












-
抗生素因其具有抗菌谱广、抗菌力强、结构简单、活性强等特点,在医疗业、畜牧业及水产养殖业发挥着重要的作用。但抗生素不能被生物体有效的吸收代谢[1],约70%—80%的抗生素会进入水生系统[2],且在环境中长期存在,除了会造成化学药物污染外,长期接触抗生素也会对生物体造成慢性毒性影响[3],诱导细菌产生抗生素抗性基因(ARGs),造成一系列生态问题[4]。对生态系统和人类健康存在不利的影响。环丙沙星(CIP)是一种典型的氟喹诺酮类抗生素,也是世界范围内应用最广泛的抗生素之一。由于环丙沙星在水环境中广泛残留,近年来,在中国的地表水源、淡水养殖环境、海水养殖环境和养殖生物中均被检出[1, 5-9],由于其卤代的杂环结构,在常规的废水处理工艺中很难被去除。因此,亟需找到一种能够高效去除废水中环丙沙星的技术。
高级氧化工艺(AOPs)对于难降解药物和个人护理产品(PPCPs)的降解非常有效,紫外/氯组合工艺作为作为一种新兴的高级氧化工艺,是一种很有应用前景的水处理技术。该工艺主要借助紫外线辐照氯从而激发产生的·OH和活性氯物种(RCS)(包括氯自由基Cl·、氧化氯·ClO、二氯自由基·Cl2等)来氧化降解有机污染物,其中,HO·是一种非选择性氧化剂,氧化电位约为2.80 V[10],能够与有机物快速反应;活性氯物种(Cl·、·ClO、·
Cl−2 )的氧化还原电位分别为2.47 V、1.5—1.8 V、2.0 V[10-12],活性氯是选择性的氧化剂,可通过单电子氧化、吸氢和不饱和碳碳键与富电子部分反应[13]。上述这些活性自由基在氧化反应中具有协同增效的作用,共同参与降解有机污染物[14-15]。近年来,紫外/氯高级氧化工艺被广泛用于去除水中的新型有机污染物,如磺胺甲恶唑[16]、氯贝酸[17]、美托洛尔[14]、二碘乙酰胺[18]、甲硝唑[19]等。作为水环境中常见有机污染物,近年来有许多利用高级氧化方法去除环丙沙星的研究,包括中压紫外线激活过氧单硫酸盐(MPUV/PMS)[20],紫外臭氧(UV/O3)[21]、紫外激活过硫酸盐(UV/PS)[22]等,但这些关于环丙沙星的去除研究主要集中于饮用水,系统研究紫外/氯高级氧化组合工艺降解废水中环丙沙星报道较少。因此,本文采用模拟的环丙沙星废水为研究对象,开展紫外/氯高级氧化工艺降解环丙沙星效能研究,分别从影响因素、反应动力学、降解产物、毒性效应等方面阐述废水中环丙沙星的降解效能与机理,并对降解工艺进行经济成本分析,进而为紫外/氯组合工艺用于废水中环丙沙星的去除提供理论基础与数据支撑。
-
环丙沙星(CIP,纯度98%)、腐植酸(透明质酸,脂肪酸≥90%)、牛血清蛋白(BSA)、碳酸氢钠、氯化钠、高效液相色谱级的乙腈和甲醇均购自麦克林生化科技有限公司(中国上海)。次氯酸钠(NaOCl,有效氯< 5%,游离碱10%—20%)购自阿拉丁生化科技股份有限公司(中国上海),硫代硫酸钠(Na2S2O3)购自国药化学试剂有限公司(中国上海)。实验所用试剂均采用超纯水配置,所用超纯水(电导率为18.25 MΩ·cm)通过优普纯水机(UPT-II-10T)制备,实验中使用的实际水体经0.45 μm水相滤膜过滤后保存备用。
-
在装有一个11 W低压汞灯(波长= 254 nm)的光化学反应器中进行,垂直光筒下放置容积为 100 mL的玻璃平皿,平皿置于磁力搅拌器上,用磁力搅拌器将反应溶液完全混合。实验开始前对紫外灯进行30 min的预热。降解实验过程中使用10 mmol·L−1的磷酸盐进行缓冲,反应溶液的pH分别采用0.1 mol·L−1NaOH和0.1 mol·L−1H2SO4进行调节,除特殊说明外,反应溶液的pH值均为7,反应体积为100 mL。实验中向含有CIP的溶液中加入特定量的氧化剂,并开始紫外线照射,在预定的时间间隔取1 mL反应液加入50 μL硫代硫酸钠的液相小瓶中进行猝灭,随后立即进行检测分析。
-
毒性实验中的小球藻采用BG-11培养基并置于光照培养箱中培养,进入对数生长期中后期后,使用250 mL锥形瓶分装,向分装的小球藻液中分别投加相同体积的CIP溶液和降解后的反应溶液,按照原始培养条件在光照培养箱中继续培养,每隔24 h取样,取样前将样品摇匀,稀释后用浮游植物荧光分析仪测定光合系统Ⅱ(PSⅡ)的最大光合效率(Fv/Fm),所有实验均重复至少2次以上。
-
CIP的浓度采用高效液相色谱(Agilent 1260, USA)测定,色谱柱采用Poroshell 120 SB (2.1 mm ×150 mm, 2.7 μm, Agilent),流动相为甲醇和0.10%甲酸溶液(V∶V ,50∶50),流速为0.8 mL·min−1,柱温为40 ℃; 荧光检测器(FLD)的激活波长为278 nm,发射波长为450 nm, 进样量为20 μL。NaOCl溶液中的有效游离氯浓度采用DPD分光光度法测定。溶液中有机物的荧光强度使用日立F-2700荧光分光光度计进行分析,激发波长设置为250—400 nm,发射波长设置为 270—500 nm,扫描间隔为5 nm,激发光和发射光的狭缝为10 nm;扫描速度为1200 nm·min−1。反应过程中的降解产物采用Thermo Fisher UltiMate 3000 液相色谱串联Thermo Scientific Q Exactive组合型四极杆-Orbitrap 质谱仪测定,选择全扫模式,扫描范围的m/z为100—1000,正离子模式(ESI+)。毒性实验中小球藻的最大光合效率(Fv/Fm)利用浮游植物荧光分析仪(Phyto-PAM, Walz, 德国)测定,综合生态毒性分析通过ECOSAR模型计算得出。
-
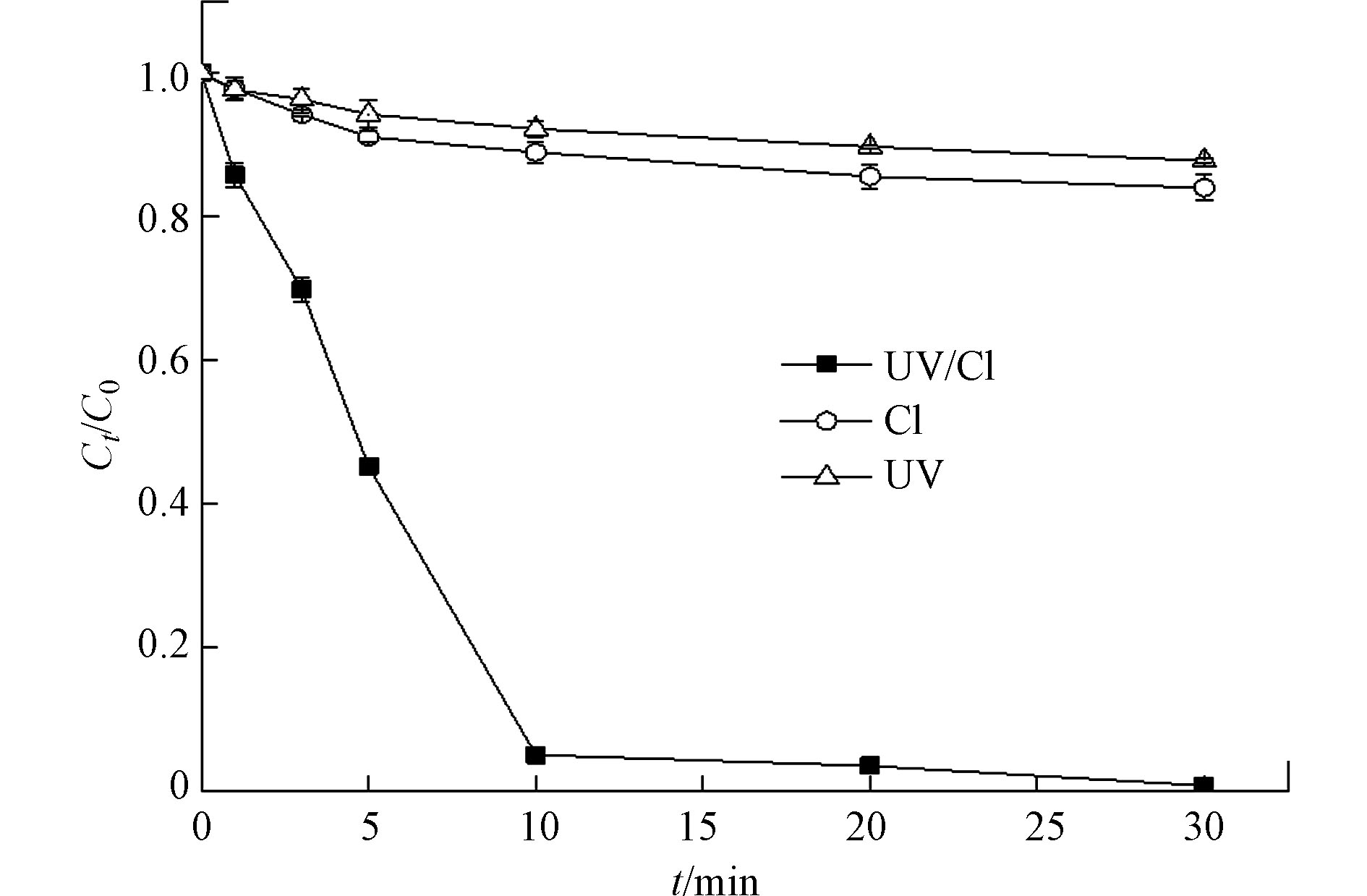
实验控制CIP的初始浓度为30 μmol·L−1,NaClO的初始浓度为0.28 mmol·L−1,反应体系但初始pH值为7.0,温度为25 ℃,CIP在单独紫外线照射、单独氯化和紫外/氯工艺过程中的降解效果如图1所示,单独紫外光照射和单独氯化对CIP的降解效果不明显,30 min内分别只有12%和16%的去除率,而紫外光照射和氯化组合工艺对CIP的降解表现出协同效应,经过30 min,CIP的去除率高达99%,分别是单独紫外照射和单独氯化的7.75倍和5.81倍。上述结果可以归结于紫外/氯组合工艺中生成了强氧化性羟基自由基(∙OH)和活性氯(RCS)。反应如式(1)—(2)。
-
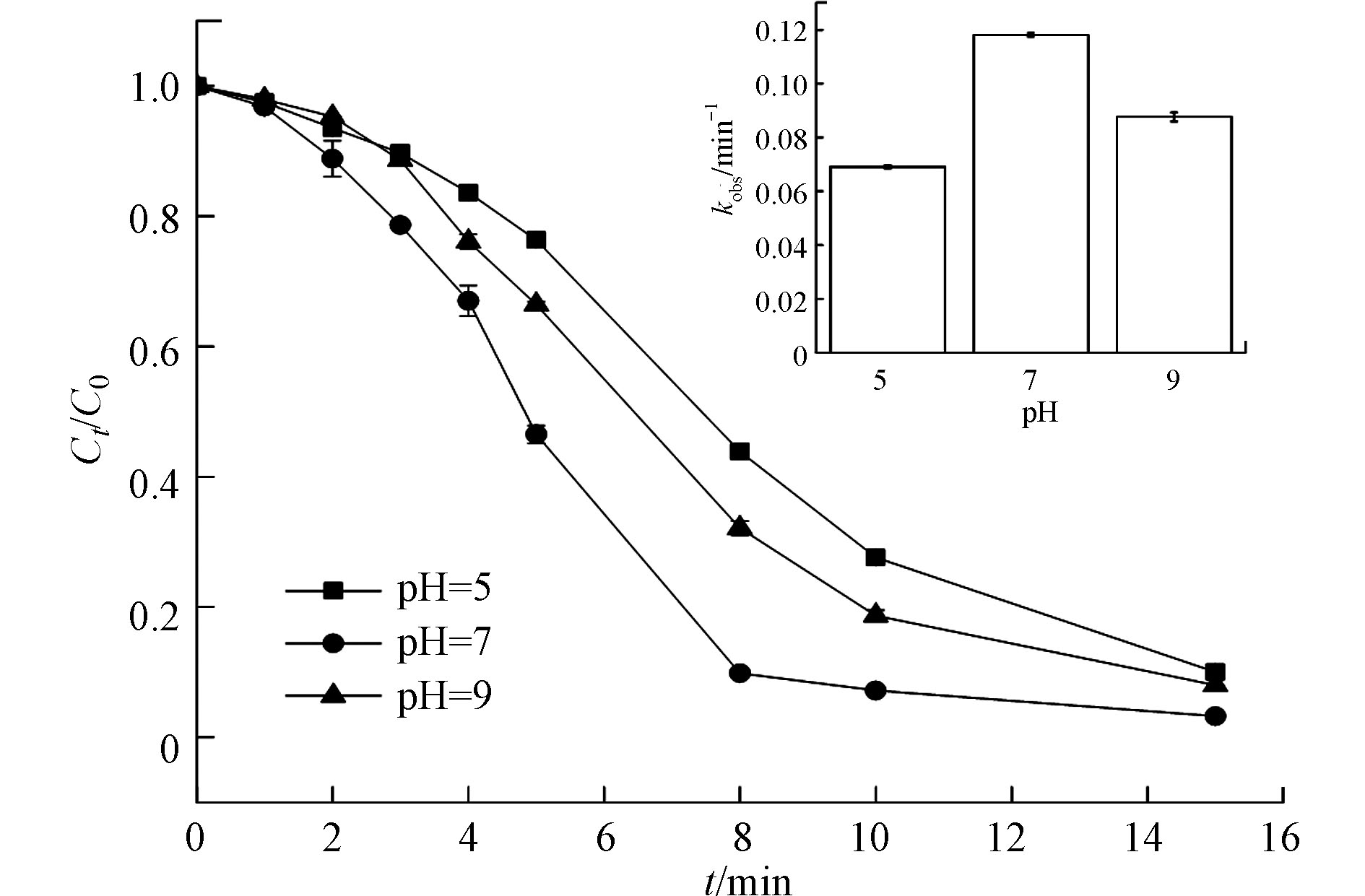
控制CIP的初始浓度为30 μmol·L−1,NaClO的初始浓度为0.28 mmol·L−1,紫外光辐照强度为0.2 mW·cm−2,温度为25 ℃,利用 0.1 mol·L−1NaOH和0.1 mol·L−1H2SO4调节溶液初始pH值分别为 5.0、7.0、9.0,探究不同的溶液初始pH值对CIP降解效能的影响,结果如图2所示。在pH=5、pH=7和pH=9时,CIP降解率8 min后分别达到57 %、91 %和68 %。其一级反应速率常数依次为0.069 min−1、0.118 min−1、0.088 min−1,由此可知,环丙沙星在酸性条件(pH=5)下降解最慢,在中性条件(pH=7)下降解最快。
这一现象与紫外/氯组合工艺降解双酚A[23]、苯甲酮[24]的趋势一致。有研究指出,紫外线对有机物的降解并不表现出pH依赖性[25-26],而pH对有机物降解效率的差异主要归因于两方面,第一,pH影响HOCl/ClO−的量子产率和溶液基质的自由基清除效果[27-28],如式3所示,次氯酸可以分解为次氯酸根,酸碱度影响HOCl/ ClO−的解离(pKa=7.5),相比于pH=9的碱性条件下,pH=7时,HClO为优势物种,254 nm紫外光照射下,HClO的量子产率高于ClO−(1.45 mol Es−1>0.95 mol Es−1)[29],可以形成更多的HO·和Cl·。溶液pH的增加导致OCl−的比例增加,降低了羟基和氯自由基的数量。同时,HClO对羟基和氯的自由基清除作用也较低[29-30],HOCl与羟基和氯自由基的速率常数分别为2.0×109 L·(mol·s)−1和3.0×109 L·(mol·s)−1,而OCl−与羟基和氯自由基的速率常数分别为8.8×109 L·(mol·s)−1和8.2×109 L·(mol·s)−1 [31-33]。当酸碱度从7.0下降到5.0,CIP去除率下降的原因可能是,由于pH值不同,CIP的形态不同(pKa= 6.2)。在酸性条件下,质子化形式的CIP(CIP3+,CIP2+和CIP1+)占主要优势,质子化的程度随着酸碱度的增加而降低;在酸碱度为7—8左右,CIP以中性/两性离子形式存在(CIP0),在碱性溶液中,CIP以去质子化形式存在(CIP−1),质子化、中性和去质子化形式的CIP均可以与HOCl反应,二级速率常数分别为4.3 × 103 L·(mol·s)−1、3.8 × 105 L·(mol·s)−1和4.9×107 L·(mol·s)−1 [34],有研究者在一项比较实验中发现,氟喹诺酮化合物(诺氟沙星、氧氟沙星和恩诺沙星)在不同的pH条件下显示出相似的直接光解降解动力学:最高反应速率常数均是目标分子的中性形式,其次是去质子化形式和质子化形式[35] ,pH=5时,虽然HOCl是主要的形式,但是由于静电相互作用(pKa= 6.2),在pH =5的质子化形式的CIP与HOCl之间的反应的kobs(4.3×103 L·(mol·s)−1)是3种CIP形式中最低的,故在中性条件下,CIP的降解效率高于酸性和碱性条件。
-
控制CIP的初始浓度30 μmol·L−1, 紫外光强为0.2 mW·cm−2,pH为7.0,投加不同浓度的次氯酸钠,研究不同氧化剂投加量对CIP的去除效果的影响,结果如图3所示,增加次氯酸纳的剂量能够显著提高环丙沙星的去除。随着游离氯量从0.07 mmol·L−1增加到0.42 mmol·L−1,10 min后CIP的去除率从76.9%增加到98.5%,反应速率常数由0.078 min−1增加至0.106 min−1。
化学氧化中,氧化剂的剂量在自由基的产生过程中起着至关重要的作用。随着氧化剂用量从0.07 mmol·L−1增加到0.28 mmol·L−1,加速了游离氯的光解,能够促进水中Cl·和HO·的产生,环丙沙星的降解速率明显增加。而随着氯用量从0.28 mmol·L−1增加到0.42 mmol·L−1,环丙沙星的降解速率没有明显的提高,这种现象可能与过量的HOCl和OCl−的清除作用有关[32, 36],如(4)—(7)所示,HOCl能清除羟基和氯自由基,速率常数分别为2.0×109 L·(mol·s)−1和3.0×109 L·(mol·s)−1,OCl−也能清除羟基和氯自由基,速率常数分别为8.8×109 L·(mol·s)−1和8.2×109 L·(mol·s)−1 [31-33],在最近的研究中,当将紫外/氯工艺应用于美托洛尔[14]、环柠檬醛[37]、阿米替利[38]、非那西丁[39]、布洛芬[15]去除时,也观察到了类似的趋势。
-
水体中广泛存在着一些无机阴离子,其中硝酸根离子的浓度高达数百毫克升,是水体中常见的阴离子,可与·OH反应而消耗部分自由基,氯离子也是一种常见的阴离子,广泛存在于自然环境中。本实验研究了在中性环境下不同浓度的氯离子和硝酸根离子对紫外/氯组合工艺去除环丙沙星的影响,结果如图4所示。
如图4a所示,当氯离子的浓度从0增加到10 mg·L−1,CIP的降解效果的几乎保持不变,可以看出,氯离子的存在对CIP降解的影响很小,其主要的原因是Cl−能与HO·和C·反应生成弱氧化性自由基[40-42],如
ClOH⋅− 和Cl⋅−2 ,然而,这些弱氧化能力的自由基可以再次分解为HO·和Cl·,ClOH⋅− 的解离反应的速率常数也达到了6.1×109 L·(mol·s)−1,自由基的数量基本保持不变,如反应(8)—(11)[41, 43]所示。与氯离子不同,由图4b可以看出,NO−3 的存在一定程度上降低了CIP的降解效率,造成这种现象的一种可能是由于NO−3 被还原成NO−2 ,NO−2 消耗了HO·形成低氧化能力的自由基,如中反应(11)—(13)所列[44],从而抑制了CIP的降解。 -
天然有机物广泛存在于各类水体中,对水处理过程会产生一定的影响。选取腐殖酸(FA)作为天然有机物的代表,从图5可以看出,1 mg·L−1的腐殖酸对降解的抑制作用不明显,随着腐殖酸的浓度增加到5.0 mg·L−1,CIP降解受到明显的抑制,经过10 min的氧化降解,CIP降解效率从93.8%减小至59.9%。当腐殖酸的浓度增加至10.0 mg·L−1,CIP降解率下降至45.8%。
造成这一现象的主要原因是天然有机物的紫外过滤、光子吸收和自由基抑制作用。首先,天然有机物(NOM)可以作为自由基清除剂消耗自由基[32, 45],研究表明,NOM可以以1.3×104 L·(mol·s)−1 [32]和2.5×104 L·(mol·s)−1 [45]的二级反应速率与Cl·和·OH反应,在消耗活性自由基的方面与CIP形成竞争关系,导致了CIP降解的抑制;其次,在相同的体系中,NOM可以通过氯化降解,其拟一级动力学常数为3×10−5 s−1[46],造成一定比例氯的消耗;最后,NOM可以作为一种内部过滤器,过滤吸收254 nm处的紫外线并发生光解作用,改变紫外光通量,减少了氯和CIP对紫外线的吸收,减少了自由基的产生[47]。
废水中不仅含有大量无机离子,而且还含有许多有机大分子物质,包括多种蛋白质,本文选取牛血清蛋白(BSA)作为蛋白类有机物代表,研究了不同浓度的BSA对CIP降解的影响,结果如图5b所示,1 mg·L−1的BSA对降解有一定的抑制作用,8 min时CIP降解效率从90.7%下降至82.4%。随着BSA浓度持续增加,10.0 mg·L−1BAS存在时,CIP的降解效率仅为42.2%。在化学结构上BSA是由不同的氨基酸组成,研究表明,特定的氨基酸与臭氧分子和HO·都有较高的反应速率[48],因此 BSA会与环丙沙星竞争HO·等其他活性自由基,进而抑制 CIP的降解效果。在对同时含有FA和CIP的水体与同时含有BSA和CIP的水体进行了氧化反应前后的三维荧光分析,结果如图6,环丙沙星、FA和BAS均具有显著的荧光特征,FA主要存在两个征吸收峰,范围在Ex/Em = 310—360/370—450 nm、Ex/Em = 240—270/370—440 nm,CIP也存在两个荧光峰Ex/Em = 270/420 nm、Ex/Em = 315/420 nm,与FA和BSA部分荧光特征峰的范围重叠,经紫外/氯工艺处理20 min后,体系荧光强度迅速下降,可以看出紫外/氯工艺组合除了对CIP有较好的去除效果,也可以有效去除水中的FA和BSA,同时也间接证实了FA和BSA与CIP之间具有争夺自由基的竞争关系。
-
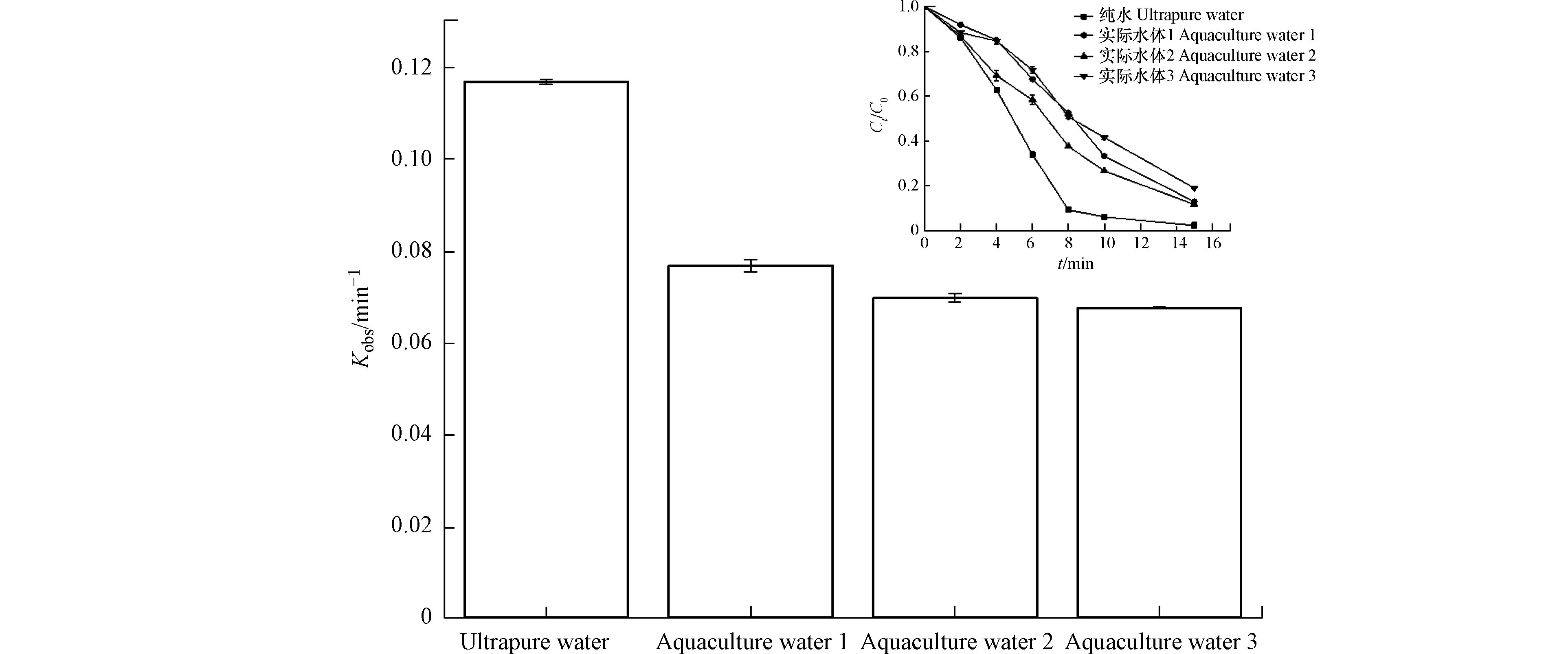
养殖废水是水环境中环丙沙星的一个重要来源,因此,实验选取了3种养殖废水开展废水中CIP降解研究,水质参数见表1,降解结果如图7所示,与超纯水相比,实际的养殖水体中的CIP降解速率常数均有所降低,造成这一现象的原因可能有两个,第一,废水中存在可能影响光降解的溶解和悬浮物质,其中,溶解有机物(DOM)可以消耗氯氧化剂,其消光系数为3.15 L·m−1·g−1[32],会降低光降解效率;第二,水体中的无机碳(
HCO−3 /CO23 )和DOM对紫外/氯工艺中污染物的去除有不利影响[15, 42, 49],HCO−3 将与CIP竞争,与⋅OH 和Cl⋅ 反应[50],NOM也会消耗⋅OH 和Cl⋅ ,其二级速率常数分别为2.5×104 L·(mol·s)−1和1.3×104 L·(mol·s)−1 [51],因此只有少量的加标氯作为游离氯参与反应。尽管CIP在实际废水中的降解速率常数低于超纯水,然而,由于多种活性物质的协同作用,其氧化降解速率仍保持较高水平,表明紫外/氯组合工艺处理环丙沙星废水的潜力。 -
通过液相色谱串联质谱法对紫外/氯降解CIP过程中的产物进行检测分析,结合报道的CIP氧化降解途径结果提出了CIP在紫外/氯工艺中降解路径[52-54],如图8所示。
紫外/氯组合工艺氧化降解CIP主要有三个途径。CIP中的两个主要反应位点是哌嗪环中的芳族叔胺基团和脂族仲胺基团[55],因此除了碳氟键的取代反应和喹诺酮部分的氧化,从而导致脱氟和羟基取代反应以外,CIP的大多数氧化反应发生在哌嗪环上。在路径Ⅰ中,环丙沙星失去羰基形成C-1,C-1脱羧基和脱氟后,形成C-2和C-3。碳氟键的键能高于碳碳键和碳氮键,而紫外线照射可以为此提供能量,在紫外线/过硫酸盐和紫外线/臭氧的研究中下也观察到了碳氟键的断裂[21]。在路径Ⅲ中,哌嗪环上的仲胺首先被攻击,可以产生中间产物C-6,两个羰基断裂,形成C-7和C-4;而在路径Ⅱ中,自由基攻击CIP的哌嗪环,碳氮键被打破,哌嗪环开环裂解,则会直接形成产物C-4脱乙烯环丙沙星,去乙烯环丙沙星的进一步氧化,通过氨基的损失产生带有羰基的C-8,哌嗪环被自由基进一步氧化,哌嗪取代基被完全破坏,形成苯胺,出现产物C-5,碳氮键断裂,环丙烷氧化断裂,形成C-9,随后进行脱羧基反应,形成C-10,这些产物可能进一步降解为CO2、H2O、F−和其他小分子化合物。
-
小球藻的毒性实验结果如图9所示,纵坐标为小球藻光合系统Ⅱ(PSⅡ)的最大光合效率(Fv/Fm),横坐标为投加CIP溶液和氧化降解处理后溶液的两组小球藻的培养时间。添加降解反应后溶液的小球藻组在48 h内,Fv/Fm值随着时间的增长呈现下降趋势,由0.57下降至0.41,培养48 h后趋于稳定,而只添加CIP溶液的小球藻组的Fv/Fm值没有明显变化,说明CIP降解产生的产物具有一定的急性毒性,抑制了小球藻的光合能力,限制了光合作用中的能量捕获,阻断了初级反应中的电子传递链,造成小球藻的最大光合效率下降。此外,采用了ECOSAR(V2.0)综合评价了CIP及其降解产物的毒性,结果如表2所示,LC50、EC50和ChV值分别代表半致死浓度、半有效浓度和慢性毒性。在推测的降解路径中,产物C-4, C-6, C-7外,已鉴定的中间产物的半数致死浓度、半有效浓度和慢性毒性值均高于CIP本身,说明在降解过程中,CIP产生了具有更高生态风险的中间产物,也例证了小球藻实验中,反应后的溶液会导致小球藻的最大光合效率下降的现象。此外,在反应结束后,毒性较大的产物C-3仍被检出,这意味着导致小球藻的最大光合效率下降急性毒性可能来自产物C-3。
-
为了进一步研究紫外/氯组合工艺处理实际废水的可行性,采用电能消耗率(EE/O)对其电能成本进行了经济成本分析。电能消耗率(EE/O)是指单位体积的水中污染物降解一个数量级所需的电能[56],单位为千瓦时(kWh),可通过如下公式计算得出:
其中,P为总电功率(kW),t是时间(h),V是溶液的体积(m3),C0和Ct是污染物的初始和最终浓度,取总电能消耗的45%作为维护费。
当初始CIP浓度为30 μmol·L−1时,温度 25 ℃,pH=7时,计算出的电能电耗值为0.85 kWh·m−3,经查阅文献,整理其他基于紫外的高级氧化工艺降解CIP的EE/O值于表3,可以观察到,与其他工艺相比,紫外/氯工艺的电能消耗显著降低,仅为紫外/过氧化氢、紫外/臭氧工艺的1/20和1/5,进一步证实紫外/氯组合工艺用于降解环丙沙星废水的可行性。
-
(1)紫外与氯组合工艺对CIP的去除具有协同增效的效果,10 min去除率高达95%。其降解速率随着氧化剂浓度的增高而增高,中性反应条件有利于环丙沙星的氧化降解;氯离子对降解效率影响不大,硝酸根离子抑制反应进行,降低CIP的降解率;天然有机物和牛血清蛋白浓度的浓度越高,其对CIP降解的抑制作用越明显。
(2)紫外/氯组合工艺氧化降解CIP的机理包括哌嗪环的氧化开环和喹诺酮氧化导致的脱羧基。小球藻毒性试验和ECOSAR分析结果均表明CIP氧化过程中生成了毒性更高的具有高生态风险的产物。
(3)实际废水环境中,CIP的降解受到一定程度的抑制,但仍保持较高的降解速率。相比于基于紫外的其他氧化工艺,紫外/氯组合工艺的电能消耗率较低,表明该工艺降解实际CIP废水的极大潜力。
紫外/氯组合工艺降解环丙沙星废水的效能、机理及毒性
UV/chlorine as an advanced oxidation process for the degradation of ciprofloxacin:Degradation efficiency, mechanism and toxicity evaluation
-
摘要: 环丙沙星是一种被广泛使用的氟喹诺酮类抗生素,在水体环境中经常被检测到,常规废水处理工艺对其去除效果有限,对水环境安全构成潜在威胁。本文拟采用紫外/氯组合工艺处理环丙沙星废水,系统研究其降解环丙沙星的效能、机理及毒性效应。考察了不同氯投加量、溶液pH、常见阴离子、腐殖酸浓度等环境因素对环丙沙星(CIP)降解效能的影响,并探究了CIP的降解路径以及产物毒性。结果表明,单独氯化和单独紫外对CIP的去除有限,而紫外/氯组合工艺对CIP的去除效果较好,去除率高达99.31%。当氯投加量由0.07 mmol·L−1增加至0.42 mmol·L−1,CIP的降解速率由0.078 min−1增加至0.106 min−1。中性反应条件更有利于CIP氧化降解。水体中存在的硝酸根离子、天然有机物和牛血清蛋白不同程度抑制CIP的降解。CIP降解过程中,共鉴定出9种氧化产物,阐述了CIP的几种降解途径。小球藻毒性试验和ECOSAR分析结果表明CIP氧化降解过程中产生了毒性较高的中间产物。经济成本分析结果表明紫外/氯组合工艺可用于处理环丙沙星废水。Abstract: Ciprofloxacin is a widely used fluoroquinolone antibiotic frequently detected in the aqueous environment and poses a threat to environmental safety. To effectively remove the antibiotic ciprofloxacin, which is difficult to remove by conventional water treatment processes, the degradation of Ciprofloxacin (CIP) by UV/ chlorine processes was evaluated. The efficacy of the process in degrading ciprofloxacin was investigated, and the degradation effect of ciprofloxacin was studied under different conditions of chlorine concentration, pH, common anion concentration and humic acid concentration, The degradation pathway of CIP and the toxicity of CIP products were explored. The results showed that compared with direct photolysis and Individual chlorination, UV/chlorine has a more pronounced CIP degradation efficiency. Remarkable CIP degradation was observed in the UV/chlorine system. the removal percentage of CIP can be reached to 99. 31%. The increase of oxidant dosing could promote the degradation of CIP, When the oxidant concentration increases from 0.07 mmol·L−1 to 0.42 mmol·L−1, the degradation rate constant reduces from 0.078 min−1 to 0.106 min−1.The CIP degradation could be improved under neutral conditions, while the presence of nitrate ions, natural organicmatter(NOM) and Bovine albumin (BSA) in the water inhibited the degradation of CIP. intermediates were identified by high performance liquid chromatography–tandem mass spectrometry. The possible degradation pathways were proposed based on the intermediate products. The results of the Chlorella toxicity test and ECOSAR analysis indicate that highly toxic intermediates are produced during the oxidative degradation of CIP. The electrical energy per order (EE/O) analysis showed that UV/chlorine process was a less energy consumption process, providing a feasible method for the treatment of ciprofloxacin in wastewater.
-
氯代苯酚类化合物具有高毒性、难降解和生物累积性的特征,是一种常见的持久性有机污染物[1-3],且广泛存在于土壤、沉积物、地表和地下水中。为了降低对受纳水体的污染,氯酚废水的处理至关重要。目前对于氯酚类废水的处理方法主要有生物降解法[4-6]、物理吸附法[7-8]、高级氧化法[9-11]等。近年来,高级氧化技术以其氧化能力强、反应速度快、限制少和处理效率高等优点受到众多学者的青睐[12]。臭氧氧化法是目前最具代表性且最有效的高级氧化法之一。众所周知,臭氧氧化有机物主要的两种方式:一是臭氧分子与有机物的直接反应;二是臭氧分解产生的羟基自由基(OH·)与有机物的间接反应[13]。纳米二氧化锰作为催化剂能有效促进臭氧催化氧化过程中羟基自由基等活性物质的形成,使其对难降解有机污染物的去除更加迅速、彻底,是催化臭氧化中最常使用的催化剂之一[14-17]。
目前,很多学者已经对催化臭氧化的降解效率及降解动力学等[18-19]进行了研究,但鲜有研究其降解过程中的生物毒性变化及其关键的致毒因子。已有研究发现,由于臭氧的部分氧化特性,生成的大量降解中间产物的生物毒性可能大于母体污染物,造成母体污染物浓度降低而总体生物毒性升高的现象[20].
本研究以3-CP作为模型污染物,探究二氧化锰催化臭氧化中生物毒性的变化规律,并识别高毒性的中间产物。在此基础上,研究催化臭氧化体系处理各类有机化合物过程中自由氯浓度的变化,以揭示高毒性中间产物的产生机理。
1. 材料与方法(Materials and methods)
1.1 试剂
3-CP(分析纯)购自中国能源化工有限公司;一水合硫酸锰(MnSO4∙H2O,分析纯)和叔丁醇(t-BuOH,分析纯)由迈瑞尔化学技术有限公司提供;高锰酸钾(KMnO4,分析纯)购于上海斯信生物科技有限公司。配制模拟湖水所用药品(CaCl2、NaHCO3、KCl、CH3COONa和MgSO4)和亚硫酸钠(Na2SO3)等均购自国药集团化学试剂有限公司且均为分析纯。
1.2 纳米二氧化锰的制备
以高锰酸钾和一水合硫酸锰为原料,采用水化学沉积法制备了MnO2纳米粒子[21-22],其反应方程式为式(1)。取物质的量比为2:3的KMnO4和MnSO4∙H2O在80 ℃下分别溶解于125 mL去离子水中;将上述两溶液缓慢滴入装有250 mL去离子水的三口瓶中,并在油浴锅中恒温(80 ℃)缓慢搅拌反应2 h;将得到的黑色或棕色悬浮液自然冷却1 h,随后超声分散10 min;将制备好的悬浮液进行抽滤,去离子水洗涤,重复3次;将洗涤抽滤后的产物在80 ℃下干燥过夜至恒重,制得纳米二氧化锰样品。
2MnO−4+3Mn2++2H2O⟶5MnO2↓+4H+ (1) 1.3 实验装置
在有效容积为2 L的玻璃反应器中进行半静态催化臭氧氧化实验,实验装置如图1所示。实验以纯氧为气源在臭氧发生器(中国青岛国林环保科技有限公司,CF-G-3-10G)中产生臭氧,使用在线检测仪(中国朗科公司,LT-200B)测定气相中臭氧浓度。臭氧与氧气的混合物以0.5 L∙min−1的流速,通过固定在反应器底部的烧结曝气头分散到溶液中,臭氧尾气用KI溶液进行吸收。3-CP的初始浓度为40 mg∙L−1,臭氧浓度为28 mg∙L−1,MnO2投加量为1 g∙L−1,反应溶液pH值为1—12。不同pH的氯酚溶液用10 mmol∙L−1磷酸缓冲液配制,并使用1 mol∙L−1 H2SO4和1 mol∙L−1 NaOH调节至相应的pH。反应过程中以固定的时间间隔取样,在取样后,立即用氮气吹脱臭氧化水样[23],以消除溶解在溶液中未反应的臭氧,然后再进行下一步操作。
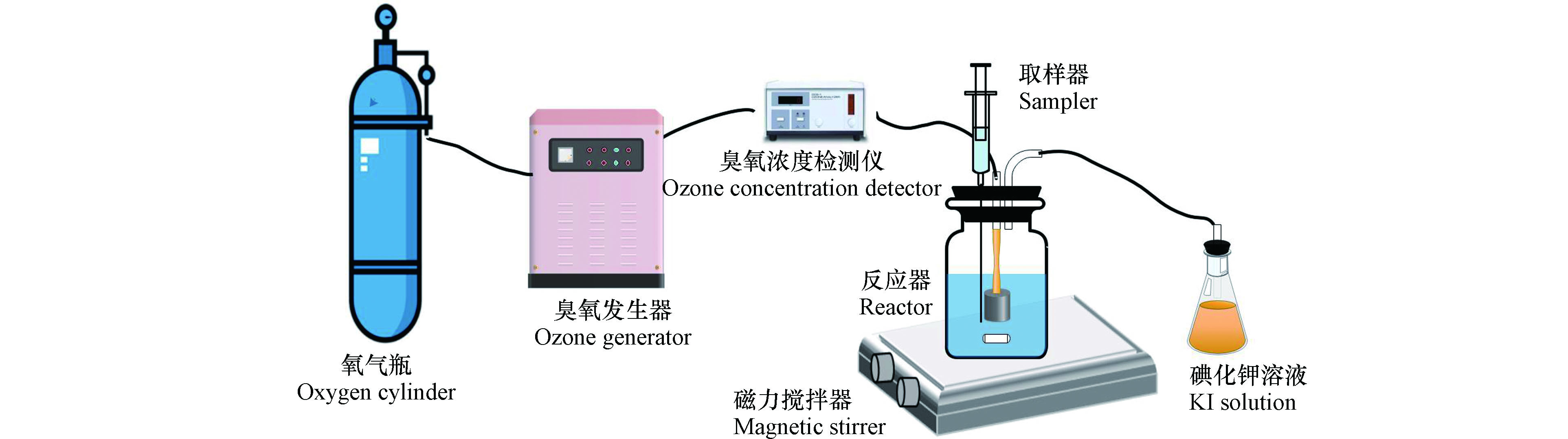 图 1 MnO2催化臭氧氧化3-CP装置示意图Figure 1. Schematic diagram of catalytic ozonation of 3-CP with MnO2
图 1 MnO2催化臭氧氧化3-CP装置示意图Figure 1. Schematic diagram of catalytic ozonation of 3-CP with MnO21.4 急性毒性的测定
使用灵敏度高的发光细菌青海弧菌(Vibrio qinghaiensis sp.NovQ67)[24],进行发光菌发光抑制实验测定急性生物毒性。具体的测试方法根据已有的研究方法[25-27]进行了适当的调整。
取-80 ℃条件下保存的Q67菌种(1 mL菌液)接入装有液体培养基的锥形瓶,于22 ℃条件下,振荡(180 r∙min−1)培养16—18 h。取100 μL菌液于96孔板测定发光强度(RLU),离心(200 r∙min−1,10 min)后收集菌体,用模拟湖水将菌体制成菌悬液。调整菌悬液的密度,使其发光强度在20万—60万RLU∙mL−1之间,待用。实验前,将水样pH值调至7,并用人工配制的模拟湖水将样品稀释成一系列的浓度梯度。样品的相对浓度以稀释倍数的倒数表示。将180 μL水样和20 μL调整后的菌液先后加入96孔板中,模拟湖水设置空白对照,每个浓度设置3个平行。振荡15 min后,用酶标仪(Ifinite-200,Tecan,瑞士)测定各孔的发光强度。使用公式(2)计算发光抑制率IR(%):
IR(%)=RLIBC−RLISRLIBC 式中,RLIBC表示空白对照组中发光细菌发出的相对发光强度,RLIS表示原始和稀释的样品中发光细菌发出的相对发光强度。
剂量-效应浓度曲线和最大半抑制浓度利用Origin软件的logistic模型来进行拟合和计算:
y=Bottom+Top - Bottom1+(IC50x)Slope (3) 式中,x表示稀释溶液中初始样品的百分比;y表示发光抑制率;Slope为斜率参数;IC50表示相对半数效应浓度。
根据公式(4)计算毒性单元:
TU=TOCIC50 (4) 式中:TU表示毒性单元;TOC表示总有机碳浓度。
1.5 分析测试
纳米二氧化锰的晶相结构采用XRD表征,仪器型号为D8ADVANCED,最大输出功率≥2.2 kV,最大管流80 mA,靶材为铜靶,扫描方式θ/θ或θ/2θ速,测量仪最小度数0.0001°。场发射扫描电镜(FESEM)(FEI Quanta 250F,美国)用于表征纳米二氧化锰的表面形貌。采用Zeta电位仪(Zeta Sizer 3000型,Malvern,英国)对纳米二氧化锰的表面零点电荷(pHzpc)进行测定。总有机碳分析仪(Multi N/C 3100,Elementar,德国)测定TOC浓度。自由氯由哈希余氯/总氯水质测试检测仪(PC-II,美国)测定。3-CP浓度通过配备有UV检测器(SPD-M20A,波长为180 nm至800 nm)的高效液相色谱仪(岛津LC-20A,日本)测得。在改良的C18柱(Lnertsil ODS-SP,250 mm×4.6 mm,5 μm)上进行分析,3-CP的计算波长为275 nm;进样体积为10 μL,流动相为流速1 mL∙min−1的甲醇与水的混合液,比例为6.5:3.5,柱温保持在40 ℃,保留时间约为7.0 min。
2. 结果与讨论 (Results and discussion)
2.1 纳米二氧化锰的表征
图2(a)为催化剂样品的X射线衍射光谱谱图,催化剂在2θ为12.6°、18.2°、28.8°、37.5°、41.9°、49.8°、56.4°、60.2°和69.7°处分别表现出了对应α-MnO2的(110)、(200)、(310)、(211)、(301)、(411)、(600)、(521)和(541)晶面的特征衍射峰[28],与α-MnO2(JCPDS 44-0141)匹配度高。图2(b)为纳米二氧化锰的FESEM图,可以看出所制备的纳米二氧化锰有团簇现象,但主要为松散的纳米线状,其直径约50—100 nm,长约0.2—1 μm。
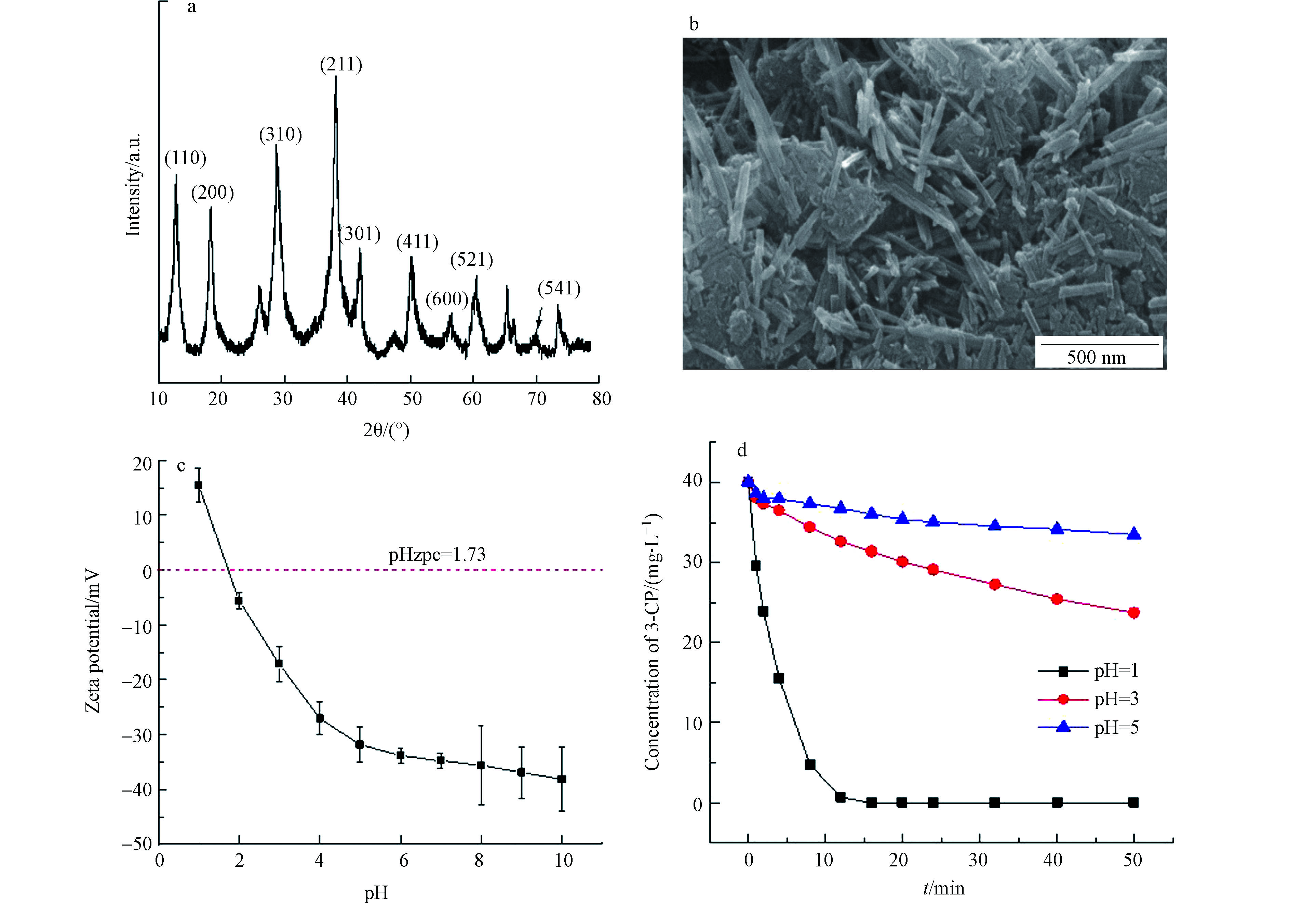 图 2 纳米二氧化锰的XRD、FESEM、Zeta电位及吸附性能表征Figure 2. Characterizations of nano manganese dioxide by XRD, FESEM, Zeta potential and adsorption properties
图 2 纳米二氧化锰的XRD、FESEM、Zeta电位及吸附性能表征Figure 2. Characterizations of nano manganese dioxide by XRD, FESEM, Zeta potential and adsorption properties如图2(c)所示,通过表面的电势的测量,催化剂的pHzpc=1.73(R2=0.998),与参考文献接近[29]。所以pH<1.73时,催化剂表面带正电;当pH>1.73时,催化剂表面带负电。如图2(d),纳米MnO2对3-CP的吸附反应进行8 min(pH=1、3、5)时,对3-CP的吸附率分别为89%、15%和5%,即随着本体溶液pH的升高,纳米MnO2催化剂对3-CP的吸附能力逐渐减弱。而3-CP的pKa为8.8—9.1[30],酸性条件下以未解离的分子形式存在。因而静电作用是导致3-CP在纳米二氧化锰表面吸附的主要原因。
2.2 催化臭氧化实验
本实验在不同pH(pH=1、3、5、7、10、12)下,考察了催化臭氧氧化3-CP过程中急性毒性、3-CP浓度、TOC浓度和自由氯浓度的变化。实验结果如图3所示。
2.2.1 不同pH下纳米二氧化锰的催化降解效果
从图3(a)中可以看出,当pH=1时,在1 min内3-CP降解率接近100%,且TOC浓度迅速下降,随着反应时间推移,TOC含量保持在5 mg∙L−1左右。3-CP的迅速去除一方面是由于催化剂MnO2在pH=1时有较强吸附能力,另一方面,酸性条件下MnO2能够催化臭氧对3-CP的氧化降解,但并未被完全矿化,而是生成了其它中间产物。
如图3(b)—(e)所示,当pH=3—12时,3-CP浓度和TOC浓度逐渐降低。pH=3时,3-CP完全降解需要约32 min,随着pH逐渐增大,3-CP完全降解所需时间随之减少。一方面,随着pH的升高,臭氧分子更容易分解生成氧化性更强的羟基自由基[30]。另一方面,Poznyak和Vivero[31]指出,氯酚类物质在高pH下发生解离,生成的离子形式与氧化剂臭氧分子和羟基自由基的反应速率大于分子形式。
2.2.2 不同pH下催化降解过程中毒性变化
如图3(a)所示,当pH=1时,实验过程中生物毒性没有升高,这可能是因为3-CP的去除主要是MnO2的吸附作用,高毒性中间产物没有累积。如图3(b)—3(e)所示,当pH=3—10时,生物毒性都呈现先升高后下降的变化,且生物毒性的最高值随pH的增加呈下降的趋势。当pH=3时,生物毒性最高值约为初始值的26倍,而当pH=12时,生物毒性没有明显升高的现象发生。造成这一现象的原因可能随着pH的升高,臭氧分解生成的大量羟基自由基能够和高毒性中间产物迅速反应,减少了中间产物的累积。
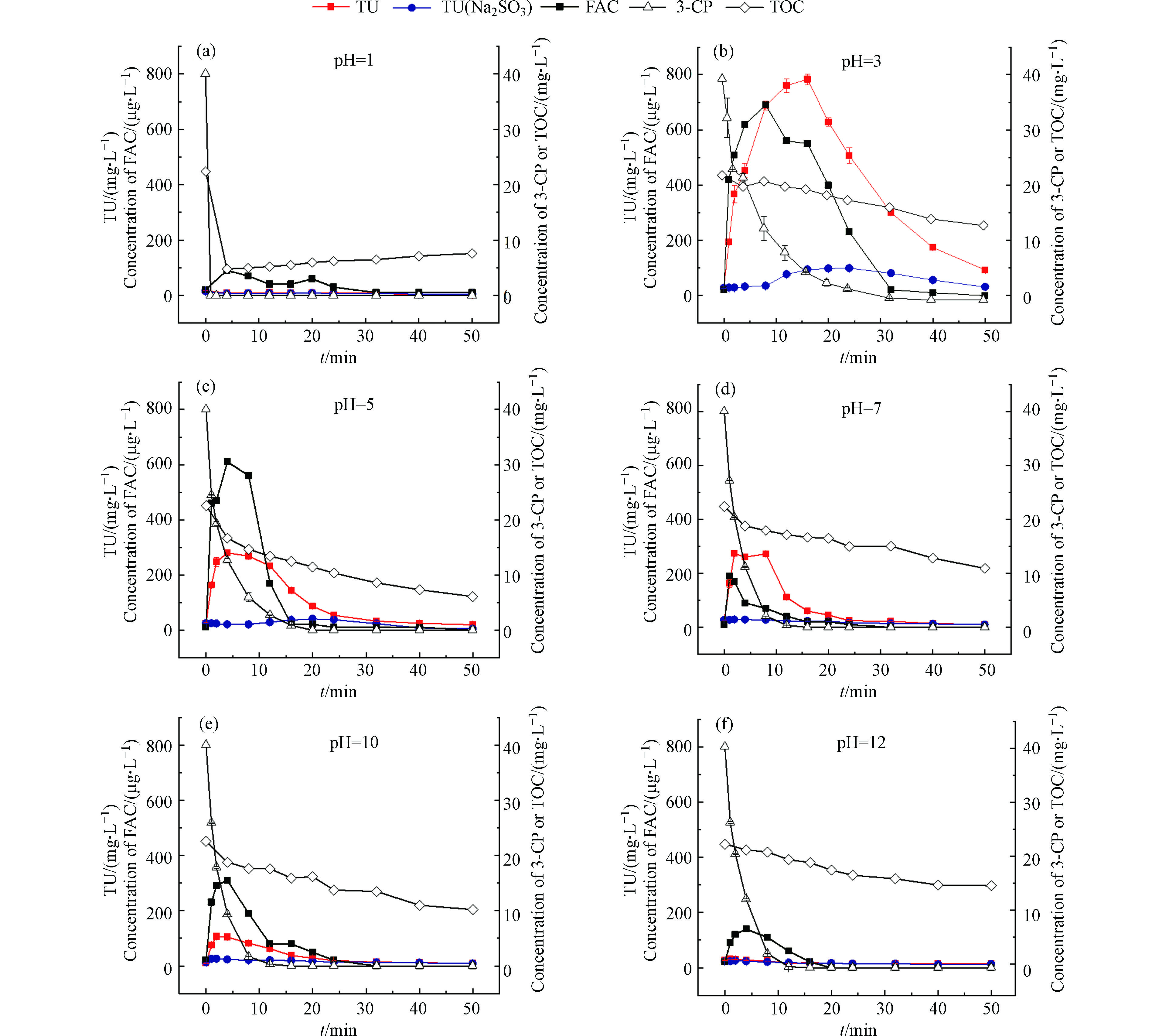 图 3 不同pH条件下纳米MnO2催化臭氧化3-CPFigure 3. Ozonation of 3-CP catalyzed by nano-MnO2 at different pH
图 3 不同pH条件下纳米MnO2催化臭氧化3-CPFigure 3. Ozonation of 3-CP catalyzed by nano-MnO2 at different pH为了确定体系中的MnO2催化剂是否是造成毒性升高的原因,在pH=3的情况下,研究了MnO2催化臭氧化3-CP、吸附3-CP以及臭氧化MnO2过程中的生物毒性变化。如图4所示,当反应体系中不通入臭氧或不加入污染物3-CP时,没有明显的生物毒性升高的现象发生。由此可以得出结论,在3-氯酚的催化臭氧氧化过程中,MnO2催化剂本身对生物毒性没有影响,仅作催化作用。
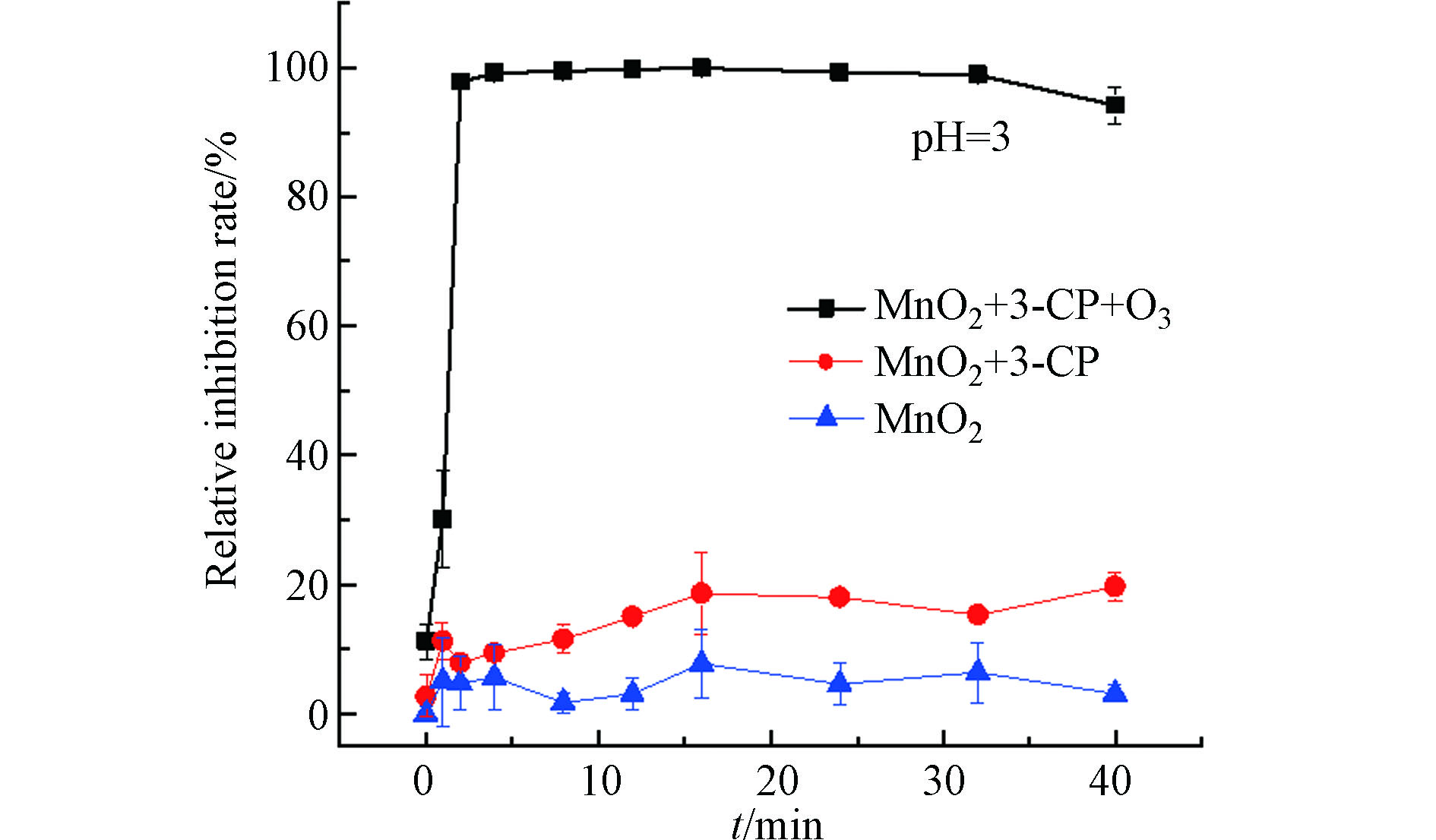 图 4 MnO2催化臭氧化3-CP、吸附3-CP以及臭氧化MnO2后毒性的变化Figure 4. Changes of toxicity of MnO2 catalytic ozonation/adsorption 3-CP and ozonation of MnO2
图 4 MnO2催化臭氧化3-CP、吸附3-CP以及臭氧化MnO2后毒性的变化Figure 4. Changes of toxicity of MnO2 catalytic ozonation/adsorption 3-CP and ozonation of MnO22.2.3 毒性与常规指标相关性分析
为了识别催化臭氧化中的关键致毒因子,监测了反应过程中自由氯的浓度变化,如图3(a)—(f),自由氯浓度的变化与生物毒性的变化一致,在pH=3—10时也呈现了先升高后下降的趋势。为了说明生物毒性与各指标的相关性,通过皮尔森相关系数法,对生物毒性和3-CP残留浓度、TOC浓度及自由氯浓度的相关性进行了分析,分析结果如表1所示。
表 1 急性毒性与各指标的相关性Table 1. Correlation between acute toxicity and each index常规指标Conventional index 皮尔森相关系数Pearson correlation coefficient (r) pH=1 pH=3 pH=5 pH=7 pH=10 pH=12 3-CP浓度 0.957** −0.344 0.118 0.232 0.175 0.822** TOC 0.963** 0.267 0.503 0.542 0.600* 0.924** 自由氯 −0.282 0.764** 0.872** 0.759** 0.969** 0.760** 注:* P<0.05;** P<0.01。 Note: * means P<0.05; ** means P<0.01. | Show Table DownLoad:
CSV
DownLoad:
CSV
从表1中可以看出,在生物毒性显著增加的4组(pH=3、5、7和10)中,生物毒性和自由氯浓度在0.01水平上显著相关,生物毒性与3-CP残留浓度及TOC基本不存在相关性。另外,已有研究发现,在相同浓度下,自由氯对发光细菌的毒性比3-CP大得多[32]。由此推断,催化臭氧化3-CP中生成的高毒性中间产物可能为自由氯。为了验证自由氯在体系生物毒性变化中的作用,取反应后的溶液1.2 mL,加入10 mmol∙L−1亚硫酸钠溶液36 μL,以研究自由氯去除后生物毒性的变化规律。如图3(a)—(f)所示,加入自由氯去除剂亚硫酸钠后,生物毒性显著下降。在pH=3—10时,生物毒性的去除率最高达到100%。但在pH=3时,加入亚硫酸钠后的水样生物毒性仍显著高于初始值。有研究表明[33],在臭氧氧化氯酚类物质中能够生成某些有机中间转化产物,如氯代邻二酚、氯代己二烯二酸等,它们可能是除无机高毒性中间产物自由氯外,会造成催化/臭氧化过程中生物毒性升高的物质。因此,MnO2催化臭氧化3-氯酚中生成的自由氯是急性毒性升高的关键致毒因子。
2.3 自由氯生成机制研究
自由氯作为一种强氧化剂,长期以来一直是抑制市政和工业废水中微生物生长的有效消毒剂[32]。然而,如果经处理后的废水中含有自由氯,则可能对水生生物具有不良影响,引起一定的健康和安全问题。以往研究表明,使用Fenton处理含氯酚类的焦化废水时,自由氯会导致急性毒性增加[24]。高级氧化处理中自由氯的生成并导致处理后生物毒性升高需要进一步重视,这对高级氧化处理工艺的安全运行至关重要。在2.2催化臭氧化实验所有实验组中,当pH=3时生物毒性最高值最大,约为初始值的26倍。因此,将进一步探究pH=3时自由氯的生成机制。
2.3.1 氯离子对自由氯生成的影响
氯酚在臭氧化中会脱氯,形成氯离子[32]。臭氧氧化氯离子过程中主要的氧化剂有两种:一是羟基自由基,羟基自由基在酸性条件下,与氯离子反应生成氯自由基,最终生成氯气;二是臭氧分子,臭氧分子与氯离子反应,生成次氯酸根,在酸性条件下,再生成次氯酸,次氯酸分解产生氯气;在碱性条件下,次氯酸根被氧化成亚氯酸根和氯酸根,反应方程式及速率常数如表2所示。但O3对氯离子的氧化作用一般可以忽略,因为反应速率常数小于10−3 L·mol−1·s−1[32]。为了确定单独催化臭氧化氯离子对自由氯生成的影响,在pH=3的条件下,对浓度为2、5、10、50、100 mg·L−1的氯离子溶液进行催化臭氧化实验。如图5(a)所示,实验过程中没有明显的自由氯的生成。因此,催化臭氧化氯酚中自由氯的生成,不是通过单独的催化臭氧化氯酚脱氯形成的氯离子。
表 2 催化臭氧化氯离子过程反应方程式及速率常数Table 2. Reaction equation and rate constant of catalytic ozonation of chloride ion编号Number 反应方程式Reaction equation 速率常数Rate constant (k) 参考文献Reference (1) OH⋅+Cl−⟶ClOH−⋅ k1=(4.2±0.2)×109 L·mol−1·s−1 [35] (2) ClOH−⋅+H+⟶Cl−+OH⋅ k2=(2.4±0.4)×1010 L·mol−1·s−1 [35] (3) Cl⋅+Cl−⟶Cl−2⋅ k3=(7.8±0.8)×109 L·mol−1·s−1 [35] (4) 2Cl−2⋅⟶Cl−+Cl2↑ k4=(3.5±2.7)×109 L·mol−1·s−1 [35] (5) O3+Cl−⟶ClO−+O2 k5=2.48×10−3 L·mol−1·s−1 [32] (6) ClO−+H+⟶HClO k6=5.0×1010 L·mol−1·s−1 [36] (7) HClO+H++Cl−⟶Cl2↑+H2O k7=2.14×104 L2·mol−2·s−1 [36] 注:反应(1)—(4)温度为297±2 K,反应(5)—(7)温度为298 K. Note: The temperature of reactions (1)—(4) is 297 ± 2 K, and reactions (5)—(7) is 298 K. | Show TableDownLoad:
CSV
 图 5 氯离子、羟基自由基和共存有机物对FAC的影响Figure 5. Effects of (a) chloride ion, (b) hydroxyl radical and (c) coexisting organic compounds on free chlorine
图 5 氯离子、羟基自由基和共存有机物对FAC的影响Figure 5. Effects of (a) chloride ion, (b) hydroxyl radical and (c) coexisting organic compounds on free chlorine2.3.2 羟基自由基对自由氯生成的影响
如图5(b),为了探究自由氯生成中起作用的氧化剂,在催化臭氧氧化3-CP的体系中加入20 mL羟基自由基淬灭剂叔丁醇(TBA)后,发现反应过程中自由氯浓度明显升高。同时,臭氧氧化含氯离子的苯酚溶液实验结果与臭氧化3-CP类似,检测到明显的自由氯的生成;且加入TBA后,自由氯浓度也明显升高。如体系中不存在TBA,羟基自由基会与氯离子反应最终生成氯气,消耗一部分氯离子。在加入TBA后,羟基自由基被迅速反应,增加了直接与臭氧分子反应的氯离子,因而造成自由氯浓度增大。因此,在催化臭氧氧化3-CP的体系中,自由氯生成的主要氧化剂是臭氧分子。
2.3.3 共存有机物对自由氯生成的影响
为探究有机物共存对自由氯生成的影响,研究了不同有机物和氯离子共存时催化臭氧化中自由氯浓度的变化。其中氯离子浓度均为11 mg·L−1,有机物分别为乙醇(14.3 mg·L−1)、乙酸(18.7 mg·L−1)、富马酸(36.1 mg·L−1)、苯(24.3 mg·L−1)、苯酚(29.3 mg·L−1)和对苯二酚(34.3 mg·L−1)。
如图5(c)所示,小分子酸乙酸,不饱和酸富马酸,以及苯加氯离子的体系中没有检测到明显的自由氯。苯酚和苯二酚加氯离子的体系中检测到自由氯明显升高,且在同等摩尔浓度下,自由氯在苯二酚加氯离子的体系中浓度约为苯酚加氯离子体系中的两倍。在氯含量相同的情况下,苯酚加氯离子的催化臭氧臭氧化实验中,自由氯生成的最大值与3-氯酚催化臭氧化相当。因此,可以推测出催化臭氧化氯离子与一些酚类有机物的混合体系中会产生自由氯。
2.3.4 催化臭氧化中自由氯的生成机制
基于以上分析,推测催化臭氧氧化中自由氯生成机理,如图6所示。MnO2催化剂带负电,具有很强的吸附性能,能够吸附污染物3-CP和氧化剂臭氧分子。一部分被吸附的臭氧分子能够直接与污染物发生反应,生成有机中间产物及脱氯生成氯离子。另一部分臭氧分子可以在催化剂表面分解,生成高氧化性的羟基自由基。羟基自由基能够氧化氯离子,生成氯气,如表2(1)—(4)反应,由于反应体系连续曝气,氯气逸出,不会造成反应体系生物毒性的明显升高。
臭氧分子直接与氯离子反应,在pH<7.5时,主要生成HClO[36],如反应(5)和(6),但由于臭氧分子与氯离子反应常数仅为2.48×10−3 L∙mol−1∙s−1[32],因而不会造成自由氯的明显升高。根据2.3.1—2.3.3节,当酚类有机污染物及其中间产物与氯离子共存时,在催化剂MnO2的作用下,臭氧分子氧化污染物时共氧化氯离子,提高了反应(5)的反应速率,生成次氯酸,造成自由氯升高,因而生物毒性升高。如反应(7),生成的次氯酸与氯离子及氢离子反应生成氯气,在体系连续曝气的条件下,氯气逸出,进而使得生物毒性随着反应时间增加而下降。实验中,在尾气吸收装置碘化钾溶液中检测到氯离子的存在,间接证明了自由氯生成机制推测的正确性。
3. 结论(Conclusion)
MnO2催化臭氧化3-氯酚时,生物毒性不随母体污染物和TOC浓度同比例降低而是出现了先升高后下降的现象,尤其是当pH=3时,生物毒性最高值约为初始值的26倍。通过生物毒性与常规指标的相关性分析发现,催化臭氧化3-CP过程中生物毒性的大小和自由氯浓度在pH=3—10时显著相关。催化臭氧化溶液中加入自由氯去除剂亚硫酸钠后,生物毒性几乎全部去除,最高点生物毒性去除率达到100%。故造成生物毒性升高的关键致毒因子是自由氯。在MnO2催化剂的作用下,3-氯酚发生脱氯作用生成氯离子。当与酚类有机污染物及其中间产物共存时,臭氧分子能够迅速共氧化氯离子,生成次氯酸,造成生物毒性升高。
-
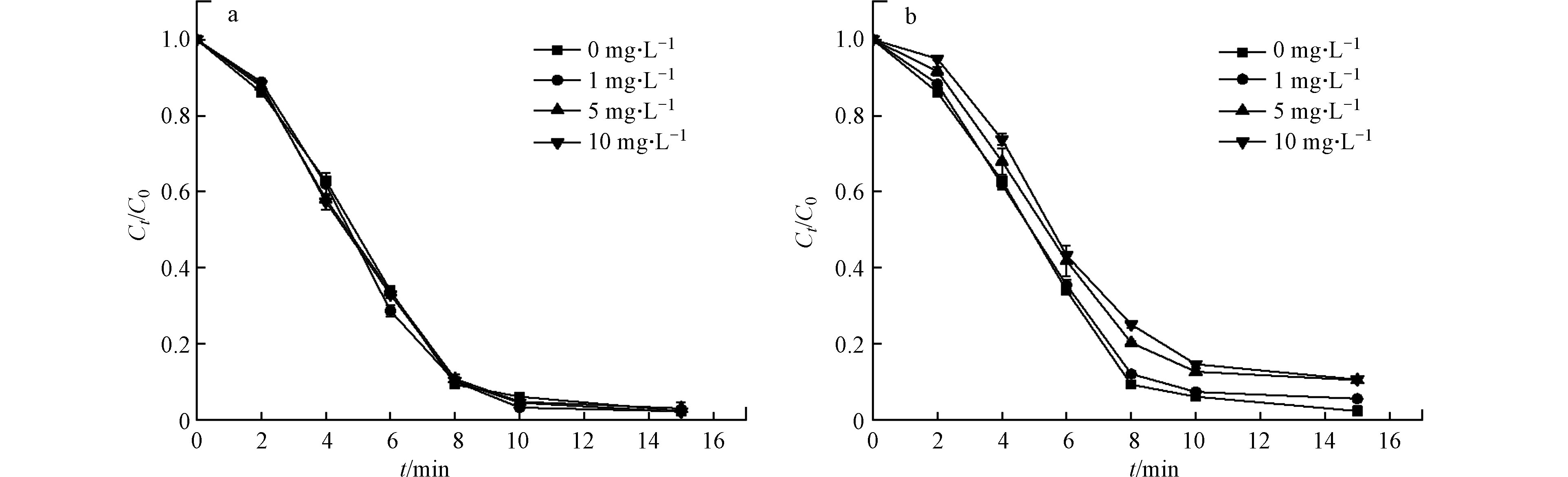
图 4 水中常见阴离子(a.氯离子; b.硝酸根离子)对CIP的降解效果的影响
Figure 4. Effect of common anions (a. Cl−1; b.
NO−3 ) on the degradation of CIP in water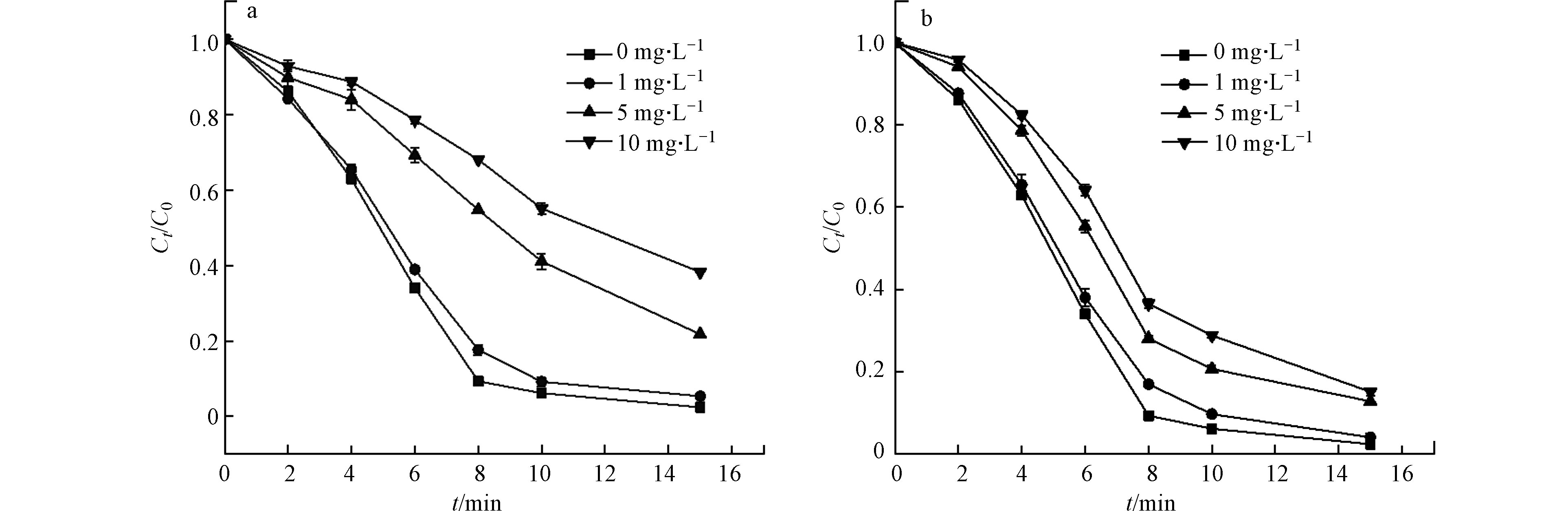
图 5 养殖水中常见有机物(a.腐殖酸 b.牛血清蛋白)对CIP的降解效果的影响
Figure 5. Effect of common organic substance (a. natural organic matter b. Bovine albumin) on the degradation of CIP in aquaculture water
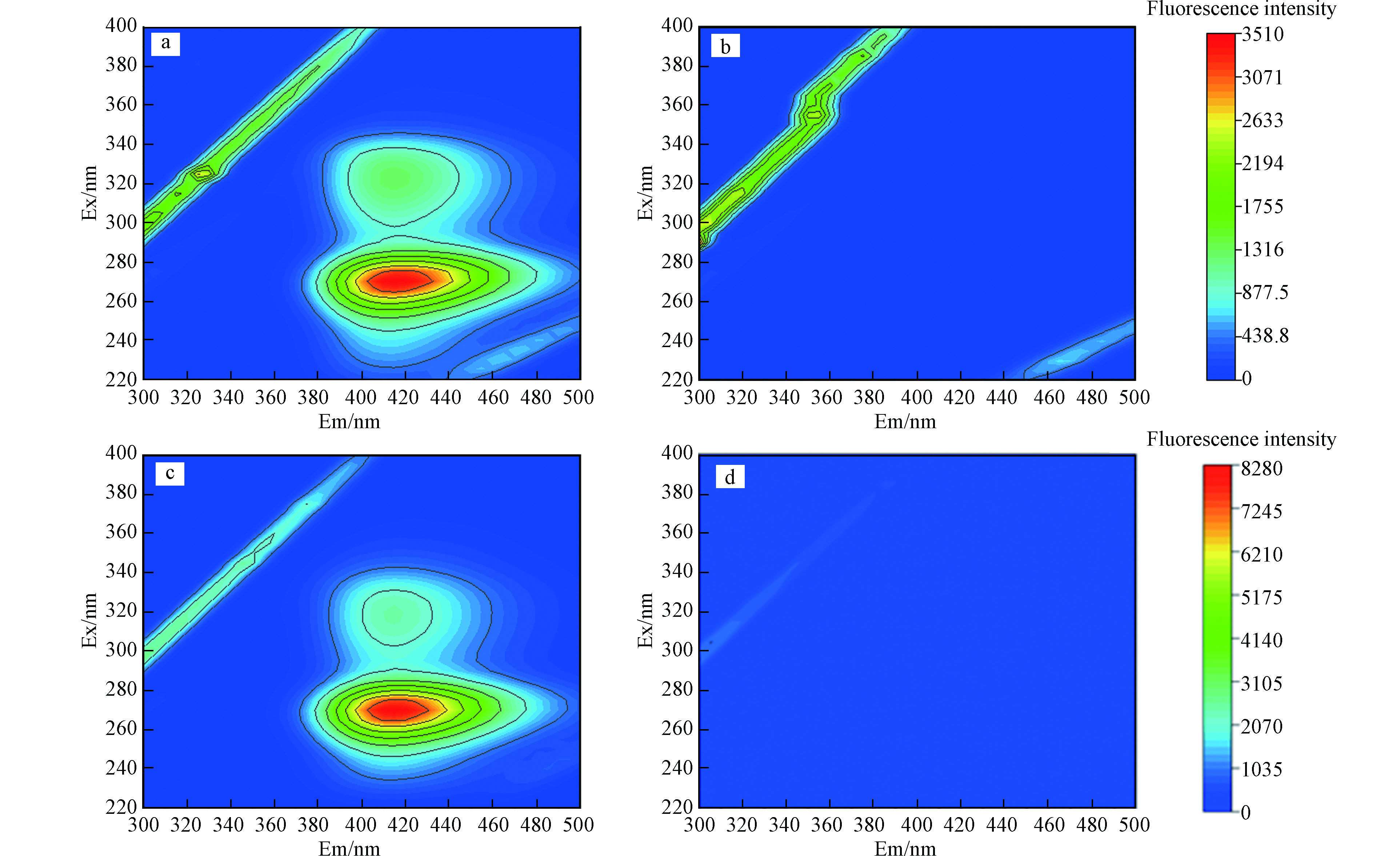
图 6 三维荧光图(a、b分别为FA+CIP复合物氧化前后 c、d 分别为BSA+CIP氧化前后)
Figure 6. Fluorescence spectra for NOMs in ultrapure water and BSA in ultrapure water(a、b:Before and after oxidation reaction of FA and CIP synthetic wastewater c、d:Before and after oxidation reaction of BSA and CIP synthetic wastewater)
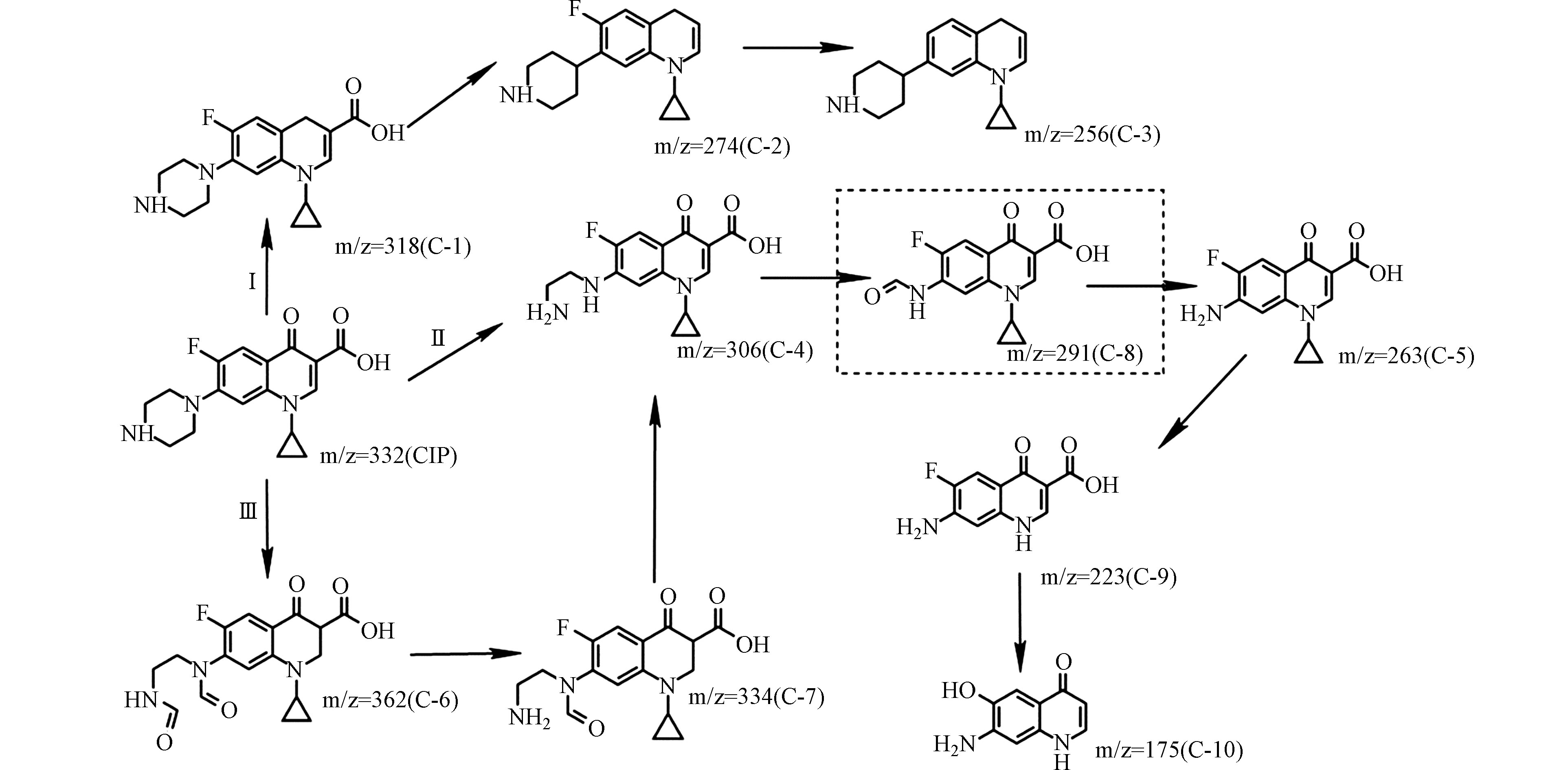
图 8 CIP产物路径图
Figure 8. Proposed CIP transformation pathways under UV/Cl oxidation system
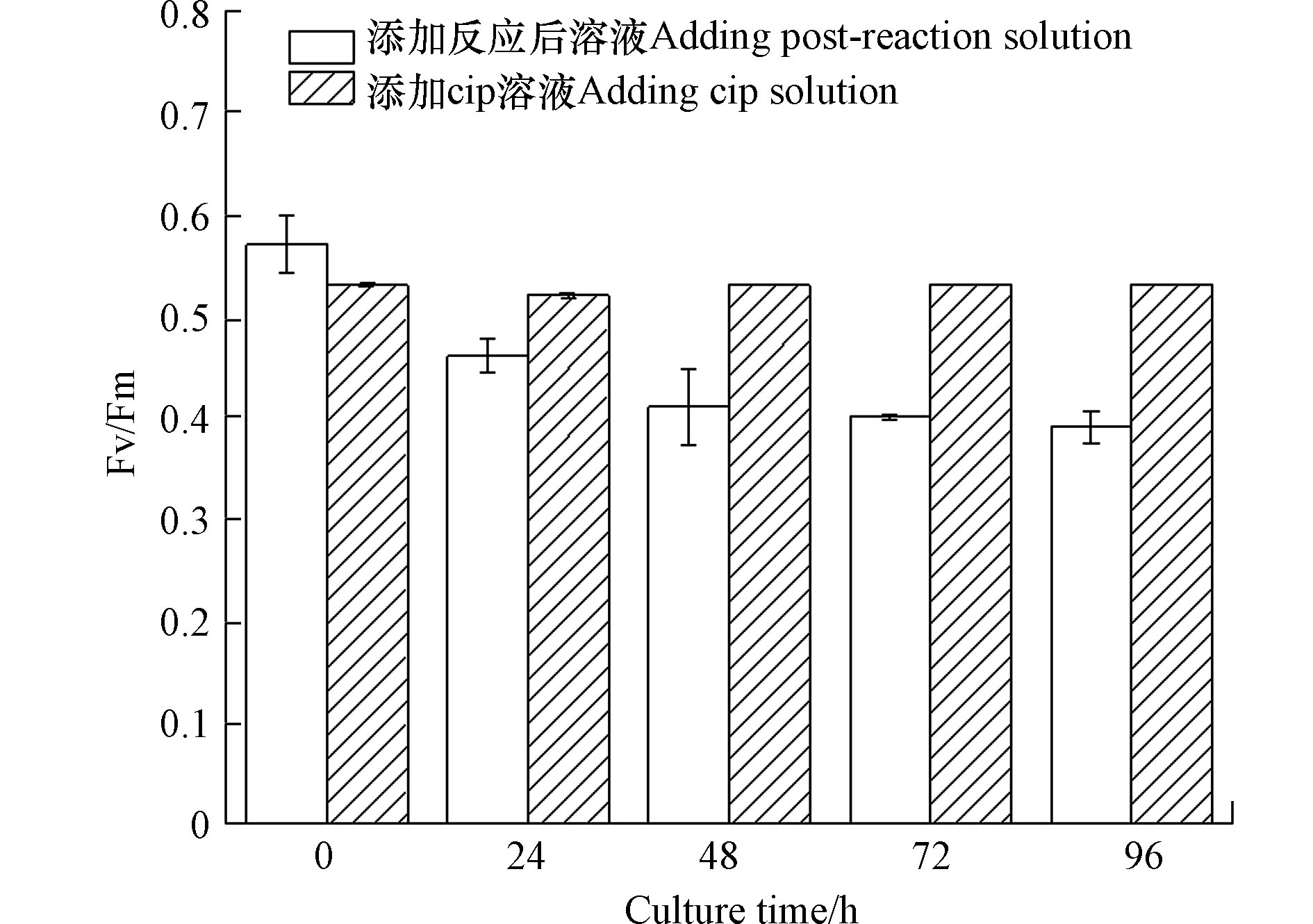
图 9 投加反应后溶液和CIP溶液后小球藻最大光合效率变化图
Figure 9. Changes in the maximal photochemical efficiency of PSⅡ in the darkafter dosing with post reaction solution and CIP solution
表 1 实际水体水质参数
Table 1. Water quality parameter of actual water bodies
水质参数Water quality parameter 实际水体1Aquaculture water 1 实际水体2Aquaculture water 2 实际水体3Aquaculture water 3 pH 9.06 8.55 9.47 NH4+-N/(mg·L−1) 1.16 0.89 1.36 NO3-N/(mg·L−1) 3.84 2.68 7.85 TP/(mg·L−1) 0.11 0.28 0.49 CODMn/(mg·L−1) 15.65 13.65 17.68
下载: 导出CSV
表 2 基于ECOSAR软件分析评估CIP及主要氧化产物的急性毒性结果(mg ·L−1)
Table 2. Estimated acute toxicity of CIP and its intermediates using ECOSAR software (mg · L−1)
物质 Compouds 鱼Fish 水蚤Daphnia 绿藻Green algae 96h-LC50 ChV 48h-LC50 ChV 96h-EC50 ChV CIP 13100 1550 1240 81.3 1620 455 C-1 1070 122 102 6.78 130 36.9 C-2 6.04 0.212 0.850 0.082 0.505 0.189 C-3 8.17 0.313 1.12 0.105 0.702 0.258 C-4 47300 7700 4020 238 6470 1680 C-5 899 12.4 42.8 0.488 128 47.9 C-6 237000 194000 330000 30500 121000 14400 C-7 141000 29000 11100 609 20800 5120
下载: 导出CSV
表 3 基于紫外的高级氧化工艺降解CIP过程中的单位电能消耗量比较
Table 3. Comparison of EE/O in CIP degradation by different UV-based advanced oxidation processes
下载: 导出CSV
-
[1] SU H, XU W, HU X, et al. Spatiotemporal variations and source tracking of antibiotics in an ecological aquaculture farm in Southern China [J]. Sci Total Environ, 2021, 763: 143022. [2] BURRIDGE L, WEIS J S, CABELLO F, et al. Chemical use in salmon aquaculture: A review of current practices and possible environmental effects [J]. Aquaculture, 2010, 306(1-4): 7-23. doi: 10.1016/j.aquaculture.2010.05.020 [3] YANG Y, SONG W, LIN H, et al. Antibiotics and antibiotic resistance genes in global lakes: A review and meta-analysis [J]. Environ Int, 2018, 116: 60-73. doi: 10.1016/j.envint.2018.04.011 [4] HE Z, CHENG X, KYZAS G Z, et al. Pharmaceuticals pollution of aquaculture and its management in China [J]. Journal of Molecular Liquids, 2016, 223: 781-789. doi: 10.1016/j.molliq.2016.09.005 [5] LIU X, STEELE J C, MENG X Z. Usage, residue, and human health risk of antibiotics in Chinese aquaculture: A review [J]. Environ Pollut, 2017, 223: 161-169. doi: 10.1016/j.envpol.2017.01.003 [6] CHEN H, LIU S, XU X R, et al. Antibiotics in typical marine aquaculture farms surrounding Hailing Island, South China: occurrence, bioaccumulation and human dietary exposure [J]. Mar Pollut Bull, 2015, 90(1-2): 181-187. doi: 10.1016/j.marpolbul.2014.10.053 [7] 胡冠九, 穆肃, 赵永刚, 等 南京典型县区饮用水源抗生素含量特征 [J]. 环境化学, 2015, 37(1): 192-193. HU G J, MU S , ZHAO Y G et al. Characteristics of antibiotic content in drinking water sources in typical counties of Nanjing, China [J]. Environmental Chemistry, 2015, 37(1): 192-193(in Chinese).
[8] 李兆新, 董晓, 吴蒙蒙, 等 黄海桑沟湾养殖区海水中喹诺酮类抗生素的残留状况 [J]. 海洋环境科学 2018, 37(2): 182-192. LI Z X, DONG X, WU M M, et al. Quinolone residues in seawater of aquaculture area, Sanggou bay, Yellow Sea, China[J]. Marine Environmental Science 2018, 37(2): 182-192(in Chinese).
[9] 刘晓晖. 洞庭湖流域水环境中典型抗生素污染特征, 来源及风险评估 [D]. 济南, 山东师范大学, 2017. LIU D H, Pollution level, source and ecological risk of typical antibiotics in the Dongting Lake, China [D]. Jinan, Shandong Normal University(in Chinese).
[10] LEE Y, GUNTEN U V. Oxidative transformation of micropollutants during municipal wastewater treatment: comparison of kinetic aspects of selective (chlorine, chlorine dioxide, ferrate Ⅵ, and ozone) and non-selective oxidants (hydroxyl radical) [J]. Water Research, 2010, 44(2): 555-566. doi: 10.1016/j.watres.2009.11.045 [11] ALFASSI Z B, HUIE R E, MOSSERI S, et al. Kinetics of one-electron oxidation by the ClO radical [J]. J. Phys. Chem, 1988, 32(1): 3888-3891. [12] GUO K, WU Z, SHANG C, et al. Radical Chemistry and Structural Relationships of PPCP Degradation by UV/Chlorine Treatment in Simulated Drinking Water [J]. Environmental Science & Technology 2017, 51(18): 10431-10439. [13] GREBEI J E, PIGNATELLO J I, MITCH W A, et al. Effect of halide ions and carbonates on organic contaminant degradation by hydroxyl radical-based advanced oxidation processes in saline waters [J]. Environmental Science & Technology 2010, 44(17): 6822-6828. [14] GAO Y Q, ZHANG J, LI C, et al. Comparative evaluation of metoprolol degradation by UV/chlorine and UV/H2O2 processes [J]. Chemosphere, 2020, 243: 125325, 10.1016/j. chemosphere. 2019.125325. [15] XIANG Y, FANG J, SHANG C. Kinetics and pathways of ibuprofen degradation by the UV/chlorine advanced oxidation process [J]. Water Research, 2016, 90: 301-308. doi: 10.1016/j.watres.2015.11.069 [16] KWON M, YOON Y, KIM S, et al. Removal of sulfamethoxazole, ibuprofen and nitrobenzene by UV and UV/chlorine processes: A comparative evaluation of 275nm LED-UV and 254nm LP-UV [J]. Sci Total Environ, 2018, 637-638: 1351-1357. doi: 10.1016/j.scitotenv.2018.05.080 [17] LU X, SHAO Y, GAO N, et al. Investigation of clofibric acid removal by UV/persulfate and UV/chlorine processes: Kinetics and formation of disinfection byproducts during subsequent chlor(am)ination [J]. Chemical Engineering Journal, 2018, 331: 364-371. doi: 10.1016/j.cej.2017.08.117 [18] LIU Z, LIN Y L, XU B, et al. Degradation of diiodoacetamide in water by UV/chlorination: Kinetics, efficiency, influence factors and toxicity evaluation [J]. Chemosphere, 2020, 240: 124761. [19] PAN Y, LI X, FU K, et al. Degradation of metronidazole by UV/chlorine treatment: Efficiency, mechanism, pathways and DBPs formation [J]. Chemosphere, 2019, 224: 228-236. doi: 10.1016/j.chemosphere.2019.02.081 [20] AO X, LIU W, SUN W, et al. Medium pressure UV-activated peroxymonosulfate for ciprofloxacin degradation: Kinetics, mechanism, and genotoxicity [J]. Chemical Engineering Journal, 2018, 345: 87-97. doi: 10.1016/j.cej.2018.03.133 [21] LIU H, GAO Y, WANG J, et al. The application of UV/O3 process on ciprofloxacin wastewater containing high salinity: Performance and its degradation mechanism [J]. Chemosphere, 2021, 276: 130220. doi: 10.1016/j.chemosphere.2021.130220 [22] HM A, XYA B, DC B, et al. Degradation of ciprofloxacin using UV-based advanced removal processes: comparison of persulfate-based advanced oxidation and sulfite-based advanced reduction processes [J]. Science of The Total Environment, 2020, 764: 144510. [23] CHAVES F P, GOMES G, DELLA-FLORA A, et al. Comparative endocrine disrupting compound removal from real wastewater by UV/Cl and UV/H2O2: Effect of pH, estrogenic activity, transformation products and toxicity [J]. Sci Total Environ, 2020, 746: 141041. doi: 10.1016/j.scitotenv.2020.141041 [24] LEE Y M, LEE G, KIM M K, et al. Kinetics and degradation mechanism of Benzophenone-3 in chlorination and UV/chlorination reactions [J]. Chemical Engineering Journal, 2020, 393: 124780. doi: 10.1016/j.cej.2020.124780 [25] KONG X, JIANG J, MA J, et al. Degradation of atrazine by UV/chlorine: Efficiency, influencing factors, and products [J]. Water Research, 2016, 90: 15-23. doi: 10.1016/j.watres.2015.11.068 [26] KHAN J A, HE X, SHAH N S, et al. Kinetic and mechanism investigation on the photochemical degradation of atrazine with activated H2O2, S2O8 and HSO5 [J]. Chemical Engineering Journal, 2014, 252: 393-403. doi: 10.1016/j.cej.2014.04.104 [27] STUMM W, MORGAN J J . Aquatic chemistry : Chemical equilibria and rates in naturalwaters [M]. New York : Wiley, 1995. [28] WATTS M J, LINDEN K G. Chlorine photolysis and subsequent OH radical production during UV treatment of chlorinated water [J]. Water Research, 2007, 41(13): 2871-2878. doi: 10.1016/j.watres.2007.03.032 [29] NOWELL L H, HOIGNÉ L N J. Photolysis of aqueous chlorine at sunlight and ultraviolet wavelengths—II. Hydroxyl radical production [J]. Water Research, 1992, 26: 599-660. doi: 10.1016/0043-1354(92)90233-T [30] DEBORDE M, GUNTEN U V . Reactions of chlorine with inorganic and organic compounds during water treatment--kinetics and mechanisms: A critical review [J]. Water Research. 2008, 42(1-2): 13-51. [31] BUXTON G V, GREENSTOCK C L, HELMAN W P, et al. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals ·OH/·O− in Aqueous Solution [J]. Journal of Physical & Chemical Reference Data 1988, 17: 536-635. [32] FANG J, YUN F, SHANG C, et al. The roles of reactive species in micropollutant degradation in the UV/free chlorine system [J]. Environmental Science & Technology, 2014, 48(3): 1859-1868. [33] MATTHEW B M, ANASTASIO C. A chemical probe technique for the determination of reactive halogen species in aqueous solution: Part 1 – bromide solutions [J]. Atmospheric Chemistry and Physics, 2006, 6(1): 899-940. [34] DODD M C, SHAH A D, GUNTEN U V, et al. Interactions of fluoroquinolone antibacterial agents with aqueous chlorine: Reaction kinetics, mechanisms, and transformation pathways [J]. Environmental Science & Technology, 2005, 39: 7065-7076. [35] WAMMER K H, KORTE A R, LUNDEEN R A, et al. Direct photochemistry of three fluoroquinolone antibacterials: Norfloxacin, ofloxacin, and enrofloxacin [J]. Water Research, 2013, 47(1): 439-448. doi: 10.1016/j.watres.2012.10.025 [36] HUANG N, WANG T, WANG W L, et al. UV/chlorine as an advanced oxidation process for the degradation of benzalkonium chloride: Synergistic effect, transformation products and toxicity evaluation [J]. Water Research, 2017, 114: 246-253. doi: 10.1016/j.watres.2017.02.015 [37] XIANG H, SHAO Y, GAO N, et al. Removal of β-cyclocitral by UV/persulfate and UV/chlorine process: Degradation kinetics and DBPs formation [J]. Chemical Engineering Journal, 2020, 382: 122659. doi: 10.1016/j.cej.2019.122659 [38] YANG W, TANG Y, LIU L, et al. Chemical behaviors and toxic effects of ametryn during the UV/chlorine process [J]. Chemosphere, 2020, 240: 124941. doi: 10.1016/j.chemosphere.2019.124941 [39] ZHU Y, WU M, GAO N, et al. Degradation of phenacetin by the UV/chlorine advanced oxidation process: Kinetics, pathways, and toxicity evaluation [J]. Chemical Engineering Journal, 2018, 335: 520-529. doi: 10.1016/j.cej.2017.10.070 [40] BUXTON G V, BYDDER M, SALMON G A. The reactivity of chlorine atoms in aqueous solution [J]. Physical Chemistry, 1999, 1(1): 269-273. [41] SUN P, LEE W N, ZHANG R, et al. Degradation of DEET and caffeine under UV/chlorine and simulated sunlight/chlorine conditions [J]. Environmental Science & Technology, 2016, 50(24): 13265-13273. [42] WU Z, FANG J, XIANG Y, et al. Roles of reactive chlorine species in trimethoprim degradation in the UV/chlorine process: Kinetics and transformation pathways [J]. Water Research, 2016, 104(NOV.1): 272-282. [43] ABDALRHMAN A S, WANG C, HOW Z T, et al. Degradation of cyclohexanecarboxylic acid as a model naphthenic acid by the UV/chlorine process: Kinetics and by-products identification [J]. J Hazard Mater, 2020, 402: 123476, 10.1016/j. jhazmat. 2020.123476. [44] ZHANG T Y, LIN Y L, XU B, et al. Effect of UV irradiation on the proportion of organic chloramines in total chlorine in subsequent chlorination [J]. Chemosphere, 2016, 144(FEB.): 940-947. [45] LEE C, YOON J, GUNTEN U V. Oxidative degradation of N-nitrosodimethylamine by conventional ozonation and the advanced oxidation process ozone/hydrogen peroxide [J]. Water Res, 2007, 41(3): 581-590. doi: 10.1016/j.watres.2006.10.033 [46] A P W, B P C, C H M. Reactivity of natural organic matter with aqueous chlorine and bromine [J]. Water Res, 2004, 38(6): 1502-1513. doi: 10.1016/j.watres.2003.12.014 [47] XUE T, SHI, YONG Z, et al. Kinetics and pathways of Bezafibrate degradation in UV/chlorine process [J]. Environmental Science and Pollution Research, 2017, 25(1): 672-682. [48] BUFFLE M O, GUNTEN U V. Phenols and amine induced HO· generation during the initial phase of natural water ozonation [J]. Environmental Science & Technology, 2006, 40(9): 3057-3063. [49] LU X, SHAO Y, GAO N, et al. Degradation of diclofenac by UV-activated persulfate process: Kinetic studies, degradation pathways and toxicity assessments [J]. Ecotoxicology and Environmental Safety, 2017, 141: 139-147. [50] YANG Y, JIANG J, LU X, et al. Production of sulfate radical and hydroxyl radical by reaction of ozone with peroxymonosulfate: A novel advanced oxidation process [J]. Environmental Science & Technology, 2015, 49(12): 7330-7339. [51] KATSOYIANNIS I A, CANONICA S, GUNTEN U V. Efficiency and energy requirements for the transformation of organic micropollutants by ozone, O3/H2O2 and UV/H2O2 [J]. Water Research, 2011, 45(13): 3811-3822. doi: 10.1016/j.watres.2011.04.038 [52] DENG J, XU M, FENG S, et al. Iron-doped ordered mesoporous Co3O4 activation of peroxymonosulfate for ciprofloxacin degradation: Performance, mechanism and degradation pathway [J]. Science of The Total Environment, 2018, 658(MAR.25): 343-356. [53] DING X, YANG X, WANG P, et al. Oxygen vacancy boosted photocatalytic decomposition of ciprofloxacin over Bi2MoO6: Oxygen vacancy engineering, biotoxicity evaluation and mechanism study [J]. Journal of Hazardous Materials, 2019, 364: 691-699. doi: 10.1016/j.jhazmat.2018.10.063 [54] LUO K, YANG Q, PANG Y, et al. Unveiling the mechanism of biochar-activated hydrogen peroxide on the degradation of ciprofloxacin [J]. Chemical Engineering Journal, 2019, 374: 520-530. doi: 10.1016/j.cej.2019.05.204 [55] HU L, STEMIG A M, WAMMER K H, et al. Oxidation of antibiotics during water treatment with potassium permanganate: Reaction pathways and deactivation [J]. Environmental Science & Technology, 2011, 45(8): 3635-3642. [56] MIKLOS D B, REMY C, JEKEL M, et al. Evaluation of advanced oxidation processes for water and wastewater treatment - A critical review [J]. Water Research, 2018, 139: 118-131. doi: 10.1016/j.watres.2018.03.042 [57] LESTER Y, AVISAR D, GOZLAN I, et al. Removal of pharmaceuticals using combination of UV/H2O2/O3 advanced oxidation process [J]. Water Sci Technol, 2011, 64(11): 2230-2238. doi: 10.2166/wst.2011.079 [58] MAHDI AHMED M, CHIRON S. Ciprofloxacin oxidation by UV-C activated peroxymonosulfate in wastewater [J]. J Hazard Mater, 2014, 265: 41-46. doi: 10.1016/j.jhazmat.2013.11.034 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5398
- HTML全文浏览数: 5398
- PDF下载数: 120
- 施引文献: 0















