


-
多环芳烃衍生物(SPAHs)是多环芳烃(PAHs)芳香环上的氢被硝基(—NO2)、羟基(—OH)或氯原子等取代的一类衍生物[1 − 2],在大气[3 − 4]、水体[5 − 6]及冰雪环境[7 − 8]中被广泛检出. 羟基多环芳烃(OH-PAHs)[9 − 10]、硝基多环芳烃(N-PAHs)[11 − 12]与氯代多环芳烃(Cl-PAHs)[13]是目前国内外研究较为广泛的典型SPAHs. SPAHs在自然过程中的来源主要为微生物过程以及燃料、煤和生物质燃烧[1]. 除此之外,PAHs通过光化学(紫外线)反应、自由基(如·OH、·NO2)和活性氧物种(如1O2)介导的反应,也可转化为SPAHs[14]. 目前,水环境及高纬度的冰雪环境中SPAHs的存在状况已有报道,例如,太湖水体中10种N-PAHs浓度水平为8—243 ng·L−1[15],日本河水中5种N-PAHs高达604 ng·L−1[16],北京污水处理厂出水中3种SPAHs浓度为75—584 ng·L−1[17],北极格陵兰岛积雪中3种PAHs浓度为1.9 ng·kg−1[18]. SPAHs与PAHs结构相似,具有“三致”效应,其毒性甚至比母体PAHs更强,对动植物及人体具有一定的风险,成为一类倍受关注的新污染物[19 − 20]. 因此,深入揭示SPAHs的环境行为和风险具有重要意义.
研究表明,表层水体中光化学反应是SPAHs的重要转化方式[21]. 水环境中含有的SPAHs类物质会因为结冰作用而封存在冰中,大气中的SPAHs也可能被吸附在冰雪表面[22]. 在高纬度高海拔的冰雪环境中,由于温度低,微生物作用减弱,紫外线较强,所以光降解是其中SPAHs的主要转化方式[9, 23]. 水/冰中SPAHs可以通过光化学反应,生成毒性更大的光转化产物,并表现为光修饰毒性[24 − 25]. 尤其是在冰中,由于结冰过程产生局部浓缩效应,污染物可能通过光致聚合生成毒性更大的产物. 对于SPAHs,GE等[9, 23]研究了水中和冰中4种OH-PAHs的光化学转化机制,发现其生成了对发光细菌(Vibrio fischeri)毒性更大的产物. 冰中2-和4-氯酚对Vibrio fischeri也表现为光修饰毒性[26 − 27]. 水/冰等环境介质中N-PAHs及Cl-PAHs均可以发生光化学转化[28 − 29],但其光修饰毒性未见报道. 由于SPAHs类化合物的光化学转化表现出多路径的特点,其光转化产物可能具有比母体化合物更高的毒性. 因此有必要深入探究水/冰环境中此类化合物的光化学转化和光修饰毒性,并揭示其光修饰毒性的变化规律及原因.
PAHs系列污染物中,萘作为最简单的稠环芳烃,环境浓度最高;芴、菲、芘的浓度水平也较高[30 − 31]. 由于PAHs可通过生物、化学反应转化为相应的SPAHs,所以萘、芴、菲、芘的衍生物更值得关注. 本文选取4种OH-PAHs (羟基萘系列物)、3种N-PAHs (硝基取代的芴、菲、芘)和3种Cl-PAHs (氯代的芴、菲、芘)为模型化合物,考察其在模拟日光照射下水中与冰中的光降解情况,并深入探究这些介质中光反应溶液体系对发光细菌(Vibrio fischeri)的光修饰毒性及其差异,从而理解高纬度寒冷地区SPAHs光化学转化的行为与风险.
-
所选取的4种OH-PAHs (羟基萘系列物)为1-羟基萘(1-OHNap)、2-羟基萘(2-OHNap)、4-硝基-1-羟基萘(4-N-1-OHNap)、4-氯-1-羟基萘(4-Cl-1-OHNap);3种N-PAHs为2-硝基芴(2-NFlu)、9-硝基菲(9-NPhe)、1-硝基芘(1-NPyr);3种Cl-PAHs为2-氯芴(2-ClFlu)、9-氯菲(9-ClPhe)、1-氯芘(1-ClPyr). 这些模型化合物纯度均大于95%,购自于百灵威公司(北京)、Aldrich公司(美国)、TCI公司(日本). 乙腈为质谱纯;NaCl、ZnSO4等试剂为分析纯. 超纯水由美国Millipore-Milli Q系统制备. 发光细菌(Vibrio fischeri)冻干粉由浙江清华长三角研究院提供.
-
配制10种不同SPAHs的纯水溶液(OH-PAHs初始浓度C0 = 2 μmol·L−1;N-PAHs,Cl-PAHs C0 = 0.2 μmol·L−1,均使用5%乙腈作助溶剂),将每种溶液分装于2只相同的石英试管(V=40 mL)中. 进行水中的光解实验时,将其中1份溶液置于旋转式光化学反应仪中,使用置于Pyrex冷却套管中的500 W高压汞灯作为光源(λ>290 nm),模拟太阳光,对样品进行光照. 使用UV-A紫外辐照计(北京师范大学光电仪器厂)测量试管前后的光强,取平均值,得到反应溶液365 nm处的光强为8.09 mW·cm−2;420 nm处的光强为8.03 mW·cm−2. 对于另1份溶液,将其置于−20 ℃冷冻至完全结冰;并将相同的光化学反应仪和光源移到冷冻箱[(−5±1) ℃]中,进行冰中的光解实验. 每批实验设置平行与空白,同时进行暗对照,定时取样,其中冰中的样品取样后置于暗室融化,待测.
使用美国Agilent
1260 HPLC分析SPAHs浓度,色谱柱为SB C18柱(2.1 mm×100 mm, 1.8 μm),柱温25 ℃,流动相为超纯水-乙腈(30:70,体积比),流速0.3 mL·min−1,进样量10 μL. 优化后各SPAHs的检测条件与保留时间如表1所示.Agilent
1260 HPLC-MS/MS三重四极杆质谱仪(美国)用于光转化产物的分析. 采用梯度洗脱,流动相(乙腈和超纯水)中乙腈的比例设置为:0—12 min,10%—80%;12—15 min,80%—100%;15—16 min,100%;16—23 min,100%—10%;23 min,10%. 其他液相色谱参数同上. 质谱采用电喷雾电离源,负离子模式,监测模式为Scan与Sim-Scan;离子源温度300 ℃;干燥气(N2) 250 ℃、7.0 L·min−1;离子源喷射电压3000 V;雾化气(N2)压强25 psi;碎片电压优化为120—138 V;质荷比m/z的扫描范围20—500 amu. -
根据国际标准方法ISO
11348 -3-2007,将测试条件进行合理优化. 以发光细菌(Vibrio fischeri)作为指示生物,使用美国HACH Eclox快速水质检测仪中的“Luminescent Bacteria Toxicity Testing”模式,在避光条件下,对光照前后的纯水与纯水冰SPAHs溶液进行15 min急性毒性实验,同时设置空白对照与平行实验. 以发光细菌的平均相对抑制率(I%)表示其光修饰毒性大小:式中,I0为样品在0 min时的发光强度,样品加入菌液后立即混匀、测量,单位为相对发光强度;I15为放置15 min后的发光强度.
使用SPSS 26.0统计分析软件中的“单因素方差分析”对结果进行显著性检验,将不同光照时间样品的发光抑制率与未光照样品进行比较,若光照后样品的发光抑制率显著增加(P<0.05),则该模型化合物具有光修饰毒性.
-
暗对照中,所考察的模型化合物无明显降解. 10种SPAHs未光照时对发光细菌Vibrio fischeri的发光抑制率(I%)如表2所示. 与OH-PAHs与N-PAHs相比,Cl-PAHs对Vibrio fischeri的I%较大,具有较高风险,这与前人对氯代化合物毒性风险的研究结果一致[32 − 33].
在模拟日光(λ>290 nm)照射下,模型化合物的母体浓度均有下降. 1-OHNap等4种OH-PAHs在水中的光降解半减期(t1/2)为24.50—95.06 min;在冰中的t1/2为14.25—198.69 min. 随着母体浓度的降低,纯水体系反应溶液在不同光照时间对发光细菌Vibrio fischeri的发光抑制率如图1所示. 1-OHNap [图1 (a)]对Vibrio fischeri存在发光抑制作用,光转化过程中其I%随着光照时间的延长(35—110 min)呈现明显上升趋势(P<0.05),说明在该过程中,虽然母体浓度逐渐降低,但生成的光转化产物与中间体具有比母体物质更高的毒性. 4-N-1-OHNap [图1 (c)]在光照过程中(155—200 min),对Vibrio fischeri的I%具有微弱上升的趋势(P<0.05),表现出较小的光修饰毒性. 相较于其它3种OH-PAHs,4-Cl-1-OHNap [图1 (d)]光照过程生成的产物具有最高的I% (64.51%),在光照后期(75—110 min),反应体系的I%呈现下降趋势(P<0.05),这可能是因为生成的产物也具有光反应活性,可以发生光转化. 尽管如此,光照110 min时,母体浓度已降至95%以上,但产物仍对Vibrio fischeri具有较高的I% (37.15%),表现出明显的光修饰毒性. 2-OHNap [图1 (b)]光照过程中对Vibrio fischeri的I%变化不显著(P>0.05),没有表现出明显的光修饰毒性.
进一步研究了冰中4种OH-PAHs对Vibrio fischeri的I%随光照时间的变化(图2). 发现随着母体浓度的降低,1-OHNap [图2 (a)]在冰中对Vibrio fischeri仍然具有抑制作用(35.05%),说明在水中与冰中1-OHNap都能够生成具有更高毒性的光转化产物. 与水中不同的是,2-OHNap [图2 (b)]在光照前期(10—50 min)的I%出现显著上升(P<0.05),说明在冰中2-OHNap反应溶液对Vibrio fischeri表现出一定的抑制作用,这可能是由于在水与冰中光转化产物不同引起的;4-Cl-1-OHNap [图2 (d)]的I%出现先下降后上升的趋势,这可能是由于结冰过程使得4-Cl-1-OHNap具有明显的冷冻浓缩效应[27],导致在冰中的直接光解与水中的差异. 4-N-1-OHNap [图2 (c)]在冰中没有表现出显著的光修饰毒性(P>0.05).
-
2-NFlu等3种N-PAHs在水中的光降解t1/2为47.73—250.90 min;在冰中的光降解t1/2为50.97—533.19 min. 随着母体浓度的降低,水中反应溶液在不同光照时间对Vibrio fischeri的发光抑制率如图3所示. 发现光照后2-NFlu [图3 (a)]和9-NPhe [图3 (b)]反应溶液体系对Vibrio fischeri的I%小于光照前母体化合物本身,说明其在光照过程中没有表现出光修饰毒性. 但值得注意的是,9-NPhe在光照后期(60—180 min)出现了I%回升的现象,这可能是由于9-NPhe光转化产物的累积. 1-NPyr [图3 (c)]反应溶液在光照中期(150—240 min)时I%稍有增加(P<0.05),表现出较小的光修饰毒性.
对比研究了冰中3种N-PAHs对Vibrio fischeri的发光抑制率随光照时间的变化(图4). 研究发现,2-NFlu [图4 (a)]在光照过程(60—510 min)中反应体系对Vibrio fischeri的I% (10.0%—14.8%)高于未光照体系(5.4%),说明冰中2-NFlu可能生成了与水中不同的光照产物. 9-NPhe [图4 (b)]在冰中没有表现出显著的光修饰毒性. 而冰中1-NPyr [图4 (c)]在光照后期,尤其是在光照时间510 min时,发光抑制率显著上升(P<0.05),表明光照后期1-NPyr反应溶液仍对Vibrio fischeri具有较高的I%,且与水中相比,冰中I%演变趋势明显不同.
-
2-ClFlu等3种Cl-PAHs在水中的光降解t1/2为14.53—133.14 min;在冰中的光降解t1/2为57.20—309.23 min. 随着母体浓度的降低,水中反应溶液在不同光照时间对Vibrio fischeri的I%如图5所示. 发现,与其它2类SPAHs相比,Cl-PAHs母体具有较高的I% (35.7%—81.0%),这与含氯的化合物具有较高的毒性风险相一致[32 − 33]. 2-ClFlu [图5 (a)]与9-ClPhe [图5 (b)]反应溶液的I%总体呈现下降的趋势,光照使得这2种Cl-PAHs毒性减小,这可能是由于—Cl基团在光照过程中被活性氧物种氧化或被其它基团取代. 而1-ClPyr [图5 (c)]在光照的中期(120 min)与末期(360 min)对Vibrio fischeri的I%显著上升(P<0.05),这可能是由于在光照中期生成的光转化产物具有比母体化合物更高的毒性,光照末期则是由于生成的光转化产物与光照中期不同,或是光转化产物的累积.
对比探究了3种Cl-PAHs在冰中对Vibrio fischeri的I%随光照时间的变化(图6). 发现与水中不同的是,冰中2-ClFlu [图6 (a)]与1-ClPyr [图6 (c)]的反应溶液对发光细菌的I%高于其母体化合物(P<0.01),在冰中表现出光修饰毒性. 并且1-ClPyr在光照末期(720 min),母体化合物浓度已降至70%以上,但反应体系的I%再次上升,这说明光转化产物或次级光转化产物同样具有比母体更高的毒性. 而对于冰中9-ClPhe [图6 (b)],母体化合物对Vibrio fischeri的I%为负(-17.06%),说明未光照时9-ClPhe对Vibrio fischeri具有生物应激效应,而光照后反应体系对Vibrio fischeri的I%显著上升(P<0.01),这说明9-ClPhe在冰中与水中发生了不同的光化学行为,可能生成了不同的光转化产物,从而仅在冰中表现出较高的光修饰毒性.
-
选取2种-NO2或-Cl取代的OH-PAHs,分析其光转化产物. 结合光照前后的总离子流图、MS和MS2图,鉴定了产物的分子量Mw和结构式,并进一步推测了光转化的途径(图7). 4-N-1-OHNap与4-Cl-1-OHNap分别生成了3种主要的产物,其中产物P189与P190 (Mw分别为189与190)仅在冰中被检测到. 根据这2种OH-PAHs的光转化途径(图7)可知,水中与冰中OH-PAHs先脱除NO2或Cl取代基,经氧化生成酮类产物;继而,苯环发生羟基化与多重羟基化反应,生成羟基取代产物(P174). GE等[10]在探究水中与冰中羟基芴的光转化过程时,也发现了羟基化产物. 但在冰中,还容易生成硝基化或多重羟基化产物(P189与P190). 这可能是由于溶质的冷冻浓缩效应引起的,冰相中的污染物往往浓缩富集在冰晶边缘或间隙处,浓度较高,可能易于发生自由基(如·OH、·NO2)参与的自敏化反应[23, 34 − 35]. 水中和冰中光转化产物和路径的不同是两相中光修饰毒性具有差异的根本原因[10, 25, 27].
-
在高纬度与高海拔地区,大部分面积被冰雪覆盖,紫外光强烈. 表3显示了夏季中午南极长城站、北极黄河站,以及冬季中午西安、大连的平均太阳光光强[9, 23],可见南北极地区的平均光强比西安、大连还要高,尤其是在紫外光区365 nm处. 这表明光降解可能是极地等高纬度地区水体与冰雪表面SPAHs等有机污染物的主要转化方式. GE等[9, 23]实验发现,南极和北极冰雪表面典型OH-PAHs的环境表观光降解t1/2为6.0 min—5.4 h. 鉴于光转化过程对寒冷地区污染物消减的重要性,研究光化学转化产物的毒性,就显得尤为重要. 本研究发现3类SPAHs的光修饰毒性在水中与冰中的表现规律不同,冰雪环境中SPAHs、PAHs等有机污染物的光修饰毒性值得进一步关注.
-
多环芳烃衍生物(SPAHs)是一类来源广泛、风险隐蔽性的新污染物. 本文选取4种OH-PAHs、3种N-PAHs和3种Cl-PAHs共10种典型的SPAHs,研究了其在模拟日光(λ>290 nm)照射下水中与冰中反应溶液对发光细菌Vibrio fischeri的光修饰毒性,得到如下结论:
(1) 水和冰中,3类SPAHs的光转化可以生成具有较高风险的中间产物,对发光细菌Vibrio fischeri表现为光修饰毒性,并且部分SPAHs在水中与冰中表现出不同的毒性演变趋势. 对于Cl-PAHs,水中仅1-ClPyr对Vibrio fischeri的发光抑制率显著上升(P<0.05),而在冰中3种Cl-PAHs的发光抑制率均显著上升(P<0.01).
(2) 水中与冰中OH-PAHs可以通过取代基脱除、光氧化和苯环羟基化等光化学反应生成酮类和羟基代产物,更容易对发光细菌Vibrio fischeri产生毒性. 水中和冰中光转化产物和路径的不同是两相中光修饰毒性具有差异的根本原因.
(3) 本研究揭示了寒冷地区水中和结冰环境中SPAHs的光修饰毒性,有助于更准确评价这些典型新污染物的环境风险.
水中与冰中多环芳烃衍生物对发光细菌(Vibrio fischeri)的光修饰毒性
Photo-modified toxicity of polycyclic aromatic hydrocarbon derivatives to Vibrio fischeri in water and ice
-
摘要: 多环芳烃衍生物(SPAHs)是一类普遍存在、来源广泛、风险隐蔽的新污染物,可由多环芳烃(PAHs)通过微生物及化学作用转化生成,其毒性甚至比母体PAHs更强,研究其环境行为和毒性效应具有重要意义. 本研究选取羟基多环芳烃(OH-PAHs)、硝基多环芳烃(N-PAHs)和氯代多环芳烃(Cl-PAHs)共10种模型化合物,考察了在模拟日光(λ>290 nm)照射下,水中与冰中这些典型SPAHs对发光细菌Vibrio fischeri的光修饰毒性. 毒性实验表明,对于OH-PAHs,1-羟基萘、4-氯-1-羟基萘在水中与冰中均表现出显著的光修饰毒性(P<0.05),而4-硝基-1-羟基萘仅在水相表现出较小的光修饰毒性;对于N-PAHs,1-硝基芘在水中与冰中均表现出光修饰毒性,2-硝基芴仅在冰中表现出光修饰毒性;Cl-PAHs中,仅1-氯芘在水中表现出光修饰毒性,而3种Cl-PAHs在冰中均表现出显著的光修饰毒性. 通过HPLC-MS/MS分析,OH-PAHs光转化主要涉及的反应路径为取代基脱除、光氧化与苯环羟基化. 以上结果阐明了SPAHs通过光化学转化可生成具有较高毒性的中间产物,对Vibrio fischeri表现为光修饰毒性,且水中与冰中的光修饰毒性具有差异,这有助于更准确评价寒冷地区这些典型新污染物的环境风险.Abstract: Polycyclic aromatic hydrocarbon derivatives (SPAHs) are a class of ubiquitous and new pollutants with various sources and unclear risks. SPAHs can be transformed from polycyclic aromatic hydrocarbons (PAHs) through microbial and chemical reactions, and they are even more toxic than their parent PAHs. It is of great significance to investigate the environmental behavior and toxic effects of SPAHs. In this study, 10 model compounds, including hydroxy-PAHs (OH-PAHs), nitro-PAHs (N-PAHs) and chlorinated PAHs (Cl-PAHs), were selected. The photo-modified toxicity of these typical SPAHs to Vibrio fischeri under simulated sunlight (λ>290 nm) was investigated in water and ice systems. Toxicity tests indicated that the two OH-PAHs 1-hydroxynaphthalene and 4-chloro-1-hydroxynaphthalene showed significant photo-modified toxicity (P<0.05) in water and ice, while 4-nitro-1-hydroxynaphthalene only exhibited minor photo-modified toxicity in the water phase. For N-PAHs, 1-nitropyrene showed photo-modified toxicity in both water and ice phases, while the photo-modified toxicity of 2-nitrofluorene to Vibrio fischeri was observed only in ice. Among Cl-PAHs, only 1-chloropyrene presented photo-modified toxicity in water, while all showed significant photo-modified toxicity in ice. Based on HPLC-MS/MS, the main reaction pathways involved in the phototransformation of OH-PAHs were substituents removal, oxidization and hydroxylation. These results indicate that SPAHs underwent a photochemical transformation and produced intermediates with high toxicity, showing photo-modified toxicity to Vibrio fischeri, which could facilitate more accurate assessment of the environmental risks of these typical new pollutants in cold regions.
-
聚羟基脂肪酸酯(polyhydroxyalkanoates,PHAs)是微生物体内一种天然的高分子聚合物,具有优良的生物可降解性、生产能耗低、无害化和资源化等特点[1],可有效解决难降解的塑料产品垃圾造成的“白色污染”的问题[2]。传统PHAs生产方法多采用纯培养菌种合成,原料成本高,限制了PHAs的大规模生产和应用[3]。若利用活性污泥混合菌群合成PHAs,既能减少污水处理过程中的剩余污泥排放量,还能适应多种不同底物类型,可显著地降低PHAs的生产成本[4]。因此,利用活性污泥合成PHAs已成为近年来广大研究者研究的热点。研究表明,活性污泥既能在厌氧又能在好氧条件下积累PHAs[5]。但根据相关研究[6-7]可知,有关活性污泥合成PHAs的研究,大多利用乙酸钠等碳源经过长时间驯化积累高产PHAs菌种,成本高且不易进行控制。
目前,工业化生产PHAs的纯菌种工艺所使用的碳源都是纯物质[8]。这种生产出来的PHAs成本高,其中底物成本是大规模生产PHAs产品时的主要成本,据估计约40%的PHAs成本来自于底物[9],因此,最经济有效的PHAs生产工艺应该包括廉价底物的应用,并且微生物能够有效利用这种底物高效率的合成PHAs。花生渣是一种可再生的,价格低廉的食物副产品,据调查,我国是花生总产量和花生油总产量最大的国家[10],因此,其作为底物来源可大大降低生产PHAs产品的成本,同时可进一步开发花生渣的应用价值。本文将讨论未经驯化的剩余污泥利用花生渣厌氧发酵产生的VFAs(挥发性脂肪酸)作为有机碳源,合成PHAs的工况优化问题。
本研究采用同步亚硝化反硝化脱氮除磷系统在连续流中的二沉池剩余污泥和A2O工艺运行的实际水厂二沉池剩余污泥,有研究[11]表明,活性污泥在COD≤800 mg·L−1时具有较好的合成PHAs的能力,其PHAs的合成主要发生在厌氧阶段,因此,实验设计反应时间为5 h,COD为650~750 mg·L−1,在微氧的条件(DO≤0.2 mg·L−1)下,比较了2种污泥利用花生渣厌氧发酵产生的VFAs所合成PHAs的量。并在原有的微氧条件下,通过增设前置曝气的方式消耗微生物原有的PHAs,使微生物处于“饥饿”状态,从而提高微氧过程中利用VFAs中充足的碳源所合成PHAs的量。通过控制前置曝气时间、曝气气量来促进了PHAs的合成,可为活性污泥利用廉价碳源来合成PHAs提供思路,且为利用脱氮除磷工艺剩余污泥合成PHAs提供参考。
1. 材料与方法
1.1 实验原料
1)实验污泥。污泥取自广州市沥滘污水处理厂二沉池的剩余污泥和课题组连续流中亚硝化/反硝化除磷系统二沉池的剩余污泥[12]。
2)实验发酵液。采用花生渣作为发酵液原材料。花生渣碎片经48 h晒干后,通过食物搅拌机搅碎后用400目孔径过滤筛过滤,最后将花生渣加水进行发酵。
1.2 实验装置
图1为厌氧-限氧的连续流反应器和运行同步亚硝化/反硝化脱氮除磷系统。该反应器的厌氧区体积为13.8 L,厌氧区底部连接水箱。限氧区体积为39 L,限氧区设曝气盘,二沉池底部连接蠕动泵,将部分污泥回流到厌氧区,剩余污泥添加到PHAs合成装置的一个烧杯中。PHAs合成装置含有2个有效体积为2 L的烧杯,烧杯连接搅拌装置以保证反应过程中泥水混合均匀,并连接有曝气装置进行前置曝气,发酵罐中装有搅拌桨,并保持密封以维持其厌氧状态。
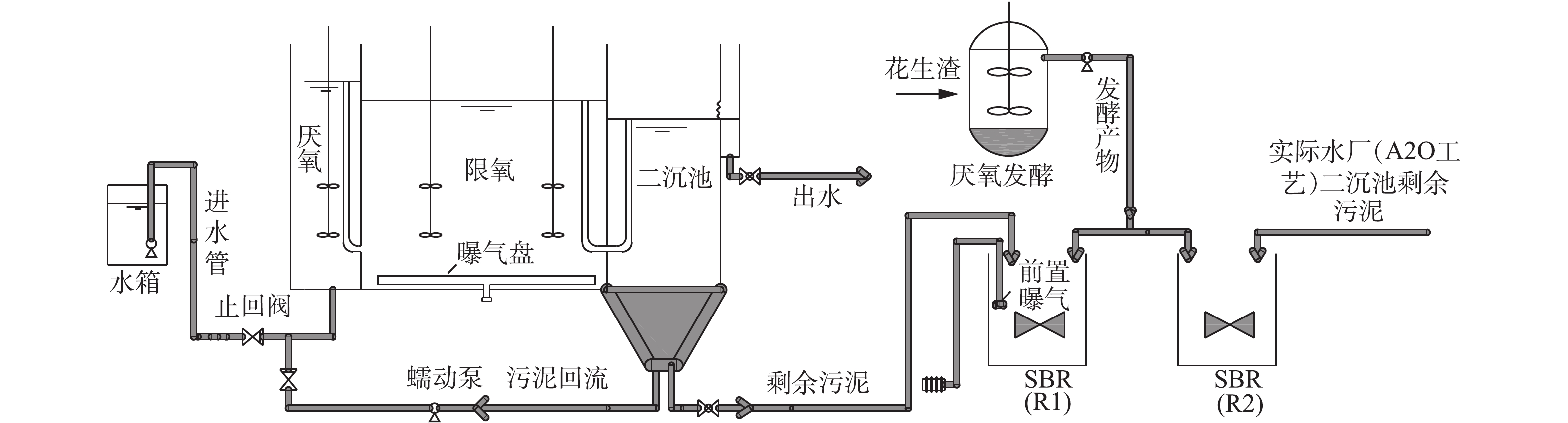 图 1 A/OLA连续流与PHA合成装置Figure 1. Diagram of A/OLA continuous flow and PHA synthesis device
图 1 A/OLA连续流与PHA合成装置Figure 1. Diagram of A/OLA continuous flow and PHA synthesis device1.3 实验方法
本研究利用花生渣厌氧发酵产生的VFAs[15],在微氧(DO≤0.2 mg·L−1)的条件下合成PHAs,实验重复2次,测得数据取其平均值。分2个阶段进行实验。
第I阶段。取适当的二沉池剩余污泥于2个2 L的烧杯中,加入人工配制的营养液及发酵液,其中发酵液中VFAs含量为20 985~21 284 mg·L−1,COD为26 000~30 000 mg·L−1,TP为100~145 mg·L−1,氨氮(NH4+-N)为3 100~3 280 mg·L−1,蛋白质为1 760~2 250 mg·L−1,糖原为199~252 mg·L−1;营养液中镁离子(MgSO4·7H2O)为120 mg·L−1,钙离子(CaCl2)为30 mg·L−1,亚铁离子(FeSO4)为3 mg·L−1,EDTA为80 mg·L−1,H3BO3为0.5 mg·L−1,ZnSO4·7H2O为0.3 mg·L−1。设定连续流中同步亚硝化反硝化脱氮除磷系统二沉池的剩余污泥为R1、采用A2O工艺的实际水厂的剩余污泥为R2,控制污泥浓度(MLSS)为3 300~3 600 mg·L−1,COD为650~750 mg·L−1,pH为7~8,污泥反应时间为5 h,每间隔0.5 h取样,测得COD及TP的变化曲线,并检测ORP的变化情况[16-17]。此阶段主要考察2种脱氮除磷工艺下剩余污泥在微氧条件下合成PHAs的情况。
第II阶段。采用第I阶段得出的具有良好PHAs合成能力的剩余污泥,在微氧前增设短时间曝气,其余操作步骤不变,其目的是消耗微生物原有的PHAs,使微生物处于“饥饿”状态,从而提高了微氧过程中PHAs积累净增量,此阶段主要考察曝气时间和曝气量对微氧阶段细胞内PHAs含量的影响。曝气实验分为2步骤。
设计曝气时间为15 min,曝气气量分别为20、30、50 L·h−1,反应时间一共5 h,每间隔30 min取样并测定其PHAs含量,在其中选出细胞内PHAs含量达到最高时所对应的曝气气量。
在上述步骤中所确定的PHAs最高合成量对应的曝气气量下,调控曝气时间对细胞内PHAs含量的影响实验。设计曝气时间分别为5、10、15和20 min,考察了曝气时间对细胞内PHAs含量的影响。
1.4 分析方法
1)常规分析方法。实验中主要的分析项目包括COD、氨氮、总磷(TP)、DO、pH和ORP等。其中DO、pH、ORP采用WTW的系列仪器进行在线检测,其余项目分析方法按国家环保局颁布的《水和废水监测分析方法》(第4版)[13]规定的标准进行,COD采用重铬酸钾消解法,TP采用钼锑抗分光光度法,氨氮(NH4+)采用纳氏试剂分光光度法,DO、pH、ORP采用德国WTW340i仪器进行在线监测。
2)PHAs检测方法。PHAs的测定采用气相色谱法[14],污泥经过离心、冷冻干燥后,取适量放于耐热管中。分别投加2 mL氯仿、2 mL体积分数为10%的硫酸的甲醇溶液、2 mL 50 mg·L−1的苯甲酸的甲醇溶液。耐热管密封后,放入100 ℃的水浴锅中加热4 h,取出耐热管冷却至室温,加入2 mL去离子水振荡10 min后,静止1 h。待溶液分层后,取下层有机相,用0.45 µm的滤膜过滤后加入进样瓶中,然后进行色谱分析(安捷伦7080A-5975C气质联用仪)。色谱柱型号DB-1,进样口温度为230 ℃,柱箱从100 ℃保持2 min以后以15 ℃·min−1升温至160 ℃,MS扫描范围为20~550。
2. 结果与讨论
2.1 不同脱氮除磷工艺下的剩余污泥在微氧条件下合成PHAs的情况
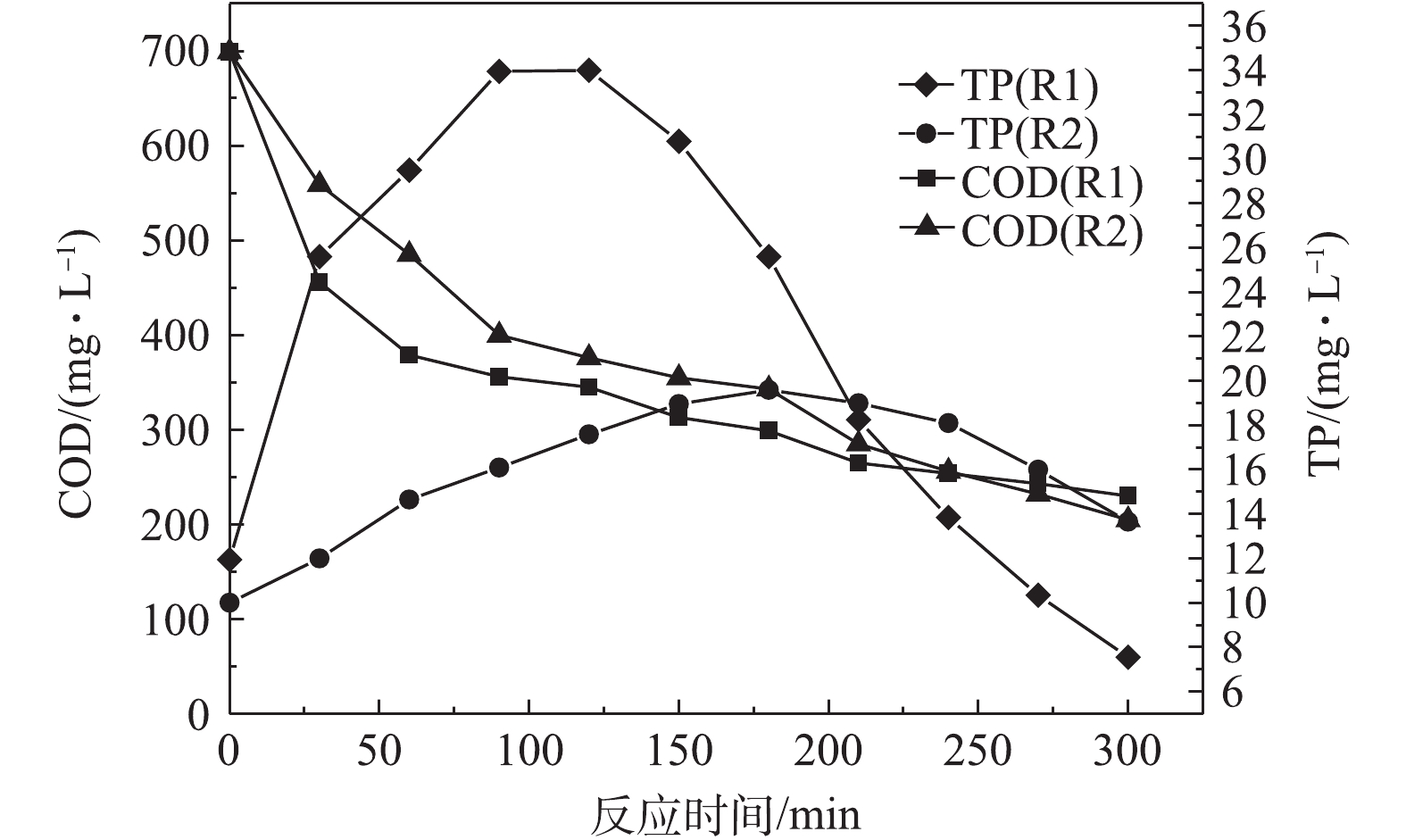
图2和图3为不同剩余污泥在微氧条件下合成PHAs量和ORP、TP、COD的变化情况。由图2可知,R1和R2中初始的PHAs含量分别为16 mg·g−1和7.015 mg·g−1,说明这2种脱氮除磷工艺下的剩余污泥内的初始PHAs含量相差较大。由图2可知,R1和R2在微氧条件下合成PHAs的量达到最高值的过程中,PHAs含量随着ORP的减小而增加,在PHAs含量最高时,ORP分别为−44.2 mV和−69.9 mV,均为最低值。随后,随着PHAs减少,ORP反而增大。这说明在微氧条件下剩余污泥利用厌氧发酵产生的VFAs合成PHAs时,ORP可作为PHAs合成量的指示参数。
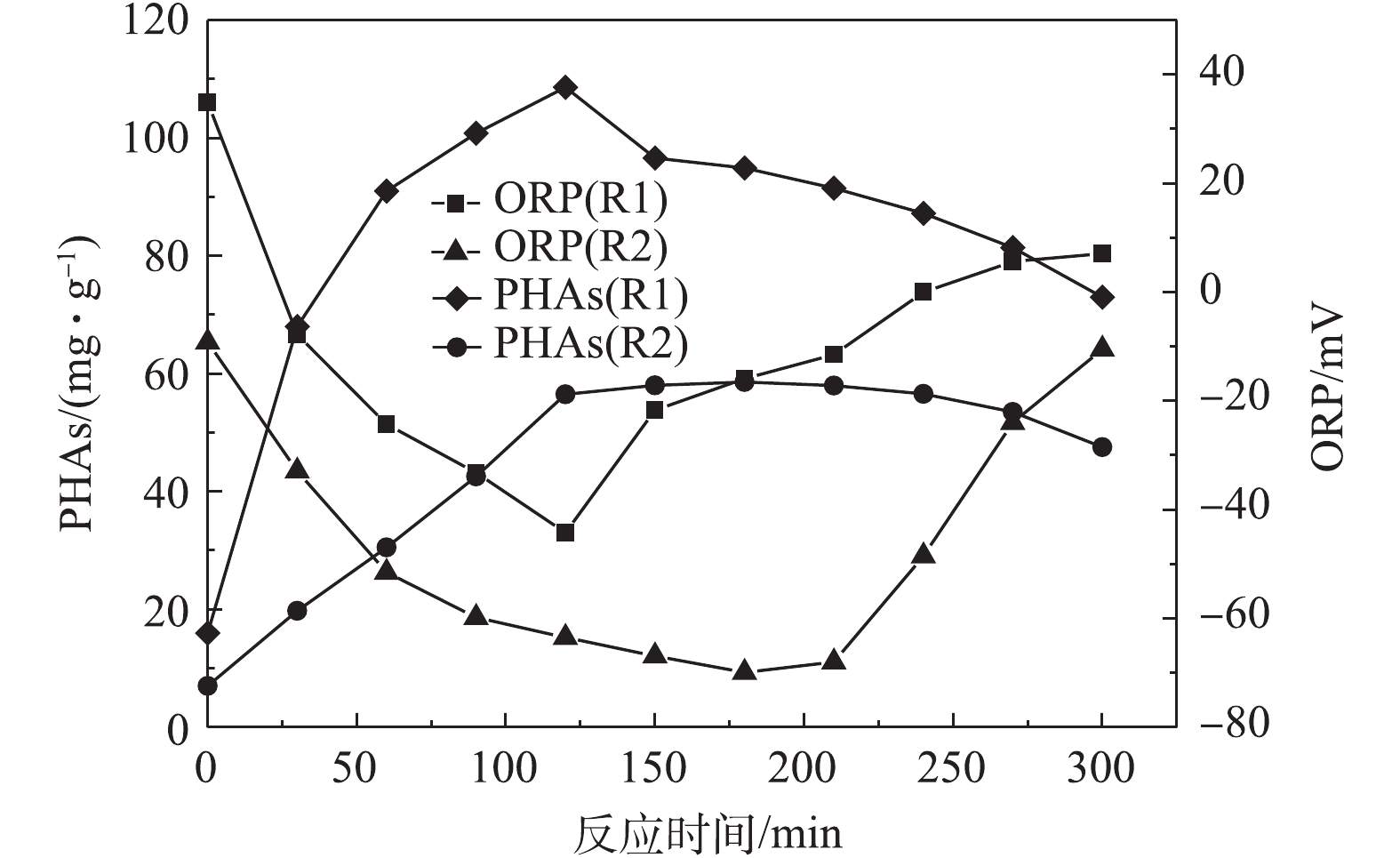 图 2 不同剩余污泥合成PHAs的量与ORP的变化Figure 2. Changes of PHAs amount synthesized from different excess activated sludge and the corresponding ORP
图 2 不同剩余污泥合成PHAs的量与ORP的变化Figure 2. Changes of PHAs amount synthesized from different excess activated sludge and the corresponding ORP由图3可知,当反应为120 min时,R1中的PHAs含量达到最大值,为108.6 mg·g−1,释磷34.0 mg·L−1,COD降低了345.0 mg·L−1,当反应为180 min时,R2中的PHAs达到最大值,为58.58 mg·g−1,释磷19.6 mg·L−1,COD下降了356 mg·L−1。与R2相比,R1中的PHAs含量在更短的时间内可达到最大值,其碳源利用率更高,释磷量更多。这是因为R1是连续流中厌氧/限氧(A/OLA)工艺的剩余污泥,PHAs作为内碳源驱动同步亚硝化反硝化脱氮除磷过程[18]。在脱氮除磷的过程中,其进水碳源偏低,则污泥微生物利用PHAs作为内碳源参与细胞新陈代谢的循环过程,在厌氧时合成PHAs,好氧时消耗PHAs,不断经历“饱食”与“饥饿”的环境。当碳源充足时,通入少量氧气形成微氧环境,R1中微生物可以较快地通过氧化有机物进行合成代谢,积累PHAs,PHAs合成的能量又来自于细胞内聚磷酸盐的水解[19],所以释磷量的变化能在一定程度上反映PHAs合成量的变化。R2中PHAs的含量较少,这是由于在R2是A2O工艺的剩余污泥,部分有机物会在R2中的微生物细胞内转化为糖原、聚磷酯,导致PHAs的积累量较少[20]。以上结果说明在2种脱氮除磷工艺下的剩余污泥,在微氧条件下合成PHAs量有所不同,其利用碳源合成PHAs量的大小与脱氮除磷的工艺有一定关系。
2.2 前置曝气对R1在微氧条件下合成PHAs的影响
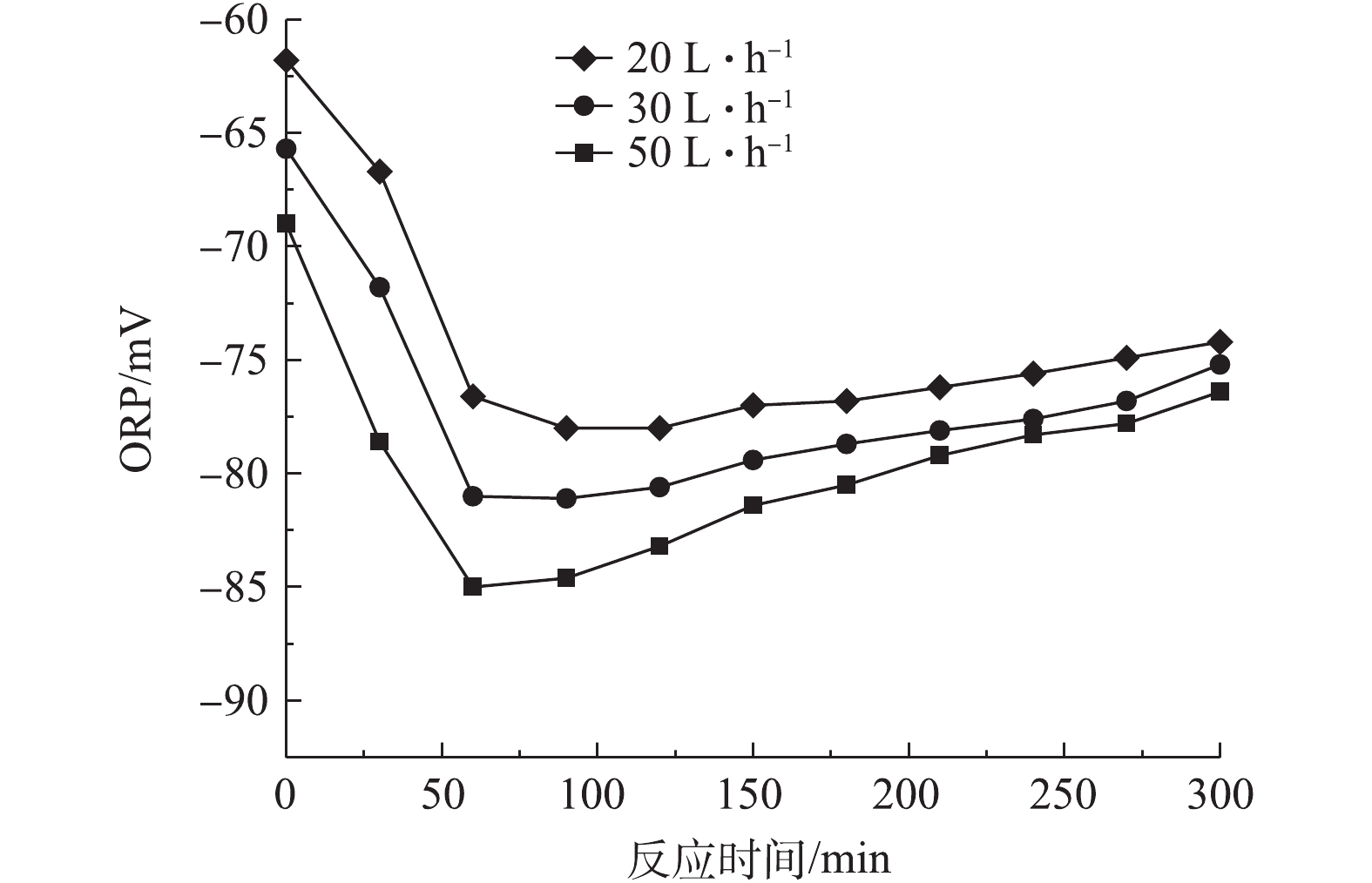
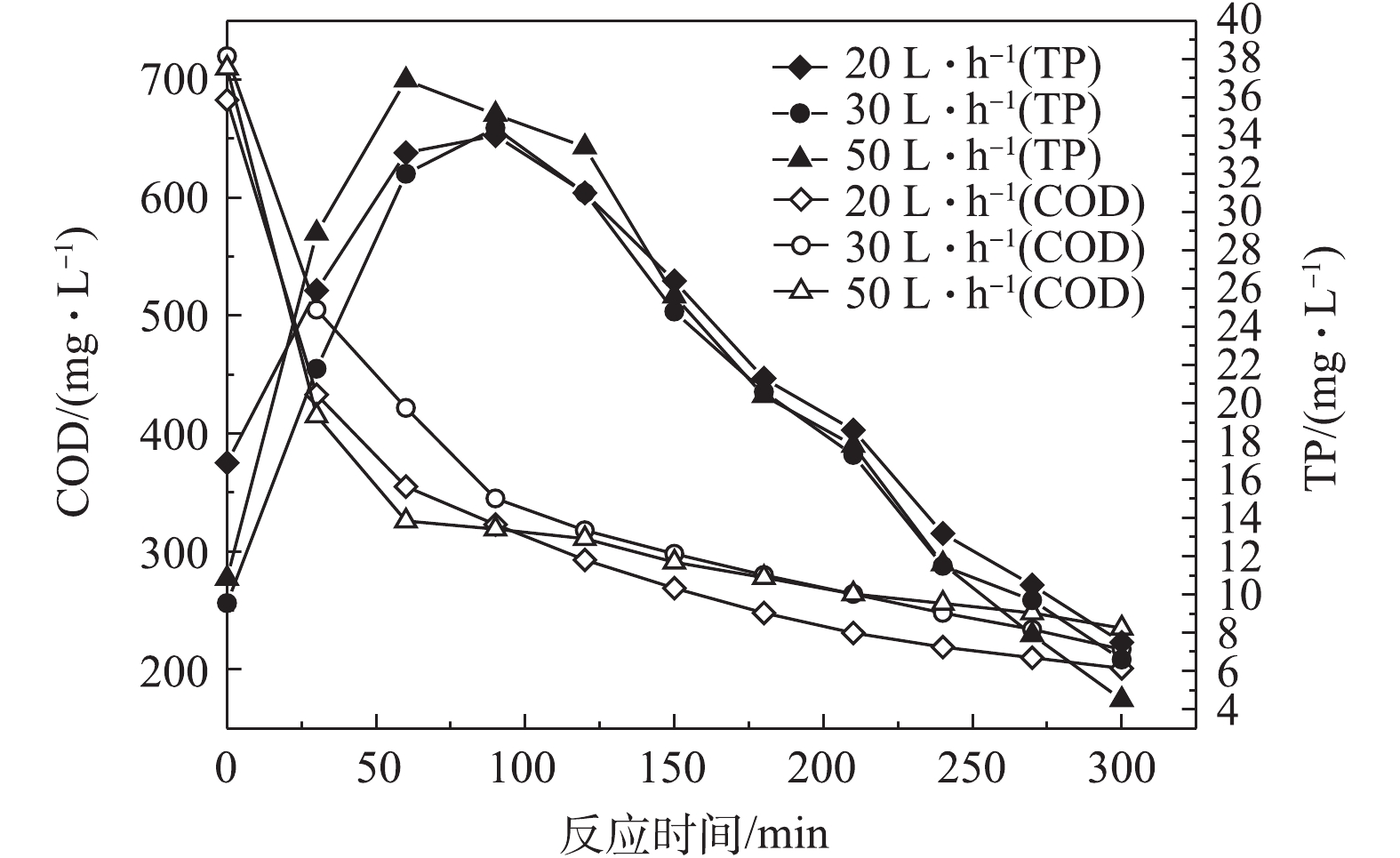
1)前置曝气气量对R1在微氧条件下合成PHAs的影响。图4为前置曝气气量对R1中PHAs含量的影响结果,图5和图6分别为不同曝气气量下R1合成PHAs过程中ORP、COD和TP的变化曲线。由图4可知,前置曝气会消耗R1中微生物细胞内的PHAs。曝气气量为20、30和50 L·h−1时,消耗细胞内PHAs的量分别为1.6、3.79、5.79 mg·g−1。但随着曝气气量的增大,R1在微氧条件下合成PHAs的量也随之增加,分别为114.46,133.58和158.32 mg·g−1。当曝气气量为50 L·h−1时,合成最高量PHAs所需的时间为60 min,较其他2种曝气量条件达到最高量所需的时间更短。这是因为在微氧条件下,曝气气量越大,消耗原有基质的速率越大,造成细胞内“饥饿”的环境,微生物的匮乏条件更为严峻,增强了微生物对碳源的吸收动力[21],待VFAs进入系统,微氧环境下提供少量的DO会为微生物吸收VFAs提供能量,具有合成PHAs能力的微生物能迅速利用外碳源合成PHAs。另外,如图5所示,同时对照图3可知,曝气前R1中ORP为34.9 mV,而在20、30、50 L·h−1曝气气量下R1中对应的ORP分别为−61.8、−65.7、−69 mV,即曝气后ORP出现了下降,推测是因为厌氧发酵产生的VFAs中含有较多的细菌,而细菌与ORP有较强的相关性[22],因此,在曝气后可能因细菌的生长繁殖导致了ORP的降低,但20、30和50 L·h−1曝气气量下最低的ORP分别为−78、−81.1和−85.0 mV,对应所需的时间分别为90、90、60 min。当ORP最小时,PHAs合成量最大,ORP仍可作为PHAs合成量的指示参数。如图6可知,当曝气气量为20、30和50 L·h−1,分别达到PHAs最高合成量时,释磷量分别为34,34.4和36.9 mg·L−1,COD下降分别为360、375和384 mg·L−1。在相同曝气时间内,随着曝气气量的增加,释磷量略有上升,但变化不大,有机物降解量呈现增长趋势,这与合成PHAs的量相符合。
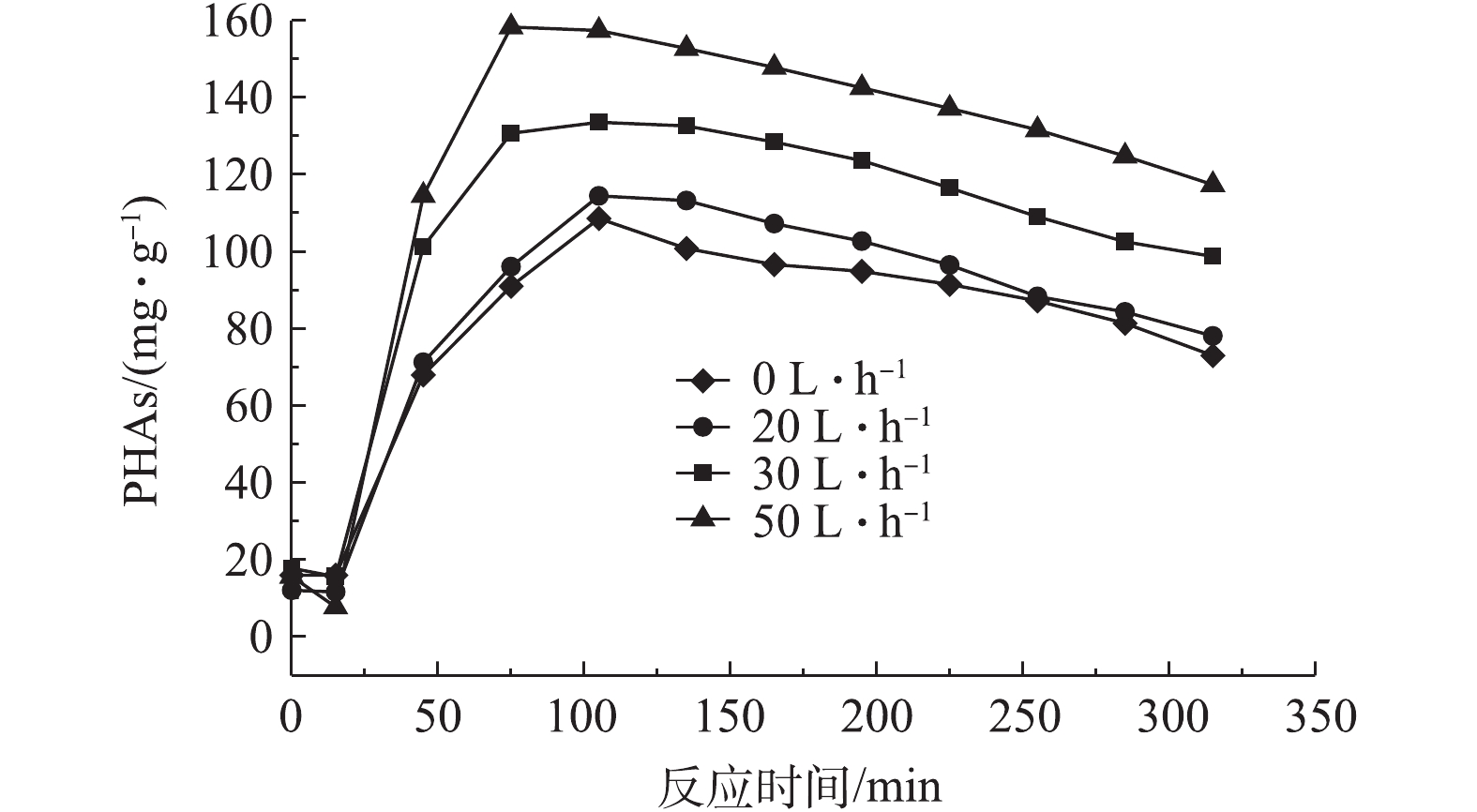 图 4 前置曝气气量对R1中PHAs含量的影响Figure 4. Effect of pre-aerated gas volume on PHAs content in R1
图 4 前置曝气气量对R1中PHAs含量的影响Figure 4. Effect of pre-aerated gas volume on PHAs content in R1以上结果表明,在微氧条件下,相同曝气时间内,曝气气量越大,消耗PHAs量也越多,致使微生物处于“饥饿”的环境下。曝气后加入有机碳源VFAs后,微生物能够利用微氧条件下提供的能量迅速吸收VFAs合成PHAs。当曝气气量为50 L·h−1时,合成PHAs的量最大,达到158.32 mg·g−1。
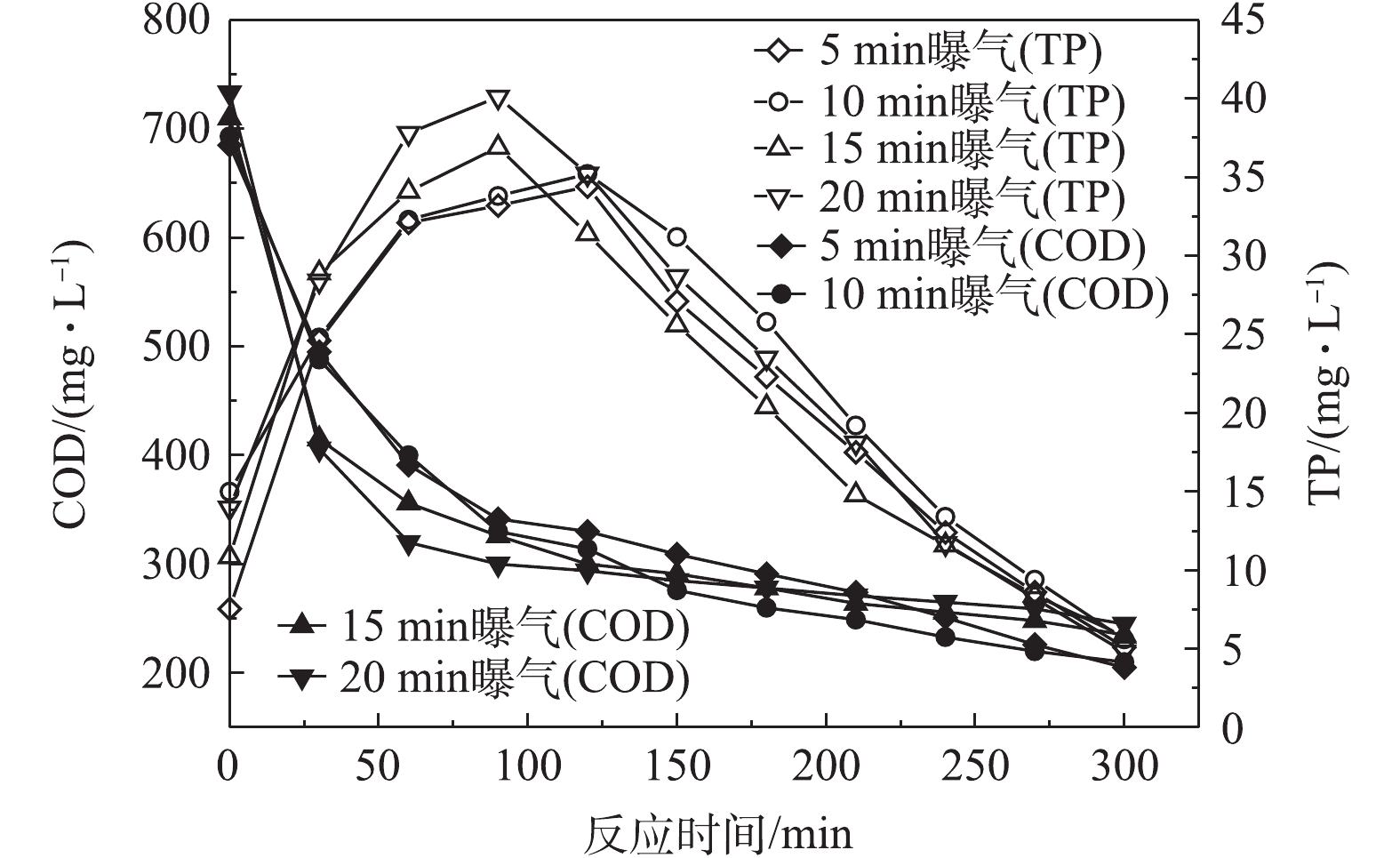
2) 前置曝气时间对R1在微氧条件下合成PHAs的影响。图7为前置曝气时间对R1合成PHAs量的影响结果,图8和图9分别反应了不同曝气时间下R1合成PHAs过程中ORP、COD和TP的变化。由图7可知,随着曝气时间的增加,消耗初始污泥中PHAs的量分别为1.2、3.81、5.79和7.6 mg·g−1,但所合成PHAs的最高量呈增长的趋势,分别为110.5、141.48、158.32和172.5 mg·g−1。相比于无前置曝气时PHAs的最高合成量,曝气时间为5、10、15和20 min时对应的PHAs最高合成量分别增长了1.7%、30.2%、45.78%和58.84%,并且随着曝气时间的增长,达到PHAs最高合成量所需的反应时间有提前的趋势,这表明增设前置曝气的方式能有效促进PHAs的合成,从而提高了PHAs最高合成量,并且能缩短合成PHAs最高量的时间,加快平均反应速率。由图8可知,随着曝气时间的增长,ORP也在下降。当ORP最低时,PHAs合成量最高,ORP的增减依旧能反应PHAs量的变化。由图9可以看出,曝气时间越长,释磷的差异并不大,但COD反应前1 h降解速度小幅度增加。这说明前置曝气不仅能加快碳源的吸收,还能提高碳源的利用率。
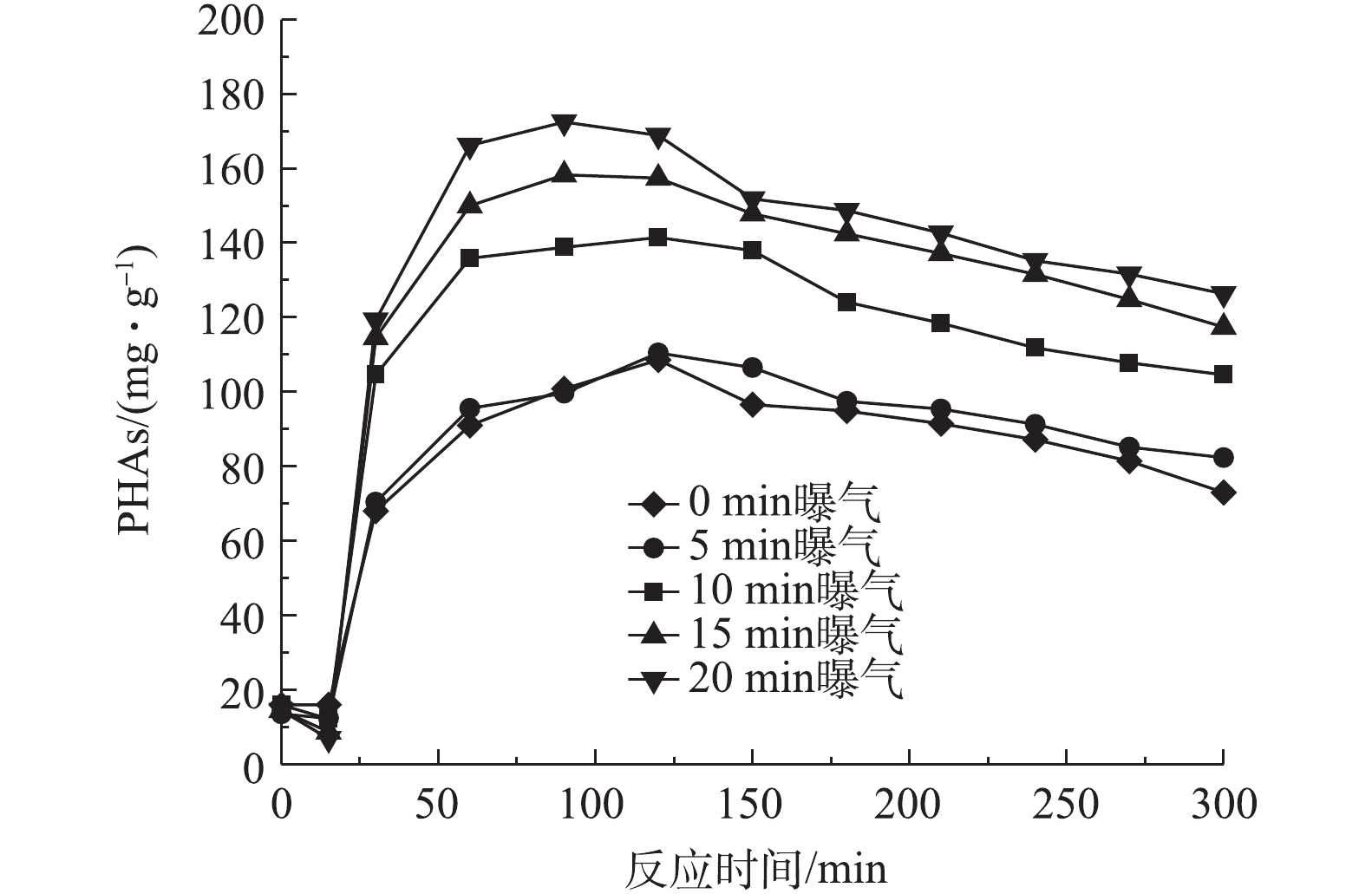 图 7 前置曝气时间对R1合成PHAs量的影响Figure 7. Influence of pre-aeration time on PHAs amount synthesized from R1
图 7 前置曝气时间对R1合成PHAs量的影响Figure 7. Influence of pre-aeration time on PHAs amount synthesized from R1以上结果表明,在微氧条件下,当曝气时间为50 L·h−1时,随着曝气时间的增加,消耗初始PHAs含量越大,合成PHAs的量也越大。当曝气时间为20 min时,PHAs最高合成量为172.5 mg·g−1。
3. 结论
1)同步亚硝化反硝化脱氮除磷系统的剩余污泥比A2O工艺的剩余污泥更具有合成PHAs的能力,其中R1中PHAs最高合成量可达到108.6 mg·g−1,R2中PHAs最高合成量可达到58.58 mg·g−1。
2)增设前置曝气时,微氧条件下曝气气量增大和曝气时间的延长,可以促进PHAs的合成,当曝气气量为50 L·s−1,曝气20 min后,PHAs的最高合成量可达到172.5 mg·g−1。
3)在120 min时,R1中的PHAs的合成量达到了最高值,为108.6 mg·g−1,此时,ORP达到了最低值,为−44.2 mV;在180 min时,R2中的PHAs的合成量达到了最高值,为58.58 mg·g−1,此时,ORP达到了最低值,为−69.9 mV。
-
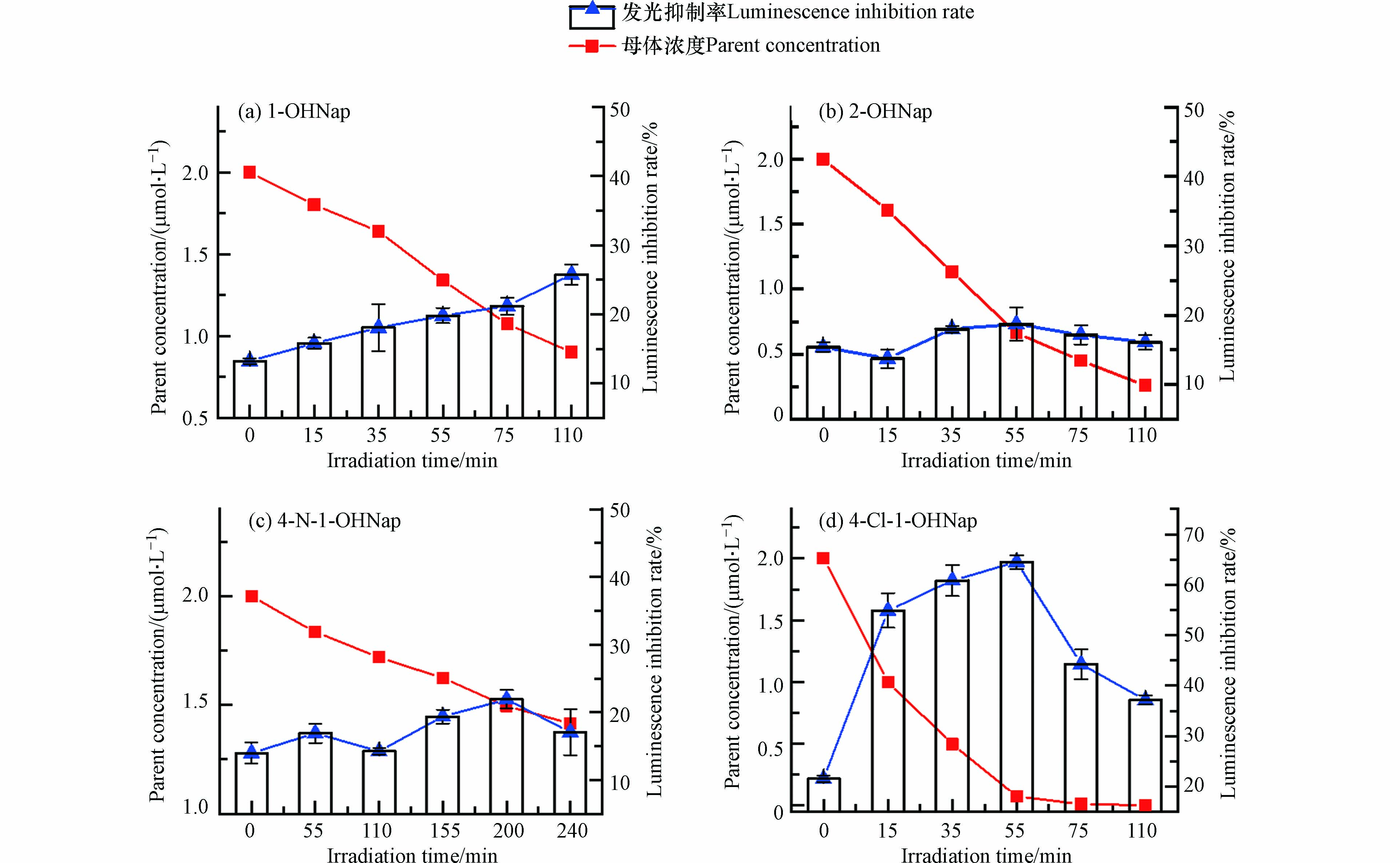
图 1 水中4种OH-PAHs母体浓度及反应溶液对Vibrio fischeri的发光抑制率随光照时间的变化
Figure 1. Changes of 4 OH-PAHs parent concentrations in water and luminescence inhibition rate of the reaction solutions to Vibrio fischeri with irradiation time

图 2 冰中4种OH-PAHs母体浓度的演变及反应溶液对Vibrio fischeri的发光抑制率随光照时间的变化
Figure 2. Evolution of parent concentrations of 4 OH-PAHs in ice, and changes in luminescence inhibition rate of the reaction solutions to Vibrio fischeri with irradiation time

图 3 水中3种N-PAHs母体浓度及反应溶液对Vibrio fischeri的发光抑制率随光照时间的变化
Figure 3. Changes of 3 N-PAHs parent concentrations in water and luminescence inhibition rate of the reaction solutions to Vibrio fischeri with irradiation time
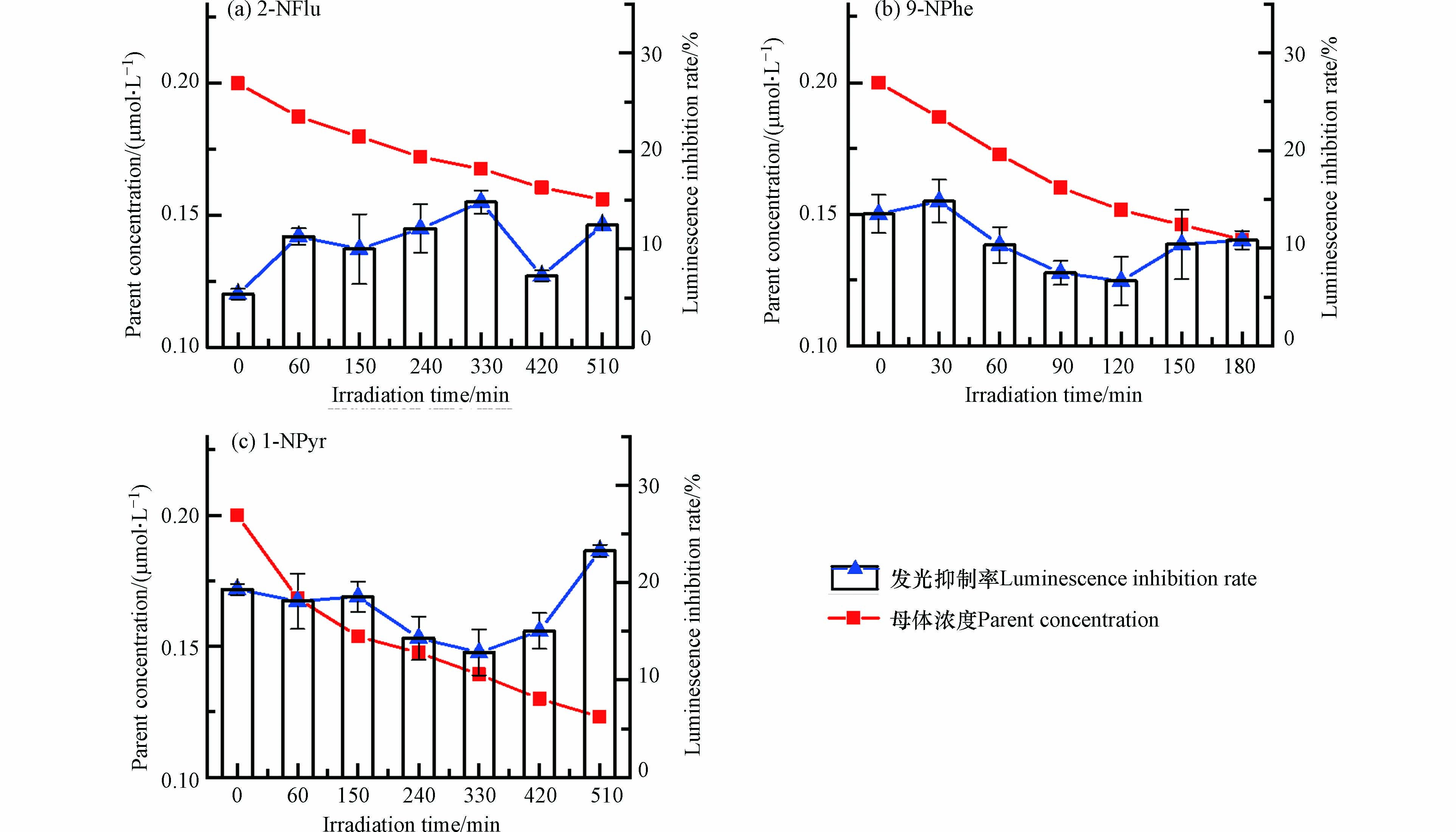
图 4 冰中3种N-PAHs母体浓度的演变及反应溶液对Vibrio fischeri的发光抑制率随光照时间的变化
Figure 4. Evolution of parent concentrations of 3 N-PAHs in ice, and changes in luminescence inhibition rate of the reaction solutions to Vibrio fischeri with irradiation time
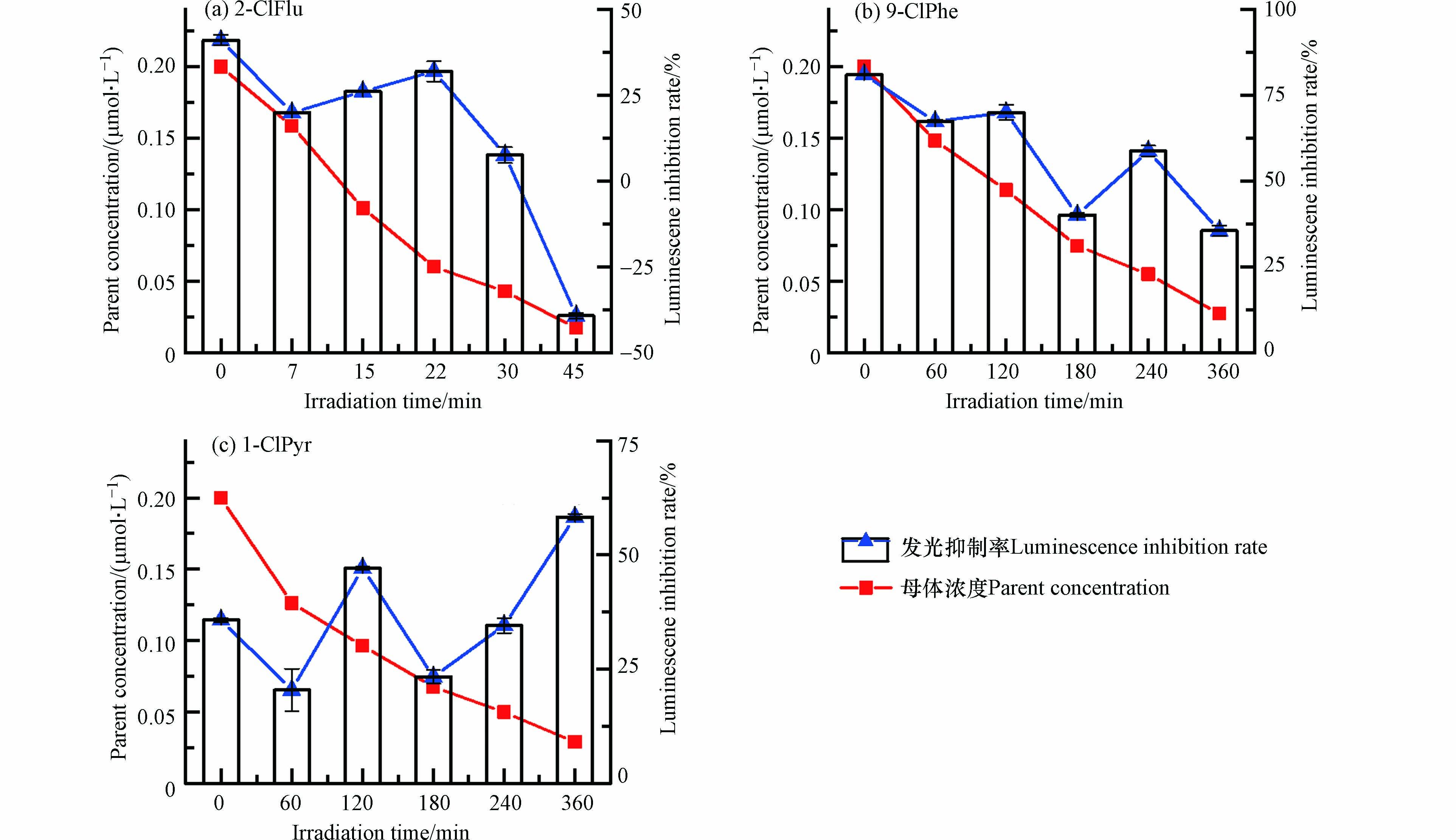
图 5 水中3种Cl-PAHs母体浓度及反应溶液对Vibrio fischeri的发光抑制率随光照时间的变化
Figure 5. Changes of 3 Cl-PAHs parent concentrations in water and luminescence inhibition rate of the reaction solutions to Vibrio fischeri with irradiation time
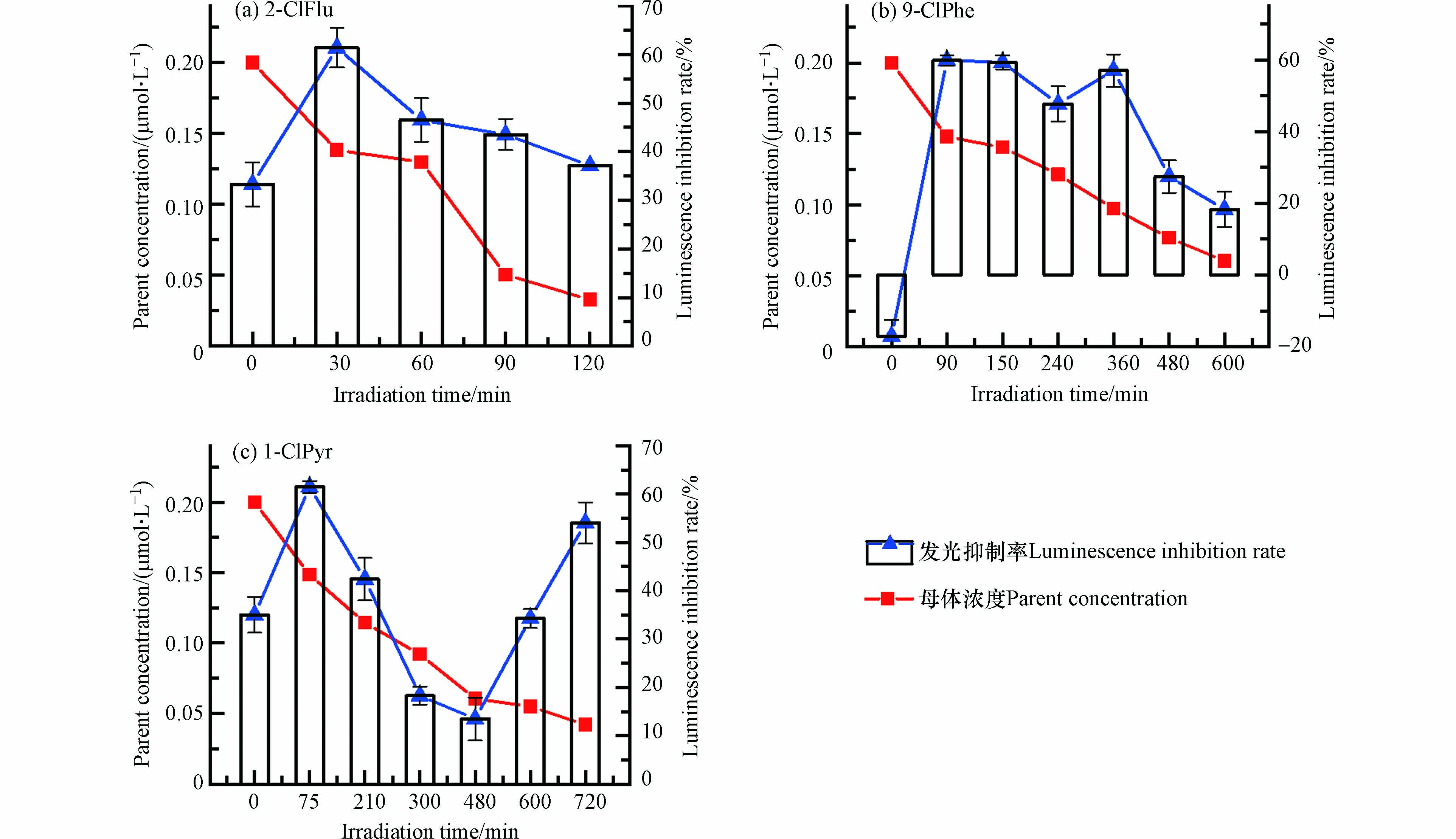
图 6 冰中3种Cl-PAHs母体浓度的演变及反应溶液对Vibrio fischeri的发光抑制率随光照时间的变化
Figure 6. Evolution of parent concentrations of 3 Cl-PAHs in ice, and changes in luminescence inhibition rate of the reaction solutions to Vibrio fischeri with irradiation time
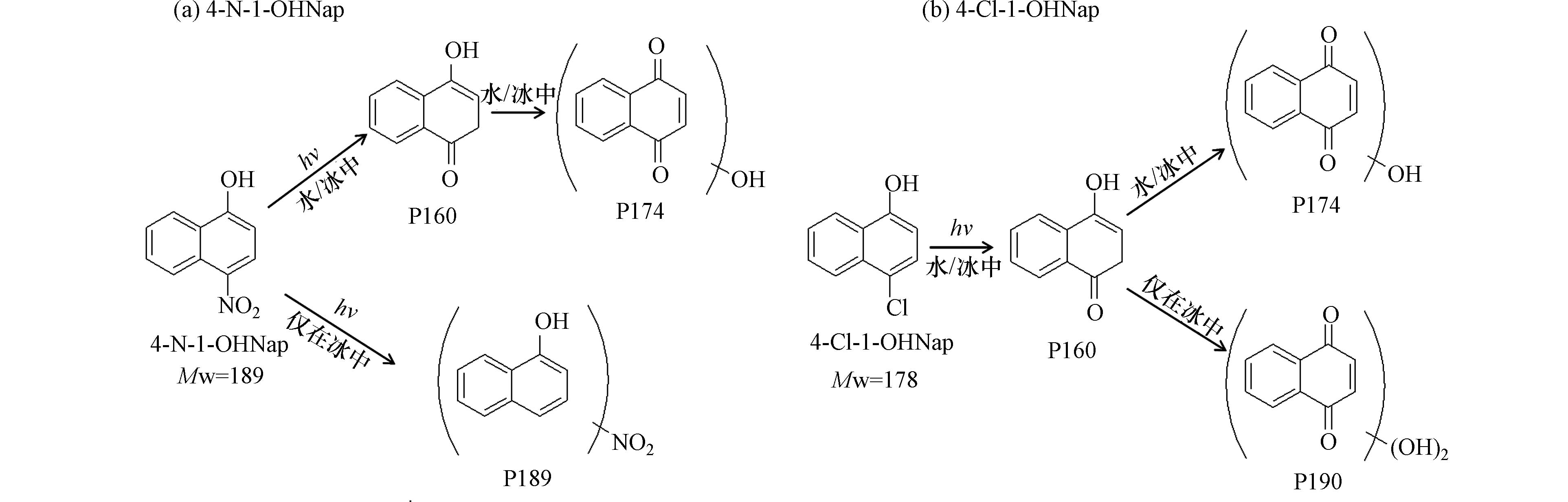
图 7 模拟日光照射(λ > 290 nm)下水中与冰中2种OH-PAHs的光化学转化途径
Figure 7. Phototransformation pathways of 2 OH-PAHs in water and ice under simulated sunlight irradiation (λ > 290 nm).
表 1 10种SPAHs的液相色谱检测参数与保留时间(tR)
Table 1. HPLC determination parameters and retention time (tR) of 10 SPAHs
化合物Compound 简称Abbreviation 检测器Detector 检测波长/nmDetection wavelength tR/min 1-羟基萘 1-OHNap 紫外检测器 232 9.0 2-羟基萘 2-OHNap 232 8.7 4-硝基-1-羟基萘 4-N-1-OHNap 237 10.3 4-氯-1-羟基萘 4-Cl-1-OHNap 231 9.1 2-氯芴 2-ClFlu 荧光检测器 λex 305; λem 353 8.1 9-氯菲 9-ClPhe λex254; λem 365 10.1 1-氯芘 1-ClPyr λex 276; λem 380 8.0 2-硝基芴 2-NFlu 紫外检测器 333 2.0 9-硝基菲 9-NPhe 247 2.2 1-硝基芘 1-NPyr 235 2.9 注:λex为激发波长;λem为发射波长(λex is the excitation wavelength; λem is the emission wavelength)  下载: 导出CSV
下载: 导出CSV
表 2 未光照时OH-PAHs (C0 = 2 μmol·L−1)、N-PAHs与Cl-PAHs (C0 = 0.2 μmol·L−1)水溶液对Vibrio fischeri的发光抑制率
Table 2. Luminescence inhibition rate (I%) of aqueous solutions of parent OH-PAHs (C0 = 2 μmol·L−1), N-PAHs and Cl-PAHs (C0 = 0.2 μmol·L−1) to Vibrio fischeri
OH-PAHs I% N-PAHs I% Cl-PAHs I% 1-OHNap 13.23 2-NFlu 22.29 2-ClFlu 41.07 2-OHNap 15.41 9-NPhe 19.26 9-ClPhe 81.02 4-N-1-OHNap 13.99 1-NPyr 24.69 1-ClPyr 35.73 4-Cl-1-OHNap 21.55
下载: 导出CSV
表 3 南北极和西安、大连晴天中午太阳光在365 nm和420 nm处的平均光强(I)
Table 3. Average light intensity (I) of sunlight at 365 nm and 420 nm at sunny noon in Antarctica, Arctic, Xi'an and Dalian
光强测定时间、地点Light-intensity measurement time and place I365 / (mW·cm−2) I420 / (mW·cm−2) 南极长城站,夏季中午(62°S,59°W;2014年1月) 2.16 3.81 北极黄河站,夏季中午(79°N,12°E;2015年7月) 1.61 3.54 中国西安,冬季中午(34°N,108°E;2021年1月) 0.58 2.64 中国大连,冬季中午(39°N,121°E;2014年1月) 1.31 2.90
下载: 导出CSV
-
[1] ABBAS I, BADRAN G, VERDIN A, et al. Polycyclic aromatic hydrocarbon derivatives in airborne particulate matter: Sources, analysis and toxicity[J]. Environmental Chemistry Letters, 2018, 16(2): 439-475. doi: 10.1007/s10311-017-0697-0 [2] WEI C, BANDOWE B A M, HAN Y M, et al. Polycyclic aromatic hydrocarbons (PAHs) and their derivatives (alkyl-PAHs, oxygenated-PAHs, nitrated-PAHs and azaarenes) in urban road dusts from Xi’an, Central China[J]. Chemosphere, 2015, 134: 512-520. doi: 10.1016/j.chemosphere.2014.11.052 [3] ZHAO T, YANG L X, HUANG Q, et al. PM2.5-bound polycyclic aromatic hydrocarbons (PAHs) and nitrated-PAHs (NPAHs) emitted by gasoline vehicles: Characterization and health risk assessment[J]. Science of the Total Environment, 2020, 727: 138631. doi: 10.1016/j.scitotenv.2020.138631 [4] LAMMEL G, KITANOVSKI Z, KUKUČKA P, et al. Oxygenated and nitrated polycyclic aromatic hydrocarbons in ambient air-levels, phase partitioning, mass size distributions, and inhalation bioaccessibility[J]. Environmental Science & Technology, 2020, 54(5): 2615-2625. [5] 曾超怡, 徐辉, 许岩, 等. 长江重点江段水体中多环芳烃及其衍生物的分布及健康风险[J]. 环境科学学报, 2021, 41(12): 4932-4941. doi: 10.13671/j.hjkxxb.2021.0201 ZENG C Y, XU H, XU Y, et al. Distribution and health risk of polycyclic aromatic hydrocarbons(PAHs)and their derivatives in surface water of the Yangtze River[J]. Acta Scientiae Circumstantiae, 2021, 41(12): 4932-4941(in Chinese). doi: 10.13671/j.hjkxxb.2021.0201
[6] 付璐婧, 乔梦, 赵旭, 等. 京津潮白河多环芳烃及其衍生物分布[J]. 生态毒理学报, 2019, 14(3): 233-239. FU L J, QIAO M, ZHAO X, et al. Distribution of polycyclic aromatic hydrocarbons and their derivatives in chaobai river of Beijing-Tianjin area[J]. Asian Journal of Ecotoxicology, 2019, 14(3): 233-239(in Chinese).
[7] NA G S, LIU C Y, WANG Z, et al. Distribution and characteristic of PAHs in snow of Fildes Peninsula[J]. Journal of Environmental Sciences, 2011, 23(9): 1445-1451. doi: 10.1016/S1001-0742(10)60605-5 [8] FUOCO R, GIANNARELLI S, ONOR M, et al. A snow/firn four-century record of polycyclic aromatic hydrocarbons (PAHs) and polychlorobiphenyls (PCBs) at Talos Dome (Antarctica)[J]. Microchemical Journal, 2012, 105: 133-141. doi: 10.1016/j.microc.2012.05.018 [9] 葛林科, 任红蕾, 霍城, 等. 冰中9-羟基芴的光化学降解[J]. 中国科学(化学), 2015, 45(6): 655-661. GE L K, REN H L, HUO C, et al. Photochemical degradation of 9-hydroxyfluorene in ices[J]. Scientia Sinica(Chimica), 2015, 45(6): 655-661(in Chinese).
[10] GE L K, CAO S K, HALSALL C, et al. Photodegradation of hydroxyfluorenes in ice and water: A comparison of kinetics, effects of water constituents, and phototransformation by-products[J]. Journal of Environmental Sciences, 2023, 124: 139-145. doi: 10.1016/j.jes.2021.11.002 [11] WANG T, ZHAO J, LIU Y, et al. PM2.5‐bound polycyclic aromatic hydrocarbons (PAHs), nitrated PAHs (NPAHs) and oxygenated PAHs (OPAHs) in typical traffic‐related receptor environments[J]. Journal of Geophysical Research:Atmospheres, 2022, 127(5): e2021JD035951. doi: 10.1029/2021JD035951 [12] 马涛, 孔继婕, 韩孟书, 等. 环境中硝基多环芳烃的污染现状及其毒性效应研究进展[J]. 环境化学, 2020, 39(9): 2430-2440. doi: 10.7524/j.issn.0254-6108.2019062907 MA T, KONG J J, HAN M S, et al. Review on the pollution status and toxicity effects of nitrated polycyclic aromatic hydrocarbons in the environment[J]. Environmental Chemistry, 2020, 39(9): 2430-2440(in Chinese). doi: 10.7524/j.issn.0254-6108.2019062907
[13] OHURA T, HORII Y, YAMASHITA N. Spatial distribution and exposure risks of ambient chlorinated polycyclic aromatic hydrocarbons in Tokyo Bay area and network approach to source impacts[J]. Environmental Pollution, 2018, 232: 367-374. doi: 10.1016/j.envpol.2017.09.037 [14] KRZYSZCZAK A, CZECH B. Occurrence and toxicity of polycyclic aromatic hydrocarbons derivatives in environmental matrices[J]. The Science of the Total Environment, 2021, 788: 147738. doi: 10.1016/j.scitotenv.2021.147738 [15] KONG J J, HAN M S, LIU Y, et al. Analysis of trace-level nitrated polycyclic aromatic hydrocarbons in water samples by solid-phase microextraction with gas chromatography and mass spectrometry[J]. Journal of Separation Science, 2018, 41(12): 2681-2687. doi: 10.1002/jssc.201701271 [16] CHONDO Y, LI Y, MAKINO F, et al. Determination of selected nitropolycyclic aromatic hydrocarbons in water samples[J]. Chemical & Pharmaceutical Bulletin, 2013, 61(12): 1269-1274. [17] 付璐婧, 李一兵, 乔梦, 等. 多环芳烃及其衍生物在北京纳污河流中的分布及健康风险[J]. 环境科学, 2019, 40(1): 256-262. doi: 10.13227/j.hjkx.201806097 FU L J, LI Y B, QIAO M, et al. Distribution and risk assemmment of polycyclic aromatic hydrocarbons and their derivatives in wastewater-receiving rivers in Beijing[J]. Environmental Science, 2019, 40(1): 256-262(in Chinese). doi: 10.13227/j.hjkx.201806097
[18] BIGOT M, HAWKER D W, CROPP R, et al. Spring melt and the redistribution of organochlorine pesticides in the sea-ice environment: A comparative study between Arctic and Antarctic regions[J]. Environmental Science & Technology, 2017, 51(16): 8944-8952. [19] PARK J, BALL L M, RICHARDSON S D, et al. Oxidative mutagenicity of polar fractions from polycyclic aromatic hydrocarbon-contaminated soils[J]. Environmental Toxicology and Chemistry, 2008, 27(11): 2207-2215. doi: 10.1897/07-572.1 [20] KUANG D, ZHANG W Z, DENG Q F, et al. Dose-response relationships of polycyclic aromatic hydrocarbons exposure and oxidative damage to DNA and lipid in coke oven workers[J]. Environmental Science & Technology, 2013, 47(13): 7446-7456. [21] SANCHES S, LEITÃO C, PENETRA A, et al. Direct photolysis of polycyclic aromatic hydrocarbons in drinking water sources[J]. Journal of Hazardous Materials, 2011, 192(3): 1458-1465. doi: 10.1016/j.jhazmat.2011.06.065 [22] 白东晓, 葛林科, 张蓬, 等. 冰雪中有机污染物的环境光化学行为[J]. 中国科学(化学), 2022, 52(1): 1-14. BAI D X, GE L K, ZHANG P, et al. Environmental photochemical behavior of organic pollutants in ice and snow[J]. Scientia Sinica(Chimica), 2022, 52(1): 1-14(in Chinese).
[23] GE L K, Li J, Na G S, et al. Photochemical degradation of hydroxy PAHs in ice: Implications for the polar areas[J]. Chemosphere, 2016, 155: 375-379. doi: 10.1016/j.chemosphere.2016.04.087 [24] 刘纪阳, 薛爽, 张营, 等. 水相和冰相中不同pH条件下溶解性有机质对苊光降解的影响[J]. 环境科学学报, 2021, 41(5): 1930-1939. doi: 10.13671/j.hjkxxb.2020.0400 LIU J Y, XUE S, ZHANG Y, et al. Effect of dissolved organic matter on photodegradation of acenaphthene under different pH conditions in water and ice[J]. Acta Scientiae Circumstantiae, 2021, 41(5): 1930-1939(in Chinese). doi: 10.13671/j.hjkxxb.2020.0400
[25] GE L K, NA G S, CHEN C E, et al. Aqueous photochemical degradation of hydroxylated PAHs: Kinetics, pathways, and multivariate effects of main water constituents[J]. The Science of the Total Environment, 2016, 547: 166-172. doi: 10.1016/j.scitotenv.2015.12.143 [26] KLÁNOVÁ J, KLÁN P, NOSEK J, et al. Environmental ice photochemistry: Monochlorophenols[J]. Environmental Science & Technology, 2003, 37(8): 1568-1574. [27] BLAHA L, KLANOVA J, KLAN P, et al. Toxicity increases in ice containing monochlorophenols upon photolysis: Environmental consequences[J]. Environmental Science & Technology, 2004, 38(10): 2873-2878. [28] NIU J F, WANG L L, YANG Z F. QSPRs on photodegradation half-lives of atmospheric chlorinated polycyclic aromatic hydrocarbons associated with particulates[J]. Ecotoxicology and Environmental Safety, 2007, 66(2): 272-277. doi: 10.1016/j.ecoenv.2006.02.014 [29] XUE H H, ZHENG N, KANG C L, et al. Photochemical degradation of nitrated PAHs in snow[J]. Atmospheric Environment, 2019, 199: 260-264. doi: 10.1016/j.atmosenv.2018.11.026 [30] WANG X, W LI Y, CAI F S, et al. Fully automatic single-drop microextraction with one-setp extraction and derivatization and its application for rapid analysis of hydroxylated polycyclic aromatic hydrocarbons in seawaters[J]. Talanta, 2017, 164: 727-734. doi: 10.1016/j.talanta.2016.06.011 [31] WANG X L, KANG H Y, WU J F. Determination of chlorinated polycyclic aromatic hydrocarbons in water by solid-phase extraction coupled with gas chromatography and mass spectrometry[J]. Journal of Separation Science, 2016, 39(9): 1742-1748. doi: 10.1002/jssc.201501286 [32] ZHAO Q, FANG Q, LIU H Y, et al. Halide-specific enhancement of photodegradation for sulfadiazine in estuarine waters: Roles of halogen radicals and main water constituents[J]. Water Research, 2019, 160: 209-216. doi: 10.1016/j.watres.2019.05.061 [33] LIU H, ZHAO H M, QUAN X, et al. Formation of chlorinated intermediate from bisphenol A in surface saline water under simulated solar light irradiation[J]. Environmental Science & Technology, 2009, 43(20): 7712-7717. [34] CHEN Z Y, ANASTASIO C. Concentrations of a triplet excited state are enhanced in illuminated ice[J]. Environmental Science. Processes & Impacts, 2017, 19(1): 12-21. [35] STATHIS A A, HENDRICKSON-STIVES A K, KAHAN T F. Photolysis kinetics of toluene, ethylbenzene, and xylenes at ice surfaces[J]. The Journal of Physical Chemistry A, 2016, 120(34): 6693-6697. doi: 10.1021/acs.jpca.6b05595 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 896
- HTML全文浏览数: 896
- PDF下载数: 74
- 施引文献: 0
