


-
我国夏季臭氧污染形势严峻[1],挥发性有机化合物(volatile organic compounds, VOCs)是近地面臭氧的重要前体物,我国大部分地区臭氧污染为VOCs控制型[2]. 近年来,我国采取了极为严格的VOCs削减控制方案,但臭氧污染形势改善不明显,有些地区臭氧污染出现反弹[3].
制定VOCs削减控制策略基于大气VOCs观测结果,准确客观的观测结果是制定精准VOCs削减策略的技术基础. 2018年起我国开始在重点地区开展环境空气VOCs的手工监测工作,关于大气VOCs污染特征、来源解析、环境影响评价的研究报道也层出不穷[4 − 6],相关监测及研究工作的开展主要基于手工采样监测结果. 其中苏玛罐采样具有可采集全组分样品、保存时间长[7 − 8]的特点,是实验室分析最常用的采样方式[9 − 11]. 大气中存在O3、OHˑ、NO3等强氧化性基体物质,在样品采集、运输、保存和分析的过程中,VOCs会与大气中的强氧化剂发生化学反应而消耗损失[12 − 13],影响样品的代表性. 影响苏玛罐采集有代表性大气VOCs样品的关键因素主要包括苏玛罐内壁的惰性化水平、密封性、洁净度,目前主要关注苏玛罐内壁对VOCs的吸附作用[14],没有考虑采集样品后,VOCs可能会继续与大气中的氧化剂发生大气化学反应而造成浓度、组成的变化. 目前大气中的臭氧对VOCs测定的影响已引起关注,但结论不统一:邵敏等[15]提出臭氧在大气VOCs样品热解吸进样时会与烯烃反应,但也有研究认为大气中臭氧不影响VOCs的测定[16],该研究是在一定浓度的标气样品中人为加入一定浓度的臭氧,研究臭氧的存在对样品保存的影响,但该研究对象未能反应真实的大气环境,大气中除有臭氧,还存在OH、NO3等强氧化自由基[17]. 客观真实的大气VOCs观测结果是制定大气VOCs精准减排政策的基础和依据,也是臭氧污染治理成效的保障,目前未见有关真实大气环境样品中大气氧化性基体对VOCs测定影响深入研究的报道. VOCs化学组成繁杂,部分物质化学反应活性强,大气寿命从几分钟到几小时[12],有研究表明活性较强的VOCs样品预先通过KI小柱或Na2SO3小柱,可有效去除臭氧等强氧化性基体,保证VOCs组分的完整性[13]. 国家环境保护标准《环境空气 醛、酮类化合物的测定 高效液相色谱法》(HJ 683-2014)规定在采样管前串联KI除臭氧小柱采集醛、酮类化合物[18],因此,可使用KI除臭氧小柱研究大气中臭氧等强氧化性基体对VOCs样品采集的影响.
本研究以大气中常见的107种VOCs为研究目标,采用罐采样-大气预浓缩-气相色谱/质谱氢火焰检测器法分析技术[19],对比苏玛罐采样口前串联和不串联KI小柱两种采样方式,研究大气中氧化性基体对VOCs测定的影响;通过臭氧生成潜势(Ozone formation potential, OFP)评价,研究不同采样方式进行环境影响评价结果的客观性. 探讨大气中氧化基体对VOCs测定结果的影响,为全国开展大气VOCs精准监测提供技术支持.
-
采样点位于济南市东部的山东省生态环境厅院内,南面为山地,植被茂密;北面为居民、商业混合区,西面为加气站,可反映区域大气整体状况. 采样时间为2022年5月—7月大气氧化性强的月份,限流阀调节采样流速,13:00—15:00采集2 h样品. 每次采样同步采集2个样品,其中一个样品直接采集,另一个在限流阀前端接KI小柱,去除大气中氧化性基体,每个样品使用一个新的KI小柱. 本次研究采集了7 d共7组样品.
-
Shimadzu QP 2020型气相色谱/质谱联用仪(日本);
3100 型自动清罐仪(带恒温加热箱)、4700 型动态稀释仪(ENTECH);新购置的内壁硅烷化处理的SUMMA罐(ENTECH),3.2 L;电子制冷大气预浓缩仪、除水仪(MARKS);挥发性有机物标气(1.0 μmol·mol−1, 四川中测标物有限公司,包括57种PAMS、47种TO15、乙醇、柠檬烯和α-蒎烯));内标标准气(1.0 μmol·mol−1, 四川中测标物有限公司,包括一溴氯甲烷、1,2-二氟苯和氯苯-d5),高纯氮气≥99.999%,高纯氦气≥99.999%;DB-624毛细管柱(60 m×0.32 mm ×1.8 μm),PLOT 毛细管柱(30 m×0.32 mm× 10 μm),KI小柱(1.4 g/2.5 cc,Agela Technologies). -
自动进样器取样200 mL,经-30 ℃的空管除水后进入-30℃吸附剂冷阱中进行捕集浓缩去除二氧化碳、氮气等环境中常量组分的干扰,以最快的速度加热至300 ℃,解吸进入色谱柱分离. 气相色谱条件:色谱柱升温程序:40 ℃保持3 min,以8 ℃·min−1升温到50 ℃保持2 min,再以8 ℃·min−1升温到150 ℃,保持10 min,再以15 ℃·min−1升温到185 ℃,保持16.5 min.
-
1.00 μmol·mol−1有证标准物质二级稀释至2.00 nmol·mol−1作为标准使用气;1.00 μmol·mol−1有证内标储备气稀释至50.0 nmol·mol−1标准使用气. 分别取50 、100 、200 、400 、800 mL标准使用气和25 mL内标使用气,建立内标标准曲线,相对响应因子法定量,每种化合物的相对响应因子的相对标准偏差≤20%,否则应进行空白检验;每20个样品进行1次中间浓度点校准,偏差≤20%,否则查找原因重新绘制曲线;记录每个样品的内标响应,与校准曲线的偏差≤30%. 为保证不同采样方式所用的苏玛罐性能的一致性,使用2个新购置苏玛罐循环使用,每个苏玛罐采样前均做空白检查和惰性化检查. 采样前,采用高纯氮气模拟样品经过KI除氧化基体小柱,采集到苏玛罐中进行定性分析,未检出挥发性有机物成分,表明KI不会引入干扰物质.
-
本研究采用最大增量反应活性(maximum incremental reactivity,MIR),计算VOCs物种O3生成的潜势OFP,评价不同化合物在不同采样方式下对O3生成的影响.
计算公式如下:
式中,OFP(j)为化合物j的臭氧生成潜势(μg·m−3);Conc.(j)为化合物j浓度(单位μg·m−3);MIR(j):单位量化合物j最大增量反应活性(单位质量VOC生成单位量的O3),具体数值引用Carter and William[20]的研究
-
为研究大气中氧化性基体对VOCs成分定性分析的影响,通过质谱全扫描模式对样品进行分析,检出率高于80%的化合物结果见表1. 从表1可以看出,去除氧化性基体后的样品共定性检出110种化合物,包括55种烷烃、23种含氧有机物、13种烯烃、8种卤代烃、7种苯系物及包括乙炔在内的4种其他化合物. 与未去除氧化性基体的样品相比,去除氧化性基体后检出的含氧化合物、长链烷烃、长链烯烃更多. 含氧化合物包括乙醛、丙醛、3-甲基丁醛等醛类化合物及2-丁基-1-辛醇、5-甲基-2-(1-异丙基)-1-己醇、2-乙基-4-甲基-1-戊醇、2-己基-1-癸醇、2-丁基-1-辛醇等长链的醇类化合物;长链烷烃化合物包括5-甲基十一烷、十五烷、十六烷等;长链烯烃类化合物包括5-甲基-1-己烯、4,5-二甲基-1-己烯、2,6-二甲基-3-庚烯、2,4-二甲基-1-庚烯、3-十四烯、1-壬烯、2-十二烯等,只有在去除氧化性基体采集的样品中有检出.
未除氧化性基体的采样方式采集的样品中定性检出的化合物种类较少,共定性检出40种化合物,绝大部分化合物与去除氧化性基体样品定性检出的种类一致,包括乙烷、乙烯、乙炔、丙烷、丙烯等空气中常见的短链VOCs;仅2-氧基丙酸乙酯、环己烷、2,4-己二炔、6-甲基庚基乙烯基醚和2-甲基丙基环己烷5种化合物只在未除氧化性基体的样品中有定性检出,占检出总量的14.3%. 未除氧化性基体的样品未检出长链烯烃、长链烷烃、长链醇类化合物,表明此类化合物在氧化基体环境中不稳定,采样后或测定过程中会继续参与氧化反应而损失. 总体看,去除氧化性基体后定性检出的化合物种类多,长链的烷烃、烯烃、醇类物质在未去除氧化性基体的样品中均未检出,大气中的部分VOCs会在样品采集、运输和保存等环节继续与氧化性基体反应而消耗损失,样品中的氧化性基体明显影响VOCs组成的分析判定.
-
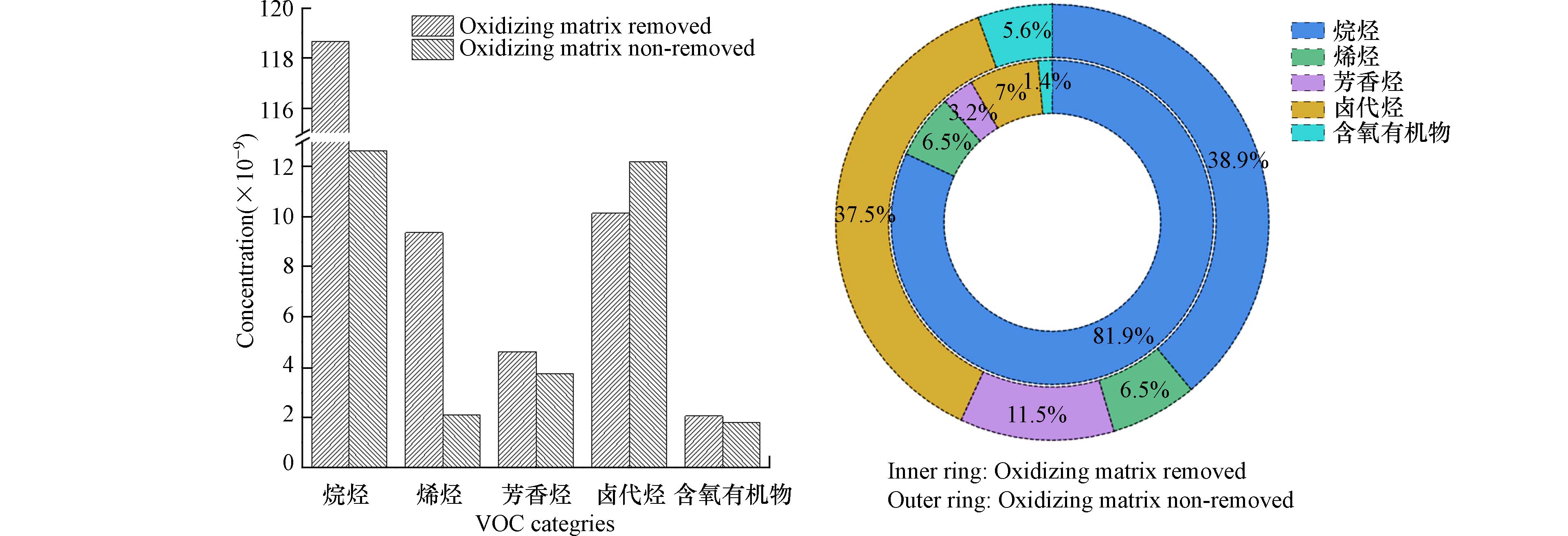
为研究大气中氧化基体对VOCs浓度测定的影响,对107种VOCs(29种烷烃,13种烯烃,35种卤代烃,10种含氧有机物,18种芳香烃、乙炔及二硫化碳)测定,分别将去除和未去除氧化性基体的7组样品的每种化合物进行浓度平均,将两种采样方式每类化合物的浓度均值分别加和得到烷烃、烯烃、芳香烃、含氧有机物和卤代烃的浓度,比较两种采样方式对每类化合物测定结果的影响(图1). 去除氧化性基体的TVOCs平均浓度为144.8 nmol·mol−1,其中烷烃类化合物浓度最高(平均浓度为118.7 nmol·mol−1),占81.9%,主要由2,2-二甲基丁烷(平均浓度15.7 nmol·mol−1)、2,3-二甲基丁烷(平均浓度12.2 nmol·mol−1)、环戊烷(平均浓度15.5 nmol·mol−1)和正己烷贡献(平均浓度28.1 nmol·mol−1);卤代烃浓度第二(平均浓度为10.1 nmol·mol−1),占7%;烯烃浓度第三(平均浓度9.36 nmol·mol−1),占6.5%;芳香烃浓度第四(平均浓度为4.63 nmol·mol−1),占3.2%;含氧有机物浓度最低(平均浓度为2.05 nmol·mol−1),占1.4%. 未去除氧化性基体样品测定的平均TVOCs浓度低(平均浓度为32.5 nmol·mol−1),烷烃(平均浓度为12.6 nmol·mol−1)和卤代烃(平均浓度为12.2 nmol·mol−1)浓度最高,分别占TVOCs的 38.9%和37.5%;芳香烃浓度第三(平均浓度为3.73 nmol·mol−1),占11.5%;烯烃浓度第四(平均浓度为2.11 nmol·mol−1),占6.5%;含氧有机物浓度最低(平均浓度为1.81 nmol·mol−1),占5.6%. 由此可以看出,样品中氧化性基体的存在对测定样品各物种的浓度及组成特征均有显著影响. 其中样品中氧化性基体对烷烃、烯烃类化合物浓度测定结果的影响最大,分别是未去除氧化性基体浓度的9.42倍和4.44倍;对芳香烃、卤代烃和含氧有机物影响小,去除与不去除氧化性基体采集样品测定浓度的相对偏差分别为10.7%、9.19%和6.31%. 因此,大气中的氧化性基体对VOCs浓度的测定及物种组成的分析都有影响,其中对烷烃和烯烃浓度测定的影响最为显著,表明烷烃、烯烃活性强,在样品采集、保存、运输和测定的过程中,会与氧化基体反应消耗损失;测定大气中的芳香烃、卤代烃及含氧有机物,受大气中强氧化性基体的影响较小.
本研究共定量检出60种目标化合物,图2为两种不同采样方式测定的化合物的平均浓度. 从图2可以看出,烷烃中乙烷、丙烷、正丁烷、异丁烷等4种C2—C4低碳烷烃受大气中氧化性基体影响小,去除氧化性基体是不除氧化性基体浓度的1.5倍左右;C5以上的支链烷烃和环状烷烃的测定受大气中氧化性基体影响大,其中对2-甲基庚烷、环戊烷和2,2-二甲基丁烷影响最大,分别是不去氧化性基体样品浓度测定值的542倍、174倍和116倍,去除氧化基体样品中2-甲基庚烷、环戊烷和2,2-二甲基丁烷的浓度范围分别为0.12—22.1 nmol·mol−1、0.10—31.7 nmol·mol−1和0.35—23.7 nmol·mol−1之间,未去除氧化氧化性基体的浓度范围分别为:未检出—0.04 nmol·mol−1,0.02—0.25 nmol·mol−1和0.03—0.25 nmol·mol−1. 大气中氧化基体对2,3-二甲基丁烷、甲基环戊烷、3-甲基庚烷及正己烷的影响次之,分别是不除氧化性基体样品浓度测定值的98倍、68倍、38倍和33倍,去除氧化基体样品中2,3-二甲基丁烷、甲基环戊烷、3-甲基庚烷和正己烷的浓度范围分别为:0.28—19.8 nmol·mol−1,0.04—24.2 nmol·mol−1,0.03—1.71 nmol·mol−1和0.40—71.8 nmol·mol−1,未去除氧化性基体的浓度范围分别为:0.03—0.25 nmol·mol−1,未检出—0.25 nmol·mol−1,未检出—0.03和0.27—1.99 nmol·mol−1. 正辛烷、正壬烷、正癸烷、十一烷和十二烷等高碳直链烷烃受大气中氧化基体影响低于支链烷烃,是不除氧化性基体的12—18倍. 烯烃受大气中氧化性基体影响不如烷烃显著,其中1-丁烯受大气中氧化性基体影响最大,是不除氧化性基体的28倍,乙烯、丙烯则是不除氧化性基体的3.3倍和5.3倍,这与1-丁烯化学活性强、反应速率快有关[21],大气中氧化基体对1-丁烯影响显著;1-己烯、柠檬烯和1-戊烯受氧化性基体影响也较大,分别是不除氧化性基体的16倍、8.8倍和3.7倍. 芳香烃中乙苯、间/对-二甲苯和邻二甲苯受大气中氧化性基体影响显著,浓度分别是不除氧化性基体的1.4倍、1.5倍和1.6倍,其余芳香烃化合物的测定基本不受大气中氧化性基体的影响. 卤代烃中仅四氯乙烯受大气中氧化性基体影响,是不除氧化性基体的1.5倍,其余卤代烃化合物不受大气中氧化性基体的影响.
由此可以看出,烷烃类化合物受大气中氧化性基体影响最大,C2—C4低碳烷烃大气化学反应活性弱,受大气中氧化性基体影响小,高碳支链烷烃受大气中氧化基体影响最大,其次是高碳直链烷烃;所有烯烃类化合物均受大气中氧化性基体的影响,其中对1-丁烯影响最大;卤代烃中仅四氯乙烯受大气中氧化性基体的影响,异丙醇、乙酸乙酯和四氢呋喃3种含氧有机物不受大气中强氧化性基体的影响.
-
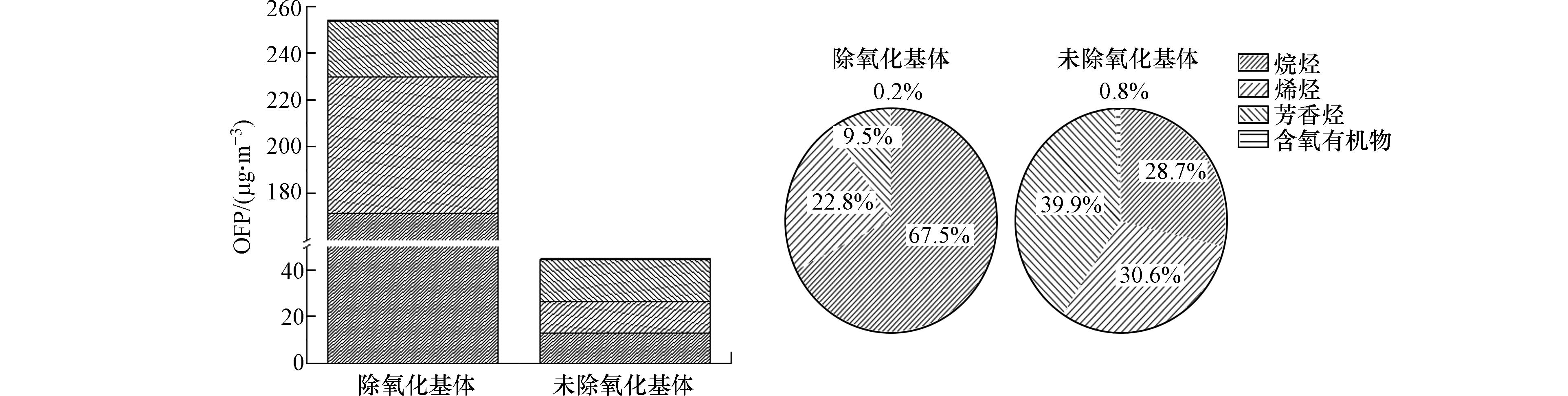
臭氧生成潜势为各VOCs化合物在最佳反应条件下对臭氧生成的最大贡献[22],定量描述VOCs化合物对大气臭氧污染的影响,一般通过研究大气中对OFP影响较大的物种及来源,采取针对性的VOCs控制措施,降低大气臭氧污染程度,是制定VOCs削减控制策略的重要依据[23]. 本文为研究大气中氧化性基体对OFP的影响,对两种采样方式的OFP结果的影响进行比较. 从图3可见,去除氧化性基体样品的OFP高于未去除氧化性基体的样品,去除氧化性基体样品的平均OFP值为254.1 μg·m−3,未去除氧化性基体样品的平均OFP值为44.6 μg·m−3. 去除氧化性基体的样品,各物种对OFP贡献大小的顺序为烷烃(67.5%)>烯烃(22.8%)>芳香烃(9.5%)>含氧有机物(0.2%);未去除氧化性基体的样品,各物种对OFP贡献大小的顺序为芳香烃(40.0%)>烯烃(30.6%)>烷烃(28.7%)>含氧有机物(0.8%). 因此,采样时是否去除氧化性基体,对于评价大气VOCs的OFP浓度及优势物种的组成均有显著影响,去除氧化性基体样品的OFP大,主要是烷烃贡献,占67.5%;未去除氧化性基体样品的OFP小,主要为芳香烃和烯烃贡献,分别贡献40.0%和30.6%. 采样时是否去除大气中氧化性基体采集的样品在进行OFP评价时获得的结论不同,在制定VOCs削减控制策略以改善臭氧污染问题时,去除氧化性基体采集的样品结果显示烷烃是主要的控制目标,未去除氧化性基体采集样品的结果显示芳香烃和烯烃是优先控制物种.
OFP为各化合物在最佳反应条件下可产生的最大臭氧量. 因气象条件不利及污染物扩散传输等因素影响,大气中VOCs不可能全部转化为臭氧,OFP理论计算值应高于臭氧实际观测浓度. 图4为大气中实际臭氧浓度及两种采样方式计算的OFP理论值. 从图中可以看出,未去除氧化性基体的采样方式,OFP理论计算值低于大气中臭氧浓度,而且OFP的变化趋势与大气臭氧实测浓度变化趋势不同;去除氧化性基体样品的OFP值高于大气臭氧实际观测浓度,而且与臭氧实测浓度变化规律具有很好的同步性,表明去除大气中氧化性基体样品OFP的理论计算值更能反映大气VOCs对臭氧生成的真实情况,去除氧化性基体采集的样品更能反映大气VOCs的客观浓度. 此外,从图4可以看出6月13日,去除氧化性基体采集样品的OFP理论计算值低于臭氧实测浓度,但仍高于未去除氧化性基体采样的OFP,这可能与6月13日为小雨天气,空气湿度大,有研究表明在高湿环境中,醛酮化合物通过KI小柱会损失[24],图5为受大气氧化基体影响较大的典型烷烃和烯烃化合物在观测期间的浓度时间序列变化,从图5可以看出,6月13日除氧化基体样品的所有烷烃化合物浓度降低,烯烃化合物浓度变化不明显,由这天臭氧浓度与其他观测期间持平,可以看出6月13日降水期间采集的去除氧化基体样品中烷烃化合物浓度被低估. 因此,高湿样品通过KI小柱时,大气VOCs会造成损失,在雨天不适合采用KI小柱采集环境空气VOCs.
-
(1)大气中氧化性基体在样品采样、运输和保存过程中,与部分VOCs化合物继续发生反应,去除大气中氧化性基体采集的样品更符合大气VOCs真实浓度水平.
(2)大气中氧化性基体影响VOCs组成,主要对醛类化合物、长链烯烃、长链烷烃和长链醇类化合物有影响,去除大气中氧化性基体采集的样品可定性检出的化合物种类更多.
(3)去除大气中氧化性基体采集样品的VOCs测定浓度高,大气氧化性基体对烷烃影响最大,其次是烯烃,对卤代烃和含氧有机物的影响最小.
(4)去除大气中氧化性基体采集样品的OFP高,高于未去除氧化性基体样品的OFP及大气臭氧实测浓度,未去除氧化性基体采集样品的OFP低于大气臭氧浓度实测值. 去除大气氧化性基体的OFP与大气中臭氧实测浓度变化规律具有很好的同步性,更能反应大气臭氧污染状况.
大气环境中氧化基体对VOCs测定的影响
Effects of oxidizing matrix on VOCs detection of atmosphere samples
-
摘要: 大气中挥发性有机物(volatile organic compounds,VOCs)种类多,活性强,部分VOCs在采样、分析过程中会与大气中的氧化基体发生反应. 为研究大气中氧化基体对VOCs测定的影响,选择苏玛罐与KI小柱联用,采集去除氧化性基体的样品,与未去除氧化性基体的样品比较,探讨大气中氧化性基体对VOCs测定的影响. 结果表明,去除氧化性基体的样品定性检出的化合物种类多,共定性检出110种化合物,未去除氧化基体样品定性检出的化合物种类少,共定性检出40种化合物;氧化基体的存在影响VOCs的浓度和组成,去除氧化基体的总挥发性有机物(TVOCs)平均浓度为144.8 nmol·mol−1,其中烷烃为体积分数最大的组分,占81.9%,其次是卤代烃、烯烃、芳香烃和含氧有机物,分别占7%、6.5%、3.2%和1.4%,未去除氧化基体TVOCs平均浓度为32.5 nmol·mol−1,其中烷烃、氯代烃占比最高,分别占38.9%和37.5%,其次是芳香烃、烯烃和含氧有机物,分别占11.5%、6.5%和5.6%. 大气中氧化性基体影响臭氧生成潜势(ozone formation potential,OFP)理论计算值及对OFP贡献优势物种的判断,去除氧化性基体样品的OFP高(平均值254.1 μg·m−3),各物种对OFP贡献大小的顺序为烷烃(67.5%)>烯烃(22.8%)>芳香烃(9.5%)>含氧有机物(0.2%);未去除氧化性基体样品的OFP值低(平均值44.6 μg·m−3),各物种对OFP贡献大小的顺序为芳香烃(40.0%)>烯烃(30.6%)>烷烃(28.7%)>含氧有机物(0.8%);去除氧化基体样品的OFP高于臭氧实测浓度,且与臭氧浓度具有很好的一致性,未去除氧化性基体样品的OFP值低于臭氧浓度,去除氧化性基体后采集的样品更符合大气VOCs的真实状态,未来大气VOCs监测,采样时应考虑联用KI小柱去除大气中存在的氧化基体.Abstract: There are many kinds of Volatile organic compounds, they have strong activity and parts of them will have reactions with the oxidants exist in the atmosphere during sampling and analysis. In order to study the influence of oxidant substances on VOCs determination, two kinds of samples were collected, one(oxidant substances removed sample) was collected with a sampling method that connected Suma canister with KI column to remove oxidant substances, the other(oxidant substances non-removed sample) was collect by Suma canister as usual with oxidant substances in it, compared the determination results between two samples. The results showed that more compounds were qualitatively detected in oxidant substances removed samples, there were 110 compounds were qualitatively detected in oxidant substances removed samples, while 40 compounds were qualitatively detected in oxidant substances non-removed samples. Oxidant substances affects the concentration and composition of VOCs, the average concentration of total volatile organic compounds (TVOCs) of oxidant substances removed samples was 144.8 nmol·mol−1, alkanes were the most abundant species, accounted for 81.9%, followed by halogenated hydrocarbon, olefin, aromatic hydrocarbon and OVOCs, accounted for 7%, 6.5%, 3.2% and 1.4%, respectively; the TVOCs of oxidant substances non-removed samples was 32.5 nmol·mol−1, alkane (38.9%) and halogenated hydrocarbon (37.5%) were the most abundant species, then followed by aromatic hydrocarbon (11.5%), olefin (6.5%) and OVOCs (5.6%). The oxidizing matrix affects the ozone formation potential (OFP) and the dominant species. The OFP of the oxidizing matrix sample was 254.1 μg·m−3, alkanes contributed most (67.5%) , then followed by olefins (22.8%) , aromatics (9.5%) and oxygenated organic compounds (0.2%); the OFP of oxidant substances non-removed samples was 44.6 μg·m−3, aromatic contributed most (40.0%), then followed by olefin (30.6%), alkane (28.7%) and oxygenated organic matter (0.8%). The OFP of oxidant substances removed sample was higher than actual ozone concentration, and had a good consistency with the ozone concentration. The OFP of oxidant substances non-removed sample was lower than the actual ozone concentration. The sample collected by oxidant substances removed was more consistent with the real state of atmospheric VOCs. The removal of oxidant substances in the atmosphere with KI column should be considered during sampling.
-
Key words:
- Suma canister sampling /
- atmosphere VOCs /
- oxidant substances /
- ozone /
- OFP
-
我国夏季臭氧污染形势严峻[1],挥发性有机化合物(volatile organic compounds, VOCs)是近地面臭氧的重要前体物,我国大部分地区臭氧污染为VOCs控制型[2]. 近年来,我国采取了极为严格的VOCs削减控制方案,但臭氧污染形势改善不明显,有些地区臭氧污染出现反弹[3].
制定VOCs削减控制策略基于大气VOCs观测结果,准确客观的观测结果是制定精准VOCs削减策略的技术基础. 2018年起我国开始在重点地区开展环境空气VOCs的手工监测工作,关于大气VOCs污染特征、来源解析、环境影响评价的研究报道也层出不穷[4 − 6],相关监测及研究工作的开展主要基于手工采样监测结果. 其中苏玛罐采样具有可采集全组分样品、保存时间长[7 − 8]的特点,是实验室分析最常用的采样方式[9 − 11]. 大气中存在O3、OHˑ、NO3等强氧化性基体物质,在样品采集、运输、保存和分析的过程中,VOCs会与大气中的强氧化剂发生化学反应而消耗损失[12 − 13],影响样品的代表性. 影响苏玛罐采集有代表性大气VOCs样品的关键因素主要包括苏玛罐内壁的惰性化水平、密封性、洁净度,目前主要关注苏玛罐内壁对VOCs的吸附作用[14],没有考虑采集样品后,VOCs可能会继续与大气中的氧化剂发生大气化学反应而造成浓度、组成的变化. 目前大气中的臭氧对VOCs测定的影响已引起关注,但结论不统一:邵敏等[15]提出臭氧在大气VOCs样品热解吸进样时会与烯烃反应,但也有研究认为大气中臭氧不影响VOCs的测定[16],该研究是在一定浓度的标气样品中人为加入一定浓度的臭氧,研究臭氧的存在对样品保存的影响,但该研究对象未能反应真实的大气环境,大气中除有臭氧,还存在OH、NO3等强氧化自由基[17]. 客观真实的大气VOCs观测结果是制定大气VOCs精准减排政策的基础和依据,也是臭氧污染治理成效的保障,目前未见有关真实大气环境样品中大气氧化性基体对VOCs测定影响深入研究的报道. VOCs化学组成繁杂,部分物质化学反应活性强,大气寿命从几分钟到几小时[12],有研究表明活性较强的VOCs样品预先通过KI小柱或Na2SO3小柱,可有效去除臭氧等强氧化性基体,保证VOCs组分的完整性[13]. 国家环境保护标准《环境空气 醛、酮类化合物的测定 高效液相色谱法》(HJ 683-2014)规定在采样管前串联KI除臭氧小柱采集醛、酮类化合物[18],因此,可使用KI除臭氧小柱研究大气中臭氧等强氧化性基体对VOCs样品采集的影响.
本研究以大气中常见的107种VOCs为研究目标,采用罐采样-大气预浓缩-气相色谱/质谱氢火焰检测器法分析技术[19],对比苏玛罐采样口前串联和不串联KI小柱两种采样方式,研究大气中氧化性基体对VOCs测定的影响;通过臭氧生成潜势(Ozone formation potential, OFP)评价,研究不同采样方式进行环境影响评价结果的客观性. 探讨大气中氧化基体对VOCs测定结果的影响,为全国开展大气VOCs精准监测提供技术支持.
1. 实验部分(Environmental section)
1.1 样品采集
采样点位于济南市东部的山东省生态环境厅院内,南面为山地,植被茂密;北面为居民、商业混合区,西面为加气站,可反映区域大气整体状况. 采样时间为2022年5月—7月大气氧化性强的月份,限流阀调节采样流速,13:00—15:00采集2 h样品. 每次采样同步采集2个样品,其中一个样品直接采集,另一个在限流阀前端接KI小柱,去除大气中氧化性基体,每个样品使用一个新的KI小柱. 本次研究采集了7 d共7组样品.
1.2 仪器设备和试剂
Shimadzu QP 2020型气相色谱/质谱联用仪(日本);
3100 型自动清罐仪(带恒温加热箱)、4700 型动态稀释仪(ENTECH);新购置的内壁硅烷化处理的SUMMA罐(ENTECH),3.2 L;电子制冷大气预浓缩仪、除水仪(MARKS);挥发性有机物标气(1.0 μmol·mol−1, 四川中测标物有限公司,包括57种PAMS、47种TO15、乙醇、柠檬烯和α-蒎烯));内标标准气(1.0 μmol·mol−1, 四川中测标物有限公司,包括一溴氯甲烷、1,2-二氟苯和氯苯-d5),高纯氮气≥99.999%,高纯氦气≥99.999%;DB-624毛细管柱(60 m×0.32 mm ×1.8 μm),PLOT 毛细管柱(30 m×0.32 mm× 10 μm),KI小柱(1.4 g/2.5 cc,Agela Technologies).1.3 样品分析条件
自动进样器取样200 mL,经-30 ℃的空管除水后进入-30℃吸附剂冷阱中进行捕集浓缩去除二氧化碳、氮气等环境中常量组分的干扰,以最快的速度加热至300 ℃,解吸进入色谱柱分离. 气相色谱条件:色谱柱升温程序:40 ℃保持3 min,以8 ℃·min−1升温到50 ℃保持2 min,再以8 ℃·min−1升温到150 ℃,保持10 min,再以15 ℃·min−1升温到185 ℃,保持16.5 min.
1.4 质量保证与质量控制
1.00 μmol·mol−1有证标准物质二级稀释至2.00 nmol·mol−1作为标准使用气;1.00 μmol·mol−1有证内标储备气稀释至50.0 nmol·mol−1标准使用气. 分别取50 、100 、200 、400 、800 mL标准使用气和25 mL内标使用气,建立内标标准曲线,相对响应因子法定量,每种化合物的相对响应因子的相对标准偏差≤20%,否则应进行空白检验;每20个样品进行1次中间浓度点校准,偏差≤20%,否则查找原因重新绘制曲线;记录每个样品的内标响应,与校准曲线的偏差≤30%. 为保证不同采样方式所用的苏玛罐性能的一致性,使用2个新购置苏玛罐循环使用,每个苏玛罐采样前均做空白检查和惰性化检查. 采样前,采用高纯氮气模拟样品经过KI除氧化基体小柱,采集到苏玛罐中进行定性分析,未检出挥发性有机物成分,表明KI不会引入干扰物质.
1.5 臭氧生成潜势(ozone formation potential, OFP)计算
本研究采用最大增量反应活性(maximum incremental reactivity,MIR),计算VOCs物种O3生成的潜势OFP,评价不同化合物在不同采样方式下对O3生成的影响.
计算公式如下:
stringUtils.convertMath(!{formula.content}) 式中,OFP(j)为化合物j的臭氧生成潜势(μg·m−3);Conc.(j)为化合物j浓度(单位μg·m−3);MIR(j):单位量化合物j最大增量反应活性(单位质量VOC生成单位量的O3),具体数值引用Carter and William[20]的研究
2. 结果与讨论(Results and discussion)
2.1 氧化性基体的存在对VOCs成分定性分析的影响
为研究大气中氧化性基体对VOCs成分定性分析的影响,通过质谱全扫描模式对样品进行分析,检出率高于80%的化合物结果见表1. 从表1可以看出,去除氧化性基体后的样品共定性检出110种化合物,包括55种烷烃、23种含氧有机物、13种烯烃、8种卤代烃、7种苯系物及包括乙炔在内的4种其他化合物. 与未去除氧化性基体的样品相比,去除氧化性基体后检出的含氧化合物、长链烷烃、长链烯烃更多. 含氧化合物包括乙醛、丙醛、3-甲基丁醛等醛类化合物及2-丁基-1-辛醇、5-甲基-2-(1-异丙基)-1-己醇、2-乙基-4-甲基-1-戊醇、2-己基-1-癸醇、2-丁基-1-辛醇等长链的醇类化合物;长链烷烃化合物包括5-甲基十一烷、十五烷、十六烷等;长链烯烃类化合物包括5-甲基-1-己烯、4,5-二甲基-1-己烯、2,6-二甲基-3-庚烯、2,4-二甲基-1-庚烯、3-十四烯、1-壬烯、2-十二烯等,只有在去除氧化性基体采集的样品中有检出.
表 1 大气中强氧化性物质对化学组分的影响Table 1. The Influence of Strong Oxidizing Substances in the Atmosphere on Chemical Components采样方式Sampling mode 定性检出的化合物Qualitatively detected compounds 除氧化剂(110种) 乙烷、乙烯、乙炔、丙烷、丙烯、乙醛、2-甲基丁烷、三氯氟甲烷、戊烷、乙醇、1,3-戊二烯、丙醛、丙酮、异丙醇、二硫化碳、乙腈、1-戊烯、2,2-二甲基丁烷、2,3-二甲基丁烷、正己烷、2-甲基丙醛、三甲基硅醇、1-氯-3-甲基丁烷、甲基环戊烷、2-丁酮、三氯甲烷、2,4-二甲基戊烷、1-己烯、苯、庚烷、2,5-二甲基己烷、2,4-二甲基己烷、3-甲基丁醛、1,2-二氯丙烷、二氯甲烷、5-甲基-1-己烯、4,5-二甲基-1-己烯、2-甲基庚烷、3-甲基庚烷、乙酸己酯、二甲基二硫、2,5-二甲基-1-己烯、1,2-二甲基环己烷、甲苯、2,6-二甲基-3-庚烯、2-乙基-2,4-二甲基戊烷、庚烷、2-甲基十一烷、2-甲基戊醛、乙酸丁酯、己醛、2,4-二甲基-1-庚烯、辛烷、4,5-二甲基辛烷、2,4-二甲基庚烷、2,3-二甲基庚烷、乙苯、壬烷、间/对-二甲苯、2,4-二甲基己烷、4-乙基辛烷、邻-二甲苯、3,3-二甲基丁基苯、庚醛、2-甲基-1-戊醇、十一烷、1,1’-二氧辛烷、3-亚甲基庚烷、2-丁基-1-辛醇、2,2,4,6,6-五甲基庚烷、4-甲基癸烷、2-甲基癸烷、2-甲基辛烷、2,5-二甲基癸烷、3,3-二甲基辛烷、5-甲基-2-(1-异丙基)-1-己醇、均三甲苯、2,2,6-三甲基辛烷、2-乙基-4-甲基-1-戊醇、十二烷、5-甲基十一烷、3-甲基癸烷、1,4-二氯苯、1-氯-十六烷、3-十四烯、1-壬烯、3-甲基壬烷、4-甲基十一烷、2-甲基十一烷、2,6,11-三甲基十二烷、2-十二烯、1-碘-癸烷、十五烷、2-己基-1-癸醇、2,6-二甲基十一烷、1,2,3-三甲基环戊烷、环十二烷、2-丁基-1-辛醇、2,5-二甲基十一烷、十六烷、十七烷、十八烷、3-丁基-1-辛醇、2-甲基十四烷、2,6,10-三甲基十二烷、4-甲基十二烷、丁基环戊烷、3-甲基十四烷、十九烷、2-己基-1-硅醇 不除氧化剂(40种) 乙烷、乙烯、乙炔、丙烷、丙烯、2-甲基丁烷、三氯氟甲烷、戊烷、乙醇、1,3-戊二烯、丙酮、异丙醇、二硫化碳、乙腈、二氯甲烷、1-戊烯、1-氯-3-甲基丁烷、正己烷、2-甲基丙醛、三甲基硅醇、1-氯3-甲基丁烷、2-氧基丙酸乙酯、三氯甲烷、2,4-二甲基戊烷、环己烷、2,4-己二炔、6-甲基庚基乙烯基醚、5-甲基-2-(1-异丙基)-1-己醇、癸醛、1,2-二氯丙烷、乙酸己酯、二甲二硫、甲苯、己醛、2-甲基丙基环己烷、间/对-二甲苯、邻-二甲苯、1-氯十六烷、1,4-二氯苯、2-丁基-1-辛醇 | Show Table DownLoad:
CSV
DownLoad:
CSV
未除氧化性基体的采样方式采集的样品中定性检出的化合物种类较少,共定性检出40种化合物,绝大部分化合物与去除氧化性基体样品定性检出的种类一致,包括乙烷、乙烯、乙炔、丙烷、丙烯等空气中常见的短链VOCs;仅2-氧基丙酸乙酯、环己烷、2,4-己二炔、6-甲基庚基乙烯基醚和2-甲基丙基环己烷5种化合物只在未除氧化性基体的样品中有定性检出,占检出总量的14.3%. 未除氧化性基体的样品未检出长链烯烃、长链烷烃、长链醇类化合物,表明此类化合物在氧化基体环境中不稳定,采样后或测定过程中会继续参与氧化反应而损失. 总体看,去除氧化性基体后定性检出的化合物种类多,长链的烷烃、烯烃、醇类物质在未去除氧化性基体的样品中均未检出,大气中的部分VOCs会在样品采集、运输和保存等环节继续与氧化性基体反应而消耗损失,样品中的氧化性基体明显影响VOCs组成的分析判定.
2.2 氧化基体的存在对VOCs浓度测定的影响
为研究大气中氧化基体对VOCs浓度测定的影响,对107种VOCs(29种烷烃,13种烯烃,35种卤代烃,10种含氧有机物,18种芳香烃、乙炔及二硫化碳)测定,分别将去除和未去除氧化性基体的7组样品的每种化合物进行浓度平均,将两种采样方式每类化合物的浓度均值分别加和得到烷烃、烯烃、芳香烃、含氧有机物和卤代烃的浓度,比较两种采样方式对每类化合物测定结果的影响(图1). 去除氧化性基体的TVOCs平均浓度为144.8 nmol·mol−1,其中烷烃类化合物浓度最高(平均浓度为118.7 nmol·mol−1),占81.9%,主要由2,2-二甲基丁烷(平均浓度15.7 nmol·mol−1)、2,3-二甲基丁烷(平均浓度12.2 nmol·mol−1)、环戊烷(平均浓度15.5 nmol·mol−1)和正己烷贡献(平均浓度28.1 nmol·mol−1);卤代烃浓度第二(平均浓度为10.1 nmol·mol−1),占7%;烯烃浓度第三(平均浓度9.36 nmol·mol−1),占6.5%;芳香烃浓度第四(平均浓度为4.63 nmol·mol−1),占3.2%;含氧有机物浓度最低(平均浓度为2.05 nmol·mol−1),占1.4%. 未去除氧化性基体样品测定的平均TVOCs浓度低(平均浓度为32.5 nmol·mol−1),烷烃(平均浓度为12.6 nmol·mol−1)和卤代烃(平均浓度为12.2 nmol·mol−1)浓度最高,分别占TVOCs的 38.9%和37.5%;芳香烃浓度第三(平均浓度为3.73 nmol·mol−1),占11.5%;烯烃浓度第四(平均浓度为2.11 nmol·mol−1),占6.5%;含氧有机物浓度最低(平均浓度为1.81 nmol·mol−1),占5.6%. 由此可以看出,样品中氧化性基体的存在对测定样品各物种的浓度及组成特征均有显著影响. 其中样品中氧化性基体对烷烃、烯烃类化合物浓度测定结果的影响最大,分别是未去除氧化性基体浓度的9.42倍和4.44倍;对芳香烃、卤代烃和含氧有机物影响小,去除与不去除氧化性基体采集样品测定浓度的相对偏差分别为10.7%、9.19%和6.31%. 因此,大气中的氧化性基体对VOCs浓度的测定及物种组成的分析都有影响,其中对烷烃和烯烃浓度测定的影响最为显著,表明烷烃、烯烃活性强,在样品采集、保存、运输和测定的过程中,会与氧化基体反应消耗损失;测定大气中的芳香烃、卤代烃及含氧有机物,受大气中强氧化性基体的影响较小.
本研究共定量检出60种目标化合物,图2为两种不同采样方式测定的化合物的平均浓度. 从图2可以看出,烷烃中乙烷、丙烷、正丁烷、异丁烷等4种C2—C4低碳烷烃受大气中氧化性基体影响小,去除氧化性基体是不除氧化性基体浓度的1.5倍左右;C5以上的支链烷烃和环状烷烃的测定受大气中氧化性基体影响大,其中对2-甲基庚烷、环戊烷和2,2-二甲基丁烷影响最大,分别是不去氧化性基体样品浓度测定值的542倍、174倍和116倍,去除氧化基体样品中2-甲基庚烷、环戊烷和2,2-二甲基丁烷的浓度范围分别为0.12—22.1 nmol·mol−1、0.10—31.7 nmol·mol−1和0.35—23.7 nmol·mol−1之间,未去除氧化氧化性基体的浓度范围分别为:未检出—0.04 nmol·mol−1,0.02—0.25 nmol·mol−1和0.03—0.25 nmol·mol−1. 大气中氧化基体对2,3-二甲基丁烷、甲基环戊烷、3-甲基庚烷及正己烷的影响次之,分别是不除氧化性基体样品浓度测定值的98倍、68倍、38倍和33倍,去除氧化基体样品中2,3-二甲基丁烷、甲基环戊烷、3-甲基庚烷和正己烷的浓度范围分别为:0.28—19.8 nmol·mol−1,0.04—24.2 nmol·mol−1,0.03—1.71 nmol·mol−1和0.40—71.8 nmol·mol−1,未去除氧化性基体的浓度范围分别为:0.03—0.25 nmol·mol−1,未检出—0.25 nmol·mol−1,未检出—0.03和0.27—1.99 nmol·mol−1. 正辛烷、正壬烷、正癸烷、十一烷和十二烷等高碳直链烷烃受大气中氧化基体影响低于支链烷烃,是不除氧化性基体的12—18倍. 烯烃受大气中氧化性基体影响不如烷烃显著,其中1-丁烯受大气中氧化性基体影响最大,是不除氧化性基体的28倍,乙烯、丙烯则是不除氧化性基体的3.3倍和5.3倍,这与1-丁烯化学活性强、反应速率快有关[21],大气中氧化基体对1-丁烯影响显著;1-己烯、柠檬烯和1-戊烯受氧化性基体影响也较大,分别是不除氧化性基体的16倍、8.8倍和3.7倍. 芳香烃中乙苯、间/对-二甲苯和邻二甲苯受大气中氧化性基体影响显著,浓度分别是不除氧化性基体的1.4倍、1.5倍和1.6倍,其余芳香烃化合物的测定基本不受大气中氧化性基体的影响. 卤代烃中仅四氯乙烯受大气中氧化性基体影响,是不除氧化性基体的1.5倍,其余卤代烃化合物不受大气中氧化性基体的影响.
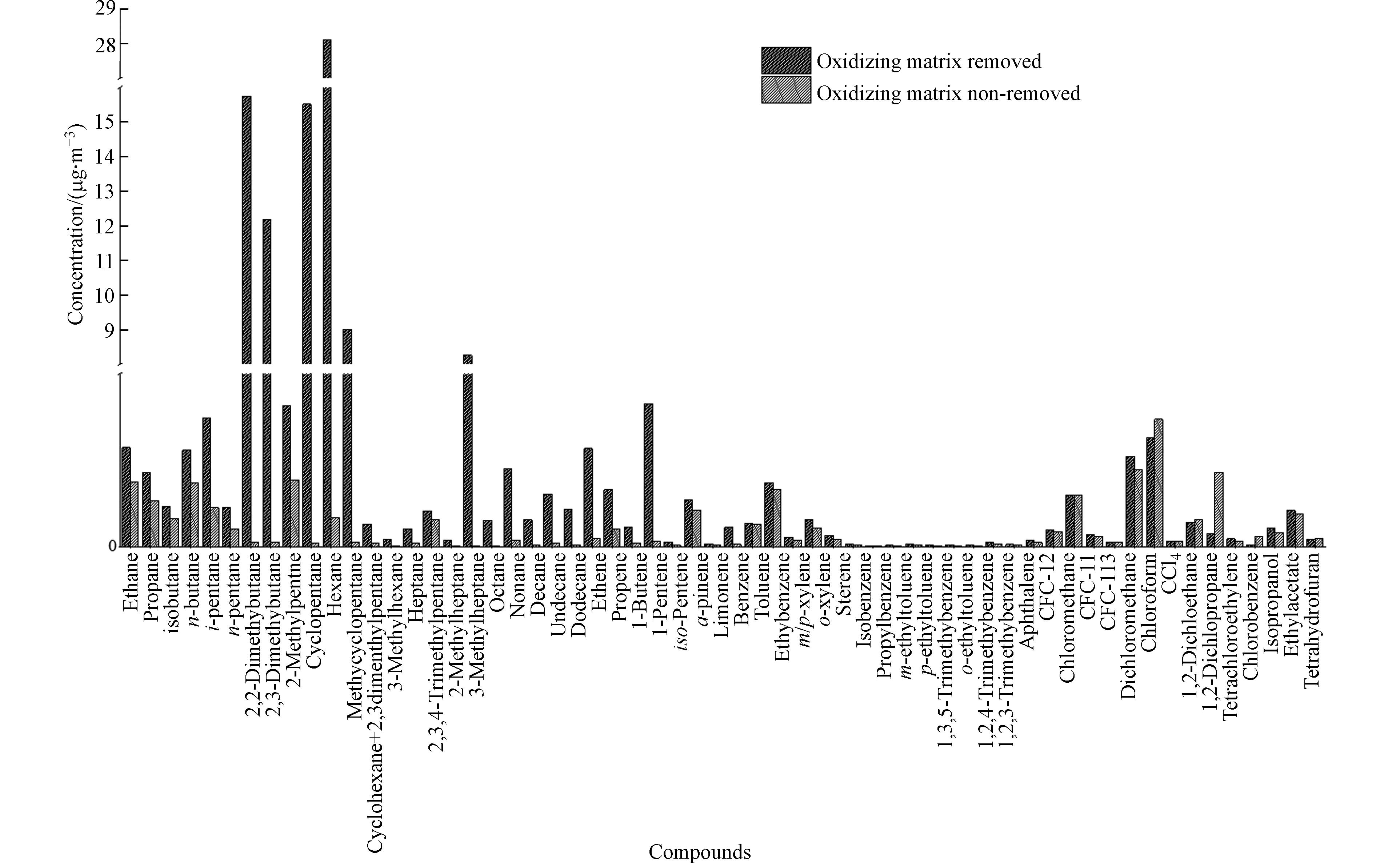 图 2 大气氧化性基体对每种化合物的影响Figure 2. The Influence of Oxidizing Matrix for Each VOCs Components
图 2 大气氧化性基体对每种化合物的影响Figure 2. The Influence of Oxidizing Matrix for Each VOCs Components由此可以看出,烷烃类化合物受大气中氧化性基体影响最大,C2—C4低碳烷烃大气化学反应活性弱,受大气中氧化性基体影响小,高碳支链烷烃受大气中氧化基体影响最大,其次是高碳直链烷烃;所有烯烃类化合物均受大气中氧化性基体的影响,其中对1-丁烯影响最大;卤代烃中仅四氯乙烯受大气中氧化性基体的影响,异丙醇、乙酸乙酯和四氢呋喃3种含氧有机物不受大气中强氧化性基体的影响.
2.3 大气氧化基体对臭氧生成潜势评价的影响
臭氧生成潜势为各VOCs化合物在最佳反应条件下对臭氧生成的最大贡献[22],定量描述VOCs化合物对大气臭氧污染的影响,一般通过研究大气中对OFP影响较大的物种及来源,采取针对性的VOCs控制措施,降低大气臭氧污染程度,是制定VOCs削减控制策略的重要依据[23]. 本文为研究大气中氧化性基体对OFP的影响,对两种采样方式的OFP结果的影响进行比较. 从图3可见,去除氧化性基体样品的OFP高于未去除氧化性基体的样品,去除氧化性基体样品的平均OFP值为254.1 μg·m−3,未去除氧化性基体样品的平均OFP值为44.6 μg·m−3. 去除氧化性基体的样品,各物种对OFP贡献大小的顺序为烷烃(67.5%)>烯烃(22.8%)>芳香烃(9.5%)>含氧有机物(0.2%);未去除氧化性基体的样品,各物种对OFP贡献大小的顺序为芳香烃(40.0%)>烯烃(30.6%)>烷烃(28.7%)>含氧有机物(0.8%). 因此,采样时是否去除氧化性基体,对于评价大气VOCs的OFP浓度及优势物种的组成均有显著影响,去除氧化性基体样品的OFP大,主要是烷烃贡献,占67.5%;未去除氧化性基体样品的OFP小,主要为芳香烃和烯烃贡献,分别贡献40.0%和30.6%. 采样时是否去除大气中氧化性基体采集的样品在进行OFP评价时获得的结论不同,在制定VOCs削减控制策略以改善臭氧污染问题时,去除氧化性基体采集的样品结果显示烷烃是主要的控制目标,未去除氧化性基体采集样品的结果显示芳香烃和烯烃是优先控制物种.
OFP为各化合物在最佳反应条件下可产生的最大臭氧量. 因气象条件不利及污染物扩散传输等因素影响,大气中VOCs不可能全部转化为臭氧,OFP理论计算值应高于臭氧实际观测浓度. 图4为大气中实际臭氧浓度及两种采样方式计算的OFP理论值. 从图中可以看出,未去除氧化性基体的采样方式,OFP理论计算值低于大气中臭氧浓度,而且OFP的变化趋势与大气臭氧实测浓度变化趋势不同;去除氧化性基体样品的OFP值高于大气臭氧实际观测浓度,而且与臭氧实测浓度变化规律具有很好的同步性,表明去除大气中氧化性基体样品OFP的理论计算值更能反映大气VOCs对臭氧生成的真实情况,去除氧化性基体采集的样品更能反映大气VOCs的客观浓度. 此外,从图4可以看出6月13日,去除氧化性基体采集样品的OFP理论计算值低于臭氧实测浓度,但仍高于未去除氧化性基体采样的OFP,这可能与6月13日为小雨天气,空气湿度大,有研究表明在高湿环境中,醛酮化合物通过KI小柱会损失[24],图5为受大气氧化基体影响较大的典型烷烃和烯烃化合物在观测期间的浓度时间序列变化,从图5可以看出,6月13日除氧化基体样品的所有烷烃化合物浓度降低,烯烃化合物浓度变化不明显,由这天臭氧浓度与其他观测期间持平,可以看出6月13日降水期间采集的去除氧化基体样品中烷烃化合物浓度被低估. 因此,高湿样品通过KI小柱时,大气VOCs会造成损失,在雨天不适合采用KI小柱采集环境空气VOCs.
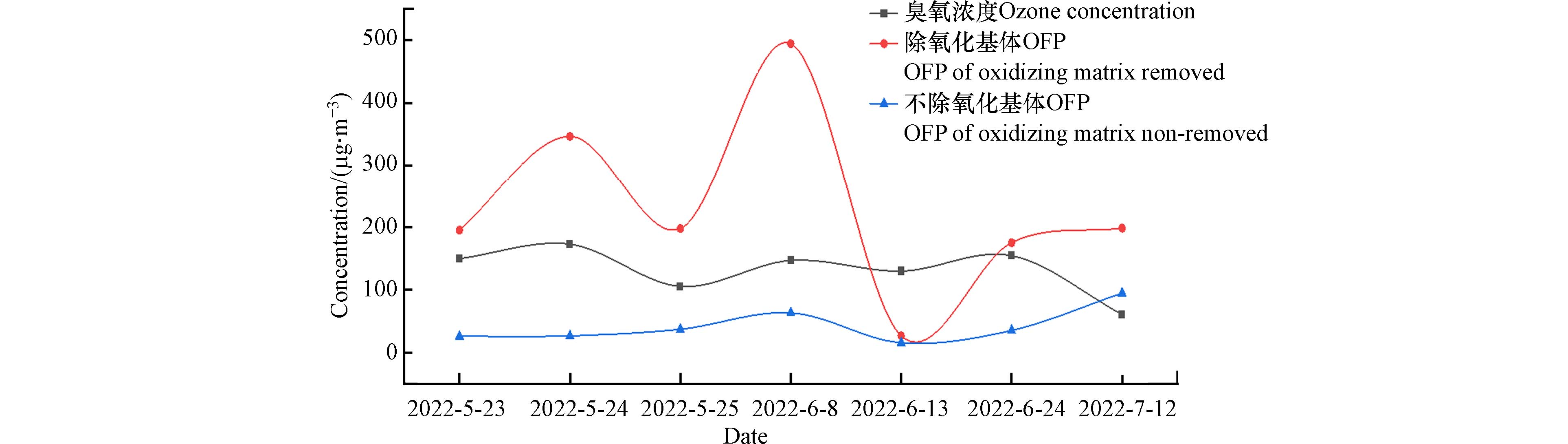 图 4 OFP理论计算值与大气臭氧实测浓度变化时间序列图Figure 4. Time Series Diagram of OFP and Measured Atmospheric O3 Concentration
图 4 OFP理论计算值与大气臭氧实测浓度变化时间序列图Figure 4. Time Series Diagram of OFP and Measured Atmospheric O3 Concentration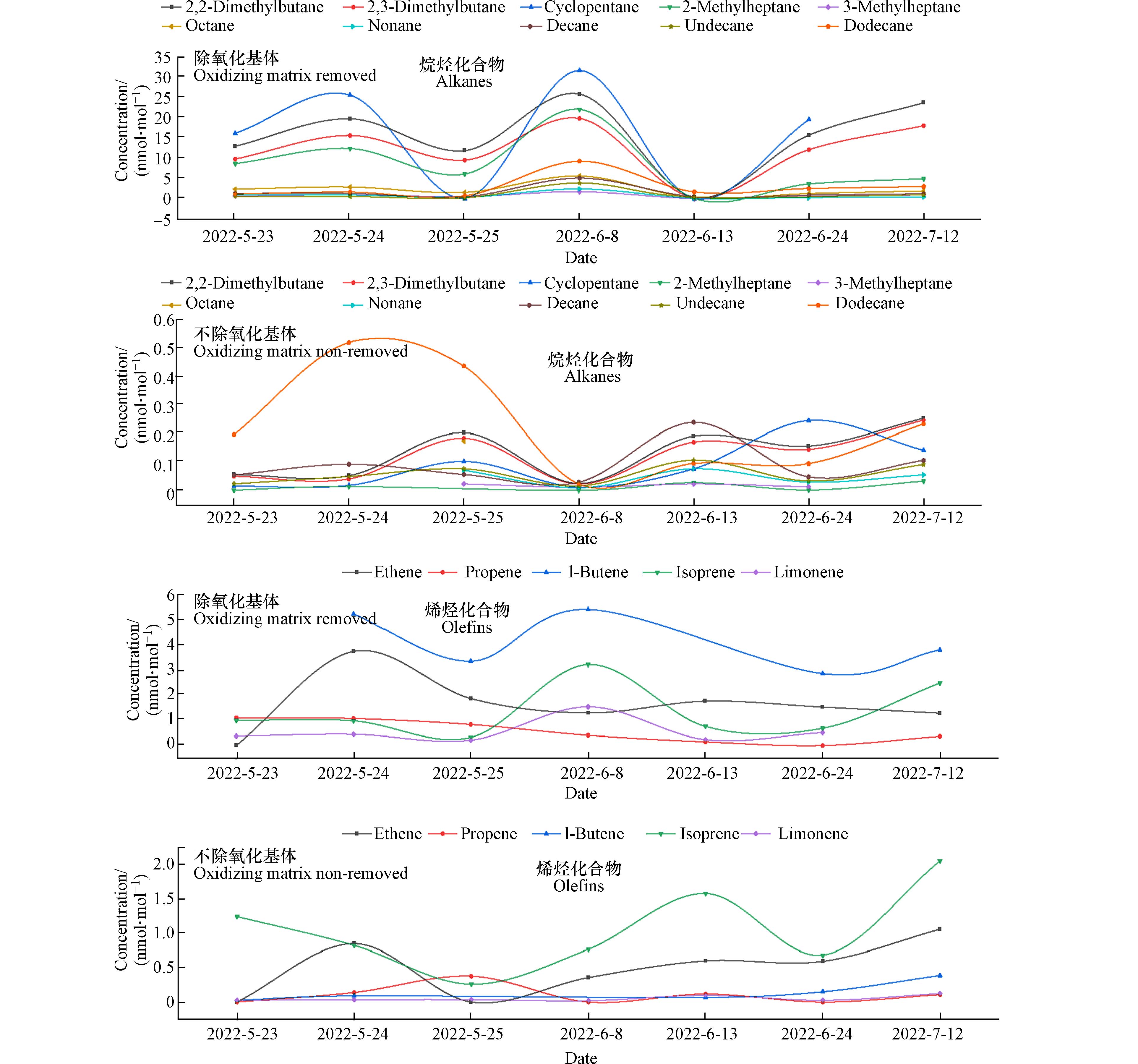 图 5 典型化合物浓度变化时间序列图Figure 5. Time series diagram of concentration of typical compoundsa:烷烃化合物;b:烯烃化合物a: alkanes; b: olefins
图 5 典型化合物浓度变化时间序列图Figure 5. Time series diagram of concentration of typical compoundsa:烷烃化合物;b:烯烃化合物a: alkanes; b: olefins3. 结论(Conclusion)
(1)大气中氧化性基体在样品采样、运输和保存过程中,与部分VOCs化合物继续发生反应,去除大气中氧化性基体采集的样品更符合大气VOCs真实浓度水平.
(2)大气中氧化性基体影响VOCs组成,主要对醛类化合物、长链烯烃、长链烷烃和长链醇类化合物有影响,去除大气中氧化性基体采集的样品可定性检出的化合物种类更多.
(3)去除大气中氧化性基体采集样品的VOCs测定浓度高,大气氧化性基体对烷烃影响最大,其次是烯烃,对卤代烃和含氧有机物的影响最小.
(4)去除大气中氧化性基体采集样品的OFP高,高于未去除氧化性基体样品的OFP及大气臭氧实测浓度,未去除氧化性基体采集样品的OFP低于大气臭氧浓度实测值. 去除大气氧化性基体的OFP与大气中臭氧实测浓度变化规律具有很好的同步性,更能反应大气臭氧污染状况.
-
图 2 大气氧化性基体对每种化合物的影响
Figure 2. The Influence of Oxidizing Matrix for Each VOCs Components
图 4 OFP理论计算值与大气臭氧实测浓度变化时间序列图
Figure 4. Time Series Diagram of OFP and Measured Atmospheric O3 Concentration
图 5 典型化合物浓度变化时间序列图
Figure 5. Time series diagram of concentration of typical compounds
表 1 大气中强氧化性物质对化学组分的影响
Table 1. The Influence of Strong Oxidizing Substances in the Atmosphere on Chemical Components
采样方式Sampling mode 定性检出的化合物Qualitatively detected compounds 除氧化剂(110种) 乙烷、乙烯、乙炔、丙烷、丙烯、乙醛、2-甲基丁烷、三氯氟甲烷、戊烷、乙醇、1,3-戊二烯、丙醛、丙酮、异丙醇、二硫化碳、乙腈、1-戊烯、2,2-二甲基丁烷、2,3-二甲基丁烷、正己烷、2-甲基丙醛、三甲基硅醇、1-氯-3-甲基丁烷、甲基环戊烷、2-丁酮、三氯甲烷、2,4-二甲基戊烷、1-己烯、苯、庚烷、2,5-二甲基己烷、2,4-二甲基己烷、3-甲基丁醛、1,2-二氯丙烷、二氯甲烷、5-甲基-1-己烯、4,5-二甲基-1-己烯、2-甲基庚烷、3-甲基庚烷、乙酸己酯、二甲基二硫、2,5-二甲基-1-己烯、1,2-二甲基环己烷、甲苯、2,6-二甲基-3-庚烯、2-乙基-2,4-二甲基戊烷、庚烷、2-甲基十一烷、2-甲基戊醛、乙酸丁酯、己醛、2,4-二甲基-1-庚烯、辛烷、4,5-二甲基辛烷、2,4-二甲基庚烷、2,3-二甲基庚烷、乙苯、壬烷、间/对-二甲苯、2,4-二甲基己烷、4-乙基辛烷、邻-二甲苯、3,3-二甲基丁基苯、庚醛、2-甲基-1-戊醇、十一烷、1,1’-二氧辛烷、3-亚甲基庚烷、2-丁基-1-辛醇、2,2,4,6,6-五甲基庚烷、4-甲基癸烷、2-甲基癸烷、2-甲基辛烷、2,5-二甲基癸烷、3,3-二甲基辛烷、5-甲基-2-(1-异丙基)-1-己醇、均三甲苯、2,2,6-三甲基辛烷、2-乙基-4-甲基-1-戊醇、十二烷、5-甲基十一烷、3-甲基癸烷、1,4-二氯苯、1-氯-十六烷、3-十四烯、1-壬烯、3-甲基壬烷、4-甲基十一烷、2-甲基十一烷、2,6,11-三甲基十二烷、2-十二烯、1-碘-癸烷、十五烷、2-己基-1-癸醇、2,6-二甲基十一烷、1,2,3-三甲基环戊烷、环十二烷、2-丁基-1-辛醇、2,5-二甲基十一烷、十六烷、十七烷、十八烷、3-丁基-1-辛醇、2-甲基十四烷、2,6,10-三甲基十二烷、4-甲基十二烷、丁基环戊烷、3-甲基十四烷、十九烷、2-己基-1-硅醇 不除氧化剂(40种) 乙烷、乙烯、乙炔、丙烷、丙烯、2-甲基丁烷、三氯氟甲烷、戊烷、乙醇、1,3-戊二烯、丙酮、异丙醇、二硫化碳、乙腈、二氯甲烷、1-戊烯、1-氯-3-甲基丁烷、正己烷、2-甲基丙醛、三甲基硅醇、1-氯3-甲基丁烷、2-氧基丙酸乙酯、三氯甲烷、2,4-二甲基戊烷、环己烷、2,4-己二炔、6-甲基庚基乙烯基醚、5-甲基-2-(1-异丙基)-1-己醇、癸醛、1,2-二氯丙烷、乙酸己酯、二甲二硫、甲苯、己醛、2-甲基丙基环己烷、间/对-二甲苯、邻-二甲苯、1-氯十六烷、1,4-二氯苯、2-丁基-1-辛醇
下载: 导出CSV
-
[1] LI K, JACOB D J, LIAO H, et al. Ozone pollution in the North China Plain spreading into the late-winter haze season[J]. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(10): e2015797118. [2] 李凯, 潘宁, 梅如波, 等. 泰安市大气挥发性有机物污染特征及来源解析[J]. 环境化学, 2022, 41(2): 482-490. LI K, PAN N , EI R B, et al. Characteristics and source apportionment of ambient volatile organic compounds in Taian[J]. Environmental Chemistry, 2022, 41(2): 482-490.
[3] WANG J, ZHANG Y L, WU Z F, et al. Ozone episodes during and after the 2018 Chinese National Day holidays in Guangzhou: Implications for the control of precursor VOCs[J]. Journal of Environmental Sciences, 2022, 114: 322-333. doi: 10.1016/j.jes.2021.09.009 [4] 朱轲欣, 刘立忠, 刘焕武, 等. 大连市夏季 VOCs化学反应活性及来源[J]. 环境科学, 2022, 43(8): 3944-3952. ZHU K X, LIU L Z, LIU H W, et al. Chemical reaction activity and source apportionment of atmospheric VOCs in summer in Dalian[J]. Environmental Science, 2022, 43(8): 3944-3952.
[5] ZHENG H, KONG S F, YAN Y Y, et al. Compositions, sources and health risks of ambient volatile organic compounds (VOCs) at a petrochemical industrial park along the Yangtze River[J]. The Science of the Total Environment, 2020, 703: 135505. doi: 10.1016/j.scitotenv.2019.135505 [6] MOZAFFAR A, ZHANG Y L, FAN M Y, et al. Characteristics of summertime ambient VOCs and their contributions to O3 and SOA formation in a suburban area of Nanjing, China[J]. Atmospheric Research, 2020, 240: 104923. doi: 10.1016/j.atmosres.2020.104923 [7] 李光辉, 蒋斌, 王思行, 等. 罐采样-预浓缩/气相色谱-质谱联用测定污染源废气中118种挥发性有机物[J]. 分析测试学报, 2020, 39(12): 1441-1450. doi: 10.3969/j.issn.1004-4957.2020.12.002 LI G H, JIANG B, WANG S H, et al. Determination of 118 volatile organic compounds in source emission by canister sampling-preconcentration/gas chromatography-mass spectrometry[J]. Journal of Instrumental Analysis, 2020, 39(12): 1441-1450. doi: 10.3969/j.issn.1004-4957.2020.12.002
[8] 何丽, 谭丽. 大气中挥发性有机物监测技术方法研究[J]. 绿色科技, 2023, 25(2): 214-219,241. doi: 10.3969/j.issn.1674-9944.2023.02.041 HE L, TAN L. Overview on analytical method of volatile organic compounds in atmospheric environment[J]. Journal of Green Science and Technology, 2023, 25(2): 214-219,241. doi: 10.3969/j.issn.1674-9944.2023.02.041
[9] LI Y D, YIN S S, YU S J, et al. Characteristics, source apportionment and health risks of ambient VOCs during high ozone period at an urban site in central plain, China[J]. Chemosphere, 2020, 250: 126283. doi: 10.1016/j.chemosphere.2020.126283 [10] ZHANG Z J, WANG H, CHEN D, et al. Emission characteristics of volatile organic compounds and their secondary organic aerosol formation potentials from a petroleum refinery in Pearl River Delta, China[J]. The Science of the Total Environment, 2017, 584/585: 1162-1174. doi: 10.1016/j.scitotenv.2017.01.179 [11] 徐洁, 唐伟, 张玮琦, 等. 大连市秋冬季环境空气中VOCs特征及臭氧生成潜势分析[J]. 中国环境监测, 2021, 37(6): 31-37. XU J, TANG W, ZHANG W Q, et al. Characteristics and ozone formation potential of atmospheric VOCs in autumn and winter in Dalian[J]. Environmental Monitoring in China, 2021, 37(6): 31-37.
[12] ATKINSON R, AREY J. Gas-phase tropospheric chemistry of biogenic volatile organic compounds: A review[J]. Atmospheric Environment, 2003, 37: 197-219. doi: 10.1016/S1352-2310(03)00391-1 [13] ZENG J Q, ZHANG Y L, ZHANG H N, et al. Design and characterization of a semi-open dynamic chamber for measuring biogenic volatile organic compounds (BVOCs) emissions from plants[J]. Atmospheric Measurement Techniques, 2022, 15(1): 79-93. doi: 10.5194/amt-15-79-2022 [14] KELLY T J, HOLDREN M W. Applicability of canisters for sample storage in the determination of hazardous air pollutants[J]. Atmospheric Environmen, 1995, 29(19): 2595-2608. doi: 10.1016/1352-2310(95)00192-2 [15] 邵敏, 袁斌, 王鸣, 等. 挥发性有机物(VOCs)来源及其大气化学作用[M]. 北京: 科学出版社, 2020. SHAO M, YUAN B, WANG M, et al. Volatile organic compounds in the atmosphere: Sources and the roles in atmospheric chemistry[M]. Beijing: Science Press, 2020.
[16] PALLUAU F, MIRABEL P, MILLET M. Influence of ozone on the sampling and storage of volatile organic compounds in canisters[J]. Environ Chem Lett, 2007, 5: 51-55. doi: 10.1007/s10311-006-0090-x [17] REN Y G, CAI M, DAELE V, et al. Rate coefficients for the reactions of OH radical and ozone with a series of unsaturated esters[J]. Atmospheric Environment, 2019, 200(Mar): 243-253. [18] 生态环境部. 环境空气 醛酮类化合物的测定 液相色谱法: HJ683-2014[S]. Ministry of Ecology and Environment of the People’s Republic of China. Ambient air Deternination of aldehyde and ketone compounds-High performance liquid chromatography: HJ683-2014[S].
[19] 张烃, 张翼翔, 杜祯宇, 等. 电子制冷预浓缩-双柱气相色谱-质谱/氢火焰检测器法测定空气中104种挥发性有机物[J]. 色谱, 2019, 37(4): 418-425. doi: 10.3724/SP.J.1123.2018.12022 ZHANG T, ZHANG Y X, DU Z Y, et al. Determination of 104 volatile organic compounds in air by double column gas chromatography-mass spectrometry/flame ionization detector coupled with electronically controlled cryo-focusing unit[J]. Chinese Journal of Chromatography, 2019, 37(4): 418-425. doi: 10.3724/SP.J.1123.2018.12022
[20] CARTER W P L. Development of Ozone Reactivity Scales for Volatile Organic Compounds[J]. Air Waste Man. Ass, 1994, 44(7): 881-899. doi: 10.1080/1073161X.1994.10467290 [21] FELTHAM E J, ALMOND M J, MARSTON G, et al. Reactions of alkenes with ozone in the gas phase: A matrix-isolation study of secondary ozonides and carbonyl-containing reaction products[J]. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2000, 56(13): 2605-2616. doi: 10.1016/S1386-1425(00)00368-1 [22] CARTER W P L. Development of the SAPRC-07 chemical mechanism[J]. Atmospheric Environment, 2010, 44(40): 5324-5335. doi: 10.1016/j.atmosenv.2010.01.026 [23] 王冰, 李光明, 马红磊, 等. 濮阳市VOCs污染特征及其臭氧生成潜势分析[J]. 中国环境监测, 2020, 36(3): 59-65. WANG B, LI G M, MA H L, et al. Analysis of VOCs pollution characteristics and ozone generation potential in Puyang[J]. Environmental Monitoring in China, 2020, 36(3): 59-65.
[24] LI J, LI L Y, WU R R, et al. Inventory of highly resolved temporal and spatial volatile organic compounds emission in China[C]. WIT Transactions on Ecology and the Environment, Air Pollution XXIV. June 20-22, 2016. Crete, Greece. Southampton UK: WIT Press, 2016: 262-270. -

 点击查看大图
点击查看大图
计量
- 文章访问数: 565
- HTML全文浏览数: 565
- PDF下载数: 12
- 施引文献: 0
