属于热传导加热,其过程是通过在垂直(或倾斜)阵列加热井中安置电加热棒,电加热元件可在高达800 ℃的条件下平稳运行,绝大部分有机污染物、重质或轻质非水相液体,将会通过蒸发、蒸馏、沸腾、氧化和高温分解等过程挥发或被处理掉[17]。电热脱附技术基本不受污染物理化性质、复杂地质及水文地质条件等因素的限制,具备应用范围广、设备安装便捷、加热过程安全、处理速度快等优势,正逐步被广泛应用于各种低渗透性、难处理有机物污染场地治理工程中[18],如ISTD曾大规模成功应用于美国新泽西泰特波罗机场附近地块等修复项目[19]。目前,国内在宁波、苏州、天津、武汉等城市的大型修复项目中已陆续开展电热脱附技术实验及应用。
本研究旨在中试的尺度下探讨电热脱附技术在现实污染场地中不同加热间距下的升温速率、加热边界温度传递范围和污染物去除率,分析了土壤导热率、土壤含水率、水文地质条件等影响因素对原位热脱附效果的影响特征,以期为我国有机污染场地热修复应用提供参考。
-
中试实验区域位于我国北方某退役化学试剂厂,为污染土壤和地下水复合区域,面积278.9 m2,污染深度2~10 m,修复土方量2 231.2 m3。场地调查结果显示,主要污染物为氯乙烯、顺-1,2-二氯乙烯、苯、氯苯等挥发性有机污染物;土壤中氯乙烯、氯苯最高浓度分别为5.4 mg·kg−1和30 mg·kg−1;地下水中氯乙烯、顺-1,2-二氯乙烯、苯、氯苯最高浓度分别为3.79×103、3.35×103、1.06×103、2.34×104 μg·kg−1。地质勘察结果表明,该区域可分为5个工程地质层,细分为8个工程地质亚层,地层岩性分布见表1。实验区地表以下0~10.00 m大致分为4类:0~2.00 m为填土层,多为黏性土夹较多碎石和砖块等建筑垃圾;2.00~3.20 m为不透水层,以粉质黏土为主;3.20~4.70 m为潜水含水层,以砂质粉土为主;4.70~10.00 m大多为粉质黏土、粉质黏土夹粉土薄层团块。同时,对实验区内各土层理化性质进行了测试,结果如表2所示。
-
实验系统由加热单元、抽提单元、温度监测单元和电气控制单元4部分组成。工艺装置如图1所示。首先,在土壤中安置电加热棒并通电升温,电加热棒通过热辐射的形式将热量传递到与土壤接触的密封金属套管上,通过热传导使得周围污染土壤逐渐升温至目标温度。目标污染物发生挥发和裂解反应后,含有污染物的蒸汽和水通过多相抽提井被抽提至地表,再经各自的输送管道运至尾水/尾气处理设备并进行无害化处置,最后达标排放。在整个加热过程中,利用温度、压力监测井实时监控整个修复区域,并通过智能化、自动化控制系统对每一个加热设备的工作温度进行实时调控。
-
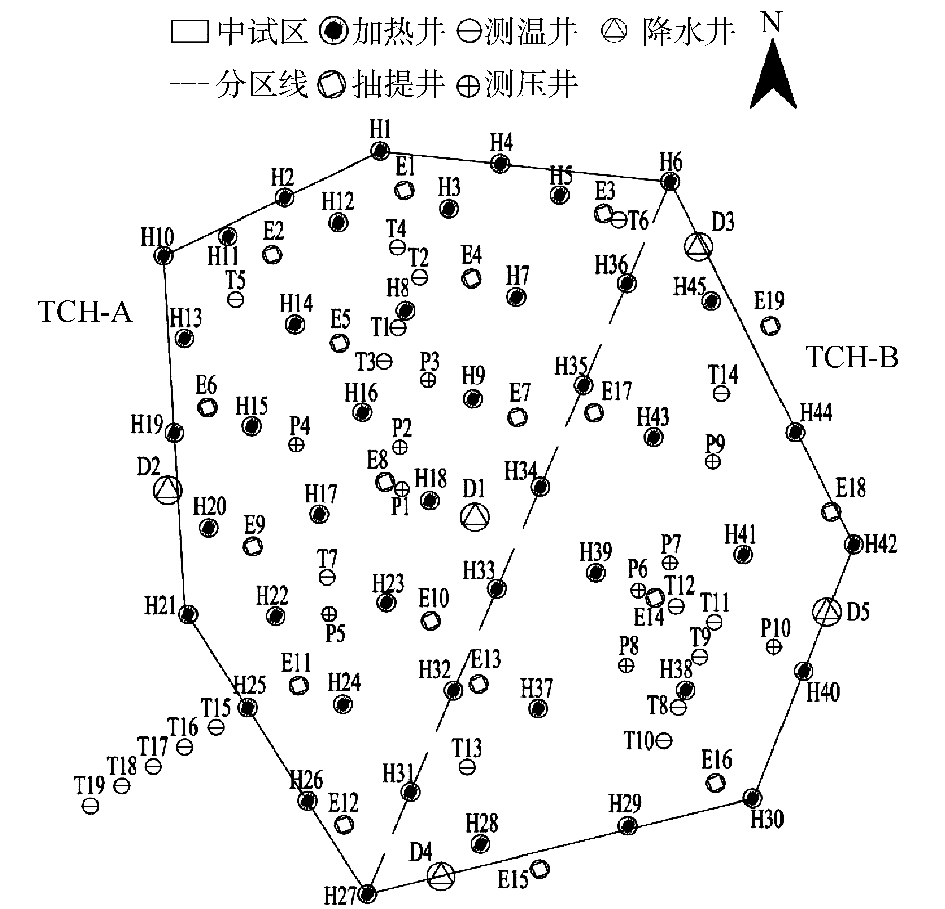
实验区域内土层下0~2 m污染土壤采用异位方式处置,将其清挖至暂存车间内,边坡放坡系数1∶0.9。然后平整清挖后的基坑,为后续钻井做准备。为防止周边污染对实验区域的影响,在区域四周设置一圈双排三轴水泥搅拌桩止水帷幕,深度15.5 m,厚度1.45 m,长度94.0 m。同时,利用降水井将止水帷幕内地下水抽干,尽可能地降低加热过程中热能的损耗。最后,根据场地水文地质情况及计算机辅助模拟计算结果,在本实验场地共设计加热井45个,抽提井19个,测温井19个,降水井5个,压力监测井10个。实验区井位布置如图2所示。
-
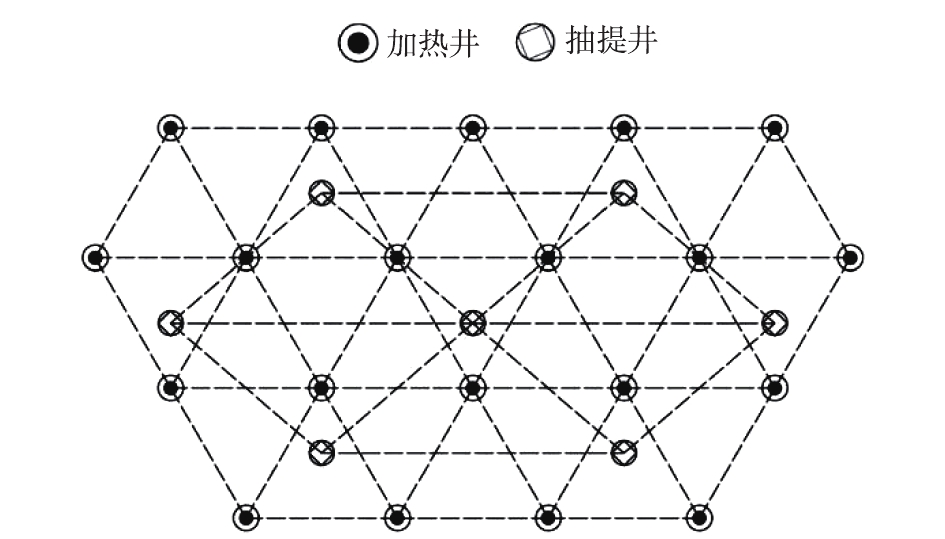
本实验采用正三角形布点方法进行加热井布点,抽提井均匀分布在加热井周围,如图3所示。加热井与抽提井总体数量比约为2∶1,污染范围边界的加热井适当加密布点。借鉴已有项目的工程经验,结合本场地地层分布特征及其渗透性,为研究不同加热间距下区域温度变化情况,分别设计加热间距为3.0 m(TCH-A)和 4.0 m(TCH-B)2个分区。加热深度为10.0 m,加热井深度为10.5 m。
-
为研究不同加热间距的实验分区内单井有效加热范围,在分区TCH-A、TCH-B内各选择1个加热井,在距热源不同距离的水平方向上设置一系列测温井(图2),以获得温度监测数据,从而分析单一加热井在有效加热范围内的温度变化规律。测温井具体位置为:分区TCH-A距离单井热源分别为0.5、1.0、1.5、1.73 m(最冷点),对应编号为T1~T4;分区TCH-B距离单井热源分别为0.5、1.0、1.5、2.0、2.3 m(最冷点),对应编号为T8~T12。测温深度为9.0 m,测温井深度为10.5 m,如图4所示。
为研究加热区域边界外温度变化与热传递范围,保证水泥搅拌桩不会受热出现裂缝而影响止水效果,选择1个边界加热井,在远离热源不同距离的水平方向上设置一系列测温井。测温井距离单井热源分别为0.5、1.0、1.5、2.0、2.5 m,对应编号为T15~T19。测温深度为9.0 m,测温井深度为10.5 m,具体位置见图2。为监测中试实验运行过程中整个加热区域内的温度变化情况,每个分区增设测温井。测温井具体位置为:分区TCH-A增加3个,对应编号T5~T7;分区TCH-B增加2个,对应编号T13~T14。此类测温井布设在最冷点,即加热井布点正三角形的质心位置。
-
在本场地中试实验前,考虑到实验区域内不同分区、距离热源不同位置受热不均,可能存在处理效果不一的情况,因此,在实验区域内共布设了4个土壤采样点。其中,在2个分区各布设2个采样点,均布设在3个加热井热源连线三角形的质心位置(最冷点),分别记为S01~S04;每个采样点垂直方向上按深度每隔1.5 m(即土层下2.0~3.5、3.5~5.0、5.0~6.5、6.5~8.0、8.0~9.5 m处)采集一个土壤样品;同时,在靠近抽提井位置布设3个地下水采样点,编号为W01~W03。具体采样点位布置如图5所示。本实验中,土壤及地下水采样点的布设能够大致代表整个中试实验区域的污染情况与修复情况。
在整个实验过程中,共采集2个批次土壤及地下水样品,通电加热前为第1批次采样;待整个实验区域平均温度维持在100~110 ℃,持续1周后停止加热,保持抽提系统正常运行,此时进行第2批次采样。样品采集后立即放入0~4 ℃的冷藏箱内保存,并于48 h内送往实验室进行污染物测定。实验室检测指标为目标污染物中的超标污染物:土壤检测氯乙烯和氯苯,VOCs类检测方法依据HJ 605-2011[20];地下水样品检测氯乙烯、顺-1,2-二氯乙烯、苯和氯苯,VOCs类检测方法依据GB/T 5750.8-2006[21]。
-
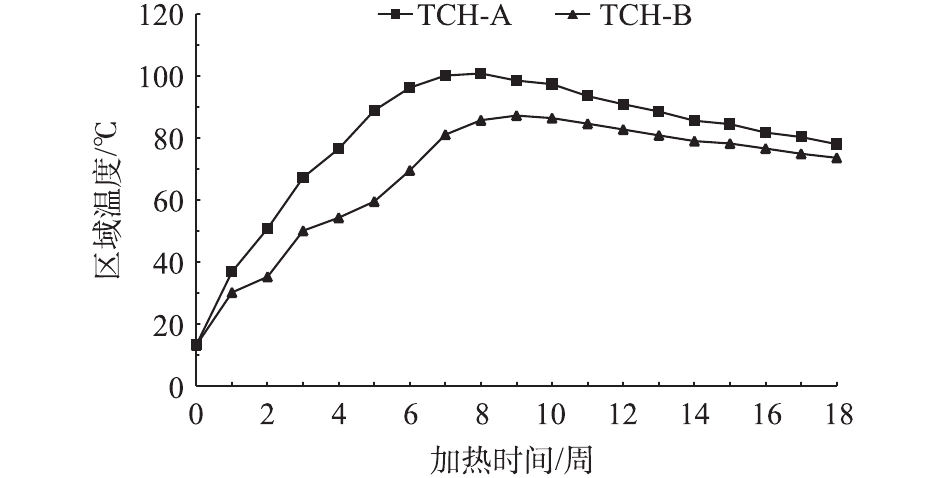
利用电热脱附技术对实验场地进行了为期8周的加热处理,区域温度随时间的变化情况见图6。加热间距的布置决定了区域升温时间和均匀升温效果。TCH-A分区平均温度升至80 ℃需要4~5周;而TCH-B分区则需要7周。实验分区TCH-B比实验分区TCH-A的升温效果滞后大约15 d。加热7周后TCH-A实验分区平均温度维持在100~110 ℃,TCH-B实验分区平均温度达到80~90 ℃,整个实验区域内部基本达到土壤及地下水中目标污染物的共沸温度。通常,对于修复苯、三氯乙烯等VOCs污染的地块,温度上升至100 ℃就能将此类污染物完全去除[6]。维持该温度1周后,停止加热,保持抽提系统正常运行,继续对实验区域温度进行监测。加热8周后停止加热,由于受到TCH-A分区高温传递的影响,TCH-B分区表现出短暂的温度上升趋势。降温过程持续至12周后,整个实验区域平均温度降至80~90 ℃;降温过程持续18周后,平均温度降至70~80 ℃,降温速度减慢并长时间维持。
为确定本场地最佳加热间距,进一步研究了不同分区内单井热源有效加热范围,获取设置在单井热源周边测温点的温度数据并进行了分析。如图7(a)所示,在实验分区TCH-A距离单井热源0.5 m处,加热2周,温度可升至106.8 ℃;在距离单井热源1.0、1.5 m处,加热5周,温度分别升至110.1 ℃和100.6 ℃;在加热井布置三角形质心位置(最冷点),距离单井热源1.73 m处,加热8周,温度可升至99.5 ℃。由此可知,TCH-A分区整体加热8周后,整个区域平均温度均可达到100 ℃左右,加热井布置三角形质心最冷点位置温度也可达到99.5 ℃。如图7(b)所示,与实验分区TCH-A对比,在TCH-B距离单井热源0.5 m处,加热7周,温度升至106.4 ℃;在距离单井热源1.0 m处,加热8周,温度升至102.2 ℃;在距离单井热源1.5、2.0 m处以及加热井布置三角形质心位置(最冷点位置),直到停止加热1周后,即第9周温度才达到峰值,分别升至89.4、82.6、81.7 ℃。由此可知,TCH-B分区在加热8周后,只有距离单井热源0.5、1.0 m处,温度升至100 ℃以上,1.5、2.0 m处以及加热井布置三角形质心位置(最冷点位置)均未达到100 ℃。停止加热1周后,距离加热井较远位置继续升温,这是由于受到距离加热井较近位置持续高温传递的影响,低温位置表现出短暂的温度上升趋势。实验分区TCH-A与TCH-B距离单井热源不同测温位置的温度均在停止加热1周后,即第9周开始相互趋近,并随着降温过程进行,整个实验区域温度趋于均衡。在实验进行至第18周时,实验分区TCH-A距离单井热源不同测温位置的平均温度为81.8 ℃;实验分区TCH-B距离单井热源不同测温位置的平均温度为73.8 ℃。
已有的工程经验[5]表明,热传导热脱附技术加热井间距设置要求一般为1.5~4.0 m。本研究中加热间距3.0 m,相较于4.0 m,材料投入虽有所增加,但可缩短升温时间而节省工期,同时,在相同加热周期内可保证修复区域具有更均匀的升温效果。
-
为防止周边污染对实验区域的影响,同时为提升升温速率,进行有效除水,在加热区域外3.0 m处,设置水泥搅拌桩止水帷幕。为研究加热区域边界温度变化与热传递范围,即高温传递是否会到达水泥搅拌桩位置从而影响其强度和止水效果,最终确定适合本场地原位热解吸工艺顺利实施的最佳止水帷幕与加热边界的间距。整个实验周期内加热边界温度变化与热传递范围如图8所示。在距离边界加热井0.5 m处,最高温出现在实验进行11周之后,温度为67.7 ℃;在距离边界加热井1.0、1.5、2.0、2.5 m处,最高温出现在实验进行15周之后,温度分别为49.1、39.7、33.4、29.1 ℃;在实验进行18周后,距边界加热井0.5、1.0、1.5、2.0、2.5 m处,温度分别降至55.3、47.0、38.9、32.8、28.0 ℃。
在前8周的加热期内,距离边界加热井0.5、1.0、1.5、2.0、2.5 m处的5个测温点温度均不断上升,且距离越近,升温越快。距边界加热井2.0 m甚至更远处温度不会超过30 ℃,即使延长一定的热传递时间,实验进行15周后,距边界加热井2.0 m处的最高温度为33.4 ℃。虽然从第9周开始已停止加热,但加热区内温度仍相对较高,向四周的热传递并未停止,降温过程中区域内外温差在逐渐缩小。若在8周后继续加热并维持区域温度,可能距边界加热井2.0 m处温度会不止30 ℃。
由此可知,为避免因热传递温度升高影响水泥搅拌桩的强度从而影响止水效果,随着加热时间的延长,可进一步增加止水帷幕与加热边界的间距。
-
由表3可知,实验前土壤中不同目标污染物在2.0~10.0 m土层内纵向分布不均匀。氯乙烯主要分布在3.5 m以下,其中S01采样点6.5~8.0 m处浓度最高,为5.4 mg·kg−1;氯苯在2.0~10.0 m均有分布,在3.5 m以下,污染物浓度普遍较高,其中S01采样点6.5~8.0 m处浓度最高,为30 mg·kg−1。实验进行18周后,再次采样发现,土壤中目标污染物含量均表现为下降。S01~S04采样点不同深度氯乙烯全部未检出,表明土壤中氯乙烯已处理干净。S01~S04采样点不同深度氯苯检出浓度最高,可达0.21 mg·kg−1,这表明土壤中氯苯绝大部分已被抽提出来。由污染物去除率计算可知,3.5~5.0、5.0~6.5、6.5~8.0、8.0~9.5 m处氯苯的平均去除率分别为95%、99%、99%、99%。进一步对比DB11/T 811-2011[22]污染场地土壤(住宅用地)中规定的氯乙烯、氯苯的筛选值,其值分别为0.25 mg·kg−1和41 mg·kg−1。由此可知,电热脱附后土壤中污染物绝大部分已向液相和气相转化并被有效抽提,处理效果优异。
-
由表4可知,实验前地下水中检出目标污染物分别为氯乙烯、顺-1,2-二氯乙烯、苯、氯苯等4种。W01采样点氯乙烯、苯、氯苯浓度最高,分别为3.79×103、1.06×103、2.34×104 μg·kg−1;W03采样点顺-1,2-二氯乙烯浓度最高,为3.35×103 μg·kg−1。实验18周后再次采样发现,地下水中目标污染物含量均表现为下降。W01采样点4种目标污染物氯乙烯、顺-1,2-二氯乙烯、苯、氯苯检出浓度最高,分别为417、198、37.2、101 μg·kg−1。通过计算去除率可知,地下水中氯乙烯、顺-1,2-二氯乙烯、苯、氯苯的平均去除率分别为90.5%、93.5%、96.4%、99.3%。进一步对比DB11/T 1278-2015[23]地下水(居住)中规定的氯乙烯、顺-1,2-二氯乙烯、苯、氯苯的筛选值,其值分别为20、70、10、300 μg·kg−1。由此可知,实验期内虽未完全达到筛选值标准,但地下水中污染物大部分已挥发出来并被有效抽提,处理效果尚好。因此,在该技术实际工程应用中为实现达标,可考虑增加热脱附停留时间。
-
电热脱附技术的成功应用取决于加热温度、停留时间、土壤含水率和土壤孔隙率等影响因素。在本研究中,结合加热升温过程、污染物去除率等研究结果对各影响因素进行了逐一分析与讨论。
-
由实验前后区域污染物含量变化可知,加热温度是影响原位热脱附效果的主要因素[24-26]。HERON等[27]研究表明,利用电热脱附技术处理三氯乙烯污染土壤,在23 ℃条件下,脱附效果很差且处理周期持续1年之久;但在100 ℃条件下,仅需37 d,去除率即可达到99.8%;加热区域温度越高,热脱附效果也就越好。实验区内土壤及地下水中目标污染物包括氯乙烯、顺-1,2-二氯乙烯、苯、氯苯,沸点分别为-13.9、60.0、80.1、131.7 ℃。加热进行2周后,仅加热井附近温度较高,TCH-A分区中0.5 m处升至100 ℃以上,TCH-B分区中0.5 m处温度升至60 ℃以上。此时,区域内氯乙烯、顺-1,2-二氯乙烯等低沸点的有机污染物开始逐渐向气相转化。加热进行7周后,整个区域温度均升至80 ℃以上,TCH-A分区平均温度升至100 ℃以上,TCH-B分区平均温度升至80 ℃以上。此时区域内苯、氯苯等高沸点的有机污染物开始大量向气相转化。从土壤中目标污染物含量的变化可以看出,氯乙烯去除完全时,氯苯还有少量残留。这是由于氯苯从土壤中脱附所需的温度更高,开始向气相转化的时间点相对滞后。综上所述,加热目标温度可直接影响污染物的去除效果。当然,为最终确定适合场地污染特征的最佳加热目标温度,同时也需综合考虑能耗及工期。
-
由实验前后各区域污染物含量变化可知,加热至目标温度后,持续热脱附停留时间同样是影响热脱附效果的关键因素。在热脱附处理加热升温前期,温度起主导作用;而在温度相对稳定的中后期,时间则是主要影响因素。张攀等[28]研究表明,在300 ℃条件下热脱附硝基苯,加热30 min,脱附效率为86.93%;维持温度20 min后,脱附效率为91.03%;加热至目标温度后的持续时间越长,热脱附效果越好。在本研究中,加热7周后,继续加热,维持该温度持续1周,土壤及地下水中目标污染物的去除效果均较为理想。其中,土壤中氯乙烯全部去除,氯苯平均去除率达到99%;地下水中氯乙烯、顺-1,2-二氯乙烯、苯、氯苯的平均去除率分别为90.5%、93.5%、96.4%、99.3%。但实验中各区域内仍有少量污染物残留,这可能是由于维持高温阶段持续时间短。若将加热至目标温度后持续停留时间再延长,则污染物去除率可能会进一步提升。因此,在原位热脱附实际应用时,须结合加热温度与热脱附停留时间2个重要影响因素,综合考虑材料、能耗、工期等因素,通过实验获得最佳的搭配方式。
-
土壤含水率的高低直接影响着热脱附效率和工程成本。孙磊等[29]的研究表明,较低或较高的土壤含水率会降低五氯酚在土壤中的热脱附效率。对于电热脱附技术而言,较高的土壤含水率,土壤热容量高,升温至目标温度需要花费更长的时间,消耗更多的能源。因此,为提高升温效率,缩短工期,在加热开始前,须尽可能地对止水帷幕内的区域采取工程降排水措施。在加热升温过程中,利用多相抽提井也可抽提出一部分水量。对于干土,其导热系数、导温系数更小,可能会减小土壤热传导的效率。但对于本场地条件而言,因加热目标温度在100~110 ℃即可去除绝大多数污染物,不会出现更高的温度,故修复完成后各区域内仍然有一定量的土壤水,不会出现干土的工况。因此,本研究暂不对干土的工况展开讨论。
-
土壤渗透性的高低影响着有机污染物的抽提效率。尽管原位热脱附技术对于土壤渗透性低的污染地块仍然适用,但对于渗透性较好的土壤,经电热脱附后,土壤气相中的VOCs、SVOCs更容易被抽提,去除率更高[30-31]。高国龙等[32]的研究表明,对于土壤结构紧实或黏性较大的土壤,热脱附效率较低。本实验场地3~7 m以砂质粉土为主,属于潜水含水层;7~10 m为粉质黏土、淤泥质粉质黏土层。污染物主要分布在含水层的中下部以及隔水层的顶部,因此,在设计加热、抽提深度时,须覆盖污染在地层中的主要分布位置。
综上所述,应用电热脱附技术去除土壤中有机污染物须综合考虑加热目标温度、热脱附停留时间、土壤含水率、土壤渗透性等影响因素。加热目标温度越高,热脱附停留时间越长,热脱附效果越好;土壤含水率较低、渗透性较好的土壤热脱附效果也更好。此外,为防止修复过程中产生二次污染,对于抽提废气的有效收集和无害化处理也是至关重要的[33],应遵循“先启抽提、后启加热,先关加热、后关抽提”的运行原则。
-
1)不同加热间距,相应的实验分区升温效果不同。加热间距为3.0 m,分区升温时间短,其均匀升温效果优于加热间距为4.0 m的分区,但2种间距布设方式均可达到去除区域污染的最终目标。
2)实验期内,加热边界热传递有效范围在2.0 m左右。针对本场地,可保守考虑,将止水帷幕设置在距离加热边界3.0 m及更远的位置。
3)电热脱附原位修复技术对实验区内土壤及地下水中目标污染物去除效果良好。土壤中目标污染物氯乙烯、氯苯平均去除率为100%、99%;地下水中目标污染物氯乙烯、顺-1,2-二氯乙烯、苯、氯苯的平均去除率分别为90.5%、93.5%、96.4%、99.3%。
4)电热脱附原位修复技术主要受加热目标温度、热脱附停留时间的影响,土壤含水率、土壤渗透性等因素也影响着热脱附效果。加热目标温度越高,热脱附停留时间越长,热脱附效果越好;对于含水率较低且渗透性较好的土壤,热脱附效果也更好。
5)电热脱附原位修复技术处理氯代烃类有机物污染场地效果良好,能够进行大规模的实际应用。在工程设计时,须综合考虑污染特征、地质及水文地质条件、材料投入、能耗成本、施工工期等因素,通过计算寻找最佳的搭配方式。
原位电热脱附技术在某有机污染场地修复中的应用效果
Application effect of in-situ electric thermal desorption technology used in remediation at an organics-contaminated site
-
摘要: 以某退役化学试剂厂土壤及地下水中氯乙烯、顺-1,2-二氯乙烯、苯、氯苯为目标污染物,基于电热脱附技术开展了中试规模的修复研究。结果表明:经电热脱附处理后,土壤中氯乙烯、氯苯的平均去除率分别达到100%、99%,均低于北京市《场地土壤环境风险评价筛选值》中污染场地(住宅用地)中土壤筛选值;地下水中氯乙烯、顺-1,2-二氯乙烯、苯、氯苯的平均去除率分别为90.5%、93.5%、96.4%、99.3%。此外,加热井设计间距对土壤温度变化有明显影响,间距为3.0 m的加热井布设方案下的升温时间短且升温效果好,优于间距为4.0 m的加热井布设方案,但两者均可达到去除污染物的目标;加热边界有效热传递范围可达2.0 m;止水帷幕与加热边界的最佳间距至少为3.0 m;目标温度越高,热脱附时间越长,热脱附效率则越高。同时,还讨论了土壤含水率及渗透性等因素对脱附效果的影响。电热脱附技术对修复氯代烃类有机物污染场地具有良好的效果,可进行大规模的工程应用。Abstract: In this study, vinyl chloride, cis-1, 2-dichloroethylene, benzene in the chlorobenzene-contaminated soil and the corresponding groundwater from a retired chemical reagent factory were taken as the objects, the pilot test of in-situ electric thermal desorption technique was conducted to treat them. The results revealed that the average removal rates of vinyl chloride and chlorobenzene from the treated soil with in-situ electric thermal desorption reached 100% and 99%, respectively, the corresponding residual contents in the treated soils were lower than the screening levels for soil environmental risk assessment of sites (residential land) in Beijing. The average removal rates of vinyl chloride, cis-1, 2-dichloroethylene, benzene and chlorobenzene from groundwater were 90.5%, 93.5%, 96.4% and 99.3%, respectively. The distance between heating wells had the effect on the temperature variation. At the distance of 3.0 m, the heating time was shorter and the heating effect was better than that at the distance of 4.0 m, while both of them could achieve the goal of contaminants removal. The effective heat transfer range of the heating boundary reached 2.0 m, and the optimal spacing between the water-resisting curtain and the heating boundary was determined at least 3.0 m. The removal efficiency was positively correlated with heating temperature and thermal desorption time. Both longer time and higher efficiency for the thermal desorption occurred when higher target temperature was set. Meanwhile, the effects of soil moisture and porosity on the removal efficiency were discussed. The results indicate that the in-situ electric thermal desorption technology can be applied to remediate the site contaminated by chlorinated hydrocarbon organic pollutants in the large-scale practice.
-
近年来,随着城市化进程的加速,许多原本位于城区的化工企业被迫从城市中心地带迁出,从而遗留了大量的污染场地[1-3]。其中,有机污染场地中的土壤和地下水污染问题较为突出,由此带来的环境和健康风险严重威胁着人们的生产和生活。因此,对其中污染严重的场地遗址进行修复已刻不容缓[4-6]。
针对有机污染场地的修复,近些年来,国内应用异位热脱附技术较多,且积累了大量的工程经验及成功案例。但是,应用异位热脱附技术修复有机污染场地的过程中尚存在着二次污染难控制、社会影响较大等问题。尤其涉及污染较深的场地,不适合使用异位热脱附技术[7-9]。原位热脱附技术(in-situ thermal desorption,ISTD)是通过对土壤直接或间接加热,促使有机污染物挥发,由固相、液相向气相转化,进而对其进一步收集并去除的技术[9]。ISTD作为修复土壤和地下水中有机污染的一项重要技术,在修复低渗透性土壤、污染深度较深且有机物污染严重的场地中具有很大的优势和应用前景[10-11]。
ISTD按照不同的加热方式可分为蒸汽加热[12-13]、电阻加热[14-15]和热传导加热[16]。电热脱附技术属于热传导加热,其过程是通过在垂直(或倾斜)阵列加热井中安置电加热棒,电加热元件可在高达800 ℃的条件下平稳运行,绝大部分有机污染物、重质或轻质非水相液体,将会通过蒸发、蒸馏、沸腾、氧化和高温分解等过程挥发或被处理掉[17]。电热脱附技术基本不受污染物理化性质、复杂地质及水文地质条件等因素的限制,具备应用范围广、设备安装便捷、加热过程安全、处理速度快等优势,正逐步被广泛应用于各种低渗透性、难处理有机物污染场地治理工程中[18],如ISTD曾大规模成功应用于美国新泽西泰特波罗机场附近地块等修复项目[19]。目前,国内在宁波、苏州、天津、武汉等城市的大型修复项目中已陆续开展电热脱附技术实验及应用。
本研究旨在中试的尺度下探讨电热脱附技术在现实污染场地中不同加热间距下的升温速率、加热边界温度传递范围和污染物去除率,分析了土壤导热率、土壤含水率、水文地质条件等影响因素对原位热脱附效果的影响特征,以期为我国有机污染场地热修复应用提供参考。
1. 实验部分
1.1 污染区域
中试实验区域位于我国北方某退役化学试剂厂,为污染土壤和地下水复合区域,面积278.9 m2,污染深度2~10 m,修复土方量2 231.2 m3。场地调查结果显示,主要污染物为氯乙烯、顺-1,2-二氯乙烯、苯、氯苯等挥发性有机污染物;土壤中氯乙烯、氯苯最高浓度分别为5.4 mg·kg−1和30 mg·kg−1;地下水中氯乙烯、顺-1,2-二氯乙烯、苯、氯苯最高浓度分别为3.79×103、3.35×103、1.06×103、2.34×104 μg·kg−1。地质勘察结果表明,该区域可分为5个工程地质层,细分为8个工程地质亚层,地层岩性分布见表1。实验区地表以下0~10.00 m大致分为4类:0~2.00 m为填土层,多为黏性土夹较多碎石和砖块等建筑垃圾;2.00~3.20 m为不透水层,以粉质黏土为主;3.20~4.70 m为潜水含水层,以砂质粉土为主;4.70~10.00 m大多为粉质黏土、粉质黏土夹粉土薄层团块。同时,对实验区内各土层理化性质进行了测试,结果如表2所示。
表 1 实验区地层岩性分布Table 1. Stratigraphic lithologic distribution of the experimental area成因 土层名称 标准层号 层厚/m 层底标高/m 性质 人工堆积① 杂填土1 ①1 0.40~0.60 2.50~2.30 均匀性差 人工堆积① 黏质粉土素填土2 ①2 0.40~1.60 2.00~0.90 均匀性差 坑底淤积② 粉质黏土1 ②1 0.60~1.00 0.90~−0.30 属不透水层 坑底淤积② 砂质粉土2 ②2 1.50~3.30 −1.80~−3.60 属弱透水层 古河道、洼淀冲积③ 粉质黏土1 ③1 1.30~3.10 −4.90~−5.10 属不透水层 古河道、洼淀冲积③ 黏质粉土-砂质粉土2 ③2 5.00~5.20 −10.10~−10.60 垂直向为微透水层,水平向为弱透水层 河床~河漫滩相沉积④ 粉质黏土 ④ 4.00~4.50 −14.40~−15.10 属微透水层 湖沼相沉积④ 砂质粉土 ⑤ 3.00~6.50 −18.10~−21.60 属弱透水层 注:场地地表标高为2.90 m。 | Show Table DownLoad:
CSV
表 2 实验区各土层理化性质Table 2. Physical and chemical properties of every soil in experimental area
DownLoad:
CSV
表 2 实验区各土层理化性质Table 2. Physical and chemical properties of every soil in experimental area标准层号 土层名称 含水率/% 容重/(g·cm−3) 孔隙率/% 初始温度/℃ 导热系数/(W·(m·K)−1) 比热容/(kJ·(kg·K)−1) 导温系数/(m2·h−1) ①1 杂填土 25.9 1.92 44.4 18.1 1.87 1.79 1.86×10−3 ①2 黏质粉土素填土 25.9 1.92 44.4 18.1 1.87 1.79 1.86×10−3 ②1 粉质黏土 24.5 1.96 42.0 13.3 1.76 1.71 1.90×10−3 ②2 砂质粉土 25.4 1.98 42.0 11.6 1.87 1.83 1.88×10−3 ③1 粉质黏土 39.4 1.80 52.4 14.1 1.59 1.58 2.03×10−3 ③2 黏质粉土-砂质粉土 28.0 1.94 43.5 14.5 1.75 1.70 1.95×10−3 ④ 粉质黏土 21.7 2.03 38.6 14.4 1.88 1.86 1.80×10−3 ⑤ 砂质粉土 22.6 2.01 39.1 — — — — 注:由于第⑤层土层深度已超过污染深度,未检测相关理化性质,“—”表示未检测。 | Show TableDownLoad:
CSV
1.2 实验系统
实验系统由加热单元、抽提单元、温度监测单元和电气控制单元4部分组成。工艺装置如图1所示。首先,在土壤中安置电加热棒并通电升温,电加热棒通过热辐射的形式将热量传递到与土壤接触的密封金属套管上,通过热传导使得周围污染土壤逐渐升温至目标温度。目标污染物发生挥发和裂解反应后,含有污染物的蒸汽和水通过多相抽提井被抽提至地表,再经各自的输送管道运至尾水/尾气处理设备并进行无害化处置,最后达标排放。在整个加热过程中,利用温度、压力监测井实时监控整个修复区域,并通过智能化、自动化控制系统对每一个加热设备的工作温度进行实时调控。
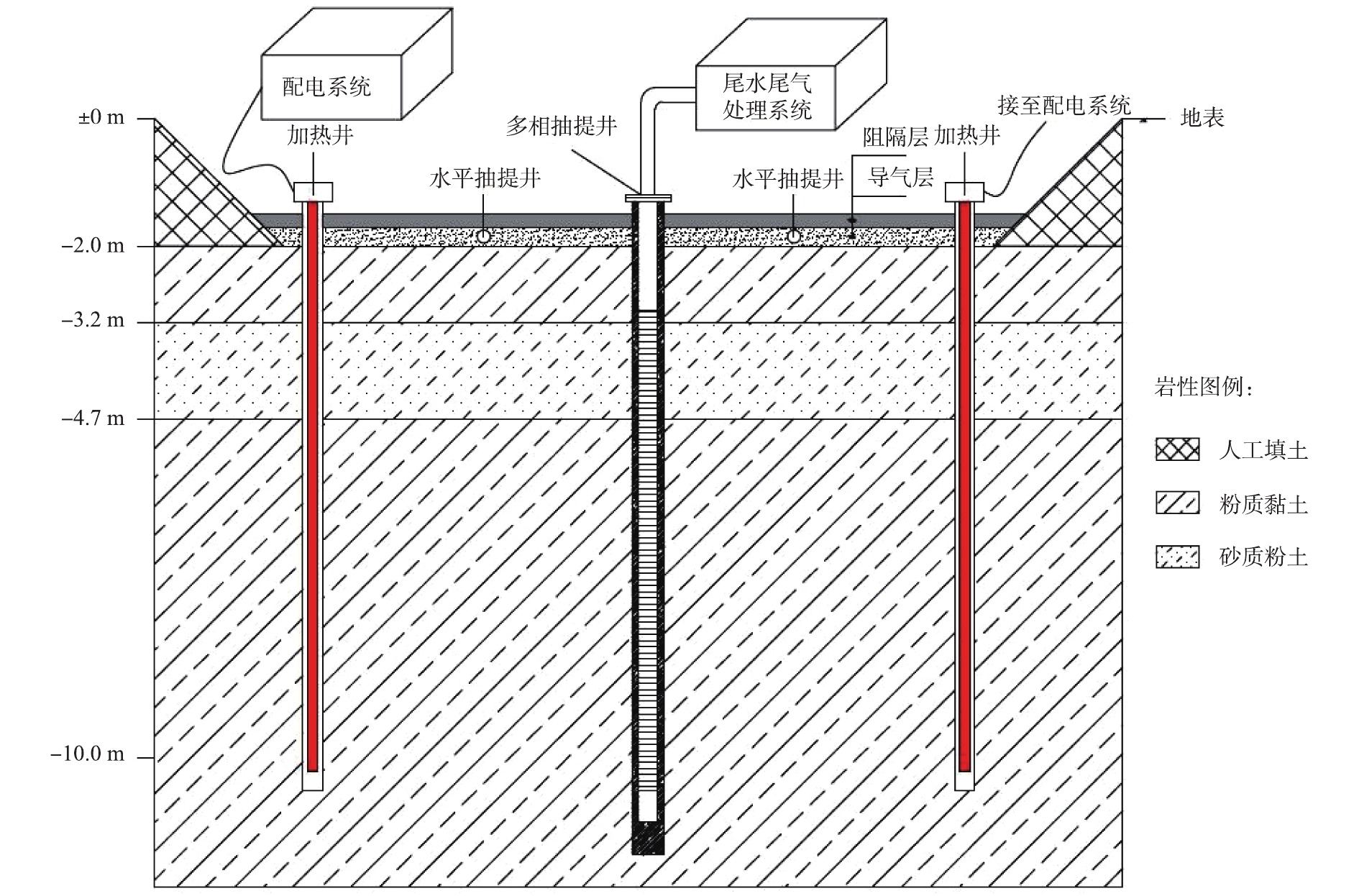 图 1 电热脱附技术设计原理Figure 1. Design principle profile of electric thermal desorption technology
图 1 电热脱附技术设计原理Figure 1. Design principle profile of electric thermal desorption technology1.3 场地井位设计
实验区域内土层下0~2 m污染土壤采用异位方式处置,将其清挖至暂存车间内,边坡放坡系数1∶0.9。然后平整清挖后的基坑,为后续钻井做准备。为防止周边污染对实验区域的影响,在区域四周设置一圈双排三轴水泥搅拌桩止水帷幕,深度15.5 m,厚度1.45 m,长度94.0 m。同时,利用降水井将止水帷幕内地下水抽干,尽可能地降低加热过程中热能的损耗。最后,根据场地水文地质情况及计算机辅助模拟计算结果,在本实验场地共设计加热井45个,抽提井19个,测温井19个,降水井5个,压力监测井10个。实验区井位布置如图2所示。
1.3.1 加热井、抽提井布点设计
本实验采用正三角形布点方法进行加热井布点,抽提井均匀分布在加热井周围,如图3所示。加热井与抽提井总体数量比约为2∶1,污染范围边界的加热井适当加密布点。借鉴已有项目的工程经验,结合本场地地层分布特征及其渗透性,为研究不同加热间距下区域温度变化情况,分别设计加热间距为3.0 m(TCH-A)和 4.0 m(TCH-B)2个分区。加热深度为10.0 m,加热井深度为10.5 m。
1.3.2 测温井布点设计
为研究不同加热间距的实验分区内单井有效加热范围,在分区TCH-A、TCH-B内各选择1个加热井,在距热源不同距离的水平方向上设置一系列测温井(图2),以获得温度监测数据,从而分析单一加热井在有效加热范围内的温度变化规律。测温井具体位置为:分区TCH-A距离单井热源分别为0.5、1.0、1.5、1.73 m(最冷点),对应编号为T1~T4;分区TCH-B距离单井热源分别为0.5、1.0、1.5、2.0、2.3 m(最冷点),对应编号为T8~T12。测温深度为9.0 m,测温井深度为10.5 m,如图4所示。
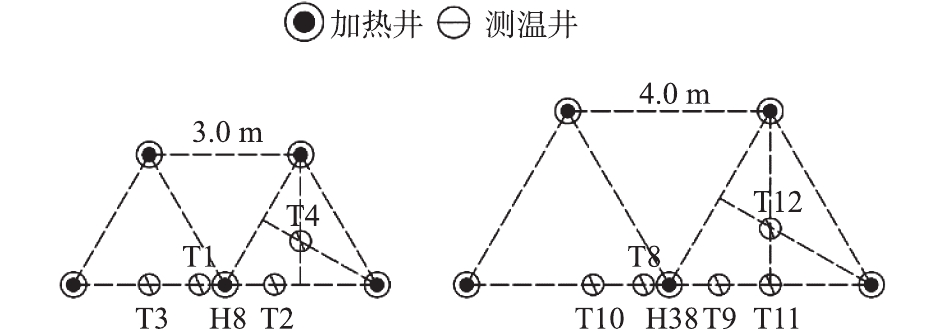 图 4 不同分区距离单井热源的测温井布点设计Figure 4. Design of temperature measuring wells from single well heat source in different divisions
图 4 不同分区距离单井热源的测温井布点设计Figure 4. Design of temperature measuring wells from single well heat source in different divisions为研究加热区域边界外温度变化与热传递范围,保证水泥搅拌桩不会受热出现裂缝而影响止水效果,选择1个边界加热井,在远离热源不同距离的水平方向上设置一系列测温井。测温井距离单井热源分别为0.5、1.0、1.5、2.0、2.5 m,对应编号为T15~T19。测温深度为9.0 m,测温井深度为10.5 m,具体位置见图2。为监测中试实验运行过程中整个加热区域内的温度变化情况,每个分区增设测温井。测温井具体位置为:分区TCH-A增加3个,对应编号T5~T7;分区TCH-B增加2个,对应编号T13~T14。此类测温井布设在最冷点,即加热井布点正三角形的质心位置。
1.4 采样与分析
在本场地中试实验前,考虑到实验区域内不同分区、距离热源不同位置受热不均,可能存在处理效果不一的情况,因此,在实验区域内共布设了4个土壤采样点。其中,在2个分区各布设2个采样点,均布设在3个加热井热源连线三角形的质心位置(最冷点),分别记为S01~S04;每个采样点垂直方向上按深度每隔1.5 m(即土层下2.0~3.5、3.5~5.0、5.0~6.5、6.5~8.0、8.0~9.5 m处)采集一个土壤样品;同时,在靠近抽提井位置布设3个地下水采样点,编号为W01~W03。具体采样点位布置如图5所示。本实验中,土壤及地下水采样点的布设能够大致代表整个中试实验区域的污染情况与修复情况。
在整个实验过程中,共采集2个批次土壤及地下水样品,通电加热前为第1批次采样;待整个实验区域平均温度维持在100~110 ℃,持续1周后停止加热,保持抽提系统正常运行,此时进行第2批次采样。样品采集后立即放入0~4 ℃的冷藏箱内保存,并于48 h内送往实验室进行污染物测定。实验室检测指标为目标污染物中的超标污染物:土壤检测氯乙烯和氯苯,VOCs类检测方法依据HJ 605-2011[20];地下水样品检测氯乙烯、顺-1,2-二氯乙烯、苯和氯苯,VOCs类检测方法依据GB/T 5750.8-2006[21]。
2. 结果与分析
2.1 实验前后不同加热分区温度变化对比
利用电热脱附技术对实验场地进行了为期8周的加热处理,区域温度随时间的变化情况见图6。加热间距的布置决定了区域升温时间和均匀升温效果。TCH-A分区平均温度升至80 ℃需要4~5周;而TCH-B分区则需要7周。实验分区TCH-B比实验分区TCH-A的升温效果滞后大约15 d。加热7周后TCH-A实验分区平均温度维持在100~110 ℃,TCH-B实验分区平均温度达到80~90 ℃,整个实验区域内部基本达到土壤及地下水中目标污染物的共沸温度。通常,对于修复苯、三氯乙烯等VOCs污染的地块,温度上升至100 ℃就能将此类污染物完全去除[6]。维持该温度1周后,停止加热,保持抽提系统正常运行,继续对实验区域温度进行监测。加热8周后停止加热,由于受到TCH-A分区高温传递的影响,TCH-B分区表现出短暂的温度上升趋势。降温过程持续至12周后,整个实验区域平均温度降至80~90 ℃;降温过程持续18周后,平均温度降至70~80 ℃,降温速度减慢并长时间维持。
为确定本场地最佳加热间距,进一步研究了不同分区内单井热源有效加热范围,获取设置在单井热源周边测温点的温度数据并进行了分析。如图7(a)所示,在实验分区TCH-A距离单井热源0.5 m处,加热2周,温度可升至106.8 ℃;在距离单井热源1.0、1.5 m处,加热5周,温度分别升至110.1 ℃和100.6 ℃;在加热井布置三角形质心位置(最冷点),距离单井热源1.73 m处,加热8周,温度可升至99.5 ℃。由此可知,TCH-A分区整体加热8周后,整个区域平均温度均可达到100 ℃左右,加热井布置三角形质心最冷点位置温度也可达到99.5 ℃。如图7(b)所示,与实验分区TCH-A对比,在TCH-B距离单井热源0.5 m处,加热7周,温度升至106.4 ℃;在距离单井热源1.0 m处,加热8周,温度升至102.2 ℃;在距离单井热源1.5、2.0 m处以及加热井布置三角形质心位置(最冷点位置),直到停止加热1周后,即第9周温度才达到峰值,分别升至89.4、82.6、81.7 ℃。由此可知,TCH-B分区在加热8周后,只有距离单井热源0.5、1.0 m处,温度升至100 ℃以上,1.5、2.0 m处以及加热井布置三角形质心位置(最冷点位置)均未达到100 ℃。停止加热1周后,距离加热井较远位置继续升温,这是由于受到距离加热井较近位置持续高温传递的影响,低温位置表现出短暂的温度上升趋势。实验分区TCH-A与TCH-B距离单井热源不同测温位置的温度均在停止加热1周后,即第9周开始相互趋近,并随着降温过程进行,整个实验区域温度趋于均衡。在实验进行至第18周时,实验分区TCH-A距离单井热源不同测温位置的平均温度为81.8 ℃;实验分区TCH-B距离单井热源不同测温位置的平均温度为73.8 ℃。
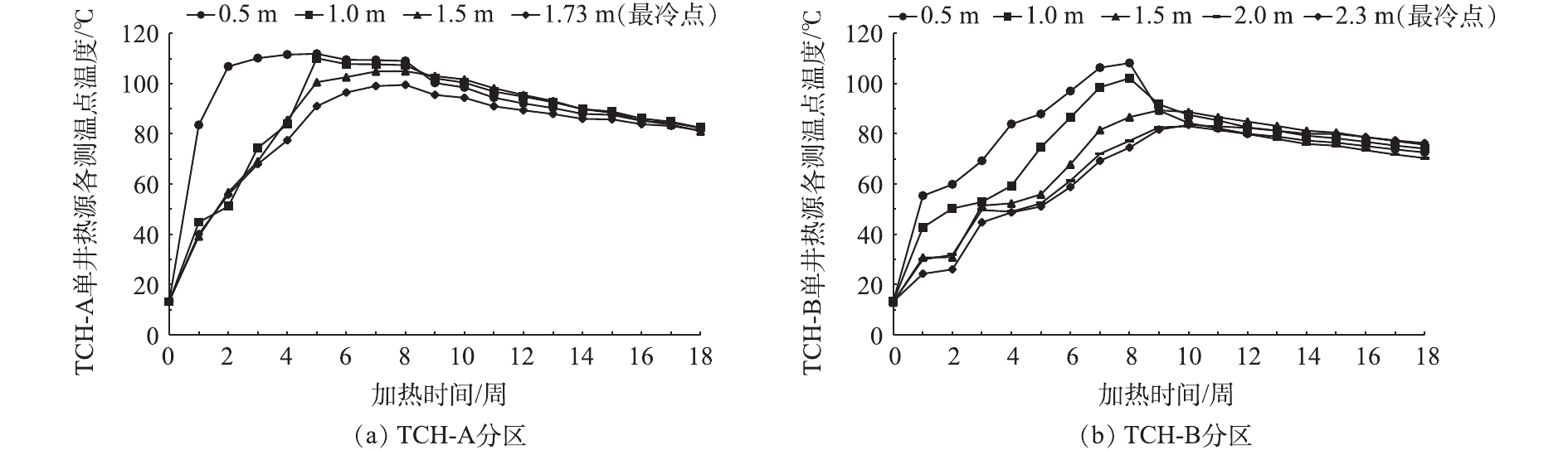 图 7 不同分区距离单井热源不同位置的温度变化Figure 7. Changes of temperature at different measuring points from single well heat source in different area
图 7 不同分区距离单井热源不同位置的温度变化Figure 7. Changes of temperature at different measuring points from single well heat source in different area已有的工程经验[5]表明,热传导热脱附技术加热井间距设置要求一般为1.5~4.0 m。本研究中加热间距3.0 m,相较于4.0 m,材料投入虽有所增加,但可缩短升温时间而节省工期,同时,在相同加热周期内可保证修复区域具有更均匀的升温效果。
2.2 实验期内加热边界热传递范围
为防止周边污染对实验区域的影响,同时为提升升温速率,进行有效除水,在加热区域外3.0 m处,设置水泥搅拌桩止水帷幕。为研究加热区域边界温度变化与热传递范围,即高温传递是否会到达水泥搅拌桩位置从而影响其强度和止水效果,最终确定适合本场地原位热解吸工艺顺利实施的最佳止水帷幕与加热边界的间距。整个实验周期内加热边界温度变化与热传递范围如图8所示。在距离边界加热井0.5 m处,最高温出现在实验进行11周之后,温度为67.7 ℃;在距离边界加热井1.0、1.5、2.0、2.5 m处,最高温出现在实验进行15周之后,温度分别为49.1、39.7、33.4、29.1 ℃;在实验进行18周后,距边界加热井0.5、1.0、1.5、2.0、2.5 m处,温度分别降至55.3、47.0、38.9、32.8、28.0 ℃。
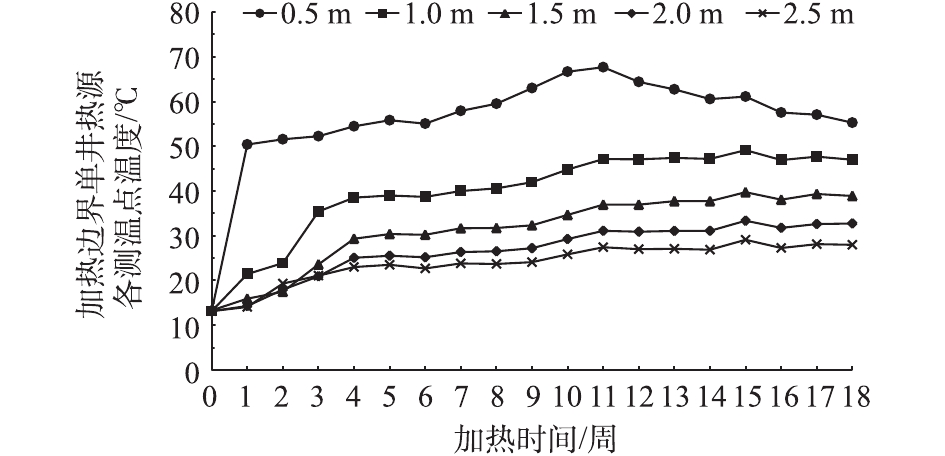 图 8 加热区边界不同距离温度的变化Figure 8. Changes of temperature at different measuring points on the boundary of the heating area
图 8 加热区边界不同距离温度的变化Figure 8. Changes of temperature at different measuring points on the boundary of the heating area在前8周的加热期内,距离边界加热井0.5、1.0、1.5、2.0、2.5 m处的5个测温点温度均不断上升,且距离越近,升温越快。距边界加热井2.0 m甚至更远处温度不会超过30 ℃,即使延长一定的热传递时间,实验进行15周后,距边界加热井2.0 m处的最高温度为33.4 ℃。虽然从第9周开始已停止加热,但加热区内温度仍相对较高,向四周的热传递并未停止,降温过程中区域内外温差在逐渐缩小。若在8周后继续加热并维持区域温度,可能距边界加热井2.0 m处温度会不止30 ℃。
由此可知,为避免因热传递温度升高影响水泥搅拌桩的强度从而影响止水效果,随着加热时间的延长,可进一步增加止水帷幕与加热边界的间距。
2.3 实验前后各区域污染物含量变化
2.3.1 实验区土壤中目标污染物含量变化
由表3可知,实验前土壤中不同目标污染物在2.0~10.0 m土层内纵向分布不均匀。氯乙烯主要分布在3.5 m以下,其中S01采样点6.5~8.0 m处浓度最高,为5.4 mg·kg−1;氯苯在2.0~10.0 m均有分布,在3.5 m以下,污染物浓度普遍较高,其中S01采样点6.5~8.0 m处浓度最高,为30 mg·kg−1。实验进行18周后,再次采样发现,土壤中目标污染物含量均表现为下降。S01~S04采样点不同深度氯乙烯全部未检出,表明土壤中氯乙烯已处理干净。S01~S04采样点不同深度氯苯检出浓度最高,可达0.21 mg·kg−1,这表明土壤中氯苯绝大部分已被抽提出来。由污染物去除率计算可知,3.5~5.0、5.0~6.5、6.5~8.0、8.0~9.5 m处氯苯的平均去除率分别为95%、99%、99%、99%。进一步对比DB11/T 811-2011[22]污染场地土壤(住宅用地)中规定的氯乙烯、氯苯的筛选值,其值分别为0.25 mg·kg−1和41 mg·kg−1。由此可知,电热脱附后土壤中污染物绝大部分已向液相和气相转化并被有效抽提,处理效果优异。
表 3 土壤中目标污染物含量变化Table 3. Concentration variation of target pollutants in soil污染物种类 编号 目标污染物浓度/(mg·kg−1) 2.0~3.5 m 3.5~5.0 m 5.0~6.5 m 6.5~8.0 m 8.0~9.5 m 实验前 实验后 实验前 实验后 实验前 实验后 实验前 实验后 实验前 实验后 氯乙烯 S01 <0.10 <0.10 0.81 <0.10 <0.10 <0.10 5.4 <0.10 <0.10 <0.10 S02 <0.10 <0.10 2.5 <0.10 <0.10 <0.10 <0.10 <0.10 0.68 <0.10 S03 <0.10 <0.10 0.33 <0.10 0.18 <0.10 0.19 <0.10 <0.10 <0.10 S04 <0.10 <0.10 <0.10 <0.10 0.74 <0.10 <0.10 <0.10 <0.10 <0.10 氯苯 S01 0.89 <0.05 1.9 <0.05 4.2 <0.05 30 <0.05 5.2 <0.05 S02 0.22 <0.05 0.65 0.18 12 0.21 2.0 0.18 7.9 0.11 S03 0.12 <0.05 4.8 0.13 0.11 <0.05 <0.05 <0.05 <0.05 <0.05 S04 0.48 <0.05 1.8 0.14 12 0.21 0.12 <0.05 <0.05 <0.05 | Show TableDownLoad:
CSV
2.3.2 实验区地下水中目标污染物含量变化
由表4可知,实验前地下水中检出目标污染物分别为氯乙烯、顺-1,2-二氯乙烯、苯、氯苯等4种。W01采样点氯乙烯、苯、氯苯浓度最高,分别为3.79×103、1.06×103、2.34×104 μg·kg−1;W03采样点顺-1,2-二氯乙烯浓度最高,为3.35×103 μg·kg−1。实验18周后再次采样发现,地下水中目标污染物含量均表现为下降。W01采样点4种目标污染物氯乙烯、顺-1,2-二氯乙烯、苯、氯苯检出浓度最高,分别为417、198、37.2、101 μg·kg−1。通过计算去除率可知,地下水中氯乙烯、顺-1,2-二氯乙烯、苯、氯苯的平均去除率分别为90.5%、93.5%、96.4%、99.3%。进一步对比DB11/T 1278-2015[23]地下水(居住)中规定的氯乙烯、顺-1,2-二氯乙烯、苯、氯苯的筛选值,其值分别为20、70、10、300 μg·kg−1。由此可知,实验期内虽未完全达到筛选值标准,但地下水中污染物大部分已挥发出来并被有效抽提,处理效果尚好。因此,在该技术实际工程应用中为实现达标,可考虑增加热脱附停留时间。
表 4 地下水中目标污染物含量变化Table 4. Concentration variation of target pollutants in groundwater污染物种类 编号 目标污染物浓度/(μg·L−1) 实验前 实验后 氯乙烯 W01 3.79×103 417 W02 2.58×103 219 W03 2.28×103 190 顺-1,2-二氯乙烯 W01 2.62×103 198 W02 2.59×103 167 W02 3.35×103 192 苯 W01 1.06×103 37.2 W02 389 10.8 W03 18.6 4.30 氯苯 W01 2.34×104 101 W02 7.95×103 61.0 W03 734 54.0 | Show TableDownLoad:
CSV
3. 影响因素分析
电热脱附技术的成功应用取决于加热温度、停留时间、土壤含水率和土壤孔隙率等影响因素。在本研究中,结合加热升温过程、污染物去除率等研究结果对各影响因素进行了逐一分析与讨论。
3.1 热脱附温度
由实验前后区域污染物含量变化可知,加热温度是影响原位热脱附效果的主要因素[24-26]。HERON等[27]研究表明,利用电热脱附技术处理三氯乙烯污染土壤,在23 ℃条件下,脱附效果很差且处理周期持续1年之久;但在100 ℃条件下,仅需37 d,去除率即可达到99.8%;加热区域温度越高,热脱附效果也就越好。实验区内土壤及地下水中目标污染物包括氯乙烯、顺-1,2-二氯乙烯、苯、氯苯,沸点分别为-13.9、60.0、80.1、131.7 ℃。加热进行2周后,仅加热井附近温度较高,TCH-A分区中0.5 m处升至100 ℃以上,TCH-B分区中0.5 m处温度升至60 ℃以上。此时,区域内氯乙烯、顺-1,2-二氯乙烯等低沸点的有机污染物开始逐渐向气相转化。加热进行7周后,整个区域温度均升至80 ℃以上,TCH-A分区平均温度升至100 ℃以上,TCH-B分区平均温度升至80 ℃以上。此时区域内苯、氯苯等高沸点的有机污染物开始大量向气相转化。从土壤中目标污染物含量的变化可以看出,氯乙烯去除完全时,氯苯还有少量残留。这是由于氯苯从土壤中脱附所需的温度更高,开始向气相转化的时间点相对滞后。综上所述,加热目标温度可直接影响污染物的去除效果。当然,为最终确定适合场地污染特征的最佳加热目标温度,同时也需综合考虑能耗及工期。
3.2 热脱附停留时间
由实验前后各区域污染物含量变化可知,加热至目标温度后,持续热脱附停留时间同样是影响热脱附效果的关键因素。在热脱附处理加热升温前期,温度起主导作用;而在温度相对稳定的中后期,时间则是主要影响因素。张攀等[28]研究表明,在300 ℃条件下热脱附硝基苯,加热30 min,脱附效率为86.93%;维持温度20 min后,脱附效率为91.03%;加热至目标温度后的持续时间越长,热脱附效果越好。在本研究中,加热7周后,继续加热,维持该温度持续1周,土壤及地下水中目标污染物的去除效果均较为理想。其中,土壤中氯乙烯全部去除,氯苯平均去除率达到99%;地下水中氯乙烯、顺-1,2-二氯乙烯、苯、氯苯的平均去除率分别为90.5%、93.5%、96.4%、99.3%。但实验中各区域内仍有少量污染物残留,这可能是由于维持高温阶段持续时间短。若将加热至目标温度后持续停留时间再延长,则污染物去除率可能会进一步提升。因此,在原位热脱附实际应用时,须结合加热温度与热脱附停留时间2个重要影响因素,综合考虑材料、能耗、工期等因素,通过实验获得最佳的搭配方式。
3.3 土壤含水率
土壤含水率的高低直接影响着热脱附效率和工程成本。孙磊等[29]的研究表明,较低或较高的土壤含水率会降低五氯酚在土壤中的热脱附效率。对于电热脱附技术而言,较高的土壤含水率,土壤热容量高,升温至目标温度需要花费更长的时间,消耗更多的能源。因此,为提高升温效率,缩短工期,在加热开始前,须尽可能地对止水帷幕内的区域采取工程降排水措施。在加热升温过程中,利用多相抽提井也可抽提出一部分水量。对于干土,其导热系数、导温系数更小,可能会减小土壤热传导的效率。但对于本场地条件而言,因加热目标温度在100~110 ℃即可去除绝大多数污染物,不会出现更高的温度,故修复完成后各区域内仍然有一定量的土壤水,不会出现干土的工况。因此,本研究暂不对干土的工况展开讨论。
3.4 土壤渗透性
土壤渗透性的高低影响着有机污染物的抽提效率。尽管原位热脱附技术对于土壤渗透性低的污染地块仍然适用,但对于渗透性较好的土壤,经电热脱附后,土壤气相中的VOCs、SVOCs更容易被抽提,去除率更高[30-31]。高国龙等[32]的研究表明,对于土壤结构紧实或黏性较大的土壤,热脱附效率较低。本实验场地3~7 m以砂质粉土为主,属于潜水含水层;7~10 m为粉质黏土、淤泥质粉质黏土层。污染物主要分布在含水层的中下部以及隔水层的顶部,因此,在设计加热、抽提深度时,须覆盖污染在地层中的主要分布位置。
综上所述,应用电热脱附技术去除土壤中有机污染物须综合考虑加热目标温度、热脱附停留时间、土壤含水率、土壤渗透性等影响因素。加热目标温度越高,热脱附停留时间越长,热脱附效果越好;土壤含水率较低、渗透性较好的土壤热脱附效果也更好。此外,为防止修复过程中产生二次污染,对于抽提废气的有效收集和无害化处理也是至关重要的[33],应遵循“先启抽提、后启加热,先关加热、后关抽提”的运行原则。
4. 结论
1)不同加热间距,相应的实验分区升温效果不同。加热间距为3.0 m,分区升温时间短,其均匀升温效果优于加热间距为4.0 m的分区,但2种间距布设方式均可达到去除区域污染的最终目标。
2)实验期内,加热边界热传递有效范围在2.0 m左右。针对本场地,可保守考虑,将止水帷幕设置在距离加热边界3.0 m及更远的位置。
3)电热脱附原位修复技术对实验区内土壤及地下水中目标污染物去除效果良好。土壤中目标污染物氯乙烯、氯苯平均去除率为100%、99%;地下水中目标污染物氯乙烯、顺-1,2-二氯乙烯、苯、氯苯的平均去除率分别为90.5%、93.5%、96.4%、99.3%。
4)电热脱附原位修复技术主要受加热目标温度、热脱附停留时间的影响,土壤含水率、土壤渗透性等因素也影响着热脱附效果。加热目标温度越高,热脱附停留时间越长,热脱附效果越好;对于含水率较低且渗透性较好的土壤,热脱附效果也更好。
5)电热脱附原位修复技术处理氯代烃类有机物污染场地效果良好,能够进行大规模的实际应用。在工程设计时,须综合考虑污染特征、地质及水文地质条件、材料投入、能耗成本、施工工期等因素,通过计算寻找最佳的搭配方式。
-
图 1 电热脱附技术设计原理
Figure 1. Design principle profile of electric thermal desorption technology
图 4 不同分区距离单井热源的测温井布点设计
Figure 4. Design of temperature measuring wells from single well heat source in different divisions
图 7 不同分区距离单井热源不同位置的温度变化
Figure 7. Changes of temperature at different measuring points from single well heat source in different area
图 8 加热区边界不同距离温度的变化
Figure 8. Changes of temperature at different measuring points on the boundary of the heating area
表 1 实验区地层岩性分布
Table 1. Stratigraphic lithologic distribution of the experimental area
成因 土层名称 标准层号 层厚/m 层底标高/m 性质 人工堆积① 杂填土1 ①1 0.40~0.60 2.50~2.30 均匀性差 人工堆积① 黏质粉土素填土2 ①2 0.40~1.60 2.00~0.90 均匀性差 坑底淤积② 粉质黏土1 ②1 0.60~1.00 0.90~−0.30 属不透水层 坑底淤积② 砂质粉土2 ②2 1.50~3.30 −1.80~−3.60 属弱透水层 古河道、洼淀冲积③ 粉质黏土1 ③1 1.30~3.10 −4.90~−5.10 属不透水层 古河道、洼淀冲积③ 黏质粉土-砂质粉土2 ③2 5.00~5.20 −10.10~−10.60 垂直向为微透水层,水平向为弱透水层 河床~河漫滩相沉积④ 粉质黏土 ④ 4.00~4.50 −14.40~−15.10 属微透水层 湖沼相沉积④ 砂质粉土 ⑤ 3.00~6.50 −18.10~−21.60 属弱透水层 注:场地地表标高为2.90 m。
下载: 导出CSV
表 2 实验区各土层理化性质
Table 2. Physical and chemical properties of every soil in experimental area
标准层号 土层名称 含水率/% 容重/(g·cm−3) 孔隙率/% 初始温度/℃ 导热系数/(W·(m·K)−1) 比热容/(kJ·(kg·K)−1) 导温系数/(m2·h−1) ①1 杂填土 25.9 1.92 44.4 18.1 1.87 1.79 1.86×10−3 ①2 黏质粉土素填土 25.9 1.92 44.4 18.1 1.87 1.79 1.86×10−3 ②1 粉质黏土 24.5 1.96 42.0 13.3 1.76 1.71 1.90×10−3 ②2 砂质粉土 25.4 1.98 42.0 11.6 1.87 1.83 1.88×10−3 ③1 粉质黏土 39.4 1.80 52.4 14.1 1.59 1.58 2.03×10−3 ③2 黏质粉土-砂质粉土 28.0 1.94 43.5 14.5 1.75 1.70 1.95×10−3 ④ 粉质黏土 21.7 2.03 38.6 14.4 1.88 1.86 1.80×10−3 ⑤ 砂质粉土 22.6 2.01 39.1 — — — — 注:由于第⑤层土层深度已超过污染深度,未检测相关理化性质,“—”表示未检测。
下载: 导出CSV
表 3 土壤中目标污染物含量变化
Table 3. Concentration variation of target pollutants in soil
污染物种类 编号 目标污染物浓度/(mg·kg−1) 2.0~3.5 m 3.5~5.0 m 5.0~6.5 m 6.5~8.0 m 8.0~9.5 m 实验前 实验后 实验前 实验后 实验前 实验后 实验前 实验后 实验前 实验后 氯乙烯 S01 <0.10 <0.10 0.81 <0.10 <0.10 <0.10 5.4 <0.10 <0.10 <0.10 S02 <0.10 <0.10 2.5 <0.10 <0.10 <0.10 <0.10 <0.10 0.68 <0.10 S03 <0.10 <0.10 0.33 <0.10 0.18 <0.10 0.19 <0.10 <0.10 <0.10 S04 <0.10 <0.10 <0.10 <0.10 0.74 <0.10 <0.10 <0.10 <0.10 <0.10 氯苯 S01 0.89 <0.05 1.9 <0.05 4.2 <0.05 30 <0.05 5.2 <0.05 S02 0.22 <0.05 0.65 0.18 12 0.21 2.0 0.18 7.9 0.11 S03 0.12 <0.05 4.8 0.13 0.11 <0.05 <0.05 <0.05 <0.05 <0.05 S04 0.48 <0.05 1.8 0.14 12 0.21 0.12 <0.05 <0.05 <0.05
下载: 导出CSV
表 4 地下水中目标污染物含量变化
Table 4. Concentration variation of target pollutants in groundwater
污染物种类 编号 目标污染物浓度/(μg·L−1) 实验前 实验后 氯乙烯 W01 3.79×103 417 W02 2.58×103 219 W03 2.28×103 190 顺-1,2-二氯乙烯 W01 2.62×103 198 W02 2.59×103 167 W02 3.35×103 192 苯 W01 1.06×103 37.2 W02 389 10.8 W03 18.6 4.30 氯苯 W01 2.34×104 101 W02 7.95×103 61.0 W03 734 54.0
下载: 导出CSV
-
[1] 吴嘉茵, 方战强, 薛成杰, 等. 我国有机物污染场地土壤修复技术的专利计量分析[J]. 环境工程学报, 2019, 13(8): 2015-2024. [2] 骆永明. 中国污染场地修复的研究进展、问题与展望[J]. 环境监测管理与技术, 2011, 23(3): 1-6. doi: 10.3969/j.issn.1006-2009.2011.03.002 [3] 王艳伟, 李书鹏, 康绍果, 等. 中国工业污染场地修复发展状况分析[J]. 环境工程, 2017, 35(10): 175-178. [4] 宋昕, 林娜, 殷鹏华. 中国污染场地修复现状及产业前景分析[J]. 土壤, 2015, 47(1): 1-7. doi: 10.11766/trxb201405210245 [5] YANG H, HUANG X, THOMPSON J R, et al. China’s soil pollution: Urban brownfields[J]. Science, 2014, 344(6185): 691-692. [6] 张学良, 廖鹏辉, 李群, 等. 复杂有机物污染地块原位热脱附修复技术的研究[J]. 土壤通报, 2018, 49(4): 243-250. [7] 白利平, 罗云, 刘俐, 等. 污染场地修复技术筛选方法及应用[J]. 环境科学, 2015, 36(11): 4218-4224. [8] 陶欢, 廖晓勇, 阎秀兰, 等. 应用多属性决策分析法筛选污染场地土壤修复技术[J]. 环境工程学报, 2017, 11(8): 4850-4860. doi: 10.12030/j.cjee.201707165 [9] 康绍果, 李书鹏, 范云. 污染地块原位加热处理技术研究现状与发展趋势[J]. 化工进展, 2017, 36(7): 2621-2631. [10] MYERS K F, KARN R A, ENG D Y, et al. In situ thermal desorption of VOCs in vadose zone soils[J]. Field Analytical Chemistry & Technology, 2015, 2(3): 163-171. [11] KUNKEL A M, SEIBERT J J, ELLIOTT L J, et al. Remediation of elemental mercury using in situ thermal desorption (ISTD)[J]. Environmental Science & Technology, 2006, 40(7): 2384-2389. [12] WANG N, PENG S, CHEN J J. Steam and air co-injection in removing TCE in 2D-sand box[J]. Environmental Science, 2014, 35(7): 2785. [13] BETZ C, FARBERL A, GREEN C M, et al. Removing volatile and semi-volatile contaminants from the unsaturated zone by injection of a steam/air-mixture[C]//University of Edinburgh. Contaminated Soil’98. Proceedings of the Sixth International FZK/TNO Conference on Contaminated Soil, Edinburgh, 1998: 575-584. [14] OBERLE D, CROWNOVER E, KLUGER M. In situ remediation of 1, 4-dioxane using electrical resistance heating[J]. Remediation Journal, 2015, 25(2): 35-42. doi: 10.1002/rem.2015.25.issue-2 [15] BEYKE G, FLEMING D. In situ thermal remediation of DNAPL and LNAPL using electrical resistance heating[J]. Remediation Journal, 2010, 15(3): 5-22. [16] IBEN I E T, EDELSTEIN W A, SHELDON R B, et al. Thermal blanket for in-situ remediation of surficial contamination: A pilot test[J]. Environmental Science & Technology, 1996, 30(11): 3144-3154. [17] BAKER R S, LACHANCE J, HERON G. In-pile thermal desorption of PAHs, PCBs and dioxins/furans in soil and sediment[J]. Land Contamination & Reclamation, 2006, 14(2): 620-624. [18] HERON G, LACHANCE J, BAKER R. Removal of PCE DNAPL from tight clays using in situ thermal desorption[J]. Groundwater Monitoring & Remediation, 2013, 33(4): 31-43. [19] HERON G, PARKER K, FOURNIER S, et al. World’s largest in situ thermal desorption project: Challenges and solutions[J]. Ground Water Monitoring & Remediation, 2015, 35(3): 89-100. [20] 中华人民共和国环境保护部. 土壤和沉积物挥发性有机物的测定吹扫捕集/气相色谱-质谱法: HJ 605-2011[S]. 北京: 中国环境科学出版社, 2011. [21] 中华人民共和国卫生部, 中国国家标准化管理委员会. 生活饮用水标准检验方法有机物指标: GB/T 5750.8-2006[S]. 北京: 中国标准出版社, 2006. [22] 北京市质量技术监督局. 场地土壤环境风险评价筛选值: DB11/T 811-2011[S]. 北京, 2011. [23] 北京市质量技术监督局. 污染场地挥发性有机物调查与风险评估技术导则: DB11/T 1278-2015[S]. 北京, 2015. [24] HERON G, PARKER K, GALLIGAN J, et al. Thermal treatment of eight CVOC source zones to near nondetect concentrations[J]. Ground Water Monitoring and Remediation, 2009, 29(3): 56-65. doi: 10.1111/gwmr.2009.29.issue-3 [25] 赵涛, 马刚平, 周宇, 等. 多环芳烃类污染土壤热脱附修复技术应用研究[J]. 环境工程, 2017, 35(11): 178-181. [26] 王瑛, 李扬, 黄启飞, 等. 温度和停留时间对DDT污染土壤热脱附效果的影响[J]. 环境工程, 2012, 30(1): 116-120. [27] HERON G, VAN ZUTPHEN M, CHRISTENSEN T H, et al. Soil heating for enhanced remediation of chlorinated solvents: A laboratory study on resistive heating and vapor extraction in a silty, low-permeable soil contaminated with trichloroethylene[J]. Environmental Science & Technology, 1998, 32(10): 1474-1481. [28] 张攀, 高彦征, 孔火良. 污染土壤中硝基苯热脱附研究[J]. 土壤, 2012, 44(5): 801-806. doi: 10.3969/j.issn.0253-9829.2012.05.015 [29] 孙磊, 蒋新, 周健民, 等. 五氯酚污染土壤的热修复初探[J]. 土壤学报, 2004, 41(3): 462-465. doi: 10.3321/j.issn:0564-3929.2004.03.021 [30] FISCHER U, SCHULIN R, KELLER M. Experimental and numerical investigation of soil vapor extraction[J]. Water Resources Research, 1996, 32(12): 3413-3427. doi: 10.1029/95WR02668 [31] ALBERGARIA J T, DELERUE-MATOS C. Soil vapor extraction in sandy soils: Influence of airflow rate[J]. Chemosphere, 2008, 73(9): 1557-1561. doi: 10.1016/j.chemosphere.2008.07.080 [32] 高国龙, 蒋建国, 李梦露. 有机物污染土壤热脱附技术研究与应用[J]. 环境工程, 2012, 30(1): 128-131. [33] 王奕文, 马福俊, 张倩, 等. 热脱附尾气处理技术研究进展[J]. 环境工程技术学报, 2017, 7(1): 52-58. doi: 10.3969/j.issn.1674-991X.2017.01.008 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5000
- HTML全文浏览数: 5000
- PDF下载数: 207
- 施引文献: 0
