


湿式空气氧化法处理TNT红水的影响因素
Research on influencing factors of TNT wastewater treatment by wet air oxidation
-
摘要: 使用湿式空气氧化技术与生物技术联合处理TNT精制阶段所产生的红水,研究了湿式空气氧化过程中反应温度、反应时间、初始压强和pH值对流出物的COD值与流出物可生化性的影响规律。研究结果表明,随着反应温度和反应初始压强的升高、反应时间的延长、催化剂投加量的增加和反应pH值的降低,出水COD值均有所下降。在温度为300℃、初始压强为14 MPa、反应时间为2 h、pH值为5.3、铁盐催化剂添加量为2 g的条件下,出水COD值为498 mg/L,COD去除率可达到99.27%。在温度为300℃、初始压强为11 MPAbstract: The wet air oxidation integrated with bio-treatment processes was used to treat the TNT redwater from the TNT purifying stage. WAO experiments were carried out under different temperatures,initial pressures,pH and reaction times,in order to optimize reaction conditions for the COD and biodegradability of the effluent. The results show that,the COD removal rate significantly increased with higher temperature,longer reaction time,higher initial pressure and lower pH. The COD of effluent is 498 mg/L and the removal of COD can achieve 99.27% when the reaction temperature,initial pressure,reaction time,pH,and quantity of ferric salt are 300℃,14 MPa,2 h,5.3,and 2 g,respectively. When the temperature,initial pressure,reaction time,pH,and quantity of ferric salt are chosen to be 300℃,11 MPa,1 h,3,and 1 g,respectively,the BOD5/COD of effluent is 0.301,indicating that the biodegradability of the effluent is improved. According to the biological experiment,the WAO effluent of the TNT redwater treated with the reaction temperature higher than 300℃ is available for the secondary treatment using the biological method.
-
低温等离子体(non-thermal plasma, NTP)具有反应条件温和(常温常压)、适应性广、反应快速等优点,通过产生大量活性物种(O、·OH、O3等)将VOCs降解,受到了广泛关注,然而高的能耗及大量副产物生成限制了该技术工业化应用. 为了克服活性物种寿命短而导致的NTP降解VOCs效率不高、副产物生成的缺点,近年来,研究者开发了多种具有多孔结构的催化剂与NTP协同降解VOCs,通过延长污染物在放电区的停留时间,有效利用副产物O3产生原子氧、过氧自由基等,达到减少副产物的产生、提高能量密度、降低能耗、提高碳平衡等效果[1-7]. 已经研究的与NTP联合的催化剂包括铁电体材料、半导体催化剂、贵金属催化剂以及分子筛[8-11].
金属有机骨架(metal organic frameworks, MOFs)是一种由有机配体和金属离子或金属簇组合成的多孔催化吸附材料,由于其具有可调规整的孔道、大的比表面积和高的孔隙率,应用前景广泛. MOFs材料在获得一定能量时,由于轨道离域而呈现出半导体的特性[12],介质阻挡放电(dielectric barrier discharge, DBD)等离子体中高能电子的能量可达1—20 eV,可以提供足够的能量活化MOFs产生新的活性自由基(电子-空穴对),具有吸“拟光催化过程”和催化效果,从而促进污染物的降解[13];另外,MOFs材料高的比表面积使其具有优异的吸附性能,可有效延长VOCs在反应区的停留时间,提高处理效率,逐渐应用在等离子体和气体吸附中. 例如,Wang等[14]通过水热法制备了MIL-101(Cr)并用于吸附苯表现出良好的吸附性能;Bahri等[15]采用等离子体协同MIL-101、MIL-53材料降解甲苯,发现MOFs材料的加入可以提高甲苯的降解效果、降低副产物O3的生成. 因此,研制适合DBD体系的高性能MOFs催化剂,提高两者的协同作用效果,对于推动该技术工业化应用具有重要的意义.
MOF-74由二价过渡金属(硝酸盐、醋酸盐)和配体2,5-二羟基-对苯二甲酸合成(DOT),具有一维六角形孔洞结构和高密度“开放”金属位点,具有高比表面积、有机配体和金属离子可调性剂以及很好的稳定性,具有优异的电化学性能及光催化特性,在吸附分离、光电催化产氢、化学传感、烟气脱硝等领域受到广泛关注[16-18]. 例如,Chen等[16]采用微波辅助法合成的Ni-MOF-74对CO2吸附性能好:Feng等[8]将MOF-74(Mn-Co-Ni)催化剂与NTP协同应用于甲苯降解,相同条件下甲苯去除率提高了42.9%,并有效控制了副产物O3生成. 目前,将MOF-74材料与DBD协同降解VOCs还鲜有报道[8],催化剂引入DBD后在等离子体内较低温度下O2是否参与吸附活化为O-和O2-等参与催化反应的机理还不明晰.
本研究采用溶剂热法合成制备了Mn基MOF-74(Mn-MOF-74)材料,并通过改变配体为1, 4-苯二甲酸(TPA)制备了Mn-TPA-DMF多孔材料,将两种催化剂引入DBD等离子体降解甲苯气体. 采用XRD、FTIR、SEM、BET和XPS等表征技术对催化剂的结构进行分析,对比了两种Mn基MOFs材料加入DBD后甲苯的降解效果、副产物的形成,推测了DBD催化降解甲苯的反应机理.
1. 实验部分(Experimental section)
1.1 材料
四水合硝酸锰(Mn(NO3)2·4H2O)、2,5-二羟基对苯二甲酸、1,4-苯二甲酸(TPA)、N,N-二甲基甲酰胺(DMF)、乙醇和丙醇等,所有试剂均为分析纯,不需经进一步纯化可以直接使用.
1.2 催化剂的制备
采用溶剂热合成法制备催化剂. 准确称取2.84 mL Mn(NO3)2·4H2O和0.66 g DOT,溶解在15:1:1(V:V:V)的DMF-乙醇-水混合物中,反应混合物超声处理5 min,然后转移到高压釜中,放置在120 ℃热空气烘箱内24 h. 然后取出高压釜在室温下冷却,将获得的棕色晶体用乙醇洗涤后去离子水洗涤多次,然后将样品置于60 ℃烘箱中干燥,制备Mn-MOF-74催化剂.
另外,称取0.92 g Mn(NO3)2·4H2O 和0.90 g TPA,同时溶解在50 mL DMF中,超声处理10 min后,将所得溶液转移到高压釜中,在120 ℃ 的热空气烘箱中保持24 h. 将反应混合物冷却至室温后,得到浅白色结晶化合物,用DMF、甲醇和水洗涤多次,获得的产物在130 ℃下真空干燥,制备Mn-TPA-DMF催化剂.
1.3 催化剂的表征
使用X射线衍射仪(XRD,PANalytical,荷兰),获得制备的催化剂的晶体结构. 催化剂样品的形态通过来扫描电子显微镜(SEM,ZESS,德国的Gemini 300)表征,相应的元素分布通过能量色散X射线光谱仪(EDS)分析系统获得. 使用氮气吸附-脱附实验测定样品的孔隙信息,BET和BJH方程中的吸附数据计算催化剂的比表面积、孔径分布和孔体积. X射线光电子能谱(XPS)通过具有Al-Kα辐射源的Thermo Scientific K α谱仪测量,用C1s为284.8 eV的数据校准测试的元素的结合能数据.
1.4 催化剂的性能评价
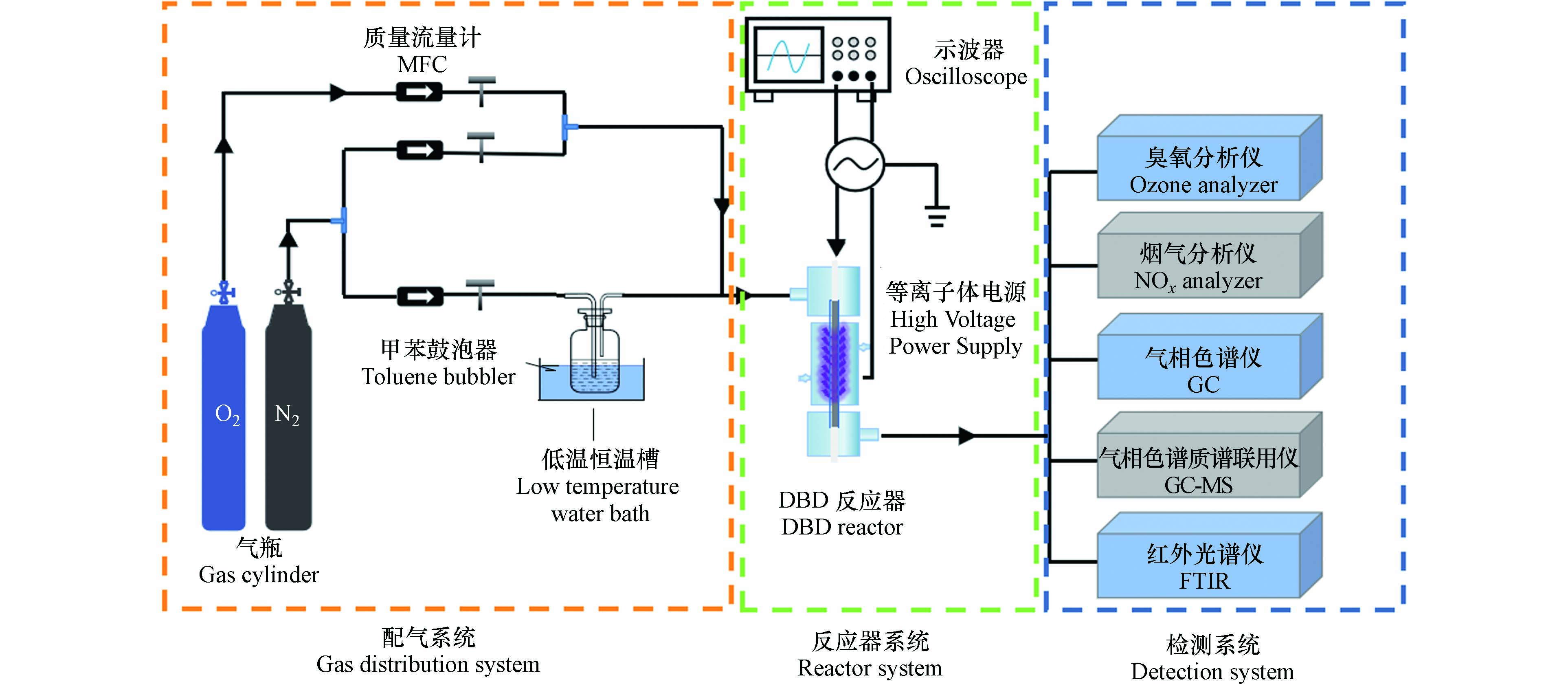
DBD催化降解甲苯气体的工艺流程如图1所示. 该系统由三部分组成:甲苯气体发生器系统、DBD等离子体反应器系统和废气检测系统. 氮气通过装有液体甲苯的气体鼓泡器发生器来生成一定浓度的含甲苯气体. 使用3个质量流量控制器(MFC)分别控制氮气、氧气的流量,得到所需的甲苯初始浓度. 实验时,甲苯初始浓度1368 mg·m−3,气体流量1 L·min−1,氧含量4%(体积分数).
DBD反应器为50 mm放电长度、3 mm放电间隙的同轴结构(图1),采用石英管作为介质层,使用循环水作为接地电极,内管放置1根不锈钢棒作为高压电极. 高压电源为南京苏曼等离子体科技有限公司生产的CTP-2000 K实验电源,输出频率范围为5—20 kHz. DBD催化实验时,将0.3 g催化剂(40—60目)与1.5 g石英砂混合,装入DBD反应器中. 使用高压探头和电流探头测定电压和电流,并通过数字示波器(Tektronix TBS 1000 C)记录放电波形,通过利萨如(V-Q Lissajous)方法计算DBD放电功率P(方程1),借助方程(2)计算能量密度(SED). 本文气体流量(Q)为1 L·min−1,通过调节放电电压来实现SED的变化.
stringUtils.convertMath(!{formula.content}) (1) stringUtils.convertMath(!{formula.content}) (2) 式中,f为放电频率(9.6 kHz),C为电容(0.47 μF),A为利萨如图面积,P为放电功率(W),Q为气体流量
(L⋅min−1) 降解前后的甲苯、CO和CO2的浓度均使用气相色谱仪(GC9790型,浙江福立分析仪器有限公司)在线测定,其中CO和CO2通过Ni转化炉转化为甲烷后由FID检测器测定. O3和NOx(NO2和NO)的浓度分别用O3分析仪(2B Model 106-M,美国)和烟气分析仪(MGA 6-plus,德国)测定. 采用气相色谱-质谱联用仪(GC-MS)对尾气中的有机副产物进行了分析. 尾气用正己烷吸收20 min,再超声1 h. 样品进样至GC-MS中,通过自动进样器进行分析. 甲苯降解率(η)、矿化度(Md)、CO2选择性(
SCO2 stringUtils.convertMath(!{formula.content}) (3) stringUtils.convertMath(!{formula.content}) (4) stringUtils.convertMath(!{formula.content}) (5) stringUtils.convertMath(!{formula.content}) (6) 式中,Cin和Cout分别为甲苯进口和出口的浓度(mg·m−3),M为甲苯的分子量(g·mol−1),[CO2]和[CO]分别为出口CO2和CO的浓度(mol·m−3),Ey为反应器的能量效率(g·kWh−1).
2. 结果与讨论(Results and discussion)
2.1 催化剂的表征分析
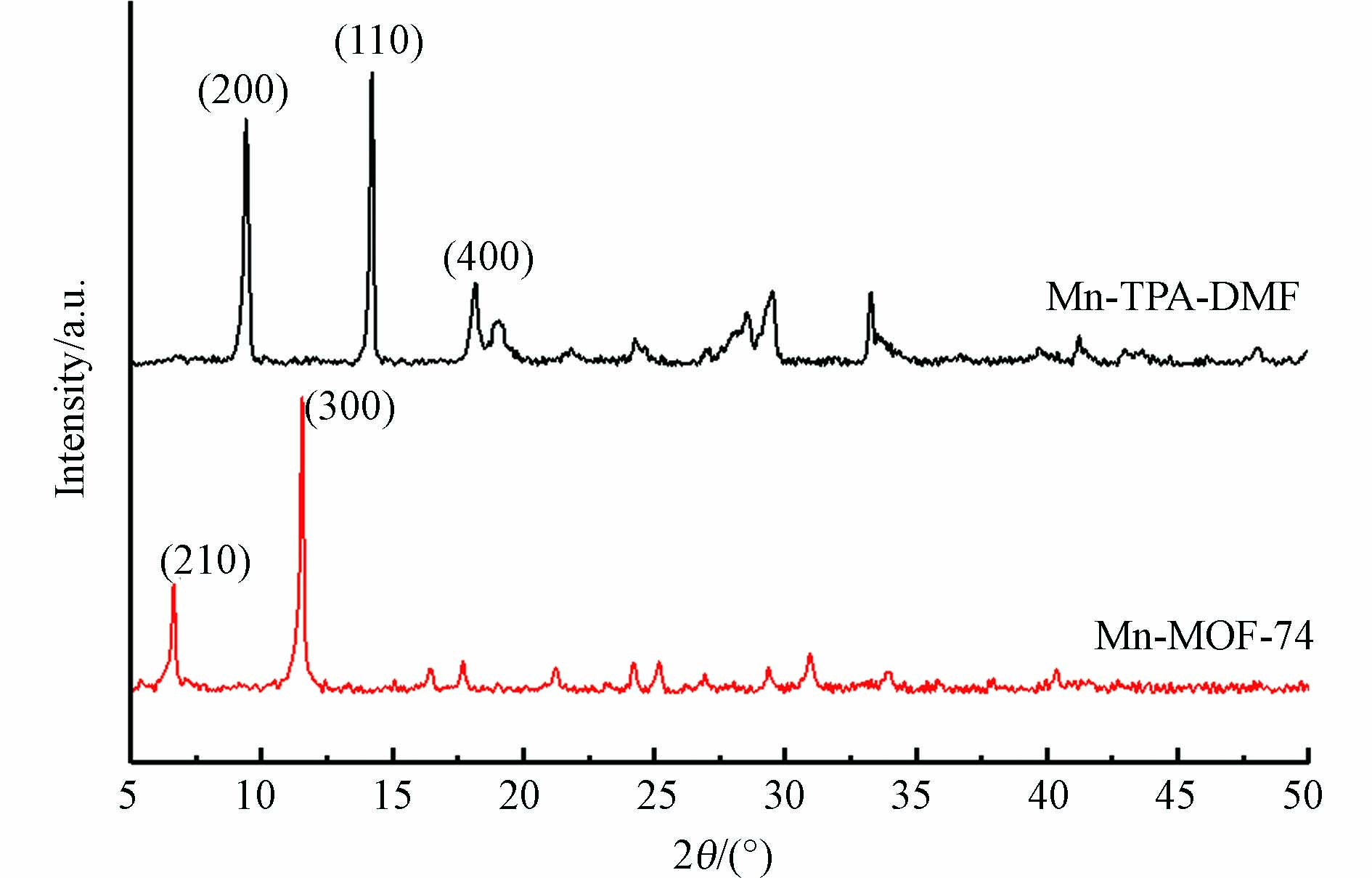
如图2所示,为了确定材料的晶体结构,对制备的样品进行了XRD表征. Mn-MOF-74材料在2θ=6.8°和11.8°附近有两个很强的衍射峰,分别对应于Mn-MOF-74的(210)和(300)晶面[18],其他位置的衍射峰都相对较弱. Mn-TPA-DMF材料在2θ=9.41°、14.22°、18.17°附近的3个衍射峰分别对应材料的(200)、(110)和(400)晶面[19]. 本研究中合成的Mn-MOF-74和Mn-TPA-DMF的XRD谱图的峰位置、强度及顺序与先前文献报道的相一致[20-21],表明材料合成成功.
图3(a)为Mn-MOF-74使用前后的FTIR图. 3422 cm−1附近的特征峰为OH或H2O的伸缩振动吸收峰,表明水或其他溶剂分子存在于Mn-MOF-74通道中[22]. 1567 cm−1和1401 cm−1附近的峰为MOF-74中COO-的不对称和对称伸缩振动峰[23],1240 cm−1附近的峰为C—N键振动吸收峰,说明溶剂DMF吸附在催化剂表面或者孔道内[24],1196 cm−1处的特征峰为C—O单键伸缩振动峰,886 cm−1和806 cm−1处的特征峰为苯环C—H键面内和面外摇摆振动峰,574 cm−1处的特征峰归属于Mn—O伸缩振动峰[25],表明Mn-MOF-74的成功合成.
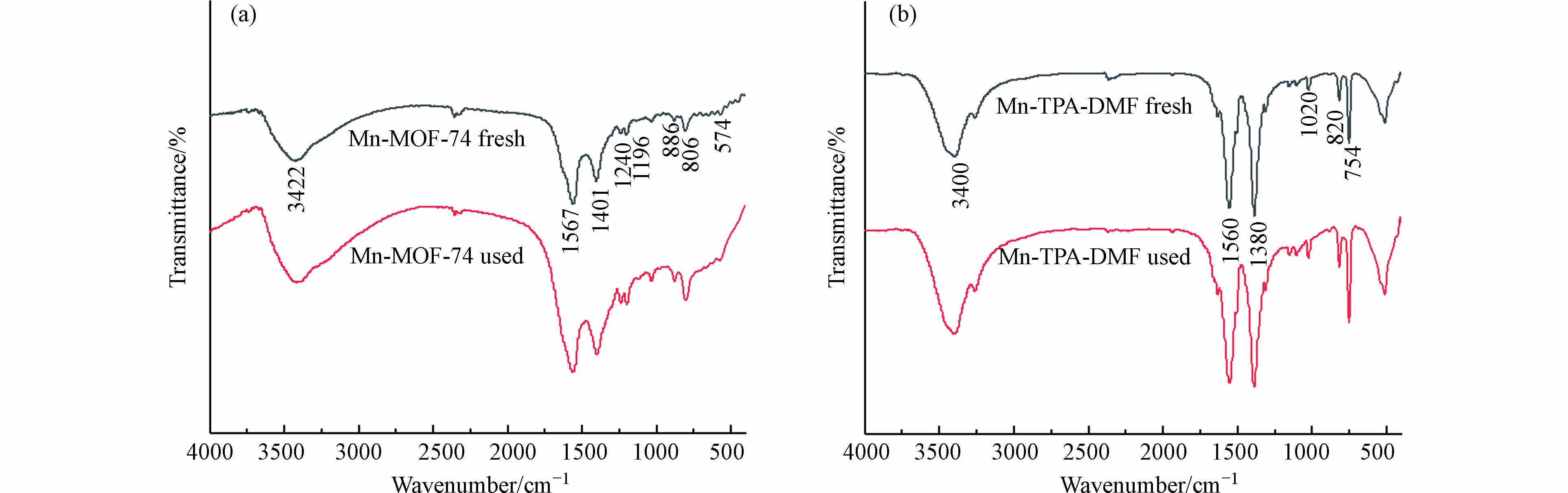 图 3 Mn-MOF-74和Mn-TPA-DMF反应前后的红外图谱Figure 3. FTIR spectra of Mn-MOF-74 and Mn-TPA-DMF before and after catalysis reaction
图 3 Mn-MOF-74和Mn-TPA-DMF反应前后的红外图谱Figure 3. FTIR spectra of Mn-MOF-74 and Mn-TPA-DMF before and after catalysis reaction图3(b)为Mn-TPA-DMF使用前后(fresh 和 used表示)的FTIR图,具有与Mn-MOF-74相似的特征峰,但特征峰向低频(波数)轻微偏移,这可能是由于Mn与2,5-二羟基对苯二甲酸和1,4-苯二甲酸两种不同的有机配体结合引起的. 此外,通过反应前后的红外图谱对比可知,Mn-MOF-74和Mn-TPA-DMF在反应前后没有发生结构上的改变,表明其具有良好的稳定性,适合实际工业应用.
本研究通过SEM分析了催化剂的形貌结构,如图4所示. Mn-MOF-74和Mn-TPA-DMF均呈现出不规则的二维片状结构,表面上有细小的块状颗粒附着. 此外,催化剂的元素映射图表明了Mn、O、C元素的存在,且呈均匀分布.
 图 4 (a—c)Mn-MOF-74和(d—f)Mn-TPA-DMF的SEM和元素映射Figure 4. SEM image and element mapping of (a—c) Mn-MOF-74 and (d—f) Mn-TPA-DMF
图 4 (a—c)Mn-MOF-74和(d—f)Mn-TPA-DMF的SEM和元素映射Figure 4. SEM image and element mapping of (a—c) Mn-MOF-74 and (d—f) Mn-TPA-DMF图5为催化剂的N2吸附-脱附曲线,根据曲线信息计算出的BET比表面积和孔隙体积如表1所示. Mn-MOF-74和Mn-TPA-DMF的N2吸附-脱附曲线均为IV型,并在高的压力下表现出脱附滞后,形成回滞环,说明两种材料都是含有中孔(2—50 nm)结构的材料. 与Mn-TPA-DMF相比(比表面积和孔体积分别为29.123 m2·g−1和0.02421 cm3·g−1),Mn-MOF-74呈现更高的比表面积及孔体积,分别为83.368 m2·g−1和0.17630 cm3·g−1,本文合成的Mn-MOF-74的比表面积高于相似的方法合成的Mg-MOF-74催化剂(18.04 m2·g−1)[26],可以提供更多的表面活性位点.
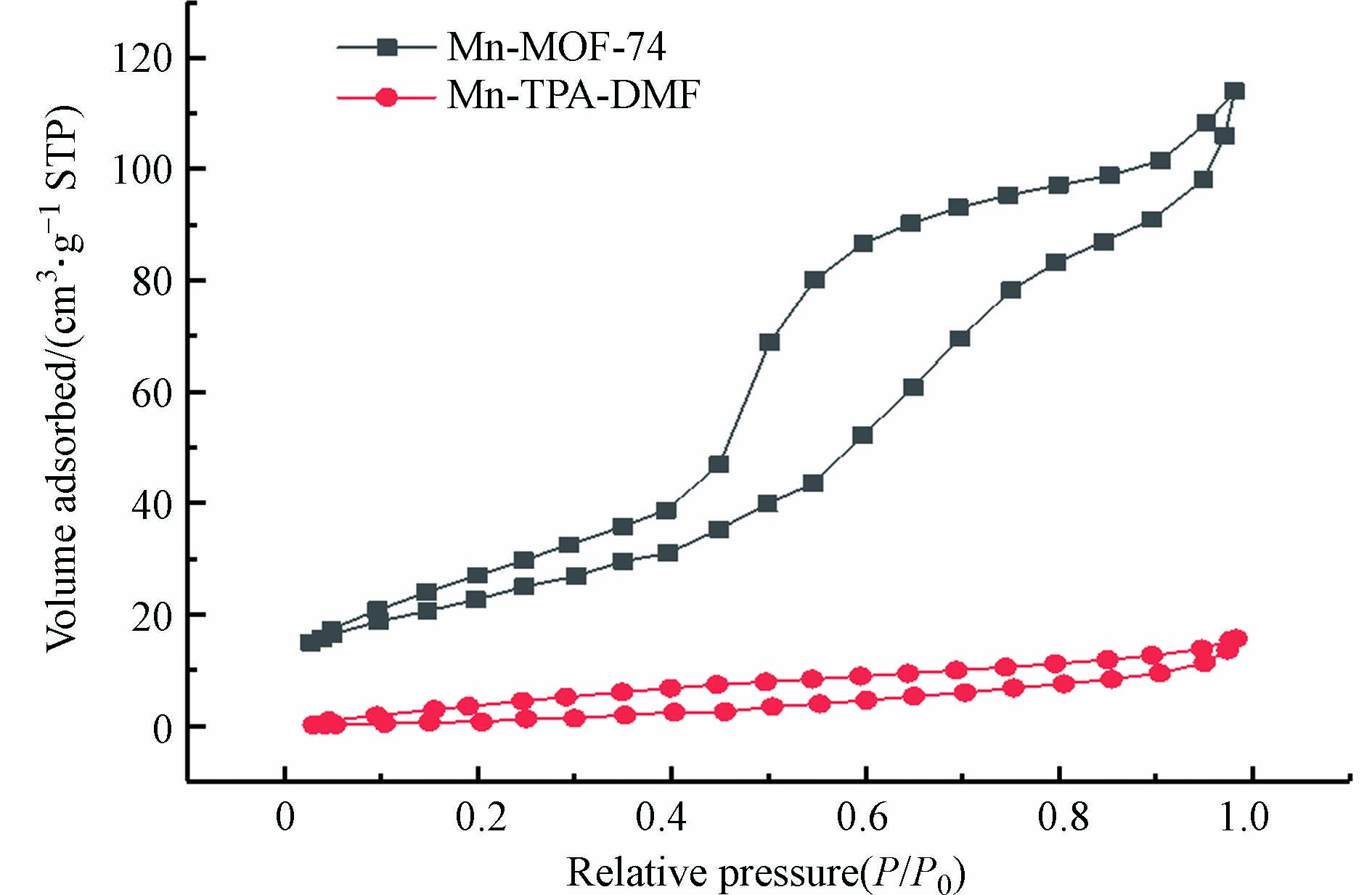 图 5 Mn-MOF-74和Mn-TPA-DMF的氮气吸附-脱附曲线Figure 5. Nitrogen adsorption and desorption curves of Mn-MOF-74 and Mn-TPA-DMF表 1 Mn-MOF-74和Mn-TPA-DMF的比表面积和孔容分析Table 1. BET surface area and pore volume analysis of Mn-MOF-74 and Mn-TPA-DMF
图 5 Mn-MOF-74和Mn-TPA-DMF的氮气吸附-脱附曲线Figure 5. Nitrogen adsorption and desorption curves of Mn-MOF-74 and Mn-TPA-DMF表 1 Mn-MOF-74和Mn-TPA-DMF的比表面积和孔容分析Table 1. BET surface area and pore volume analysis of Mn-MOF-74 and Mn-TPA-DMF催化剂 Catalyst 比表面积/(m2·g−1) Specific surface area 孔容/(cm3·g−1) Pore volume 平均孔径/nm Average diameter Mn-MOF-74 83.368 0.17630 8.460 Mn-TPA-DMF 29.123 0.02421 3.326 | Show Table DownLoad:
CSV
DownLoad:
CSV
图6所示为Mn-MOF-74和Mn-TPA-DMF在甲苯催化反应前后的XPS分析. 图6(a)的全谱分析中显示有Mn、C、N和O元素的存在,表明MOFs材料的成功合成,N元素的存在,说明DMF分子也参与了Mn-MOF-74的配位[27]. 图6(b)为Mn-MOF-74的Mn 2p XPS光谱,位于640.7 eV和652.0 eV的峰分别归因于Mn 2p3/2和Mn 2p1/2的特征峰. 将Mn 2p3/2进行分峰拟合,在结合能为639.3、640.7 、644.7 eV处的峰分别归属于Mn2+、Mn3+和Mn4+[10, 28]. 以上结果表明,Mn以多价态存在,反应过程中可以相互转换,有利于催化反应的进行. 气相中的O2、O3和电子能够借助Mn2+、Mn3+、Mn4+之间的表面转换,被吸附和转移到催化剂表面的甲苯和中间产物上,产生活性粒子(OH、·O等),从而将甲苯进一步矿化为CO2. 如表2所示,通过Mn 2p光谱的定量分析可知,两种Mn基催化剂反应前Mn3+占比都较高,Mn3+的存在能够增加氧空穴,有利于将O2和O3激活为活性氧,进而参与反应;反应后Mn3+占比变化不大,说明Mn3+一直参与甲苯的氧化反应,比较稳定. Mn3+略有下降的原因可能为反应后氧被激活,消耗了部分Mn3+. 在多价态的锰中,高价态的锰可以氧化甲苯,低价态的锰可以通过等离子体催化过程中臭氧的活化而被再氧化. 此外,位于656.0 eV附近的峰归因于Mn 2p特征峰的卫星峰. 反应后Mn的特征峰的强度变低且位置向高结合能的位置轻微偏移,这可能是由于催化剂在放电过程中参与甲苯氧化反应引起的.
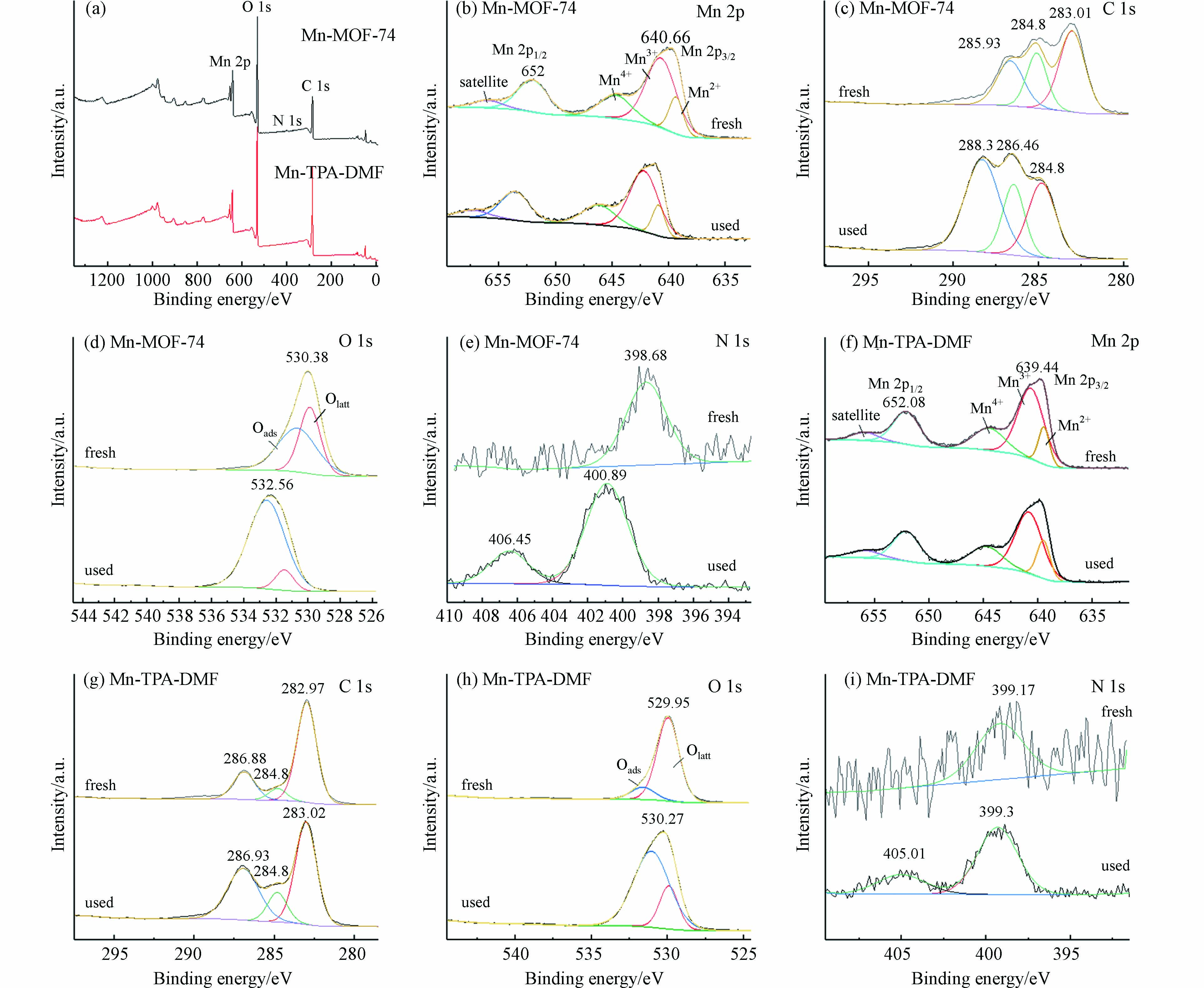 图 6 Mn-MOF-74和Mn-TPA-DMF催化反应前后的XPS分析Figure 6. XPS analysis of Mn-MOF-74 and Mn-TPA-DMF before and after catalytic reaction(a)全谱分析,(b-e)Mn-MOF-74的Mn 2p, C 1s, O 1s, N 1s XPS光谱,(f-i)Mn-TPA-DMF的Mn 2p, C 1s, O 1s, N 1s XPS光谱(a) Full spectrum analysis, and (b-e) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-Muf-74, and(f-i) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-TPA-DMF表 2 Mn-MOF-74和Mn-TPA-DMF各元素价态的组成Table 2. Valence composition of the elements of Mn-MOF-74 and Mn-TPA-DMF
图 6 Mn-MOF-74和Mn-TPA-DMF催化反应前后的XPS分析Figure 6. XPS analysis of Mn-MOF-74 and Mn-TPA-DMF before and after catalytic reaction(a)全谱分析,(b-e)Mn-MOF-74的Mn 2p, C 1s, O 1s, N 1s XPS光谱,(f-i)Mn-TPA-DMF的Mn 2p, C 1s, O 1s, N 1s XPS光谱(a) Full spectrum analysis, and (b-e) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-Muf-74, and(f-i) Mn 2p, C 1s, O 1s, N 1s XPS spectra of Mn-TPA-DMF表 2 Mn-MOF-74和Mn-TPA-DMF各元素价态的组成Table 2. Valence composition of the elements of Mn-MOF-74 and Mn-TPA-DMF催化剂 Catalyst 表面元素比Mn3+/(∑Mnn+) Surface element ratio Mn3+/(∑Mnn+) Oads/Olatt Before After Before After Mn-MOF-74 0.63 0.62 1.23 8.60 Mn-TPA-DMF 0.62 0.60 0.16 6.28 | Show TableDownLoad:
CSV
图6(c)为Mn-MOF-74的C 1s XPS谱,在283.01 eV、285.1 eV和286.65 eV处分峰拟合为3个峰,分别对应于C—C,C—O 和O—C=O[10],反应后特征峰的位置向高结合能的位置轻微偏移,这可能是由于等离子放电或与甲苯及中间产物的结合.
图6(d)为Mn-MOF-74的O 1s XPS光谱,通过分峰拟合可以分为两个特征峰. 位于约529.9 eV处的峰可归因于材料中的晶格氧(O2-)(标记为Olatt),位于约530.7 eV处的峰则归因于表面吸附氧[10, 29]. O2在催化剂上吸附后通常会经过以下变化:吸附氧Oads→O2-(ads)→O-(ads)→Olatt. 一般认为,源自氧空位的Oads比Olatt具有更高的迁移率,并且在VOCs催化氧化过程中比Olatt更有效[30, 31]. 通过表2可知,反应前吸附氧(Oads)的含量比较少,反应后Oads含量明显升高,说明反应过程中催化剂能活化氧物种产生大量Oads,使Oads参与了甲苯的氧化反应. 其中,Mn-MOF-74催化剂显示出较高的Oads/Olatt摩尔比,这表明在Mn-MOF-74催化剂表面上有较高含量的Oads,即表面氧空位密度越高,O2分子越容易在催化剂表面吸附和活化,催化性能越好. 图6(d)表明,发生DBD催化反应后Oads的占比显著增加,表明Mn-MOF-74可以活化氧物种参与甲苯的氧化反应.
图6(e)为Mn-MOF-74的N 1s XPS光谱,在398.7 eV 的峰归属于N(C)3,是由残留DMF引起的. 反应后406.5 eV处出现了1个特征峰,是由等离子体电荷效应引起,说明有少量NOx副产物产生[32].
图6(f-i)为Mn-TPA-DMF的Mn 2p, C 1s, O 1s, N 1s XPS光谱,均呈现和Mn-MOF-74相似的特征峰,从图中也可以分析得出,Mn和表面活性氧物种参与了甲苯的氧化反应.
2.2 DBD催化降解甲苯的效果分析
如图7(a)显示了甲苯在不同DBD催化体系中的降解行为. 由图7可知,甲苯的降解效率随SED的增加也就是放电电压的升高而增加,DBD-催化体系比单独DBD体系具有更高的甲苯去除效率. SED为605.97 J·L−1时,DBD、DBD+Mn-TPA-DMF和DBD+Mn-MOF-74的3个体系中甲苯去除率分别为74.67%、91.23%和94.91%. 能量密度越高,产生的电流脉冲的数量和强度越大,形成的微放电越多,从而通过高能电子和大量活性物质改善了VOCs的降解效果[33-34]. 换句话说,SED升高,增大了高能电子的绝对数量及与气体分子的碰撞几率,导致体系中活性粒子数增加,同时可能诱导催化剂表面发生反应,改变催化剂的特性和微孔放电特性. 此外,两种催化剂相比,Mn-MOF-74比Mn-TPA-DMF表现出更好的催化协同性能. 从表2中可以看出,Mn-TPA-DMF和Mn-MOF-74的金属元素的价态及含量差别不大,但与Mn-TPA-DMF相比,Mn-MOF-74呈现更高的比表面积及孔体积. 结合催化活性测试结果即Mn-MOF-74与等离子体结合时甲苯降解效果更好,说明比表面积和孔体积是影响催化剂活性的主要因素,这与以往的文献报道一致[35].
 图 7 DBD催化体系中不同SED甲苯的去除效率和能量效率Figure 7. (a) Toluene removal efficiency and (b) energy efficiency under different SED in DBD-catalytic systems
图 7 DBD催化体系中不同SED甲苯的去除效率和能量效率Figure 7. (a) Toluene removal efficiency and (b) energy efficiency under different SED in DBD-catalytic systems能量效率可以定义为单位电耗降解甲苯的量,可以说明等离子体催化过程的能耗. 图7(b)可以观察到,能量效率都随着能量密度的升高而降低,说明从经济的角度能量密度越小越好. 与单独DBD相比,相同的SED下DBD催化能量效率有所提高,说明能耗降低了. 当SED为263.68 J·L−1时,单独使用DBD系统的能量效率值为7.43 g·kWh−1,DBD+Mn-MOF-74的能量效率值为11.22 g·kWh−1,说明等离子体与催化剂之间可能存在协同效应. 因此,DBD与催化剂的组合可以提高能量效率,有效降低甲苯降解的能耗,提高DBD系统的经济性.
为了进一步评价DBD-催化体系的性能,进一步研究了CO/CO2浓度、CO2选择性和甲苯矿化度,如图8所示. 如图8(a)所示,在所有情况下,COx浓度都随SED的增加而升高. 此外,DBD催化时生成的CO和CO2的浓度与单独DBD降解时浓度相比显著增加. 当SED为605.87 J·L−1时,DBD+Mn-MOF-74中CO2和CO的浓度分别为1375 mg·m−3和1019.13 mg·m−3,DBD单独体系中为785.71 mg·m−3和712.75 mg·m−3,CO2和CO生成量为单独DBD的175%和143%.
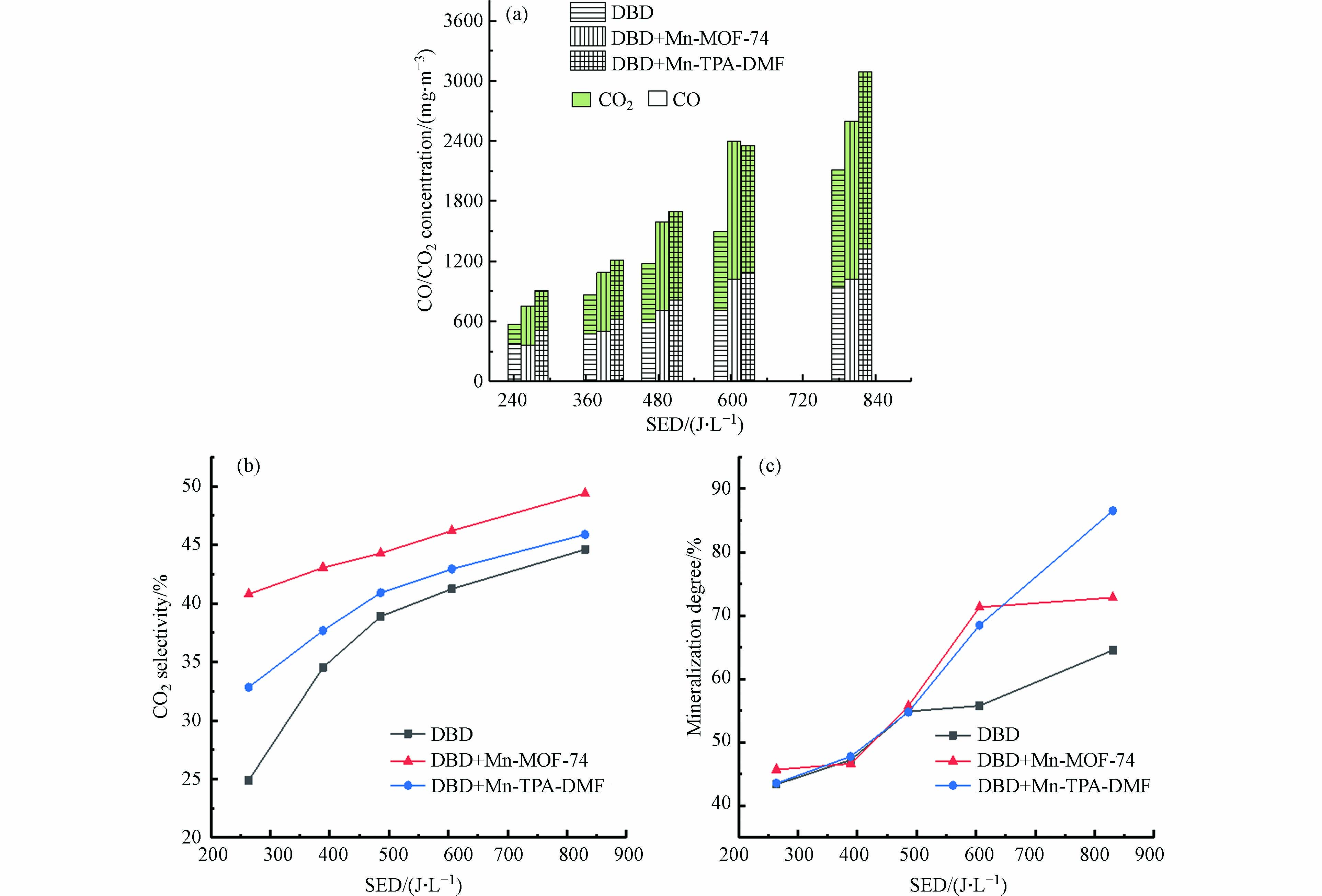 图 8 DBD催化体系(a) CO和CO2浓度(b) CO2选择性和(c)矿化度随SED的变化Figure 8. Changes in (a) CO and CO2 concentration, (b) CO2 selectivity and (c) mineralization degree with SED under DBD catalyst systems
图 8 DBD催化体系(a) CO和CO2浓度(b) CO2选择性和(c)矿化度随SED的变化Figure 8. Changes in (a) CO and CO2 concentration, (b) CO2 selectivity and (c) mineralization degree with SED under DBD catalyst systems图8(b)显示DBD+Mn-TPA-DMF、DBD+Mn-MOF-74和单独DBD体系的CO2选择性随着能量密度的增加而增加. DBD+Mn-MOF-74体系的CO2选择性在能量密度为263.68 J·L−1时为40.79%,比单独DBD体系(24.89%)提高了15.9%.
如图8(c)所示,随着SED的增加,DBD单独体系和DBD-催化剂体系的矿化度都增加. 当能量密度为605.87 J·L−1时,DBD+Mn-TPA-DMF和DBD+Mn-MOF-74体系的矿化率分别为68.48%和71.31%,比单独DBD体系(55.78%)分别提高了12.7%和15.53%. 当SED为830.57 J·L−1时,DBD+Mn-TPA-DMF体系甲苯的矿化度达到最高值86.53%,表明DBD+Mn-TPA-DMF在较高的SED下更有利于甲苯彻底降解为CO2.
NOx和O3是DBD降解VOCs的主要副产物. 图9显示了单独DBD和DBD催化系统中副产物O3和NOx浓度. 在图9(a)中,O3浓度随着SED的增加而降低,这可能是由于O3参与了COx和NOx等其他活性物种的生成[36]. 外加Mn基MOFs催化材料后,DBD降解甲苯气体中副产物O3浓度显著下降,例如,当SED为388.75 J·L−1时,单独DBD系统中O3浓度约为390.64 mg·m−3,而在DBD+Mn-MOF-74和DBD+Mn-TPA-DMF体系中O3浓度分别降至264.64 mg·m−3和303.64 mg·m−3,O3浓度的降低可归因于O3在催化剂表面分解成O2并形成活性原子氧物质(O* )[37]. 就挥发性有机化合物氧化的反应性而言,O* 是比O3更具化学活性的物质,因此,将O3分解为原子氧的能力是等离子体催化过程中挥发性有机化合物降解的重要因素[38]. 此外,Mn2+/Mn3+和Mn4+/Mn3+的氧化还原循环也可以加速表面氧物种的释放,有利于甲苯的氧化[39]. 与Mn-TPA-DMF催化剂相比,Mn-MOF-74催化剂具有较大的比表面积,更有利于O3的吸附和分解,在催化剂表面形成更多的活性氧,提高了催化剂的去除效率和CO2选择性[40].
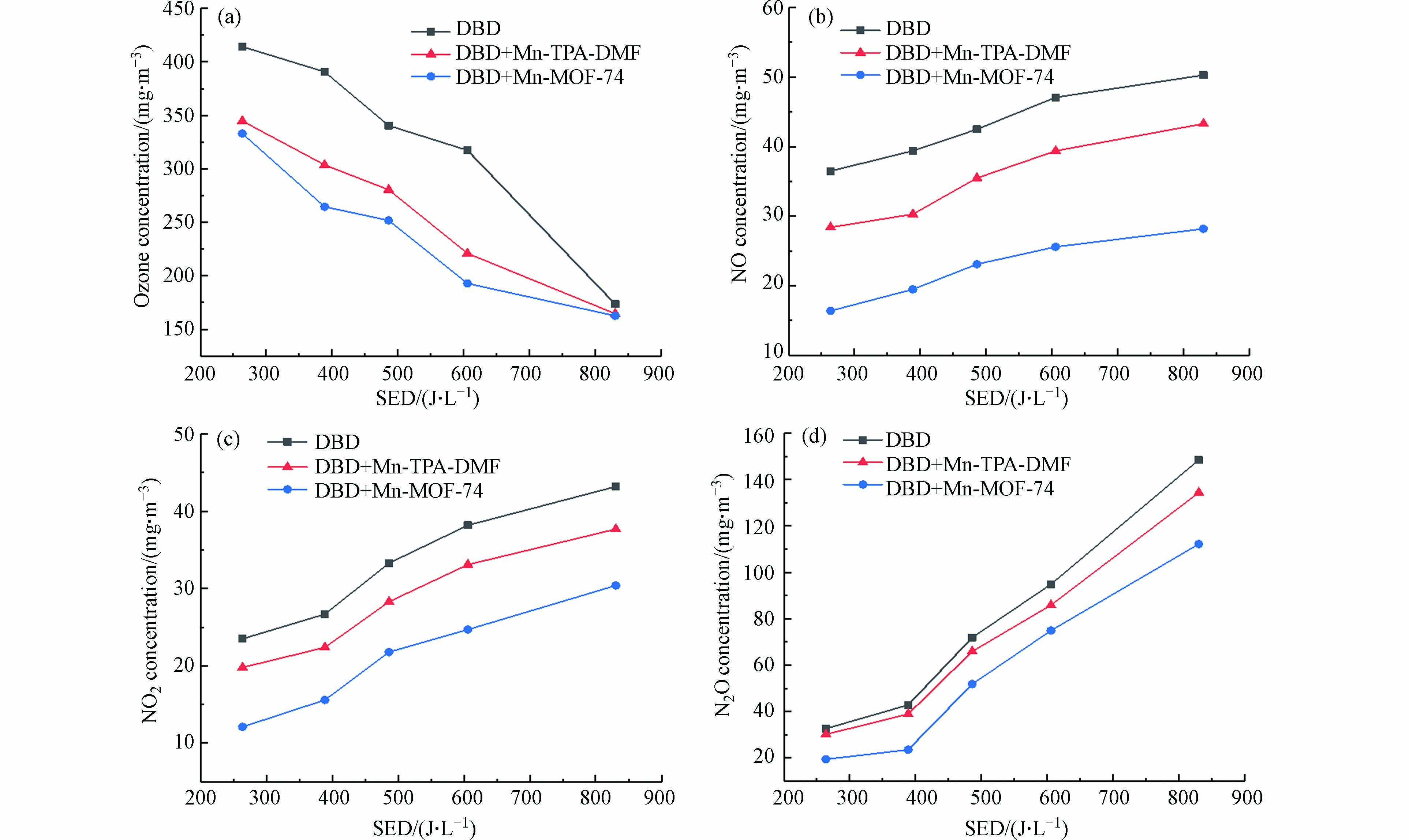 图 9 DBD、DBD+Mn-MOFs催化降解甲苯(a)O3浓度和(b)、(c)、(d) NOx浓度随能量密度的变化Figure 9. DBD and catalyst added (a) O3 concentration and (b) , (c) , (d) NOx concentration as a function of energy density
图 9 DBD、DBD+Mn-MOFs催化降解甲苯(a)O3浓度和(b)、(c)、(d) NOx浓度随能量密度的变化Figure 9. DBD and catalyst added (a) O3 concentration and (b) , (c) , (d) NOx concentration as a function of energy density采用烟气分析仪检测了DBD和DBD-催化体系中3种NOx(NO、N2O和NO2)的浓度,得到NOx生成浓度与SED的关系(图9 b-d). 如图9b-d所示,NO、NO2和N2O的浓度均随SED的增大而增大,与单独DBD系统相比,DBD-催化体系中生成的NOx浓度明显降低,且Mn-MOF-74催化剂的NOx生成量低于Mn-TPA-DMF,表明Mn-MOF-74催化剂更有利于抑制NOx的生成. 例如,当SED为263.68 J·L−1时,单独DBD系统产生的NO2浓度为26.7 mg·m−3,DBD+Mn-TPA-DMF和DBD+Mn-MOF-74体系分别降至22.4 mg·m−3和12.1 mg·m−3. 通过上述结果可知,催化剂可以提高反应的选择性,进而抑制副产物NOx等的生成. 其生成量降低的原因在于Mn基催化剂的引入有利于O3在催化剂的表面生成氧气和表面活性氧,生成的活性氧进而参与甲苯的降解反应,避免了与N2生成NOx. 此外,催化剂的加入还有利于高能电子传输,并降低NOx的解离能进而可以使部分NOx解离为N2[41- 42].
2.3 DBD/Mn-MOFs催化降解甲苯的机理分析
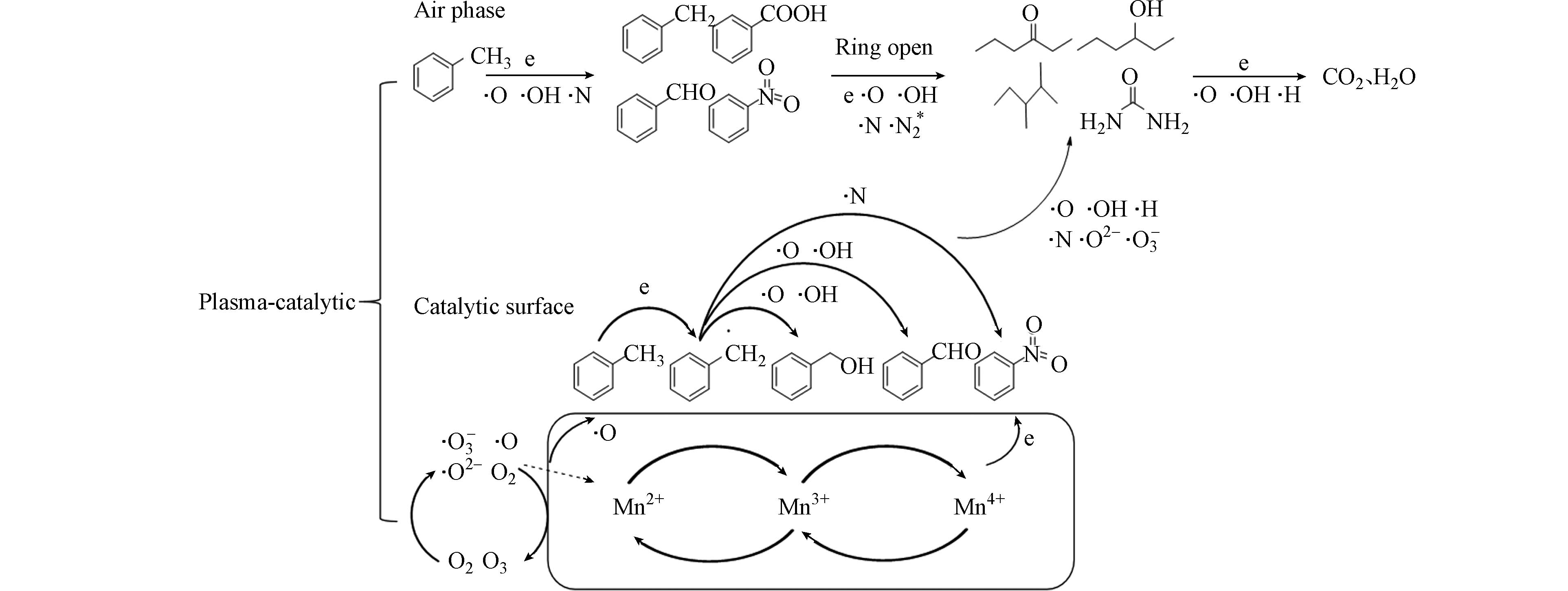
采用气相色谱-质谱联用技术(GC-MS)对DBD及DBD复合催化剂的有机副产物进行了分析. 将反应气体用正己烷吸收20 min,然后将吸收液注入GC-MS仪器进行检测. 结合降解前后材料表征结果及甲苯降解效果,推测DBD/Mn-MOFs催化降解甲苯的机理如下图:由图10可知,等离子体Mn-MOFs催化降解甲苯主要包括3个路径. 等离子体电场内具有丰富的高能电子,高能电子轰击甲苯分子导致甲苯降解是一个主要途径;高能电子与空气中的O2、N2等产生活性粒子(·OH, N, N2*, ·O, N2*, O2- ),活性粒子降解甲苯是第二条途径;再次,催化剂表面的反应取决于甲苯及中间产物的化学吸附、吸附氧的数量及Mn2+、Mn3+、Mn4+之间的转换. 气相中的O2、O3和电子能够借助Mn2+、Mn3+、Mn4+之间的表面转换,被吸附和转移到催化剂表面的甲苯和中间产物上,进一步产生活性粒子(OH、·O等),从而将甲苯进一步矿化为CO2. XPS结果显示催化反应后催化剂的Oads的占比显著增加,也进一步证实Mn基MOFs表面氧空位作为表面缺陷成为催化反应的活性位点,导致O3在催化剂表面分解产生更多的表明氧物种(O和O2),促进甲苯和中间产物的深度氧化[10,31]. 当然,Mn基MOFs催化剂的引入,也会使得等离子体中表面局部场强增大,改变了等离子体放电特性,对降解也会产生影响.
3. 结论(Conclusion)
本文制备了Mn-MOF-74和Mn-TPA-DMF两种Mn基MOFs催化材料,放置在DBD等离子体反应器对甲苯进行催化降解实验. 通过材料表征、降解效果分析等,探索了DBD催化降解机理,结论如下:
1) 与单独DBD相比,DBD与Mn基MOFs催化剂联用显著提高了甲苯的去除率、CO2选择性、矿化度和能量效率,并显著抑制了O3和NOx副产物的生成. DBD+Mn-MOF-74催化降解甲苯效果最好,当SED为605.87 J·L−1时,甲苯降解率达94.91%、CO2选择性达46.2%、矿化度达71.3%,与单独DBD相比,副产物O3和NO生成量下降到50%左右.
2) Mn-MOF-74与Mn-TPA-DMF催化剂相比,表现出更优异的催化活性,这个与Mn-MOF-74具有更高的比表面积、更多的Oads含量、更强的氧化还原性能相关.
3) XPS材料表征表明,Mn基修饰的MOFs材料中Mn作为电荷转移媒介,促进了电荷的循环转移. 通过DBD催化反应前后的催化剂的FTIR图谱可知,Mn-MOF-74和Mn-TPA-DMF在反应前后没有发生结构上的改变,说明催化剂性能稳定.
期刊类型引用(38)
1. 徐敏,吴鹏,展漫军,汪贞. 成组生物毒性测试法对复合有机污染场地修复后土壤进行生态毒性评价. 环境生态学. 2025(03): 10-16+24 .  百度学术
百度学术
2. 赵英凯. 原位热脱附技术修复农药污染场地时的尾气治理工艺探究. 上海化工. 2024(03): 21-26 . 百度学术
3. 姬大伟,张生东,张烨,王进卿,徐旭,詹明秀. 石油烃污染土壤阴燃反应特性及修复效果研究. 环境科学研究. 2024(08): 1789-1797 . 百度学术
4. 高卫国,钮国耀,邢绍文. 热脱附修复有机污染土壤节能降耗研究进展. 当代化工研究. 2023(01): 5-7 . 百度学术
5. 姜春旭,冯俊小,黄显模,黄志峰,张志涛. 基于原位热脱附技术修复污染土壤的加热井数值优化. 环境工程. 2023(03): 163-171 . 百度学术
6. 来珊珊,李雪娇,贺艳娟. 石油污染土壤热脱附修复的应用研究进展. 石油化工应用. 2023(02): 1-7 . 百度学术
7. 魏伟,李鸿炫,梁冠富,刘宇,刘丽平. 原位电加热技术修复有机磷农药污染场地的中试研究. 中国资源综合利用. 2023(06): 23-27 . 百度学术
8. 凃啸宇,王静,李晓雪. 安徽省某退役地块污染土壤修复工程实例. 工程与建设. 2023(03): 954-958 . 百度学术
9. 范婷婷,靳德成,刘鹏,王祥,赵远超,邓绍坡,张胜田,刘泽权,万金忠. 原位热脱附能效评价方法构建及应用研究. 生态与农村环境学报. 2023(09): 1213-1220 . 百度学术
10. 吴东海,韩俊楠,刘斌,袁曾路,毛竹. 污染土壤热脱附技术的碳达峰碳中和路径初探. 环境工程. 2023(S2): 701-706 . 百度学术
11. 朱煜. 原位热脱附修复技术节能降耗措施研究进展. 能源与环保. 2023(10): 186-193+200 . 百度学术
12. 王啟华,黄文涛,陈建,成德久. 地下水修复技术及止水帷幕技术在污染场地项目中的运用. 科技创新与应用. 2022(01): 178-180+183 . 百度学术
13. 徐馨宇,胡楠,范利武. 土壤原位热传导修复水-汽-热耦合输运模拟. 浙江大学学报(工学版). 2022(01): 144-151+160 . 百度学术
14. 刘立朋,徐梦奔,顾海林,詹明秀,徐旭,焦文涛,籍龙杰,田汪洋,张涛,吕韬,池作和. 回填材料对土壤原位热传导修复的影响及数值模拟. 环境工程学报. 2022(02): 555-564 . 本站查看
15. 陈俊宇. 污染场地修复中复杂有机物的原位热处理技术筛选. 皮革制作与环保科技. 2022(03): 166-168 . 百度学术
16. 杨顺美,焦文涛,刘峰,陈芒,王安宇,李烜桢,蒲生彦. 电阻加热修复佳乐麝香污染土壤的工艺优化. 环境工程学报. 2022(04): 1284-1293 . 本站查看
17. 詹明秀,刘立朋,顾海林,张涛,韦金义,徐旭,焦文涛,籍龙杰,田汪洋,池作和. 原位热修复过程中土壤内热质传递研究现状与展望. 环境工程学报. 2022(04): 1272-1283 . 本站查看
18. 籍龙杰,沈宗泽,刘鹏,迟克宇,郭丽莉,李书鹏,秦之瑞,岳勇,韩自玉,焦文涛. 有机污染场地原位热脱附工程尾水尾气的处理技术进展. 环境工程学报. 2022(05): 1407-1415 . 本站查看
19. 梁晶,伍海兵,张浪. 上海市搬迁地土壤团聚体的分布特征. 水土保持通报. 2022(02): 59-66 . 百度学术
20. 施维林,罗王捷. 有机物污染土壤修复技术研究与应用进展. 苏州科技大学学报(自然科学版). 2022(02): 1-8 . 百度学术
21. 黄文涛,汪军,邓呈逊,成德久,王啟华. 蒸汽注射技术原位修复非水相液体污染土壤的研究进展. 环境科学与管理. 2022(07): 92-97 . 百度学术
22. 陆玥,刘艺超,徐红霞,孙媛媛,吴吉春. 裂隙介质中流速及倾斜度对四氯乙烯的运移影响研究. 高校地质学报. 2022(04): 554-564 . 百度学术
23. 籍龙杰,陈芒,张维琦,李承志,余韬,李书鹏,詹明秀,焦文涛. 原位热脱附去除土壤有机污染机理及技术研究. 能源环境保护. 2022(05): 46-52 . 百度学术
24. 张海静,殷瑶,姜文超. 土壤原位燃气热脱附二次回流加热系统研究. 环境污染与防治. 2022(10): 1308-1313+1356 . 百度学术
25. 杨洁,叶春梅,司马菁珂,黄沈发,周栋. “双碳”目标下污染场地原位热处理技术发展趋势. 环境工程学报. 2022(11): 3517-3529 . 本站查看
26. 丁露. 间接热脱附技术在长三角地区土壤修复中的应用研究. 广东化工. 2021(12): 316-318 . 百度学术
27. 赵涛,周宇,马刚平,李世青,曹学龙. 焦化类污染场地原位热脱附修复效果试验. 环境工程. 2021(04): 201-205 . 百度学术
28. 刘莹莹. 原位燃气热脱附技术在有机污染土壤修复工程中的应用. 中国资源综合利用. 2021(06): 56-58 . 百度学术
29. 陈聪聪. 浅谈原位热脱附技术在我国污染场地的应用及其研究进展. 绿色环保建材. 2021(09): 15-17 . 百度学术
30. 谢炳坤,姜祖明,曾俊,籍龙杰,刘鹏,李书鹏,韩进,田齐东. 多环芳烃类污染场地应用原位电热脱附技术的能效分析. 环境工程. 2021(08): 173-178+187 . 百度学术
31. 孙玉超,刘耀,杨咸睿,刘志阳,曾跃春. 原位燃气热脱附技术在某退役焦化厂污染场地修复中的应用. 工业技术创新. 2021(05): 45-49 . 百度学术
32. 张圳,张晨,郭爱红. 焦化行业PAHs污染土壤的修复技术. 河北环境工程学院学报. 2021(06): 64-68 . 百度学术
33. 许丹芸,张亚宁,朱玲,桑义敏. 基于COMSOL模拟的有机污染土壤ERH修复工艺优化. 环境工程学报. 2021(11): 3642-3650 . 本站查看
34. 吴宇豪,尹立普,王晴,范利武,俞自涛. 有机污染黏壤土热脱附后热导率的变化特性. 环境工程学报. 2021(12): 3967-3973 . 本站查看
35. 黄菀,耿竹凝,李广贺,张芳. 热修复过程中重质非水相液体(DNAPL)共沸实验. 中国环境科学. 2020(09): 3903-3910 . 百度学术
36. 熊樱,蔡云,王永敏,杨晓东. 原位燃气热脱附技术在有机污染土壤修复工程的应用. 化工管理. 2020(31): 87-90 . 百度学术
37. 韩伟,黄少辰,叶渊,李彦希,申屠雷吉,王子剑. 基于风险防控的土壤氟污染特征及修复目标探讨. 环境保护科学. 2020(06): 160-166 . 百度学术
38. 韩伟,叶渊,焦文涛,张海涛,李彦希,申屠雷吉. 污染场地修复中原位热脱附技术与其他相关技术耦合联用的意义、效果及展望. 环境工程学报. 2019(10): 2302-2310 . 本站查看
其他类型引用(14)
-



 点击查看大图
点击查看大图
计量
- 文章访问数: 1896
- HTML全文浏览数: 1134
- PDF下载数: 837
- 施引文献: 52
