





$</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M13">$\left[{\mathrm{K}\mathrm{M}\mathrm{n}\mathrm{O} }_{4}\right]/(\mathrm{m}\mathrm{o}\mathrm{l}\cdot\mathrm{L}^{-1})$</tex-math><alternatives><img class="graphic" src="2022072801_M13.jpg"><img class="graphic" src="2022072801_M13.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">回归方程 Regression equation</td><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M14">$ {k}_{{\rm{obs}}}/{\mathrm{m}\mathrm{i}\mathrm{n}}^{-1} $</tex-math><alternatives><img class="graphic" src="2022072801_M14.jpg"><img class="graphic" src="2022072801_M14.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">线性相关系数<i>R</i><sup>2</sup> Linearly dependent coefficient</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">3.165×10<sup>−6</sup></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M15">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.1488{t}+0.0173$</tex-math><alternatives><img class="graphic" src="2022072801_M15.jpg"><img class="graphic" src="2022072801_M15.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">0.1488</td><td class="table_top_border2" align="center" valign="middle">0.9956</td></tr><tr><td align="center" valign="middle">6.329×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M16">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.3165{t}+0.0247$</tex-math><alternatives><img class="graphic" src="2022072801_M16.jpg"><img class="graphic" src="2022072801_M16.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.3165</td><td align="center" valign="middle">0.9979</td></tr><tr><td align="center" valign="middle">9.494×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M17">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.4867{t}+0.0685$</tex-math><alternatives><img class="graphic" src="2022072801_M17.jpg"><img class="graphic" src="2022072801_M17.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.4867</td><td align="center" valign="middle">0.9952</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">1.266×10<sup>−5</sup></td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M18">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.6457{t}+0.0168$</tex-math><alternatives><img class="graphic" src="2022072801_M18.jpg"><img class="graphic" src="2022072801_M18.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.6457</td><td class="table_bottom_border" align="center" valign="middle">0.9951</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td class="table_top_border" align="center" valign="middle">回归方程 Regression equation</td><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M14">$ {k}_{{\rm{obs}}}/{\mathrm{m}\mathrm{i}\mathrm{n}}^{-1} $</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M13">$\left[{\mathrm{K}\mathrm{M}\mathrm{n}\mathrm{O} }_{4}\right]/(\mathrm{m}\mathrm{o}\mathrm{l}\cdot\mathrm{L}^{-1})$</tex-math><alternatives><img class="graphic" src="2022072801_M13.jpg"><img class="graphic" src="2022072801_M13.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">回归方程 Regression equation</td><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M14">$ {k}_{{\rm{obs}}}/{\mathrm{m}\mathrm{i}\mathrm{n}}^{-1} $</tex-math><alternatives><img class="graphic" src="2022072801_M14.jpg"><img class="graphic" src="2022072801_M14.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">线性相关系数<i>R</i><sup>2</sup> Linearly dependent coefficient</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">3.165×10<sup>−6</sup></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M15">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.1488{t}+0.0173$</tex-math><alternatives><img class="graphic" src="2022072801_M15.jpg"><img class="graphic" src="2022072801_M15.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">0.1488</td><td class="table_top_border2" align="center" valign="middle">0.9956</td></tr><tr><td align="center" valign="middle">6.329×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M16">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.3165{t}+0.0247$</tex-math><alternatives><img class="graphic" src="2022072801_M16.jpg"><img class="graphic" src="2022072801_M16.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.3165</td><td align="center" valign="middle">0.9979</td></tr><tr><td align="center" valign="middle">9.494×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M17">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.4867{t}+0.0685$</tex-math><alternatives><img class="graphic" src="2022072801_M17.jpg"><img class="graphic" src="2022072801_M17.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.4867</td><td align="center" valign="middle">0.9952</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">1.266×10<sup>−5</sup></td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M18">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.6457{t}+0.0168$</tex-math><alternatives><img class="graphic" src="2022072801_M18.jpg"><img class="graphic" src="2022072801_M18.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.6457</td><td class="table_bottom_border" align="center" valign="middle">0.9951</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td class="table_top_border" align="center" valign="middle">线性相关系数<i>R</i><sup>2</sup> Linearly dependent coefficient</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">3.165×10<sup>−6</sup></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M15">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.1488{t}+0.0173$</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M13">$\left[{\mathrm{K}\mathrm{M}\mathrm{n}\mathrm{O} }_{4}\right]/(\mathrm{m}\mathrm{o}\mathrm{l}\cdot\mathrm{L}^{-1})$</tex-math><alternatives><img class="graphic" src="2022072801_M13.jpg"><img class="graphic" src="2022072801_M13.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">回归方程 Regression equation</td><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M14">$ {k}_{{\rm{obs}}}/{\mathrm{m}\mathrm{i}\mathrm{n}}^{-1} $</tex-math><alternatives><img class="graphic" src="2022072801_M14.jpg"><img class="graphic" src="2022072801_M14.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">线性相关系数<i>R</i><sup>2</sup> Linearly dependent coefficient</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">3.165×10<sup>−6</sup></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M15">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.1488{t}+0.0173$</tex-math><alternatives><img class="graphic" src="2022072801_M15.jpg"><img class="graphic" src="2022072801_M15.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">0.1488</td><td class="table_top_border2" align="center" valign="middle">0.9956</td></tr><tr><td align="center" valign="middle">6.329×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M16">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.3165{t}+0.0247$</tex-math><alternatives><img class="graphic" src="2022072801_M16.jpg"><img class="graphic" src="2022072801_M16.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.3165</td><td align="center" valign="middle">0.9979</td></tr><tr><td align="center" valign="middle">9.494×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M17">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.4867{t}+0.0685$</tex-math><alternatives><img class="graphic" src="2022072801_M17.jpg"><img class="graphic" src="2022072801_M17.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.4867</td><td align="center" valign="middle">0.9952</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">1.266×10<sup>−5</sup></td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M18">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.6457{t}+0.0168$</tex-math><alternatives><img class="graphic" src="2022072801_M18.jpg"><img class="graphic" src="2022072801_M18.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.6457</td><td class="table_bottom_border" align="center" valign="middle">0.9951</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td class="table_top_border2" align="center" valign="middle">0.1488</td><td class="table_top_border2" align="center" valign="middle">0.9956</td></tr><tr><td align="center" valign="middle">6.329×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M16">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.3165{t}+0.0247$</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M13">$\left[{\mathrm{K}\mathrm{M}\mathrm{n}\mathrm{O} }_{4}\right]/(\mathrm{m}\mathrm{o}\mathrm{l}\cdot\mathrm{L}^{-1})$</tex-math><alternatives><img class="graphic" src="2022072801_M13.jpg"><img class="graphic" src="2022072801_M13.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">回归方程 Regression equation</td><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M14">$ {k}_{{\rm{obs}}}/{\mathrm{m}\mathrm{i}\mathrm{n}}^{-1} $</tex-math><alternatives><img class="graphic" src="2022072801_M14.jpg"><img class="graphic" src="2022072801_M14.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">线性相关系数<i>R</i><sup>2</sup> Linearly dependent coefficient</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">3.165×10<sup>−6</sup></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M15">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.1488{t}+0.0173$</tex-math><alternatives><img class="graphic" src="2022072801_M15.jpg"><img class="graphic" src="2022072801_M15.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">0.1488</td><td class="table_top_border2" align="center" valign="middle">0.9956</td></tr><tr><td align="center" valign="middle">6.329×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M16">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.3165{t}+0.0247$</tex-math><alternatives><img class="graphic" src="2022072801_M16.jpg"><img class="graphic" src="2022072801_M16.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.3165</td><td align="center" valign="middle">0.9979</td></tr><tr><td align="center" valign="middle">9.494×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M17">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.4867{t}+0.0685$</tex-math><alternatives><img class="graphic" src="2022072801_M17.jpg"><img class="graphic" src="2022072801_M17.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.4867</td><td align="center" valign="middle">0.9952</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">1.266×10<sup>−5</sup></td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M18">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.6457{t}+0.0168$</tex-math><alternatives><img class="graphic" src="2022072801_M18.jpg"><img class="graphic" src="2022072801_M18.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.6457</td><td class="table_bottom_border" align="center" valign="middle">0.9951</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td align="center" valign="middle">0.3165</td><td align="center" valign="middle">0.9979</td></tr><tr><td align="center" valign="middle">9.494×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M17">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.4867{t}+0.0685$</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M13">$\left[{\mathrm{K}\mathrm{M}\mathrm{n}\mathrm{O} }_{4}\right]/(\mathrm{m}\mathrm{o}\mathrm{l}\cdot\mathrm{L}^{-1})$</tex-math><alternatives><img class="graphic" src="2022072801_M13.jpg"><img class="graphic" src="2022072801_M13.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">回归方程 Regression equation</td><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M14">$ {k}_{{\rm{obs}}}/{\mathrm{m}\mathrm{i}\mathrm{n}}^{-1} $</tex-math><alternatives><img class="graphic" src="2022072801_M14.jpg"><img class="graphic" src="2022072801_M14.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">线性相关系数<i>R</i><sup>2</sup> Linearly dependent coefficient</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">3.165×10<sup>−6</sup></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M15">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.1488{t}+0.0173$</tex-math><alternatives><img class="graphic" src="2022072801_M15.jpg"><img class="graphic" src="2022072801_M15.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">0.1488</td><td class="table_top_border2" align="center" valign="middle">0.9956</td></tr><tr><td align="center" valign="middle">6.329×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M16">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.3165{t}+0.0247$</tex-math><alternatives><img class="graphic" src="2022072801_M16.jpg"><img class="graphic" src="2022072801_M16.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.3165</td><td align="center" valign="middle">0.9979</td></tr><tr><td align="center" valign="middle">9.494×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M17">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.4867{t}+0.0685$</tex-math><alternatives><img class="graphic" src="2022072801_M17.jpg"><img class="graphic" src="2022072801_M17.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.4867</td><td align="center" valign="middle">0.9952</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">1.266×10<sup>−5</sup></td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M18">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.6457{t}+0.0168$</tex-math><alternatives><img class="graphic" src="2022072801_M18.jpg"><img class="graphic" src="2022072801_M18.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.6457</td><td class="table_bottom_border" align="center" valign="middle">0.9951</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td align="center" valign="middle">0.4867</td><td align="center" valign="middle">0.9952</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">1.266×10<sup>−5</sup></td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M18">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.6457{t}+0.0168$</tex-math><img class="inline-formula" style="display:none;" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M13">$\left[{\mathrm{K}\mathrm{M}\mathrm{n}\mathrm{O} }_{4}\right]/(\mathrm{m}\mathrm{o}\mathrm{l}\cdot\mathrm{L}^{-1})$</tex-math><alternatives><img class="graphic" src="2022072801_M13.jpg"><img class="graphic" src="2022072801_M13.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">回归方程 Regression equation</td><td class="table_top_border" align="center" valign="middle"><inline-formula><tex-math id="M14">$ {k}_{{\rm{obs}}}/{\mathrm{m}\mathrm{i}\mathrm{n}}^{-1} $</tex-math><alternatives><img class="graphic" src="2022072801_M14.jpg"><img class="graphic" src="2022072801_M14.png"></alternatives></inline-formula></td><td class="table_top_border" align="center" valign="middle">线性相关系数<i>R</i><sup>2</sup> Linearly dependent coefficient</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">3.165×10<sup>−6</sup></td><td class="table_top_border2" align="center" valign="middle"><inline-formula><tex-math id="M15">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.1488{t}+0.0173$</tex-math><alternatives><img class="graphic" src="2022072801_M15.jpg"><img class="graphic" src="2022072801_M15.png"></alternatives></inline-formula></td><td class="table_top_border2" align="center" valign="middle">0.1488</td><td class="table_top_border2" align="center" valign="middle">0.9956</td></tr><tr><td align="center" valign="middle">6.329×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M16">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.3165{t}+0.0247$</tex-math><alternatives><img class="graphic" src="2022072801_M16.jpg"><img class="graphic" src="2022072801_M16.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.3165</td><td align="center" valign="middle">0.9979</td></tr><tr><td align="center" valign="middle">9.494×10<sup>−6</sup></td><td align="center" valign="middle"><inline-formula><tex-math id="M17">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.4867{t}+0.0685$</tex-math><alternatives><img class="graphic" src="2022072801_M17.jpg"><img class="graphic" src="2022072801_M17.png"></alternatives></inline-formula></td><td align="center" valign="middle">0.4867</td><td align="center" valign="middle">0.9952</td></tr><tr><td class="table_bottom_border" align="center" valign="middle">1.266×10<sup>−5</sup></td><td class="table_bottom_border" align="center" valign="middle"><inline-formula><tex-math id="M18">$\mathrm{ln}\dfrac{C}{ {C}_{0} }=-0.6457{t}+0.0168$</tex-math><alternatives><img class="graphic" src="2022072801_M18.jpg"><img class="graphic" src="2022072801_M18.png"></alternatives></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.6457</td><td class="table_bottom_border" align="center" valign="middle">0.9951</td></tr></tbody>
</table></div></foreignObject></svg>"></inline-formula></td><td class="table_bottom_border" align="center" valign="middle">0.6457</td><td class="table_bottom_border" align="center" valign="middle">0.9951</td></tr></tbody>
</table></div></foreignObject></svg>)
-
我国北方黄河沿岸的水库水源发生了较为严重的鱼腥味问题,引起了人们的心理恐慌[1],继而加大水厂的水处理压力. 研究发现鱼腥味主要由一些胺类物质和不饱和醛类物质引起[2],在低温、贫营养化水体中,随着一些藻类如锥囊藻、针杆藻的大量生长和腐败,产生的中等强度的鱼腥味和土霉味[3],令人不适. 国内外报道的主要鱼腥味嗅味物质有三甲胺、二甲胺、2,4-庚二烯醛、2,4-癸二烯醛、2,4,7-三烯醛、2,6-壬二烯醛等[4-9]. 庚二烯醛和癸二烯醛等是多元不饱和烯醛类物质,可以由水生生物体内的多元不饱和脂肪酸先后经过脂肪氧合酶和脂氢过氧化物裂解酶的催化作用产生[10];反,反- 2,4-癸二烯醛是水体中硅藻脂氧合裂解产物[11]. 目前自来水厂混凝、沉淀、过滤等常规处理方法对以上引起水中鱼腥味的醛类物质的去除效果十分有限,增加合适的预处理或深度处理环节显得尤为必要[2].
传统水厂多采用氧化剂对原水进行预处理,分解有机物,降低CODMn等指标. 醛类嗅味物质的氧化处理技术,现阶段大多数研究都集中于β-环柠檬醛的氧化去除. 根据Jüttner等的实验[12],β-环柠檬醛在富营养化水体中主要是由微囊藻的细胞分裂过程中产生的胡萝卜素(β - carotene)氧化分解产生的,可引起水体产生草木异嗅味. 张可佳、高乃云等[13]进行了高锰酸钾氧化去除水中β-环柠檬醛的研究,并建立了相关的动力学模型,结果表明高锰酸钾氧化β-环柠檬醛的效果良好,在氧化后30 min内去除率达到90%,并且高锰酸钾与β-环柠檬醛的反应符合二级动力学反应,二级动力学常数为107.2 L−1·mol−1·s−1. 刘禧文[14]等研究发现高锰酸钾对1-辛烯-3-醇、β-环柠檬醛和2,4,6-三氯苯甲醚这3种嗅味物质均有一定去除效果,去除率在40%—55%. 饮用水处理中关于常见氧化剂对其他醛类嗅味物质去除效能的报道并不多见,值得进一步深入研究.
本研究以呼和浩特市JH饮用水厂检出频率和浓度均较高的5种醛类物质——反,反-2,4-庚二烯醛(tt24hept)、反-2-辛烯醛(t2oa)、反,反-2,4-辛二烯醛(tt24oda)、反,反-2,4-癸二烯醛(tt24dda)和 β-环柠檬醛(β-cyclo)为研究对象,选择实际水厂运用较多的高锰酸钾为氧化剂,从去除率、氧化时间等方面对氧化效果进行评价;同时开展高锰酸钾氧化各醛类嗅味物质的反应动力学研究,利用理论反应方程拟合动力学反应过程,得到理论速率常数,继而通过JH水厂原水加标实验验证高锰酸钾氧化醛类嗅味物质的效果与过程,希望能够为实际生产提供指导和经验支持.
-
实验所用试剂:反,反-2,4-庚二烯醛、反,反- 2,4 -辛二烯醛、反,反-2,4-癸二烯醛购自德国CNW公司,β-环柠檬醛、反-2-辛烯醛购自英国Alfa Aesar公司,均为色谱纯,结构式、嗅阈值等信息见表1;甲醇(CH3OH)购自Thermo Fisher Scientific公司,色谱纯;高锰酸钾(KMnO4)、五水合硫代硫酸钠(Na2S2O3·5H2O)、氯化钠(NaCl)购自北京化工厂,均为分析纯;磷酸二氢钠(NaH2PO4)购自国药集团化学试剂有限公司,分析纯;进厂原水取自呼和浩特市JH自来水厂,其主要水质指标测定如下DOC:2—3 mg·L−1,CODMn:3—4 mg·L−1,pH:6.9—7.4.
耗材与仪器:气相色谱-质谱联用仪购自日本岛津公司,型号GCMS-QP 2010 Plus;自动进样器购自德国Gerstel公司,型号MPS 2;50/30 μm DVB/CAR/PDMS SPME萃取头购自美国Supelco公司,型号SAAB-57329U;磁力搅拌器购自金坛市荣华仪器制造有限公司,78-1型;超纯水制备仪器购自法国Milli-Q公司,型号Integral 5 purification system.
-
嗅味物质初始浓度在表1中给出. 考虑到对饮用水嗅味事件的有效控制,所研究目标嗅味物质的初始浓度设定在各自嗅阈值的5 ~ 20倍范围内,同时可以保证被分析仪器稳定定量检测. tt24hept、t2oa和β-cyclo均在设定范围内. 但是tt24oda和tt24dda初始浓度为各自嗅阈值20倍时仍无法被定量检测,所以对这两种物质的初始浓度有所提高.
氧化实验:准备若干顶空瓶,称量2 g NaCl(450 ℃烘2 h)并加入1 mL 0.1 mol·L−1反应终止剂硫代硫酸钠溶液(Na2S2O3·5H2O, aq),待用. 在含有纯水的250 mL磨口锥形瓶中加入设定初始浓度(见表1)的嗅味物质,置于磁力搅拌器搅拌10 min,移取4 mL样品于顶空瓶中,立即用带有PTFE涂层的硅胶橡胶垫的瓶盖密封,待测,此样品为0 min时的样品. 然后加入2 mg·L−1氧化剂高锰酸钾(KMnO4),开始计时,分别在30、60 、120 min取样待测(个别物质在15 min内也取样). 取样完成后,采用顶空-固相微萃取(HS-SPME)进行萃取,并采用色谱-质谱联用仪(GC-MS)测定各样品的嗅味物质的浓度. 氧化实验结束后,根据各目标物的氧化效果,开展氧化动力学实验.
动力学实验:方法与氧化实验的过程相同,但取样时间均设置在反应开始后10 min以内.
氧化机理探讨实验:在5个含有纯水的250 mL磨口锥形瓶中分别加入10 μg·L−1的各嗅味物质和过量反应终止剂Na2S2O3·5H2O(aq),然后加入1 mg·L−1 KMnO4,搅拌2 h后取样分析.
实验设2组平行,均在20 ℃,pH=7(磷酸缓冲溶液调节)的条件下进行.
-
HS-SPME条件为:温度65 ℃,加热3 min,萃取30 min,解吸附3 min.
气相色谱(GC)条件:载气为高纯氦气(纯度大于99.999%),压强为50.1 kPa;流量控制方式为线速度为36.2 cm·s−1;总流量为21.1 mL·min−1;柱流量为1.01 mL·min−1;不分流进样;柱初始温度为40 ℃,保持3 min,以8 ℃·min−1升温至120 ℃,以5 ℃·min−1升温至130 ℃,再以15 ℃·min−1升温至250 ℃,保持5 min;进样口温度为240 ℃.
质谱(MS)条件:电子轰击源(EI);电子能量为70 eV;离子源温度为230 ℃;接口温度为230 ℃. 使用全扫描模式(SCAN)定性,再选择合适的特征离子,采用扫描离子模式(SIM)定量. SIM模式时5种醛类嗅味物质特征离子及保留时间见表2. 目标嗅味物质采用外标法进行定量.
-
将2 mg·L−1的KMnO4与各醛类嗅味物质反应2 h后,氧化效果如图1(a)、(b)所示,分别展示了KMnO4氧化各不同醛类嗅味物质的氧化趋势和去除率. 实验设置了空白组,从图1(a)空白组结果可以看出,反应2 h内5种醛类嗅味物质没有挥发损失. KMnO4与t2oa和tt24oda的反应中,还测定了1、3、5 min时的t2oa的浓度以及4,8,15 min时tt24oda的浓度,由图1(a)可知,反应10 min内,t2oa和tt24oda被氧化50%以上,浓度下降趋势明显,去除效果佳;在30 min后4种烯醛类嗅味物质浓度逐渐趋向平衡,而环状醛 β-cyclo浓度在60 min时趋近平衡. 图1(b)表示反应平衡后,KMnO4对5种醛类嗅味物质的氧化去除率均>75%,其中t2oa、tt24dda和 β-cyclo的去除率均>90%.
综上,KMnO4可以在短时间内去除75%以上这5种醛类嗅味物质,因此,KMnO4可以用于应对由此类醛类嗅味物质引发的突发嗅味问题.
-
由于KMnO4对5种醛类嗅味物质均具有高去除率,本研究继而开展了氧化动力学研究. 这里以KMnO4氧化tt24hept为例,详细介绍氧化动力学的计算过程. KMnO4与大多数有机物的反应为伪二级反应[13][15-16],所以先假设KMnO4氧化tt24hept的反应为伪二级反应,则其过程可以用式(1)表示.
式中,
k 为反应速率常数,单位为L⋅mol−1⋅min−1 ;[KMnO4] 为反应体系中KMnO4的浓度,单位为mol·L−1;C 为反应体系中tt24hept的浓度,单位为mol·L−1.为使浓度成为反应速率方程的唯一变量,使其中一种反应物的浓度远远大于另一反应物的浓度. 实验过程中KMnO4的初始浓度远高于目标嗅味物质浓度(≥8倍),因此,KMnO4的浓度可视为常数,令
kobs 为KMnO4与醛类嗅味物质反应的伪一级反应速率常数,单位为min−1 . 把式(2)带入式(1),作移项变形后等式两边求定积分,运算得式(3):式中,t表示反应开始后进行到t时刻,单位为s;
C(t) 表示在t时刻tt24hept的浓度,单位为mol·L−1;C0 表示tt24hept的初始浓度,单位为mol·L−1.由式(3)可知,只要测定出对应时刻t对应的tt24hept浓度,即可通过线性回归求得
kobs 的值.图2展示了0.5—2 mg·L−1 KMnO4浓度下氧化tt24hept的伪一级拟合,具体参数如表3所示. 在KMnO4过量的情况下,反应体系内剩余tt24hept浓度与初始浓度比值的自然对数和其对应时刻呈现良好的线性相关关系,说明高锰酸钾氧化tt24hept的反应对tt24hept是一级反应.
根据表3,以
kobs 为纵坐标,[KMnO4] 为横坐标关于式(2)进行拟合,结果如图3所示. 从图3看出KMnO4浓度与伪一级反应速率常数存在良好的线性关系,故高锰酸钾氧化tt24hept的反应对高锰酸钾的反应级数也是一级. 由此,此氧化反应整体是二级反应,二级反应速率常数k=5.248×104 L·mol−1·min−1.根据上述计算,同理可得KMnO4氧化t2oa、tt24oda、tt24dda、β-cyclo的伪二级反应动力学常数(见表4). 由表4可知烯醛类的嗅味物质反应速率常数比β-cyclo高一个数量级,3种二烯醛(tt24hept,tt24oda,tt24dda)反应速率常数均高于一烯醛(t2oa).
-
KMnO4对中性天然水源水中各种有机物氧化去除效果均很好,无论是低分子量、低沸点还是高分子量、高沸点有机污染物,剩余的有机污染物浓度很低[17]. KMnO4可以通过直接进行氧原子的转移而与碳碳双键(C=C)反应[18-19],很好地去除醇、醛、酚等有机污染物和致突变物质[20-21]. 本实验研究的对象为烯醛类和环醛嗅味物质,含有双键、环等不饱和键,易被KMnO4迅速氧化,且可以达到很高的去除率,如t2oa和tt24oda在10 min内被氧化去除50%. 氧化动力学实验中,通过对实验数据进行回归分析并结合研究对象中的4个烯醛类物质的结构与反应速率常数,可以发现:tt24oda比t2oa多1个碳碳双键,其反应速率常数也比t2oa的大,碳碳双键是1个化学性质活泼的原子团,由此醛类物质结构中含碳碳双键数目越多,KMnO4与它的反应速率常数就越大;tt24hept比tt24oda少1个亚甲基(—CH2—),其反应速率常数比tt24oda大,由于亚甲基是一个化学性质稳定的原子团,因此醛类物质结构中含亚甲基数目越少,KMnO4与它的反应速率常数就越大. 在判断其它烯醛类嗅味物质的氧化可处理性时,可以利用此规律进行初步估算.
其次,在pH中性条件下,KMnO4能被水中的还原性物质还原成新生态二氧化锰(MnO2)(式4):
根据MnO2的性质推测其在高锰酸钾氧化醛类嗅味物质中的作用机制:一是生成的新生态MnO2具有自催化作用,可很好的催化高锰酸钾氧化过程;二是MnO2具有较大的比表面积,可有效地吸附水中的有机物[22-23]. 以往的研究也可以证明这一推测的合理性,庞素艳等[24]发现KMnO4氧化降解酚类化合物的过程中存在着明显的自催化现象,并推测有机物吸附在MnO2表面形成络合物,比存在于溶液中更易被高锰酸钾氧化,是一种表面吸附络合催化作用.
本研究中为了验证MnO2在氧化中是否产生一定协同作用,提前在反应体系加入了过量的硫代硫酸钠,投加1 mg·L−1 KMnO4后,高锰酸钾被迅速充分转化为新生态MnO2. 如图4所示,新生态MnO2反应2h后5种醛类物质的去除率为13.73% — 37.66%,且二烯醛tt24hept、tt24oda、tt24dda的去除率明显高于其他物质,这表示在反应过程中除了KMnO4氧化去除,MnO2也起到了一定催化或吸附作用. 更详细的协同作用机制仍有待深入探讨和剖析.
-
综上,实验得到KMnO4氧化五种醛类嗅味物质的氧化动力学常数,因此可以根据动力学指导生产,将式(3)变形可得式(5),即可求得不同浓度下KMnO4氧化醛类嗅味物质的理论动力学反应方程:
以tt24hept为例,KMnO4投加量为2 mg·L−1时,其氧化tt24hept的方程式如下(式6),并可根据此方程做出理论反应曲线.
本研究选取呼和浩特市JH饮用水厂进厂原水加标实验考察理论反应曲线是否适用于水厂条件下的氧化反应. 如图5,反应开始5 min时,tt24hept仅剩余4.4%,整个氧化反应在前5 min内已基本完成. 除0时刻外,各时刻的tt24hept浓度比均高于经验反应曲线上相同时刻对应的tt24hept浓度比,且随着反应时间的增加,此差距越来越小. 这说明原水中有其它还原剂(如腐殖酸和氨氮等有机物)存在与tt24hept竞争,而后随着其它还原剂的氧化减少,KMnO4氧化tt24hept的反应受其影响减小,实际反应曲线表现为越来越接近理论反应曲线. 因此理论反应方程可以模拟原水在实际反应中tt24hept的浓度比随时间的变化,进而指导实践. 在后续的实验中发现原水条件下,理论方程对tt24oda、tt24dda和β-cyclo等3种醛类特征嗅味物质实际氧化过程也有很好的拟合度.
对于可用理论反应方程模拟KMnO4氧化醛类特征嗅味物质的过程,根据式(5),欲使水中嗅味物质降至嗅阈值(或有关标准)OTC以下,则有
所以
由不等式(7)可知,氧化剂的投加量
[KMnO4] 与氧化时间t 和进厂原水中嗅味物质的浓度[C]0 有关. 根据水厂条件确定氧化时间t 和进厂原水中的[C]0 ,即可得到水厂面临相应问题时的适宜投加量. 当饮用水中存在多种嗅味物质时,应当先找出最适合的氧化剂(能有效去除最多种类的嗅味物质),再分别计算出去除对应嗅味物质的投加量,然后取最大值,得到适宜投加量.由式(7)可得到KMnO4关于不同反应时间和氧化剂投加量对有关醛类特征嗅味物质的去除率. 以本研究所设定的各嗅味物质初始浓度为例,得到使各嗅味物质浓度降至嗅阈值以下所使用的最低KMnO4投加量及对应的反应时间,见表5.
-
本研究开展了高锰酸钾氧化5种醛类嗅味物质的研究,得到如下结论:
(1) KMnO4对反,反-2,4-庚二烯醛(tt24hept)、反-2-辛烯醛(t2oa)、反,反-2,4-辛二烯醛(tt24oda)、反,反-2,4-癸二烯醛(tt24dda)和β-环柠檬醛(β-cyclo)这5种醛类特征嗅味物质均有较好的氧化效果:反应30 min左右,氧化去除率均>75%.
(2)通过回归分析发现,在pH=7,20℃的条件下,KMnO4氧化与上述5种醛类特征嗅味物质的反应均符合伪二级动力学模型,并通过计算得到KMnO4与5种醛类嗅味物质反应的伪二级动力学常数:5.25×104、2.66×104、4.50×104、2.71×104 、5.37×103 L·mol−1·min−1.
(3)KMnO4氧化过程中会生成新生态MnO2,对醛类嗅味有机物也有一定的去除效果,去除率在13.73%— 37.66%.
(4)理论反应方程可以很好地模拟原水条件下,KMnO4分别氧化tt24hept、tt24oda、tt24dda和 β-cyclo 这4种醛类特征嗅味物质的反应过程. 而对于KMnO4氧化t2oa的反应,理论方程的拟合程度不高,需要进一步研究影响反应的因素,才能更好的指导生产. 同时可以根据研究结果,在确定氧化时间t和原水中嗅味物质初始浓度
C0 的情况下,即可得到控制目标嗅味物质达到嗅阈值以下的适宜高锰酸钾投加量.
高锰酸钾氧化饮用水中醛类嗅味物质的效果及动力学研究
Study on the effect and kinetics of aldehydes oxidation by potassium permanganate in drinking water
-
摘要: 我国北方呼和浩特市以黄河为水源的JH饮用水厂近年来冬季经常有醛类嗅味物质检出,常规处理工艺如混凝、沉淀等对其去除效果有限,需要对其进行其它处理工艺的探究. 本文选择高锰酸钾对水厂检出频率和浓度均较高的反,反-2,4-庚二烯醛(tt24hept)、反-2-辛烯醛(t2oa)、反,反-2,4-辛二烯醛(tt24oda)、反,反-2,4-癸二烯醛(tt24dda)和β-环柠檬醛(β-cyclo)5种醛类嗅味物质进行氧化控制研究,探究其去除效果、氧化动力学和氧化机理. 结果表明,20 ℃,pH=7时,2 mg·L-1高锰酸钾氧化5种醛类嗅味物质30 min后,去除率达75%以上. 根据动力学分析可知,高锰酸钾氧化5种醛类嗅味物质属于伪二级动力学过程,其伪二级反应速率常数分别为5.25×104、2.66×104、4.50×104、2.71×104、5.37×103 L·mol−1·min-1,醛类嗅味物质结构中含碳碳双键数目越多、含亚甲基数目越少,反应速率常数越大. 同时,氧化过程会产生新生态二氧化锰,促进高锰酸钾对嗅味物质的控制效果. 最后,通过水厂原水加标实验效果验证,理论反应方程可为饮用水厂应对醛类物质嗅味问题提供相应的理论依据并指导生产.Abstract: Aldehyde odor substances have always been detected in recent years in winter for JH drinking water treatment plant in Hohhot city of northern China, which with the resource water from Yellow River. Conventional treatment processes such as coagulation, sedimentation and other effects on its removal are limited to control the aldehyde odorants, other treatment processes need to explore. In this paper, potassium permanganate was selected for the oxidation of five aldehyde odorants, trans,trans-2,4-heptadienal (tt24hept), trans-2-octenal (t2oa), trans,trans-2,4-octadienal (tt24oda), trans,trans-2,4-decadienal (tt24dda) and β-cyclocitral (β-cyclo), which were detected with high frequency and concentration in JH plants. The oxidation removal, kinetics and mechanism were investigated. The results showed that at 20 °C and pH=7, the removal of the five aldehyde odorants was over 75% after 30 min of oxidation by 2 mg·L−1 potassium permanganate. According to the kinetic analysis, the oxidation of the five aldehyde odorants by potassium permanganate belongs to the pseudo-second-order dynamic process, and its pseudo-second-order rate constants were 5.25×104, 2.66×104, 4.50×104, 2.71×104 and 5.37×103 L·mol-1·min−1, respectively. The higher number of carbon-carbon double bond and the lower amount of methylene presented in the structure of an aldehyde odor substance cause the higher reaction rate constant. At the same time, the oxidation process will produce new in-site manganese dioxide, which promoting the oxidation effect of potassium permanganate on the aldehyde odorants. Finally, through the spiked recovery experiments by the raw water of JH plant, it was verified that the theoretical reaction equation could provide the corresponding theoretical basis, and guide the production of the problem of aldehydes in drinking water treatment plants.
-
Key words:
- potassium permanganate /
- aldehyde odorants /
- oxidation kinetics /
- manganese dioxide.
-
电化学厌氧消化(electrochemical anaerobic digestion,EAD)是一种利用微生物电解池(microbial electrolysis cell,MEC)的耦合厌氧消化(anaerobic digestion,AD)的新技术,其原理是利用MEC的制氢功能,对系统内的二氧化碳(CO2)进行固定和转化,生成更多的CH4,从而减少CO2的排放[1],并极大地提高了CH4的转化率和沼气的品质[2],所以,EAD作为一种新型的沼气制备技术,因其具有甲烷转化率高、能量回收率高、能耗低、适用于多种有机废弃物和废水的降解等优势而受到了广泛的关注,是目前AD领域的最新发现[3-5].
但不管是AD,还是EAD,绝大多数相关研究仍然停留在提高消化能力或者副产物的产率等方面[6-8],很少涉及代谢过程的研究. 代谢通量分析(metabolic flux analysis,MFA)是一项研究胞内代谢状态和分析系统代谢能力的强有力技术,通过MFA:(1)可以确定细胞或者系统内存在的不同代谢途径,明确物质的流向和流量[8];(2)可以计算胞内的中间产物的通量,对未知途径加以辨别[9];(3)可以识别代谢节点的刚柔性[10],和对环境扰动的影响做出评价,从而对代谢途径中具有特殊地位的途径实施有效调控[11](如优化目标产物代谢途径、计算最大理论转化率等[12]). 因此,本文将利用MFA中的通过通量平衡分析(flux balance analysis,FBA)的方法,构建EAD代谢网络,研究EAD代谢网络中的相关代谢途径的通量分布信息.
在代谢通量分析中,通量是反映各代谢产物在整个代谢网络中参与程度的重要参数[7],可以依据通量的大小评价各代谢途径在整个代谢网络中所发挥的作用[13]. 然而由于EAD代谢网络中的相关代谢途径,往往是在相应的酶催化下进行的,在EAD代谢途径中,乙酸的形成和转化是EAD产甲烷过程中最为关键的节点. 乙酸的形成主要的途径是乙酰辅酶A通过磷酸转乙酰酶(PTA)转化为乙酰基-P,然后再通过乙酸激酶(AK)从乙酰基-P转化为乙酰基-P,AK催化反应产生ATP[14]. 厌氧消化EMP途径所产生的丙酮酸转化为乙醛和二氧化碳,然后乙醛通过ADH还原成乙醇[15]. 丁酸的产生则与磷酸转丁酰酶(PTB)与丁酸激酶相关. 此外,ADP/ATP转运酶也发挥着重要的作用. ADP/ATP转运酶又被称为腺嘌呤核苷酸易位子或ADP/ATP载体蛋白,ADP/ATP转运酶通过ADP 与ATP相互转化产生能量从而为细胞提供能量[16]. 而在乙酸转化中,CoF420参与产甲烷菌中的氧化还原反应,充当电子载体. CoF430是甲基辅酶M还原酶的辅助因子,在甲烷生成的最后一步释放甲烷. 而辅酶M是古菌产甲烷菌代谢中甲基转移反应所需的辅酶.
综上所述,利用FBA不仅可以对EAD系统的代谢过程进行表征,还能对代谢通量进行分析和直观地了解代谢网络中不同代谢途径的产物生成情况以及代谢关键节点处的酶活性变化水平.
1. 实验部分(Experimental section)
1.1 实验装置
EAD实验装置由 电解电流及电压记录单元、 EAD反应单元和排水集气单元组成,EAD反应单元主要包含400 mL的发酵罐,其中电极距离为3.0 cm,一端的电极片完全地浸于发酵液中,另一端通过导线与宽屏无纸记录仪(SIN-R7000A,中国)相连;在EAD系统中,阳极为石墨电极,阴极为铂电极,参比电极为饱和的Ag/AgCl电极. EAD实验装置如图1所示.
 图 1 EAD实验装置Figure 1. EAD experimental setupa. 电解电流及电压记录单元,b. EAD反应单元,c. 排水集气单元a. electrolysis current and voltage recording unit, b. EAD reaction unit, c. drainage gas gathering unit.
图 1 EAD实验装置Figure 1. EAD experimental setupa. 电解电流及电压记录单元,b. EAD反应单元,c. 排水集气单元a. electrolysis current and voltage recording unit, b. EAD reaction unit, c. drainage gas gathering unit.1.2 接种物、缓冲营养液和微量元素液
厌氧活性污泥来自云南师范大学昆明实验室通过猪粪厌氧发酵. 经检测,pH值为8.07,TS为17.11%,VS为10.08%.
缓冲营养液配比:NaH2PO4∙2H2O(5.54 g·L−1)、Na2HPO4∙12H2O(23.09 g·L−1)、KCl(0.26 g·L−1)和NH4Cl(0.62 g·L−1).
微量元素液配比:Na2WO4∙2H2O(0.03 g·L−1)、NaCl(1 g·L−1)、FeCl2∙7H2O(0.07 g·L−1)、CuSO4∙5H2O(0.01 g·L−1)、NiCl2∙6H2O(0.02 g·L−1)、C6H9NO6(1.50 g·L−1)、MgSO4(3.00 g·L−1)、AlK(SO4)2∙12H2O(0.01 g·L−1)、ZnCl2(0.13 g·L−1)、CaCl2∙2H2O(0.10 g·L−1)、H3BO3(0.01 g·L−1)、MnCl2∙4H2O(0.60 g·L−1)、CoCl2∙6H2O(0.10 g·L−1)、Na2MoO4(0.03 g·L−1)和复合维生素(1 粒·L−1).
1.3 实验方法
EAD实验设置3个实验组,每组3个平行. 首先在每个实验组的发酵罐中添加:葡萄糖1.0 g,接种物120.0 g,缓冲营养液100.0 mL,微量元素液10.0 mL,再补加蒸馏水使物料的总质量为360.0 g;其次将密封好的发酵罐连接气体收集装置,再利用高纯氮排出体系内的溶解氧和空气(通气时间为3 min),使EAD体系处于厌氧状态;最后施加恒定电压1.0 V,进行培养.
培养阶段:将发酵罐置于恒温磁力搅拌器中,设置温度为30.0 ℃;每天监测pH值变化,待其恢复到7.0左右,立即添加等量的葡萄糖将体系培养至稳定.
扰动阶段:培养阶段结束后,测定发酵液中底物和代谢产物的浓度,同时记下该时间点为扰动的初始点,随后添加1.0 g的葡萄糖,施加扰动后,通过监测体系中底物的消耗量和代谢产物的生成量,然后进行代谢通量分析.
1.4 常规检测分析
日产气量:采用排水法法,每天定时记录产气量. TS和VS:在(105±5)℃下干燥并在(550±10)℃下马弗炉燃烧1 h后,测量污泥的. pH值:采用pH计(HAOSHI H-1008A,中国).
VFAs:样品经离心分离,取上清液,采用气相色谱仪(FULI, GC9790II,中国),以FID为检测器进行测量[17]. 色谱柱为KB- FFAP(30 m×0. 32 mm×0. 25 μm)毛细管柱.
气体含量:采用气相色谱仪(FULI, GC9790II,中国),以TCD 检测器对H2、CH4和CO2含量进行测量. 色谱柱为TDX-01填充柱. 葡萄糖含量:采用3,5-二硝基水杨酸比色法测定[18].
1.5 酶活测量与分析
酶活测量采用改良的双抗体夹心酶联免疫吸附测定(ELISA)方法[19-21]在测量过程中,以ADP/ATP转运酶、磷酸转乙酰酶(PTA)、乙酸激酶(AK)、磷酸转丁酰酶(PTB)、丁酸激酶(BK)、辅酶M(CoM)、甲基丙二酰CoA变位酶(MCM)、辅酶F420(F420)、辅酶F430(F430)为标准酶. 检测时在酶标包被板上设定空白孔、标准孔、待测样品孔. 空白孔不加样品及标准酶,作为对照,标准孔加标准酶,待测样品孔中先加酶试剂盒中的样品稀释液,然后再加待测样品. 按照酶试剂盒说明书进行操作,最后采用Epoch酶标仪在450 nm波长下进行测定,以空白孔调零依序测量各孔的吸光度(OD值),并通过标准曲线计算样品中酶的活性.
1.6 通量分析
通量分析采用通量平衡分析(FBA)的方法,采用代谢通量分析软件Cell Net Analyzer进行代谢网络的构建及分析. FBA是MFA中基于质量守恒的代谢过程研究常用的一种数学建模分析法[22],在拟稳态条件下,模型中代谢产物处于平衡状态,此时,可将代谢物的通量描述为一个线性方程,只需对底物和相关产物的浓度变化进行测量,再采用线性规划或者计量矩阵分析方法则可确定每种产物的代谢通量[23].
2. 结果与讨论 (Results and discussion)
2.1 EAD培养阶段的产气及含量分析
EAD反应过程中产气特性的变化是表征反应器效能的重要因素. 图2为培养过程中的产气变化情况,从图2可以发现,在反应器启动阶段,气体中以CO2为主,随着反应器的运行,产甲烷菌逐渐生长富集. 在稳定阶段,其甲烷含量最高可以达到55%左右,葡萄糖厌氧消化中理论产甲烷含量在50%左右,也就是说,MEC的引入明显提升了葡萄糖的理论产甲烷率,而且此时CO2的含量降低到11%左右,明显的降低了CO2的排放. 在反应器稳定后,日产气量也得到了明显的提升,从最初的50 mL·d−1左右增加到了200 mL·d−1左右. 对于氢气含量,除启动阶段有氢气产生外,反应中后期基本没有氢气产生,这可能是由于前期pH较低,适于产氢菌的生长,中后期pH值恢复较快,可以迅速恢复至已6.5以上,不利于产氢菌的生长,导致氢气含量极低. 此外,也可能是由于电化学辅助促进了H2还原CO2途径产甲烷,使得反应器中的H2迅速被噬氢产甲烷菌消耗,从而提高了CH4的含量,降低了H2的含量.
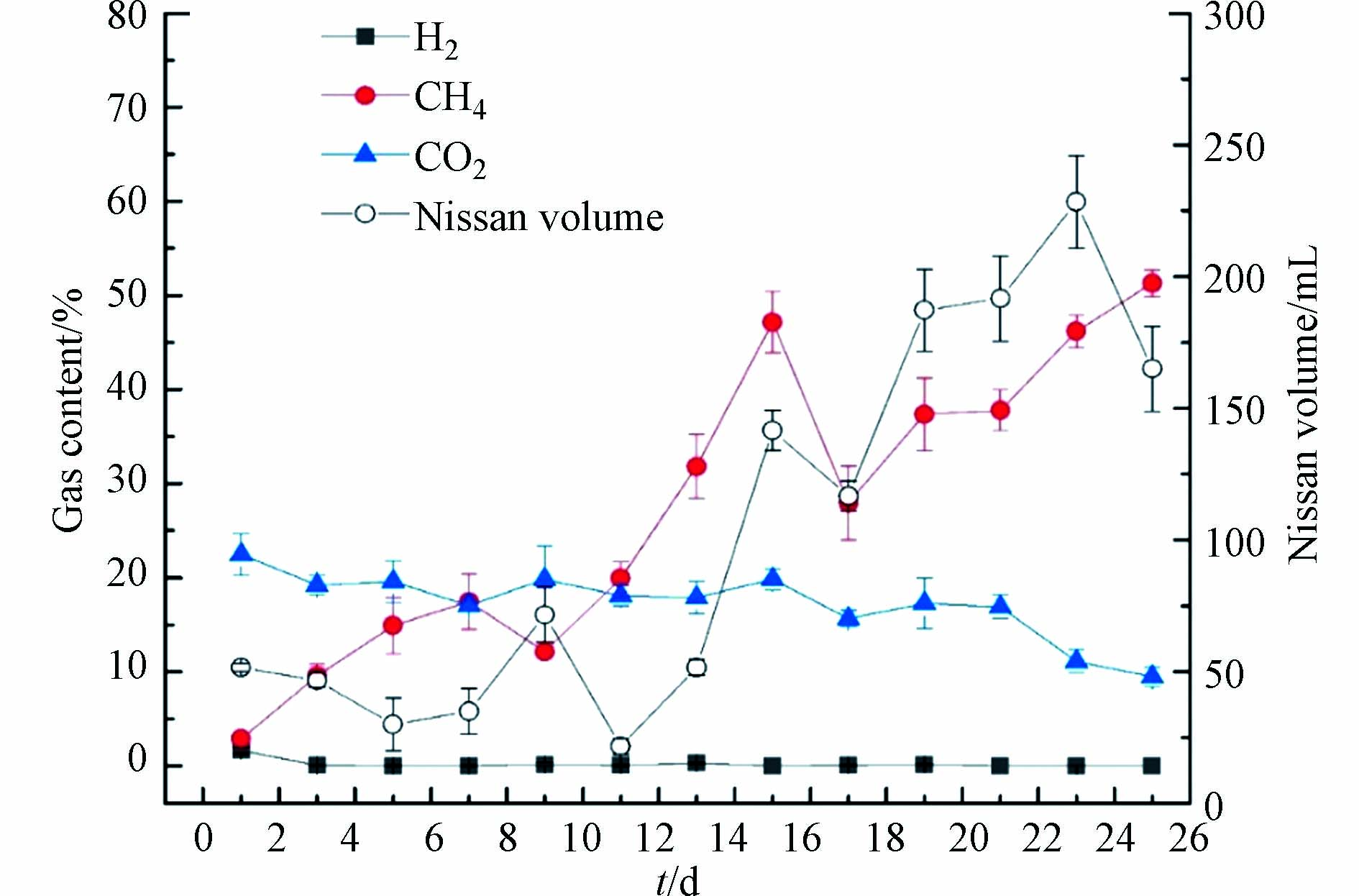 图 2 EAD培养阶段中产气体积及气体组分含量Figure 2. Gas production volume and gas component content in EAD operation stage
图 2 EAD培养阶段中产气体积及气体组分含量Figure 2. Gas production volume and gas component content in EAD operation stage2.2 EAD培养阶段的VFAs的分析
VFAs是厌氧消化中重要的中间产物,其产生和分解过程受多种因素影响,同时也关系到EAD产甲烷过程的稳定性. EAD体系中 VFAs的浓度一旦超过 200 mmol·L−1 就有可能发生酸抑制问题[24],并对产甲烷过程产生严重的影响,然而本次实验研究并未出现VFAs累积的现象. 图3显示了EAD培养过程中各短链有机酸的变化情况. 从图3可以看出,各短链有机酸的变化与底物补充时间相关,培养前期VFAs波动较大且浓度较高,但15 d后,因阳极上形成了电活性微生物膜,致使VFAs能够在阳极表面降解,尽管进料时间间隔缩短为3 d,也没有产生VFAs累积,表明EAD具有较强的抗酸化的能力.
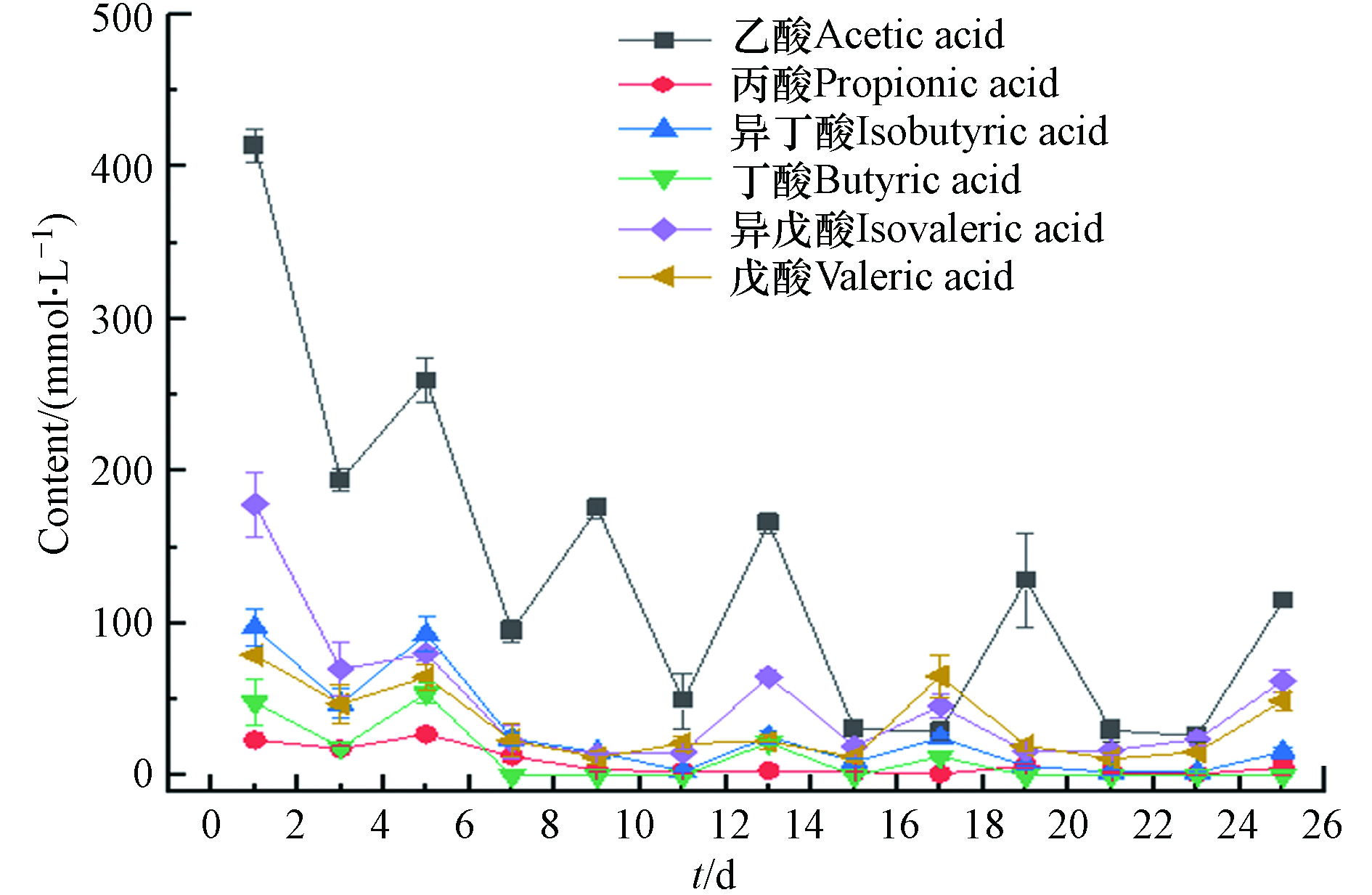 图 3 EAD运行阶段中VFAs的含量变化Figure 3. Changes in the content of VFAs during the EAD operation stage
图 3 EAD运行阶段中VFAs的含量变化Figure 3. Changes in the content of VFAs during the EAD operation stage在厌氧消化中丁酸累积是抑制厌氧消化的重要因素之一,由于丁酸的产氢产乙酸过程必须依赖氢还原CO2产甲烷过程,否则本身不能自发进行,一旦产氢产乙酸和产甲烷过程失衡,将会产生严重的丁酸抑制作用。然而在EAD中丁酸的降解除了依赖氢还原CO2产甲烷过程外,还可以在电活性微生物的作用下,在电极表面降解. 在这条途径中,丁酸分解为CO2、H+和电子, H+向阴极迁移在阴极上获得电子而形成H2,这部分H2同样被氢营养型产甲烷菌利用还原CO2,加强H2还原CO2产甲烷途径.
2.3 代谢网络构建
对于纯培养体系而言,由于体系内菌种单一,代谢产物和途径比较明确,构建与之相对应的代谢网络相对比较容易. 但对于复杂的混合培养而言,特别是像EAD这种由多种微生物构成的体系,变量较多,对于代谢网络的构建及分析难度较大[25],为了使代谢通量分析能适用于混合体系,提出了“统一微生物”这一个概念[26],同时国内外一些学者已经在部分混合体系中作了应用上的尝试,并且成功地建立了发酵制氢体系的代谢网络[27],还有在新型多级厌氧甲烷反应器中也进行了FBA的研究[28],但还需进一步地完善,才能使其适用于EAD的混合培养体系中. EAD是在AD中引入MEC,由于MEC的引入,使H+向阴极迁移,在阴极上生成H2,这在AD基础上增加了一条重要的产氢途径. 根据相关研究表明[9, 12],在严格的厌氧条件下,丙酮酸几乎不会流向三羧酸循环,甲酸也不是所有严格厌氧菌都能产生. 结合相关研究[29-33],构建了如图4所示的EAD代谢网络该网络包含了37个主要的代谢反应式,每一步的代谢的具体生化反应过程如表1所示.
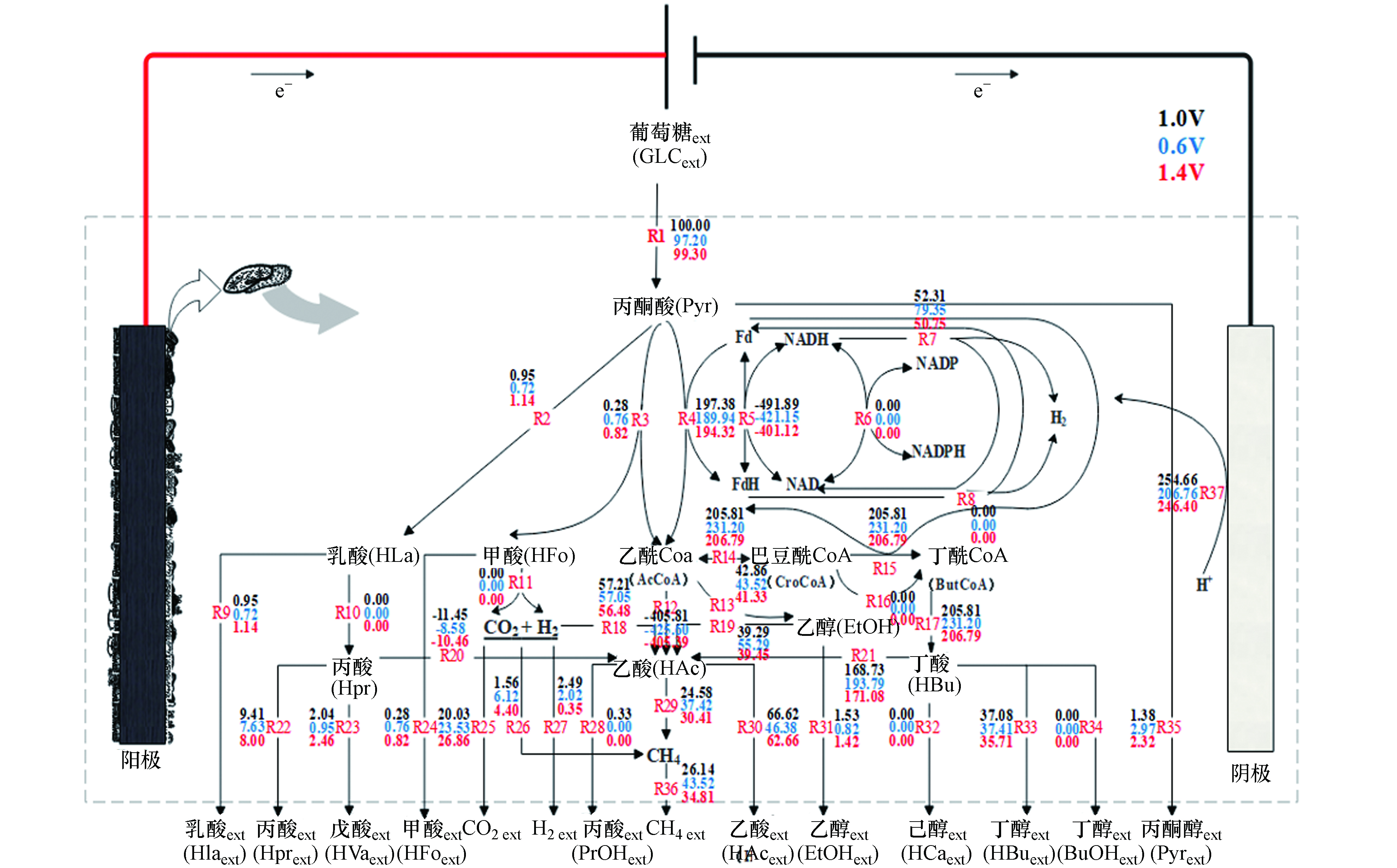 表 1 EAD体系葡萄糖代谢过程包含的主要生化反应Table 1. The main biochemical reactions included in the glucose metabolism process of the EAD system
表 1 EAD体系葡萄糖代谢过程包含的主要生化反应Table 1. The main biochemical reactions included in the glucose metabolism process of the EAD systemNO. 反应式 Equation NO. 反应式 Equation R1 GLC+PEP ∀ G6P + Pyr R20 HPr + 2 H2O ∀ HAc + CO2 +3 H2 R2 NADH + Pyr ∀ HLa + NAD R21 HBu + 2 H2O ∀ 2 HAc + 2 H2 R3 Pyr + NADH ∀ NAD + HFo +AcCoA R22 HPr = HPr (ext) R4 2 Fd + Pyr + CoA ∀ CO2 + AcCoA + 2 FdH R23 EtOH+ HPr ∀ Hva (ext) + H2O R5 2 Fd + NADH = 2 FdH + NAD R24 HFo = HFo (ext) R6 NADPH + NAD ⇌ NADH + NADP R25 CO2 = CO2 (ext) R7 2 NADH = H2 + 2 NAD R26 CO2 + 4 H2 = CH4 + 2 H2O R8 2 FdH = H2 + 2 Fd R27 H2 = H2 (ext) R9 HLa = HLa (ext) R28 2 AcCoA + H2 ∀ PrOH(ext) +2 CoA + CO2 R10 HLa + NADH ∀ HPr + NAD R29 HAc ∀ CO2 + CH4 R11 HFo ∀ CO2 + H2 R30 HAc = HAc (ext) R12 AcCoA + ADP + iP ∀ATP + HAc + CoA R31 EtOH = EtOH (ext) R13 AcCoA + 2 NADH ∀ EtOH + CoA + 2 NAD R32 HBu + EtOH ∀ HCa (ext) + H2O R14 2 AcCoA + NADH ⇌ CoA + H2O + CroCoA + NAD R33 HBu = HBu (ext) R15 CroCoA + 2 Fd + NADH ∀ ButCoA + 2 FdH + NAD R34 HBu + 2 NADH ∀ BuOH(ext) + CoA + 2 NAD R16 CroCoA + NADH ∀ ButCoA + NAD R35 Pyr ⇌ Pyr (ext) R17 ButCoA + ADP + iP ∀ HBu + ATP + CoA R36 CH4 = CH4 (ext) R18 2 CO2 + 4 H2 ∀ HAc +2 H2O R37 2 H+ + 2 e- = H2 R19 EtOH + 2H2O ∀ 4 H+ + HAc 注:iP为磷酸根离子,CoA为辅酶A,H+为氢离子,e-为转移电子或者电解协助装置提供的电子. Note: iP is the phosphate ion, CoA is the coenzyme A, H+ is the hydrogen ion, and e- is the electron provided by the transfer electron or electrolysis assistance device. | Show Table DownLoad:
CSV
DownLoad:
CSV
2.4 电压扰动对EAD产甲烷代谢通量的影响
在培养阶段中,当EAD运行稳定后,从取样口取样,检测残余葡萄糖和相关代谢物的含量,然后添加1 g的葡萄糖,以此时的状态为FBA的初始状态,实施电压扰动. 在对照组电压为1.0 V的基础上,扰动组1电压降低至0.6 V,而扰动组2增加至1.4 V. 14 h后,再次检测残余葡萄糖和相关代谢物,产气量和气体含量. 经过计算得到EAD扰动前后的发酵液中代谢产物的净生成速率,如表2所示.
表 2 EAD体系中关键代谢物的净生成速率①(mmol·L−1·h−1)Table 2. Net formation rate of key metabolites in EAD system①(mmol·L−1·h−1)代谢物Metabolites 对照组1.0 VControl group 1.0 V 扰动组0.6 VDisturbance group 0.6 V 扰动组1.4 VDisturbance group 1.4 V 葡萄糖② 1.1274 ± 0.0154 1.0958 ± 0.0055 1.1195 ± 0.0175 甲 酸 0.0032± 0.0000 0.0086 ± 0.0021 0.0093± 0.0026 乙 酸 0.7511 ± 0.1440 0.5229 ± 0.0770 0.7064 ± 0.1202 丙 酸 0.1061 ± 0.0096 0.0860 ± 0.0049 0.0902 ± 0.0023 丁 酸③ 0.4180± 0.0335 0.4218± 0.0391 0.4026 ± 0.0201 乳 酸 0.0107 ± 0.0046 0.0081 ± 0.0035 0.0128 ± 0.0056 丙酮酸 0.0156 ± 0.0037 0.0335± 0.0074 0.0261 ± 0.0171 戊 酸④ 0.0230 ± 0.0107 0.0107 ± 0.0046 0.0277 ± 0.0093 己 酸 0.0000 ± 0.0000 0.0000 ± 0.0000 0.0000 ± 0.0000 乙 醇 0.0173 ± 0.0014 0.0092 ± 0.0019 0.0160 ± 0.0067 丙 醇 0.0000 ± 0.0000 0.0000 ± 0.0000 0.0000 ± 0.0000 丁 醇 0.0000 ± 0.0000 0.0000 ± 0.0000 0.0000 ± 0.0000 CO2 0.2258 ± 0.0229 0.2255 ± 0.0376 0.2716 ± 0.0071 CH4 0.2947 ± 0.0127 0.4909 ± 0.0515 0.3924 ± 0.0293 H2 0.0281 ± 0.0231 0.0228 ± 0.0137 0.0039 ± 0.0024 注:表2中的实验数据①为电压扰动实验组的主要代谢物的净生成速率;葡萄糖②的测量值代表底物的净摄取速率;丁酸③为正丁酸和异丁酸的总和,戊酸④为正戊酸和异戊酸的总和. Note: The experimental data① in Table 2 are the net production rates of major metabolites in the voltage disturbance group; The measured value of glucose② represents the net uptake rate of substrate; Butyrate③ is the sum of n-butyrate and isobutyrate, valerate④ is the sum of n-valerate and isovalerate. | Show TableDownLoad:
CSV
根据表2中的净生成速率,采用运行Cell Net Analyzer软件,获得EAD代谢通量分布信息,如图4所示,并以H2、CO2、HAc和CH4代谢物进行评价.
在0.6 V扰动下,NADH通量明显优于1.4 V电压扰动,且此条件下最终产物甲烷通量达到43.52,明显高于1.0 V和1.4 V下的26.14和34.81. 由图4可知,H2的代谢途径主要形成途径为:R7、R20、R21和R37. 由通量值可知,EAD中H2主要来源于电极反应R37,和丁酸降解为乙酸的途径R21,其次是NADH平衡调节产氢R7. CH4为最终的目标代谢物,主要由H2还原CO2途径R26和乙酸转化途径R29两条途径形成. EAD体系中CH4的合成途径是以乙酸转化途径占主导地位,其次为H2还原CO2途径,1.0 V下EAD体系的这两条途径的占CH4产生总量的64.95%和35.05%. 0.6 V和1.4 V扰动下,通过H2还原CO2产生的产甲烷(R26)分别占13.44%和12.76%,相比对照组5.97%均有所提高,表明电压扰动使得EAD产甲烷代谢途径偏向了氢营养型产甲烷. 值得注意的EAD产甲烷系统明显产生了甲酸,但根据代谢通量,CH4和H2并无一来源于甲酸,说明EAD反应体系中可能没有噬甲酸型产甲烷菌存在. 最终产物H2大多由EAD中的阴极产生,但在最终产物中,氢气通量明显下降,说明H2还原为CH4(R26)的途径较为活跃. 本次从发酵液代谢物中未检测到己酸、丙醇和己酸盐. 乙酸的来源有R12、R18、R19、R20和R21途径. 通量值表明乙酰辅酶A转化是乙酸最主要的来源,其次是丁酸转化、H2还原CO2、乙醇转化和丙酸转化. 其通量大小受多个中间产物的影响,但在电压扰动(0.6 V和1.4 V)下,其通量明显增大,其中以0.6 V扰动增幅最大,说明反应器中的产酸微生物在此时相对较活跃. 乳酸和乙醇的产生不利于H2的生产,图中0.6 V扰动下的乳酸通量较高,这也可能是导致了H2较低的原因之一.
2.5 酶活水平变化
表3为葡萄糖代谢过程中关键酶活性的变化. 其中ADP/ATP转运酶的活性,0.6 V和1.4 V扰动时的酶活性均高于对照组1.0 V 的239.28 IU·L−1. ADP/ATP是能量代谢的载体,为微生物生长和繁殖提供能量基础. 当电压扰动时,需要大量能量来使微生物群落发生转变,因此ADP/ATP转运酶酶活性增加. 在乙酸代谢途径中首要的是乙酸的活化,它需要由乙酸激酶AK和磷酸转乙酰酶PTA的先后催化来进行. 在ATP参与下由乙酸激酶AK催化,乙酸被磷酸化为乙酰磷酸,然后乙酰磷酸再在磷酸转乙酰酶PTA催化下生成乙酰辅酶A,进行乙酸代谢[34-35]. 但从图5中可以看出,所产生的乙酸大部分都R12途径逆反应生成乙酰辅酶A,其中当进行0.6 V扰动时,R12逆反应通量最大,这与在0.6 V时PTA活性达到115.69 U·L−1检测结果相吻合. 当进行电压扰动时,EAD体系内在乙酸代谢中PTA和AK的酶活调节现象相似,有研究表明[36]当谷氨酸棒杆菌生长在以乙酸(取代葡萄糖)为碳源的培养基上时,PTA和AK酶活性明显提高,该现象是微生物碳代谢中葡萄糖效应的具体体现,表明酶在基因表达环节上可能存在葡萄糖基质上的负调控效应或乙酸基质上的正调控效应. 降低电压,PTA与AK酶活性增加. PTB和BK酶与丁酸产生有关,由图5代谢通量可知,丁酸的产生是从乙酰基通过PTB与BK的共同催化所形成. 在丁酸代谢中,PTB催化反应产生大量的丁酰辅酶A,但产生的丁酸大部分却通过R21转化为乙酸. 在0.6 V扰动下,PTB的酶活性352.19 U·L−1,明显高于对照组310.19 U·L−1和1.4 V的338.04 U·L−1,但BK酶活性,对照组1.0 V的酶活性52.59 U·L−1,要略高于0.6 V的50.92 U·L−1. PTB酶活性高于BK,这是由于PTB需要先将丁酰辅酶A转化为在丁酰磷酸,最终再由BK催生与ADP反应形成丁酸和ATP. 而BK在1.0 V与0.6 V酶活性差异不大,这是可能是由于电压BK影响不大. 辅酶M在甲烷形成的最终反应步骤中充当甲基的载体. CoM的化学结构虽然很简单,但在甲基还原酶的反应体系中却有高度的专一性,因此在进行电压扰动时,辅酶M的酶活性变化差异不大. 值得一提是,MCM酶活性变化与CoM酶活性变化相似. 在丙酮酸转化为丙酸代谢过程中,关键酶为MCM,在电压扰动下,酶活性变化差异较小. 结合图6中的产丙酸代谢通量图可以看出,丙酸通量变化也差异较小,说明酶活性与所构建的代谢通量有较好的相关性. CoF420在产甲烷途径中电子载体,而CoF430产甲烷古菌形成甲烷最后步骤作为甲基载体的四吡咯化合物,因此产甲烷的总通量应当取决于CoF420与CoF430共同酶活性,在0.6 V电压扰动下,总酶活性为63.01 IU·L−1,对照组为56.94 IU·L−1,1.4 V扰动为59.57 IU·L−1,这与图5中产甲烷代谢通量中,0.6 V时产甲烷最多,1.4 V次之,对照组最少结果相吻合.
表 3 代谢途径中关键酶活性水平的变化Table 3. Changes in activity levels of key enzymes in metabolic pathways酶名称Enzyme 对照组1.0 VControl group 1.0 V 扰动组0.6 VDisturbance group 0.6 V 扰动组1.4 VDisturbance group 1.4 V 单位Unit ADP/ATP转运酶 239.28±23.54 337.04±7.88 344.23±7.54 IU·L−1 磷酸转乙酰酶(PTA) 99.78±6.39 115.69±11.32 97.69±7.50 U·L−1 乙酸激酶(AK) 106.26±24.23 151.38±28.81 120.25±28.93 U·L−1 磷酸转丁酰酶(PTB) 310.19±1.29 352.19±12.66 338.04±22.08 U·L−1 丁酸激酶(BK) 52.59±6.63 50.92±0.90 36.08±4.89 U·L−1 辅酶M(CoM) 173.84±17.97 191.01±23.53 176.85±12.82 U·L−1 甲基丙二酰CoA变位酶(MCM) 58.17±4.06 51.71±2.71 54.20±3.09 U·L−1 辅酶F420(CoF420) 40.38±2.22 43.72±4.37 36.86±6.48 IU·L−1 辅酶F430(CoF430) 16.56±2.05 19.29±1.55 22.71±2.14 IU·L−1 | Show TableDownLoad:
CSV
3. 结论(Conclusion)
本研究通过外加电压对EAD体系扰动过程中代谢通量的影响,对EAD代谢网络通量上的变化结合关键酶活性的变化,进一步分析电压扰动对EAD系统代谢产物生成上的提高或者降低提供理论依据,由通量分析结果可知,EAD体系中并没有很高的H2生成,相反,EAD体系中产甲烷能力得到提高. 原因可能是由于电解协助装置的引入,极大地促进H2的产生,随后,所产生的H2被用来还原CO2产乙酸和产甲烷的代谢过程,进而提升了EAD系统的产甲烷能力. 结合关键节点处酶活性变化,酶活性与代谢通量有着较好的相关性. 在进行电压扰动时,ADP/ATP转运酶、PTA与AK、PTB与BK,这些酶活性均随电压变化而产生较大的差异. 而CoM与MCM酶活性,具有高度专一性的酶,随电压扰动变化不大. 在0.6 V电压扰动下,CoF420与CoF430酶活性与产甲烷代谢通量变化相吻合.
-

图 1 KMnO4氧化5种醛类嗅味物质的趋势图(a)和2 h去除率(b)(嗅味物质初始浓度:tt24hept 100 μg·L−1,t2oa 50 μg·L−1,tt24oda 250 μg·L−1,tt24dda 50 μg·L−1,β-cyclo 20 μg·L−1;KMnO4浓度:2 mg·L−1)
Figure 1. Oxidation trend diagram (a) and 2 h removal rate (b) of 5 aldehydes odorous substances oxidated by KMnO4 (Initial concentration of odorous substances:tt24hept 100 μg·L−1,t2oa 50 μg·L−1,tt24oda 250 μg·L−1, tt24dda 50 μg·L−1, β-cyclo 20 μg·L−1;KMnO4:2 mg·L−1)

图 2 各KMnO4浓度下氧化tt24hept的伪一级动力学回归曲线
Figure 2. Pseudo-first-order kinetic regression curves of tt24hept oxidation by different dosage of KMnO4
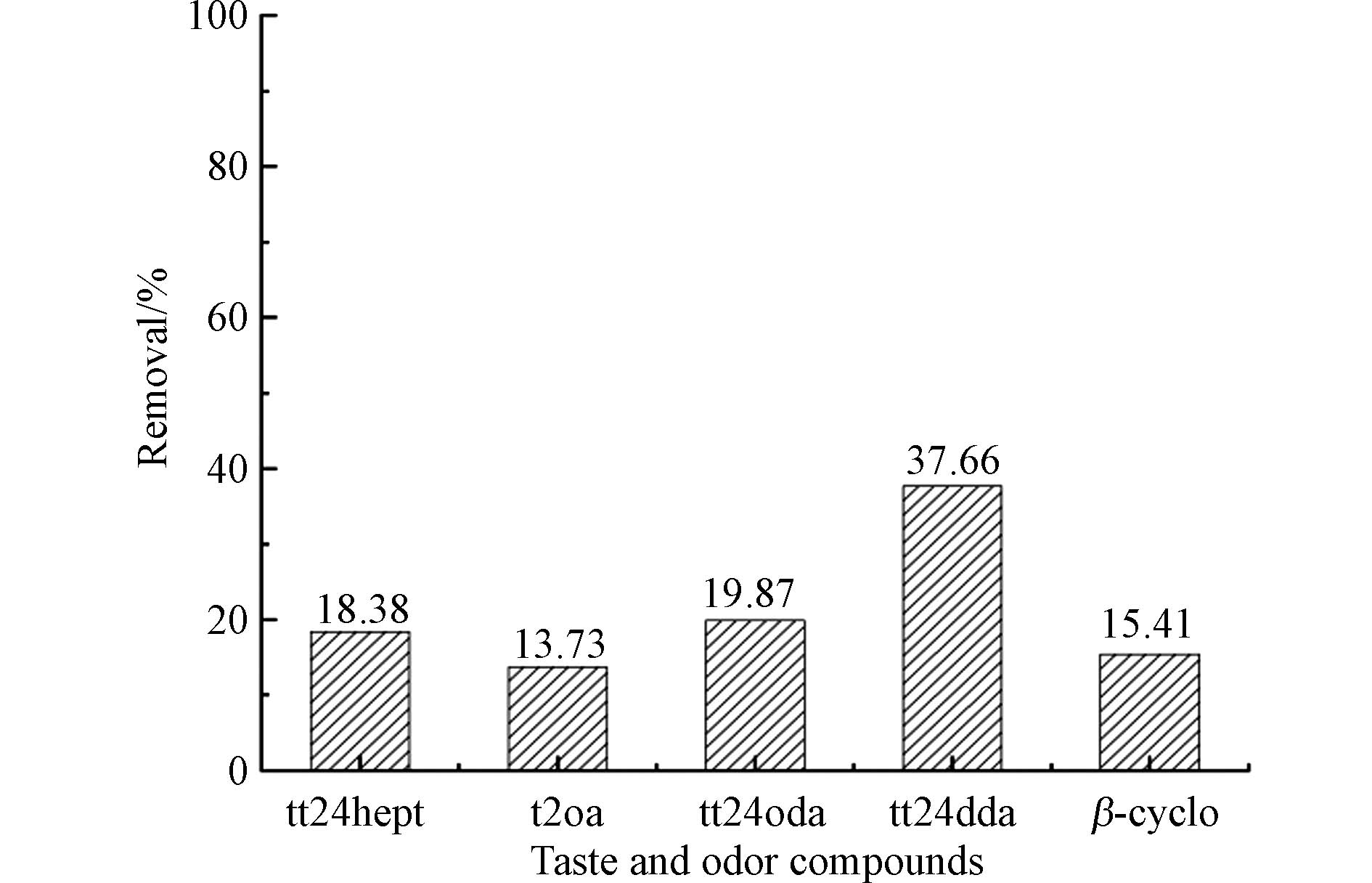
图 4 KMnO4(1mg·L−1)充分转化为新生态MnO2反应2 h后5种醛类物质的去除率
Figure 4. The removal of five aldehydes by the in-site MnO2 for 2 h
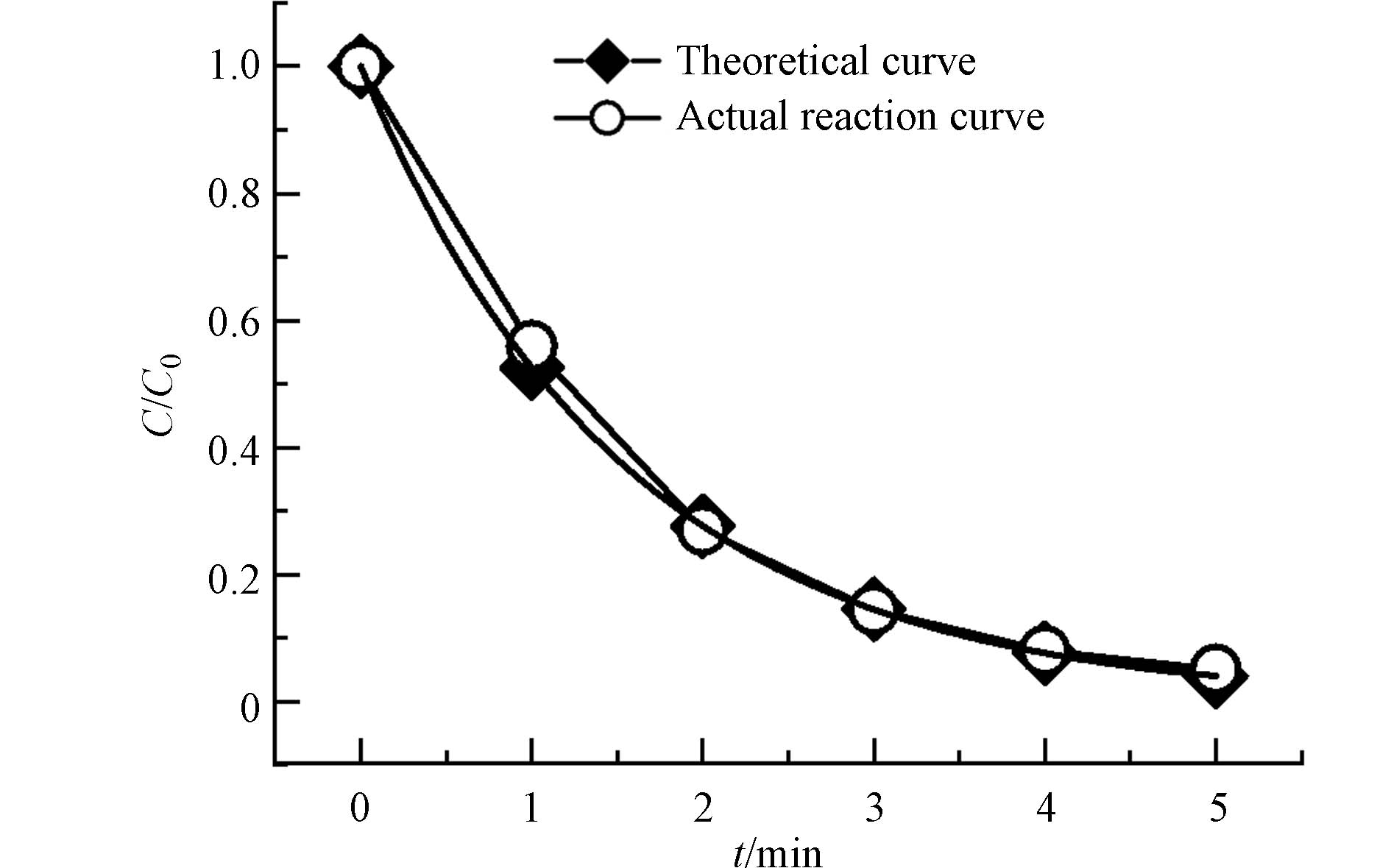
图 5 原水条件下,0—5 min内KMnO4氧化tt24hept浓度比随时间变化的理论曲线和实际反应曲线
Figure 5. Theoretical curve and actual reaction curve of KMnO4 oxidation tt24hept concentration ratio with time in 0~5 min under raw water condition
表 1 5种醛类嗅味物质基本信息
Table 1. Basic information of five aldehyde odorants
物质名称Substance name 英文名English name 结构式Constitutional formula 嗅阈值/(μg·L−1)(OTC) CAS 初始浓度/(μg·L−1)Initial concentration 反,反-2,4-庚二烯醛 trans,trans-2,4-heptadienal(tt24hept) (OTC)</td><td class="table_top_border" style="width:10%" align="center" valign="middle">CAS</td><td class="table_top_border" style="width:20%" align="center" valign="middle">初始浓度/(μg·L<sup>−1</sup>)Initial concentration</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">反,反-2,4-庚二烯醛</td><td class="table_top_border2" align="center" valign="middle">trans,trans-2,4-heptadienal(tt24hept)</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">物质名称Substance name</td><td class="table_top_border" align="center" valign="middle">英文名English name</td><td class="table_top_border" align="center" valign="middle">结构式Constitutional formula</td><td class="table_top_border" align="center" valign="middle">嗅阈值/(μg·L<sup>−1</sup>)(OTC)</td><td class="table_top_border" style="width:10%" align="center" valign="middle">CAS</td><td class="table_top_border" style="width:20%" align="center" valign="middle">初始浓度/(μg·L<sup>−1</sup>)Initial concentration</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">反,反-2,4-庚二烯醛</td><td class="table_top_border2" align="center" valign="middle">trans,trans-2,4-heptadienal(tt24hept)</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">5</td><td class="table_top_border2" align="center" valign="middle">4313-03-5</td><td class="table_top_border2" align="center" valign="middle">100</td></tr><tr><td align="center" valign="middle">反-2-辛烯醛</td><td align="center" valign="middle">trans-2-octenal(t2oa)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">3</td><td align="center" valign="middle">2548-87-0</td><td align="center" valign="middle">50</td></tr><tr><td align="center" valign="middle">反,反-2,4-辛二烯醛</td><td align="center" valign="middle">trans,trans-2,4-octadienal(tt24oda)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">10</td><td align="center" valign="middle">30361-28-5</td><td align="center" valign="middle">250</td></tr><tr><td align="center" valign="middle">反,反-2,4-癸二烯醛</td><td align="center" valign="middle">trans,trans-2,4-decadienal(tt24dda)</td><td align="center" valign="middle"><styled-content style="width:2.82cm"><img class="graphic" src="2022072801-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">0.3</td><td align="center" valign="middle">25152-84-5</td><td align="center" valign="middle">50</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-环柠檬醛</td><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-cyclocitral(<i>β</i>-cyclo)</td><td class="table_bottom_border" style="padding-bottom:1pt;" align="center" valign="middle"><styled-content style="width:1.89cm"><img class="graphic" src="2022072801-Tab1-T5.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">3</td><td class="table_bottom_border" align="center" valign="middle">432-25-7</td><td class="table_bottom_border" align="center" valign="middle">20</td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td><td class="table_top_border2" align="center" valign="middle">5</td><td class="table_top_border2" align="center" valign="middle">4313-03-5</td><td class="table_top_border2" align="center" valign="middle">100</td></tr><tr><td align="center" valign="middle">反-2-辛烯醛</td><td align="center" valign="middle">trans-2-octenal(t2oa)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">物质名称Substance name</td><td class="table_top_border" align="center" valign="middle">英文名English name</td><td class="table_top_border" align="center" valign="middle">结构式Constitutional formula</td><td class="table_top_border" align="center" valign="middle">嗅阈值/(μg·L<sup>−1</sup>)(OTC)</td><td class="table_top_border" style="width:10%" align="center" valign="middle">CAS</td><td class="table_top_border" style="width:20%" align="center" valign="middle">初始浓度/(μg·L<sup>−1</sup>)Initial concentration</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">反,反-2,4-庚二烯醛</td><td class="table_top_border2" align="center" valign="middle">trans,trans-2,4-heptadienal(tt24hept)</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">5</td><td class="table_top_border2" align="center" valign="middle">4313-03-5</td><td class="table_top_border2" align="center" valign="middle">100</td></tr><tr><td align="center" valign="middle">反-2-辛烯醛</td><td align="center" valign="middle">trans-2-octenal(t2oa)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">3</td><td align="center" valign="middle">2548-87-0</td><td align="center" valign="middle">50</td></tr><tr><td align="center" valign="middle">反,反-2,4-辛二烯醛</td><td align="center" valign="middle">trans,trans-2,4-octadienal(tt24oda)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">10</td><td align="center" valign="middle">30361-28-5</td><td align="center" valign="middle">250</td></tr><tr><td align="center" valign="middle">反,反-2,4-癸二烯醛</td><td align="center" valign="middle">trans,trans-2,4-decadienal(tt24dda)</td><td align="center" valign="middle"><styled-content style="width:2.82cm"><img class="graphic" src="2022072801-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">0.3</td><td align="center" valign="middle">25152-84-5</td><td align="center" valign="middle">50</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-环柠檬醛</td><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-cyclocitral(<i>β</i>-cyclo)</td><td class="table_bottom_border" style="padding-bottom:1pt;" align="center" valign="middle"><styled-content style="width:1.89cm"><img class="graphic" src="2022072801-Tab1-T5.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">3</td><td class="table_bottom_border" align="center" valign="middle">432-25-7</td><td class="table_bottom_border" align="center" valign="middle">20</td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">3</td><td align="center" valign="middle">2548-87-0</td><td align="center" valign="middle">50</td></tr><tr><td align="center" valign="middle">反,反-2,4-辛二烯醛</td><td align="center" valign="middle">trans,trans-2,4-octadienal(tt24oda)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">物质名称Substance name</td><td class="table_top_border" align="center" valign="middle">英文名English name</td><td class="table_top_border" align="center" valign="middle">结构式Constitutional formula</td><td class="table_top_border" align="center" valign="middle">嗅阈值/(μg·L<sup>−1</sup>)(OTC)</td><td class="table_top_border" style="width:10%" align="center" valign="middle">CAS</td><td class="table_top_border" style="width:20%" align="center" valign="middle">初始浓度/(μg·L<sup>−1</sup>)Initial concentration</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">反,反-2,4-庚二烯醛</td><td class="table_top_border2" align="center" valign="middle">trans,trans-2,4-heptadienal(tt24hept)</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">5</td><td class="table_top_border2" align="center" valign="middle">4313-03-5</td><td class="table_top_border2" align="center" valign="middle">100</td></tr><tr><td align="center" valign="middle">反-2-辛烯醛</td><td align="center" valign="middle">trans-2-octenal(t2oa)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">3</td><td align="center" valign="middle">2548-87-0</td><td align="center" valign="middle">50</td></tr><tr><td align="center" valign="middle">反,反-2,4-辛二烯醛</td><td align="center" valign="middle">trans,trans-2,4-octadienal(tt24oda)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">10</td><td align="center" valign="middle">30361-28-5</td><td align="center" valign="middle">250</td></tr><tr><td align="center" valign="middle">反,反-2,4-癸二烯醛</td><td align="center" valign="middle">trans,trans-2,4-decadienal(tt24dda)</td><td align="center" valign="middle"><styled-content style="width:2.82cm"><img class="graphic" src="2022072801-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">0.3</td><td align="center" valign="middle">25152-84-5</td><td align="center" valign="middle">50</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-环柠檬醛</td><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-cyclocitral(<i>β</i>-cyclo)</td><td class="table_bottom_border" style="padding-bottom:1pt;" align="center" valign="middle"><styled-content style="width:1.89cm"><img class="graphic" src="2022072801-Tab1-T5.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">3</td><td class="table_bottom_border" align="center" valign="middle">432-25-7</td><td class="table_bottom_border" align="center" valign="middle">20</td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">10</td><td align="center" valign="middle">30361-28-5</td><td align="center" valign="middle">250</td></tr><tr><td align="center" valign="middle">反,反-2,4-癸二烯醛</td><td align="center" valign="middle">trans,trans-2,4-decadienal(tt24dda)</td><td align="center" valign="middle"><styled-content style="width:2.82cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">物质名称Substance name</td><td class="table_top_border" align="center" valign="middle">英文名English name</td><td class="table_top_border" align="center" valign="middle">结构式Constitutional formula</td><td class="table_top_border" align="center" valign="middle">嗅阈值/(μg·L<sup>−1</sup>)(OTC)</td><td class="table_top_border" style="width:10%" align="center" valign="middle">CAS</td><td class="table_top_border" style="width:20%" align="center" valign="middle">初始浓度/(μg·L<sup>−1</sup>)Initial concentration</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">反,反-2,4-庚二烯醛</td><td class="table_top_border2" align="center" valign="middle">trans,trans-2,4-heptadienal(tt24hept)</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">5</td><td class="table_top_border2" align="center" valign="middle">4313-03-5</td><td class="table_top_border2" align="center" valign="middle">100</td></tr><tr><td align="center" valign="middle">反-2-辛烯醛</td><td align="center" valign="middle">trans-2-octenal(t2oa)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">3</td><td align="center" valign="middle">2548-87-0</td><td align="center" valign="middle">50</td></tr><tr><td align="center" valign="middle">反,反-2,4-辛二烯醛</td><td align="center" valign="middle">trans,trans-2,4-octadienal(tt24oda)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">10</td><td align="center" valign="middle">30361-28-5</td><td align="center" valign="middle">250</td></tr><tr><td align="center" valign="middle">反,反-2,4-癸二烯醛</td><td align="center" valign="middle">trans,trans-2,4-decadienal(tt24dda)</td><td align="center" valign="middle"><styled-content style="width:2.82cm"><img class="graphic" src="2022072801-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">0.3</td><td align="center" valign="middle">25152-84-5</td><td align="center" valign="middle">50</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-环柠檬醛</td><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-cyclocitral(<i>β</i>-cyclo)</td><td class="table_bottom_border" style="padding-bottom:1pt;" align="center" valign="middle"><styled-content style="width:1.89cm"><img class="graphic" src="2022072801-Tab1-T5.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">3</td><td class="table_bottom_border" align="center" valign="middle">432-25-7</td><td class="table_bottom_border" align="center" valign="middle">20</td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td><td align="center" valign="middle">0.3</td><td align="center" valign="middle">25152-84-5</td><td align="center" valign="middle">50</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-环柠檬醛</td><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-cyclocitral(<i>β</i>-cyclo)</td><td class="table_bottom_border" style="padding-bottom:1pt;" align="center" valign="middle"><styled-content style="width:1.89cm"><img class="graphic" src="data:image/svg+xml,<svg xmlns='http://www.w3.org/2000/svg' width='350' height='600'><foreignObject width='2000' height='100%'><div xmlns='http://www.w3.org/1999/xhtml' style='font-size:16px;'><table>
<thead><tr><td class="table_top_border" align="center" valign="middle">物质名称Substance name</td><td class="table_top_border" align="center" valign="middle">英文名English name</td><td class="table_top_border" align="center" valign="middle">结构式Constitutional formula</td><td class="table_top_border" align="center" valign="middle">嗅阈值/(μg·L<sup>−1</sup>)(OTC)</td><td class="table_top_border" style="width:10%" align="center" valign="middle">CAS</td><td class="table_top_border" style="width:20%" align="center" valign="middle">初始浓度/(μg·L<sup>−1</sup>)Initial concentration</td></tr></thead>
<tbody><tr><td class="table_top_border2" align="center" valign="middle">反,反-2,4-庚二烯醛</td><td class="table_top_border2" align="center" valign="middle">trans,trans-2,4-heptadienal(tt24hept)</td><td class="table_top_border2" align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T1.jpg"></styled-content></td><td class="table_top_border2" align="center" valign="middle">5</td><td class="table_top_border2" align="center" valign="middle">4313-03-5</td><td class="table_top_border2" align="center" valign="middle">100</td></tr><tr><td align="center" valign="middle">反-2-辛烯醛</td><td align="center" valign="middle">trans-2-octenal(t2oa)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T2.jpg"></styled-content></td><td align="center" valign="middle">3</td><td align="center" valign="middle">2548-87-0</td><td align="center" valign="middle">50</td></tr><tr><td align="center" valign="middle">反,反-2,4-辛二烯醛</td><td align="center" valign="middle">trans,trans-2,4-octadienal(tt24oda)</td><td align="center" valign="middle"><styled-content style="width:3cm"><img class="graphic" src="2022072801-Tab1-T3.jpg"></styled-content></td><td align="center" valign="middle">10</td><td align="center" valign="middle">30361-28-5</td><td align="center" valign="middle">250</td></tr><tr><td align="center" valign="middle">反,反-2,4-癸二烯醛</td><td align="center" valign="middle">trans,trans-2,4-decadienal(tt24dda)</td><td align="center" valign="middle"><styled-content style="width:2.82cm"><img class="graphic" src="2022072801-Tab1-T4.jpg"></styled-content></td><td align="center" valign="middle">0.3</td><td align="center" valign="middle">25152-84-5</td><td align="center" valign="middle">50</td></tr><tr><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-环柠檬醛</td><td class="table_bottom_border" align="center" valign="middle"><i>β</i>-cyclocitral(<i>β</i>-cyclo)</td><td class="table_bottom_border" style="padding-bottom:1pt;" align="center" valign="middle"><styled-content style="width:1.89cm"><img class="graphic" src="2022072801-Tab1-T5.jpg"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">3</td><td class="table_bottom_border" align="center" valign="middle">432-25-7</td><td class="table_bottom_border" align="center" valign="middle">20</td></tr></tbody>
</table></div></foreignObject></svg>"></styled-content></td><td class="table_bottom_border" align="center" valign="middle">3</td><td class="table_bottom_border" align="center" valign="middle">432-25-7</td><td class="table_bottom_border" align="center" valign="middle">20</td></tr></tbody>
</table></div></foreignObject></svg>)
5 4313-03-5 100 反-2-辛烯醛 trans-2-octenal(t2oa) 3 2548-87-0 50 反,反-2,4-辛二烯醛 trans,trans-2,4-octadienal(tt24oda) 10 30361-28-5 250 反,反-2,4-癸二烯醛 trans,trans-2,4-decadienal(tt24dda) 0.3 25152-84-5 50 β-环柠檬醛 β-cyclocitral(β-cyclo) 3 432-25-7 20
下载: 导出CSV
表 2 5种醛类嗅味物质的特征离子及保留时间
Table 2. Characteristic ions and retention times of five aldehyde odorants
嗅味物质 Taste and odor compounds 质荷比 m/z 开始时间/min Start time 结束时间/min Terminal time 保留时间/min Retention time tt24hept 81*, 53, 39 16.41 17.77 16.895 t2oa 41*, 55, 70 15.00 20.00 16.660 tt24oda 81*, 39, 41 12.00 14.00 12.660 tt24dda 81*, 41, 67 22.83 24.26 23.335 β-cyclo 137*, 152, 123 12.00 13.50 12.767 注:*为特征离子,其余为参考离子. Note: * the characteristic ion, and the rest are reference ions.
下载: 导出CSV
表 3 各KMnO4浓度下氧化tt24hept的伪一级动力学参数
Table 3. Pseudo-first-order kinetic parameters of tt24hept oxidation by different dosage of KMnO4
[KMnO4]/(mol⋅L−1) 回归方程 Regression equation kobs/min−1 线性相关系数R2 Linearly dependent coefficient 3.165×10−6 lnCC0=−0.1488t+0.0173 0.1488 0.9956 6.329×10−6 lnCC0=−0.3165t+0.0247 0.3165 0.9979 9.494×10−6 lnCC0=−0.4867t+0.0685 0.4867 0.9952 1.266×10−5 lnCC0=−0.6457t+0.0168 0.6457 0.9951
下载: 导出CSV
表 4 KMnO4氧化5种醛类嗅味物质的伪二级反应动力学常数
Table 4. Kinetic constants of pseudo-second-order reaction of KMnO4 oxidation of five aldehydes odorants
嗅味物质Taste and odor compounds k/(L·mol−1·min−1) tt24hept 5.25×104 t2oa 2.66×104 tt24oda 4.50×104 tt24dda 2.71×104 β-cyclo 5.37×103
下载: 导出CSV
表 5 KMnO4去除四种醛类特征嗅味物质的投加量参考
Table 5. Reference of KMnO4 to remove four aldehydes
嗅味物质Taste and odor compounds 初始浓度/(μg·L−1)Initial concentration 氧化时间/minOxidation time 氧化剂量/(mg·L−1)Oxidative dosage tt24hept 100 40 0.50 tt24oda 250 60 0.50 tt24dda 50 70 0.50 β-cyclo 20 120 1.00
下载: 导出CSV
-
[1] ZHAO Y Y, YU J W, SU M, et al. A fishy odor episode in a North China reservoir: Occurrence, origin, and possible odor causing compounds [J]. Journal of Environmental Sciences (China), 2013, 25(12): 2361-2366. doi: 10.1016/S1001-0742(12)60317-9 [2] SUN D L, YU J W, AN W, et al. Identification of causative compounds and microorganisms for musty odor occurrence in the Huangpu River, China [J]. Journal of Environmental Sciences (China), 2013, 25(3): 460-465. doi: 10.1016/S1001-0742(12)60012-6 [3] LI X, YU J W, GUO Q Y, et al. Source-water odor during winter in the Yellow River area of China: Occurrence and diagnosis [J]. Environmental Pollution(Barking, Essex:1987), 2016, 218: 252-258. doi: 10.1016/j.envpol.2016.06.069 [4] 李霞, 魏魏, 乔莉, 等. 低温期黄河水源鱼腥味问题的预处理技术应用探讨[J]. 给水排水, 2015, 51(S1): 25-29. LI X, WEI W, QIAO L, et al. Discussion on the application of pretreatment technology for fish smell in Yellow River water source during low temperature period[J]. Water & Wastewater Engineering, 2015, 51(Sup 1): 25-29(in Chinese).
[5] WU W, TAO N P, GU S Q. Characterization of the key odor-active compounds in steamed meat of Coilia ectenes from Yangtze River by GC–MS–O [J]. European Food Research and Technology, 2014, 238(2): 237-245. doi: 10.1007/s00217-013-2098-3 [6] WU N, GU S Q, TAO N P, et al. Characterization of important odorants in steamed male Chinese mitten crab (Eriocheir sinensis) using gas chromatography-mass spectrometry-olfactometry [J]. Journal of Food Science, 2014, 79(7): C1250-C1259. doi: 10.1111/1750-3841.12511 [7] WATSON S, SATCHWLLL T. Chrysophyte odour production: Resource-mediated changes at the cell and population levels [J]. Phycologia, 2003, 42: 393-405. doi: 10.2216/i0031-8884-42-4-393.1 [8] WENDEL T, JÜTTNER F. Lipoxygenase-mediated formation of hydrocarbons and unsaturated aldehydes in freshwater diatoms [J]. Phytochemistry, 1996, 41(6): 1445-1449. doi: 10.1016/0031-9422(95)00828-4 [9] WEE J L, HARRIS S A, SMITH J P, et al. Production of the taste/odor-causing compound, trans-2, Cis-6-nonadienal, within the synurophyceae [J]. Journal of Applied Phycology, 1994, 6(4): 365-369. doi: 10.1007/BF02182152 [10] RIBALET F, WICHARD T, POHNERT G, et al. Age and nutrient limitation enhance polyunsaturated aldehyde production in marine diatoms [J]. Phytochemistry, 2007, 68(15): 2059-2067. doi: 10.1016/j.phytochem.2007.05.012 [11] MIRALTO A, BARONE G, ROMANO G, et al. The insidious effect of diatoms on copepod reproduction [J]. Nature, 1999, 402(6758): 173-176. doi: 10.1038/46023 [12] JÜTTNER F, WATSON S B, von ELERT E, et al. Β-cyclocitral, a grazer defence signal unique to the cyanobacterium Microcystis [J]. Journal of Chemical Ecology, 2010, 36(12): 1387-1397. doi: 10.1007/s10886-010-9877-0 [13] 张可佳, 高乃云, 黎雷. 高锰酸钾氧化嗅味物质β-环柠檬醛的动力学 [J]. 中南大学学报(自然科学版), 2011, 42(4): 1161-1166. ZHANG K J, GAO N Y, LI L. Kinetics of oxidation of odorant β-cyclocitral by potassium permanganate [J]. Journal of Central South University (Science and Technology), 2011, 42(4): 1161-1166(in Chinese).
[14] 刘禧文, 闫慧敏, 韩正双, 等. 水中8种典型嗅味物质的氧化去除研究 [J]. 供水技术, 2020, 14(4): 1-7. LIU X W, YAN H M, HAN Z S, et al. Research on the removal of 8 typical taste and odor compounds from source water by oxidation methods [J]. Water Technology, 2020, 14(4): 1-7(in Chinese).
[15] 廖宇, 张慧鑫, 李璐玮, 等. 饮用水中两种硫醚类嗅味物质的氧化去除 [J]. 环境化学, 2020, 39(5): 1254-1261. doi: 10.7524/j.issn.0254-6108.2019081303 LIAO Y, ZHANG H X, LI L W, et al. Removal of two typical sulfides odorants by different oxidants in drinking water [J]. Environmental chemistry, 2020, 39(5): 1254-1261(in Chinese). doi: 10.7524/j.issn.0254-6108.2019081303
[16] 徐勇鹏, 杨静琨, 王在刚. 高锰酸钾氧化去除水中三氯生动力学研究 [J]. 哈尔滨工业大学学报, 2011, 43(12): 48-52. XU Y P, YANG J K, WANG Z G. Kinetics on triclosan oxidation by potassium permanganate in drinking water [J]. Journal of Harbin Institute of Technology, 2011, 43(12): 48-52(in Chinese).
[17] 马军, 李圭白, 李晓东. 高锰酸钾除微污染效能-GC/MS分析 [J]. 中国给水排水, 1999, 15(5): 13-15. MA J, LI G B, LI X D. Removal of organic micropollutants from water by permanganate pre-oxidation-GC/MS analysis [J]. China Water & Wastewater, 1999, 15(5): 13-15(in Chinese).
[18] YAN Y E, SCHWARTZ F. Oxidative degradation and kinetics of chlorinated ethylenes by potassium permanganate [J]. Journal of Contaminant Hydrology, 1999, 37: 343-365. doi: 10.1016/S0169-7722(98)00166-1 [19] WIBERG K B, SAEGEBARTH K A. The mechanisms of permanganate oxidation. IV. hydroxylation of olefins and related reactions [J]. Journal of the American Chemical Society, 1957, 79(11): 2822-2824. doi: 10.1021/ja01568a042 [20] BECK C B. Physicochemical processes for water quality control, Walter J. Weber, Jr. (with eight contributors), Interscience, New York(1972). 640 pages. $19.95 [J]. AIChE Journal, 1973, 19(2): 413. [21] SUFFET I H, BAKER R J, YOHE T L. Pretreatment of drinking water to control organic contaminants and taste and odor[C]//Pretreatment in Chemical Water and Wastewater Treatment, 1988: 15-39. [22] 庞雅丽. 高锰酸钾与粉末活性炭联用去除水中嗅味物质[D]. 北京: 北京工业大学, 2011. PANG Y L. Study on the use of potassium permanganate in combination with powder activated carbon for the removal of taste and odor in drinking water[D]. Beijing: Beijing University of Technology, 2011(in Chinese).
[23] VEGA E, MARTIN M J, GONZALEZ-OLMOS R. Integration of advanced oxidation processes at mild conditions in wet scrubbers for odourous sulphur compounds treatment [J]. Chemosphere, 2014, 109: 113-119. doi: 10.1016/j.chemosphere.2014.02.061 [24] 庞素艳, 江进, 马军, 等. MnO2催化KMnO4氧化降解酚类化合物 [J]. 环境科学, 2010, 31(10): 2331-2335. PANG S Y, JIANG J, MA J, et al. Oxidation of phenolic compounds with permanganate catalyzed by manganese dioxide [J]. Environmental Science, 2010, 31(10): 2331-2335(in Chinese).
-

 点击查看大图
点击查看大图
计量
- 文章访问数: 2096
- HTML全文浏览数: 2096
- PDF下载数: 38
- 施引文献: 0

















