

-
塑料是现代生产生活中不可或缺的重要材料,全球的塑料年产量约为3.6亿t,大量未得到妥善处理处置的塑料固体废物对全球生态环境安全造成了巨大的冲击[1 − 4]. 据统计,目前废弃塑料的回收率仅约为9%,绝大多数塑料废物经填埋、焚烧处理消纳或直接被丢弃进入到自然环境中,极大地造成了资源的浪费和对环境的破坏[5 − 10].
目前超过99%的回收塑料通过机械回收(Mechanical recycling)方法处理,其再生产物的质量与机械性能往往会出现衰减;其余约1%的回收塑料经热解(Pyrolysis)或气化(Gasification)等方法处理,用于生产热解油中的轻质馏分或 “富氢气体”[3, 11]. 分析表明,现行的塑料回收转化技术难以应对日益严峻的塑料污染危机[12],如何更好地挖掘塑料本身的潜在资源价值,提高转化产物的质量与应用空间,成为当下塑料升级转化技术探索的研究热点[13].
石墨烯与碳纳米管作为研究应用最广泛的两类低维碳纳米材料,得益其优异的化学-热稳定性、机械性能、光电性能与丰富的微结构与孔特征,被广泛用于能源、复合材料、电子器件、环境、检测、催化等领域[14 − 15]. 如图1所示,近年来国内外研究者在借鉴传统低维碳纳米材料制备方法的基础上,开发出一系列基于热解(Pyrolysis)-化学气相沉积方法(chemical vapour deposition, CVD)、等离子体增强化学气相沉积方法(plasma-enhanced CVD)、或热化学方法(激光、焦耳加热、电弧法)的塑料衍生碳纳米材料制备方法,将多种类型的塑料前驱体转化为石墨烯、碳纳米管及其衍生材料,代表了一条重要的塑料升级转化路径[16 − 17].
为更好地系统梳理塑料升级转化制备低维碳纳米材料的合成方法与研究进展,本文以石墨烯与碳纳米管这两类典型低维碳纳米材料为研究对象,分类简要总结了每种材料制备所使用的方法、装置构型、反应条件和材料转化机制. 在此基础上,结合每种特定方法对应终产物的材料特征与物化性能,细化了两类低维碳纳米材料的产物类型,包括层状石墨烯(Layered graphene)、多孔石墨烯(Porous graphene)、粉体石墨烯(Graphene powders)和多壁碳纳米管(Multiwalled carbon nanotubes, MWCNTs),并介绍了各种型材料当前的应用场景及其性能.
-
石墨烯凭借其独特的晶体与能带结构赋予的优异电学、光学、热学和力学等特性,被广泛应用于能源、材料、催化和环境等诸多领域[18]. 除了严格意义上的由按六方晶格排列的二维sp2杂化碳原子层构成的单层石墨烯(Monolayer graphene)以外,层状石墨烯、三维石墨烯和功能化石墨烯等石墨烯衍生材料也被广泛制备研究,极大的丰富了该类型材料的应用空间[19].
如图2a所示,近年来研究人员利用生产使用量最高的(占比约57%)以聚乙烯(polyethylene, PE)和聚丙烯(polypropylene, PP)为代表的聚烯烃类塑料、以聚对苯二甲酸乙二醇酯 (polyethylene terephthalate, PET)为代表的聚酯类塑料、聚氯乙烯(polyvinyl chloride, PVC)和聚苯乙烯(polystyrene, PS)等为固态前驱体,基于“自下而上”的材料制备策略,通过控制反应条件,借助不同的生长与组装机制,制备出一系列具有不同形貌结构及其物化性能特征的石墨烯及其衍生材料. 期间形成了以传统化学气相沉积方法及其衍生形式和以基于激光或焦耳加热的两种热化学方法为代表的三种塑料衍生石墨烯材料制备模式. 下面本文将分类梳理上述三种塑料衍生石墨烯的制备方法.
-
国内外研究人员通过借鉴传统低维碳纳米材料制备工艺中的化学气相沉积方法,利用多种类型的原生塑料或废弃塑料作为固态碳源,替代高纯低分子碳氢化合物气源或甲醇、乙醇等易挥发液态碳源,制备出一系列层状石墨烯及其衍生材料. CVD是利用气态或蒸气态的物质,在气相或气固界面上发生反应,生成固态沉积物的过程[20],如图2b所示,按照塑料作为碳源被热解转化为气态前驱体的位置与材料生长位置的相对关系,目前研究工作中基于CVD方法制备石墨烯材料的反应构型可被分为“单区热解‐CVD”与“异位热解‐CVD”两种模式.
如图2所示,“单区热解‐CVD”模式中,塑料底物在反应衬底上借助高温辐射热提供的能量直接热解,通过气相中的均相反应或生长衬底表面的异相反应形成活性更高的含碳活性基团,再通过石墨烯成核、生长、拼接等一系列基元反应步骤,根据具体的反应条件与生长环境,在衬底上形成特定的石墨烯材料. 如表1所示,Tour等利用PS作为碳源,在Cu箔上于1050 ℃的Ar/H2气氛中合成出低缺陷、约97%光透过率、方阻1.5—3.0 kΩ·sq−1的单层石墨烯材料,并通过一系列表征手段验证了该材料具有单层石墨烯特有的组成、结构和光谱特征 [21]. 在此案例中,PS本身相对于CH4等常规前驱体碳源物质具有更低的C—H键键能(约292—305 kJ·mol−1),在具有催化活性的Cu衬底表面PS可以更高效地脱氢、裂解并最终产生石墨烯成核生长所需的碳活性物种;同时Cu较低的碳溶量限制了碳活性物种向金属体相的扩散,最终使其在Cu衬底表面经“自限制生长模式”形成单层石墨烯产物[25]. 类似的,Joshi等借助Ni箔做为生长衬底与催化剂,在常压900 ℃的N2反应气氛中,利用PET作为固态前驱体,也同样合成出高结晶度的单层石墨烯材料[22]. 此案例中,作者应使用了两种不同厚度的Ni箔分别置于塑料底物上方与底部(如表1所示),共同作为催化剂参与PET衍生石墨烯的生长过程,Ni具有2个未配对的3d电子,反应过程中C—H键的电子可以转移到未充满的d轨道上,从而有效降低C—H键的断裂能垒,以此提高碳源裂解的效率[26]. 此外,由于Ni材料具有较高的碳溶解度,碳活性物种(完全脱氢的活性碳原子)生成后会溶解进入到Ni金属体相中,在降温阶段活性碳原子过饱和发生偏析进而于衬底上形成石墨烯材料,遵循“偏析生长机制”. 因此在利用Ni作为催化剂和衬底制备单层石墨烯材料的过程中要精确控制降温速率,防止大量溶解碳剧烈析出进而形成多层石墨烯结构,影响产物的质量与理化性能[27].
除了上述塑料衍生石墨烯制备案例中所使用的Cu与Ni以外,诸如与Cu同处ⅠB-ⅡB族的Au,与Ni同处ⅧB族的Rh、Ru、Co、Fe、Ir,以及ⅣB-ⅥB族的Cr、Mo、W、Ti、V等过渡金属也可以被用作衬底实现石墨烯的生长. 不同类型的金属对前驱体催化裂解的能力、碳溶量及相互作用强度等均具有差异,相应的石墨烯生长行为也不尽相同[27]. 例如ⅠB-ⅡB族过渡金属的碳溶量及催化活性普遍较低,石墨烯生长过程中往往无金属碳化物形成;ⅧB族过渡金属催化活性与碳溶量较高,石墨烯生长会伴随亚稳态金属碳化物的形成,在高温退火阶段通过原子转移或原子替换实现金属碳化物向石墨烯的转变;在ⅣB-ⅥB族过渡金属介导的石墨烯生长过程中,催化活性随着金属碳化物的形成显著提高,并且金属碳化物具有较高的稳定性,能够在降温过程中有效抑制体相碳原子的偏析,减少多层石墨烯出现的概率[28]. 为缓解单一过渡金属介导石墨烯生长的局限性,Ni-Cu或Ni-Mo等多种类型的二元合金催化剂也常常被构建使用:其旨在保持较高碳源催化裂解效率的同时,减少或稳定进入金属体相的碳原子,进而降低偏析过程的强度,保证单层石墨烯的制备质量与效率[29]. 此外,金属衬底的化学稳定性、晶面取向、与石墨烯晶格的失配度等因素也会对石墨烯生长过程及产物特征有显著影响,对于塑料衍生单层石墨烯的制备而言,合理地选择和处理衬底是保证产物质量的先提条件.
除了使用结构规则的金属作为生长衬底和催化界面以外,非金属材料也可以被用来转化塑料获取石墨烯材料,目前非金属衬底介导的墨烯生长机理仍不明确,并且非晶态衬底的表面结构形貌复杂,缺陷密度大,实际应用中往往难以利用其实现对石墨烯产物结构与形貌的精确调控[30]. 例如,有研究者利用有机改性蒙脱土为生长模板与催化剂,与废弃PP颗粒混合后在750 ℃反应条件下以最高质量分数为86.6%的产率制备出几百纳米到几微米长的具有多层结构的叶片状石墨烯材料,HRTEM表征发现堆叠构成层状石墨烯的碳纳米层具有典型的褶皱形态和弯曲边缘,碳层中出现多种无规则碳环结构并含有杂原子,材料存在大量缺陷,碳产物品质相对较低[23]. 类似的,有研究者利用纳米膨润黏土为衬底,以废弃PE、PP与PET为碳源,在N2气氛中经两次热解(450 ℃,945 ℃)制备得到具有少层结构的层状石墨烯材料[31]. 利用该石墨烯产物作为电极材料组装而成的超级电容器具有较高的比电容(398 F·g−1)与功率以及能量密度(1009.74 W·kg−1,38 Wh·kg−1);此外利用该材料作为半导体层构建组成的染料敏化薄膜电池同样也具有较高的开路电压(0.77 V)和短路电流密度(0.3 mA·cm−2).
赋予石墨烯材料三维宏观形态的同时保留其单层或少层石墨烯的理化性能,可以克服单层石墨烯材料自支撑性差、材料强度不足的问题,使其更好地在污染控制、催化或能源储存等应用领域发挥材料本身的优异属性[19]. 在CVD过程中,生长衬底作为“宏观形态模板”很大程度上决定了石墨烯产物的宏观结构特征;石墨烯在生长过程中可以复刻“模板”的三维结构并保持畴区的连续拼接,通过刻蚀或材料的自分解过程去除相应模板材料后,可以便捷地获取具有丰富结构特征的三维石墨烯材料[19, 32]. 除了应用广泛的泡沫及颗粒金属材料,金属氧化物(MgO、ZnO)、金属盐(过渡金属盐、碱金属盐)及SiO2等也可以作为三维石墨烯生长的“硬模板”[33 − 34],这些硬模板材料在石墨烯生长过程中通常物化性质会发生一定的改变. 例如金属盐(硝酸盐、碳酸盐)在高温还原性气氛中会发生分解和还原,转化为催化活性更高的金属催化剂辅助底物裂解及石墨烯生长,同时利用材料分解产生的气体赋予石墨烯产物独特的三维结构[35]. 硬模板材料在反应结束后往往需要额外的刻蚀过程去除,这不可避免的增加了环境足迹并在一定程度上降低了三维石墨烯材料的质量[36]. 相对的,以铵盐(NH4Cl、(NH4)2CO3)和尿素为代表的“软模板”可以在石墨烯生长过程中发生自分解完全转化为气体(NH3/HCl/CO2)[37 − 38],在赋予石墨烯产物“气泡”结构的同时实现与石墨烯产物的“自发分离”,这个过程需要对软模板材料的分解过程进行精细的控制以提高对石墨烯产物结构的控制效果[39].
基于以上三维石墨烯制备策略,研究者利用Ni网做为生长模板,于900 ℃的真空环境(~1.0 mTorr)中,利用含有少量石蜡成分的PE膜作为碳源,制备出具有中空少层石墨烯微纳结构特征的宏观三维网状石墨烯材料. 该材料具有良好的机械柔韧性、自支撑能力,并展现出较好的电化学性能、可控的透光率、优异的超疏水性和自清洁能力[24]. 为评估材料的应用性能,该PE衍生石墨烯网状材料被组装成为固态超级电容器电极,其中空的结构显著地提高了电极界面的面积,经测试与同样基于CVD方法制备的致密层状石墨烯组装成的固态电容器相比,比电容提高了20—30倍;此外,研究人员还测试了该材料分离回收泄露油脂的性能,以及作为抗污纱窗等产品的自清洁能力,凭借材料本身的超疏水性及高表面粗糙度,该材料在上述测试中均展现出优秀的实际应用性能(油脂分离回收效率约90.9%)[24]. 类似的,有研究者利用三维胶体硅作为三维石墨烯宏观结构模板,以FeCl3作为微观结构模板,聚乙烯醇(poly vinyl alcohol)为碳源,在1000 ℃的Ar/H2还原气氛中退火处理30 min,成功制备出具有优异导电性(52 S·cm−1)和较大比表面积(1025 m2·g−1)的三维网状石墨烯材料,利用该材料组装的双层电容器具有优异的性能(比电容245 F·g−1)和循环稳定性,在纳米电子器件和能源储存领域具有一定应用潜力[40].
相较于装置构型与反应条件相对简单的“单区热解-CVD”模式,“异位热解-CVD”模式将塑料底物热解区域与碳纳米材料生长区域分离开来(如图2),从而可以实现对每块区域反应条件的独立控制,如施加不同的温度设置、填充不同类型的催化剂;此外该构型还引入了可以动态调整的载气模块,能够更好地实现对碳纳米材料生长过程和产物特征的调控. 例如在制备单层大晶畴石墨烯材料时,控制石墨烯成核密度成为关键的调控手段,低成核密度可以削减晶格取向不同的石墨烯畴区之间拼接的概率,进而降低晶界密度,增大石墨烯畴区尺寸[41]. 通过控制石墨烯成核阶段碳活性物种的浓度,可以很好地实现对成核密度的控制[42]. 基于上述理论,如表2所示,研究者借助“异位热解-CVD”系统,将底物PS的热解区域与石墨烯生长区域分离,利用载气(Ar/H2)主导热解产物至CVD单元的传质过程,仅在CVD腔室中使用Cu箔作为单层石墨烯生长所需的催化衬底,为保证石墨烯的生长速率与质量,CVD区域温度保持在1050 ℃,而热解区域的温度则采用动态变化的控制形式[25]. 反应初期,为使碳活性物种达到过饱和进而进入成核阶段,研究人员将热解区域温度迅速提升至205 ℃,同时载气的流量控制在300 cm3·min−1,一旦石墨烯晶核形成,立刻将热解温度降至临界值(195 ℃)以下从而抑制新的成核过程,成功将成核密度控制在每平方厘米100核;此后缓慢将载气流量与热解温度分别提升至600 cm3·min−1与215 ℃,以此提高石墨烯生长的速率,最终经过80min制取得到具有高达1.2 mm畴区尺寸的单层石墨烯产物[25]. 相似的,Tanemura等借助“异位热解-CVD”系统,通过精确控制混合塑料底物(PP+PS)热解的升温速度(1.5 ℃·min−1)来调控成核密度及石墨烯单晶畴区的生长速率,最终在多晶Cu箔上制备出六边形或圆形的晶畴尺寸最高达90—100 μm的单层石墨烯产物,相对的采用更快的热解升温速率则只能获取到双层或少层的石墨烯产物[43].
同样的,“异位热解-CVD”模式也适用于注重塑料转化效率及产物直接应用能力等方面,而对产物形貌结构调控需求不高的塑料衍生石墨烯制备场景. 例如有研究人利用“异位热解-CVD”构型在1050 ℃的Ar/H2气氛中,使用多种原生或废弃塑料(PET、PE、PVC、PP、PS)作为固态碳源,使用Ni箔作为衬底得到了结构与理化性质相似的多层石墨烯薄膜[44]. 此案例中由不同种类塑料转化制备得到的石墨烯薄膜均具有良好的柔韧性、自支撑能力、拉伸强度((55.17 ± 5.1) MPa)以及较高的电导率(3824 S·cm−1),利用该类型材料替代商业化石墨材料组装得到的锂离子电池和柔性锂电池,具有较高的电池容量(381.4 mAh·g−1)、库伦效率(> 99%)和优异的柔韧性(可弯折180°),有望作为天然石墨的替代材料应用于能源储存等场景[45]. 此外,该石墨烯产物还具备优异的电热性能,在施加5 V电压的工况下可以迅速均匀升温至322.6 ℃,快速的温度响应特性令其有望被开发做为可穿戴加热器件使用.
-
2014年,研究人员使用CO2激光器在空气中辐照聚酰亚胺(polyimide, PI)薄膜,将其直接转化为具有三维孔结构的石墨烯材料(图2c). 该多孔石墨烯具有较高的比表面积(340 m2·g−1)与热稳定性(> 900 ℃)以及优异的导电能力(5—25 S·cm−1)[46]. 利用该激光介导的热化学方法可以实现在多种聚合物基材表面直接“打印”各种复杂形状甚至图案的多孔石墨烯薄层,进而形成复合材料[47]. 通过简单地调整激光参数、反应气氛或底物种类,可实现对石墨烯孔结构特征、组成和形貌的调控. 这种精确可控且简便快捷的多孔石墨烯制备方法令“激光石墨烯”成为了多种应用场景的材料基础,在空气净化[48]、电催化[49]、能源转化及储能[50 − 52]、检测[53 − 55]、抗污[56]、微流控[57]等领域均取得优异的性能表现. 然而,能够直接借助基于激光辐照的热化学过程转化为多孔石墨烯材料的塑料种类十分有限. 常见的聚烯烃类塑料在光热作用下极易转化为挥发性低分子碳氢化合物从而逸出反应界面,其较低的熔点也会导致材料在转化过程中形态发生急剧变化. 在研究初期,仅PI和聚醚酰亚胺(polyetherimide, PEI)这两类结构相近的合成聚合物可以被用作“激光石墨烯”的前驱体[47]. 基于上述问题,研究人员提出基于多重激光辐照方式的反应优化策略(如表3所示),通过使用格栅激光束或简单地调整激光器与基材平面z轴的距离,使基材透过低通量重叠激光束的辐射,将可作为激光石墨烯前驱体的合成聚合物种类由早期的高温工程塑料(PI、 PEI、聚对苯二甲酰对苯二胺、聚苯硫醚等)扩展到交联热固性聚合物(酚醛树脂、环氧树脂、交联聚苯乙烯)[58]. 区别于传统石墨烯柔性复合材料“制备+转移”的复杂工序,利用该方法可以非常便捷且精确地在各种规则平整的基材表面刻蚀出几十至几百微米的多孔石墨烯薄层,非常适合作为柔性电子器件、微流控芯片等先进复合材料的制备方法[60 − 61]. 虽然理论上所有可以被转化为无定形碳的富碳前驱体都可以通过激光辐照的方式被深度石墨化处理进而形成石墨烯材料,但是目前生产使用量最高的聚烯烃类塑料仍无法高效利用该激光介导的热化学方法进行转化[47],并且激光介导的光热/光化学反应界面深度非常有限,一定程度上限制了其作为塑料衍生低维碳纳米材料宏量化制备方法的发展潜力.
-
如图2d所示,近年来研究人员利用焦耳加热(Joule heating)的方式,在不借助催化剂的情况下,成功将多种低值富碳前驱体(混合塑料废物、塑料热解产物、废轮胎等)转化为粉体石墨烯[16, 62, 63]. 该方法利用焦耳热效应,通过添加导电助剂(炭黑、粉状石墨烯)赋予混合反应底物一定的导电能力,借助强脉冲电流以105 K·s−1的升温速率瞬间将反应物加热至约3000 K的高温,将底物中的无定形碳组分转化为高度石墨化的“闪焦石墨烯”(Flash graphene)[16]. 值得注意的是,在非稳态的转化过程中,石墨烯层与层间排列并没有形成稳定的AB堆垛形式,而是呈现二维乱层堆垛的情况. 闪焦石墨烯层间距约0.345 nm,大于典型AB堆垛石墨烯的层间距(0.337 nm)[64 − 65],这样的排布特征也赋予粉体石墨烯产物更好的分散能力,经测试其在溶液中的分散浓度可达4 g·L−1. 实际应用时,添加质量分数为0.05%的闪焦粉体石墨烯即可将水泥复合材料的抗压强度提升25%,作为聚二甲基硅氧烷的添加剂时可将复合材料的抗压强度提升250%(质量分数0.1% 填加量). 在能源相关应用领域,闪焦石墨烯也被报道可以作为储能设备的电极材料[16]. 早期焦耳加热介导的混合塑料(PET、HDPE、LDPE、 PVC、 PP、PS、聚丙烯腈(PAN))热化学转化过程中,杂原子(O, N, Cl等)的引入容易使石墨烯产物中出现较高的缺陷结构. 研究人员通过优化焦耳加热的反应构型,采用交流-直流(低压-高压)序列焦耳加热的形式(表3),先利用较长时间(8 s)的低压交流焦耳加热过程去除部分挥发性组分得到中间碳产物,再通过500 ms的高压直流脉冲电流继续提高产物的石墨化程度,最终显著降低了塑料衍生石墨烯产物中缺陷的含量并提升了产物的质量[59]. 此外,该石墨烯产物同样具备良好的分散能力(层间距0.345 nm),降低了使用过程中发生团聚的概率[59].
除了制备形貌结构不规则的粉体石墨烯,通过引入“模板法”,基于焦耳加热的热化学转化过程同样也可以赋予产物特定的宏观结构. 例如有研究者利用Ca(OAc)2作为“硬模板”材料,与塑料均匀混合后,在焦耳加热过程中将模板材料两步分解为丙酮与二氧化碳,通过物理鼓吹、化学刻蚀和残留CaO的“硬模板”作用,赋予石墨烯产物丰富的2D片层孔结构和3D层间孔道,最终得到的多孔粉体石墨烯比表面积高达874 m2·g−1 [66]. 该产物在CO2吸附测试实验中,相较于常规的粉体石墨烯,其吸附量提升了10倍. 此外在电催化及能源储存应用场景测试中,该多孔粉体石墨烯同样展现出优秀的性能表现[66]. 例如研究人员利用该多孔石墨烯作为电化学析氢催化剂,取得了较好的析氢性能、更低的过电位、更高的电流密度和优异的循环稳定性;在将多孔粉体石墨烯用作锂离子电池电极材料时,相较于常规的石墨烯电极和Cu电极,其具有更低的成核过电势和更好的电化学性能.
粉体石墨烯是目前最可能实现规模化制备的塑料衍生碳纳米材料,能够满足复合材料、能源储存、环境修复、热管理等诸多领域的应用需求[16, 65, 67],然而目前由焦耳加热介导的热化学转化方法C转化率仍然较低(22%—27%),产物中存在大量气态热解物质(约60 %),目前仍然需要深入探索更有效的反应调控方法[59].
-
碳纳米管(carbon nanotubes, CNTs)特殊的结构特征赋予其优异的机械强度和良好的导电与导热性能,在环境、能源、催化、纳电子器件、复合材料等众多领域展现出巨大的应用潜力[68]. CNTs典型的制备方法目前有3种,分别是CVD、电弧法(Arc-discharge)与激光烧蚀法(Laser ablation),其中CVD方法在CNTs的精细结构控制、特定取向生长及宏观形貌调控等方面具有显著的优势;电弧法的产物成分通常比较复杂,除了CNTs以外往往还混杂其他形态的碳纳米微粒,并且在较高的电弧温度(3000—3700 ℃)下多种形态的碳材料组分易发生烧结,目标产物分离纯化困难;激光烧蚀法则不适合规模化制备,与前文所述的激光介导的多孔石墨烯制备工艺存在类似的问题[69].
与近十年来才开始兴起且尚停留在实验室阶段的塑料衍生石墨烯制备技术发展历程相比,利用塑料作为固态碳源制备CNTs的工作最早可被追溯到1997年,研究者利用PE颗粒为固态碳源,利用镍基催化剂在He氛围中于420—450 ℃温度下制备出直径分布10—40 nm的MWCNTs[70]. 此后,塑料衍生CNTs的制备其底物种类与来源、装置构型、催化剂类型与反应条件等发展出庞大的变化组合形式(如图3所示). 然而,绝大多数塑料转化制备CNTs的方法仍基于CVD方法或其衍生形式,即通过与塑料衍生石墨烯材料制备方法高度相似的 “热解-CVD”过程,获取以MWCNTs为主的产物性能差异相对较小的碳纳米材料. CNTs的化学气相沉积生长机理与制备方法同石墨烯材料有着高度的相似度,原因在于两种材料具有相似的基本结构单元和生长过程. 催化剂尺寸、分布及反应过程中碳产物形态变化特征是划分这两种碳同素异形体不同形貌结构演化过程的关键. 下面本文将分类展述利用CVD方法及其衍生形式(如等离子体增强化学气相沉积方法)和基于电弧法制备塑料衍生CNTs的底物类型、反应条件、产物形貌结构及应用性能特征.
-
如表4所示,研究人员使用多种原生或废弃塑料(PP、PE、PS、PVC、PET等),采用“单区热解-CVD”与“异位热解-CVD”模式,成功“批式”或“连续”制备出直径分布4—338 nm不等的WMCNTs [71 − 73]. 利用CVD方法制备塑料衍生碳纳米管的过程中,过渡金属类(同本文“1.1”节所述)、金属氧化物类与非金属类催化剂(SiC、C60、纳米金刚石等)均被广泛应用尝试[72, 76]. 其中金属氧化物类催化剂因具有较高的热稳定性,可以有效缓解催化剂团聚等现象导致的碳纳米材料结构不可控的问题[77];而全碳催化的使用可以避免金属对碳产物的污染,并可以为碳纳米管生长提供具有特定结构特征的外延模板[78]. “热解-CVD”的反应温度范围通常为400—1000 ℃,温度对于塑料衍生碳纳米管的生长和结构演变过程起到重要的影响[79],一方面温度影响着碳源分子的裂解,塑料中稳定的C—C键和C—H键活化通常需要在较高的反应温度中才能高效进行;另一方面温度直接影响碳活性物种在催化剂表面或体相中的迁移速率,温度越高,碳的迁移速率越快,可以加速碳纳米管成核和生长的过程. 此外温度还对催化剂的分散、聚集和扩散行为有显著影响,需要根据催化的负载特征及热稳定性等综合选取合适的碳纳米管生长温度,避免过高温度导致的催化剂聚集或奥斯特瓦尔德熟化(Ostwald ripening)[80 − 82].
同前文1.1节案例类似的,相较于利用“单区热解-CVD”模式制备塑料衍生碳纳米管的过程,“异位热解-CVD”模式同样具备更好的过程调控和产物优化能力;此外“异位热解-CVD”模式还可以通过耦合额外的预分离处理单元,降低混合塑料热解产物中杂质或有害成分对催化剂寿命及碳纳米管质量的影响. 例如在热解与CVD反应区域间加设常压精馏塔或吸附装置,通过剥离热解产物中的重油组分及有害物质(HCl等),有效缓解催化剂结焦或中毒失活等问题[83].
目前,上述利用“热解-CVD”方法转化塑料制备碳纳米管材料的过程中,仍面临C转化率低(2.2%—57.7%)、产物结构均一性差、催化剂易结焦失活等问题的挑战[72]. 潜在原因之一在于传统CVD方法转化塑料制备CNTs的过程通常利用热辐射作为能量输入的方式,反应过程不均匀的热场分布可能会导致不同分区塑料热解产物分布、气态前驱体深度裂解和碳纳米材料催化生长速率等难于统一.
等离子体增强化学气相沉积方法,即借助微波或射频提供部分碳源热解的能量,使含碳前驱体电离形成等离子体,进一步的驱动其在催化剂界面发生化学组装反应生长得到碳产物的方法,近年来被证实可以利用其等离子体与催化剂的协同作用,提高碳纳米材料的制备效率与产物质量[84, 85],并有望一定程度缓解上述问题. 例如,研究人员借助等离子体增强化学气相沉积方法,以微波为能量源在催化界面构建热场并形成局域等离子体环境,强化热解产物深度裂解过程,提高碳纳米管在催化界面的生长动力[86]. 该方式可将C转化率提升至57.6%—73.1%,并且得到形貌与尺寸分布均匀的MWCNTs产物(内径约6 nm,外径10—20 nm)[74]. 如表4所示,利用PE、PP、PS及其混合物为转化底物,使用FeAlOx为微波吸收材料与催化剂,经过多次循环转化过程碳材料的平均产量可达到620 mg·g−1. 类似的,研究者利用铁基催化剂在催化剂界面同时构造热场及强电场,利用催化界面的等离子体环境强化碳源的裂解及碳活性物种(CH、C2等)的产生,以PE、PP及PS为底物制备出形貌均匀的MWCNTs(内径约5.4 nm,外径约13.9 nm)[71]. 然而,由于微波穿透深度有限,且目前对等离子体增强化学气相沉积方法转化塑料制备CNTs的反应机制尚缺乏统一清晰的认识,该转化模式还需要开展更深入的催化机制研究与反应器优化工作.
-
作为传统CNTs制备工艺之一的电弧法(典型装置构型如图3b所示),近来年也被用于转化塑料制备CNTs. Prathap等利用电弧法将PET热解固体产物转化为具有多种结构特征的包含MWCNTs、纳米纤维、纳米碳球、富勒烯等材料的混合纳米碳产物[75]. 反应过程参数及产物分布特征如表4所示,在N2气氛中PET经高温裂解得到的焦化产物经研磨后填入中空高纯石墨棒作为阳极,通过电弧放电在阳极末端与旋转阴极间产生高温热场,利用复合阳极中石墨与PET热解固体产物中的无定形碳的共蒸发,在阴极沉积出MWCNTs等低维碳纳米产物. 根据反应位置的不同,旋转阴极沉积物与位于阳极中孔内部不同深度的混合碳产物的组成、形貌、石墨化程度均存在显著差异. 其中,阴极沉积物中的MWCNTs石墨化程度虽然相对较高,直径分布集中且管壁数量与弯曲盘绕结构更少,但是仍掺杂有多种类型的碳纳米材料并且产率较低(< 15%),很大程度限制了目标产物的质量与该方法的规模化潜力.
-
作为塑料升级转化路径中重要的分支,多种类型的塑料已经成功通过热解-化学气相沉积方法、基于激光、焦耳加热和电弧法的热化学方法或等离子体增强化学气相沉积方法被升级转化为一系列具有独特形貌、结构、性能及应用特征的低维碳纳米材料,如层状石墨烯、粉体石墨烯、多孔石墨烯及MWCNTs等. 如何提升塑料升级转化碳纳米材料产物的“质”和“量” 是该领域的研究重点. 就产物“质”而言,目前以材料性能为导向的塑料升级转化过程,如利用“异位热解-CVD”模式制备单层石墨烯材料,可以获得同传统利用高纯单一碳源(甲烷等)制备石墨烯材料过程一致的、具有相似结构与性能特征的低缺陷和大晶畴单层石墨烯产物. 就产物“量”而言,以塑料规模化升级转化制备低维碳纳米材料为导向的方法路径,如以焦耳加热介导的热化学方法快速制备粉体石墨烯,以 “异位热解-CVD”模式连续制备MWCNTs,均具有潜力在将来形成一条可以规模化制备碳纳米材料的工艺路径,实现塑料向导电添加剂、复合材料等高值产品的升级转化. 然而,需要明确的是目前塑料升级转化制备低维碳纳米材料的基本反应机制与产物结构性能调控手段仍然不够系统明确,反应所需能耗较高,催化剂介导的过程稳定性较差,产物转化率较低,规模化转化工艺及装备仍未成型,未来仍需要更多的科研力量与产业资源集中到此领域中开展深入和广泛的研究.
塑料升级转化制备低维碳纳米材料的方法及研究进展
Research progress in preparation of low-dimensional carbon nanomaterials derived from plastics
-
摘要: 将塑料转化为具有优异性能的低维碳纳米材料,近年来已经发展成为一支重要的塑料升级转化路径并取得阶段性的研究进展. 热解-化学气相沉积方法、等离子体增强化学气相沉积方法、和基于激光、焦耳加热及电弧法的热化学方法已被开发利用,将多种类型的塑料转化为石墨烯和碳纳米管材料. 为更好地追踪本领域的研究进展,本文梳理了以上两类典型塑料衍生低维碳纳米材料的制备方法、装置构型、反应条件、材料转化机制、产物应用场景及性能,并对当前的机遇及未来的挑战进行了展望.Abstract: In recent years, the conversion of plastics into low-dimensional carbon nanomaterials with exceptional properties has emerged as a pivotal pathway for plastic upcycling, and it has made significant strides. A range of preparation methods, including pyrolysis-chemical vapor deposition, plasma-enhanced chemical vapor deposition, and thermochemical processes induced by laser, Joule heating, and arc discharge, have been developed and utilized to convert various types of plastics into graphene and carbon nanotubes. To better track the research progress, the preparation methods, reactor configurations, reaction conditions, material conversion mechanisms, product application scenarios and performance of the two typical plastic-derived low-dimensional carbon nanomaterials were summarized. Furthermore, we conclude with our perspective on the ongoing challenge and opportunities.
-
Key words:
- plastic waste /
- graphene /
- carbon nanotubes /
- low-dimensional nanomaterials.
-
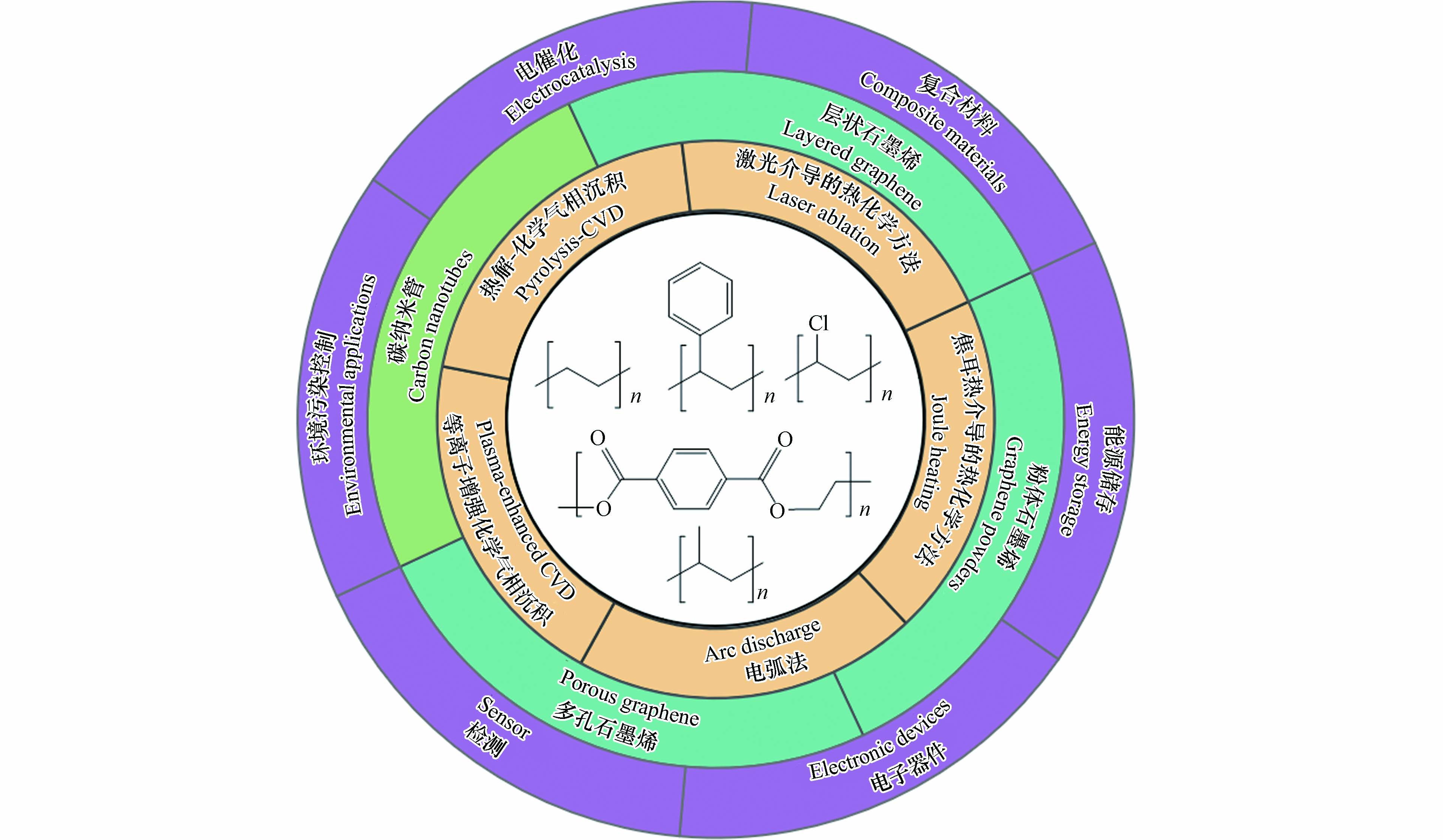
图 1 塑料升级转化制备低维碳纳米材料的底物种类、制备方法、产物类型与应用场景概述
Figure 1. Substrate types, preparation methods, product types, and application scenarios of plastic-derived low-dimensional carbon nanomaterials
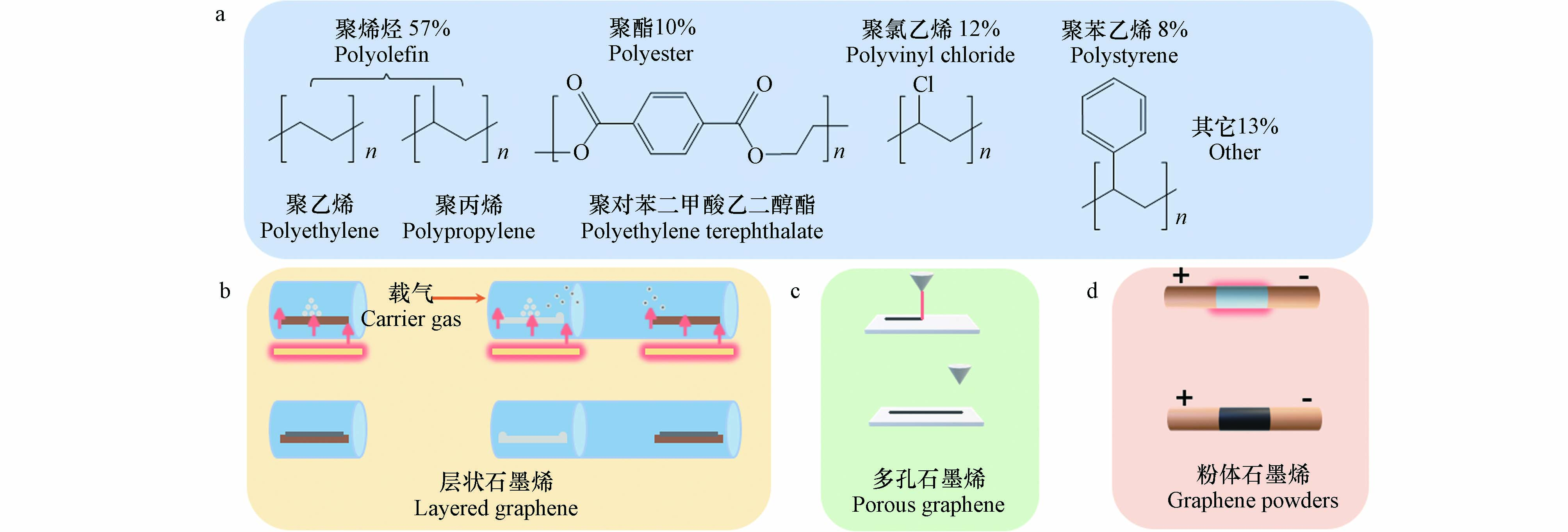
图 2 塑料升级转化制备石墨烯材料的底物种类(a)、制备方法(b)及产物类型(c、d)
Figure 2. Substrate types, preparation methods, and product types for the preparation of plastics-derived graphene
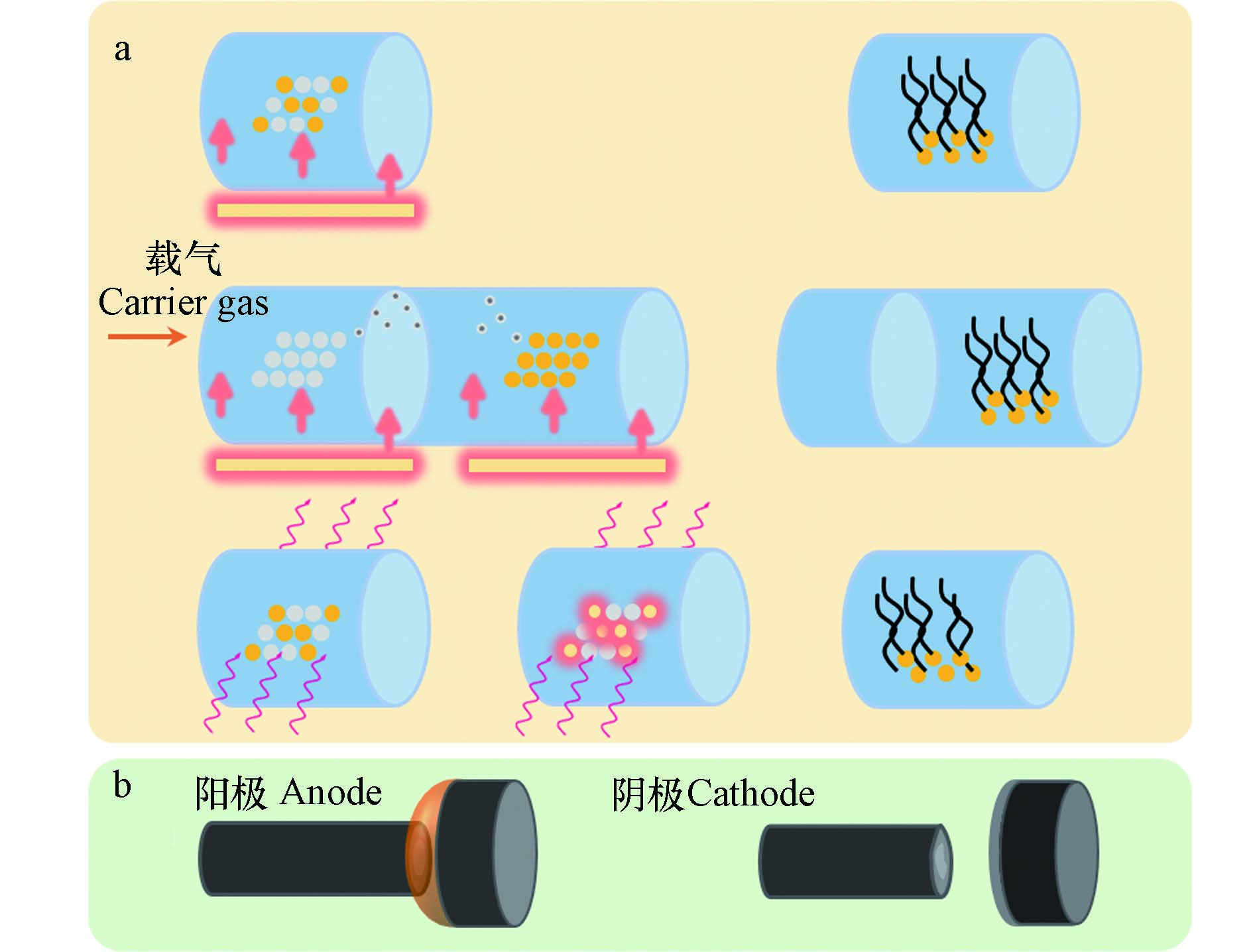
图 3 塑料升级转化制备石墨烯材料的制备方法(a)及装置构型(b)
Figure 3. Preparation methods and reactor configurations for the preparation of plastics-derived CNTs
表 1 单区热解-CVD模式升级转化塑料制备石墨烯材料的反应条件及产物特征
Table 1. Reaction conditions and product properties of single-stage pyrolysis-CVD mode for the preparation of plastics-derived graphene
方法 Method 底物 Precursors 产物 Products 反应条件 Reaction conditions 材料特征 Properties 应用 Applications 文献 Ref. 热解-CVD PS 单层石墨烯 装置构型:单室CVD,铜箔衬底,底物质量:0.01 g,温度1050 ℃;压力1.24 kPa; 反应气氛:Ar (500 cm3·min−1),H2 (100 cm3·min−1) 反应时间:15 min IG /2D < 0.56, ID/G < 0.1; 方阻:1.5–3.0 kΩ·sq−1 未报导 [21] 热解-CVD PET 单层石墨烯 装置构型:单室CVD;上方200 μm镍箔 + 底部 10 μm镍箔衬底 底物质量:0.1 g,温度900 ℃;压力101.3 kPa;降温速率5.8 ℃·min−1反应气氛:N2 (1000 cm3·min−1) 反应时间:5 min IG/2D: ~0.38 未报导 [22] 热解-CVD PE, PP, PET 多层石墨烯 装置构型:单室CVD,衬底:有机改性蒙脱土,纳米膨润土 底物质量:15 g (OMMT/PP); 温度750—945 ℃ 反应时间:15—45 min 形貌结构:片状石墨,石墨烯层数较多,且多褶皱与缺陷结构,石墨化程度较低 超级电容器,太阳能电池 [23] 热解-CVD PE 三维层状石墨烯 装置构型:单室CVD,镍网衬底,底物过量填充,温度900 ℃;压力 ~1.0 mTorr;降温速率90 ℃·min−1反应气氛:无外源气体 反应时间:8 min 形貌结构:中空多层石墨烯网 层数:6—8 厚度:2.5—3.5 nm; 方阻:< 4.2 Ω·sq−1 超级电容器,自清洁纱窗, 油水分离 [24]  下载: 导出CSV
下载: 导出CSV
表 2 异位热解-CVD模式升级转化塑料制备石墨烯材料的反应条件及产物特征
Table 2. Reaction conditions and product properties for the preparation of plastics-derived graphene via two-stage pyrolysis-CVD mode
方法 Method 底物 Precursors 产物 Products 反应条件 Reaction Conditions 材料特征 Properties 应用 Applications 文献 Ref. 热解-CVD PS 单层石墨烯 装置构型:双区热解+CVD系统 热解条件:脉冲加热梯度升温,温度80—280 ℃;压力101.3 kPa;反应气氛:300—600 cm3·min−1 (Ar+H2 (0—10 cm3·min−1));时间30—80 min CVD条件:温度950—1050 ℃;压力101.3 kPa;时间30—80 min;反应气氛:300—600 cm3·min−1 (Ar+H2 (0—10 cm3·min−1));铜箔衬底 石墨烯畴区尺寸1.2 mm 未报导 [22] 热解-CVD PE 86% wt; PS 14% wt 单层石墨烯 装置构型:双区热解+CVD系统 热解条件:温度500 ℃;压力101.3 kPa,时间 90 min;升温速率1.5 ℃·min−1;底物质量30 mg CVD条件:温度1020 ℃;压力101.3 kPa;时间90 min;降温速率16 ℃ min−1;反应气氛:Ar (98 cm3·min−1),H2 (2.5 cm3·min−1);铜箔衬底 六边形或圆形单晶石墨烯,晶畴尺寸90— 100 μm 未报导 [43] 裂解-CVD 原生或废弃PET、PE、PVC、PP、PS, 多层石墨烯 装置构型:双区热解+CVD系统 热解条件:温度1050 ℃; CVD条件:温度1050 ℃; 时间120 min;反应气氛:Ar (150 cm3·min−1),H2 (25 cm3·min−1);镍箔衬底 石墨化程度较高的紧密堆叠的多层石墨烯, ID/G: 0.03—0.65; 电导率: 3824 S·cm−1 可折叠锂离子电池柔性电极;柔性电热元器件 [44]
下载: 导出CSV
表 3 激光和焦耳加热介导的热化学方法升级转化塑料制备石墨烯材料的反应条件及产物特征
Table 3. Reaction conditions and product properties for the preparation of plastics-derived graphene via laser or Joule heating induced thermochemical processes
方法 Method 底物 Precursors 产物 Products 反应条件 Reaction Conditions 材料特征 Properties 应用 Applications 文献 Ref. 激光介导 高温工程塑料,交联热固性塑料 多孔 石墨烯 装置构型:计算机控制的CO2脉冲激光系统 激光波长:10.6 μm/9.3 μm 扫速:2—58 cm·s−1辐照密度:10—1000 p.p.i. 激光占空比:1%—5% 电导率:25 S·cm−1方阻:<15 Ω·sq−1层间距:0.34 nm 具有较高缺陷:含有大量五元环与七元环 空气净化、电催化、能源转化与储能、检测、抗污、微流控 [46, 58] 焦耳加热介导 PP, PET, PE, PS, PVC, PAN 粉体 石墨烯 装置构型:交流(120 V, 60 Hz, 8 s)+脉冲直流(60 mF, 110 V, 500 ms)序列焦耳加热系统,低电流(208 V, 0.1—25 A, 50 s)+ 高脉冲电流 (60 mF, 100 V, 250 ms) 导电助剂:质量分数为5 % 炭黑 层间距: 0.345—0.351 nm 比表面积: 650—874 m2·g−1 储能、电催化、复合材料添加剂 [16, 59]
下载: 导出CSV
表 4 CVD及电弧法介导的热化学方法升级转化塑料制备碳纳米管的反应条件及产物特征
Table 4. Reaction conditions and product properties for the preparation of plastics-derived CNTs via CVD or arc-discharge induced thermochemical processes
方法 Method 底物 Precursors 产物 Products 反应条件 Reaction Conditions 材料特征 Properties 应用 Applications 文献Ref. 热解-CVD PP, PE, PS, PVC, PET MWCNTs 装置构型:单室固定床CVD反应器;双段固定床反应器;连续热解流化床-固定催化床CVD反应器;连续热解-连续催化床CVD反应器 热解温度:400—950 ℃ CVD温度:450—1000 ℃ 产物多为MWCNTs,直径4—338 nm不等,长度0.55—50 μm不等 膜分离、电催化、水处理、能源转化与储能等 [72- 73] 等离子体增强化学气相沉积 PP, PE, PS MWCNTs 装置构型:单室石英反应腔+微波发生源 案例1:底物与催化剂配置:0.3—0.4 g塑料+同等质量催化剂(FeAlOx);微波功率:1000 W, 3—5 min 案例2:底物与催化剂配置:0.3 g塑料+0.6 g催化剂(Fe/AC);微波功率:700 W,10 min; 案例1:WCNTs内径约 6 nm,外径10–20 nm,管壁间距 0.32–0.34 nm. 案例2: WCNTs(内径约5.4 nm,外径约13.9 nm) 未报导 [71, 74] 电弧法 PET MWCNTs 混合物 热解条件:815 ℃,20 min 底物:10 g, 10×10 cm2反应气氛:N2, 10 kPa 电弧法条件:DC电源 100 A, 36 V;1 min 阳极:150 mm中空石墨棒(99%纯度,8 mm内径,12 mm外径);旋转阴极: 底物:5 g PET裂解产物粉末 反应气氛:N2, 500 Torr 阴极沉积产物:20 nm MWCNTs与其他纳米碳材料(富勒烯,多面体纳米颗粒等)的混合物; 阳极产物:高温区 (2600 ℃)为纳米通道超细碳管与MWCNTs(约95 nm)混合物,相对阴极产物石墨化较低 未报导 [75]
下载: 导出CSV
-
[1] SARDON H, DOVE A P. Plastics recycling with a difference[J]. Science, 2018, 360(6387): 380-381. doi: 10.1126/science.aat4997 [2] JAMBECK J R, GEYER R, WILCOX C, et al. Marine pollution. Plastic waste inputs from land into the ocean[J]. Science, 2015, 347(6223): 768-771. doi: 10.1126/science.1260352 [3] GIBB B C. Plastics are forever[J]. Nature Chemistry, 2019, 11(5): 394-395. doi: 10.1038/s41557-019-0260-7 [4] MIN K, CUIFFI J D, MATHERS R T. Ranking environmental degradation trends of plastic marine debris based on physical properties and molecular structure[J]. Nature Communications, 2020, 11: 727. doi: 10.1038/s41467-020-14538-z [5] GEYER R, JAMBECK J R, LAW K L. Production, use, and fate of all plastics ever made[J]. Science Advances, 2017, 3(7): e1700782. doi: 10.1126/sciadv.1700782 [6] MU J L, ZHANG S F, QU L, et al. Microplastics abundance and characteristics in surface waters from the Northwest Pacific, the Bering Sea, and the Chukchi Sea[J]. Marine Pollution Bulletin, 2019, 143: 58-65. doi: 10.1016/j.marpolbul.2019.04.023 [7] SCHWAB F, ROTHEN-RUTISHAUSER B, PETRI-FINK A. When plants and plastic interact[J]. Nature Nanotechnology, 2020, 15(9): 729-730. doi: 10.1038/s41565-020-0762-x [8] PEEKEN I, PRIMPKE S, BEYER B, et al. Arctic sea ice is an important temporal sink and means of transport for microplastic[J]. Nature Communications, 2018, 9: 1505. doi: 10.1038/s41467-018-03825-5 [9] ISOBE A, IWASAKI S, UCHIDA K, et al. Abundance of non-conservative microplastics in the upper ocean from 1957 to 2066[J]. Nature Communications, 2019, 10: 417. doi: 10.1038/s41467-019-08316-9 [10] LEBRETON L C M, van der ZWET J, DAMSTEEG J W, et al. River plastic emissions to the world’s oceans[J]. Nature Communications, 2017, 8: 15611. doi: 10.1038/ncomms15611 [11] GARCIA J M, ROBERTSON M L. The future of plastics recycling[J]. Science, 2017, 358(6365): 870-872. doi: 10.1126/science.aaq0324 [12] LAU W W Y, SHIRAN Y, BAILEY R M, et al. Evaluating scenarios toward zero plastic pollution[J]. Science, 2020, 369(6510): 1455-1461. doi: 10.1126/science.aba9475 [13] ZHENG J J, SUH S. Strategies to reduce the global carbon footprint of plastics[J]. Nature Climate Change, 2019, 9(5): 374-378. doi: 10.1038/s41558-019-0459-z [14] YADAV T P, AWASTHI K. Carbon nanomaterials: Fullerene to graphene[J]. Transactions of the Indian National Academy of Engineering, 2022, 7(3): 715-737. doi: 10.1007/s41403-022-00348-w [15] NOVOSELOV K S, GEIM A K, MOROZOV S V, et al. Electric field effect in atomically thin carbon films[J]. Science, 2004, 306(5696): 666-669. doi: 10.1126/science.1102896 [16] LUONG D X, BETS K V, ALI ALGOZEEB W, et al. Gram-scale bottom-up flash graphene synthesis[J]. Nature, 2020, 577(7792): 647-651. doi: 10.1038/s41586-020-1938-0 [17] HOU Q D, ZHEN M N, QIAN H L, et al. Upcycling and catalytic degradation of plastic wastes[J]. Cell Reports Physical Science, 2021, 2(8): 100514. doi: 10.1016/j.xcrp.2021.100514 [18] NOVOSELOV K S, FAL′KO V I, COLOMBO L, et al. A roadmap for graphene[J]. Nature, 2012, 490(7419): 192-200. doi: 10.1038/nature11458 [19] SUN Z X, FANG S Y, HU Y H. 3D graphene materials: From understanding to design and synthesis control[J]. Chemical Reviews, 2020, 120(18): 10336-10453. doi: 10.1021/acs.chemrev.0c00083 [20] SUN L Z, YUAN G W, GAO L B, et al. Chemical vapour deposition[J]. Nature Reviews Methods Primers, 2021, 1: 5. doi: 10.1038/s43586-020-00005-y [21] RUAN G D, SUN Z Z, PENG Z W, et al. Growth of graphene from food, insects, and waste[J]. ACS Nano, 2011, 5(9): 7601-7607. doi: 10.1021/nn202625c [22] YOU Y, MAYYAS M, XU S, et al. Growth of NiO nanorods, SiC nanowires and monolayer graphene via a CVD method[J]. Green Chemistry, 2017, 19(23): 5599-5607. doi: 10.1039/C7GC02523H [23] GONG J A, LIU J E, WEN X, et al. Upcycling waste polypropylene into graphene flakes on organically modified montmorillonite[J]. Industrial & Engineering Chemistry Research, 2014, 53(11): 4173-4181. [24] NGUYEN D D, HSIEH P Y, TSAI M T, et al. Hollow few-layer graphene-based structures from parafilm waste for flexible transparent supercapacitors and oil spill cleanup[J]. ACS Applied Materials & Interfaces, 2017, 9(46): 40645-40654. [25] WU T R, DING G Q, SHEN H L, et al. Triggering the continuous growth of graphene toward millimeter-sized grains[J]. Advanced Functional Materials, 2013, 23(2): 198-203. doi: 10.1002/adfm.201201577 [26] WANG Y L, HU P, YANG J, et al. C—H bond activation in light alkanes: A theoretical perspective[J]. Chemical Society Reviews, 2021, 50(7): 4299-4358. doi: 10.1039/D0CS01262A [27] LIN L, DENG B, SUN J Y, et al. Bridging the gap between reality and ideal in chemical vapor deposition growth of graphene[J]. Chemical Reviews, 2018, 118(18): 9281-9343. doi: 10.1021/acs.chemrev.8b00325 [28] SHU H B, TAO X M, DING F. What are the active carbon species during graphene chemical vapor deposition growth?[J]. Nanoscale, 2015, 7(5): 1627-1634. doi: 10.1039/C4NR05590J [29] CHEN S S, CAI W W, PINER R D, et al. Synthesis and characterization of large-area graphene and graphite films on commercial Cu-Ni alloy foils[J]. Nano Letters, 2011, 11(9): 3519-3525. doi: 10.1021/nl201699j [30] LIAN Y M, UTETIWABO W, ZHOU Y D, et al. From upcycled waste polyethylene plastic to graphene/mesoporous carbon for high-voltage supercapacitors[J]. Journal of Colloid and Interface Science, 2019, 557: 55-64. doi: 10.1016/j.jcis.2019.09.003 [31] PANDEY S, KARAKOTI M, SURANA K, et al. Graphene nanosheets derived from plastic waste for the application of DSSCs and supercapacitors[J]. Scientific Reports, 2021, 11: 3916. doi: 10.1038/s41598-021-83483-8 [32] XU Y X, LIN Z Y, ZHONG X, et al. Holey graphene frameworks for highly efficient capacitive energy storage[J]. Nature Communications, 2014, 5: 4554. doi: 10.1038/ncomms5554 [33] WU D Y, ZHU C, SHI Y T, et al. Biomass-derived multilayer-graphene-encapsulated cobalt nanoparticles as efficient electrocatalyst for versatile renewable energy applications[J]. ACS Sustainable Chemistry & Engineering, 2019, 7(1): 1137-1145. [34] XIA J S, ZHANG N, CHONG S K, et al. Three-dimensional porous graphene-like sheets synthesized from biocarbon via low-temperature graphitization for a supercapacitor[J]. Green Chemistry, 2018, 20(3): 694-700. doi: 10.1039/C7GC03426A [35] MAHMOUDIAN L, RASHIDI A, DEHGHANI H, et al. Single-step scalable synthesis of three-dimensional highly porous graphene with favorable methane adsorption[J]. Chemical Engineering Journal, 2016, 304: 784-792. doi: 10.1016/j.cej.2016.07.015 [36] HE Y M, CHEN W J, LI X D, et al. Freestanding three-dimensional graphene/MnO2 composite networks As ultralight and flexible supercapacitor electrodes[J]. ACS Nano, 2013, 7(1): 174-182. doi: 10.1021/nn304833s [37] WANG X B, ZHANG Y J, ZHI C Y, et al. Three-dimensional strutted graphene grown by substrate-free sugar blowing for high-power-density supercapacitors[J]. Nature Communications, 2013, 4: 2905. doi: 10.1038/ncomms3905 [38] ZHENG X J, WU J, CAO X C, et al. N-, P-, and S-doped graphene-like carbon catalysts derived from onium salts with enhanced oxygen chemisorption for Zn-air battery cathodes[J]. Applied Catalysis B: Environmental, 2019, 241: 442-451. doi: 10.1016/j.apcatb.2018.09.054 [39] JIANG X F, WANG X B, DAI P C, et al. High-throughput fabrication of strutted graphene by ammonium-assisted chemical blowing for high-performance supercapacitors[J]. Nano Energy, 2015, 16: 81-90. doi: 10.1016/j.nanoen.2015.06.008 [40] YOON J C, LEE J S, KIM S I, et al. Three-dimensional graphene nano-networks with high quality and mass production capability via precursor-assisted chemical vapor deposition[J]. Scientific Reports, 2013, 3: 1788. doi: 10.1038/srep01788 [41] LI X S, MAGNUSON C W, VENUGOPAL A, et al. Graphene films with large domain size by a two-step chemical vapor deposition process[J]. Nano Letters, 2010, 10(11): 4328-4334. doi: 10.1021/nl101629g [42] YU Q K, JAUREGUI L A, WU W, et al. Control and characterization of individual grains and grain boundaries in graphene grown by chemical vapour deposition[J]. Nature Materials, 2011, 10(6): 443-449. doi: 10.1038/nmat3010 [43] SHARMA S, KALITA G, HIRANO R, et al. Synthesis of graphene crystals from solid waste plastic by chemical vapor deposition[J]. Carbon, 2014, 72: 66-73. doi: 10.1016/j.carbon.2014.01.051 [44] CUI L F, WANG X P, CHEN N, et al. Trash to treasure: Converting plastic waste into a useful graphene foil[J]. Nanoscale, 2017, 9(26): 9089-9094. doi: 10.1039/C7NR03580B [45] FROMM O, HECKMANN A, RODEHORST U C, et al. Carbons from biomass precursors as anode materials for lithium ion batteries: New insights into carbonization and graphitization behavior and into their correlation to electrochemical performance[J]. Carbon, 2018, 128: 147-163. doi: 10.1016/j.carbon.2017.11.065 [46] LIN J, PENG Z W, LIU Y Y, et al. Laser-induced porous graphene films from commercial polymers[J]. Nature Communications, 2014, 5: 5714. doi: 10.1038/ncomms6714 [47] YE R Q, JAMES D K, TOUR J M. Laser-induced graphene: From discovery to translation[J]. Advanced Materials, 2019, 31(1): 1803621. doi: 10.1002/adma.201803621 [48] STANFORD M G, LI J T, CHEN Y D, et al. Self-sterilizing laser-induced graphene bacterial air filter[J]. ACS Nano, 2019, 13(10): 11912-11920. doi: 10.1021/acsnano.9b05983 [49] ZHANG J B, ZHANG C H, SHA J W, et al. Efficient water-splitting electrodes based on laser-induced graphene[J]. ACS Applied Materials & Interfaces, 2017, 9(32): 26840-26847. [50] SONG W X, ZHU J X, GAN B H, et al. Flexible, stretchable, and transparent planar microsupercapacitors based on 3D porous laser-induced graphene[J]. Small, 2018, 14(1): 1702249. doi: 10.1002/smll.201702249 [51] CLERICI F, FONTANA M, BIANCO S, et al. in situ MoS2 decoration of laser-induced graphene as flexible supercapacitor electrodes[J]. ACS Applied Materials & Interfaces, 2016, 8(16): 10459-10465. [52] ZHANG Z C, SONG M M, HAO J X, et al. Visible light laser-induced graphene from phenolic resin: A new approach for directly writing graphene-based electrochemical devices on various substrates[J]. Carbon, 2018, 127: 287-296. doi: 10.1016/j.carbon.2017.11.014 [53] TAO L Q, TIAN H, LIU Y, et al. An intelligent artificial throat with sound-sensing ability based on laser induced graphene[J]. Nature Communications, 2017, 8: 14579. doi: 10.1038/ncomms14579 [54] FENZL C, NAYAK P, HIRSCH T, et al. Laser-scribed graphene electrodes for aptamer-based biosensing[J]. ACS Sensors, 2017, 2(5): 616-620. doi: 10.1021/acssensors.7b00066 [55] NAYAK P, KURRA N, XIA C, et al. Highly efficient laser scribed graphene electrodes for on-chip electrochemical sensing applications[J]. Advanced Electronic Materials, 2016, 2(10): 1600185. doi: 10.1002/aelm.201600185 [56] SINGH S P, LI Y L, BE’ER A, et al. Laser-induced graphene layers and electrodes prevents microbial fouling and exerts antimicrobial action[J]. ACS Applied Materials & Interfaces, 2017, 9(21): 18238-18247. [57] TAN K W, JUNG B, WERNER J, et al. Transient laser heating induced hierarchical porous structures from block copolymer–directed self-assembly[J]. Science, 2015, 349: 54-58. doi: 10.1126/science.aab0492 [58] CHYAN Y, YE R Q, LI Y L, et al. Laser-induced graphene by multiple lasing: Toward electronics on cloth, paper, and food[J]. ACS Nano, 2018, 12(3): 2176-2183. doi: 10.1021/acsnano.7b08539 [59] ALGOZEEB W A, SAVAS P E, LUONG D X, et al. Flash graphene from plastic waste[J]. ACS Nano, 2020, 14(11): 15595-15604. doi: 10.1021/acsnano.0c06328 [60] BECKHAM J L, LI J T, STANFORD M G, et al. High-resolution laser-induced graphene from photoresist[J]. ACS Nano, 2021, 15(5): 8976-8983. doi: 10.1021/acsnano.1c01843 [61] LI J T, STANFORD M G, CHEN W Y, et al. Laminated laser-induced graphene composites[J]. ACS Nano, 2020, 14(7): 7911-7919. doi: 10.1021/acsnano.0c02835 [62] WYSS K M, de KLEINE R D, COUVREUR R L, et al. Upcycling end-of-life vehicle waste plastic into flash graphene[J]. Communications Engineering, 2022, 1: 3. doi: 10.1038/s44172-022-00006-7 [63] WYSS K M, BECKHAM J L, CHEN W Y, et al. Converting plastic waste pyrolysis ash into flash graphene[J]. Carbon, 2021, 174: 430-438. doi: 10.1016/j.carbon.2020.12.063 [64] STANFORD M G, BETS K V, LUONG D X, et al. Flash graphene morphologies[J]. ACS Nano, 2020, 14(10): 13691-13699. doi: 10.1021/acsnano.0c05900 [65] CHEN W Y, LI J T, WANG Z, et al. Ultrafast and controllable phase evolution by flash joule heating[J]. ACS Nano, 2021, 15(7): 11158-11167. doi: 10.1021/acsnano.1c03536 [66] WYSS K M, CHEN W Y, BECKHAM J L, et al. Holey and wrinkled flash graphene from mixed plastic waste[J]. ACS Nano, 2022, 16(5): 7804-7815. doi: 10.1021/acsnano.2c00379 [67] BARBHUIYA N H, KUMAR A, SINGH A, et al. The future of flash graphene for the sustainable management of solid waste[J]. ACS Nano, 2021, 15(10): 15461-15470. doi: 10.1021/acsnano.1c07571 [68] ZHANG Y S, ZHU H L, YAO D D, et al. Thermo-chemical conversion of carbonaceous wastes for CNT and hydrogen production: A review[J]. Sustainable Energy & Fuels, 2021, 5(17): 4173-4208. [69] ZHANG R F, ZHANG Y Y, WEI F. Horizontally aligned carbon nanotube arrays: Growth mechanism, controlled synthesis, characterization, properties and applications[J]. Chemical Society Reviews, 2017, 46(12): 3661-3715. doi: 10.1039/C7CS00104E [70] KUKOVITSKII E F, CHERNOZATONSKII L A, L'VOV S G, et al. Carbon nanotubes of polyethylene[J]. Chemical Physics Letters, 1997, 266(3/4): 323-328. [71] ZHANG P, LIANG C, WU M D, et al. High-efficient microwave plasma discharging initiated conversion of waste plastics into hydrogen and carbon nanotubes[J]. Energy Conversion and Management, 2022, 268: 116017. doi: 10.1016/j.enconman.2022.116017 [72] DAI L L, KARAKAS O, CHENG Y L, et al. A review on carbon materials production from plastic wastes[J]. Chemical Engineering Journal, 2023, 453: 139725. doi: 10.1016/j.cej.2022.139725 [73] PENG Y J, WANG Y P, KE L Y, et al. A review on catalytic pyrolysis of plastic wastes to high-value products[J]. Energy Conversion and Management, 2022, 254: 115243. doi: 10.1016/j.enconman.2022.115243 [74] JIE X Y, LI W S, SLOCOMBE D, et al. Microwave-initiated catalytic deconstruction of plastic waste into hydrogen and high-value carbons[J]. Nature Catalysis, 2020, 3(11): 902-912. doi: 10.1038/s41929-020-00518-5 [75] JOSEPH BERKMANS A, JAGANNATHAM M, PRIYANKA S, et al. Synthesis of branched, nano channeled, ultrafine and nano carbon tubes from PET wastes using the arc discharge method[J]. Waste Management, 2014, 34(11): 2139-2145. doi: 10.1016/j.wasman.2014.07.004 [76] LIU B L, REN W C, GAO L B, et al. Metal-catalyst-free growth of single-walled carbon nanotubes[J]. Journal of the American Chemical Society, 2009, 131(6): 2082-2083. doi: 10.1021/ja8093907 [77] KANG L X, HU Y E, LIU L L, et al. Growth of close-packed semiconducting single-walled carbon nanotube arrays using oxygen-deficient TiO2 nanoparticles as catalysts[J]. Nano Letters, 2015, 15(1): 403-409. doi: 10.1021/nl5037325 [78] YAO Y G, FENG C Q, ZHANG J, et al. “cloning” of single-walled carbon nanotubes via open-end growth mechanism[J]. Nano Letters, 2009, 9(4): 1673-1677. doi: 10.1021/nl900207v [79] POUDEL Y R, LI W Z. Synthesis, properties, and applications of carbon nanotubes filled with foreign materials: A review[J]. Materials Today Physics, 2018, 7: 7-34. doi: 10.1016/j.mtphys.2018.10.002 [80] YAGLIOGLU O, CAO A Y, HART A J, et al. Wide range control of microstructure and mechanical properties of carbon nanotube forests: A comparison between fixed and floating catalyst CVD techniques[J]. Advanced Functional Materials, 2012, 22(23): 5028-5037. doi: 10.1002/adfm.201200852 [81] TANG T, CHEN X C, MENG X Y, et al. Synthesis of multiwalled carbon nanotubes by catalytic combustion of polypropylene[J]. Angewandte Chemie International Edition, 2005, 44(10): 1517-1520. doi: 10.1002/anie.200461506 [82] GUL O T. Decoupling the catalyst reduction and annealing for suppressing Ostwald ripening in carbon nanotube growth[J]. Applied Physics A, 2021, 127(10): 1-11. [83] AZARA A, BELBESSAI S, ABATZOGLOU N. A review of filamentous carbon nanomaterial synthesis via catalytic conversion of waste plastic pyrolysis products[J]. Journal of Environmental Chemical Engineering, 2022, 10(1): 107049. doi: 10.1016/j.jece.2021.107049 [84] OSTRIKOV K K, MEHDIPOUR H. Thin single-walled carbon nanotubes with narrow chirality distribution: Constructive interplay of plasma and gibbs–thomson effects[J]. ACS Nano, 2011, 5(10): 8372-8382. doi: 10.1021/nn2030989 [85] GHORANNEVIS Z, KATO T, KANEKO T, et al. Narrow-chirality distributed single-walled carbon nanotube growth from nonmagnetic catalyst[J]. Journal of the American Chemical Society, 2010, 132(28): 9570-9572. doi: 10.1021/ja103362j [86] NEYTS E C, OSTRIKOV K K, SUNKARA M K, et al. Plasma catalysis: Synergistic effects at the nanoscale[J]. Chemical Reviews, 2015, 115(24): 13408-13446. doi: 10.1021/acs.chemrev.5b00362 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2987
- HTML全文浏览数: 2987
- PDF下载数: 71
- 施引文献: 0
