



-
近年来,新污染物在水环境中频繁检出,对水生态和人类健康造成了很大的威胁[1 − 2]. 其中,作为一种广谱类抗生素药物,氟苯尼考(FLO)被广泛应用于水产养殖业中[3],未经有效处理直接排放等都将造成水体污染,文献报道其在河流等水体中被检出[4]. 由于新污染物FLO化学结构稳定、生物降解性差导致传统水处理工艺难以高效降解去除[5],因此开发安全高效的去除抗生素的废水处理技术十分关键.
高级氧化技术(AOPs)能够产生高氧化活性的自由基,从而将目标污染物转化为小分子,以实现大多数有机污染物的降解[6]. 而基于活化过一硫酸盐(PMS)的AOPs,由于效率、活性高等特点引起研究者的广泛关注[7]. 其具有的稳定化学性能、易于储存和运输等优点,可显著降低废水处理的成本[8]. 而单独的PMS对于抗生素污染物的降解能力通常有限[9],可通过过渡金属离子等均相反应来高效活化PMS[10]. 同时,相较于均相体系,以过渡金属氧化物作为催化剂的非均相体系更具有可回收性、较高的热稳定性等优势,被广泛研究[11]. 具有多种价态的金属元素锰,在各种过渡金属氧化物中,其锰氧化物(MnOx)是一种高反应活性的金属氧化物,被应用于处理污染物,但是MnOx易团簇可能抑制其催化活性[12]. 为克服这一缺点,目前已有研究将MnOx负载到活性炭、生物炭、碳纳米管等材料上,用于提高MnOx的分散性、稳定性、和催化活性[13 − 14]. 海藻酸钠(SA)作为一种天然多糖,可以与多种金属阳离子(如铁离子,锰离子等)交联形成性能稳定的凝胶[15],将其作为煅烧前驱体,形成的碳复合材料可以有效避免金属氧化物的团聚[16],促进金属氧化物颗粒的稳定分散[17],有效提高了催化活性.
因此,本研究采用海藻酸钠与锰离子交联,煅烧制备了锰氧化物/碳(MnO/C)材料,并对其进行X-射线衍射(XRD)、扫描电镜(SEM)表征,将其应用于活化PMS降解FLO. 同时,分别考察了MnO/C材料和PMS的投加量,以及FLO溶液的pH值对其降解FLO性能的影响,并通过淬灭实验、电子顺磁共振(EPR)测试来分析体系中存在的自由基种类,利用液相色谱-质谱联用(LC-MS)技术解析FLO的降解路径. 最后,将其应用于降解大环内酯类的红霉素(ERY)、磺胺类的磺胺甲恶唑(SMX)和磺胺甲氧嘧啶(SDM)以及喹诺酮类的氧氟沙星(OFL)等典型抗生素以检查其适用性.
-
实验中所用的四水合氯化锰、海藻酸钠、盐酸普萘洛尔、氟苯尼考、组氨酸、盐酸羟胺、磺胺甲恶唑、2,2,6,6-四甲基哌啶、二氧化锰、一氧化锰、红霉素、磺胺甲氧嘧啶、氧氟沙星均购自上海麦克林生化股份有限公司,甲醇、乙腈、乙酸铵、过硫酸氢钾复合盐、氢氧化钠、硫酸、叔丁醇均由西陇科学股份有限公司提供. 实验过程所有用水均为超纯水,所用试剂均为分析纯.
-
将海藻酸钠粉分散在去离子水中,均匀搅拌直至透明黏状,得到2% W/V的海藻酸钠溶液. 将其均匀滴加至浓度为0.1 mol·L−1的四水合氯化锰(MnCl2·4H2O)溶液中,交联形成海藻酸锰水凝胶小球,固化水凝胶小球24 h后用纯水洗净,在−20 ℃下冷冻,得到海藻酸锰气凝胶(Mn-SA). 将Mn-SA在900 ℃氮气氛围中以10 ℃·min−1的加热速率煅烧1 h,冷却至室温后,研磨得到MnO/C材料. 另外,将Mn-SA在空气氛围下煅烧1 h得到对比材料MnO,并将购入的MnO和MnO2命名为MnO-B和MnO2-B作为锰氧化物的性能对比材料.
-
在D8 ADVANVE上进行X射线粉末衍射(XRD)测试,操作电压与电流分别为40 mA和40 kV,扫描速度为5 (°)·min−1,步长0.02°,衍射角范围2θ = 10°−80°;采用场发射扫描电镜(SEM,Regulus
8100 )观察形貌;以2,2,6,6-四甲基哌啶(TEMP)和5,5-二甲基-1吡咯啉(DMPO)为捕获剂,在MS 5000X上进行电子顺磁共振(EPR)测试,鉴定体系中存在的自由基种类,其中测定超氧自由基时采用甲醇作为体系溶剂,使用有机元素分析仪(EA)在氧气和氮气条件下对材料中碳的含量进行测定.样品均采用高效液相色谱法(LC1260, Agilent)检测目标污染物中FLO的残余浓度,配备紫外检测器和Agilent Zorbax SB-C18色谱柱(4.6 mm×150 mm, 5 μm),其中紫外检测波长为224 nm,以甲醇和超纯水(4:6)为流动相,进样量为10 μL,柱温为30 ℃. 磺胺甲氧嘧啶(SDM)、红霉素(ERY)、氧氟沙星(OFL)、磺胺甲恶唑(SMX)均以乙腈和乙酸铵(3:7)为流动相,进样量为10 μL,柱温为35 ℃,其中,SDM、ERY、OFL、SMX的检测波长分别为:270 nm、210 nm、294 nm、270 nm.
FLO降解的中间产物的鉴定是在超高效液相色谱-串联质谱(UPLC-MS)系统(AB SciexTripleTOF
5600 )上进行的,该系统配备了负离子扫描模式下的四极飞行时间(QTOF)检测器. 分析物的分离是在反相C18柱上以0.8 mL·min−1的流速进行的,流动相为甲醇和水,二者的比例为3:7. 最后,使用Qualitative Analysis 10.0软件分析确认降解产物.进行数据分析时,使用修正后的动力学速率常数(kvalue)对活化PMS去除FLO的降解动力学进行评价[18]. 通过将拟合后的有机污染物的速率常数(k)除以催化剂的剂量(d)和氧化剂浓度(C1),再乘以有机污染物浓度(C0),计算出修正kvalue,计算公式如下:
-
在FLO溶液中加入一定量的催化剂和一定浓度的PMS在室温条件下进行降解反应,并在相同条件下探究溶液pH对降解体系的影响,通过降解不同抗生素以证明该体系具有普适性. 实验过程中,每隔一段时间(0、5、10、15、30、45、60 min),提取1 mL溶液经过0.22 μm聚四氟乙烯过滤器过滤后,立即加入盐酸羟胺终止反应,待测.
具体实验条件为:在10 mg·L−1 FLO、0.2 mmol·L−1 PMS、0.2 g·L−1 MnO/C、溶液pH = 7条件下探究其降解FLO的性能;在实验条件为0.2 g·L−1 MnO/C、溶液pH = 7的条件下,将PMS浓度设置为0.1、0.2、0.3、0.4 mmol·L−1,以探究PMS的最优条件;在0.3 mmol·L−1 PMS、溶液pH = 7的条件下,分别向体系中投加0、0.1、0.2 g·L−1 MnO/C材料,用于研究材料用量对降解FLO的影响;除此之外,在0.3 mmol·L−1 PMS、0.2 g·L−1 MnO/C的条件下,用稀硫酸和氢氧化钠调节体系溶液pH值,分别为3、5、7、9、11,以探究溶液pH值对降解污染物的影响;在实验条件为:0.2 g·L−1 MnO/C、0.3 mmol·L−1 PMS、溶液pH = 7的条件下探究其对不同抗生素的降解效果,使用的抗生素浓度均为10 mg·L−1. 淬灭实验中采用分别添加10 mmol·L−1的组氨酸(His)和叔丁醇(TBA)对MnO/C-PMS体系进行淬灭.
-
制备的材料的XRD表征结果如图1(a)所示. 由图1(a)可见,MnO/C和MnO在2θ = 34.20°、39.69°、57.39°、68.52°和72.03°均出现了明显的衍射峰,分别对应MnO(PDF标准卡片 ICDD 065-0638)的(111)、(200)、(220)、(311)和(222)晶面. 在MnO/C的XRD图谱上观察到在2θ = 24°出现鼓包峰,这可能是因为MnO/C材料中存在无定形碳[19]. 通过有机元素分析仪(EA)测定其可能的元素组分,其中碳在MnO/C的占比为35.7%,证明该材料中含有碳. 图1(b)通过扫描电子显微镜(SEM)观察样品MnO/C的形貌,发现MnO/C材料为细碎颗粒状,大小不一,表面粗糙不平,这可能使得其具有更大的反应面积和更密集的接触面积有利于提供反应活性位点[20].
-
首先,探究MnO/C是否能够活化PMS降解FLO. 如图2(a)所示,仅PMS体系对FLO的降解效果不理想,60 min时仅为2%. 在仅MnO/C体系下,15 min时FLO的去除效率达到34%,60 min时提高至69%,这可能是由于MnO/C复合材料具有较大的比表面积(361.5 m2·g−1),其对污染物具有一定的吸附能力[14]. 而在PMS和MnO/C共存的体系中,FLO去除的速率显著提升,15 min内达到了70%,并达到平衡.
探究PMS浓度对MnO/C性能的影响. 如图2(b)所示,当PMS浓度由0.1 mmol·L−1增加到0.2 mmol·L−1时反应速率明显增加,并在15 min时迅速达到平衡. 对不同PMS浓度体系进行拟合,对应的反应速率常数如图2(c)所示,分别为:k0.3(0.143 min−1)>k0.4(0.102 min−1)>k0.2(0.075 min−1)>k0.1(0.042 min−1),在PMS为0.3 mmol·L−1下,其去除FLO的速率最快,当PMS的浓度超过该浓度后,体系去除FLO的效率不再提升. 这可能是因为吸附在MnO/C材料表面的PMS分子趋于饱和[21],过量将不再提高FLO的降解率. 因此,选用0.3 mmol·L−1 PMS作为实验的最佳用量. 进一步探究MnO/C用量对FLO降解效率的影响,由图2(d)可知,未投加MnO/C的体系中,相较于图2(a),PMS从0.2 mmol·L−1提升至0.3 mmol·L−1,FLO去除效率仅提高3%,少量去除可能是因为PMS自身分解生成的活性氧物种增加所致[22]. 而在PMS和MnO/C共存的体系中,MnO/C用量为0.2 g·L−1时,其降解FLO的速率和效率明显优于MnO/C用量为0.1 g·L−1时,在15 min内降解85%的FLO. 这可能是由于随着MnO/C的用量提升,可供激活PMS的活性位点也在增加[23],从而提升了FLO的降解效率. 因此,选择0.2 g·L−1的MnO/C用量作为实验最佳用量.
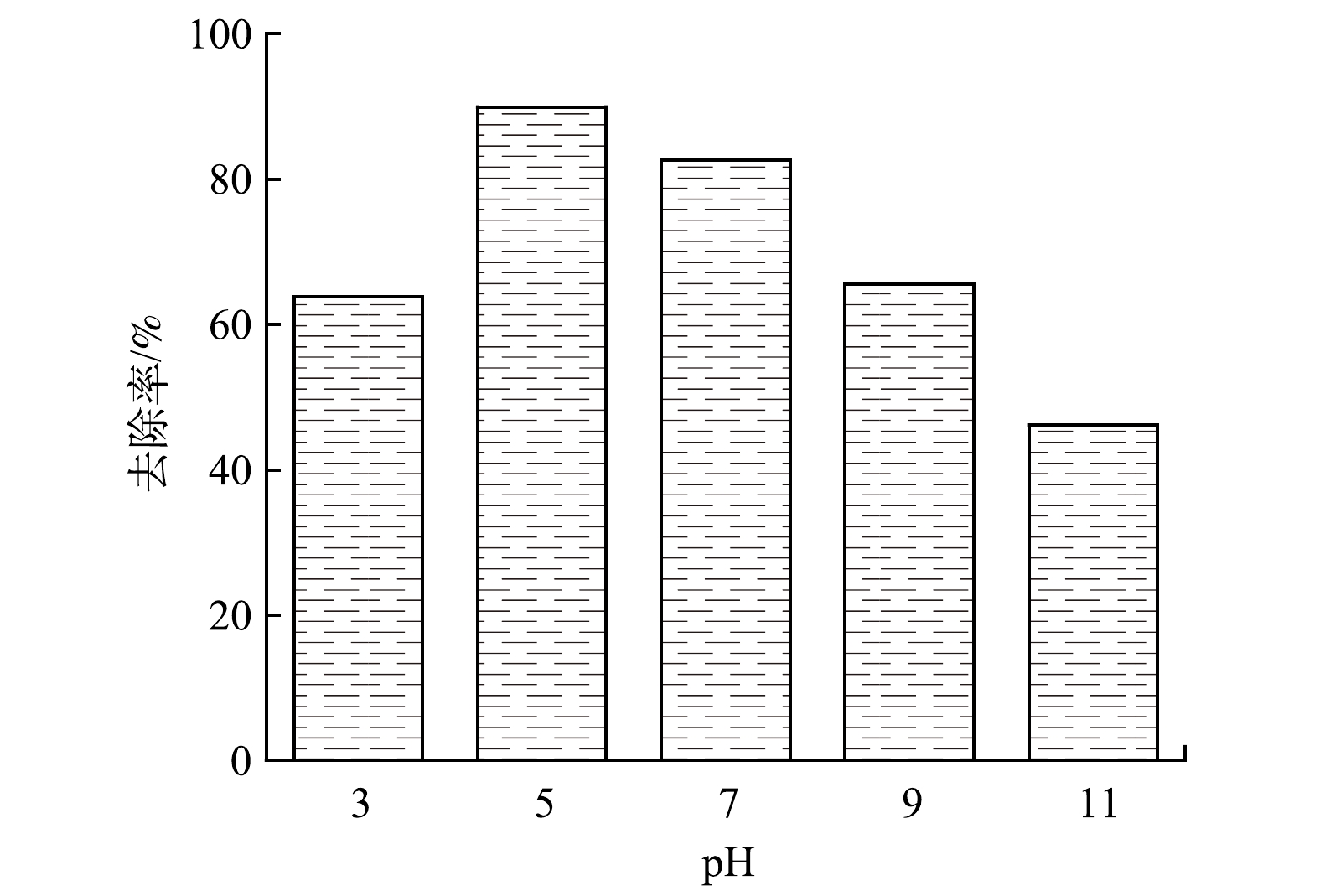
溶液pH值对污染物的去除具有重要影响,因此,探究不同pH值对MnO/C-PMS体系降解FLO的影响. 如图2(e)所示,体系的pH值会对MnO/C催化剂降解FLO的实验产生较大的影响. 在MnO/C-PMS体系中,pH值为7时反应15 min降解了8.5 mg·g−1的FLO,经计算其kvalue值为23.83 min−1·mol·L−1. 同时,在酸性条件下(pH值为3和5时),MnO/C也高效活化了PMS,30 min内FLO的降解率均可达85%以上. 在碱性条件下,FLO的降解效率和速率虽仍有效果,但显著下降,在30 min内去除率仅为50%左右. 实验结果表明,反应速率随着反应体系pH的升高而下降,这可能是由于锰氧化物的催化效果受pH的影响较大. 以Mn2+形式存在的锰在酸性环境下更容易被氧化成Mn3+[24]. 该转化过程可能有利于促进水分子与Mn3+反应产生单线态氧(1O2),从而提高催化活性. 而在碱性条件下,锰离子的氧化还原反应可能受到抑制,这可能是因为Mn2+在碱性环境中会形成氢氧化锰沉淀,Mn2+的浓度下降影响了Mn3+的生成,从而导致活性氧的生成效率降低[25]. 综上,MnO/C-PMS体系能够在pH范围为3—7的条件下有效活化PMS,实现对FLO的高效去除. 结合实际水体条件以及MnO/C的性能测试,选用pH = 7用于后续机理研究.
为了判别所制备MnO/C材料的优异性能,进一步对比同等反应条件下,MnO(空气煅烧Mn-SA制得,作为同条件下制的未含碳的MnO)、MnO/C-B及MnO2-B活化PMS降解FLO的效果,结果如图2(f)所示,MnO-B-PMS体系、MnO2-B-PMS和MnO-PMS体系对FLO几乎没有去除效果,在60 min内降解率均低于17%,这可能是因为单一金属氧化物颗粒团聚,使锰氧化物体系降解污染物效率不高[14,26]. 表明了以水凝胶作为前驱体制备的MnO/C材料,在煅烧碳化过程中,金属可以有效分散在碳上. 该锰与碳相结合的策略可以有效提高锰氧化物的催化活性. 进一步对比已报道的催化剂活化PMS去除FLO研究可知,相较于LaMBC(kvalue=0.41 min-1·mol·L−1)[27]、Co3O4-BC(kvalue=0.99 min−1·mol·L−1)[28],本文的kvalue值达到了23.83 min−1·mol·L−1,MnO/C展现出了较大的性能优势,表明该材料能够在较低的材料用量(0.2 g·L−1)和PMS浓度(0.3 mmol·L−1)下高效活化PMS降解FLO.
-
通过EPR的测试鉴定体系中的活性氧物种,结果如图3(a、b)所示,以DMPO为捕获剂时,未检测到DMPO-·OH、DMPO-SO4·-和DMPO-· O2−的特征信号峰,表明该体系过程中可能产生的·OH、SO4·-和·O2−较少或极易被转化而未检测到. 而以TEMP为捕获剂时,该体系检测到1O2,说明催化剂MnO/C活化PMS过程中生成了1O2. 因此,认为·OH、SO4·-和·O2−并不是该体系中的主要活性自由基,而生成的1O2在去除FLO的过程中可能发挥主要作用[27]. 为进一步探究MnO/C催化剂活化PMS的机理,进行自由基的淬灭实验. 其中,叔丁醇(TBA)用于淬灭体系中可能存在的·OH和SO4·-[29],其结果如图3(c)所示. TBA的引入使FLO的降解速率下降,k值由0.143 min−1变化为0.049 min−1,但在60 min内仍保持85%的去除效率. 这可能是由于反应过程中生成少量的·OH和SO4·-,对反应有一定影响,但并不是影响FLO去除的主要活性自由基. 将作为1O2淬灭剂的组氨酸[30]引入到体系中,在60 min时FLO的降解率仅为13%. 对比未加入淬灭剂的体系(85%),组氨酸的引入显著抑制了FLO的降解. 因此,结合EPR的测试结果(图3(a)),认为在MnO/C-PMS体系中,1O2可能是主导FLO降解过程的主要活性物种.
综上所述,以Mn-SA为前驱体煅烧制备的MnO/C复合材料中锰作为催化活性中心,能够有效激活PMS(反应方程式1、2),形成的SO5−能够与水反应进一步生成1O2(反应方程式3),形成的1O2用于降解FLO等新兴有机污染物. 另外,过程中形成的SO4−·会与水作用,转变为其他物质(反应方程式4)[31]. 对比不同的锰氧化物和已报道的文献可知,Mn-SA作为煅烧前驱体可以有效促进金属氧化物颗粒稳定分散在碳上,该复合材料提高了MnO的催化活性,更有利于活化PMS产生1O2,具体机理如图4所示.
测定反应过程生成的中间产物可以进一步分析FLO的降解路径,本文采用LC-MS检测MnO/C-PMS体系中,FLO降解生成的中间产物. 反应过程中,除了FLO之外,分别检出了R1、R2、R3、R4、R5、R6等6种物质(图5),进一步推测FLO的降解路径,如图5所示. FLO的降解路径包括水解、羟基化、脱氟、脱氯氧化[32 − 35]. 其中,路径1是通过活性氧的作用实现脱氯、氧化,产生小分子产物及氯离子[32],而在路径2中,R2-R3的过程是由于C—S键受到攻击断裂,而后实现脱氯氧化,进一步促进脱氟,形成分子量更小的产物[33]. 路径3中,C—C键先行断裂,产生R4,而后进一步脱氯产生R5[34]. 从R5到R6的过程主要是由于FLO完全脱氯后会产生的自发水解反应,使得产物进一步的分解[35]. 该实验结果证明,MnO/C-PMS体系选择性产生的1O2可以有效帮助FLO进行脱卤和氧化,产生分子量更小的产物.
-
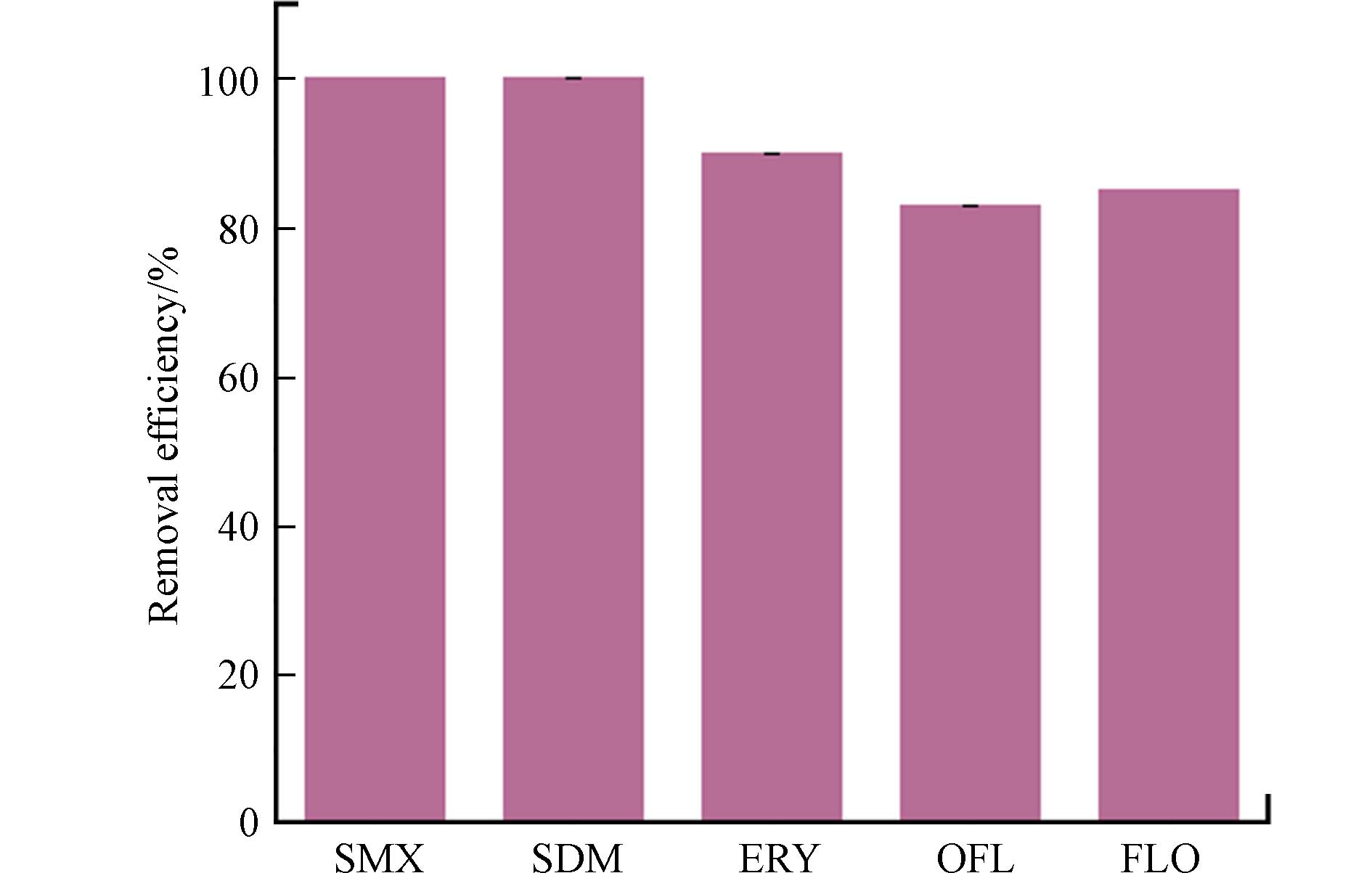
为了测试MnO/C-PMS体系对不同污染物的降解效果,选取了大环内酯类的ERY(红霉素)、磺胺类的SMX(磺胺甲恶唑)和SDM(磺胺甲氧嘧啶)以及喹诺酮类的OFL(氧氟沙星)作为不同种类的抗生素进行MnO/C活化PMS性能测试研究. 如图6所示,该体系对SDM、SMX均有较好的去除效果,降解效率均达到了100%,ERY的降解率为90%,OFL的降解率为83%. 由结果可知,该体系对新污染物均具有良好的降解效果(均>83%),能够满足不同类型的抗生素的处理要求,对新污染物的去除具有良好的应用潜力和适用性.
-
采用绿色环保的海藻酸锰水凝胶小球作为前驱体,并冷却干燥煅烧衍生制备碳负载一氧化锰(MnO/C)的复合材料,可以有效将MnO分散在碳上,使其表现出较高的催化活性,通过活化PMS可以高效去除抗生素等新污染物. MnO/C-PMS体系在15 min内降解了8.5 mg·g−1 (kvalue=23.83 min−1·mol·L−1)的FLO. 其机理主要为选择性生成1O2的非自由基路径实现降解,对FLO的降解机理主要包括脱卤和氧化过程. 同时,该体系对磺胺类的磺胺甲氧嘧啶(SDM)、大环内酯类的红霉素(ERY)和喹诺酮类的氧氟沙星(OFL)等典型新污染物均具有良好的降解效果. 本文中的金属锰与海藻酸钠交联煅烧形成的MnO/C复合材料为高级氧化水处理的绿色过程提供了策略,具有潜在的应用价值.
锰氧化物/碳活化过硫酸盐选择性生成单线态氧降解水中氟苯尼考
Research on the selective generation of singlet oxygen from manganese oxide/carbon activated persulfate for the degradation of florfenicol in water
-
摘要: 为实现养殖废水中的新污染物氟苯尼考(FLO)的高效降解,本研究制备了锰氧化物/碳材料(MnO/C)用于活化过一硫酸盐(PMS)去除FLO. 首先,考察了PMS用量、MnO/C用量、溶液pH值等对水中FLO去除的影响. 结果表明,在PMS浓度为0.3 mmol·L−1、MnO/C用量为0.2 g·L−1、FLO浓度为10 mg·L−1,pH值为7时,15 min内降解了8.5 mg·g−1 (kvalue=23.83 min−1·mol·L−1)的FLO. 并通过自由基淬灭实验及电子顺磁共振测试(EPR)分析表明,MnO/C-PMS体系中主要以单线态氧(1O2)的非自由基路径催化降解FLO,液质分析结果可知,MnO/C-PMS对FLO的降解机理主要包括脱卤和氧化. 且该体系对磺胺类的磺胺甲氧嘧啶(SDM)、大环内酯类的红霉素(ERY)和喹诺酮类的氧氟沙星(OFL)等典型新污染物均具有良好的降解效果(均>83%). 因此,MnO/C在活化PMS上表现出对新污染物良好的降解能力,具有潜在应用性.
-
关键词:
- 氟苯尼考 /
- 锰氧化物/碳复合材料 /
- 过一硫酸盐 /
- 非自由基路径 /
- 高级氧化
Abstract: In order to efficiently degrade the new pollutant florfenicol (FLO) in aquaculture wastewater, this study prepared carbon-coated manganese oxide materials (MnO/C). Firstly, the effects of PMS concentration, MnO/C concentration, solution pH value on the removal of FLO were discussed, respectively. The degradation rate of FLO was 8.5 mg·g−1 in 15 min (kvalue=23.83 min−1·mol·L−1), under the experimental conditions: FLO concentration was 10 mg·L−1, PMS concentration was 0.3 mmol·L−1, MnO/C concentration was 0.2 g·L−1 and pH = 7. It was demonstrated by radical scavenger experiment, electron paramagnetic resonance (EPR) tests, and liquid chromatography-mass spectrometry (LC-MS) analysis. The non-radical pathways (1O2 oxidation) dominated in the MnO/C-PMS and FLO are primarily dehalogenation and oxidized. Subsequently, The system also demonstrates effective degradation of typical emerging contaminants such as sulfamethoxypyridazine (SDM), erythromycin (ERY), and ofloxacin (OFL), with degradation rates all exceeding 83%. Therefore, MnO/C exhibits strong potential for the activation of PMS and degradation of organic pollutants, showing promising application prospects. -
全球纺织业每年的染料总消耗量超过10 000 t,并且每年约有100 t的染料被排放到自然水体中[1]。染料废水具有色度高、毒性大、成分复杂且难生物降解的特点,对环境和人体都有危害。罗丹明B(RhB)是一种在轻工业中广泛使用的阳离子型染料,具有致癌性。
处理染料废水的常用方法包括物理法、化学法和生物法[2]。近年来,随着新能源的开发与利用,具有绿色环保特征的光催化技术备受人们的关注[3]。光催化降解有机染料被认为是一种清洁高效的染料废水处理方法,该方法能够高效地将多种污染物转化为毒性较小的中间产物,或是将其彻底降解为CO2和H2O,达到无害化处理的要求。
近年来,金属有机框架(MOF)材料在光催化领域受到越来越多的关注[4-7]。MOF材料是一类由金属离子(簇)和有机配体配位而成的杂化材料[8],具有稳定性高[9]、比表面积大、孔隙度高以及结构灵活可调的典型特征[10-12]。MOF材料在异相催化等领域均有优异的表现[13-14],其出众的吸附性能[15]与异相催化潜力为传统的染料废水处理提供了新的解决方案[16]。但是,大多数 MOF材料的宽带隙只能吸收紫外光,而对可见光吸收率低[17],并且纯MOF材料存在光生电子-空穴对复合快的问题[18],导致其光催化活性并不理想。因此,许多研究致力于采用配体修饰[19-20]、掺杂金属[21-22]等方法对MOF材料进行改性,以提升其光催化活性。目前,氨基修饰是增强MOF材料可见光吸收的一种常用方法[23-25]。
本研究评估了氨基修饰Fe/Cu-MOF对RhB的光催化降解性能,考察了非氨基配体/氨基配体的配比、初始条件对催化效果的影响,并基于以上结果提出了氨基修饰Fe/Cu-MOF光催化降解RhB的反应机制,包括光捕获和电子转移路径,以及主要活性物种的生成过程。
1. 材料与方法
1.1 实验原料
氯化铁(FeCl3·6H2O)、对苯二甲酸(H2BDC)、2-氨基对苯二甲酸(NH2-BDC)、对苯醌(BQ)购于上海安耐吉化学有限公司,乙二胺四乙酸二钠(EDTA-2Na)购于上海易恩化学技术有限公司,氯化铜(CuCl2 ·2H2O)、N,N-二甲基甲酰胺(DMF)、异丙醇(IPA)、无水甲醇、无水乙醇、盐酸购于国药集团化学试剂有限公司,氢氧化铵购于天津市百世化工有限公司,罗丹明B购于天津市北联精细化学品公司,纳米磁性氧化铁购于阿拉丁试剂,以上试剂均为分析纯。
1.2 催化剂制备方法
1) Fe-MOF的合成[26-27]。称取10 mmol的FeCl3·6H2O和5 mmol的H2BDC混合溶解于60 mL DMF溶剂,转移至100 mL聚四氟乙烯内衬反应釜中,在110 ℃下反应24 h。反应完成后待自然冷却至室温,产物用DMF和无水甲醇分别洗涤3次,在80 ℃下真空干燥24 h,得到产物Fe-MOF。
2) Fe/Cu-MOF的合成。称取9.5 mmol的FeCl3·6H2O,0.5 mmol的CuCl2 ·2H2O和5 mmol的H2BDC混合溶解于60 mL DMF溶剂,其余步骤同1),得到产物为Fe/Cu-MOF。
3)不同H2BDC/NH2-BDC配比的Fe/Cu-MOF-NH2的合成。采用NH2-BDC部分代替上述H2BDC,其余步骤同2)。控制NH2-BDC的加入量可制得不同摩尔比(0∶1、1∶1、2∶1、3∶1、4∶1)的混合配体Fe/Cu-MOF-NH2。
1.3 催化剂表征方法
通过X射线衍射(XRD,X/Pert PRO MPD,帕纳科分析仪器有限公司,荷兰)对催化剂的晶体结构进行表征;采用扫描电子显微镜(SEM,MIRA 3 LMH,泰思肯有限公司,美国)对催化剂的微观形貌进行分析;光学吸收性质采用紫外可见漫反射光谱仪(DRS,SolidSpec-3700DUV,岛津公司,日本)进行测试;采用精微高博JW-BK100全自动比表面积及孔径分析仪测定目标材料的N2等温吸脱附曲线;使用单点BET和BJH方法计算材料的比表面积、孔体积以及孔径分布;通过元素分析仪(EA, elementar vario EL cube, 德国)对催化剂的元素组成进行分析。
1.4 光催化降解实验方法
取50 mg催化剂,将其分散于50 mL初始质量浓度为300 mg·L−1的RhB溶液中,在暗吸附30 min后开启氙灯,控制电流I为15 A。每隔1 h取样进行离心分离,然后使用紫外-可见分光光度计对离心分离后的上清液进行吸光度测定和光谱扫描。本文中的所有实验均为3次平行实验结果,误差均在5%以内。
2. 结果与讨论
2.1 氨基修饰前后Fe/Cu-MOF的吸附和光催化性能对比
在RhB初始质量浓度为300 mg·L−1,催化剂质量浓度为1 mg·mL−1的条件下,Fe/Cu-MOF和Fe/Cu-MOF-NH2的光催化降解RhB以及暗吸附反应的性能如图1所示。其中,入射光波长采用UV-vis全波段。当RhB的初始质量浓度为300 mg·L−1时,Fe/Cu-MOF 对RhB的吸附去除率仍可达到85%以上,但光照下的去除率无明显提升。这说明Fe/Cu-MOF的光催化活性较弱。而在光照条件下,Fe/Cu-MOF-NH2对RhB的去除率较暗反应条件下有明显提升。在同等光照条件下,商用TiO2对RhB的去除率仅为20.32%,而本研究中制备的Fe/Cu-MOF-NH2对RhB的去除率约为商用TiO2的4.7倍,Fe/Cu-MOF-NH2具有更高的反应速率;在同等条件下,不添加催化剂而仅使用氙灯光源照射并不能引起RhB的降解。Fe/Cu-MOF-NH2对RhB的去除率比Fe/Cu-MOF更高,性能更优良。这表明,氨基的引入能显著提升Fe/Cu-MOF的光催化效率。
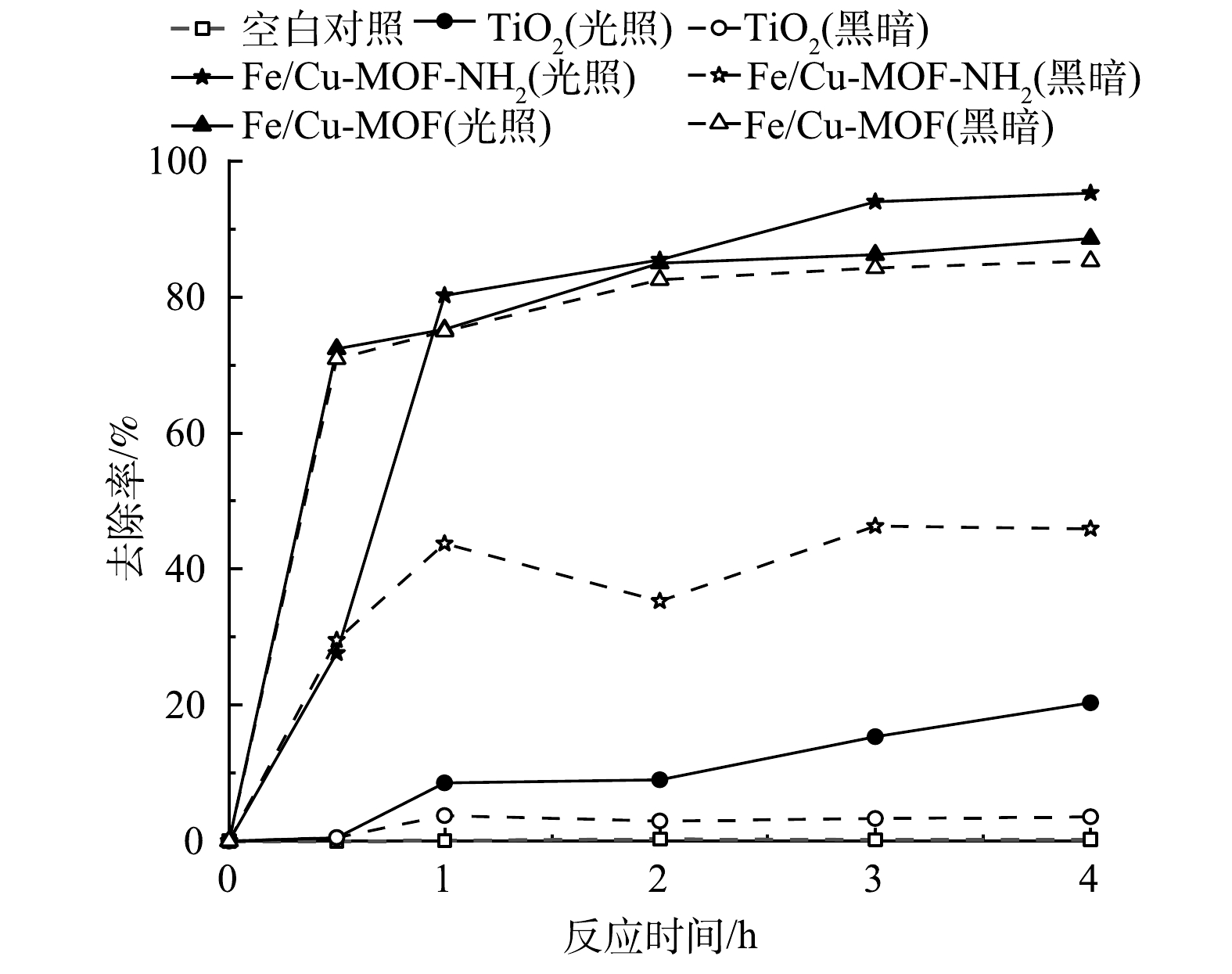 图 1 氨基修饰前后Fe/Cu-MOF对RhB的光催化降解及暗吸附性能对比Figure 1. Comparison of photocatalytic degradation and dark adsorption of RhB by Fe/Cu-MOF before and after amino modification
图 1 氨基修饰前后Fe/Cu-MOF对RhB的光催化降解及暗吸附性能对比Figure 1. Comparison of photocatalytic degradation and dark adsorption of RhB by Fe/Cu-MOF before and after amino modification2.2 催化剂表征
1)N2吸附-脱附分析。为探究Fe/Cu-MOF-NH2在暗反应下对RhB吸附性能比Fe/Cu-MOF弱的原因,在−196 ℃下测试了2种材料的N2等温吸脱附曲线(图2),比表面积与孔容孔径的数据见表1。表1 的数据说明,Fe/Cu-MOF和Fe/Cu-MOF-NH2均属于介孔材料,引入氨基后BET比表面积大幅降低,孔容积也相对减小,而对应的平均孔径增大。已有研究[28-29]表明,氨基的引入可能会造成材料比表面积和孔容的减小。大孔径孔结构的增加必然会牺牲一部分较小孔径所占的空间,由于含氨基配体的材料在合成过程中可能存在穿插结构或因小分子团簇造成的孔道堵塞,导致孔容明显减小;而BET比表面积的减小则是因为孔隙度的降低或者由于孔道堵塞造成的孔径内比表面积的降低。最明显的影响就是Fe/Cu-MOF- NH2在暗反应下对RhB的吸附效果明显低于Fe/Cu-MOF。
 图 2 Fe/Cu-MOF与Fe/Cu-MOF- NH2的N2吸脱附等温线Figure 2. Nitrogen adsorption-desorption isotherms of Fe/Cu-MOF and Fe/Cu-MOF-NH2表 1 Fe/Cu-MOF与Fe/Cu-MOF- NH2的比表面积、孔容与孔径Table 1. Specific surface area, pore volume and average pore size of Fe/Cu-MOF and Fe/Cu-MOF-NH2
图 2 Fe/Cu-MOF与Fe/Cu-MOF- NH2的N2吸脱附等温线Figure 2. Nitrogen adsorption-desorption isotherms of Fe/Cu-MOF and Fe/Cu-MOF-NH2表 1 Fe/Cu-MOF与Fe/Cu-MOF- NH2的比表面积、孔容与孔径Table 1. Specific surface area, pore volume and average pore size of Fe/Cu-MOF and Fe/Cu-MOF-NH2样品 比表面积/(m2·g−1) 孔容/(cm3·g−1) 平均孔径/nm Fe/Cu-MOF 248.3 0.211 3.401 Fe/Cu-MOF-NH2 21.11 0.120 22.805 | Show Table DownLoad:
CSV
DownLoad:
CSV
2) UV-vis分析。图3表明,Fe/Cu-MOF-NH2具有良好的可见光响应活性,在采用全光谱照射或可见光波段照射时,Fe/Cu-MOF-NH2所表现出的光催化活性相近。图4也说明Fe/Cu-MOF-NH2比Fe/Cu-MOF在可见光区域具有更强的吸收能力。
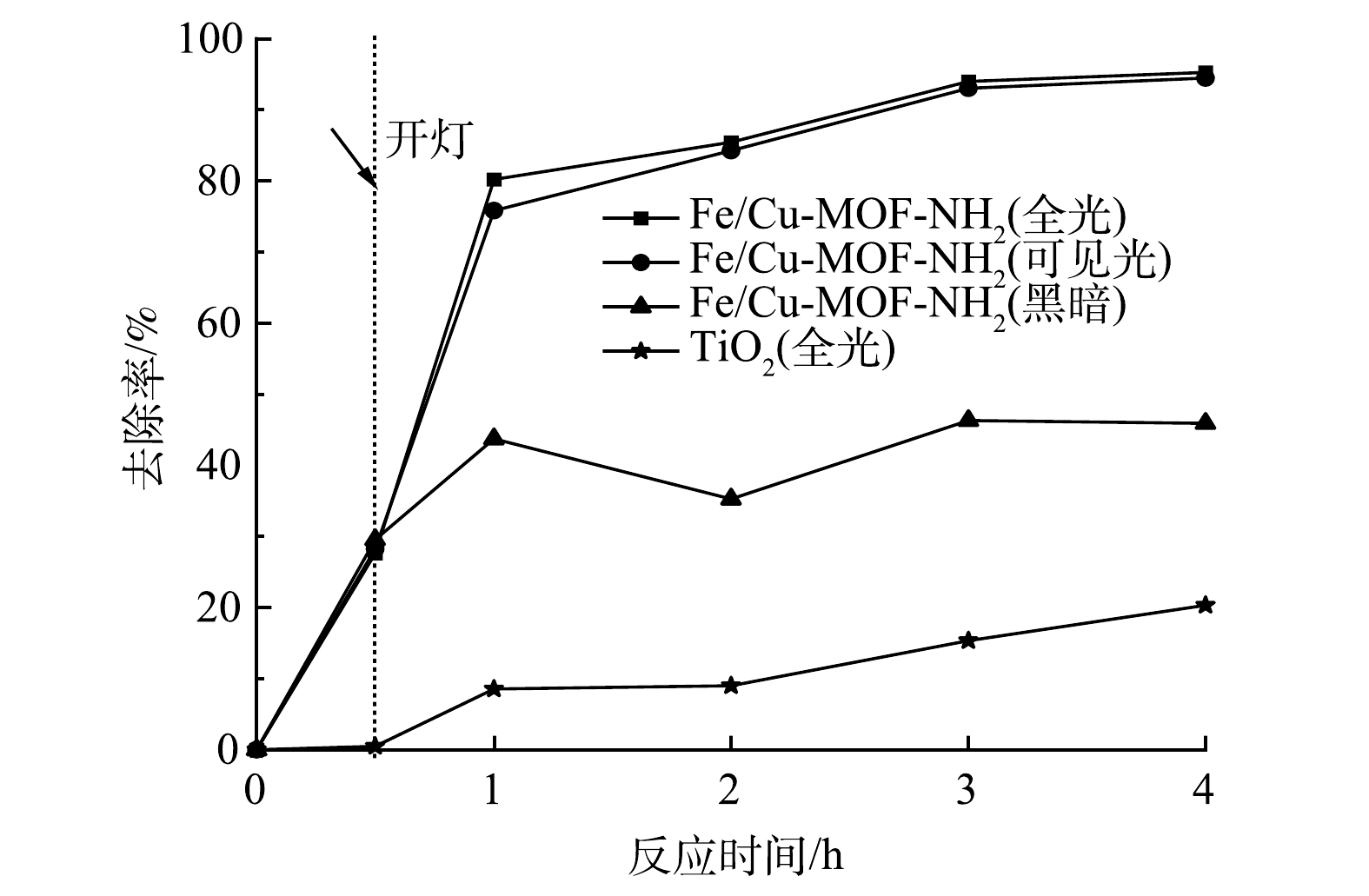 图 3 不同波段入射光下Fe/Cu-MOF- NH2对RhB的去除率Figure 3. Removal rate of RhB by Fe/Cu-MOF-NH2 under different incident light bands
图 3 不同波段入射光下Fe/Cu-MOF- NH2对RhB的去除率Figure 3. Removal rate of RhB by Fe/Cu-MOF-NH2 under different incident light bands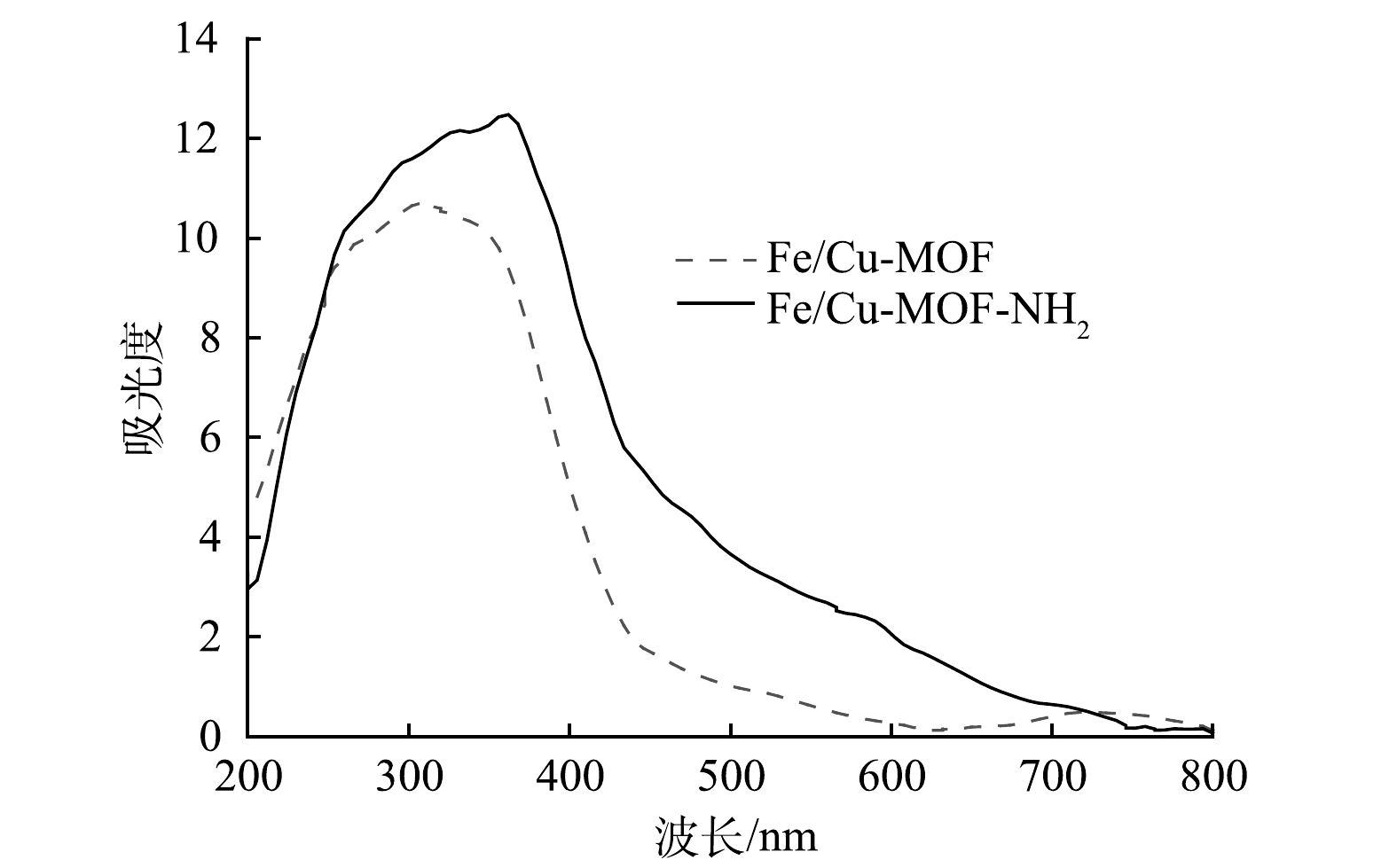 图 4 Fe/Cu-MOF与Fe/Cu-MOF- NH2的紫外-可见吸收光谱Figure 4. UV-vis absorption spectra of Fe/Cu-MOF and Fe/Cu-MOF-NH2
图 4 Fe/Cu-MOF与Fe/Cu-MOF- NH2的紫外-可见吸收光谱Figure 4. UV-vis absorption spectra of Fe/Cu-MOF and Fe/Cu-MOF-NH23)SEM分析。图5反映了Fe/Cu-MOF-NH2光催化反应前后的微观形貌。可以看出,材料在反应后微观形貌几乎没有变化,说明其具有良好的稳定性。
 图 5 Fe/Cu-MOF- NH2光催化反应前后的SEM图Figure 5. Fe/Cu-MOF-NH2 before and after photocatalytic reaction
图 5 Fe/Cu-MOF- NH2光催化反应前后的SEM图Figure 5. Fe/Cu-MOF-NH2 before and after photocatalytic reaction4)XRD分析。图6为Fe/Cu-MOF光催化前及Fe/Cu-MOF-NH2光催化反应前后的XRD图谱。其中Fe/Cu-MOF-NH2的主要衍射峰出现的位置在9.0°、10.2°、16.4°,分别对应(100)、(101)、(110)晶面,与文献报道结果[26]一致。此外,Fe/Cu-MOF-NH2光催化反应前后的衍射图谱结构相似,表明其具有良好的稳定性。
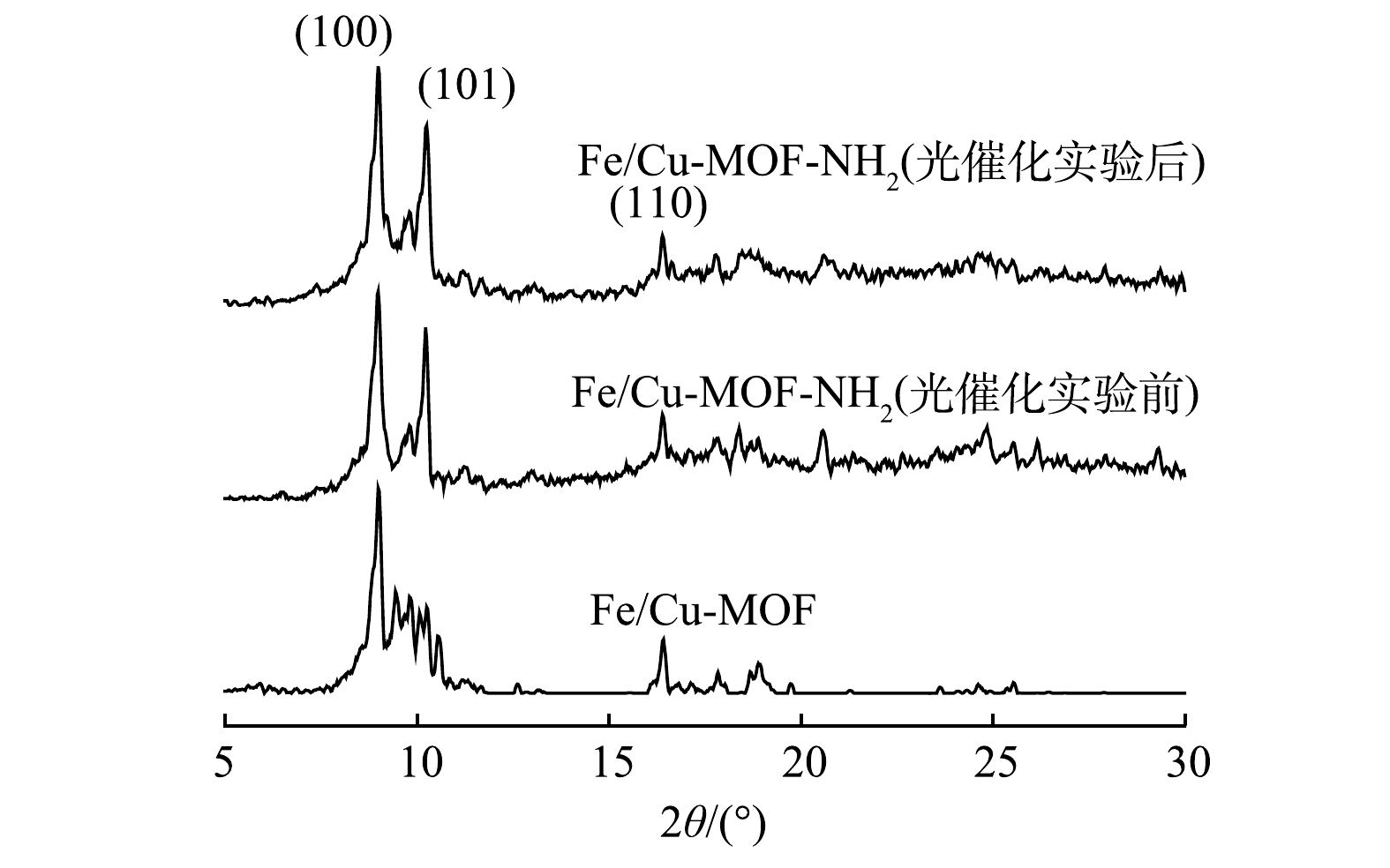 图 6 Fe/Cu-MOF-NH2光催化反应前后的XRD图谱Figure 6. Fe/Cu-MOF-NH2 before and after photocatalytic reaction by XRD
图 6 Fe/Cu-MOF-NH2光催化反应前后的XRD图谱Figure 6. Fe/Cu-MOF-NH2 before and after photocatalytic reaction by XRD5)元素分析。使用元素分析仪测试Fe/Cu-MOF和Fe/Cu-MOF-NH2中各元素的含量(表2)。Fe/Cu-MOF中不含N元素,检测出的微量N元素可能是由于纯化过程中使用DMF进行洗涤时带入的;而Fe/Cu-MOF-NH2中N元素含量明显增加,说明成功引入了氨基。根据已有研究[26]可推测,氨基以自由的非缔合形式存在。
表 2 Fe/Cu-MOF和Fe/Cu-MOF-NH2的元素含量分析Table 2. Elemental analysis of Fe/Cu-MOF and Fe/Cu-MOF-NH2% 样品 N C H Fe/Cu-MOF 1.21 30.67 4.696 Fe/Cu-MOF-NH2 4.02 31.59 5.511 | Show TableDownLoad:
CSV
2.3 Cu掺杂对铁基MOF材料稳定性的增强效果
图7(a)反映了放置不同时间后的Fe-MOF和Fe/Cu-MOF对RhB的吸附去除效果。可以看出,Fe/Cu-MOF在自然放置条件下依然保持较好的吸附性能,14 d后对RhB的去除率为82.36%,能达到新制备Fe/Cu-MOF同等条件下对RhB去除率(85.27%)的96%以上。铜离子掺杂后的材料在不采取特殊存放措施(真空密闭环境)下虽然仍存在吸附性能衰减的情况,但可缓解其衰减的进程,Fe/Cu-MOF自身结构的稳定性较Fe-MOF有所增强。图7(b)反映了Fe-MOF和Fe/Cu-MOF多次循环使用过程中吸附性能的变化情况。相较于Fe-MOF,3次循环之后Fe/Cu-MOF对RhB的去除率仍达70%以上,说明其具有良好的稳定性。这是因为:相对于Fe(Ⅲ),金属铜离子的惰性取代[30]增强了Fe/Cu-MOF的稳定性;同时,金属离子的掺杂对MOF表面产生扰动,阻碍了孔隙表面水簇的形成,从而提高MOF的水稳定性[31]。
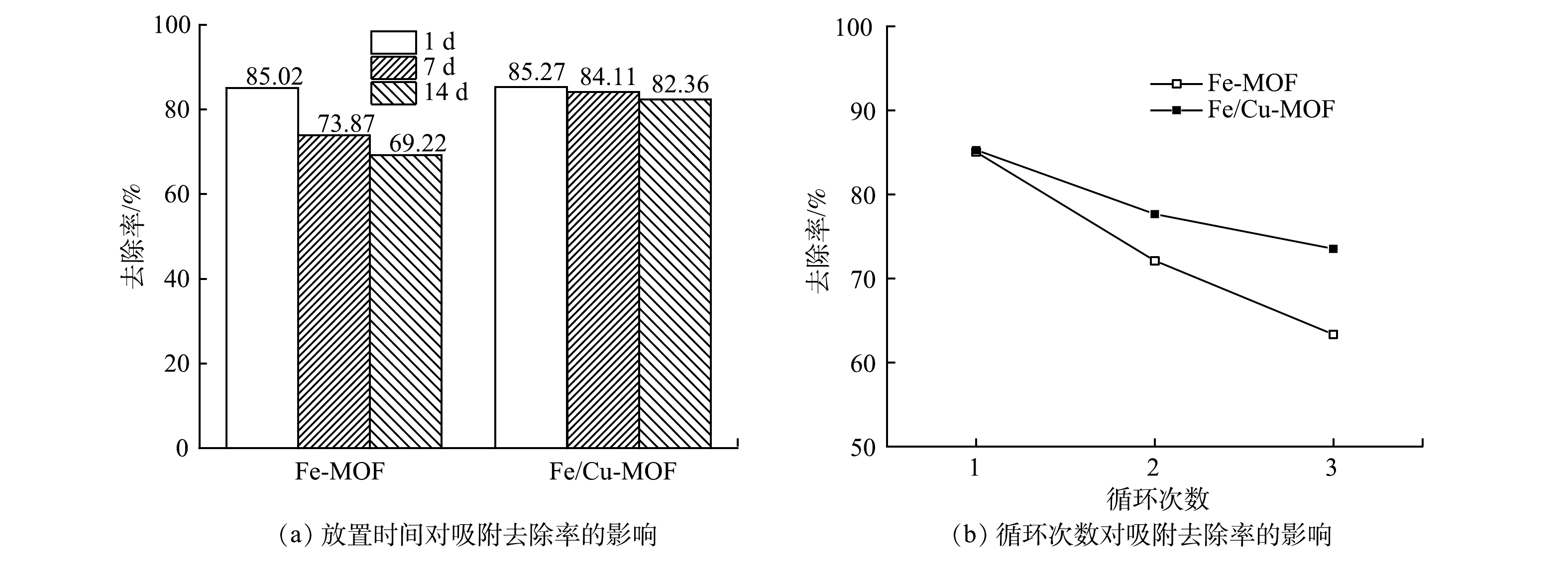 图 7 放置时间和循环次数对吸附去除率的影响Figure 7. Effect of standing time and cycles on adsorption removal efficiency
图 7 放置时间和循环次数对吸附去除率的影响Figure 7. Effect of standing time and cycles on adsorption removal efficiency2.4 初始条件对光催化降解RhB效果的影响
图8(a)和图8(b)分别反映了RhB初始质量浓度和催化剂浓度在Fe/Cu-MOF-NH2光/暗反应4 h后对RhB去除效果的影响。在催化剂质量浓度为1 mg·mL−1时,RhB的初始质量浓度越大,Fe/Cu-MOF-NH2在暗反应下对RhB的去除效果越差,而光反应下的去除效果未受到明显影响。这说明Fe/Cu-MOF-NH2的光催化活性较强。保持RhB初始质量浓度为300 mg·mL−1,当催化剂质量浓度分别为1/3、1/2、1 mg·mL−1时,催化剂质量浓度越高,反应的位点越丰富,同时染料分子与催化剂表面的有效碰撞次数也会增加,因而对RhB的降解率也越高。当RhB初始质量浓度较高时,催化剂质量浓度的降低会使Fe/Cu-MOF-NH2的光反应和暗反应下对RhB的去除率均迅速降低,但单位质量的催化剂对RhB的去除量仍呈上升趋势。这也表明Fe/Cu-MOF-NH2具有较好的催化性能。
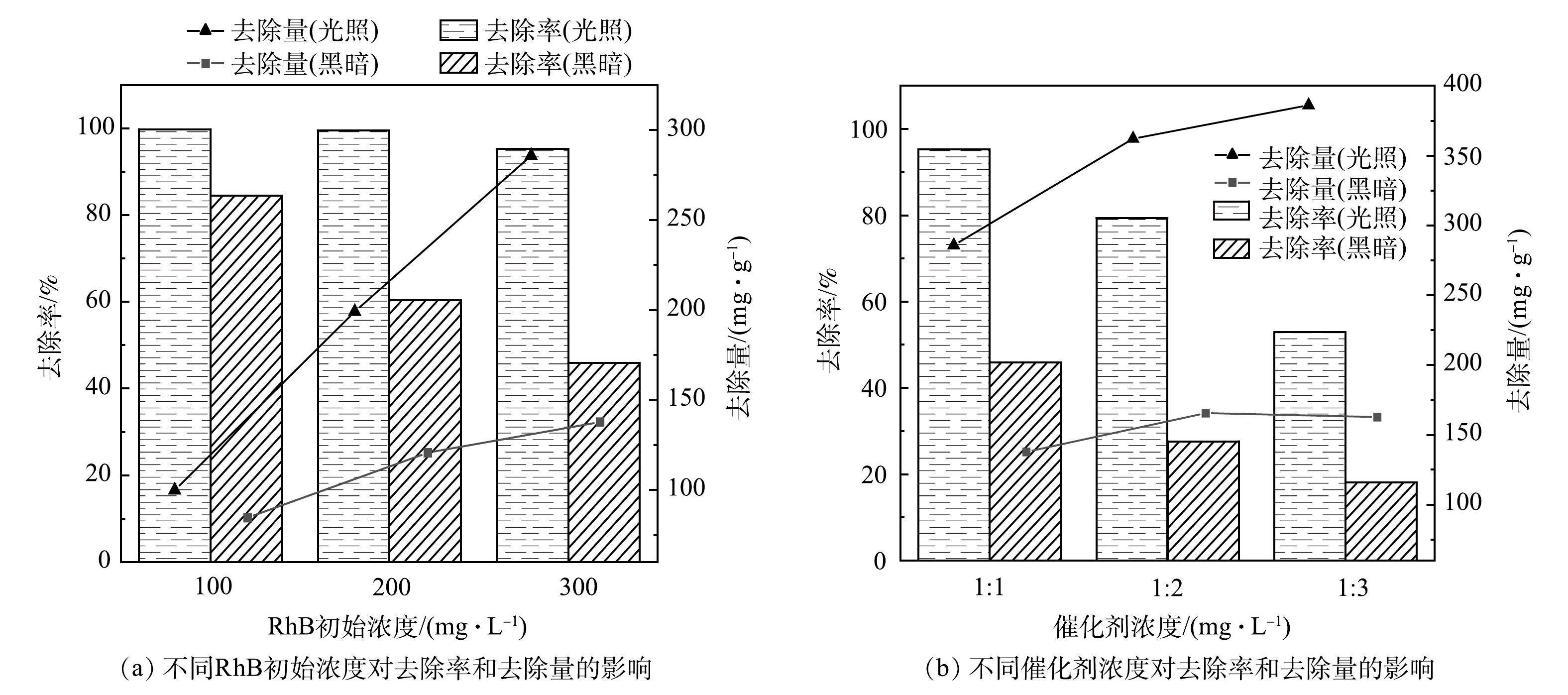 图 8 不同RhB初始浓度和催化剂浓度对去除率和去除量的影响Figure 8. Effect of RhB initial concentration and catalyst concentration on the removal rate and amount
图 8 不同RhB初始浓度和催化剂浓度对去除率和去除量的影响Figure 8. Effect of RhB initial concentration and catalyst concentration on the removal rate and amount图9反映了光照条件下在pH为3.0~11.0内测定的Fe/Cu-MOF-NH2对RhB的去除率(RhB初始质量浓度为300 mg·L−1,催化剂质量浓度为1 mg·mL−1,反应4 h)。结果表明,当pH为5~7时,Fe/Cu-MOF-NH2能保持较好的光催化性能;而当pH为3和11时,Fe/Cu-MOF-NH2对RhB去除效果下降。这是因为:较强的酸性和碱性环境均不利于MOF的稳定存在,并且随着pH的增大,溶液中OH−浓度不断增加,与脱落的金属离子形成沉淀,覆盖于催化剂表面并阻碍了反应的进行[32]。由于RhB溶液的pH稳定在4.2±0.1,且Fe/Cu-MOF-NH2在该pH条件下对RhB具有较高的去除率。因此,后续实验不再调整溶液的pH。
2.5 氨基引入量对去除RhB效果的影响
有研究[33-35]表明,氨基的引入可以实现带隙的灵活控制,混合配体(H2BDC与NH2-BDC共同参与催化剂的合成)与金属中心联接得到的MOF材料会比其中任何一种单一配体参与合成的MOF材料具备更好的光催化性能。因此,可通过改变合成材料时H2BDC与NH2-BDC的摩尔比制备得到含有不同配比混合有机配体的MOF材料,并用以探究非氨基配体/氨基配体的配比对光催化效果的影响。在RhB初始质量浓度为300 mg·L−1、催化剂质量浓度为1 mg·mL−1、pH为4.2的条件下,催化剂光催化降解RhB的结果如图10所示。配体修饰前的Fe/Cu-MOF也具备光催化降解RhB的能力,但活性较弱。氨基的引入能够提升光催化性能,在吸附效果弱于Fe/Cu-MOF的前提下,Fe/Cu-MOF-NH2光催化降解RhB的去除率仍然明显高于Fe/Cu-MOF。当H2BDC∶NH2-BDC的摩尔比为1∶1时催化剂对RhB的去除率更高。
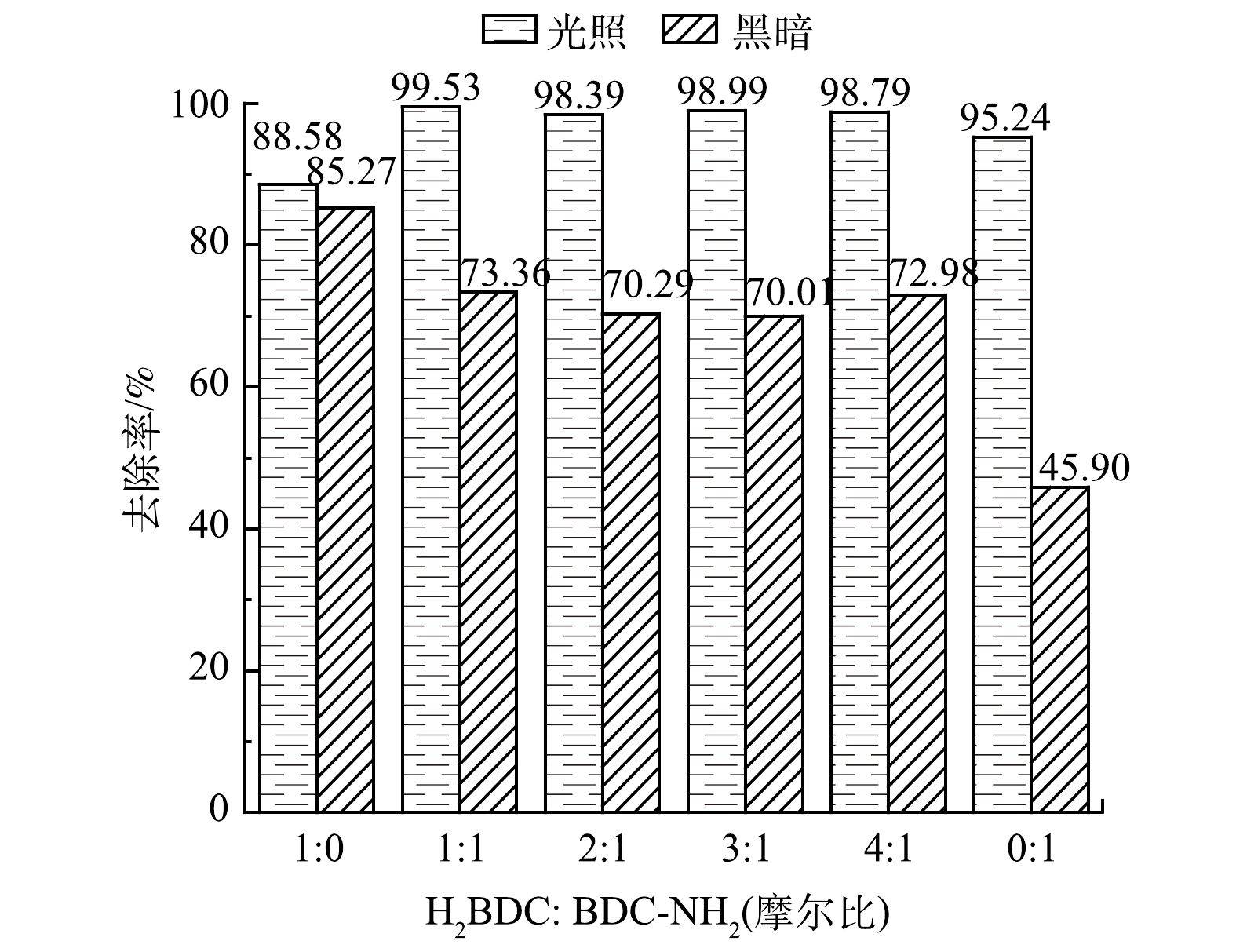 图 10 不同H2BDC:NH2-BDC(摩尔比)对RhB去除率的影响Figure 10. Effect of different H2BDC: NH2-BDC (molar ratio) on the removal rate of RhB
图 10 不同H2BDC:NH2-BDC(摩尔比)对RhB去除率的影响Figure 10. Effect of different H2BDC: NH2-BDC (molar ratio) on the removal rate of RhB此外,引入氨基后的所有样品在暗反应下的吸附性能均有不同程度的降低,其中纯NH2-BDC配体合成的样品吸附效果最弱。导致此结果的可能原因是,由于形成了更大孔径的孔,从而牺牲了部分小孔和整体孔隙度,在材料合成过程中出现的穿插结构或者小分子团簇造成的孔道堵塞均会使孔容积减小,因此,导致Fe/Cu-MOF-NH2在暗反应下对RhB的吸附效果明显低于Fe/Cu-MOF。
2.6 光催化活性机理
1)煅烧温度的影响。以H2BDC∶BDC-NH2=1∶1(摩尔比)的混合配体材料为原始材料,在空气氛下对材料在200 ℃或300 ℃下煅烧后,测试其光催化降解性能。如表3所示,N2吸脱附等温线测试结果表明BET比表面积显著降低。图11也表明,材料在暗反应下的吸附性能显著降低,而光催化活性受到的影响相对较小。这是因为:框架结构坍塌对材料的光响应活性中心Fe-O簇的破坏较小,高温煅烧后,材料生成金属氧化物Fe2O3,仍存在部分Fe-O中心。配体改变了Fe-O簇与连接体形成网状结构的孔径大小以及比表面积大小,从而影响了材料在暗反应下对RhB的吸附性能。另外,相比于同等剂量的商用磁性氧化铁,MOF材料表现出更好的光催化活性。这说明配体发挥了良好的传递电子的作用。
表 3 不同温度下煅烧的Fe/Cu-MOF-NH2的比表面积、孔容与孔径Table 3. Specific surface area, pore volume and average pore size of Fe/Cu-MOF-NH2 calcined at different temperatures样品 比表面积/(m2·g−1) 孔容/(cm3·g−1) 孔径/nm Fe/Cu-MOF-NH2 21.11 0.120 22.805 Fe/Cu-MOF-NH2-200 ℃ 18.369 0.091 23.373 Fe/Cu-MOF-NH2-300 ℃ 15.626 0.114 24.898 | Show TableDownLoad:
CSV
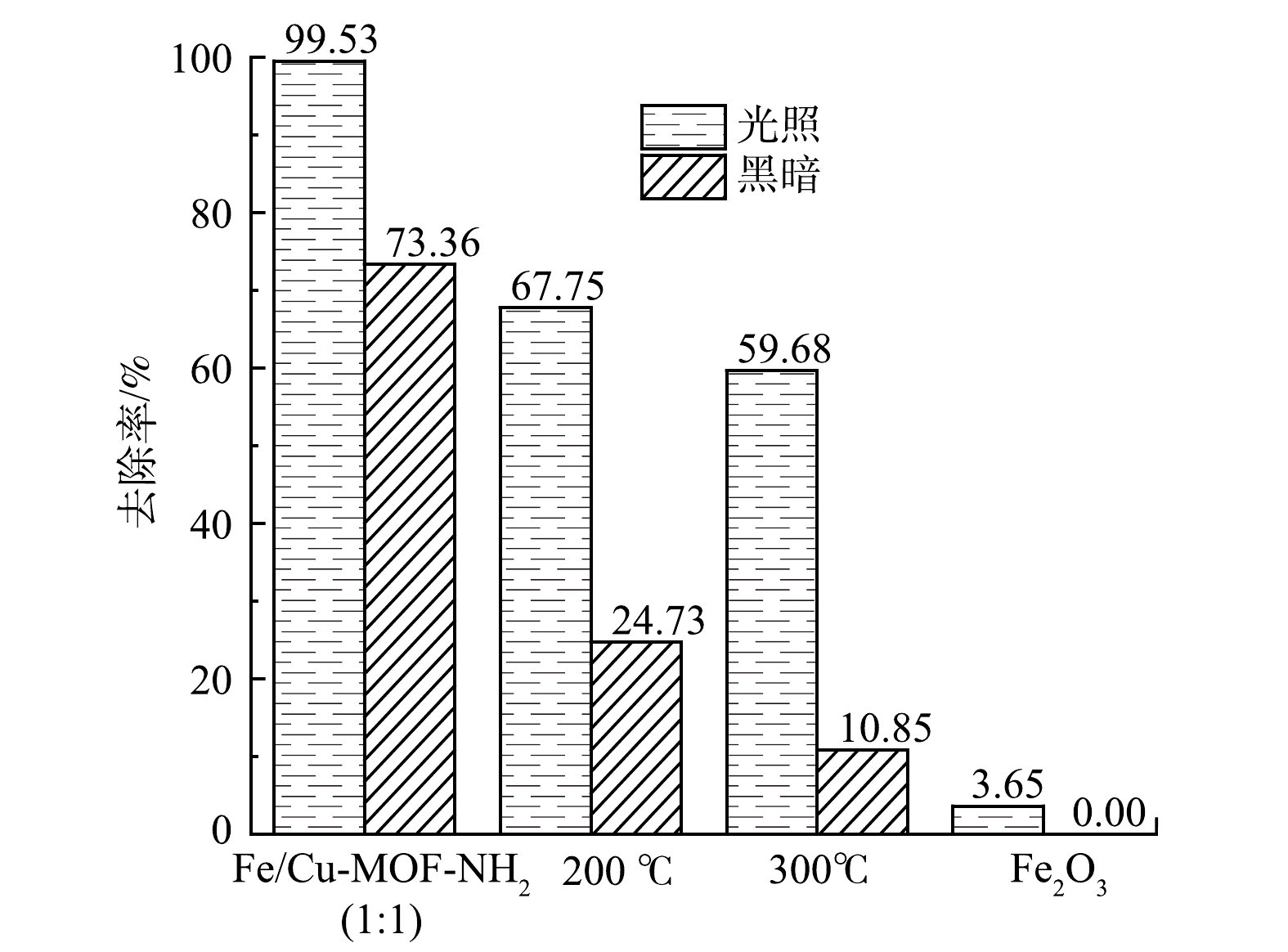 图 11 不同温度下煅烧的Fe/Cu-MOF-NH2(1∶1)对RhB去除率的影响Figure 11. Effect of Fe/Cu-MOF-NH2(1∶1) on the removal rate of RhB calcined at different temperatures
图 11 不同温度下煅烧的Fe/Cu-MOF-NH2(1∶1)对RhB去除率的影响Figure 11. Effect of Fe/Cu-MOF-NH2(1∶1) on the removal rate of RhB calcined at different temperatures2)活性物种检测。在光催化反应中,半导体受到入射光子激发产生的e-和h+通常并不直接参与反应,而是先与吸附在催化剂表面的基态分子结合,形成新的中间活性物种(·O2-、·OH等),此类高活性物种更易与目标污染物发生氧化或还原反应,从而降解去除目标污染物。为探究光催化降解RhB反应过程中的作用机制,本研究进行了Fe/Cu-MOF和 Fe/Cu-MOF-NH2在光催化反应过程中主要活性物质的淬灭实验,采用EDTA、异丙醇(IPA)、对苯醌(BQ)分别作为h+、·OH、·O2-的淬灭剂,结果如图12所示。
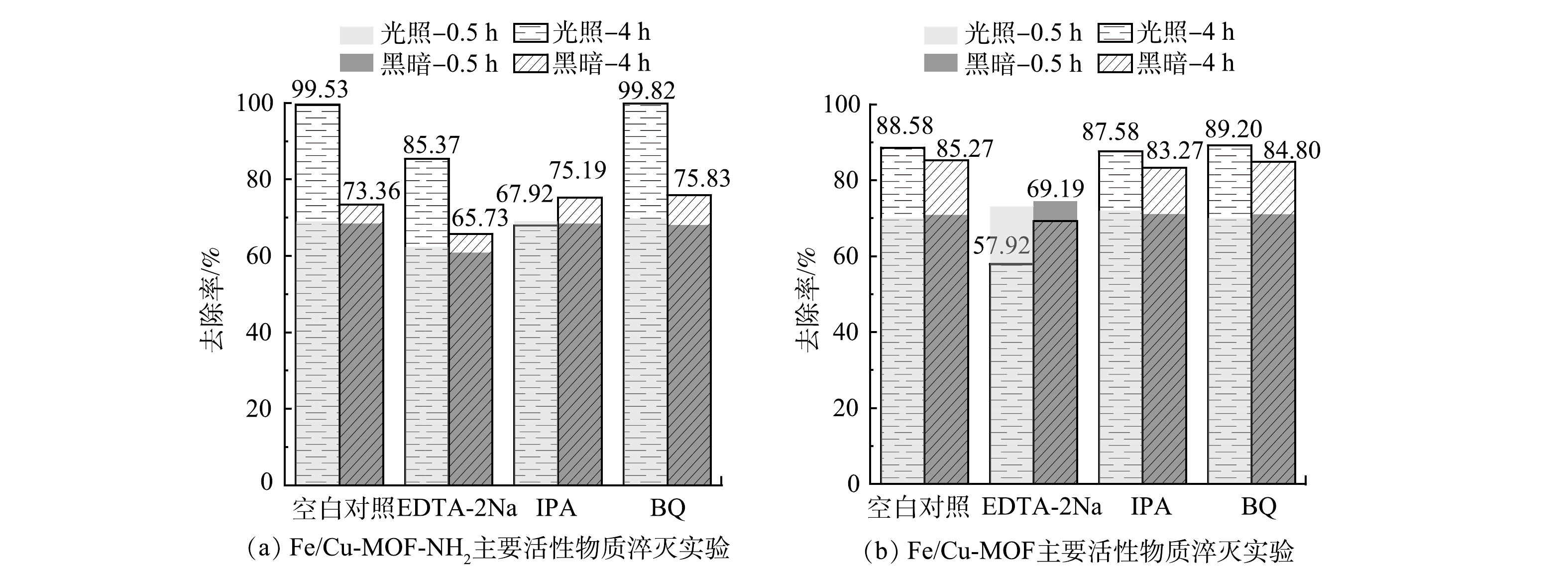 图 12 Fe/Cu-MOF- NH2和Fe/Cu-MOF主要活性物质淬灭实验Figure 12. Main active material quenching experiments for Fe/Cu-MOF-NH2 and Fe/Cu-MOF
图 12 Fe/Cu-MOF- NH2和Fe/Cu-MOF主要活性物质淬灭实验Figure 12. Main active material quenching experiments for Fe/Cu-MOF-NH2 and Fe/Cu-MOF图12反映了添加EDTA-2Na、IPA、BQ以及空白对照组在0.5 h暗吸附和4 h光反应后对RhB的去除效果。可以看出,对于Fe/Cu-MOF,淬灭剂IPA和BQ的添加对Fe/Cu-MOF在光反应下的活性几乎没有影响。这说明对应的·O2-、·OH不是Fe/Cu-MOF光催化降解RhB的主要活性物种。而EDTA-2Na的添加虽然对前期暗吸附没有造成显著影响,但后期RhB的去除率反而呈现下降的趋势,EDTA-2Na在光反应中起到了明显的抑制和破坏作用,导致先前吸附在Fe/Cu-MOF表面的染料分子脱附下来。这说明h+是Fe/Cu-MOF光催化降解RhB的主要活性物种。相比之下,RhB的氧化还原电位(1.3 V)低于Fe-MOF的VB电位[36],因此,h+氧化RhB在热力学上是完全可行的。而对于Fe/Cu-MOF- NH2,添加EDTA-2Na与IPA后光催化效果均降低,其中添加IPA对光反应的抑制更明显,说明·OH是主要活性物种。光催化反应中·OH一般来源于水中的吸附氧与水分子在氧化性物质下的共同作用。配位不饱和的铁离子作为极强的路易斯酸则很可能成为该催化反应过程的活性位点[37-38]。进一步进行除O2与添加H2O2的试验结果如图13所示。
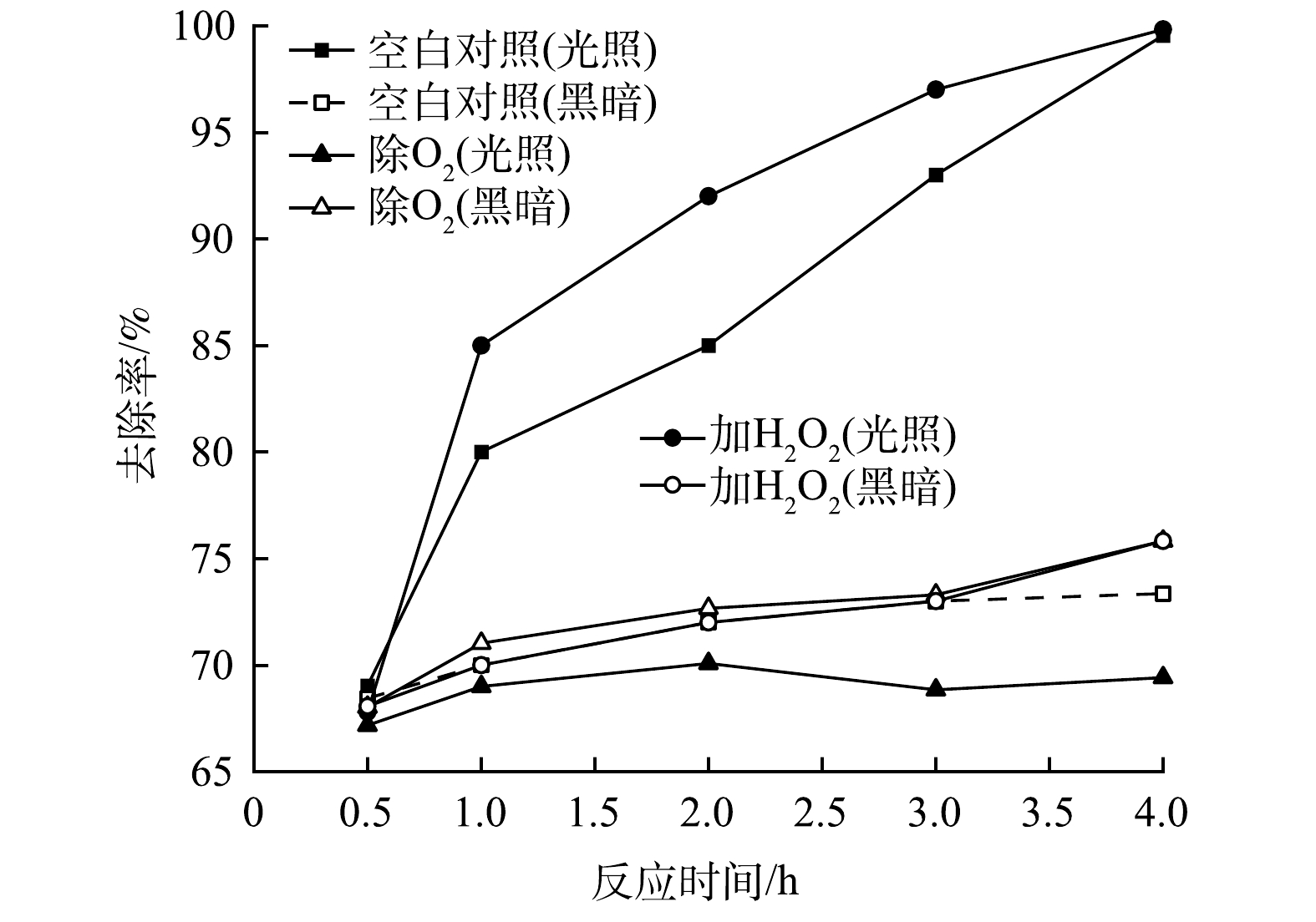 图 13 Fe/Cu-MOF- NH2除O2或添加H2O2后的光催化活性Figure 13. Photocatalytic activity of Fe/Cu-MOF-NH2 after removing O2 or adding H2O2
图 13 Fe/Cu-MOF- NH2除O2或添加H2O2后的光催化活性Figure 13. Photocatalytic activity of Fe/Cu-MOF-NH2 after removing O2 or adding H2O2通过抽除反应体系的溶解氧之后,Fe/Cu-MOF- NH2在光反应下对RhB的去除率明显降低;另外,H2O2的加入加快了RhB的光催化降解,对反应过程起到促进作用。在一种常见的催化氧化反应体系——芬顿反应中,H2O2通过与Fe(Ⅱ)发生一系列氧化还原反应生成具有高氧化性的·OH,进而完成目标物质的氧化转化。从本实验结果来看,H2O2促进了·OH的产生从而加速RhB的降解,而O2参与了·OH的生成。
图14反映了Fe/Cu-MOF与Fe/Cu-MOF- NH2在RhB初始质量浓度为300 mg·L−1,催化剂质量浓度为1 mg·mL−1,pH为4.2的条件下进行光催化降解RhB实验后剩余溶液的紫外-可见吸收光谱。RhB的特征吸收波长在554 nm,是由助色团中的孤对电子向反键轨道发生的n-π*跃迁引起的;另一个主要的吸收峰则为共轭结构中电子跃迁所致的350 nm处[39]。Fe/Cu-MOF的暗吸附反应以及Fe/Cu-MOF-NH2的光、暗反应的扫描曲线形状均与未参与反应的初始RhB溶液一致,仅有Fe/Cu-MOF的光催化反应后的溶液中出现了新的吸收峰。无氨基修饰的样品加入异丙醇后光催化活性没有受到明显抑制,说明主要活性物种不是·OH。降解后的溶液呈现淡黄色,说明单纯BDC配体合成的Fe/Cu-MOF不能实现RhB的完全氧化,而是产生了某种中间产物。
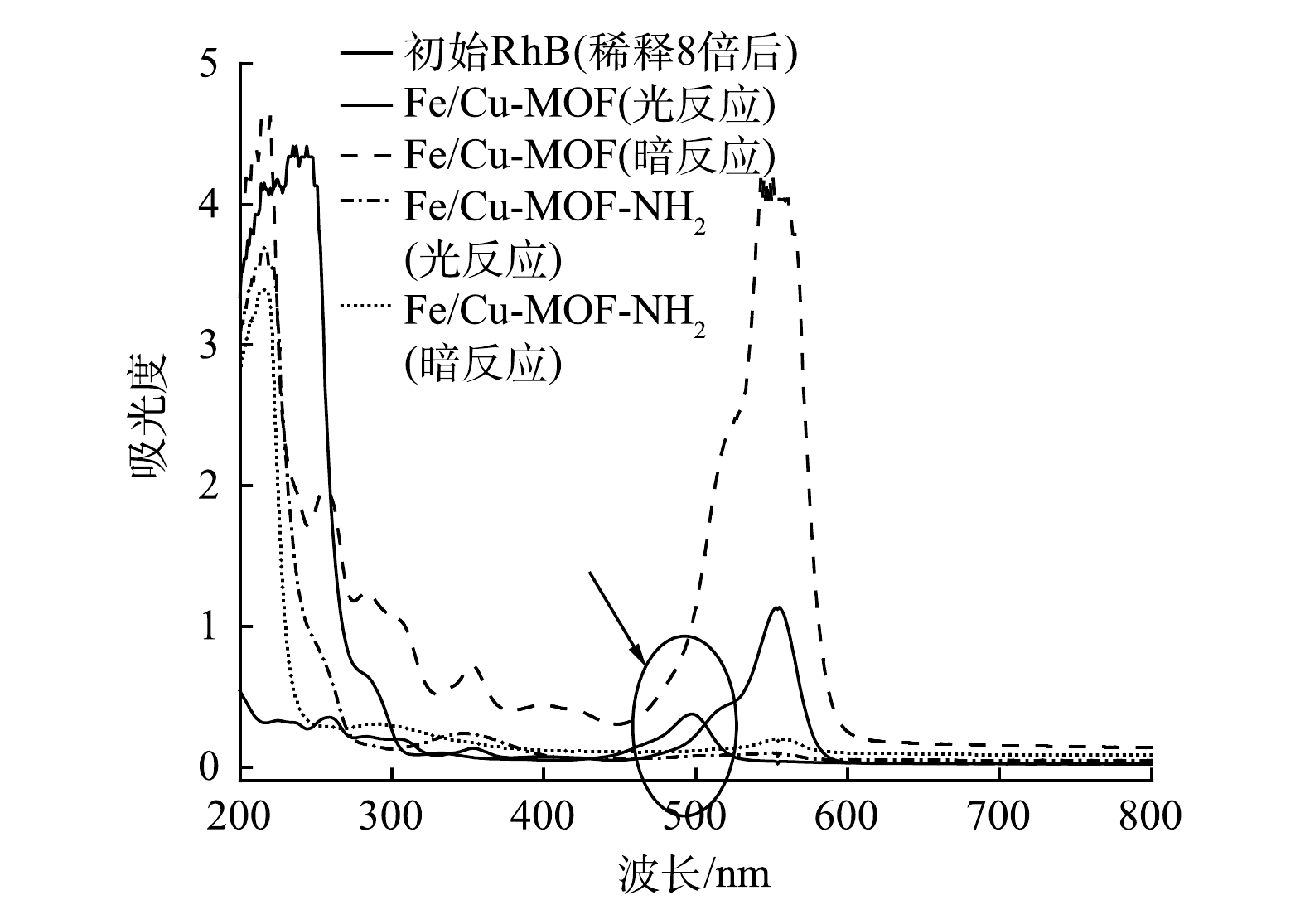 图 14 反应4 h后剩余溶液的紫外-可见吸收光谱Figure 14. UV-vis absorption spectra of the residual solution after 4 h reaction
图 14 反应4 h后剩余溶液的紫外-可见吸收光谱Figure 14. UV-vis absorption spectra of the residual solution after 4 h reaction2.7 Fe/Cu-MOF- NH2光催化氧化RhB的机理模型
Fe金属中心的MOF材料还有一个重要的特性在于Fe的变价性,类似的还有Ti,可以在Ti3+与Ti4+之间进行转换,这一性质非常有利于光电子的传输与转移。基于上述结果,提出Fe/Cu-MOF-NH2光催化降解RhB的机理模型,如图15所示。RhB与MOF中的路易斯酸位结合,即使在没有氨基的情况下也具有一定的光催化活性。在Fe/Cu-MOF-NH2中存在2种光响应中心,Fe-O簇构成的结构单元具有类金属氧化物半导体的性质,主要由O2p轨道构成的价带中的电子受光子能量激发,跃迁至导带,电子由O2-向Fe3+发生转移(FeⅢ→FeⅡ),生成的二价铁在水分子与水中溶解氧的共同作用下产生·OH。·OH具有的超高氧化还原电位可以破坏染料的生色基团,进而彻底降解染料分子。Fe-O中心是Fe/Cu-MOF-NH2光催化氧化RhB的主要活性中心。同时,氨基的引入也能提供一种新的电子转移通道。氨基受激发产生电子由配体转移至金属中心。氨基基团一旦引入配位体,其提供的孤对电子与苯环π*轨道相互作用,增加反键轨道电子密度[25],这种相互作用可以形成更高的最高占据分子轨道(HOMO)能级,使吸收边拓展至可见光区域。因此,氨基的引入加速了催化过程。
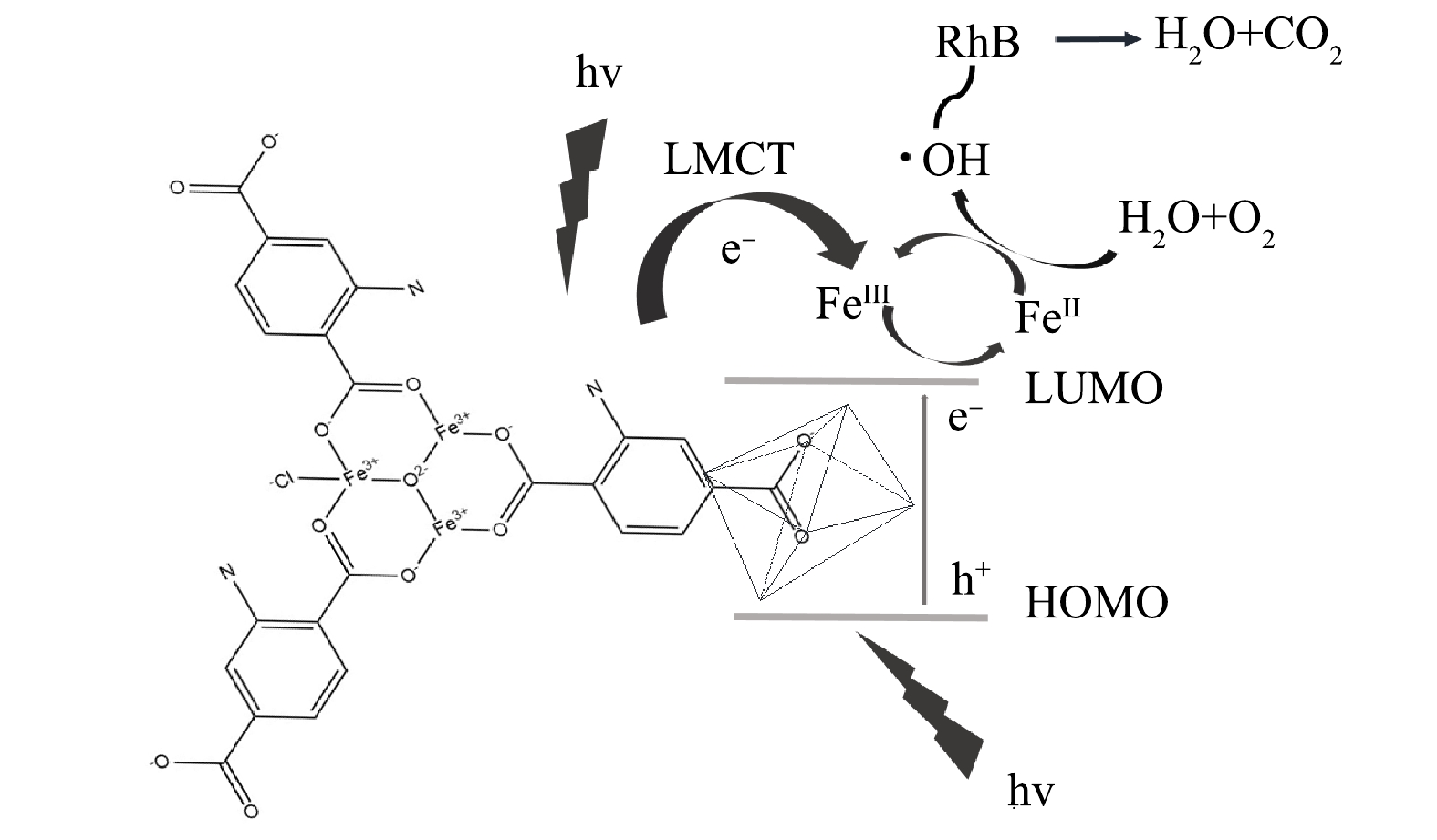 图 15 Fe/Cu-MOF-NH2光催化降解RhB过程示意图Figure 15. Schematic diagram of photocatalytic degradation of RhB by Fe/Cu-MOF-NH2
图 15 Fe/Cu-MOF-NH2光催化降解RhB过程示意图Figure 15. Schematic diagram of photocatalytic degradation of RhB by Fe/Cu-MOF-NH23. 结论
1)氨基的引入显著提升了催化剂的光催化性能。当前驱体中H2BDC∶BDC-NH2(摩尔比)为1∶1时,材料能够在保持较好吸附性能的同时达到最佳光催化活性,并在RhB初始质量浓度为300 mg·L−1,催化剂质量浓度为1 mg·L−1,pH为4.2时,反应4 h内对RhB的去除率达到99.53%。
2)氨基的引入一方面使得光吸收发生红移,另一方面促进了电子由配体转向金属中心的电荷转移机制(LMCT),从而提升了Fe/Cu-MOF-NH2的可见光催化性能。
3) Fe-O簇是Fe/Cu-MOF-NH2光催化降解RhB的主要活性中心,·OH与h+分别是 Fe/Cu-MOF-NH2与Fe/Cu-MOF在光催化过程中主要活性物种。
4) Fe/Cu-MOF-NH2光催化降解RhB的可能机制为:当受到光激发时,氨基向Fe-O簇注入电子,同时Fe-O簇中电子也会由O2p轨道跃迁至Fe的3d电子轨道;Fe(Ⅲ)接受电子转变为Fe(Ⅱ),活化氧气产生具有超高氧化还原电位的·OH,从而达到降解染料分子的目的。
-

图 1 MnO/C和MnO的X射线衍射图谱(a)和MnO/C的SEM图(b)
Figure 1. XRD patterns of MnO/C and MnO (a) and SEM images of MnO/C (b)

图 2 MnO/C-PMS反应体系降解FLO(a)、不同PMS浓度FLO降解效率的影响(b)及其拟合结果(c)、MnO/C用量对FLO降解效率的影响(d)、溶液pH对FLO降解效率影响(e)、不同材料降解FLO的性能(f)
Figure 2. Degradation of FLO in the MnO/C-PMS system (a); Effect of different PMS concentrations on FLO degradation efficiency (b) and its fitting results(c); Effect of MnO/C dosage on FLO degradation efficiency (d); Effect of initial pH on FLO degradation efficiency (e); Performance of different materials in FLO degradation (f)

图 3 MnO/C-PMS和PMS体系的EPR图谱(a、b);His和TBA对MnO/C-PMS体系降解FLO的影响(c)
Figure 3. The EPR spectra of MnO/C-PMS and PMS system (a, b);Effect of FLO removal with the addition of TBA and His in the MnO/C-PMS system (c)
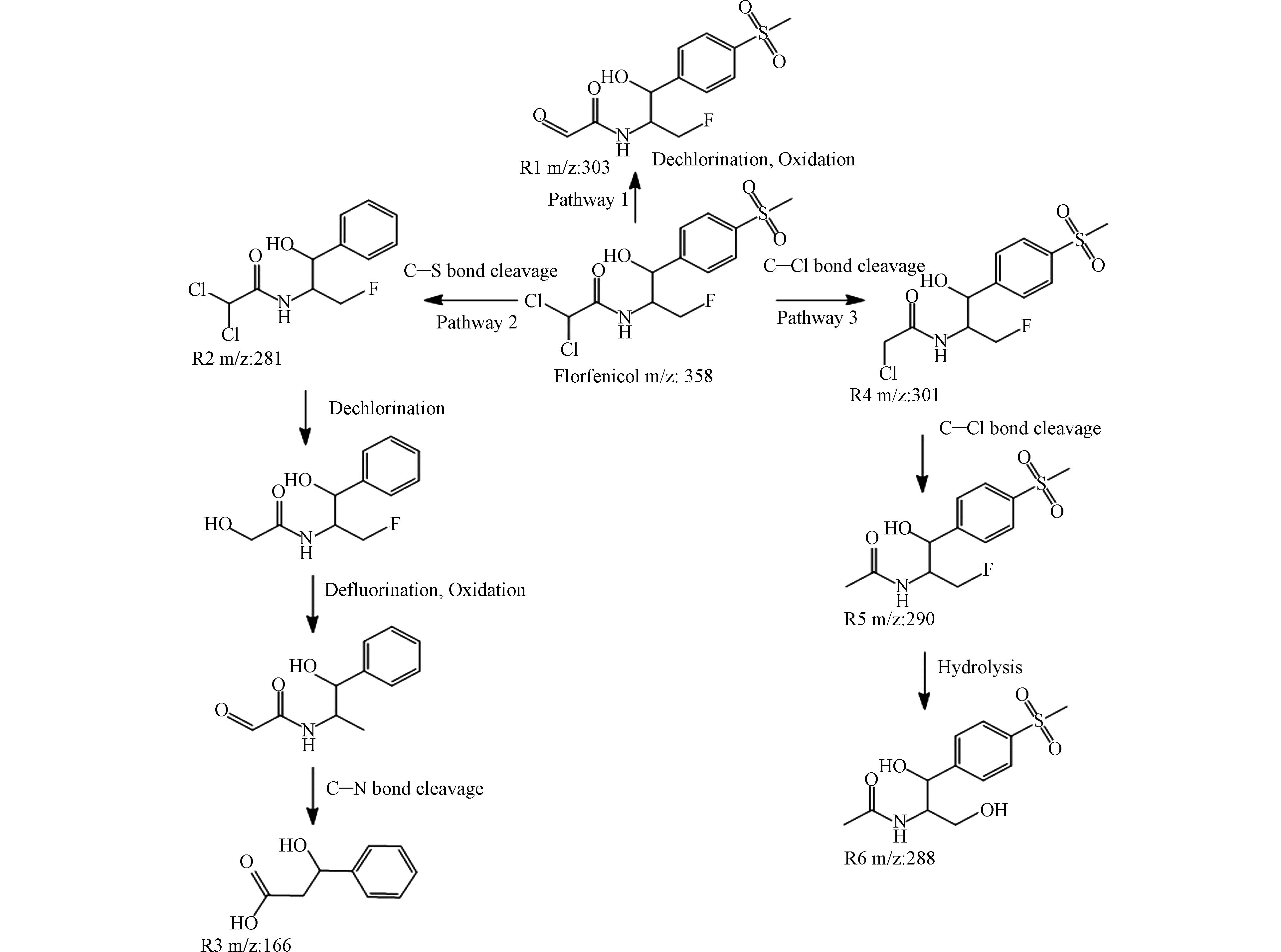
图 5 MnO/C-PMS体系降解FLO路径
Figure 5. The degradation pathway of FLO in the MnO/C-PMS system
-
[1] YU L H, ZHENG S, GAO Q. Government environmental regulation strategy for new pollutants control in mariculture[J]. Marine Policy, 2023, 150: 105545. doi: 10.1016/j.marpol.2023.105545 [2] CABELLO F C. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment[J]. Environmental Microbiology, 2006, 8(7): 1137-1144. doi: 10.1111/j.1462-2920.2006.01054.x [3] ORLANDO E A, COSTA ROQUE A G, LOSEKANN M E, et al. UPLC–MS/MS determination of florfenicol and florfenicol amine antimicrobial residues in Tilapia muscle[J]. Journal of Chromatography B, 2016, 1035: 8-15. doi: 10.1016/j.jchromb.2016.09.013 [4] OLIVEIRA A S, ALVES M, LEITÃO F, et al. Bioremediation of coastal aquaculture effluents spiked with florfenicol using microalgae-based granular sludge–a promising solution for recirculating aquaculture systems[J]. Water Research, 2023, 233: 119733. doi: 10.1016/j.watres.2023.119733 [5] ESMAILI Z, CHESHMBERAH F, SOLAIMANY NAZAR A R, et al. Treatment of florfenicol of synthetic trout fish farm wastewater through nanofiltration and photocatalyst oxidation[J]. Environmental Technology, 2017, 38(16): 2040-2047. doi: 10.1080/09593330.2016.1245359 [6] BABU PONNUSAMI A, SINHA S, ASHOKAN H, et al. Advanced oxidation process (AOP) combined biological process for wastewater treatment: A review on advancements, feasibility and practicability of combined techniques[J]. Environmental Research, 2023, 237: 116944. doi: 10.1016/j.envres.2023.116944 [7] XIAO S, CHENG M, ZHONG H, et al. Iron-mediated activation of persulfate and peroxymonosulfate in both homogeneous and heterogeneous ways: A review[J]. Chemical Engineering Journal, 2020, 384: 123265. doi: 10.1016/j.cej.2019.123265 [8] ZHAO X, AN Q D, XIAO Z Y, et al. Seaweed-derived multifunctional nitrogen/cobalt-codoped carbonaceous beads for relatively high-efficient peroxymonosulfate activation for organic pollutants degradation[J]. Chemical Engineering Journal, 2018, 353: 746-759. doi: 10.1016/j.cej.2018.07.171 [9] ANJORIN E O, ALFRED M O, SOTUNDE B, et al. Overview of the mechanism of degradation of pharmaceuticals by persulfate/peroxysulfate catalysts[J]. ChemBioEng Reviews, 2024, 11(4): e202300079. doi: 10.1002/cben.202300079 [10] YANG Y Y, ZHANG P P, HU K S, et al. Sustainable redox processes induced by peroxymonosulfate and metal doping on amorphous manganese dioxide for nonradical degradation of water contaminants[J]. Applied Catalysis B: Environmental, 2021, 286: 119903. doi: 10.1016/j.apcatb.2021.119903 [11] LIN K Y , CHEN Y C, LIN Y F. LaMO3 perovskites (M=Co, Cu, Fe and Ni) as heterogeneous catalysts for activating peroxymonosulfate in water[J]. Chemical Engineering Science, 2017, 160: 96-105. [12] WANG Y X, XIE Y B, CHEN C M, et al. Synthesis of magnetic carbon supported manganese catalysts for phenol oxidation by activation of peroxymonosulfate[J]. Catalysts, 2017, 7(1): 3. [13] ORGE C A, ÓRFÃO J J M, PEREIRA M F R. Composites of manganese oxide with carbon materials as catalysts for the ozonation of oxalic acid[J]. Journal of Hazardous Materials, 2012, 213: 133-139. [14] 李珏秀, 施启旭, 赵锐, 等. 锰基催化剂用于活化过硫酸盐降解有机废水的研究进展[J]. 环境化学, 2023, 42(11): 3861-3877. doi: 10.7524/j.issn.0254-6108.2023011704 LI J X, SHI Q X, ZHAO R, et al. Research progress on manganese based catalysts for activating persulfate degradation of organic wastewater[J]. Environmental Chemistry, 2023, 42(11): 3861-3877 (in Chinese). doi: 10.7524/j.issn.0254-6108.2023011704
[15] LIU L, LIU Z, CHEN Y, et al. In-situ synthesis of manganese oxide-carbon nanocomposite and its application in activating persulfate for bisphenol F degradation[J]. Science of the Total Environment, 2021, 772: 144953. doi: 10.1016/j.scitotenv.2021.144953 [16] DO S H, KWON Y J, BANG S J, et al. Persulfate reactivity enhanced by Fe2O3-MnO and CaO-Fe2O3-MnO composite: Identification of composite and degradation of CCl4 at various levels of pH[J]. Chemical Engineering Journal, 2013, 221: 72-80. doi: 10.1016/j.cej.2013.01.097 [17] DIAO Z H, QIAN W, GUO P R, et al. Photo-assisted degradation of bisphenol A by a novel FeS2@SiO2 microspheres activated persulphate process: Synergistic effect, pathway and mechanism[J]. Chemical Engineering Journal, 2018, 349: 683-693. doi: 10.1016/j.cej.2018.05.132 [18] ZHOU Q X, SONG C L, WANG P F, et al. Generating dual-active species by triple-atom sites through peroxymonosulfate activation for treating micropollutants in complex water[J]. Proceedings of the National Academy of Sciences of the United States of America, 2023, 120(13): e2300085120. [19] 郑佳慧, 王嘉妮, 柯佳琪, 等. 碳氮包覆纳米Fe3O4的非自由基路径光催化去除养殖废水中四环素的机制研究[J]. 环境科学研究, 2023, 36(7): 1306-1316. ZHENG J H, WANG J N, KE J Q, et al. Mechanism of tetracycline removal from aquaculture wastewater by carbon nitrogen coated nano-Fe3O4 via non-free radical photocatalysis[J]. Research of Environmental Sciences, 2023, 36(7): 1306-1316 (in Chinese).
[20] LING C Y, QIN X Z, JIANG L J, et al. Investigation of the Effect of Manganese Oxides on the Reduction of Hexavalent Chromium by Sodium Alginate-Dispersed Nano-Zero-Valent Iron and the Mechanism[J]. Water Air Soil Pollut, 2023, 234(3): 187. doi: 10.1007/s11270-023-06209-8 [21] HUANG G X, WANG C Y, YANG C W, et al. Degradation of Bisphenol A by peroxymonosulfate catalytically activated with Mn1.8Fe1.2O4 nanospheres: Synergism between Mn and Fe[J]. Environmental Science & Technology, 2017, 51(21): 12611-12618. [22] HU P D, LONG M C. Cobalt-catalyzed sulfate radical-based advanced oxidation: A review on heterogeneous catalysts and applications[J]. Applied Catalysis B: Environmental, 2016, 181: 103-117. doi: 10.1016/j.apcatb.2015.07.024 [23] YAO J Y, WU N N, TANG X S, et al. Methyl phenyl sulfoxide (PMSO) as a quenching agent for high-valent metal-oxo species in peroxymonosulfate based processes should be reconsidered[J]. Chemical Engineering Journal Advances, 2022, 12: 100378. doi: 10.1016/j.ceja.2022.100378 [24] DENG J, GE Y J, TAN C Q, et al. Degradation of ciprofloxacin using α-MnO2 activated peroxymonosulfate process: Effect of water constituents, degradation intermediates and toxicity evaluation[J]. Chemical Engineering Journal, 2017, 330: 1390-1400. doi: 10.1016/j.cej.2017.07.137 [25] HUANG J Z, ZHONG S F, DAI Y F, et al. Effect of MnO2 Phase Structure on the Oxidative Reactivity toward Bisphenol A Degradation[J]. Environmental Science & Technology, 2018, 52(19): 11309-11318. [26] XU H D, ZHANG Y C, LI J J, et al. Heterogeneous activation of peroxymonosulfate by a biochar-supported Co3O4 composite for efficient degradation of chloramphenicols[J]. Environmental Pollution, 2020, 257: 113610. doi: 10.1016/j.envpol.2019.113610 [27] PENG Y F, XUE C J, LUO J Y, et al. Lanthanum-doped magnetic biochar activating persulfate in the degradation of florfenicol[J]. Science of The Total Environment, 2024, 916: 170312. doi: 10.1016/j.scitotenv.2024.170312 [28] ZHAO Y, LI B, LI Y, et al. Synergistic activation of peroxymonosulfate between Co and MnO for bisphenol A degradation with enhanced activity and stability[J]. Journal of Colloid and Interface Science, 2022, 623: 775-786. doi: 10.1016/j.jcis.2022.05.105 [29] CHEN J S, LUO H Y, LUO D Y, et al. New insights into the degradation of nitrobenzene by activated persulfate with sulfidated nanoscale zero-valent iron: Synergistic effects of reduction and reactive oxygen species oxidation[J]. Separation and Purification Technology, 2023, 322: 124252. doi: 10.1016/j.seppur.2023.124252 [30] XU A L, WU D H, ZHANG R, et al. Bio-synthesis of Co-doped FeMnOx and its efficient activation of peroxymonosulfate for the degradation of moxifloxacin[J]. Chemical Engineering Journal, 2022, 435: 134695. doi: 10.1016/j.cej.2022.134695 [31] CHEN M J , YANG T X, ZHAO L Y, et al. Manganese oxide on activated carbon with peroxymonosulfate activation for enhanced ciprofloxacin degradation: Activation mechanism and degradation pathway[J]. Applied Surface Science, 2024, 645: 158835. [32] ZHANG Y, LI J H, ZHOU L, et al. Aqueous photodegradation of antibiotic florfenicol: kinetics and degradation pathway studies[J]. Environmental Science and Pollution Research International, 2016, 23(7): 6982-6989. doi: 10.1007/s11356-015-5897-1 [33] WANG X Y, LUO X Y, LI R, et al. Boosting peroxymonosulfate activation over partial Zn-substituted Co3O4 for florfenicol degradation: Insights into catalytic performance, degradation mechanism and routes[J]. Chemical Engineering Journal, 2024, 491: 152197. doi: 10.1016/j.cej.2024.152197 [34] TANG Z, KONG Y F, QIN Y, et al. Performance and degradation pathway of florfenicol antibiotic by nitrogen-doped biochar supported zero-valent iron and zero-valent copper: A combined experimental and DFT study[J]. Journal of Hazardous Materials, 2023, 459: 132172. doi: 10.1016/j.jhazmat.2023.132172 [35] CHEN Z H, CHEN J D, TAN S D, et al. Dechlorination Helps Defluorination: Insights into the Defluorination Mechanism of Florfenicol by S-nZVI and DFT Calculations on the Reaction Pathways[J]. Environmental Science & Technology, 2024, 58(5): 2542-2553. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 851
- HTML全文浏览数: 851
- PDF下载数: 22
- 施引文献: 0
