

 下载:
下载:
-
砷作为一种剧毒的类金属,其毒性与砷的价态有很强的相关性:As(Ⅲ) > As(Ⅴ). 研究报告表明,天然水体中的砷浓度范围很广,从小于0.5 μg·L−1到大于5000 μg·L−1,地热水中的砷浓度甚至可达50 mg·L−1,在地下水缺氧环境中,As(Ⅲ)是主要形态[1]. 全球约有9400万人至2.2亿人可能接触到高砷浓度的地下水,其中绝大多数(94%)在亚洲[2]. 美国和中国都参照世界卫生组织规定要求饮用水中砷的浓度不得高于10 μg·L−1. As(Ⅲ)对混凝剂和吸附剂的亲和力较弱,As(Ⅲ)相较于As(Ⅴ)更难去除,将As(Ⅲ)氧化为As(Ⅴ)是提高总砷去除的关键步骤[3]. 通过氯和臭氧等化学氧化手段可以将As(Ⅲ)氧化为As(Ⅴ),但很可能产生有毒副产物;通过UV/Fe[4]、UV/TiO2[5]、UV/碘化物[6]、UV/氢氧化铁[7]等方法可以将As(Ⅲ)氧化为As(Ⅴ),但引入了金属离子,不利于环境安全. H2O2在pH > 9.0时可以氧化As(Ⅲ)[8],且H2O2作为一种温和无害的氧化剂,具有绿色氧化过程,不会产生有害的副产物,但由于H2O2运输困难、不易保存,因此H2O2的原位产生技术受到了广泛关注.
MB+[9]是一种知名的噻嗪光敏剂,被大量用作染色剂、光催化剂、抗氧化剂、防腐剂和单态氧敏剂等使用,能够在光照下产生单线态氧(1O2). 除MB+外,核黄素[10]、血卟啉衍生物[11]、联吡啶钌[12]和亚甲基紫[13]等光敏剂在光照下也能够产生和MB+类似的效应. 由于MB+等噻嗪染料价格低廉,光照下对水体中污染物具有较好的氧化效果[14],受到国内外学者的广泛研究. 乙二胺四乙酸(EDTA)[15]、H2A[16]、亚硫酸盐[17]等还原剂存在下的MB+的光化学现象已有诸多报道. 当不存在还原剂时,MB+通过光敏化过程(反应1 −3)产生1O2[15]. 但还原形式的无色亚甲基蓝(LMB)只有在还原剂存在时通过反应(4)和(5)才产生,其中Rred和Rox分别表示还原剂的还原形式和氧化形式,反应(6)为反应(4)和反应(5)的总式. 此外,还原性的染料自由基(半还原性MB+·)在没有O2存在时是稳定的,在O2存在时,MB+·会失去其未成对电子,产生过氧化氢自由基(HO2·)(反应(7))[18]. 抗坏血酸(H2A)是常见的抗氧化剂,具有较强的还原性,被广泛应用于有机污染物的降解[19-20]. 已有研究表明H2A/MB+在可见光激发下能够产生抗坏血酸自由基(A·−),进而还原分子氧产生H2O2(反应(8)和反应(9))[21]. 因此,在某些还原剂存在时,MB+的光解可能会诱导分子氧的活化. MB+在许多行业和化学实验室中得到了广泛的应用(如著名的“蓝瓶子”实验). MB+排放前需要对其进行处理,将废弃MB+用来处理其它污染物不失为更好的应用.
本研究拟探索可见光/MB+/H2A体系氧化水中As(Ⅲ)的可行性与内在机理,考察了光照、pH、H2A浓度、MB+浓度、As(Ⅲ)初始浓度及水中常见阴离子(Cl−、HCO3−、NO3−)和有机质富里酸(FA)对As(Ⅲ)氧化的影响. 通过活性物种识别(自由基清除实验)和溶液光谱变化确定了可见光/MB+/H2A体系氧化As(Ⅲ)的主要活性物种及其生成机理.
-
实验中所用的盐酸、硼氢化钾、氢氧化钾、十水硼酸钠、氢氧化钠、叔丁醇(TBA)、碳酸氢钠、抗坏血酸、硝酸钠、氯化钾、磷酸等均购于国药集团化学试剂公司,亚砷酸钠购于西亚试剂有限公司,砷酸氢二钠七水化合物购于阿法埃莎化学有限公司,富里酸购于阿拉丁生化科技股份有限公司,过氧化氢酶(CAT),超氧化物歧化酶(SOD)购于源叶生物科技有限公司,实验中用水均为去离子水,所用药品均为分析纯.
-
ALB−124电子天平秤(赛多利斯仪器公司),R6200氢化物发生−原子荧光光度计(北京博晖创新光电技术股份有限公司),LC−10 ADVP液相泵(日本岛津公司),超纯水机(四川优普科技有限公司),pH 320−S精密数字酸度计(梅特勒−托利多公司),Bante 980溶解氧仪(上海班特仪器公司),801磁力搅拌装置(上海三信公司),UV-1600紫外可见分光光度计(日本岛津公司).
-
在圆柱形开放式夹套反应器(内直径100 mm)中配制500 mL含有5 μmol·L−1 As(Ⅲ)、0.1 mg·L−1 MB+、100 μmol·L−1 H2A、10 mmol·L−1不同类型缓冲液的反应液(醋酸盐缓冲液是pH=4.0、磷酸盐缓冲液是pH=6.0—8.0、硼酸盐缓冲液是pH=9.0—9.5),通过连接外部水循环恒温系统将反应温度控制在(25±1)℃,将LED灯源放置于反应器四周,通过磁力搅拌装置混匀溶液. 打开LED灯并开始计时,分别于0、10、20、30、45、60、90、120 min取4.5 mL反应液,加入0.5 mL 1:1的盐酸以猝灭反应,再进行分析.
采用液相−氢化物发生−原子荧光光度法(LC−HG−AFS)测定As(Ⅲ)和As(Ⅴ)形态的量[22],流动相为磷酸盐(45 mmol·L−1, pH=5.6)和超纯水,流速为1.3 mL·min−1,色谱柱为离子色谱柱(PRP−X100,hamilton,Switzerland),进样体积为100 μL.
本实验所用LED灯是从网上采购. 其最大发射波长为630 nm (图1). 通过紫外分光光度计在200−800 nm范围内分别扫描MB+、劳氏紫(Thionine)、天青B(Azure B)的光谱. MB+、劳氏紫和天青B的可见光区域最大吸收峰分别为660、600、600 nm(图1).
-
图2a为As(Ⅲ)在MB+/H2A体系和可见光/MB+/H2A体系中不同pH条件下的氧化. 当pH < 6.0时,As(Ⅲ)在MB+/H2A体系和可见光/MB+/H2A体系中均未发生氧化;随着pH从8.0增加至9.5,MB+/H2A体系和可见光/MB+/H2A体系中As(Ⅲ)的氧化速率分别从0、0.39 μmol·L−1·h−1增加至1.34、3.73 μmol·L−1·h−1. 由此可见,在MB+/H2A体系中加入可见光后,As(Ⅲ)的氧化速率和氧化率均明显提高.
pH会影响抗坏血酸和As(Ⅲ)的形态分布与氧化还原电位. 抗坏血酸的两个酸式解离常数分别为4.17、11.57. pH=6—10时,抗坏血酸主要以HA−形态存在,随着pH增加,A2−形态的量会进一步增加,A2−相较于HA−更容易与分子氧反应产生H2O2[8]. As(Ⅲ)的3个酸式解离常数分别为9.22、12.1、13.4,pH > 9.0时As(Ⅲ)主要以解离态存在,解离态的As(Ⅲ)在碱性条件下更容易被H2O2氧化为As(Ⅴ). LMB的3个酸式解离常数分别为1.7、4.5、5.9,因此LMB在溶液中存在四种形态(H3LMB2+、H2LMB+、HLMB、LMB−)[23]. LMB被溶解氧(DO)氧化产生MB+在中性和碱性条件下更快. 考虑到As(Ⅲ)的氧化速率和氧化率,后续反应在pH=9.5下进行.
图2b为H2A浓度对MB+/H2A体系和可见光/MB+/H2A体系中的As(Ⅲ)氧化的影响. As(Ⅲ)在MB+/H2A体系中的氧化速率随着H2A浓度增加而增加,而As(Ⅲ)在可见光/MB+/H2A体系中的氧化速率则随着H2A浓度增加先增加后趋于稳定. H2A投加量的增加一方面会促进活性物种H2O2的产生,另外一方面H2A作为还原剂,过量的H2A则会竞争消耗产生的活性物种. 在Fe(Ⅲ)/H2O2/H2A[24]和Fe(Ⅲ)/PS/H2A[25]体系中,也存在过量的抗坏血酸抑制底物降解的现象. 综上,H2A最佳投加量为300 μmol·L−1.
图2c为MB+浓度对可见光/MB+/H2A体系中的As(Ⅲ)和MB+氧化的影响. 随着MB+浓度从0.1 mg·L−1增加至5 mg·L−1,As(Ⅲ)的氧化速率提高,继续增加MB+浓度至20 mg·L−1,As(Ⅲ)的氧化速率变化不明显. 随着MB+浓度从0.1 mg·L−1增加至10 mg·L−1,MB+的氧化速率从0.02 μmol·L−1·h−1增加至0.42 μmol·L−1·h−1,继续增加MB+浓度至20 mg·L−1,MB+的氧化速率降至0.3 μmol·L−1·h−1. 这是因为随着MB+浓度的增加,MB+吸收光能增多,使得1O2浓度增加,从而有利于反应进行;但MB+达到一定浓度,足以充分使体系中氧分子转变为1O2[26],继续提高MB+浓度并不会对As(Ⅲ)和MB+的氧化产生影响. 综上,MB+最佳投加量为5 mg·L−1.
图3a和图3b分别给出了不同体系中As(Ⅲ)和MB+随时间变化结果. 在可见光/MB+/H2A体系中As(Ⅲ)和MB+的氧化率分别为90%、10%. 图3a显示As(Ⅲ)的氧化率在MB+/H2A体系和可见光/H2A体系中相同,为50%,说明pH 9.5时H2A可以活化分子氧产生H2O2氧化As(Ⅲ). 图3b显示,MB+在MB+/H2A体系中并未发生氧化,说明H2O2并不能氧化MB+. 可见光/MB+体系可以产生1O2,As(Ⅲ)和MB+在可见光/MB+体中氧化率分别为10%和15%,说明1O2对As(Ⅲ)的氧化能力很弱. 而在可见光/MB+/H2A体系中,由于溶液中的H2A会竞争消耗1O2,导致此条件下MB+氧化率低于可见光/MB+体系.
图4a为As(Ⅲ)初始浓度对可见光/MB+/H2A体系氧化As(Ⅲ)的影响. 当As(Ⅲ)浓度从5 μmol·L−1上升到50 μmol·L−1时,As(Ⅲ)的氧化率从90%降低至80%(相应的As(Ⅲ)氧化量分别为2.25 μmol和20 μmol),As(Ⅲ)的氧化率没有显著的改变,但As(Ⅲ)的氧化量显著增加. As(Ⅲ)的初始氧化速率和As(Ⅲ)的浓度成正比(图4b). 这说明可见光/MB+/H2A体系可以用来处理高浓度含砷废水.
-
As(Ⅲ)在可见光/MB+/H2A体系中的氧化可以从MB+的激发、激活的MB+(MB+*)与H2A相互作用以及反应活性物种的产生三个角度来进行说明. MB+是一种最大吸收峰为660 nm的蓝色染料,而LMB为无色且在265 nm处具有最大吸收峰[16]. 当预先用氮气曝气半小时以去除溶液中的DO,可见光照射,可以观察到MB+在660 nm处的最大吸收峰迅速消失(0—5 min),而在265 nm处出现LMB的最大吸收峰(0—120 min)(图5a). 在有氧搅拌条件下可见光照射后,反应液中DO迅速减少,溶液中部分LMB无法氧化,出现LMB的吸收峰(0—10 min),此后溶液复氧(10—120 min), LMB最大吸收峰消失(图5b). 由于实际反应是在空气氛围下进行,与N2氛围相比,MB+下降的速率较慢. 这是因为在O2存在时,MB+首先被H2A还原成LMB,然后LMB被O2氧化成MB+,这个过程非常迅速,所以在O2存在下无法观察到MB+的循环过程. 此外,当可见光照射脱氧的MB+/H2A反应溶液重新暴露于空气中,观察到无色溶液迅速变为蓝色(图6). 这也可以解释上述现象,即LMB很容易被O2氧化[17].
为了进一步理解可见光/MB+/H2A体系中的As(Ⅲ)的氧化机理,利用自由基抑制实验来捕获产生的活性物种. SOD和TBA分别作为HO2·和HO·猝灭剂,SOD与HO2·的反应速率常数为1.6×109 L·mol−1·s−1,TBA与HO·的反应速率常数为7.6×108 L·mol−1·s−1. 分别投加100 U·mL−1 SOD和2 mmol·L−1 TBA(图7),均未对As(Ⅲ)的氧化产生明显影响,表明O2·−(HO2·)或HO·不是氧化As(Ⅲ)的主要活性物种. CAT可以快速分解H2O2,因而被用于清除反应中产生的H2O2. 分别投加100 U·mL−1和250 U·mL−1的CAT,反应前30 min,As(Ⅲ)的氧化率由65%下降到25%,此后并未观察到As(Ⅲ)进一步氧化,说明反应30 min后As(Ⅲ)的氧化被完全抑制. 上述结果说明H2O2是氧化As(Ⅲ)的主要活性物种. 如图8所示,pH=9.5时,单独的H2O2能够快速氧化As(Ⅲ).
在N2氛围下,As(Ⅲ)在可见光/MB+/H2A体系中不会发生氧化(图9). 说明即便MB+和HA−之间能够电子转移,但是体系中没有分子氧,产生MB+·和A·−也不能进行后续产生H2O2的反应. 这与无氧条件下碱/H2A体系无法产生H2O2的实验结果一致[8]. 而分别通入20% 或100% O2,As(Ⅲ)的氧化效率与搅拌条件下一致,表明过多O2也没有促进体系中As(Ⅲ)的氧化.
在可见光/MB+/H2A体系中,活性物种通过以下途径产生(图10):首先,MB+通过可见光激发产生单线态1MB+(公式1),随后1MB+快速转变为三重态(3MB+)(公式2),3MB+和分子氧(3O2)反应产生1O2(公式3);其次,3MB+可以从HA−处得到一个电子生成MB+·和A·−(公式8),MB+·也可以通过歧化反应产生LMB和MB+(公式5),LMB再与O2反应产生MB+;最后,A·−与O2·−(HO2·)反应产生H2O2(公式9). HA−也可以直接与分子氧通过双电子转移产生H2O2,MB+和可见光的引入加速HA−的氧化,从而产生更多的H2O2. 在可见光/MB+/H2A体系中存在H2A可清除产生的1O2, 1O2对As(Ⅲ)的氧化贡献微乎其微,H2O2对As(Ⅲ)的氧化起主要作用.
-
HCO3−、Cl−、NO3−、FA在土壤间隙水和地下水中普遍存在,HCO3−、Cl−、NO3−、FA对As(Ⅲ)氧化的影响如图11所示,HCO3−、Cl−、NO3−对As(Ⅲ)氧化基本无影响,这是因为HCO3−、Cl−、NO3−加入反应液后并不会使MB+的吸收光谱发生变化,因此不会影响可见光/MB+/H2A体系中As(Ⅲ)的氧化,这和碱/H2A体系中环境基质对As(Ⅲ)氧化的影响一致[8]. 但FA对As(Ⅲ)氧化有一定的影响,这是因为FA加入反应液后使得MB+的吸收光谱发生了显著变化,使得MB+在660 nm处的吸收变弱,从而抑制了MB+和HA−之间的电子转移.
-
探讨了将其他噻嗪染料用于可见光/H2A体系活化分子氧氧化As(Ⅲ)的可能性. 劳氏紫和天青B也是常见的噻嗪染料,在可见光/H2A/劳氏紫体系和可见光/H2A/天青B体系中分别对测定了As(Ⅲ)和染料的浓度的变化,结果如图12所示. 实验结果表明,劳氏紫和天青B均能促进H2A活化分子氧产生H2O2氧化As(Ⅲ),但促进效果均不如MB+,这是因为相同浓度下MB+有最大的吸光度值,能更好地吸收能量. 噻嗪染料的相似行为可能是由于它们具有相同的官能团和激发状态. 推测其他光敏剂如核黄素[9]和茜素[27]都具有接受电子的能力,同样可以促进H2A活化分子氧产生H2O2氧化水中的As(Ⅲ).
-
本文从可见光/MB+/H2A活化分子氧体系出发,研究了可见光/MB+/H2A体系氧化水中As(Ⅲ)的可行性与内在机理. 其中,可见光光照、pH值、H2A与MB+的投加量对于As(Ⅲ)的氧化过程有着不同影响. 实验结果表明:
(1)可见光光照可以促进MB+/H2A体系氧化As(Ⅲ); pH越高,As(Ⅲ)氧化越快;pH=9.5下,H2A浓度的增加对As(Ⅲ)氧化呈现先促进后稳定的趋势,且O2浓度对于该体系对As(Ⅲ) 的氧化过程的影响较小. 最终得出该体系下As(Ⅲ)氧化的最佳条件:最佳H2A投加量为300 μmol·L−1;最佳MB+投加量为5 mg·L−1.
(2)通过自由基抑制剂分析验证了可见光/MB+/H2A活化分子氧体系氧化As(Ⅲ)的主要活性物种为H2O2, 1O2对As(Ⅲ)的氧化贡献微乎其微.
(3)同MB+一样,在可见光/MB+体系中添加劳氏紫和天青B这两种噻嗪染料也能促进As(Ⅲ)的氧化.
可见光/亚甲基蓝/抗坏血酸活化分子氧氧化水中的As(Ⅲ)
Oxidation of As(Ⅲ) in water by visible light/methylene blue/ascorbic acid activated molecular oxygen
-
摘要: 本文研究了可见光/亚甲基蓝(MB+)/抗坏血酸(H2A)活化分子氧体系(可见光/MB+/H2A体系)氧化水中三价砷(As(Ⅲ))的过程与机理. 考察了光照、pH、H2A浓度、MB+浓度、As(Ⅲ)初始浓度及水中常见阴离子和有机质的对As(Ⅲ)氧化效率的影响,通过自由基抑制实验和溶液光谱变化鉴定了体系中的活性物种及其生成机理. 实验结果表明,光照对As(Ⅲ)的氧化有明显促进作用;在pH=8.0—9.5范围内,As(Ⅲ)的氧化随着pH的升高而加快;pH=9.5条件下,H2A剂量的增加对As(Ⅲ)的氧化呈现先促进后趋于稳定的趋势,H2A最佳投加量为300 μmol·L−1;MB+最佳投加量为5 mg·L−1. 机理研究表明,H2A和分子氧之间的双电子反应产生的H2O2是可见光/MB+/H2A体系活化分子氧体系中氧化As(Ⅲ)的主要活性物种. MB+经可见光激发后通过促进A·−的产生进而产生H2O2. 基于同样机制,另外两种噻嗪染料(劳氏紫和天青B)在可见光/H2A体系中也能促进As(Ⅲ)的氧化.
-
关键词:
- 三价砷(As(Ⅲ)) /
- 抗坏血酸(H2A) /
- 可见光 /
- 亚甲基蓝(MB+) /
- 过氧化氢(H2O2).
Abstract: The oxidation process and mechanism for As(Ⅲ) oxidation in water with visible light/methylene blue(MB+)/ascorbic acid(H2A) activated molecular oxygen (visible light/MB+/H2A system) were systematically investigated in this work. The effects of light, pH, H2A concentration, MB+ concentration, initial concentration of As(Ⅲ), common anions and organic matter in water on the oxidation efficiency of As(Ⅲ) were examined. The active species and its formation mechanism in the system were identified by free radical inhibition assay and spectral changes. Results show that light can promote the oxidation of As(Ⅲ). In the range of pH 8.0—9.5, the oxidation rate of As(Ⅲ) increases with the increase of pH. At pH 9.5, as the dosage of H2A increase, the oxidation of As(Ⅲ) was promoted first and then remained unchanged. The optimum dosages of H2A and MB+ were 300 μmol·L−1 and 5 mg·L−1, respectively. H2O2 is the main active species for the oxidation of As(Ⅲ) in the visible light/MB+/H2A system. MB+ enhances H2O2 generation by promoting the production of A·− via visible light excitation. Based on the same mechanism, two other thiazine dyes, thionine and Azure B, can also accelerate the oxidation of As(Ⅲ) in the visible light/H2A system. -
砷作为一种剧毒的类金属,其毒性与砷的价态有很强的相关性:As(Ⅲ) > As(Ⅴ). 研究报告表明,天然水体中的砷浓度范围很广,从小于0.5 μg·L−1到大于5000 μg·L−1,地热水中的砷浓度甚至可达50 mg·L−1,在地下水缺氧环境中,As(Ⅲ)是主要形态[1]. 全球约有9400万人至2.2亿人可能接触到高砷浓度的地下水,其中绝大多数(94%)在亚洲[2]. 美国和中国都参照世界卫生组织规定要求饮用水中砷的浓度不得高于10 μg·L−1. As(Ⅲ)对混凝剂和吸附剂的亲和力较弱,As(Ⅲ)相较于As(Ⅴ)更难去除,将As(Ⅲ)氧化为As(Ⅴ)是提高总砷去除的关键步骤[3]. 通过氯和臭氧等化学氧化手段可以将As(Ⅲ)氧化为As(Ⅴ),但很可能产生有毒副产物;通过UV/Fe[4]、UV/TiO2[5]、UV/碘化物[6]、UV/氢氧化铁[7]等方法可以将As(Ⅲ)氧化为As(Ⅴ),但引入了金属离子,不利于环境安全. H2O2在pH > 9.0时可以氧化As(Ⅲ)[8],且H2O2作为一种温和无害的氧化剂,具有绿色氧化过程,不会产生有害的副产物,但由于H2O2运输困难、不易保存,因此H2O2的原位产生技术受到了广泛关注.
MB+[9]是一种知名的噻嗪光敏剂,被大量用作染色剂、光催化剂、抗氧化剂、防腐剂和单态氧敏剂等使用,能够在光照下产生单线态氧(1O2). 除MB+外,核黄素[10]、血卟啉衍生物[11]、联吡啶钌[12]和亚甲基紫[13]等光敏剂在光照下也能够产生和MB+类似的效应. 由于MB+等噻嗪染料价格低廉,光照下对水体中污染物具有较好的氧化效果[14],受到国内外学者的广泛研究. 乙二胺四乙酸(EDTA)[15]、H2A[16]、亚硫酸盐[17]等还原剂存在下的MB+的光化学现象已有诸多报道. 当不存在还原剂时,MB+通过光敏化过程(反应1 −3)产生1O2[15]. 但还原形式的无色亚甲基蓝(LMB)只有在还原剂存在时通过反应(4)和(5)才产生,其中Rred和Rox分别表示还原剂的还原形式和氧化形式,反应(6)为反应(4)和反应(5)的总式. 此外,还原性的染料自由基(半还原性MB+·)在没有O2存在时是稳定的,在O2存在时,MB+·会失去其未成对电子,产生过氧化氢自由基(HO2·)(反应(7))[18]. 抗坏血酸(H2A)是常见的抗氧化剂,具有较强的还原性,被广泛应用于有机污染物的降解[19-20]. 已有研究表明H2A/MB+在可见光激发下能够产生抗坏血酸自由基(A·−),进而还原分子氧产生H2O2(反应(8)和反应(9))[21]. 因此,在某些还原剂存在时,MB+的光解可能会诱导分子氧的活化. MB+在许多行业和化学实验室中得到了广泛的应用(如著名的“蓝瓶子”实验). MB+排放前需要对其进行处理,将废弃MB+用来处理其它污染物不失为更好的应用.
MB++hv→1MB+∗ (1) 1MB+∗→3MB+∗ (2) 3MB+∗+O2→MB++1O2 (3) 3MB+∗+Rred→MB+⋅+R⋅ox (4) 2MB+⋅→LMB+MB+ (5) 23MB+∗+2Rred→LMB+MB++2R⋅ox (6) MB+⋅+O2→MB++O⋅−2(HO⋅2) (7) 3MB+∗+HA−→MB+⋅+A⋅− (8) A⋅−+O⋅−2(HO⋅2)→DHA+H2O2 (9) 本研究拟探索可见光/MB+/H2A体系氧化水中As(Ⅲ)的可行性与内在机理,考察了光照、pH、H2A浓度、MB+浓度、As(Ⅲ)初始浓度及水中常见阴离子(Cl−、HCO3−、NO3−)和有机质富里酸(FA)对As(Ⅲ)氧化的影响. 通过活性物种识别(自由基清除实验)和溶液光谱变化确定了可见光/MB+/H2A体系氧化As(Ⅲ)的主要活性物种及其生成机理.
1. 实验部分 (Experimental section)
1.1 实验材料
实验中所用的盐酸、硼氢化钾、氢氧化钾、十水硼酸钠、氢氧化钠、叔丁醇(TBA)、碳酸氢钠、抗坏血酸、硝酸钠、氯化钾、磷酸等均购于国药集团化学试剂公司,亚砷酸钠购于西亚试剂有限公司,砷酸氢二钠七水化合物购于阿法埃莎化学有限公司,富里酸购于阿拉丁生化科技股份有限公司,过氧化氢酶(CAT),超氧化物歧化酶(SOD)购于源叶生物科技有限公司,实验中用水均为去离子水,所用药品均为分析纯.
1.2 仪器
ALB−124电子天平秤(赛多利斯仪器公司),R6200氢化物发生−原子荧光光度计(北京博晖创新光电技术股份有限公司),LC−10 ADVP液相泵(日本岛津公司),超纯水机(四川优普科技有限公司),pH 320−S精密数字酸度计(梅特勒−托利多公司),Bante 980溶解氧仪(上海班特仪器公司),801磁力搅拌装置(上海三信公司),UV-1600紫外可见分光光度计(日本岛津公司).
1.3 实验方法与分析方法
在圆柱形开放式夹套反应器(内直径100 mm)中配制500 mL含有5 μmol·L−1 As(Ⅲ)、0.1 mg·L−1 MB+、100 μmol·L−1 H2A、10 mmol·L−1不同类型缓冲液的反应液(醋酸盐缓冲液是pH=4.0、磷酸盐缓冲液是pH=6.0—8.0、硼酸盐缓冲液是pH=9.0—9.5),通过连接外部水循环恒温系统将反应温度控制在(25±1)℃,将LED灯源放置于反应器四周,通过磁力搅拌装置混匀溶液. 打开LED灯并开始计时,分别于0、10、20、30、45、60、90、120 min取4.5 mL反应液,加入0.5 mL 1:1的盐酸以猝灭反应,再进行分析.
采用液相−氢化物发生−原子荧光光度法(LC−HG−AFS)测定As(Ⅲ)和As(Ⅴ)形态的量[22],流动相为磷酸盐(45 mmol·L−1, pH=5.6)和超纯水,流速为1.3 mL·min−1,色谱柱为离子色谱柱(PRP−X100,hamilton,Switzerland),进样体积为100 μL.
本实验所用LED灯是从网上采购. 其最大发射波长为630 nm (图1). 通过紫外分光光度计在200−800 nm范围内分别扫描MB+、劳氏紫(Thionine)、天青B(Azure B)的光谱. MB+、劳氏紫和天青B的可见光区域最大吸收峰分别为660、600、600 nm(图1).
 图 1 LED灯发射光谱及MB+、劳氏紫、天青的3种噻嗪染料染料溶液的紫外可见吸收光谱Figure 1. Emission spectrum of LED lamp and UV-Vis absorption spectra of the solutions of three thiazine dyes (MB+, Thionine and Azure B).
图 1 LED灯发射光谱及MB+、劳氏紫、天青的3种噻嗪染料染料溶液的紫外可见吸收光谱Figure 1. Emission spectrum of LED lamp and UV-Vis absorption spectra of the solutions of three thiazine dyes (MB+, Thionine and Azure B).2. 结果与讨论(Results and discussion)
2.1 pH、H2A浓度、MB+和砷浓度的影响
图2a为As(Ⅲ)在MB+/H2A体系和可见光/MB+/H2A体系中不同pH条件下的氧化. 当pH < 6.0时,As(Ⅲ)在MB+/H2A体系和可见光/MB+/H2A体系中均未发生氧化;随着pH从8.0增加至9.5,MB+/H2A体系和可见光/MB+/H2A体系中As(Ⅲ)的氧化速率分别从0、0.39 μmol·L−1·h−1增加至1.34、3.73 μmol·L−1·h−1. 由此可见,在MB+/H2A体系中加入可见光后,As(Ⅲ)的氧化速率和氧化率均明显提高.
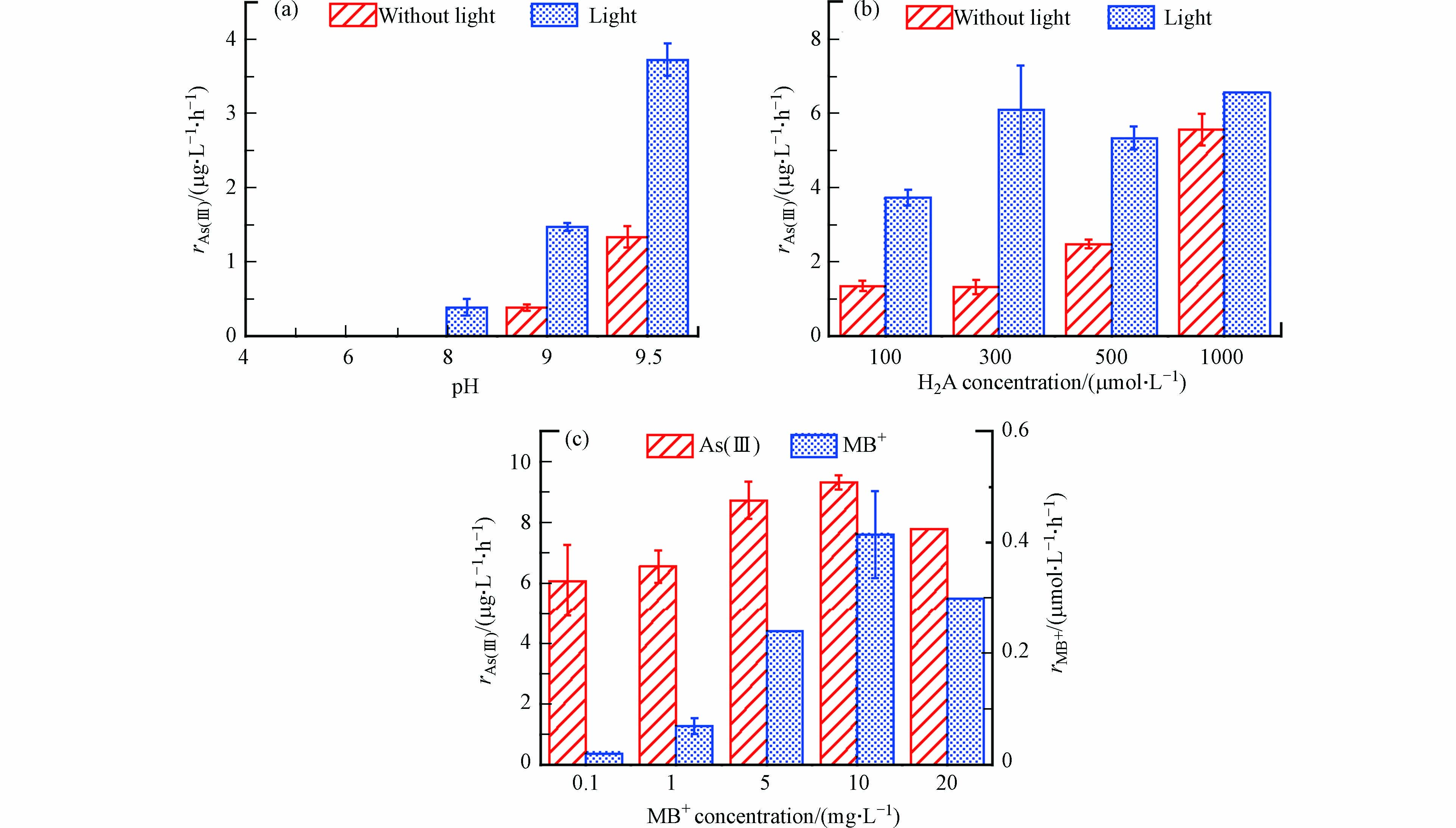 图 2 As(Ⅲ) 氧化的条件实验Figure 2. Effects of solution pH(a), concentration of H2A(b) and MB+(c)on the oxidation of As(Ⅲ).(a)溶液pH值,(b)H2A浓度,(c)MB+浓度
图 2 As(Ⅲ) 氧化的条件实验Figure 2. Effects of solution pH(a), concentration of H2A(b) and MB+(c)on the oxidation of As(Ⅲ).(a)溶液pH值,(b)H2A浓度,(c)MB+浓度pH会影响抗坏血酸和As(Ⅲ)的形态分布与氧化还原电位. 抗坏血酸的两个酸式解离常数分别为4.17、11.57. pH=6—10时,抗坏血酸主要以HA−形态存在,随着pH增加,A2−形态的量会进一步增加,A2−相较于HA−更容易与分子氧反应产生H2O2[8]. As(Ⅲ)的3个酸式解离常数分别为9.22、12.1、13.4,pH > 9.0时As(Ⅲ)主要以解离态存在,解离态的As(Ⅲ)在碱性条件下更容易被H2O2氧化为As(Ⅴ). LMB的3个酸式解离常数分别为1.7、4.5、5.9,因此LMB在溶液中存在四种形态(H3LMB2+、H2LMB+、HLMB、LMB−)[23]. LMB被溶解氧(DO)氧化产生MB+在中性和碱性条件下更快. 考虑到As(Ⅲ)的氧化速率和氧化率,后续反应在pH=9.5下进行.
图2b为H2A浓度对MB+/H2A体系和可见光/MB+/H2A体系中的As(Ⅲ)氧化的影响. As(Ⅲ)在MB+/H2A体系中的氧化速率随着H2A浓度增加而增加,而As(Ⅲ)在可见光/MB+/H2A体系中的氧化速率则随着H2A浓度增加先增加后趋于稳定. H2A投加量的增加一方面会促进活性物种H2O2的产生,另外一方面H2A作为还原剂,过量的H2A则会竞争消耗产生的活性物种. 在Fe(Ⅲ)/H2O2/H2A[24]和Fe(Ⅲ)/PS/H2A[25]体系中,也存在过量的抗坏血酸抑制底物降解的现象. 综上,H2A最佳投加量为300 μmol·L−1.
图2c为MB+浓度对可见光/MB+/H2A体系中的As(Ⅲ)和MB+氧化的影响. 随着MB+浓度从0.1 mg·L−1增加至5 mg·L−1,As(Ⅲ)的氧化速率提高,继续增加MB+浓度至20 mg·L−1,As(Ⅲ)的氧化速率变化不明显. 随着MB+浓度从0.1 mg·L−1增加至10 mg·L−1,MB+的氧化速率从0.02 μmol·L−1·h−1增加至0.42 μmol·L−1·h−1,继续增加MB+浓度至20 mg·L−1,MB+的氧化速率降至0.3 μmol·L−1·h−1. 这是因为随着MB+浓度的增加,MB+吸收光能增多,使得1O2浓度增加,从而有利于反应进行;但MB+达到一定浓度,足以充分使体系中氧分子转变为1O2[26],继续提高MB+浓度并不会对As(Ⅲ)和MB+的氧化产生影响. 综上,MB+最佳投加量为5 mg·L−1.
图3a和图3b分别给出了不同体系中As(Ⅲ)和MB+随时间变化结果. 在可见光/MB+/H2A体系中As(Ⅲ)和MB+的氧化率分别为90%、10%. 图3a显示As(Ⅲ)的氧化率在MB+/H2A体系和可见光/H2A体系中相同,为50%,说明pH 9.5时H2A可以活化分子氧产生H2O2氧化As(Ⅲ). 图3b显示,MB+在MB+/H2A体系中并未发生氧化,说明H2O2并不能氧化MB+. 可见光/MB+体系可以产生1O2,As(Ⅲ)和MB+在可见光/MB+体中氧化率分别为10%和15%,说明1O2对As(Ⅲ)的氧化能力很弱. 而在可见光/MB+/H2A体系中,由于溶液中的H2A会竞争消耗1O2,导致此条件下MB+氧化率低于可见光/MB+体系.
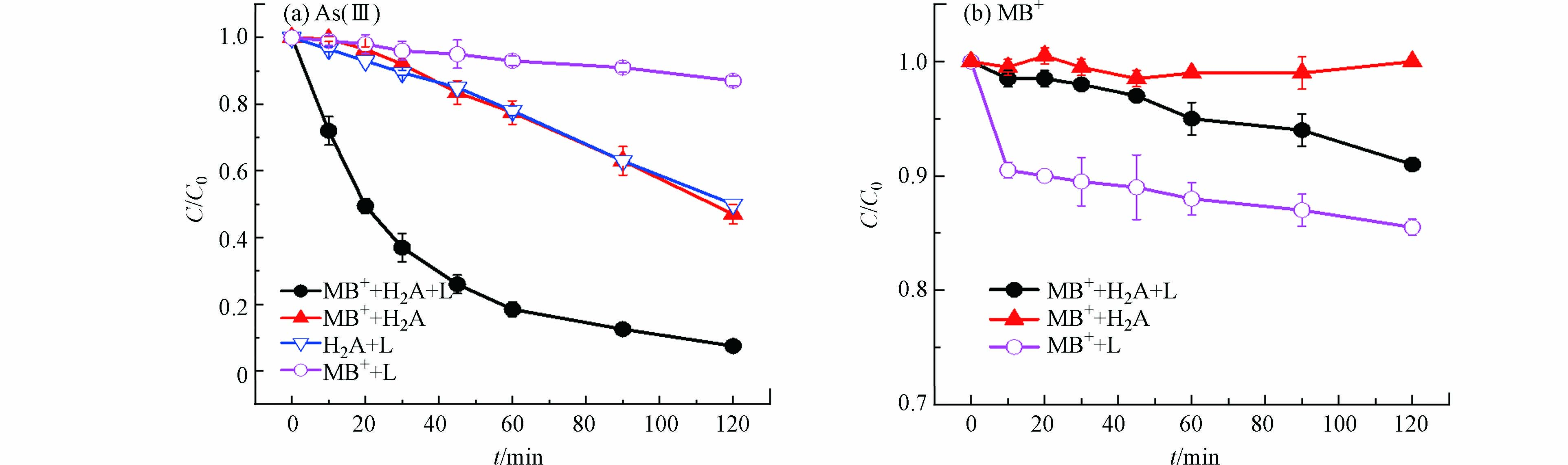 图 3 (a)As(Ⅲ)的氧化与(b)MB+的去除动力学曲线Figure 3. Kinetic curves of As(Ⅲ) oxidation(a) and MB+ removal(b)
图 3 (a)As(Ⅲ)的氧化与(b)MB+的去除动力学曲线Figure 3. Kinetic curves of As(Ⅲ) oxidation(a) and MB+ removal(b)图4a为As(Ⅲ)初始浓度对可见光/MB+/H2A体系氧化As(Ⅲ)的影响. 当As(Ⅲ)浓度从5 μmol·L−1上升到50 μmol·L−1时,As(Ⅲ)的氧化率从90%降低至80%(相应的As(Ⅲ)氧化量分别为2.25 μmol和20 μmol),As(Ⅲ)的氧化率没有显著的改变,但As(Ⅲ)的氧化量显著增加. As(Ⅲ)的初始氧化速率和As(Ⅲ)的浓度成正比(图4b). 这说明可见光/MB+/H2A体系可以用来处理高浓度含砷废水.
 图 4 可见光/MB+/H2A体系中,As(Ⅲ)初始浓度对(a)As(Ⅲ)的氧化与(b)As(Ⅲ)的氧化速率的影响Figure 4. Effect of As(Ⅲ) concentration on oxidation of As(Ⅲ)(a) and oxidation rate of As(Ⅲ)(b) in visible light/ascorbic acid/methylene blue system
图 4 可见光/MB+/H2A体系中,As(Ⅲ)初始浓度对(a)As(Ⅲ)的氧化与(b)As(Ⅲ)的氧化速率的影响Figure 4. Effect of As(Ⅲ) concentration on oxidation of As(Ⅲ)(a) and oxidation rate of As(Ⅲ)(b) in visible light/ascorbic acid/methylene blue system2.2 机理探究与氧气浓度的影响
As(Ⅲ)在可见光/MB+/H2A体系中的氧化可以从MB+的激发、激活的MB+(MB+*)与H2A相互作用以及反应活性物种的产生三个角度来进行说明. MB+是一种最大吸收峰为660 nm的蓝色染料,而LMB为无色且在265 nm处具有最大吸收峰[16]. 当预先用氮气曝气半小时以去除溶液中的DO,可见光照射,可以观察到MB+在660 nm处的最大吸收峰迅速消失(0—5 min),而在265 nm处出现LMB的最大吸收峰(0—120 min)(图5a). 在有氧搅拌条件下可见光照射后,反应液中DO迅速减少,溶液中部分LMB无法氧化,出现LMB的吸收峰(0—10 min),此后溶液复氧(10—120 min), LMB最大吸收峰消失(图5b). 由于实际反应是在空气氛围下进行,与N2氛围相比,MB+下降的速率较慢. 这是因为在O2存在时,MB+首先被H2A还原成LMB,然后LMB被O2氧化成MB+,这个过程非常迅速,所以在O2存在下无法观察到MB+的循环过程. 此外,当可见光照射脱氧的MB+/H2A反应溶液重新暴露于空气中,观察到无色溶液迅速变为蓝色(图6). 这也可以解释上述现象,即LMB很容易被O2氧化[17].
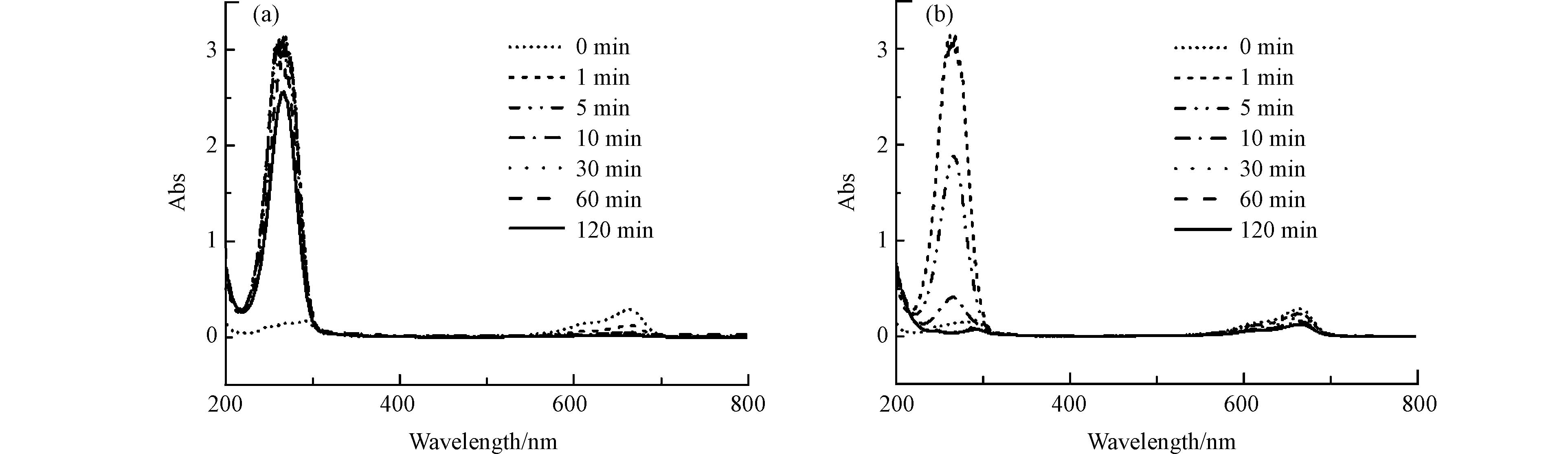 图 5 (a) 氮气条件下和(b)有氧搅拌条件下可见光/MB+/H2A体系中溶液光谱变化Figure 5. Spectral variation of the solution in a visible light/ascorbic acid/methylene blue system under nitrogen conditions(a) and stirring conditions(b)
图 5 (a) 氮气条件下和(b)有氧搅拌条件下可见光/MB+/H2A体系中溶液光谱变化Figure 5. Spectral variation of the solution in a visible light/ascorbic acid/methylene blue system under nitrogen conditions(a) and stirring conditions(b) 图 6 可见光/N2和遮光/O2条件下可见光/MB+/H2A体系中MB+吸光度变化Figure 6. In the visible light/ascorbic acid/methylene blue system under under visible light/N2 and shading/O2 conditions, the absorbance of methylene blue changes.
图 6 可见光/N2和遮光/O2条件下可见光/MB+/H2A体系中MB+吸光度变化Figure 6. In the visible light/ascorbic acid/methylene blue system under under visible light/N2 and shading/O2 conditions, the absorbance of methylene blue changes.为了进一步理解可见光/MB+/H2A体系中的As(Ⅲ)的氧化机理,利用自由基抑制实验来捕获产生的活性物种. SOD和TBA分别作为HO2·和HO·猝灭剂,SOD与HO2·的反应速率常数为1.6×109 L·mol−1·s−1,TBA与HO·的反应速率常数为7.6×108 L·mol−1·s−1. 分别投加100 U·mL−1 SOD和2 mmol·L−1 TBA(图7),均未对As(Ⅲ)的氧化产生明显影响,表明O2·−(HO2·)或HO·不是氧化As(Ⅲ)的主要活性物种. CAT可以快速分解H2O2,因而被用于清除反应中产生的H2O2. 分别投加100 U·mL−1和250 U·mL−1的CAT,反应前30 min,As(Ⅲ)的氧化率由65%下降到25%,此后并未观察到As(Ⅲ)进一步氧化,说明反应30 min后As(Ⅲ)的氧化被完全抑制. 上述结果说明H2O2是氧化As(Ⅲ)的主要活性物种. 如图8所示,pH=9.5时,单独的H2O2能够快速氧化As(Ⅲ).
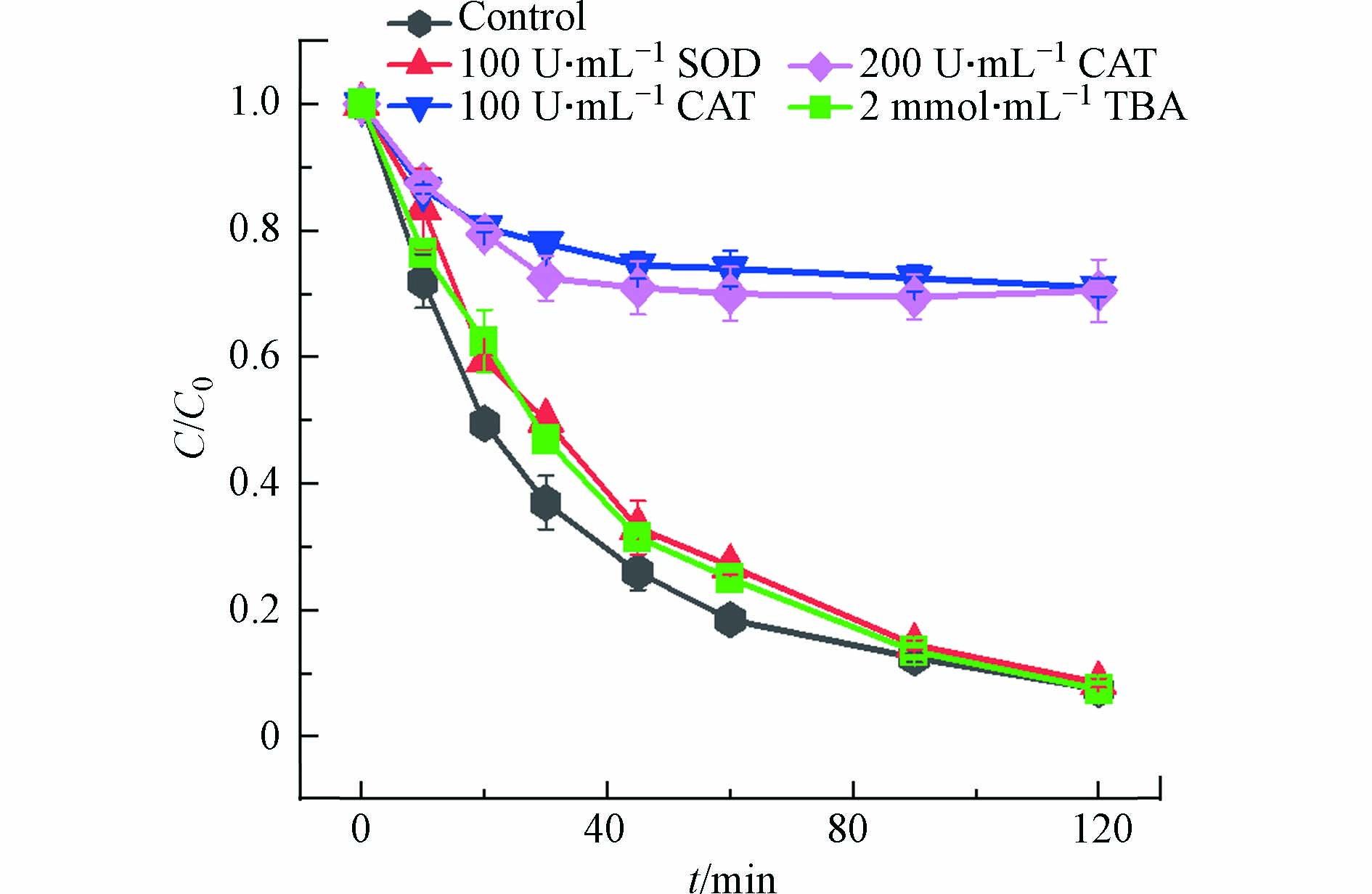 图 7 可见光/MB+/H2A 体系中自由基抑制剂对As(Ⅲ)氧化的影响Figure 7. Effect of free radical inhibitors on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue system
图 7 可见光/MB+/H2A 体系中自由基抑制剂对As(Ⅲ)氧化的影响Figure 7. Effect of free radical inhibitors on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue system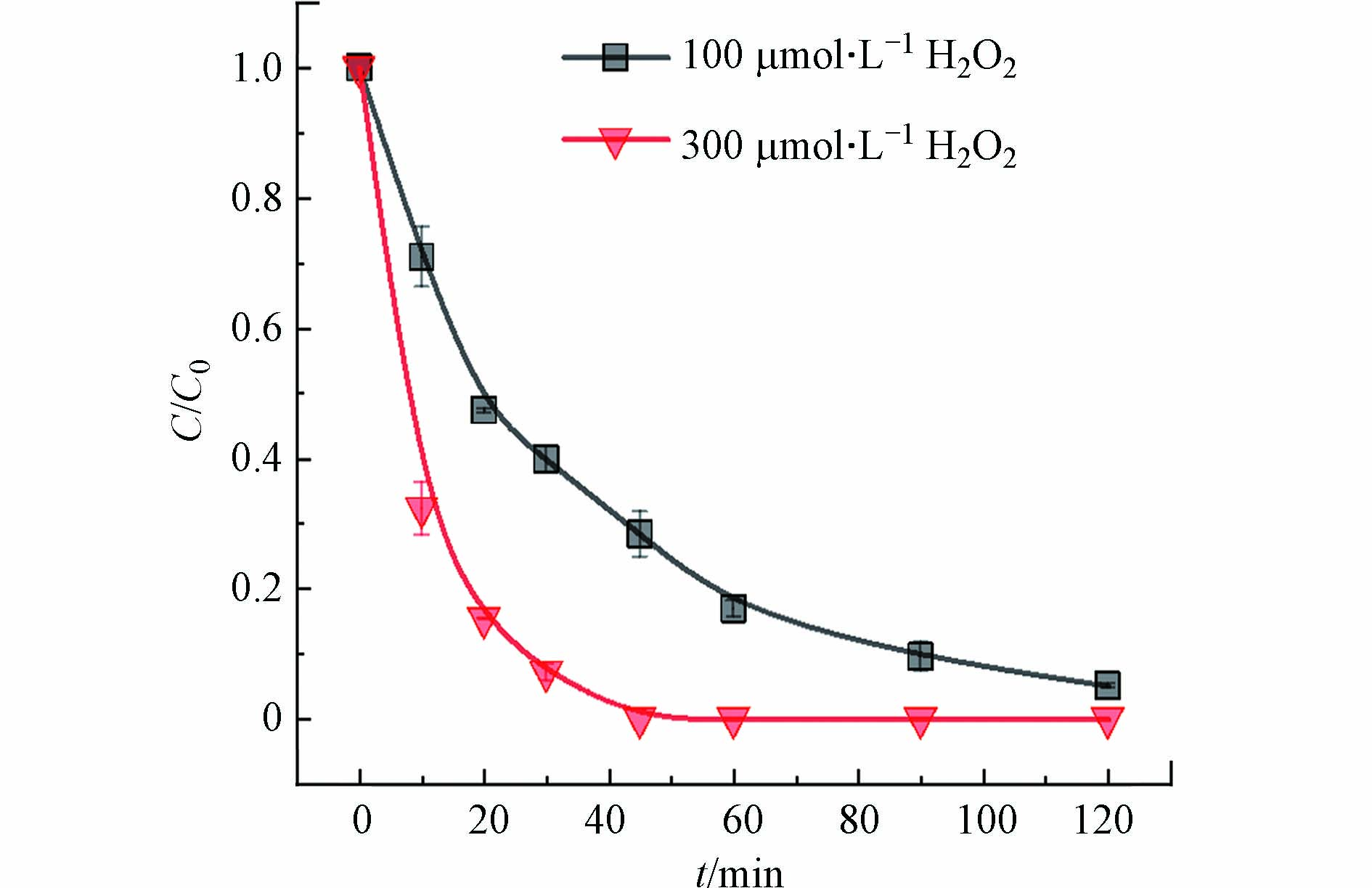 图 8 pH 9.5,H2O2 对As(Ⅲ)氧化的影响Figure 8. Effect of H2O2 on the oxidation of As(Ⅲ) at pH 9.5
图 8 pH 9.5,H2O2 对As(Ⅲ)氧化的影响Figure 8. Effect of H2O2 on the oxidation of As(Ⅲ) at pH 9.5在N2氛围下,As(Ⅲ)在可见光/MB+/H2A体系中不会发生氧化(图9). 说明即便MB+和HA−之间能够电子转移,但是体系中没有分子氧,产生MB+·和A·−也不能进行后续产生H2O2的反应. 这与无氧条件下碱/H2A体系无法产生H2O2的实验结果一致[8]. 而分别通入20% 或100% O2,As(Ⅲ)的氧化效率与搅拌条件下一致,表明过多O2也没有促进体系中As(Ⅲ)的氧化.
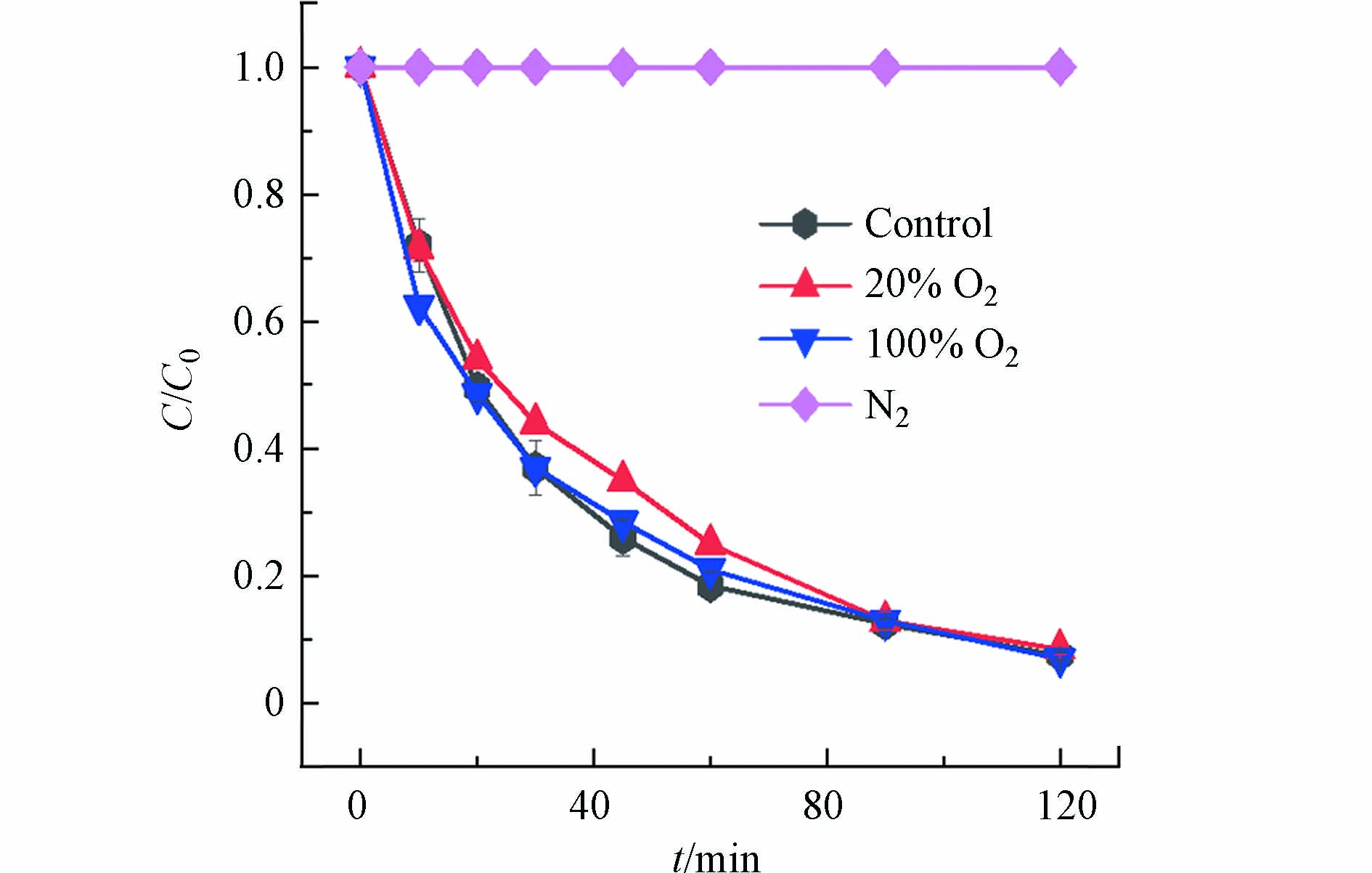 图 9 可见光/MB+/H2A体系中氧气浓度对As(Ⅲ)氧化的影响Figure 9. Effect of oxygen concentration on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.
图 9 可见光/MB+/H2A体系中氧气浓度对As(Ⅲ)氧化的影响Figure 9. Effect of oxygen concentration on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.在可见光/MB+/H2A体系中,活性物种通过以下途径产生(图10):首先,MB+通过可见光激发产生单线态1MB+(公式1),随后1MB+快速转变为三重态(3MB+)(公式2),3MB+和分子氧(3O2)反应产生1O2(公式3);其次,3MB+可以从HA−处得到一个电子生成MB+·和A·−(公式8),MB+·也可以通过歧化反应产生LMB和MB+(公式5),LMB再与O2反应产生MB+;最后,A·−与O2·−(HO2·)反应产生H2O2(公式9). HA−也可以直接与分子氧通过双电子转移产生H2O2,MB+和可见光的引入加速HA−的氧化,从而产生更多的H2O2. 在可见光/MB+/H2A体系中存在H2A可清除产生的1O2, 1O2对As(Ⅲ)的氧化贡献微乎其微,H2O2对As(Ⅲ)的氧化起主要作用.
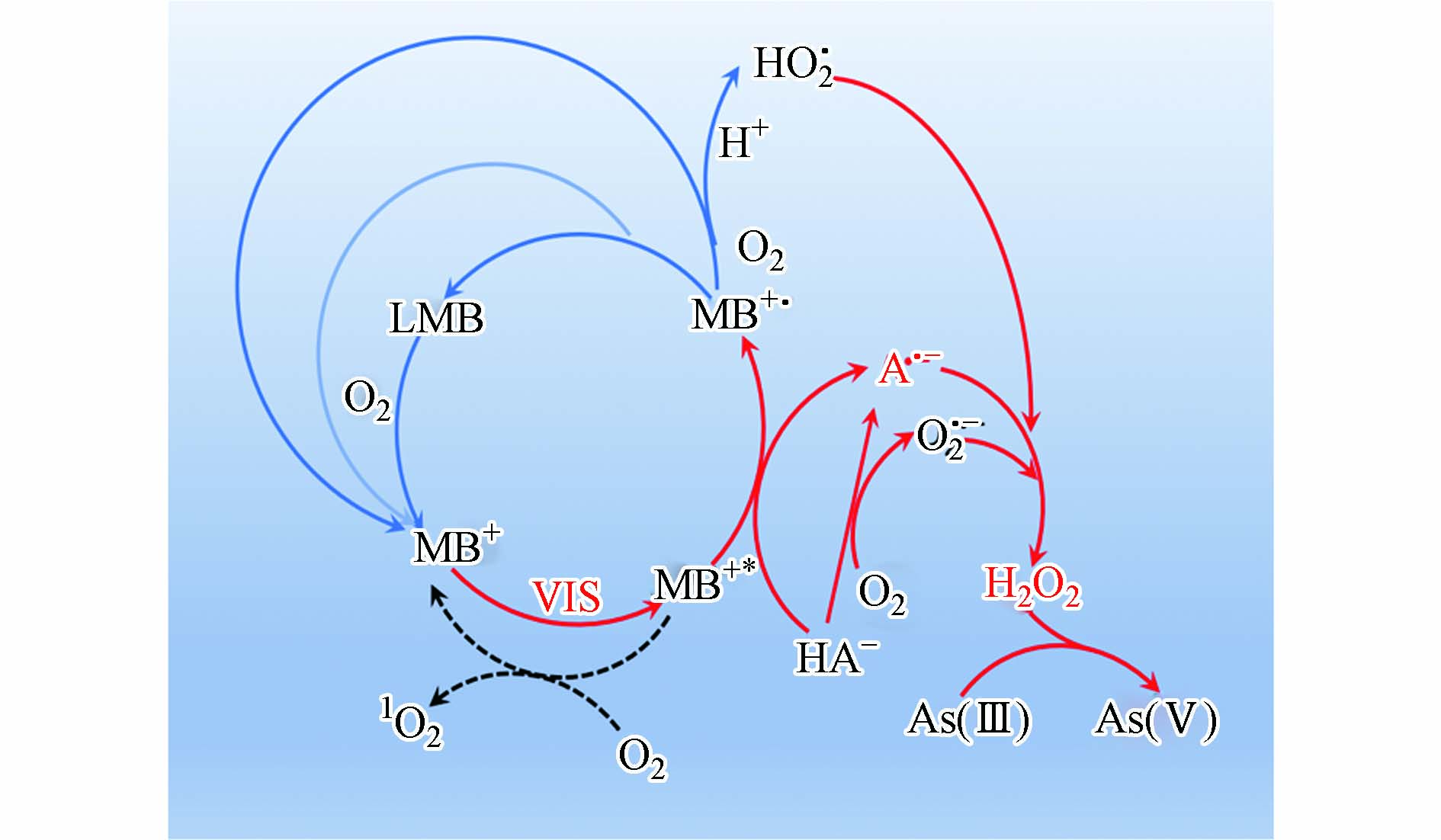 图 10 可见光/MB+/H2A体系中As(Ⅲ)氧化的机理Figure 10. Mechanism diagram of As(Ⅲ) oxidation in a visible light/ascorbic acid/methylene blue system.
图 10 可见光/MB+/H2A体系中As(Ⅲ)氧化的机理Figure 10. Mechanism diagram of As(Ⅲ) oxidation in a visible light/ascorbic acid/methylene blue system.2.3 常见阴离子与有机质的影响
HCO3−、Cl−、NO3−、FA在土壤间隙水和地下水中普遍存在,HCO3−、Cl−、NO3−、FA对As(Ⅲ)氧化的影响如图11所示,HCO3−、Cl−、NO3−对As(Ⅲ)氧化基本无影响,这是因为HCO3−、Cl−、NO3−加入反应液后并不会使MB+的吸收光谱发生变化,因此不会影响可见光/MB+/H2A体系中As(Ⅲ)的氧化,这和碱/H2A体系中环境基质对As(Ⅲ)氧化的影响一致[8]. 但FA对As(Ⅲ)氧化有一定的影响,这是因为FA加入反应液后使得MB+的吸收光谱发生了显著变化,使得MB+在660 nm处的吸收变弱,从而抑制了MB+和HA−之间的电子转移.
 图 11 可见光/MB+/H2A体系中环境基质(HCO3−、Cl−、NO3−、FA)对As(Ⅲ)氧化的影响Figure 11. Effects of environmental matrices (HCO3−、Cl−、NO3−、FAs) on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.
图 11 可见光/MB+/H2A体系中环境基质(HCO3−、Cl−、NO3−、FA)对As(Ⅲ)氧化的影响Figure 11. Effects of environmental matrices (HCO3−、Cl−、NO3−、FAs) on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.2.4 其他两种噻嗪染料的效果
探讨了将其他噻嗪染料用于可见光/H2A体系活化分子氧氧化As(Ⅲ)的可能性. 劳氏紫和天青B也是常见的噻嗪染料,在可见光/H2A/劳氏紫体系和可见光/H2A/天青B体系中分别对测定了As(Ⅲ)和染料的浓度的变化,结果如图12所示. 实验结果表明,劳氏紫和天青B均能促进H2A活化分子氧产生H2O2氧化As(Ⅲ),但促进效果均不如MB+,这是因为相同浓度下MB+有最大的吸光度值,能更好地吸收能量. 噻嗪染料的相似行为可能是由于它们具有相同的官能团和激发状态. 推测其他光敏剂如核黄素[9]和茜素[27]都具有接受电子的能力,同样可以促进H2A活化分子氧产生H2O2氧化水中的As(Ⅲ).
 图 12 可见光/H2A中噻嗪染料天青B 和劳氏紫对As(Ⅲ)的氧化及自身的降解Figure 12. As(Ⅲ) oxidation induced by thiazide dyes Thionine and Azure B, and degradation of the dyes themselves in visible light/ascorbic acid system.
图 12 可见光/H2A中噻嗪染料天青B 和劳氏紫对As(Ⅲ)的氧化及自身的降解Figure 12. As(Ⅲ) oxidation induced by thiazide dyes Thionine and Azure B, and degradation of the dyes themselves in visible light/ascorbic acid system.3. 结论(Conclusion)
本文从可见光/MB+/H2A活化分子氧体系出发,研究了可见光/MB+/H2A体系氧化水中As(Ⅲ)的可行性与内在机理. 其中,可见光光照、pH值、H2A与MB+的投加量对于As(Ⅲ)的氧化过程有着不同影响. 实验结果表明:
(1)可见光光照可以促进MB+/H2A体系氧化As(Ⅲ); pH越高,As(Ⅲ)氧化越快;pH=9.5下,H2A浓度的增加对As(Ⅲ)氧化呈现先促进后稳定的趋势,且O2浓度对于该体系对As(Ⅲ) 的氧化过程的影响较小. 最终得出该体系下As(Ⅲ)氧化的最佳条件:最佳H2A投加量为300 μmol·L−1;最佳MB+投加量为5 mg·L−1.
(2)通过自由基抑制剂分析验证了可见光/MB+/H2A活化分子氧体系氧化As(Ⅲ)的主要活性物种为H2O2, 1O2对As(Ⅲ)的氧化贡献微乎其微.
(3)同MB+一样,在可见光/MB+体系中添加劳氏紫和天青B这两种噻嗪染料也能促进As(Ⅲ)的氧化.
-
图 1 LED灯发射光谱及MB+、劳氏紫、天青的3种噻嗪染料染料溶液的紫外可见吸收光谱
Figure 1. Emission spectrum of LED lamp and UV-Vis absorption spectra of the solutions of three thiazine dyes (MB+, Thionine and Azure B).
图 2 As(Ⅲ) 氧化的条件实验
Figure 2. Effects of solution pH(a), concentration of H2A(b) and MB+(c)on the oxidation of As(Ⅲ).
图 3 (a)As(Ⅲ)的氧化与(b)MB+的去除动力学曲线
Figure 3. Kinetic curves of As(Ⅲ) oxidation(a) and MB+ removal(b)
图 4 可见光/MB+/H2A体系中,As(Ⅲ)初始浓度对(a)As(Ⅲ)的氧化与(b)As(Ⅲ)的氧化速率的影响
Figure 4. Effect of As(Ⅲ) concentration on oxidation of As(Ⅲ)(a) and oxidation rate of As(Ⅲ)(b) in visible light/ascorbic acid/methylene blue system
图 5 (a) 氮气条件下和(b)有氧搅拌条件下可见光/MB+/H2A体系中溶液光谱变化
Figure 5. Spectral variation of the solution in a visible light/ascorbic acid/methylene blue system under nitrogen conditions(a) and stirring conditions(b)
图 6 可见光/N2和遮光/O2条件下可见光/MB+/H2A体系中MB+吸光度变化
Figure 6. In the visible light/ascorbic acid/methylene blue system under under visible light/N2 and shading/O2 conditions, the absorbance of methylene blue changes.
图 7 可见光/MB+/H2A 体系中自由基抑制剂对As(Ⅲ)氧化的影响
Figure 7. Effect of free radical inhibitors on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue system
图 8 pH 9.5,H2O2 对As(Ⅲ)氧化的影响
Figure 8. Effect of H2O2 on the oxidation of As(Ⅲ) at pH 9.5
图 9 可见光/MB+/H2A体系中氧气浓度对As(Ⅲ)氧化的影响
Figure 9. Effect of oxygen concentration on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.
图 10 可见光/MB+/H2A体系中As(Ⅲ)氧化的机理
Figure 10. Mechanism diagram of As(Ⅲ) oxidation in a visible light/ascorbic acid/methylene blue system.
图 11 可见光/MB+/H2A体系中环境基质(HCO3−、Cl−、NO3−、FA)对As(Ⅲ)氧化的影响
Figure 11. Effects of environmental matrices (HCO3−、Cl−、NO3−、FAs) on As(Ⅲ) oxidation in visible light/ascorbic acid/methylene blue systems.
-
[1] SMEDLEY P L, BRITISH D G K. A review of the source, behaviour and distribution of arsenic in natural waters [J]. Applied Geochemistry, 2002, 17(10): 517-568. [2] PODGORSKI J, BERG M. Global threat of arsenic in groundwater [J]. Science, 2020, 368(6493): 845-850. doi: 10.1126/science.aba1510 [3] MARTINSON C A, REDDY K J. Adsorption of arsenic(Ⅲ) and arsenic(Ⅴ) by cupric oxide nanoparticles [J]. Journal of Colloid and Interface Science, 2009, 336(2): 406-411. doi: 10.1016/j.jcis.2009.04.075 [4] KOCAR B D, INSKEEP W P. Photochemical oxidation of As(III) in ferrioxalate solutions [J]. Environmental Science & Technology, 2003, 37(8): 1581-1588. [5] LEE H, CHOI W. Photocatalytic oxidation of arsenite in TiO2 suspension: Kinetics and mechanisms [J]. Environmental Science & Technology, 2002, 36(17): 3872-3878. [6] YEO J, CHOI W. Iodide-mediated photooxidation of arsenite under 254 nm irradiation [J]. Environmental Science & Technology, 2009, 43(10): 3784-3788. [7] SHUMLAS S L, SINGIREDDY S, THENUWARA A C, et al. Oxidation of arsenite to arsenate on birnessite in the presence of light [J]. Geochemical Transactions, 2004, 69(9): 726-732. [8] 王震华, 罗涛, 李进军, 等. 碱/抗坏血酸活化分子氧氧化水中As(Ⅲ) [J]. 环境科学学报, 2022, 42(7): 225-233. WANG Z H, LUO T, LI J J, et al. Oxidation of As(Ⅲ) in water by alkaline/ascorbic acid activated molecular oxygen [J]. Acta Scientiae Circumstantiae, 2022, 42(7): 225-233(in Chinese).
[9] KEARNS D R. Physical and chemical properties of singlet molecular oxygen [J]. Chemical Reviews, 1971, 71(4): 395-427. doi: 10.1021/cr60272a004 [10] HUANG R, CHOE E, MIN D B. Kinetics for singlet oxygen formation by riboflavin photosensitization and the reaction between riboflavin and singlet oxygen [J]. Journal of Food Science, 2006, 69: 44. [11] TANIELIAN C, HEINRICH G. Effect of aggregation on the hematoporphyrinsensitized production of singlet molecular oxygen [J]. Photochemistry and Photobiology, 1995, 61(2): 131-135. doi: 10.1111/j.1751-1097.1995.tb03950.x [12] TANIELIAN C, WOLFF C, ESCH M. Singlet oxygen production in water: Aggregation and charge-transfer effects [J]. The Journal of Physical Chemistry, 1996, 100(16): 6555-6560. doi: 10.1021/jp952107s [13] RONZANI F, TRIVELLA A, ARZOUMANIAN E, et al. Comparison of the photophysical properties of three phenothiazine derivatives: Transient detection and singlet oxygen production [J]. Photochemical & Photobiological Sciences, 2013, 12(12): 2160-2169. [14] 刘德启, 汪守建, 江飞. 均相亚甲基蓝光敏氧化法处理造纸废水研究 [J]. 水处理技术, 2004, 30(5): 273-275. LIU D Q, WANG S J, JIANG F. Treatment of paper-making wastewater by methyleneblue photochemical method [J]. Technology of Water Treatment, 2004, 30(5): 273-275(in Chinese).
[15] BONNEAU R, JOUSSOT DUBIEN J, FAURE J. Mechanism of photoreduction of thiazine dyes by edta studied by flash‐photolysis‐I [J]. Photochemistry and Photobiology, 1973, 17(5): 313-319. doi: 10.1111/j.1751-1097.1973.tb06359.x [16] LEE S K, MILLS A. Novel photochemistry of leuco-methylene blue [J]. Chemical Communications, 2003(18): 2366-2367. doi: 10.1039/b307228b [17] LUO T, WANG H, CHEN L, et al. Visible light-driven oxidation of arsenite, sulfite and thiazine dyes: A new strategy for using waste to treat waste [J]. Journal of Cleaner Production, 2021, 280: 124374. doi: 10.1016/j.jclepro.2020.124374 [18] FRIDOVICH I, HANDLER P. Detection of free radicals in illuminated dye solutions by the initiation of sulfite oxidation [J]. The Journal of Biological Chemistry, 1960, 235(6): 1835-1838. doi: 10.1016/S0021-9258(19)76891-4 [19] 韩昌序, 于海艳, 马利民. 抗坏血酸活化过硫酸盐降解水体中的阿特拉津 [J]. 环境科技, 2021, 34(2): 33-37. HAN C X, YU H Y, MA L M. Degradation of atrazine in water by ascorbic acid activated persulfate [J]. Environmental Science and Technology, 2021, 34(2): 33-37(in Chinese).
[20] CAO M H, HOU Y Z, ZHANG E, et al. Ascorbic acid induced activation of persulfate for pentachlorophenol degradation [J]. Chemosphere, 2019, 229: 200-205. doi: 10.1016/j.chemosphere.2019.04.135 [21] BUETTNER G R, DOHERTY T P, BANNISTER T D. Hydrogen peroxide and hydroxyl radical formation by methylene blue in the presence of ascorbic acid [J]. Radiation and Environmental Biophysics, 1984, 23(4): 235-243. doi: 10.1007/BF01407595 [22] LUO T, PENG Y, CHEN L, et al. Metal-free electro-activated sulfite process for As(Ⅲ) oxidation in water using graphite electrodes [J]. Environmental Science & Technology, 2020, 54(16): 10261-10269. [23] MILLS A, WANG J. Photobleaching of methylene blue sensitised by TiO2 : An ambiguous system? [J]. Journal of Photochemistry and Photobiology A:Chemistry, 1999, 127(1-3): 123-134. doi: 10.1016/S1010-6030(99)00143-4 [24] BOLOBAJEV J, TRAPIDO M, GOI A. Improvement in iron activation ability of alachlor Fenton-like oxidation by ascorbic acid [J]. Chemical Engineering Journal, 2015, 281: 566-574. doi: 10.1016/j.cej.2015.06.115 [25] LEI Y, ZHANG H, WANG J W, et al. Rapid and continuous oxidation of organic contaminants with ascorbic acid and a modified ferric/persulfate system [J]. Chemical Engineering Journal, 2015, 270: 73-79. doi: 10.1016/j.cej.2015.02.014 [26] 李苏奇, 刘子正, 王宇楠, 等. 单线态氧氧化水中对氨基苯胂酸的研究 [J]. 环境科学学报, 2016, 36(8): 2852-2858. LI S Q, LIU Z Z, WANG Y N, et al. The oxidation of p-arsanilic acid by singlet oxygen in water [J]. Acta Scientiae Circumstantiae, 2016, 36(8): 2852-2858(in Chinese).
[27] MECH J, GRELA M A, SZACIŁOWSKI K. Ground and excited state properties of alizarin and its isomers [J]. Dyes and Pigments, 2014, 103: 202-213. doi: 10.1016/j.dyepig.2013.12.009 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2474
- HTML全文浏览数: 2474
- PDF下载数: 42
- 施引文献: 0
