



-
砷元素主要经地质活动等过程释放入大气、土壤和水体等介质,造成环境砷污染[1-3]。截止2012年,全球超过2亿人处于饮水型慢性高砷暴露[4],我国有近2000万人暴露砷超过WHO安全水平(10 μg·L−1)[5]。流行病学结果显示,人群通过摄食、饮水等途径长期暴露环境砷可导致皮肤癌、膀胱癌、肺癌等恶性疾病[6-7]。此外,砷暴露还会加速糖尿病、心血管疾病等慢性疾病的病程[8-9]。美国有毒物质和疾病登记署(ATSDR)在2011年优先风险物质清单上将砷列为头号公共健康威胁,国际癌症研究机构(IARC)也将砷化合物归为一类(Group 1)致癌物[7]。可见,砷污染引起了全球的环境健康危害。然而,砷暴露致毒/癌的确切分子机制目前仍不明确。
环境介质中砷化合物主要以三价无机砷(iAsⅢ)和五价无机砷(iAsⅤ)形态存在,而无机砷在环境微生物的作用下可转化为三价二甲基砷(DMAⅢ)和一甲基砷(MMAⅢ)、五价二甲基砷(DMAⅤ)和一甲基砷(MMAⅤ)等多种有机砷形态[7]。当环境中的无机砷进入生物体后,通过甲基化、硫化等代谢过程,可进一步转化为多种甲基化和硫化形态,而不同形态砷化合物的物理化学性质差别较大,毒性也不尽相同[10-11](如表1)。一般认为,五价砷化合物的毒性低于三价砷化合物,有机砷的毒性比无机砷小。但也有例外,如DMAⅢ和MMAⅢ的毒性比iAsⅢ更强[10],五价二甲基一硫代砷酸(DMMTAV)表现出与三价无机砷和甲基砷相当的高毒性[10-11]。
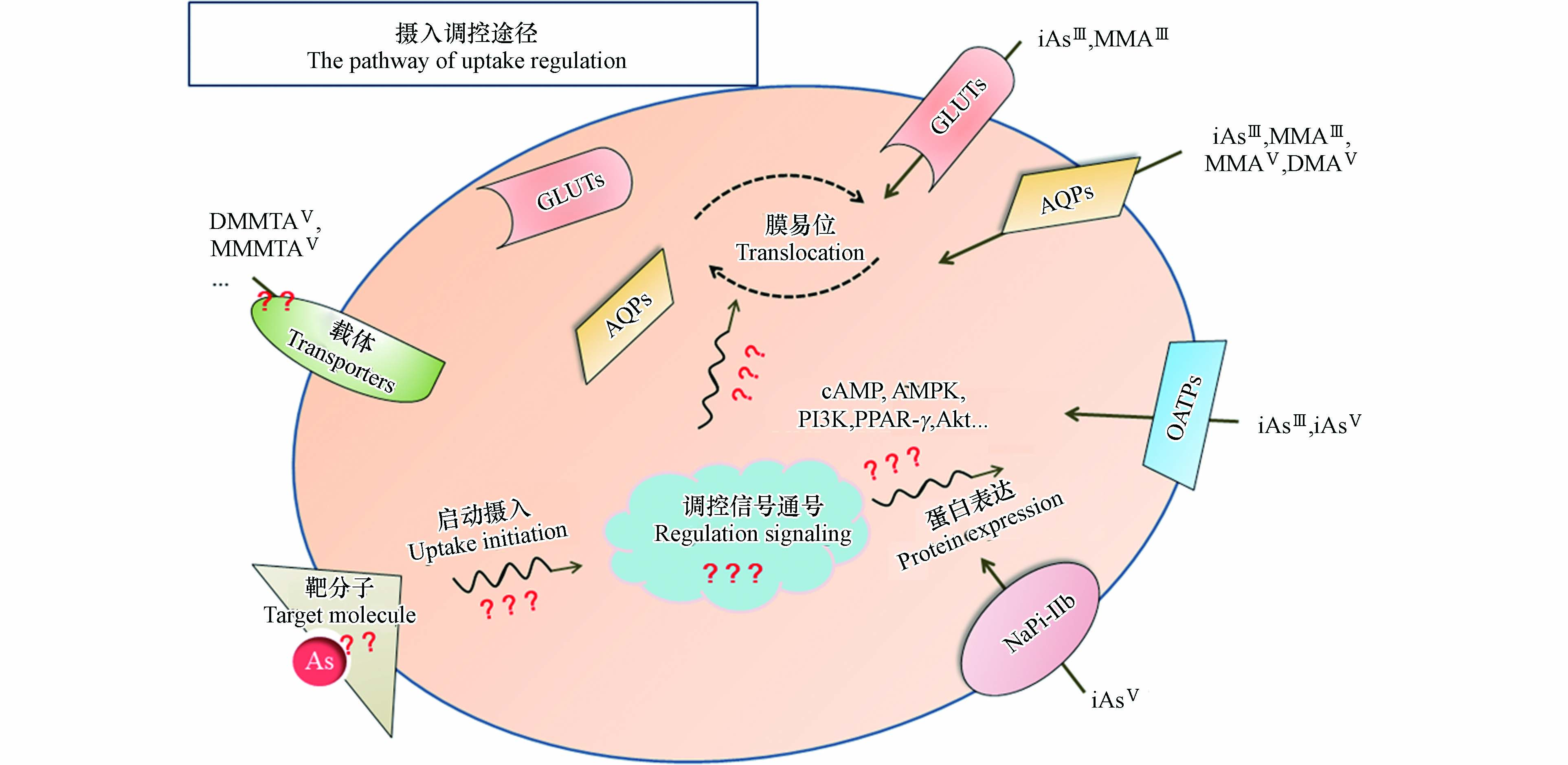
目前,关于砷暴露致毒/癌分子机制的讨论主要集中在两方面:1)砷化合物可诱导产生活性氧(ROS),引起氧化应激、能量代谢异常、DNA链发生断裂或缺失突变等,从而诱导细胞癌变[12-13]。2)砷化合物可通过半胱氨酸残基与蛋白质结合[14-15],改变蛋白质构象,影响蛋白功能[16-17]。以上推测机制只关注了砷在胞内的作用途径,并未考虑砷化合物的摄入调控过程对其毒性效应的影响。一方面,砷化合物的摄入是其与细胞作用的第一步,摄入途径、摄入剂量等都直接影响砷化合物在胞内的暴露特征(形态、分布、浓度),影响胞内的生化作用/反应;另一方面,已有文献报道部分砷形态如DMMTAV、DMAⅢ和MMAⅢ的摄入、分布与其毒性密切相关[18-19]。可见,砷的分子摄入途径及摄入形态/浓度的调控机制对其致毒/癌效应具有重要影响。因此,本文对不同砷形态的摄入行为(摄入载体类型、载体调控通路和摄入模式)和毒性效应等研究结果进行归纳,发现介导砷摄入的载体蛋白类型存在一定的形态依赖性,如iAsV主要借助钠依赖性磷酸转运蛋白NaPi-IIb进入细胞,而iAsⅢ和MMAⅢ等则主要通过水通道蛋白AQPs介导入胞。介导载体的调控则主要通过改变载体表达水平或定位状况来实现,相关调控通路涉及细胞增殖、分化、凋亡等生命过程。同时,细胞摄入砷的浓度水平、空间分布及毒性效应规律存在一致性。本文强调了砷摄入对其暴露致毒/癌的重要作用,为砷暴露致毒/癌机制研究提供了新的视角和思路,同时针对摄入调控机制中待研究的内容也提出了自己的见解。
-
砷不具备特异性的摄入系统,在生理条件下,部分砷化合物的结构与磷酸根、葡萄糖和甘油等天然底物相近,因此可借由对应的转运载体被细胞吸收[20]。截至目前,发现与砷化合物摄入相关的载体主要包括磷酸盐转运蛋白、水甘油通道蛋白、葡萄糖转运蛋白和有机阴离子转运蛋白等四大类[21],它们对砷化合物的转运呈现出形态依赖的特点。此外,部分砷形态例如iAsⅢ和三价一甲基砷的谷胱甘肽复合物[MMAⅢ(GS)2]等由于具有较高的辛醇-水分配系数和脂-水分配系数(0.04—0.13),理论上可通过简单扩散入胞,但相关研究较少,还需更多实验数据支撑[22]。
-
砷(As)和磷(P)都是第五周期主族元素,五价砷酸与磷酸的三级电离平衡常数较为接近,在生理pH值下,两者主要以二氢酸根和一氢酸根的混合形式存在。因结构类似,它们在细胞摄入时存在一定的竞争[23-24]。在非洲爪蟾卵母细胞表达系统中,发现了5种可能与iAsV摄入有关的磷酸盐转运蛋白亚型,即NaPi-Ⅱa、NaPi-Ⅱb、NaPi-Ⅱc、PiT1和PiT2。在该表达系统中,检测到小鼠、大鼠、人的NaPi-Ⅱb亚型对iAsV和P显示出相近的亲和力,对应Km值分别为57 、51、9.7 µmol·L−1[24-25]。在肠上皮细胞表达体系中,人结直肠腺癌Caco-2细胞暴露iAsV 24 h和48 h后,NaPi-Ⅱb的mRNA显著上调(NaPi-Ⅱb较NaPi-Ⅱa高2倍)[26],提示NaPi-Ⅱb很可能是人体摄入无机砷的重要途径。
-
目前在哺乳动物中已鉴定出13种水通道蛋白(AQPs),其中AQP3、AQP7、AQP9和AQP10被归为水甘油通道蛋白[27]。除水分子外,它们还可顺浓度梯度转运甘油、尿素等中性小分子[28]。生理pH值下,iAsⅢ主要以未解离的中性As(OH)3分子形式存在[29],因此,理论上也可通过AQPs介导被细胞摄入。在非洲爪蟾表达体系中,人源水甘油通道蛋白AQP3(hAQP3)、hAQP7和hAQP9对iAsⅢ的渗透效率分别是水分子的2倍、5倍和25倍[30]。近来,在人早幼粒急性白血病HL-60细胞、人慢性骨髓性白血病K562细胞和人急性T淋巴细胞白血病Jurkat细胞等10种髓系白血病和淋巴系白血病细胞中发现,AQP9的表达与iAsⅢ诱导的细胞毒性呈正相关性,其中Jurkat和HL-60细胞中AQP9的mRNA和蛋白水平相对较低,同时砷耐受性较强[31]。当AQP9转染到iAsⅢ不敏感的K562细胞和人肝癌Hep3B细胞中后,iAsⅢ诱导的细胞毒性明显增强[31],表明AQP9蛋白与iAsⅢ的摄入和毒性密切相关。在AQP7和AQP9未表达的体系中,AQP3在介导iAsⅢ摄入细胞/组织中发挥重要作用。例如人膀胱癌EJ-1细胞和人皮肤鳞癌A431细胞均未表达AQP7和AQP9,但A431细胞显著表达AQP3,在iAsⅢ相同条件暴露下,对A431引起更高毒性[10, 32]。以上研究结果均表明,AQP3、AQP7和AQP9等水通道蛋白可作为iAsⅢ的转运载体,促进不同体系对三价无机砷的摄入[33],增强砷的累积和毒性。
除AQPs外,有机阴离子转运蛋白(OATPs)也被报道与无机砷的转运有关。OATPs是SLCO基因编码的溶质载体超家族(SLC)成员[34]。人体内已发现的OATPs有11种,主要负责内、外源性溶质如胆红素、前列腺素、类固醇、药物等的运输,转运特点为顺浓度梯度的被动运输[35]。现有关于OATPs参与砷转运的报道相对较少,研究结果提示OATP1B1和OATP2B1可能在砷迁移过程中起作用,其中OATP1B1主要与iAsⅢ和iAsV的转运有关[36],而OATP2B1则参与iAsⅢ的转运[26]。
葡萄糖转运体(GLUTs)也被报道与三价无机砷的摄入相关[26]。GLUTs是SLC2A基因编码的SLC家族成员,主要负责转运包括葡萄糖、半乳糖、果糖在内的单糖入胞。已发现的人类GLUTs亚型共有14种,除HMIT/SLC2A13外,均为促进性转运载体,可介导渗透物顺浓度梯度入胞[37]。GLUT1、GLUT2和GLUT5这3种亚型与砷化合物的生物摄入相关,其中GLUT2和GLUT5与三价无机砷的转运相关。iAsⅢ可诱导Caco-2细胞中的GLUT2和GLUT5表达上调,而沉默GLUT5基因可使细胞摄入iAsⅢ的效率降低约2.5倍[26]。
-
摄入相关数据显示AQP9对三价有机砷的摄入起重要作用。在酿酒酵母表达体系中,哺乳动物源AQP9对三价有机砷MMAⅢ的转运效率几乎是iAsⅢ的3倍[38]。在表达hAQP9的非洲爪蟾卵母细胞中,AQPs抑制剂Hg2+可同时抑制iAsⅢ和MMAⅢ基于AQP9的介导入胞[39]。
GLUTs也被报道与三价有机砷的摄入相关[40]。在转染表达大鼠GLUT1(rGLUT1)的酵母菌中,iAsⅢ暴露累积量较空载对照增加1.7倍,大鼠rGLUT1对MMAⅢ的转运水平比iAsⅢ高40倍,比空载体高7倍[40]。此外,rGLUT1对MMAⅢ(Km = 1.2 mmol·L−1)的亲和力与其天然底物葡萄糖(Km = 3 mmol·L−1)相似,且两者基于GLUT1的转运表现出非竞争性抑制的特征,提示MMAⅢ利用了与葡萄糖不同的初始结合位点或转运路径。在非洲爪蟾卵母细胞表达体系中,rGLUT1对MMAⅢ的转运水平约为人源GLUT1(hGLUT1)的4倍,说明不同来源的GLUT1对于MMAⅢ转运效率差异较大。
-
水通道蛋白AQPs也可转运五价有机砷[39],非洲爪蟾卵母细胞表达hAQP9后,对MMAV和DMAV等五价有机砷表现出pH依赖的摄入行为。pH值在5.5—7.5范围内,MMAV和DMAV的细胞摄入效率随pH值升高而降低。此外,AQP9还可介导MMAV和DMAV的细胞排出[39]。目前关于五价硫代砷等新型砷形态的摄入研究还相对较少,涉及的摄入载体类型尚不明确。
砷摄入载体的相关研究主要是通过考察不同表达体系中砷化合物摄入与不同载体蛋白表达的相关关系,从而判断不同砷形态的摄入载体类型和相应摄入模式。同时,以上数据也说明了通过人为调控载体蛋白水平可以在很大程度上改变/调控砷形态的摄入效率、速率等摄入行为。
-
细胞对砷的摄入载体调控主要是通过改变载体表达量或膜定位情况两种途径来实现,调控信号通路主要涉及增殖、分化和凋亡以及氧化应激、能量代谢等细胞过程[41-42],依据载体类型将调控途径归纳如下。
-
AQP3广泛分布于肾脏、皮肤、眼结膜、脾、胃、膀胱、呼吸道[27]等器官组织中。AQP7则主要存在于人肾脏和白色脂肪组织(WAT)、大鼠/小鼠WAT和棕色脂肪组织(BAT)中[43-45]。AQP9的表达模式存在物种差异,大鼠AQP9主要分布在大脑和睾丸间质细胞内,人类AQP9存在于外周血白细胞内以及白细胞含量较高的器官如肺、脾和骨髓中,AQP9在其他动物的肝脏、附睾、皮肤等器官中也有表达[27]。
真核生物主要通过以下3种途径调节AQPs的转运。第一,在mRNA和蛋白水平调控AQPs的表达。细胞表皮生长因子(EGF)通常被认为是癌细胞中AQP3上调表达的调控因子[46],在结直肠癌[47]、卵巢癌[48]和胰腺癌等细胞[49]中,EGF对AQP3的上调诱导呈现出时间和剂量依赖的特征。除EGF外,雌激素也可通过激活AQP3基因启动子中的雌激素反应元件(ERE)上调AQP3的mRNA水平[50]。第二,通过改变AQPs构象或门控分子机制调控其活性。现有植物和微生物AQPs的相关报道表明,AQPs可以通过门控机制进行活性调节,使其构象上呈现出打开或关闭的不同状态,如菠菜水通道蛋白的磷酸化、pH和Ca2+门控分子机制[51],以及酵母水通道蛋白Aqy1的机械敏感性[52]等均可实现对AQPs介导的转运调控。第三,通过调节AQPs转运-嵌入细胞膜的易位过程,调控AQPs的介导入胞效率[53]。A类G蛋白偶联受体(GPCRs)激动剂如肾上腺素能受体(α-AR)激动剂-肾上腺素可使蛋白激酶C(PKC)磷酸化,从而增加Caco-2细胞[54]中AQP3的细胞膜易位,调控AQP3的转运。GPCR的下游信号通路分子如腺苷酸环化酶(AC)、环磷酸腺苷(cAMP)、过氧化物酶体增殖物激活受体(PPAR-γ)、磷酸腺苷酸活化蛋白激酶(AMPK)、丝氨酸-苏氨酸蛋白激酶(Akt/PKB)等可以调节AQPs的膜丰度,例如诱导AQP3和AQP7在人肝癌HuH7细胞中上调表达[55],调控AQPs的介导转运。磷酸化AMPK和Akt/PKB等还可抑制人肝母细胞瘤HepG2细胞中的AQP9基因表达[56]。可见,GPCR可通过调控下游信号通路调控AQPs转运蛋白的表达及膜定位状态,提示它有可能是砷胁迫下的生物靶分子。
-
人源GLUTs一般由500多个氨基酸残基组成,具备12个跨膜域、1个单一的N-连接糖基化位点、1个相对较大的中央细胞质连接域,它的N端和C端位于细胞质的拓扑结构域。11种GLUTs亚型均能参与葡萄糖转运。其中,GLUT1在小肠、肾脏组织中有一定表达,主要负责果糖转运;GLUT2分布在肝、胰岛、肠、肾、脑等组织,负责葡萄糖、半乳糖、果糖、甘露糖的转运;GLUT5则主要转运果糖,分布在小肠和肾脏[37]。
与AQPs介导运输类似,GLUTs的介导转运也可通过调节DNA转录表达或其膜丰度来调控。GLUT1在转录水平上受到多种机制的调控,例如转录因子骨髓细胞瘤病毒癌基因(c-Myc)和同源异型框基因(SIX1)可直接转录激活GLUT1相关基因[57-58],从而促进细胞的糖酵解过程。此外,GLUT1转运蛋白的表达和膜定位还可受与GPCRs和受体酪氨酸激酶(RTKs)等膜受体相关的信号通路,如磷脂酰肌醇3-激酶(PI3K)/雷帕霉素靶蛋白(mTOR)/Akt、cAMP、AMPK等的调控[59-60]。部分RTKs如EGFR[60]和胰岛素受体(IR)可参与GLUT1膜丰度的调控[59-60]。在EGFR突变的肺腺癌细胞中,抑制PI3K/mTOR通路,可降低糖酵解和戊糖磷酸途径的早期代谢物葡萄糖-6-磷酸和6-磷酸葡糖酸的水平,阻碍GLUT1向细胞膜转运和膜定位[59]。PI3K及下游信号通路蛋白Akt对调节GLUT1的转移、表达及活性也十分重要。在淋巴细胞中,白介素3(IL-3)可激活PI3K调节GLUT1蛋白转移至细胞膜表面,Akt的活化则可独立促进GLUT1的循环及活性,抑制GLUT1内化[61]。此外,IR的激活可促进AMPK和Akt诱导硫氧还蛋白互作蛋白(TXNIP)的磷酸化,上调GLUT1的mRNA表达,同时减少结合GLUT1,增加GLUT1的膜易位,多方面促进GLUT1的介导运输[60]。除RTKs之外,部分A类GPCR如棕色脂肪组织中的β3肾上腺素能受体(β3-AR)可通过调节下游信号分子cAMP的形成,介导GLUT1转录合成,从而增加葡萄糖分子的摄取[62]。
-
SLC家族覆盖数百种蛋白,可介导不同亲水性和亲脂性的小分子或溶质穿过细胞质膜完成跨膜运输,其中,有机阴离子药物转运蛋白OATPs在运输内、外源性药物方面起重要作用[34]。ABC超家族(ABC)或SLC超家族对药物转运的功能主要取决于质膜上载体蛋白的表达量。除了特征明确的转录修饰外,转运活性还会受到翻译后修饰的影响,从而使载体蛋白的定位和活性发生改变[63]。早期研究表明,肝脏特异性的OATP1B1和OATP1B3表达受肝细胞核因子1α(HNF-1α)控制[64]。在人肝癌HCCLM3细胞中,OATP1B3表达水平降低,OATP1B1的mRNA表达正常,但蛋白水平略有降低。在过表达肝细胞核因子3β(HNF-3β)的HCCLM3细胞中,OATP1B3转录表达受到抑制,但不影响OATP1B1转录表达,说明HNF-3β可在HCCLM3中选择性调控OATP1B3的表达[65]。此外,OATP2B1的功能还受GPCR下游信号通路PKC的介导调控,激活PKC将导致OATP2B1磷酸化增加,并降低OATP2B1的运输能力[63]。
综上,细胞可通过增殖、分化、凋亡等相关通路来调节砷摄入载体如AQPs、GLUT和OATPs的表达或易位,具体信息总结如表2。
然而,砷胁迫下的载体调控情况仍未见报道,具体调控途径尚并不明确。此外,砷摄入调控过程中的其他重要环节,例如不同砷形态的靶分子及响应方式,载体蛋白经调控对下游砷摄入行为的影响等,都有待进一步的系统研究(如图1)。
-
砷化合物的摄入、分布及毒性效应具有显著的形态依赖性。在已知砷形态中,iAsⅢ被认为是毒性最高、危害最大的形态,五价砷化合物如iAsⅤ、MMAⅤ和DMAⅤ的毒性显著低于三价砷化合物如iAsⅢ、MMAⅢ和DMAⅢ[66]。五价硫代有机砷中,五价一甲基一硫代砷酸(MMMTAV)与iAsV相似,二甲基二硫代砷酸(DMDTAV)与DMAV相似,显示出较低的细胞毒性[10-11],DMMTAV与iAsⅢ、DMAⅢ相似,与蛋白质中的硫醇基团具有较强亲和力[67],可通过结合功能蛋白,导致氧化应激,产生较高毒性[10-11]。
-
砷的体内分布和毒性高度依赖于砷化学形态及实验物种。不同动物对iAsⅢ的毒性敏感度不同,大鼠、小鼠和仓鼠等对砷的敏感性相对较高[68]。小鼠的主要砷累积器官为膀胱、肾脏和皮肤,代谢最慢的组织为皮肤,这与砷暴露致癌的靶器官相一致[69]。无机砷iAsV和iAsⅢ大多通过肠道被吸收,通过尿液被排出[70]。小鼠经口服iAsV急性暴露24 h后,肝、肾中iAsV的浓度最高[71],各组织中还检出MMAⅢ、MMAV、DMAⅢ、DMAV等砷形态,最终主要以DMAV经尿液排出体外[71]。慢性饮水暴露[73As]砷酸盐9 d后,除以上甲基砷形态外,小鼠肝脏中还检出了TMAOV形态[69]。由于肝脏内富含甲基供体S-腺苷甲硫氨酸(SAM)、砷甲基转移酶(AS3MT)[72]和多种氧化还原酶,无机砷可在肝脏内发生一系列的甲基化反应,并转化为相应形态[67],提示肝脏是砷的主要代谢器官。大鼠和仓鼠经静脉注射DMMTAV和DMDTAV后,DMMTAV与DMAⅢ两种化合物分布特征类似,主要滞留在红细胞,并大量累积于肝、肾、肌肉等器官,少部分累积于皮肤;DMDTAV的分布特征与DMAV类似,少量分布在肝、肾、皮肤,DMDTAV在肌肉的停留时间比DMAV更长,后以DMAV形态随尿排出[18-19]。仓鼠口服暴露iAsⅢ后,砷在红细胞中的累积率(<0.8%)远小于大鼠(75%)[73],同时,仓鼠体内并不存在明显的靶器官,尿液排泄砷的速率也更快,说明红细胞是砷通过代谢滞留在靶器官的关键。
慢性砷暴露会造成多种组织器官的毒性效应,而毒性症状通常首先表现在皮肤上。砷通过影响烟酰胺腺嘌呤二核苷酸磷酸氧化酶(NADPH oxidase)等负责维持氧化还原稳态的蛋白质/酶来诱发氧化应激,三价砷比五价砷诱导更多的DNA氧化损伤和更高频率的染色体畸变,表现出更强的基因毒性[12,74]。此外,三价砷等毒性较高的砷形态更易在富含角蛋白的组织如皮肤等累积[17],导致皮肤色素沉着、角化过度、鲍文氏病、鳞状细胞癌和基底细胞癌等病理表型[6]。砷暴露会影响细胞外基质中金属蛋白酶-9(MMP-9)、Clara细胞16蛋白(CC16)等蛋白分子的表达,使肺上皮沉积及发生永久性结构改变。随着砷暴露量的增加,组织炎症和肺形态变化进一步加剧,最终导致支气管炎、间质性肺病、慢性阻塞性肺疾病和肺鳞状细胞癌等疾病[75]。高毒性砷化合物如DMMTAV和DMAⅢ因较难代谢而累积在肾脏和肝脏等人体重要的代谢和解毒器官中,诱导肝酶水平升高,使丙二醛和细胞色素P450、谷胱甘肽(GSH)等水平降低,导致肝血管损伤、肝硬化等肝脏疾病[76-77]。砷还可通过对白介素8(IL-8)的表观遗传调控及其他相关细胞通路的调节增加肾脏细胞迁移和增殖能力,使其细胞周期失调并产生肾毒性[78],导致肾小管上皮细胞变性和局部矿化、尿液体积和pH值增加、电解质水平降低、尿钙水平和水消耗的增加、膀胱细胞损伤等[79]。
-
砷化合物的细胞毒性、摄入规律与其在体内暴露的相关规律基本一致。两种无机砷暴露小鼠成纤维L-A细胞时,iAsⅢ的摄入效率比iAsV高约20倍,同时iAsⅢ暴露会影响细胞增殖,降低细胞活力,增加LDH释放,刺激乳酸产生,表现出比iAsV暴露高10倍的细胞毒性[80]。甲基化以往被认为是三价无机砷iAsⅢ的主要生物解毒途径,然而,随着三价甲基砷[81]和五价硫代砷代谢物的检出,毒理学研究结果揭示三价甲基砷如DMAⅢ和五价硫代甲基砷DMMTAV具有与高毒iAsⅢ相当的细胞毒性[82,10-11]。EJ-1细胞对各砷形态的吸收量及暴露产生的细胞毒性一致,顺序为DMAⅢ≈DMMTAV>iAsⅢ>>MMAV≈DMAV≈DMDTAV[11],同时,DMMTAV暴露组细胞膜的总砷浓度最高,比DMAⅢ和iAsⅢ暴露组分别高出2倍和4倍[11],提示DMMTAV可能比三价iAsⅢ和五价甲基砷化合物具有更高的细胞膜通透性。A431细胞对三价砷iAsⅢ和DMAⅢ的摄入水平比五价砷iAsV和DMAV分别高出7倍和14倍,DMMTAV的细胞摄入量比iAsⅢ高出2倍。以上五种砷化合物的毒性大小与摄入水平一致,排序依次为DMAⅢ > iAsⅢ > DMMTAV >>iAsV > DMAV[10]。虽然DMAⅢ、DMMTAV和iAsⅢ三种形态都有较高的细胞摄入量,但各自的吸收方式并不相同。iAsⅢ和DMAⅢ暴露后,砷元素主要分布在消解后的细胞沉淀中,而DMMTAV暴露组主要分布在消解后的细胞上清中[10],提示DMMTAV可能以不同于三价砷化合物的方式/途径被细胞摄入累积。
高毒性的砷化合物如iAsⅢ、DMMTAV、DMAⅢ、MMAⅢ因细胞摄入/累积效率较高、体内滞留时间较长,同时它们易于与重要的生物大分子作用,扰乱正常的生化反应或诱发氧化应激,产生较高毒性;低毒砷化合物如DMDTAV、DMAV等因细胞摄入、累积率较小、代谢速率较快,与生物分子的相互作用也较弱,因此产生的毒性也相对较小。可见,砷化合物的摄入特征(量、效率、分布等)与其体内外毒性有正相关性,因此明确砷摄入的调控机制,对于阐明砷化合物致毒/癌的分子作用途径提供了重要理论依据。
-
综上所述,砷化合物的摄入行为(量、效率、分布等)与其体内外毒性密切相关,砷可借由其他天然底物或药物载体蛋白进入细胞,摄入行为具有显著的形态依赖性。iAsV主要借助钠依赖性磷酸盐转运蛋白NaPi-IIb进入细胞;iAsⅢ和MMAⅢ,MMAV和DMAV则主要通过水通道蛋白AQPs介导入胞;葡萄糖转运蛋白GLUTs也可辅助MMAⅢ等三价砷化合物进入细胞。相关载体表达和膜易位状况与细胞增殖、分化和凋亡以及细胞的氧化应激、能量代谢等过程的调控相关。以上调控过程都将影响砷摄入的效率、选择性以及在体内外的分布、累积和毒性效应。本文强调砷化合物的摄入调控是其致毒/癌的重要调控机制之一,为深入理解砷的致毒/癌机制提供了新的视角和启发。然而,截至目前,砷的整个摄入调控过程仍有诸多环节并不明确,例如:1)砷摄入调控的启动过程不清(生物靶点、响应/启动方式);2)新型砷形态(如含硫砷等)的转运载体蛋白未明;3)相关载体蛋白在砷胁迫下的调控机制鲜有报道等等。以上过程均为砷摄入调控的关键步骤,对于深入理解砷摄入调控机制,及进一步全面阐释砷暴露致毒/癌分子机制十分重要,值得进一步探讨研究。
砷化合物的摄入调控机制
The regulation mechanism of the arsenical uptake
-
摘要: 流行病学结果显示慢性砷暴露可导致人群罹患皮肤癌、膀胱癌、肺癌等恶性疾病,但其致毒/癌机制尚不明确。目前关于砷暴露致毒/癌机理的讨论主要集中在砷的胞内作用途径,而较少关注砷摄入调控过程对其暴露致毒/癌的贡献。在生理条件下,部分砷化合物由于结构与磷酸根、葡萄糖、甘油等天然底物相近,可借由相应的载体被细胞摄入,摄入途径和效率存在显著的砷形态依赖性。此外,砷化合物的生物毒性效应与其赋存形态直接相关。可见,砷的摄入调控对于砷的暴露致毒/癌具有重要作用。本文主要综述了在哺乳动物体系中不同砷形态的摄入载体、载体调控及对应的砷摄入分布、效率和暴露毒性,在此基础上,强调了以往在砷致毒/癌机制研究中被忽视的砷摄入调控途径。然而,砷摄入调控过程中的诸多重要环节如砷胁迫下的摄入启动和调控机制等都是空白,需进一步系统深入地研究,为深入理解砷的致毒机制提供了新的视角和研究思路。Abstract: The epidemiological results demonstrate that chronic exposure to inorganic arsenic is closely associated with the malignant diseases of skin, bladder and lung cancer, however, the molecular toxicity mechanism has not been clarified. The related studies mainly focus on the interaction of arsenicals inside cells, while the transmembrane transport of arsenicals has been overlooked. Some arsenicals can enter into cells by the transporters specific for the natural substrates of phosphate, glucose and glycerol, because of their similar structure under the physiological conditions. Furthermore, the transport pathway and efficiency are significantly dependent on the arsenic species. Meanwhile, the toxic effects upon arsenic exposure vary a lot with different species. Thus, it is speculated that the regulation of arsenical uptake plays an important role in the toxicological/carcinogenic effects. Herein, we reviewed the transporters and their regulation signaling in mammalian system for transmembrane transport of different arsenic species. And the uptake efficiency, distribution, and resultant toxicity of arsenicals in terms of species has also been summarized. In this term, the uptake regulation of arsenicals has been emphasized in the research of the toxicological/carcinogenic mechanism upon arsenic exposure. Nevertheless, several key steps in the course of arsenic uptake, like the patterns for uptake initiation and regulation are scarcely studied, which deserve further investigation in detail. This review provides new insights into the toxicity mechanism for arsenic exposure.
-
Key words:
- arsenical /
- toxicity/carcinogenesis mechanism /
- cellular uptake /
- transporter /
- regulation signaling
-

-
表 1 典型砷形态的名称及结构
Table 1. Names and structures of typical arsenicals
典型砷化合物
Typical arsenicals结构及名称
Names and structures五价砷
Pentavalent arsenic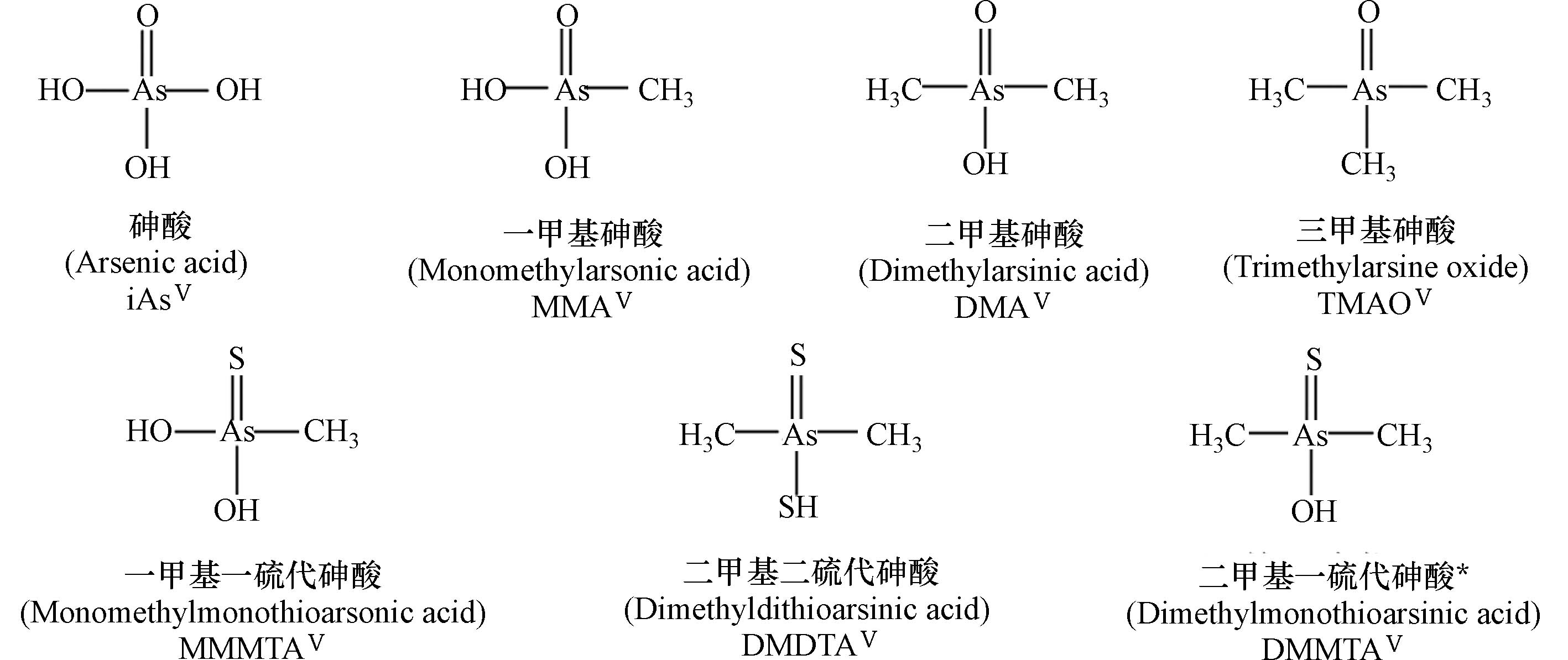
三价砷
Trivalent arsenic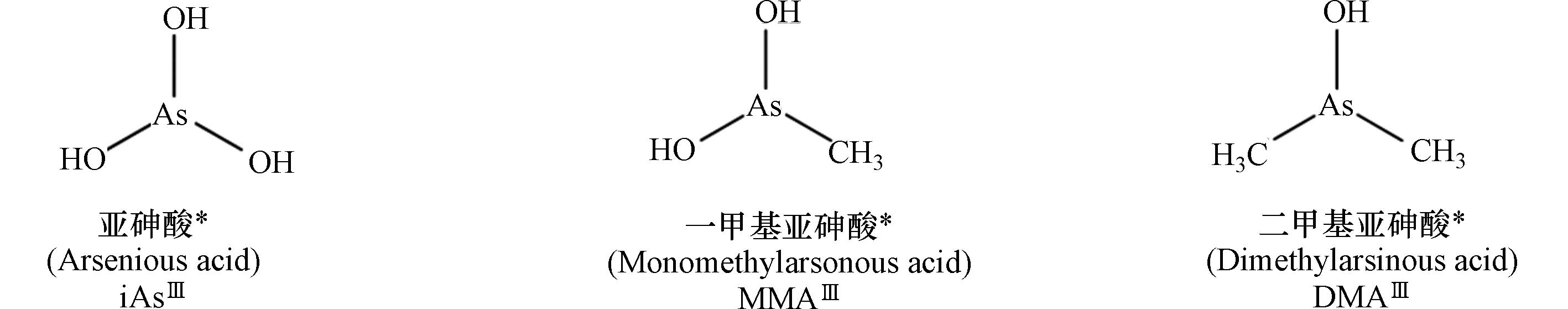
注:表中标注*砷形态具有相近的生物毒性,其中iAsⅢ被列为一类(Group 1)致癌物.
*The arsenicals exhibit similar toxicity. Among them, iAsIII has been classified as the “carcinogenic to humans” (Group 1). 下载: 导出CSV
下载: 导出CSV
表 2 砷化合物的摄入载体与载体蛋白的表达调控
Table 2. Transporters of typical arsenicals and related regulation signaling
载体蛋白类别及名称
Transporter category and name砷形态
Arsenic species载体蛋白调控通路
Regulation signaling载体蛋白调控方式
Regulation fashion磷酸盐转运蛋白 NaPi-Ⅱb iAsV[24-26] AQPs AQP3 iAsⅢ[10,30,32] EGF[47-49]、ERE[50]、AC、cAMP、
PPAR-γ、AMPK、Akt/PKB[55]mRNA、蛋白表达 PKC[54] 膜易位 AQP7 iAsIII[30] AC、cAMP、PPAR-γ、
AMPK、Akt/PKB[55]mRNA表达 AQP9 iAsⅢ[30,31,33]、MMAⅢ[38-39] MMAV[39]、DMAV[39] AMPK、Akt/PKB[56] mRNA表达 GLUTs
GLUT1MMAⅢ[40] 转录因子c-Myc[57]、SIX1[58] mRNA表达 PI3K/mTOR/Akt[59]、cAMP、
AMPK、Akt[60]mRNA、蛋白表达和
膜易位GLUT2、GLUT5 iAsⅢ[26] OATPs OATP1B1 iAsⅢ、iAsV[36] HNF-1α[64],HNF-3β[65] mRNA表达、
蛋白表达OATP2B1 iAsⅢ[26] PKC[63] 蛋白修饰
下载: 导出CSV
-
[1] DUKER A A, CARRANZA E J M, HALE M. Arsenic geochemistry and health [J]. Environment International, 2005, 31(5): 631-641. doi: 10.1016/j.envint.2004.10.020 [2] MANDAL B K, SUZUKI K T. Arsenic round the world: A review [J]. Talanta, 2002, 58(1): 201-235. doi: 10.1016/S0039-9140(02)00268-0 [3] LI Y B, DUAN Z W, LIU G L, et al. Evaluation of the possible sources and controlling factors of toxic metals/metalloids in the Florida Everglades and their potential risk of exposure [J]. Environmental Science & Technology, 2015, 49(16): 9714-9723. [4] NAUJOKAS M F, ANDERSON B, AHSAN H, et al. The broad scope of health effects from chronic arsenic exposure: Update on a worldwide public health problem [J]. Environmental Health Perspectives, 2013, 121(3): 295-302. doi: 10.1289/ehp.1205875 [5] RODRÍGUEZ-LADO L, SUN G F, BERG M, et al. Groundwater arsenic contamination throughout China [J]. Science, 2013, 341(6148): 866-868. doi: 10.1126/science.1237484 [6] HUNT K M, SRIVASTAVA R K, ELMETS C A, et al. The mechanistic basis of arsenicosis: Pathogenesis of skin cancer [J]. Cancer Letters, 2014, 354(2): 211-219. doi: 10.1016/j.canlet.2014.08.016 [7] STRAIF K, BENBRAHIM-TALLAA L, BAAN R, et al. A review of human carcinogens—Part C: Metals, arsenic, dusts, and fibres [J]. The Lancet Oncology, 2009, 10(5): 453-454. doi: 10.1016/S1470-2045(09)70134-2 [8] CHEN Y, WU F, GRAZIANO J H, et al. Arsenic exposure from drinking water, arsenic methylation capacity, and carotid intima-media thickness in Bangladesh [J]. American Journal of Epidemiology, 2013, 178(3): 372-381. doi: 10.1093/aje/kwt001 [9] MAULL E A, AHSAN H, EDWARDS J, et al. Evaluation of the association between arsenic and diabetes: A National Toxicology Program workshop review [J]. Environmental Health Perspectives, 2012, 120(12): 1658-1670. doi: 10.1289/ehp.1104579 [10] NARANMANDURA H, IBATA K, SUZUKI K T. Toxicity of dimethylmonothioarsinic acid toward human epidermoid carcinoma A431 cells [J]. Chemical Research in Toxicology, 2007, 20(8): 1120-1125. doi: 10.1021/tx700103y [11] NARANMANDURA H, CAREW M W, XU S, et al. Comparative toxicity of arsenic metabolites in human bladder cancer EJ-1 cells [J]. Chemical Research in Toxicology, 2011, 24(9): 1586-1596. doi: 10.1021/tx200291p [12] LIU F, JAN K Y. DNA damage in arsenite- and cadmium-treated bovine aortic endothelial cells [J]. Free Radical Biology and Medicine, 2000, 28(1): 55-63. doi: 10.1016/S0891-5849(99)00196-3 [13] LI Y X, HE M, CHEN B B, et al. Inhibition of arsenite methylation induces synergistic genotoxicity of arsenite and benzo(a)Pyrene diol epoxide in SCC-7 cells [J]. Metallomics, 2018, 11(1): 176-182. [14] ZHOU X X, SUN X, MOBARAK C, et al. Differential binding of monomethylarsonous acid compared to arsenite and arsenic trioxide with zinc finger peptides and proteins [J]. Chemical Research in Toxicology, 2014, 27(4): 690-698. doi: 10.1021/tx500022j [15] ZHAO L H, CHEN S M, JIA L Y, et al. Selectivity of arsenite interaction with zinc finger proteins [J]. Metallomics, 2012, 4(9): 988-994. doi: 10.1039/c2mt20090b [16] RATNAIKE R N. Acute and chronic arsenic toxicity [J]. Postgraduate Medical Journal, 2003, 79(933): 391-396. doi: 10.1136/pmj.79.933.391 [17] SHEN S W, LI X F, CULLEN W R, et al. Arsenic binding to proteins [J]. Chemical Reviews, 2013, 113(10): 7769-7792. doi: 10.1021/cr300015c [18] SUZUKI K T, IWATA K, NARANMANDURA H, et al. Metabolic differences between two dimethylthioarsenicals in rats [J]. Toxicology and Applied Pharmacology, 2007, 218(2): 166-173. doi: 10.1016/j.taap.2006.10.027 [19] NARANMANDURA H, IWATA K, SUZUKI K T, et al. Distribution and metabolism of four different dimethylated arsenicals in hamsters [J]. Toxicology and Applied Pharmacology, 2010, 245(1): 67-75. doi: 10.1016/j.taap.2010.02.001 [20] GARBINSKI L D, ROSEN B P, CHEN J. Pathways of arsenic uptake and efflux [J]. Environment International, 2019, 126: 585-597. doi: 10.1016/j.envint.2019.02.058 [21] ROGGENBECK B A, BANERJEE M, LESLIE E M. Cellular arsenic transport pathways in mammals [J]. Journal of Environmental Sciences, 2016, 49: 38-58. doi: 10.1016/j.jes.2016.10.001 [22] CHÁVEZ-CAPILLA T, MAHER W, KELLY T, et al. Evaluation of the ability of arsenic species to traverse cell membranes by simple diffusion using octanol-water and liposome-water partition coefficients [J]. Journal of Environmental Sciences, 2016, 49: 222-232. doi: 10.1016/j.jes.2016.08.007 [23] RICARDOVILLA-BELLOSTA, GIRAL H, FERRER-DUFOL A, et al. Role of phosphate transporters in the membrane transport of arsenate [J]. Toxicology Letters, 2006, 164: S150. [24] VILLA-BELLOSTA R, SORRIBAS V. Role of rat sodium/phosphate cotransporters in the cell membrane transport of arsenate [J]. Toxicology and Applied Pharmacology, 2008, 232(1): 125-134. doi: 10.1016/j.taap.2008.05.026 [25] VILLA-BELLOSTA R, SORRIBAS V. Arsenate transport by sodium/phosphate cotransporter type IIb [J]. Toxicology and Applied Pharmacology, 2010, 247(1): 36-40. doi: 10.1016/j.taap.2010.05.012 [26] CALATAYUD M, BARRIOS J A, VÉLEZ D, et al. In vitro study of transporters involved in intestinal absorption of inorganic arsenic [J]. Chemical Research in Toxicology, 2012, 25(2): 446-453. doi: 10.1021/tx200491f [27] LAFORENZA U, BOTTINO C, GASTALDI G. Mammalian aquaglyceroporin function in metabolism [J]. Biochimica et Biophysica Acta (BBA) - Biomembranes, 2016, 1858(1): 1-11. doi: 10.1016/j.bbamem.2015.10.004 [28] ROJEK A, PRAETORIUS J, FRØKIAER J, et al. A current view of the mammalian aquaglyceroporins [J]. Annual Review of Physiology, 2008, 70: 301-327. doi: 10.1146/annurev.physiol.70.113006.100452 [29] RAMÍREZ-SOLÍS A, MUKOPADHYAY R, ROSEN B P, et al. Experimental and theoretical characterization of arsenite in water: Insights into the coordination environment of As-O [J]. Inorganic Chemistry, 2004, 43(9): 2954-2959. doi: 10.1021/ic0351592 [30] LIU Z J. Roles of vertebrate aquaglyceroporins in arsenic transport and detoxification [J]. Advances in Experimental Medicine and Biology, 2010, 679: 71-81. [31] LEUNG J, PANG A N, YUEN W H, et al. Relationship of expression of aquaglyceroporin 9 with arsenic uptake and sensitivity in leukemia cells [J]. Blood, 2007, 109(2): 740-746. doi: 10.1182/blood-2006-04-019588 [32] NARANMANDURA H, OGRA Y, IWATA K, et al. Evidence for toxicity differences between inorganic arsenite and thioarsenicals in human bladder cancer cells [J]. Toxicology and Applied Pharmacology, 2009, 238(2): 133-140. doi: 10.1016/j.taap.2009.05.006 [33] LEE T C, HO I C, LU W J, et al. Enhanced expression of multidrug resistance-associated protein 2 and reduced expression of aquaglyceroporin 3 in an arsenic-resistant human cell line [J]. Journal of Biological Chemistry, 2006, 281(27): 18401-18407. doi: 10.1074/jbc.M601266200 [34] HAGENBUCH B, STIEGER B. The SLCO (former SLC21) superfamily of transporters [J]. Molecular Aspects of Medicine, 2013, 34(2/3): 396-412. [35] ROTH M, OBAIDAT A, HAGENBUCH B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies [J]. British Journal of Pharmacology, 2012, 165(5): 1260-1287. doi: 10.1111/j.1476-5381.2011.01724.x [36] LU W J, TAMAI I, NEZU J I, et al. Organic anion transporting polypeptide-C mediates arsenic uptake in HEK-293 cells [J]. Journal of Biomedical Science, 2006, 13(4): 525-533. doi: 10.1007/s11373-006-9071-0 [37] MUECKLER M, THORENS B. The SLC2 (GLUT) family of membrane transporters [J]. Molecular Aspects of Medicine, 2013, 34(2/3): 121-138. [38] LIU Z J, STYBLO M, ROSEN B P. Methylarsonous acid transport by aquaglyceroporins [J]. Environmental Health Perspectives, 2006, 114(4): 527-531. doi: 10.1289/ehp.8600 [39] MCDERMOTT J R, JIANG X, BEENE L C, et al. Pentavalent methylated arsenicals are substrates of human AQP9 [J]. BioMetals, 2010, 23(1): 119-127. doi: 10.1007/s10534-009-9273-9 [40] LIU Z J, SANCHEZ M A, JIANG X, et al. Mammalian glucose permease GLUT1 facilitates transport of arsenic trioxide and methylarsonous acid [J]. Biochemical and Biophysical Research Communications, 2006, 351(2): 424-430. doi: 10.1016/j.bbrc.2006.10.054 [41] DRUWE I L, VAILLANCOURT R R. Influence of arsenate and arsenite on signal transduction pathways: An update [J]. Archives of Toxicology, 2010, 84(8): 585-596. doi: 10.1007/s00204-010-0554-4 [42] QIAN Y, CASTRANOVA V, SHI X L. New perspectives in arsenic-induced cell signal transduction [J]. Journal of Inorganic Biochemistry, 2003, 96(2/3): 271-278. [43] KISHIDA K, SHIMOMURA I, KONDO H, et al. Genomic structure and insulin-mediated repression of the aquaporin adipose (AQPap), adipose-specific glycerol channel [J]. The Journal of Biological Chemistry, 2001, 276(39): 36251-36260. doi: 10.1074/jbc.M106040200 [44] KURIYAMA H, KAWAMOTO S, ISHIDA N, et al. Molecular cloning and expression of a novel human aquaporin from adipose tissue with glycerol permeability [J]. Biochemical and Biophysical Research Communications, 1997, 241(1): 53-58. doi: 10.1006/bbrc.1997.7769 [45] ISHIBASHI K, YAMAUCHI K, KAGEYAMA Y, et al. Molecular characterization of human Aquaporin-7 gene and its chromosomal mapping [J]. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression, 1998, 1399(1): 62-66. doi: 10.1016/S0167-4781(98)00094-3 [46] MARLAR S, JENSEN H H, LOGIN F H, et al. Aquaporin-3 in cancer [J]. International Journal of Molecular Sciences, 2017, 18(10): 2106. doi: 10.3390/ijms18102106 [47] LI A, LU D H, ZHANG Y P, et al. Critical role of aquaporin-3 in epidermal growth factor-induced migration of colorectal carcinoma cells and its clinical significance [J]. Oncology Reports, 2013, 29(2): 535-540. doi: 10.3892/or.2012.2144 [48] JI C, CAO C, LU S, et al. Curcumin attenuates EGF-induced AQP3 up-regulation and cell migration in human ovarian cancer cells [J]. Cancer Chemotherapy and Pharmacology, 2008, 62(5): 857-865. doi: 10.1007/s00280-007-0674-6 [49] LIU W J, WANG K H, GONG K M, et al. Epidermal growth factor enhances MPC-83 pancreatic cancer cell migration through the upregulation of aquaporin 3 [J]. Molecular Medicine Reports, 2012, 6(3): 607-610. doi: 10.3892/mmr.2012.966 [50] HUANG Y T, ZHOU J, SHI S, et al. Identification of estrogen response element in aquaporin-3 gene that mediates estrogen-induced cell migration and invasion in estrogen receptor-positive breast cancer [J]. Scientific Reports, 2015, 5: 12484. doi: 10.1038/srep12484 [51] Walz T, Fujiyoshi Y, Engel A. The AQP structure and functional implications [J]. Handbook of Experimental Pharmacology, 2009(190): 31-56. [52] FISCHER G, KOSINSKA-ERIKSSON U, APONTE-SANTAMARÍA C, et al. Crystal structure of a yeast aquaporin at 1.15 angstrom reveals a novel gating mechanism [J]. PLoS Biology, 2009, 7(6): e1000130. doi: 10.1371/journal.pbio.1000130 [53] CONNER A C, BILL R M, CONNER M T. An emerging consensus on aquaporin translocation as a regulatory mechanism [J]. Molecular Membrane Biology, 2013, 30(1): 101-112. doi: 10.3109/09687688.2012.743194 [54] YASUI H, KUBOTA M, IGUCHI K, et al. Membrane trafficking of aquaporin 3 induced by epinephrine [J]. Biochemical and Biophysical Research Communications, 2008, 373(4): 613-617. doi: 10.1016/j.bbrc.2008.06.086 [55] GREGOIRE F, LUCIDI V, ZERRAD-SAADI A, et al. Analysis of aquaporin expression in liver with a focus on hepatocytes [J]. Histochemistry and Cell Biology, 2015, 144(4): 347-363. doi: 10.1007/s00418-015-1341-3 [56] YOKOYAMA Y, IGUCHI K, USUI S, et al. AMP-activated protein kinase modulates the gene expression of aquaporin 9 via forkhead box a2 [J]. Archives of Biochemistry and Biophysics, 2011, 515(1/2): 80-88. [57] OSTHUS R C, SHIM H, KIM S, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-myc [J]. Journal of Biological Chemistry, 2000, 275(29): 21797-21800. doi: 10.1074/jbc.C000023200 [58] LI L, LIANG Y C, KANG L, et al. Transcriptional regulation of the Warburg effect in cancer by SIX1 [J]. Cancer Cell, 2018, 33(3): 368-385.e7. doi: 10.1016/j.ccell.2018.01.010 [59] MAKINOSHIMA H, TAKITA M, SARUWATARI K, et al. Signaling through the phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) axis is responsible for aerobic glycolysis mediated by glucose transporter in epidermal growth factor receptor (EGFR)-mutated lung adenocarcinoma [J]. The Journal of Biological Chemistry, 2015, 290(28): 17495-17504. doi: 10.1074/jbc.M115.660498 [60] ANCEY P B, CONTAT C, MEYLAN E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment [J]. The FEBS Journal, 2018, 285(16): 2926-2943. doi: 10.1111/febs.14577 [61] WIEMAN H L, WOFFORD J A, RATHMELL J C. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking [J]. Molecular Biology of the Cell, 2007, 18(4): 1437-1446. doi: 10.1091/mbc.e06-07-0593 [62] OLSEN J M, SATO M, DALLNER O S, et al. Glucose uptake in brown fat cells is dependent on mTOR complex 2-promoted GLUT1 translocation [J]. The Journal of Cell Biology, 2014, 207(3): 365-374. doi: 10.1083/jcb.201403080 [63] KÖCK K, KOENEN A, GIESE B, et al. Rapid modulation of the organic anion transporting polypeptide 2B1 (OATP2B1, SLCO2B1) function by protein kinase C-mediated internalization [J]. Journal of Biological Chemistry, 2010, 285(15): 11336-11347. doi: 10.1074/jbc.M109.056457 [64] JUNG D, HAGENBUCH B, GRESH L, et al. Characterization of the human OATP-C (SLC21A6) gene promoter and regulation of liver-specific OATP genes by hepatocyte nuclear factor 1α [J]. Journal of Biological Chemistry, 2001, 276(40): 37206-37214. doi: 10.1074/jbc.M103988200 [65] VAVRICKA S R, JUNG D, FRIED M, et al. The human organic anion transporting polypeptide 8 (SLCO1B3) gene is transcriptionally repressed by hepatocyte nuclear factor 3β in hepatocellular carcinoma [J]. Journal of Hepatology, 2004, 40(2): 212-218. doi: 10.1016/j.jhep.2003.10.008 [66] STYBLO M, del RAZO L M, VEGA L, et al. Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells [J]. Archives of Toxicology, 2000, 74(6): 289-299. doi: 10.1007/s002040000134 [67] OCHI T, KITA K, SUZUKI T, et al. Cytotoxic, genotoxic and cell-cycle disruptive effects of thio-dimethylarsinate in cultured human cells and the role of glutathione [J]. Toxicology and Applied Pharmacology, 2008, 228(1): 59-67. doi: 10.1016/j.taap.2007.11.023 [68] APOSHIAN H V. Enzymatic methylation of arsenic species and other new approaches to arsenic toxicity [J]. Annual Review of Pharmacology and Toxicology, 1997, 37: 397-419. doi: 10.1146/annurev.pharmtox.37.1.397 [69] HUGHES M F, KENYON E M, EDWARDS B C, et al. Accumulation and metabolism of arsenic in mice after repeated oral administration of arsenate [J]. Toxicology and Applied Pharmacology, 2003, 191(3): 202-210. doi: 10.1016/S0041-008X(03)00249-7 [70] BRADHAM K D, DIAMOND G L, BURGESS M, et al. In vivo and in vitro methods for evaluating soil arsenic bioavailability: Relevant to human health risk assessment [J]. Journal of Toxicology and Environmental Health, Part B, 2018, 21(2): 83-114. doi: 10.1080/10937404.2018.1440902 [71] KENYON E M, del RAZO L M, HUGHES M F. Tissue distribution and urinary excretion of inorganic arsenic and its methylated metabolites in mice following acute oral administration of arsenate [J]. Toxicological Sciences, 2005, 85(1): 468-475. doi: 10.1093/toxsci/kfi107 [72] LIN S, SHI Q, NIX F B, et al. A novel S-adenosyl-l-methionine: Arsenic(III) methyltransferase from rat liver cytosol [J]. Journal of Biological Chemistry, 2002, 277(13): 10795-10803. doi: 10.1074/jbc.M110246200 [73] NARANMANDURA H, SUZUKI N, IWATA K, et al. Arsenic metabolism and thioarsenicals in hamsters and rats [J]. Chemical Research in Toxicology, 2007, 20(4): 616-624. doi: 10.1021/tx700038x [74] WANG T S, CHUNG C H, WANG A S S, et al. Endonuclease III, formamidopyrimidine-DNA glycosylase, and proteinase K additively enhance arsenic-induced DNA strand breaks in human cells [J]. Chemical Research in Toxicology, 2002, 15(10): 1254-1258. doi: 10.1021/tx025535f [75] PARVEZ F, CHEN Y, YUNUS M, et al. Arsenic exposure and impaired lung function. Findings from a large population-based prospective cohort study [J]. American Journal of Respiratory and Critical Care Medicine, 2013, 188(7): 813-819. doi: 10.1164/rccm.201212-2282OC [76] AHMAD S, ANDERSON W L, KITCHIN K T. Dimethylarsinic acid effects on DNA damage and oxidative stress related biochemical parameters in B6C3F1 mice [J]. Cancer Letters, 1999, 139(2): 129-135. doi: 10.1016/S0304-3835(99)00022-1 [77] LIU J, ZHENG B, APOSHIAN H, et al. Chronic arsenic poisoning from burning high-arsenic-containing coal in Guizhou, China [J]. Journal of the Peripheral Nervous System, 2002, 7(3): 208. [78] SINGH R D, TIWARI R, KHAN H, et al. Arsenic exposure causes epigenetic dysregulation of IL-8 expression leading to proneoplastic changes in kidney cells [J]. Toxicology Letters, 2015, 237(1): 1-10. doi: 10.1016/j.toxlet.2015.05.014 [79] JOMOVA K, JENISOVA Z, FESZTEROVA M, et al. Arsenic: toxicity, oxidative stress and human disease [J]. Journal of Applied Toxicology, 2011, 31(2): 95-107. [80] FISCHER A B, BUCHET J P, LAUWERYS R R. Arsenic uptake, cytotoxicity and detoxification studied in mammalian cells in culture [J]. Archives of Toxicology, 1985, 57(3): 168-172. doi: 10.1007/BF00290882 [81] KLIGERMAN A D, DOERR C L, TENNANT A H, et al. Methylated trivalent arsenicals as candidate ultimate genotoxic forms of arsenic: Induction of chromosomal mutations but not gene mutations [J]. Environmental and Molecular Mutagenesis, 2003, 42(3): 192-205. doi: 10.1002/em.10192 [82] MOE B, PENG H Y, LU X F, et al. Comparative cytotoxicity of fourteen trivalent and pentavalent arsenic species determined using real-time cell sensing [J]. Journal of Environmental Sciences, 2016, 49: 113-124. doi: 10.1016/j.jes.2016.10.004 -
 点击查看大图
点击查看大图
图( 1) 表( 2)
计量
- 文章访问数: 4157
- HTML全文浏览数: 4157
- PDF下载数: 57
- 施引文献: 0
