











-
随着工业和农业的快速发展,水环境问题中的硝酸盐污染日益严重,引起了世界各国的广泛重视。硝酸盐污染物的主要来源有垃圾渗滤液、牲畜粪便、生活和工业废水等[1]。水体中的硝酸盐含量高会破坏生态环境,造成水体富营养化,影响水生生物生存[2]。用于饮用的地表水或地下水中含有过高浓度的硝酸盐会严重危害人类的健康,导致婴儿出现蓝婴综合征(高铁血红蛋白血症)、肝损伤等,世界卫生组织的国际癌症研究机构将摄入硝酸盐归类为可能对人类致癌的一类物质,特别是在促进内源性亚硝化的条件下摄入硝酸盐,硝酸盐会在体内形成N-亚硝基化合物,增大了人类患胃癌等疾病的风险[3]。因此,世界卫生组织设定饮用水中硝酸盐的最大浓度为50 mg·L−1
NO−3 [4]。中国最新生活饮用水卫生标准(GB 5749—2006)设定硝酸盐氮浓度为10 mg·L−1,地下水源限制时标准可放宽至20 mg·L−1。由于水中硝酸盐污染的严重性和危害性,人们研究出各种方法致力于去除水中的硝酸盐,主要分为生物法[5]、物理分离法[6]、催化加氢法[7]、化学还原法[8]、电催化还原法[9]。其中生物法是利用微生物在生物反应器中的反硝化作用还原硝酸盐,释放出分子态氮或一氧化二氮。该方法由于微生物培养周期较长、反应器占地面积较大、出水可能存在细菌污染,其应用存在一定的限制性[10]。离子交换法和反渗透法等物理方法操作简单,但只是将硝酸盐从污水中分离出来,并没有将污染物从根本上去除。催化加氢法需要持续投加反应所需的氢气,而氢气的易燃易爆性质使工艺存在很大危险性。电催化还原法即电催化反硝化法,是在通电的条件下,硝酸盐离子在阴极上被还原生成氮气等产物的一种脱氮方法。具有不产生污泥,占地面积小,处理效率高,安全环保、操作易控等优点[11]。1980年代,人们就已经开始将硝酸盐电化学还原法用于水处理,HORÁNYI等[12]采用金属铂作为电极材料,探究了硝酸盐氮在碱性条件下的还原反应。近年来,水中硝酸盐的电催化还原研究受到广泛关注,被认为是很有前景的处理方法。在硝酸盐的电催化还原过程中,主要在阴极上进行硝酸盐吸附和电子传递等关键步骤。电极材料、支持电解质和施加电流等对反应的动力学和产物选择性都有影响,因此,开发一种性质稳定、电催化还原活性高的电极具有重要意义。
目前,许多研究人员已将Zn[13]、Fe[14]、Cu[15]、Pt[16]、Ti[17]、碳纸[18]等材料应用于硝酸盐还原电极的制备。由于不同材料间可能存在协同效应,关于双金属、三元金属或合金电极上的硝酸盐电还原也有广泛的研究[19]。Jonoush等[20]采用Ni、Ni-Fe0和Ni-Fe0@Fe3O4电极对硝酸盐氮的去除效果以及硝酸盐氮还原反应动力学进行探究,结果表明,Ni-Fe0@Fe3O4电极的电催化还原性能最好,在电解电流密度为5 mA·cm−2、pH=6.2的条件下,对50 mg·L−1的硝酸盐溶液进行电解,240 min后硝酸盐去除率达到90.19%。Zhou等[21]以3D Pd-Cu(OH)2/CF电极为阴极,对50 mg·L−1的硝酸盐溶液进行电解,能够在45 min内将
NO−3 转化为NH+4 ,转化率为98.8%,并且在60 min内NH+4 最终被彻底氧化为氮气,反应体系的总氮去除率为98.7%。泡沫镍是一种导电导热性良好、疏松多孔、比表面积大、成本较低的电极材料[22],本研究提出了Ni foam/Cu电极的制备方法,将其作为阴极分析电催化还原硝酸盐氮反应的影响因素,采用动力学模型对硝酸盐氮还原过程进行探究。并对Ni foam/Cu电极的电催化活性和运行稳定性进行评价。
-
实验所用的电源为直流稳压恒流型,泡沫镍购自天津艾维信化工科技有限公司,硫酸铜、硝酸钠、氯化钠、硫酸钠、亚硝酸钠、氯化铵均为分析纯,购自阿拉丁试剂有限公司。
-
在电极负载前进行电极基体的预处理,取尺寸为5 cm×5 cm的泡沫镍放入10 g·L−1的90℃ NaOH溶液中加热除油20 min,然后置于0.5 mol·L−1的硫酸溶液中酸洗5 min去除表面氧化物,在去离子水中超声10 min彻底冲洗。以经过预处理后的Ni foam为阴极,Ti/RuO2-Ir2O3为阳极,进行阴极电沉积实验。配置20 mmol·L−1的CuSO4·5H2O溶液,含有0.05 mol·L−1的Na2SO4和0.04 g·L−1的十二烷基硫酸钠(C12H25SO4Na)。采用恒电流电沉积一段时间后,用去离子水冲洗。把制备好的Ni foam/Cu电极放在恒温干燥箱中干燥12 h用于后续的电解实验。
-
室温下取硝酸盐氮溶液用于电解实验,在电解液中加入0.5 g·L−1的Na2SO4作为电解质以提高溶液的导电性。每次向电解槽中加入100 mL电解液,以Ni foam/Cu电极为阴极,Ti/RuO2-Ir2O3电极为阳极进行硝酸盐氮的电催化还原实验。两个电极在溶液中的有效反应面积均为25 cm2,极板间距设置为5 mm。采用直流稳压电源在恒流模式下进行电解实验。分别设定不同的电解电流密度(2 — 12 mA·cm−2)、添加到电解液中的NaCl浓度(0.5 — 1.25 g·L−1)、
NO−3 −N的初始浓度(50 — 200 mg·L−1)进行电解实验,探究电流大小、电解质浓度、NO−3 −N的初始浓度对硝酸盐氮去除效果的影响。硝酸盐氮溶液的电化学还原过程持续2.5 h,每隔一段时间取样,分析硝酸盐氮、亚硝酸盐氮和氨氮浓度。证实Ni foam/Cu电极处理硝酸盐氮废水的可行性。 -
用扫描电子显微镜(SEM, JEOLJSM 6500F,JEOL,日本)和能量色散x射线光谱仪(EDS, Genesis 7000 X,EDAX公司,美国)分析电极的微观形貌及元素组成。通过紫外分光光度计(U-3900,日立公司,日本)用分子吸收分光光度法测定处理液的
NO−3 −N浓度[23],用N-(1-萘基)-乙二胺二盐酸盐分光光度法测定处理液的NO−2 −N浓度[24],用纳氏试剂分光光度法测定处理液的NH+4 −N浓度[25]。溶液中的镍离子和铜离子采用电感耦合等离子体原子发射光谱仪(ICP,Optima8300,PerkinElmer,美国)测定。 -
首先在20 mmol·L−1的CuSO4·5H2O电沉积溶液中,以7 mA·cm−2的电流密度沉积20 min制备Ni foam/Cu电极、不锈钢/Cu电极。对比Ni foam电极、不锈钢电极、Ni foam/Cu、不锈钢/Cu电极对100 mg·L−1硝酸盐氮电催化还原的影响。在电解电流密度为4 mA·cm−2的条件下,4个电极2.5 h的硝酸盐氮去除效果如图1(a)所示,Ni foam电极和不锈钢电极对硝酸盐氮没有明显的去除效果。与不锈钢/Cu电极的去除率相比,Ni foam/Cu电极在相同条件下的去除率提高了约34%。这是由于泡沫镍独特的三维立体结构具有较大的比表面积,为沉积金属组分Cu提供更多的活性位点,促进了硝酸盐氮电催化还原反应的进行。
然后在20 mmol·L−1的CuSO4·5H2O电沉积溶液中,以7 mA·cm−2的电流密度制备不同沉积时间的Ni foam/Cu电极。以Ni foam/Cu为阴极,Ti/RuO2-Ir2O3为阳极进行硝酸盐氮的电催化还原实验,探究电极沉积时间对溶液中100 mg·L−1的硝酸盐氮去除效果的影响。结果如图1(b)所示,随着沉积时间的增加,硝酸盐氮的去除率逐渐提高,当沉积时间超过20 min时硝酸盐氮去除率提高较为缓慢,因此选择沉积20 min作为Ni foam / Cu电极制备的条件,用于接下来的电解实验。
-
图2(a)和图2(b)表征了Ni foam / Cu电极的三维立体结构,可以看出电极具有不同大小五边形构成的网状结构,表面相对光滑。这种独特结构使电极具有较大的比表面积,提高了硝酸盐氮的电催化还原反应速率。图2(c)和图2(d)表明泡沫镍上负载了致密的Cu镀层,EDS结果显示泡沫镍上负载Cu后存在质量分数为16.71%的Cu和83.29%的Ni,证实活性金属组分铜成功地负载在泡沫镍基体上。
-
以Ni foam / Cu电极为阴极,Ti/RuO2-Ir2O3为阳极,在溶液的pH=7、电解质Na2SO4浓度为0.5 g·L−1、电解电流密度为4 mA·cm−2的条件下,分别对不同初始浓度的
NO−3 −N废水(50 — 200 mg·L−1)进行电催化还原处理。反应结果如图3(a)所示,随着硝酸盐氮初始浓度的升高,硝酸盐氮2.5 h的电解去除率先增大后降低,这与LIU等[26]的研究结果相似。50、100、150、200 mg·L−1的硝酸盐氮溶液去除率分别为71.36%、82.16%、74.72%、55.37%,溶液浓度为100 mg·L−1和150 mg·L−1时,硝酸盐氮去除率优于50 mg·L−1时硝酸盐氮的去除速率。溶液浓度为200 mg·L−1时,硝酸盐氮去除率又明显降低。这可能是由于硝酸盐氮浓度在一定范围内的增大促进硝酸盐离子在阴极表面的吸附,提高了反应速率和去除率,但是在较高的浓度下,阴极吸附量达到饱和,硝酸盐的传质被抑制,不能迅速到达电极表面,反应速率和去除率都有所降低。为了研究硝酸盐氮浓度变化的趋势,将一级动力学模型应用于硝酸盐氮还原过程,如下所述:
用拉普拉斯变换求解微分方程,得到硝酸盐氮浓度随时间的变化关系:
式中,
Ct,NO−3-N 为电解t时间时硝酸盐氮的浓度,mg·L−1;C0,NO−3-N 为硝酸盐氮的初始浓度,mg·L−1;t 为反应时间,h;k 为一级动力学反应速率常数,h−1。根据上述方程对实验结果进行非线性回归,可以得出硝酸盐氮在Ni foam /Cu电极上的电催化还原反应符合一级动力学模型,结果如图3(b)所示,硝酸盐氮初始浓度为100 mg·L−1时的动力学常数最大,其值为0.677 h−1。该反应速率常数规律对于处理不同浓度硝酸盐氮废水的工艺条件控制具有一定指导意义。
-
以Ni foam / Cu电极为阴极,Ti/RuO2-Ir2O3为阳极,两电极板间距5 mm,在含有100 mL硝酸盐氮废水模拟体系(100 mg·L−1
NO−3 −N+0.5 g·L−1 Na2SO4,pH=7)内进行电催化还原实验,进一步探究电解电流密度对硝酸盐氮还原反应的影响。当电解电流密度为2 mA·cm−2时,电解2.5 h后硝酸盐氮的去除率仅有39.73%。当电流达到8 mA·cm−2以上时,电解2.5 h后硝酸盐氮的去除率均可达到95%以上。说明当电流增大,阴极电极电位更负,电子转移速率增大,有助于电催化还原硝酸盐氮反应的进行。此外产生了更多的还原氢也使硝酸盐氮的去除率逐渐增加。同时在电解的过程中可以观察到电极表面产生的气泡也随电流的增大而增多,这是电极表面副反应产生的气体,阴极发生析氢反应,阳极发生析氧反应。说明电流增大,反应时间增长,也促进了析氧等副反应的发生。因此,在该反应体系中电解电流密度应调节在4 — 8 mA·cm−2范围内,既能达到较高的硝酸盐氮去除率,也可以避免产生过多的副反应。图4(b)表明,在该体系中电催化还原硝酸盐氮反应符合一级动力学规律,随着电解电流增大,一级反应动力学常数由0.197 h−1增大至 1.547 h−1。 -
首先以不同大小的电流对100 mg·L−1的硝酸盐氮溶液电解2.5 h,探究电解质种类对硝酸盐氮还原产物的影响。图5说明当以0.5 g·L−1硫酸钠作为电解质时,电流对氨氮和亚硝酸盐氮的生成量几乎没有影响,同时随着电解电流的增大,溶液中总氮的去除效果提升并不显著,在电解电流密度为12 mA·cm−2时达到最大,约30.93%。但当以相同浓度的氯化钠作为电解质时,氨氮生成量随着电流的增大而减少,明显提高了电催化还原体系的总氮去除率,在电解电流密度为12 mA·cm−2时,总氮去除率可以达到93.88%。
为探究NaCl电解质对电催化还原体系总氮去除效果的影响,以Ni foam / Cu电极为阴极,Ti/RuO2-Ir2O3为阳极,在溶液的pH=7、电解电流密度为8 mA·cm−2的条件下,分别加入0.5、0.75、1、1.25 g·L−1的NaCl作为电解质对100 mL含有100 mg·L−1的
NO−3 −N废水进行电催化还原处理。结果如图6所示,NaCl浓度的变化对溶液中总氮的去除有较大影响,NaCl浓度为0.5 g·L−1时总氮去除率仅41.37%,但当NaCl浓度为1.25 g·L−1时,总氮去除率提高至79.47%。随着NaCl浓度的增加,电催化还原体系的总氮去除率增大,即提高了反应的氮气选择性。亚硝酸盐氮在整个电解过程中的生成量很低,不足1 mg·L−1,几乎都参与反应生成了其它含氮物质。硝酸盐氮和总氮随着电解时间的延长均逐渐降低,氨氮呈先增加后减少的趋势,且峰值随着NaCl浓度的增加而降低,这与加入的Cl−影响
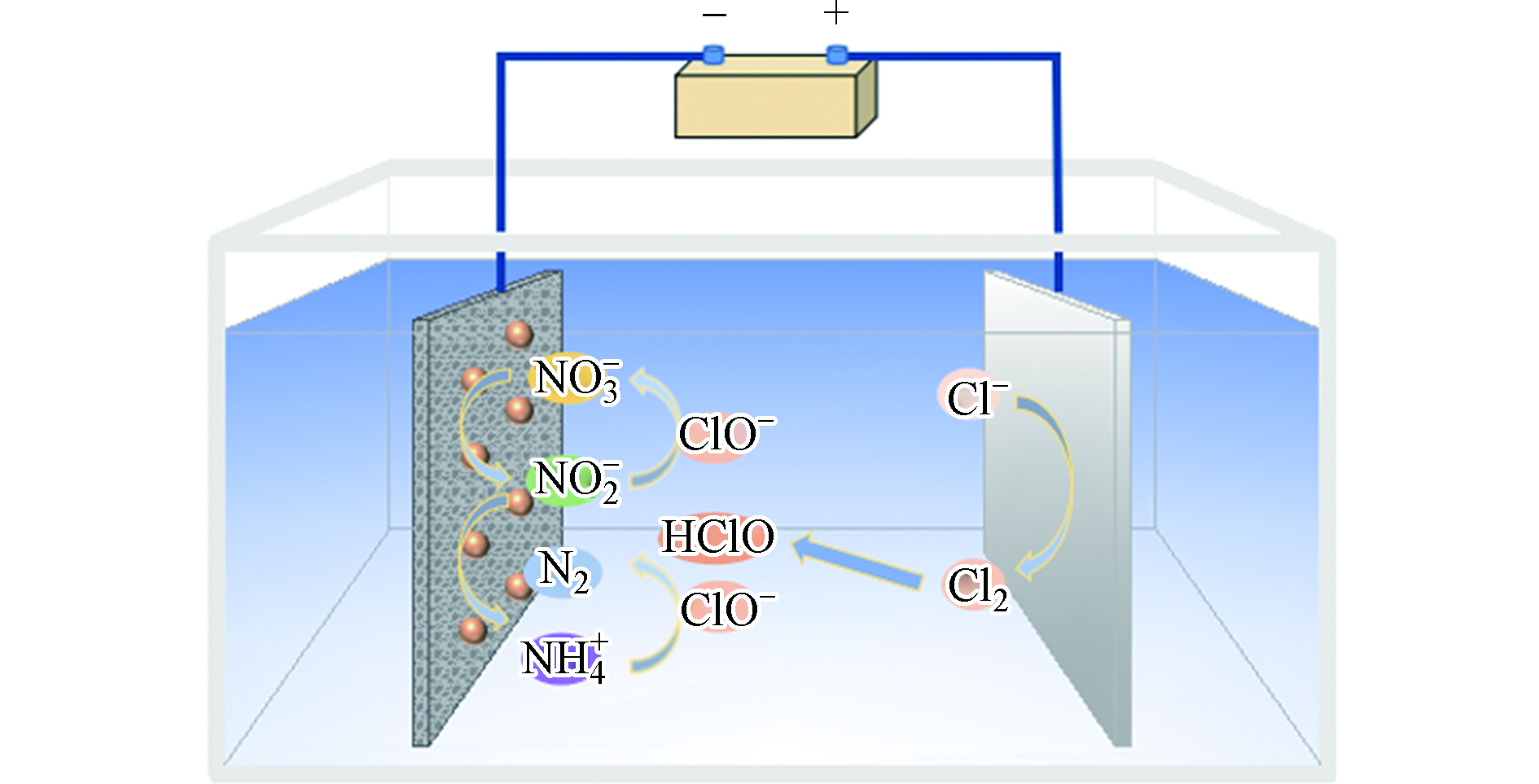
NH+4 的反应有关。向溶液中添加NaCl后,部分溶解的氯离子会被氧化为Cl2。氯气溶于水生成的HClO以及HClO电离生成ClO−均具有较强的氧化性,能够将NO−3 还原的副产物NH+4 氧化成N2。但当溶液中NaCl的含量过低时,生成的HClO量将不足以氧化所有生成的氨氮。在NaCl浓度为1.25 g·L−1的条件下,对硝酸盐氮溶液电解2.5 h后,生成氨氮的含量为2.90 mg·L−1,硝酸盐氮的去除率为82.37%,与NaCl浓度为0.5 g·L−1时相比降低了12.21%,说明如果加入NaCl的含量过多,生成过量的ClO−可能会将NO−2 氧化成NO−3 ,同时带负电荷的ClO−和Cl−与NO−3 竞争吸附会抑制硝酸盐的去除[27],具体反应过程如图7和式1—9所示。 -
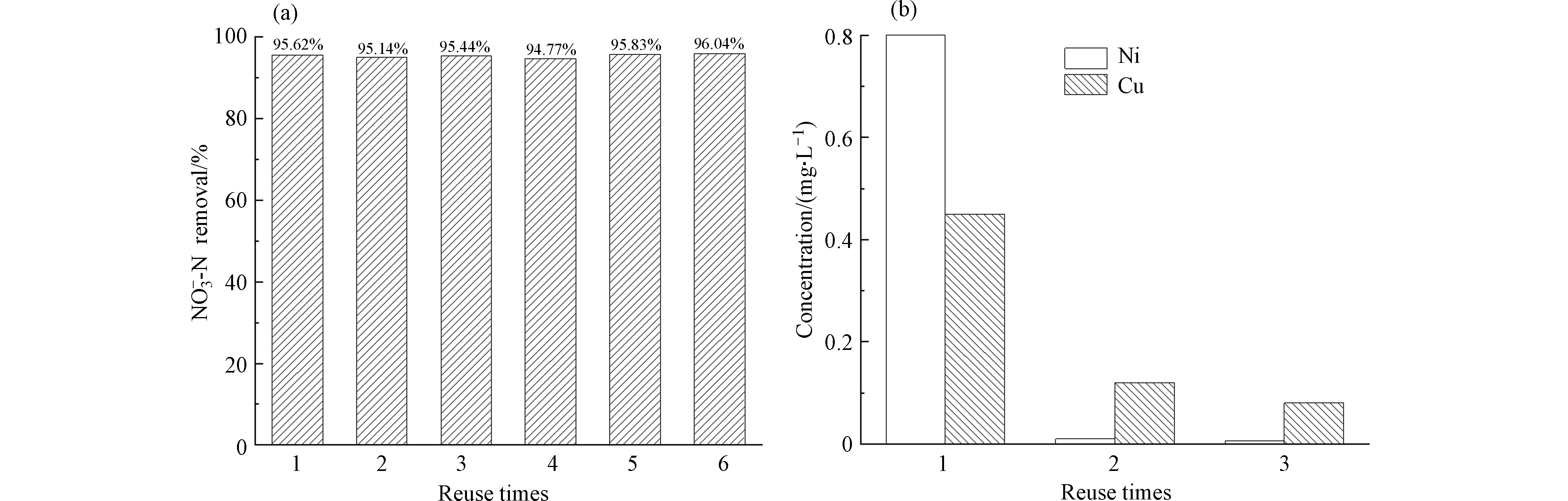
为了验证Ni foam/Cu电极电催化还原硝酸盐氮的稳定性,以Ni foam / Cu电极为阴极,Ti/RuO2-Ir2O3为阳极,在溶液pH=7、电解质NaCl浓度为0.5 g·L−1、电解电流密度为8 mA·cm−2的条件下,对100 mL含有100 mg·L−1的硝酸盐氮溶液进行了6次电解实验探究。实验结果如图8(a)所示,电解2.5 h后,硝酸盐氮的去除率均达到94.5%以上,没有呈下降趋势,并且电极表面无明显变化。
通过ICP测定3次电解反应结束后溶液中的Ni和Cu金属离子含量,结果如图8(b)所示,随着使用次数的增加,溶液中金属离子含量也随之减少,第三次电解后溶液中镍和铜的金属离子的浓度均低于0.1 mg·L−1,验证了Ni foam/Cu电极稳定性良好。
-
(1)采用恒流电沉积法制备的Ni foam / Cu电极在硝酸盐氮降解反应中具有良好的电催化还原性能和稳定性。在重复实验中硝酸盐氮的去除率均能达到94.5%以上。实验结束后溶液中镍和铜的金属离子浓度均低于1 mg·L−1,证明金属铜紧密地结合在泡沫镍基体上。
(2)增大电流有助于电催化还原硝酸盐氮反应的进行,在该电催化还原体系中电解电流密度为8 mA·cm−2时,即可以实现90%以上的硝酸盐氮去除率。
(3)在硝酸盐氮的电催化还原过程中,外加电解质NaCl浓度对产物的选择性有很大影响。在一定范围内,NaCl浓度适当增大有助于减少副产物的生成,提高总氮的去除率。当NaCl浓度为1.25 g·L−1时,氮气选择率接近100%,总氮去除率达到79.47%。
Ni foam/Cu电极电催化还原硝酸盐氮
Electrocatalytic reduction of nitrate nitrogen by Ni foam/Cu electrode
-
摘要: 采用恒流电沉积法制备了泡沫镍/铜(Ni foam/Cu)电极。以Ni foam/Cu电极为阴极、Ti/RuO2-Ir2O3电极为阳极构建电解体系,对水中硝酸盐氮进行电催化还原处理。并研究了电解质对总氮和氨氮去除率的影响和电极的稳定性。在一定范围内,增大电解质NaCl的浓度可以提高总氮和氨氮去除率。当NaCl浓度为0.5 g·L−1时,在电解电流密度为8 mA·cm−2的条件下对100 mg·L−1
NO−3 −N溶液进行6次重复电解实验,2.5 h后硝酸盐氮去除率均可以达到94.5%以上。当NaCl浓度为1.25 g·L−1时,在电解电流密度为8 mA·cm−2的条件下对100 mg·L−1NO−3 −N溶液电解2.5 h,出水中氨氮浓度只有2.90 mg·L−1,总氮去除率达到79.47%。实验结果表明,Ni foam/Cu阴极具有较高的电催化还原活性和良好的稳定性。Abstract: The Ni foam/Cu electrode was prepared by galvanostatic electrodeposition method. The Ni foam/Cu and the Ti/RuO2-Ir2O3 electrode were used as the cathode and anode respectively to construct an electrolysis system to perform electrocatalytic reduction of nitrate nitrogen in water. The effect of the electrolyte on the removal rate of total nitrogen and ammonia nitrogen and the stability of the electrode were also investigated. In a certain range, the removal rate of total nitrogen and ammonia nitrogen can be improved by increasing the electrolyte NaCl concentration. When the concentration of electrolyte NaCl increased to 0.5 g·L−1, six repeated electrolysis experiments were performed in 100 mg·L−1NO−3 -N solution for 2.5 h under the condition of 8 mA·cm−2, and the removal rate of nitrate nitrogen could reach more than 94.5%. When the concentration of electrolyte NaCl increased to 1.25 g·L−1, the concentration of ammonia nitrogen was only 2.90 mg·L−1 after 2.5 h of electrolysis under the condition of 8 mA·cm−2, and the total nitrogen removal rate reached 79.47%. The experimental results show that the Ni foam/Cu cathode has high electrocatalytic reduction activity and good stability.-
Key words:
- nitrate nitrogen /
- electrocatalytic reduction /
- Ni foam/Cu
-
受控生态生保系统(controlled ecological life support system,CELSS)通过对大气控制、温湿度控制、食物供应、水再循环和废物处理等技术整合,可保障航天员在地外环境中健康生活和有效工作,是未来地外星球基地长期稳定运行的必要保证[1]。CELSS依据地球生态圈的基本原理,在有限的密闭空间内构建了“人-植物-微生物-环境”自循环式闭路生态系统[1]。其中,植物作为关键功能部件,能够为航天员提供新鲜食物和氧气、吸收二氧化碳和净化水质。在CELSS中,通常选择小麦作为主要的粮食作物,不可避免地会产生大量的植物不可食部分,这部分固废的积累不仅会造成占用舱体空间、发酵腐败等安全卫生问题,还会造成大量资源(如水分、碳元素、氮元素、无机盐等)的浪费。如何高效处理并回收利用这类固体废物,维持CELSS中较高的物质循环利用率与闭合度,已成为CELSS中迫切需要解决的问题。
针对CELSS中小麦秸秆等固废资源化处理问题,美国和俄罗斯等国采用焚烧[2]和湿式氧化[3]等物化技术进行处理。物化技术稳定可靠、反应速率快,但存在着对设备要求高、能耗高、对系统瞬时冲击负荷大、产生氮氧化物而限制元素循环等缺点。生化处理技术则具有能耗低、反应过程温和以及能够有效实现各元素再生循环等优势。CHYNOWETH等[4]采用干式厌氧发酵工艺处理水稻秸秆、废纸和狗粮(模拟成员粪便)混合物,运行时间为23 d,有机物降解率达到了81.2%;并提出针对固废的预处理、后处理(沼渣好氧堆肥)和营养液植物栽培等方面的研究应作为未来研究的方向之一。欧洲太空局采用湿式厌氧消化工艺[5]将反应控制在水解酸化阶段而抑制产甲烷阶段,将有机底物转化为VFAs、氨氮和CO2用于后续的藻类系统和硝化系统使用。WHITAKER等[6]研制了固体高温好氧反应器用于处理志愿者产生的废物,包括粪便、厕纸、食物残渣和卫生废水等,操作温度为55~70 ℃,总固体降解率可达到74%。TIKHOMIROV等[7]通过蘑菇(真菌)培养和蚯蚓等腐生动物对植物不可食部分进行好氧堆肥处理,得到了类土壤基质并用于作物栽培。上述生化处理技术虽可一定程度上实现固废的稳定减容和资源回收,但也面临着设备尺寸较大、反应周期较长或仍需后续的好氧发酵等无害化处理的局限。而好氧堆肥技术作为无害化和资源化的处理方式,对碳氮等养分有较好的保全,可将固废转化为腐殖质,施用后能对植物生长起到促进作用,符合CELSS中物质循环再生的要求,因而受到广泛关注和研究。好氧堆肥技术是通过多种微生物的协同作用来完成物料的降解,因此,微生物的配比是影响好氧堆肥过程的关键因素[8]。有研究[9]表明,堆肥中接种微生物菌剂能使堆温快速升高,有效杀灭堆肥物料中的病原菌和杂草种子,显著促进堆肥腐熟,提高堆肥质量。另外,在CELSS内,由于微生物受到严格的控制和防护,其主要来自航天员体表和体内,种类及数量都无法满足堆肥启动要求。因此,添加一定的功能菌剂对于启动堆肥反应、促进堆肥腐熟和缩短堆制周期至关重要。目前,以微生物菌剂接种用于禽畜粪便和市政污泥相关方面的研究较多[9-10],通常添加秸秆、木屑等物质起到平衡含水率、调节C/N和通气性等作用[11],市面上也有多种针对这类固废的商业菌剂。然而,针对农业固废小麦秸秆降解处理的商用菌剂并不常见,且对于菌剂接种用于小麦秸秆堆肥降解效果的研究较少。
为实现CELSS中小麦秸秆等固废的资源化处理,提高系统物质闭合度,本研究以小麦秸秆为主要处理对象,添加厨余垃圾作为调整物料C/N比的营养调节剂,选取3种商业菌剂开展小试反应器强制通风好氧堆肥试验,探究接种菌剂对小麦秸秆好氧堆肥一次发酵阶段降解效果的影响;考察堆肥过程中各项参数变化,分析比较3种菌剂对小麦秸秆的处理效果,探讨不同菌剂在小麦秸秆好氧堆肥各个阶段的降解作用,以期为筛选研制高效降解小麦秸秆的微生物菌剂提供理论基础。
1. 材料与方法
1.1 实验原料
小麦秸秆购自江苏某农场,经机械粉碎后选取粒径为0.3~0.5 cm的麦秸待用;厨余垃圾取自某单位食堂,将其中的骨头、卫生纸、塑料袋、玉米棒芯等拣出,用粉碎机将厨余垃圾粉碎至浆糊状。堆肥所用物料的基本性质见表1。
表 1 堆肥原料的理化性质Table 1. Physical and chemical properties of the composting materials堆肥原料 含水率/% 全碳含量/% 全氮含量/% C/N比 小麦秸秆 10.11±0.01 41.54±0.38 0.93±0.03 44.67 厨余垃圾 81.09±0.11 52.64±0.46 3.69±0.08 14.27 | Show Table DownLoad:
CSV
DownLoad:
CSV
1.2 微生物菌剂
针对小麦秸秆特性,选用3种适用于秸秆腐熟的商业菌剂,代号分别为QD、DH、VT。其中,QD菌剂呈液体状,有效活菌数≥109 CFU·mL−1,主要为乳酸菌、木霉菌和芽孢杆菌等;DH菌剂呈固体粉末状,有效活菌数≥5×108 CFU·g−1,主要为枯草芽孢杆菌、米根霉、毕赤酵母菌和戊糖片球菌等;VT菌剂呈固体粉末状,有效活菌数≥5×108 CFU·g−1,主要为酵母菌、乳酸菌和芽孢杆菌等。
1.3 实验装置
本实验采用的堆肥装置如图1所示,主要由带盖塑料桶(桶有效容积为19 L,桶外壁包裹有2层保温棉,桶顶部放置有温度计,桶底部设置有物料托盘)、温度控制系统和通气系统3部分组成。
1.4 实验方法
有别于陆地生态系统,CELSS内没有自然界广泛分布的细菌、放线菌和真菌等微生物,因此,为启动堆肥反应和促进底物腐熟,接种一定的有益菌群是必须的。本实验主要考察不同菌剂对小麦秸秆堆肥过程中一次发酵阶段的降解处理效果,故未设不加菌剂的对照组实验。
实验共分为3组,分别为QD组、DH组和VT组。每组均用小麦秸秆和厨余垃圾按二者干基质量比为4:1的比例均匀混合,混合物料的C/N比控制在30∶1,并调节混合物料的水分含量在65%。接种菌剂时按物料总重的0.5%添加,即QD菌剂接种100 mL,DH菌剂和VT菌剂各接种52 g。每组混合均匀的物料等分装入3个堆肥桶内,每个堆肥桶内均含物料3.50 kg,每组设置3个重复实验。通风量设置为1 L·min−1,持续通风至堆肥结束,堆肥周期设定为30 d。
堆肥开始后分别于第1、5、9、14、19、24和29 d取样,取样前需翻堆,使物料混合均匀。采样时按照5点采样法的原则分别在堆体的上、中、下层采集鲜样共30 g,混合均匀后置于−20 ℃冰箱保存,用于各项指标的测定。
1.5 分析方法
温度采用温度计测定。将温度计插入物料中间及周围3点20 cm处测定温度,取4点温度的平均值作为最终结果,温度每隔24 h测定1次;含水率采用烘干法[12]测定。
浸提液理化性质测定。将5 g鲜样与蒸馏水按质量比1∶10混合并振荡120 min,然后在10 000 r·min−1下离心5 min,过0.45 μm滤膜后,将滤液用塑料小瓶贮存于4 ℃冰箱待用。pH用便携式pH计测定;电导率(EC)用便携式电导率仪测定;在465 nm(E4)和665 nm(E6)下的波长用紫外分光光度计[13]测定。
VS含量和C/N比分别采用灼烧法和元素分析仪法[13]测定。
2. 结果与讨论
2.1 菌剂处理下物料温度的变化特性
3种菌剂处理下物料的温度变化如图2所示。堆肥前3 d,物料中易降解的有机物如可溶性小分子有机物、多糖和脂类等开始降解,该阶段嗜温菌的活性较强,热量快速累积,温度迅速上升至50 ℃以上。3~10 d为高温期,可溶性的中间产物被继续分解转化,耐高温的放线菌数量增加,物料中有机物如淀粉、蛋白质、半纤维素和纤维素等逐步分解。QD、DH和VT处理下的最高温度分别达到了58.2、54.7和53.7 ℃,高温期分别维持了9、6和6 d。第10天后,堆体温度逐渐下降,嗜温细菌和真菌变得活跃,对残留的较难分解的有机物(如木质素)进行分解,物料表面变得疏松且颜色逐渐变为黑褐色,开始形成了腐殖酸等物质[14]。堆肥过程中分别于第5、9、14、19、24和29天对物料进行翻堆,翻堆后物料重新混合均匀,堆体温度稍有上升[15]。最终3组处理下物料的温度均稳定在31 ℃左右,与伴热带温度(发酵环境温度)趋于一致。
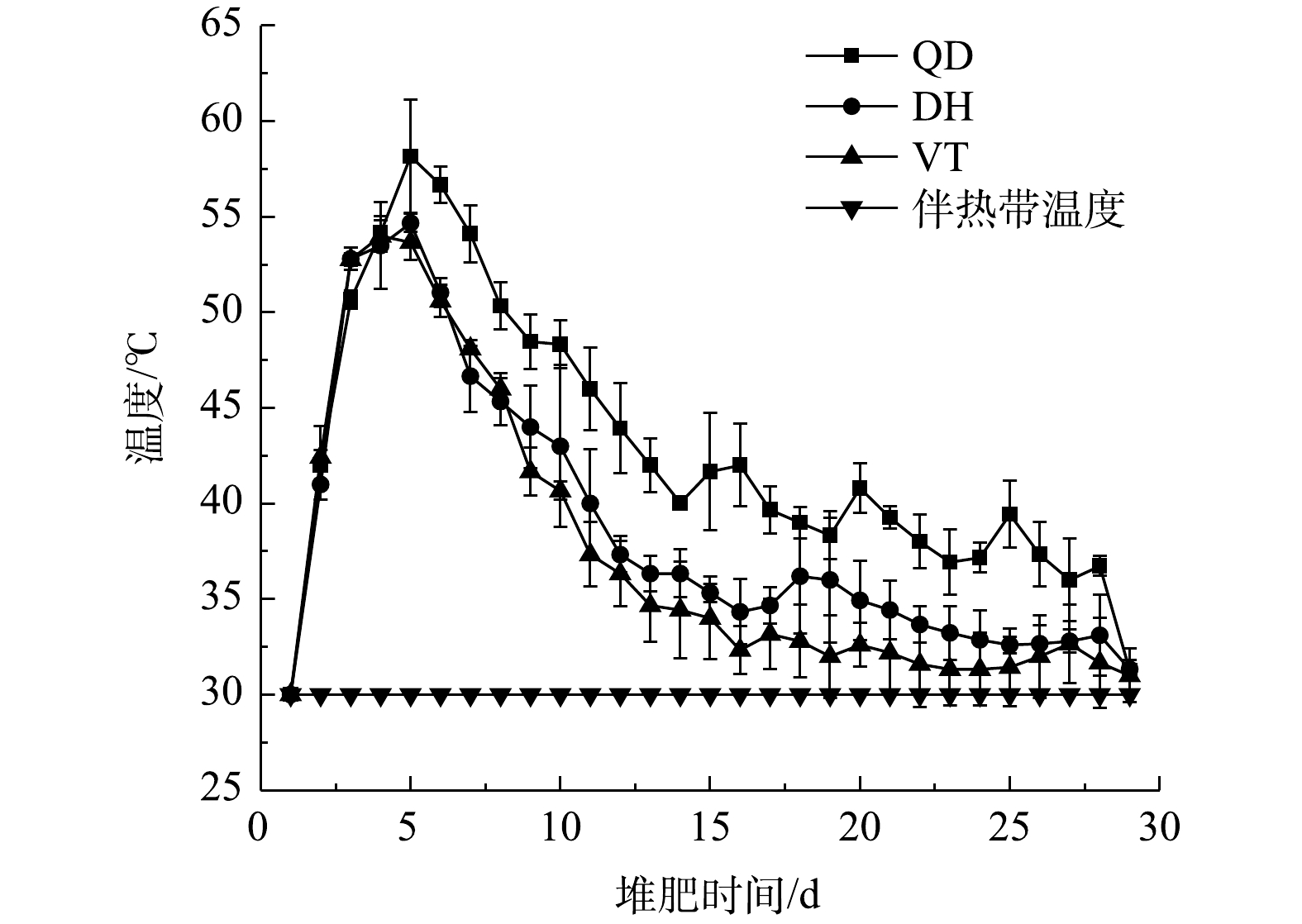 图 2 不同堆肥处理物料温度的变化Figure 2. Changes of temperature with different microbial agents during composting
图 2 不同堆肥处理物料温度的变化Figure 2. Changes of temperature with different microbial agents during composting3种菌剂处理下的物料均经历了升温、高温和降温期。在高温期维持时间的长短方面表现为QD>DH>VT,只有QD组堆体的高温期维持时间超过了7 d。在温度峰值的高低方面表现为QD>DH>VT,只有QD组堆体的最高温度超过了55 ℃,满足堆肥无害化的要求[16]。综合3组物料温度的变化情况可知,QD菌剂在堆肥过程中能使堆体温度达到55 ℃以上,在高温期持续时间较长,这说明QD菌剂中的微生物可能更多为嗜温菌和高温菌,在升温和高温期的活性更强,对堆体在前期热量的迅速增长和积累有良好的促进作用。
2.2 菌剂处理下物料含水率的变化特性
3种菌剂处理下物料含水率的变化如图3所示。堆肥物料的含水率过高或过低都会影响堆肥的质量,含水率过高会导致堆体局部厌氧,过低会导致微生物活性下降[14]。由图3可知,3组处理下物料含水率总体上均呈现先上升后下降的变化趋势。在升温-高温期物料温度迅速上升,微生物活动剧烈,物料中的有机物被强烈分解,微生物代谢产水的速率大于水分蒸发的速率,导致物料的含水率上升。QD、DH和VT处理下物料的含水率分别在第9、14和9 d达到了最高值,分别为(75.6±1.14)%、(78.9±0.93)%和(79.5±1.55)%。10 d之后,物料的温度下降,微生物活动逐渐减弱,再加上持续的通气及翻堆,物料中的水分被持续带走,微生物代谢产水的速率小于水分蒸发的速率,物料含水率逐渐降低。最终,3组处理下物料的含水率分别降至(59.73±0.13)%、(56.61±2.19)%和(57.42±0.93)%,而有机肥料腐熟的标准要求堆体含水率低于30%[16],这说明3组物料均达到了初步腐熟,完成了好氧堆肥的一次发酵阶段。后续仍需要进行二次发酵,即温度维持在中温,使物料进一步稳定,最终达到深度腐熟。
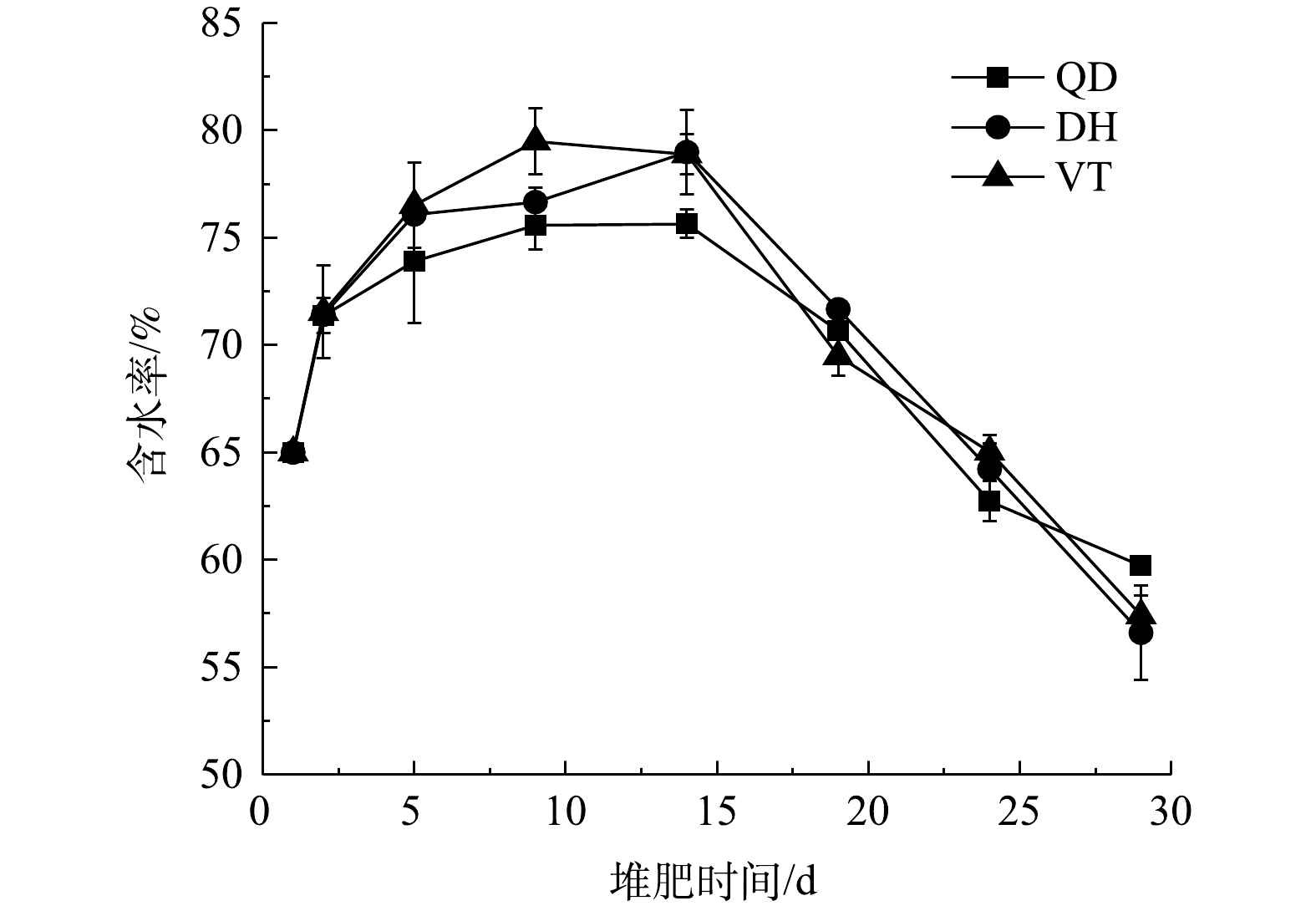 图 3 不同堆肥处理物料含水率的变化Figure 3. Changes of water content with different microbial agents during composting
图 3 不同堆肥处理物料含水率的变化Figure 3. Changes of water content with different microbial agents during composting2.3 菌剂处理下物料浸提液理化性质的变化特性
3组处理下物料浸提液理化性质的变化如图4所示。EC可以表征有机废物发酵产品中的可溶性盐含量;pH可以反映堆体所处的酸碱性环境;E4/E6可表征堆肥过程中腐殖酸的缩合度和芳构化程度[17]。由图4(a)和图4(b)可知,堆肥前期EC逐渐上升,这是由于堆体中可被微生物直接利用的物质较多,物料中易降解的物质如糖类、脂肪等被断链降解产生了VFAs和大量的无机盐离子,如
HCO−3 NO−3 NH+4  图 4 不同堆肥处理物料浸提液理化性质的变化Figure 4. Changes of physicochemical properties of the composting extracts with different microbial agents during composting
图 4 不同堆肥处理物料浸提液理化性质的变化Figure 4. Changes of physicochemical properties of the composting extracts with different microbial agents during composting2.4 菌剂处理下物料VS含量的变化特性
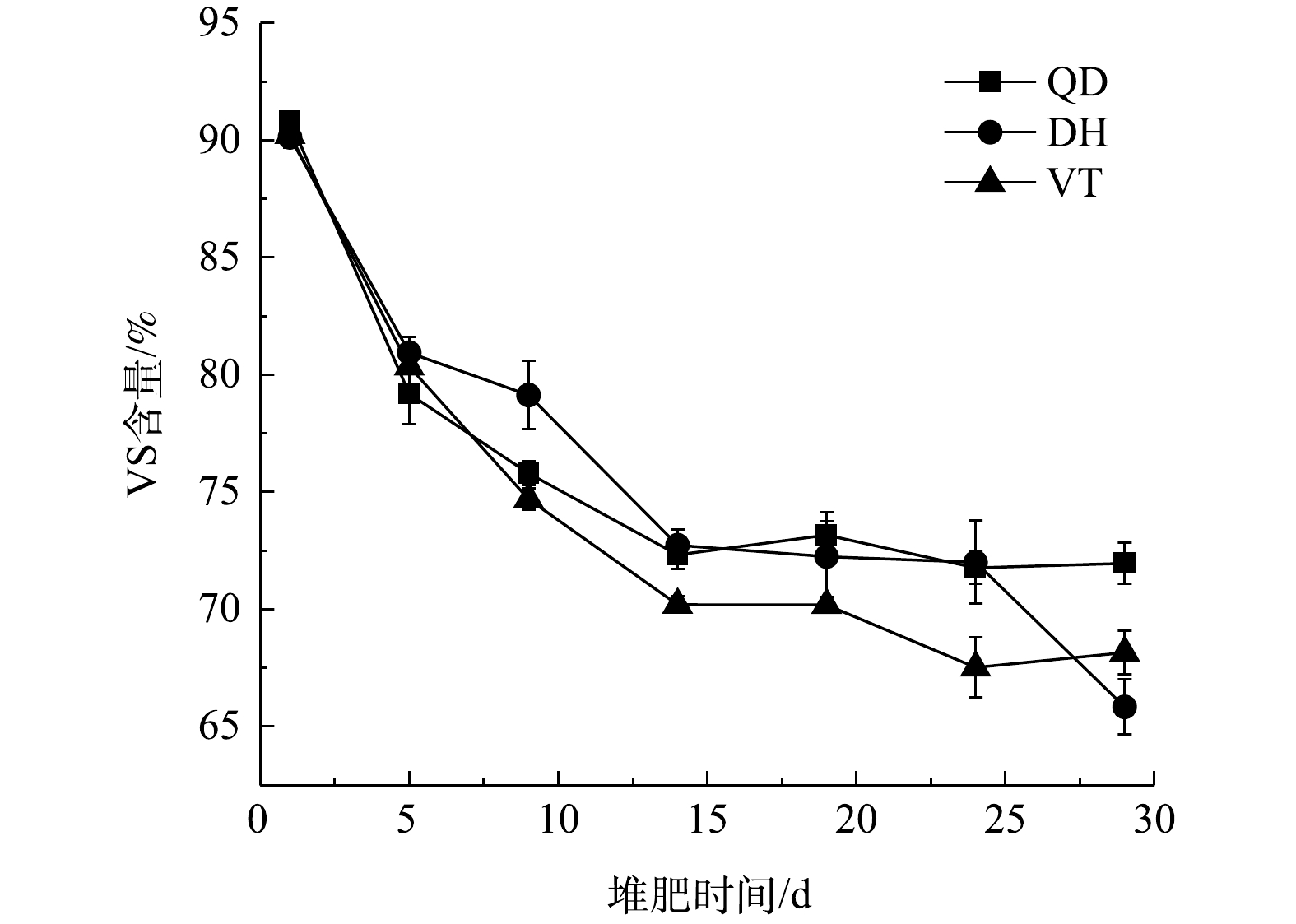
VS含量的变化反映了堆肥过程中物料有机物的降解速度和效率。3组处理下物料的VS含量变化如图5所示。由图5可知,3组处理下物料的VS含量均表现为逐渐降低的趋势,物料的初始VS含量(干基)为90%左右。在升温-高温期时,物料的温度迅速上升,微生物生命活动旺盛,物料中易降解的有机物被大量分解,碳元素主要以CO2的形式被释放,物料的VS含量迅速下降。在降温期时,物料的温度下降,此时物料内的有机物主要为难降解的木质纤维素等,有机物的降解速率变小。最终,QD、DH和VT处理下物料的VS含量分别稳定在(71.96±0.89)%、(65.84±1.19)%和(68.16±0.93)%。
3种菌剂处理下物料VS含量的减少情况如表2所示。3组处理下物料中有机物的降解效率表现为DH>VT>QD;QD、DH和VT处理下物料VS的减少量分别为(18.87±0.89)%、(24.48±1.60)%和(22.08±0.72)%。升温-高温期时,QD、DH和VT处理下物料的VS减少含量分别为(15.04±0.42)%、(10.99±1.28)%和(15.54±0.71)%,分别占VS减少总量的79.7%、45.2%和70.4%。VS含量的减少情况表明,QD和VT处理下物料中有机物的降解主要发生在升温-高温期,而DH处理下物料有机物的降解主要发生在降温期。这是因为,QD和VT菌剂中的乳酸菌和酵母菌等对糖类等物质有较强的利用能力,而DH菌剂中的枯草芽孢杆菌和米根霉能分泌纤维素酶从而对物料中的木质纤维素有着较好的降解作用[21],这说明3种菌剂对物料中有机物降解效果的差异性与菌剂中微生物的组成配比密不可分。
表 2 堆肥前后VS含量的减少情况Table 2. Reduction of VS content before and after composting% 处理组 初始VS含量 终点VS含量 升温-高温期VS减少量 VS减少总量 QD 90.83±0.18 71.96±0.89 15.04±0.42 18.87±0.89 DH 90.12±0.44 65.84±1.19 10.99±1.28 24.48±1.60 VT 90.24±0.26 68.16±0.93 15.54±0.71 22.08±0.72 | Show TableDownLoad:
CSV
2.5 菌剂处理下物料C/N比的变化特性
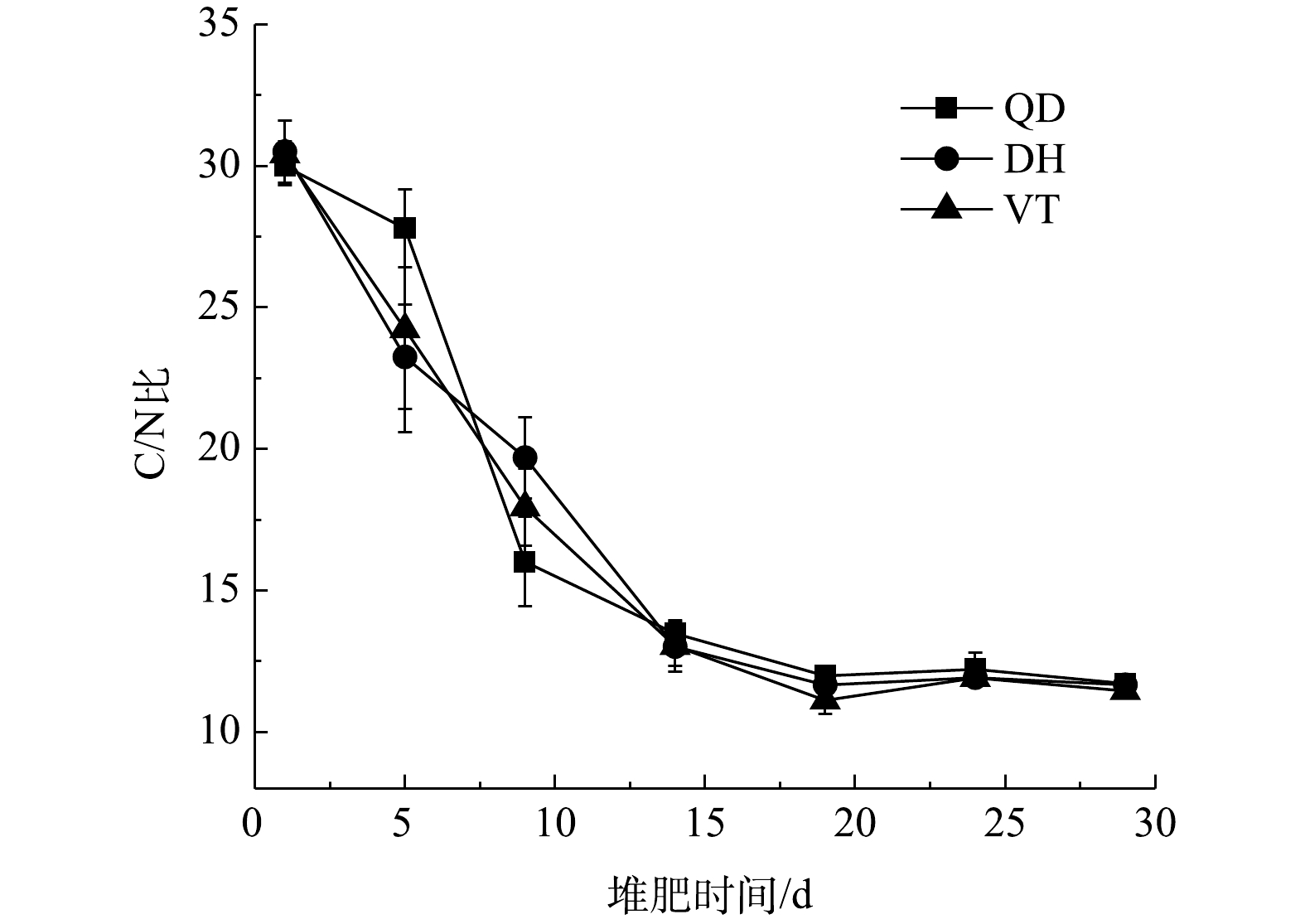
C/N比的变化可以反映堆肥过程中物料有机物矿质化和腐殖化的进程[22]。有研究[23]表明,适合微生物生长的物料C/N比范围为25∶1~30∶1。3组处理下物料C/N比的变化如图6所示,可见,3组物料的C/N比均呈现下降的趋势,变化曲线的斜率随堆肥过程的持续而逐渐降低,这与VS含量的变化情况一致。物料的初始C/N比均在30∶1左右,是适宜微生物生长的环境。堆肥前10 d堆体温度上升,微生物迅速生长繁殖。其中,易分解的含C有机物被微生物分解吸收利用,并通过呼吸作用变为CO2等气体排出堆肥系统,因而C含量逐渐变低。N素被微生物利用会以NH3的形式散失,但其下降幅度低于有机物总干物质的下降幅度,故干物质中全N含量会相对增加[22],总体则表现为C/N比迅速降低。10 d之后,物料的温度降低,微生物生命活动减弱,物料达到初步稳定腐熟,C/N比下降趋势变缓并趋于稳定。3组处理下物料的C/N比均由初始的30∶1降至12∶1以下,分别为11.71±0.16、11.67±0.20和11.45±0.16,终点C/N比与初始C/N比的比值分别为0.39、0.38和0.37,尽管满足堆肥腐熟时终点C/N比与初始C/N比的比值不超过0.5的要求[24],然而在实际应用中应该参照其他指标,如生物活性和植物毒性等,对堆肥的腐熟程度进行综合评价。
3. 结论
1) QD菌剂可以提高堆肥温度至58.2 ℃,堆体的高温期为9 d,满足堆肥无害化要求;DH菌剂可以促进物料中有机物的降解,降解率可达24.48%;3种菌剂对堆肥中腐殖质的形成和积累均有一定的促进作用。
2) 3组处理下的堆体进入降温期后均开始形成腐殖质,物料达到初步腐熟,即完成了一次发酵。后续仍需要进行二次发酵处理,使堆体达到完全腐熟,即可作为土壤改良剂或有机肥施用。
3)微生物配比不同是导致小麦秸秆好氧堆肥降解效果存在差异的重要因素。后续需分析堆肥过程中的微生物种群,进一步明确功能菌群和功能基因,考察微生物在小麦秸秆堆腐过程中的作用机理。
-

图 1 不同电极对还原硝酸盐氮的影响
Figure 1. Influence of different electrodes on reduction of nitrate nitrogen

图 2 Ni foam / Cu电极扫描电镜和EDS
Figure 2. SEM images and EDS profile of Ni foam/Cu electrode

图 3 硝酸盐氮初始浓度对还原硝酸盐氮的影响
Figure 3. Influence of initial concentration of nitrate nitrogen on reduction of nitrate nitrogen

图 4 电解电流密度对还原硝酸盐氮的影响
Figure 4. Influence of electrolytic current density on reduction of nitrate nitrogen
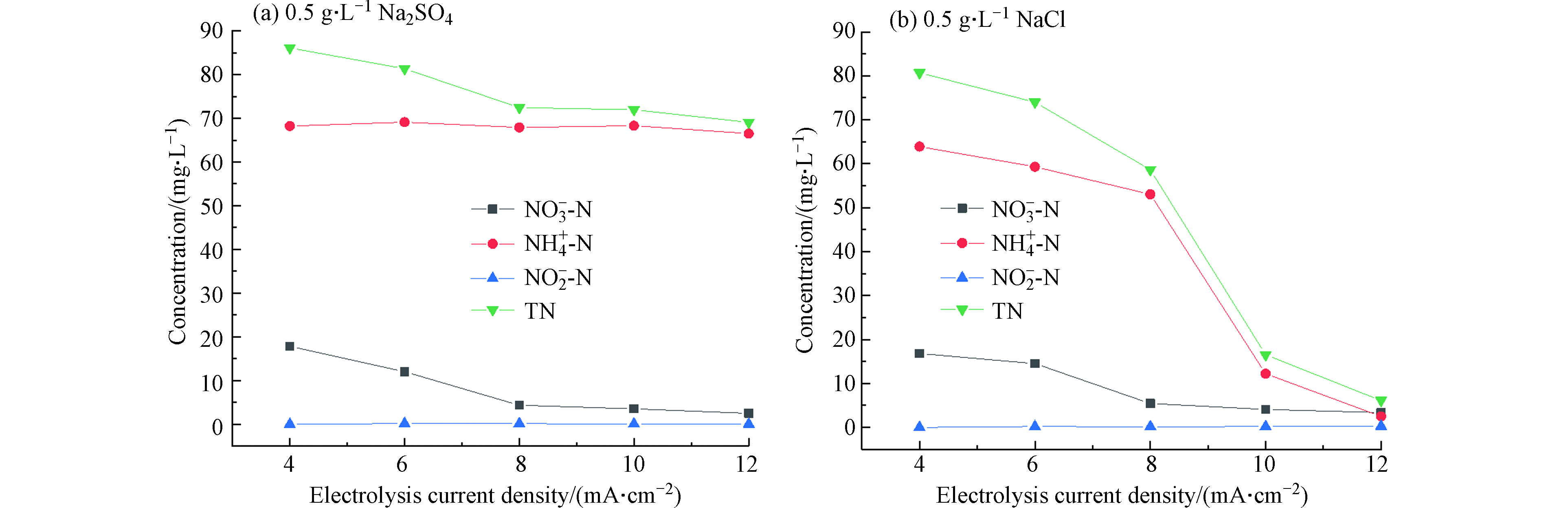
图 5 电解质种类对还原硝酸盐氮的影响
Figure 5. Influence of electrolyte types on reduction of nitrate nitrogen

图 6 电解质浓度对还原硝酸盐氮的影响
Figure 6. Effect of electrolyte concentration on reduction of nitrate nitrogen
-
[1] 陈帅, 房宁, 唐亮. 光催化法还原水中硝酸盐氮研究进展 [J]. 环境与发展, 2018, 30(9): 105-106. CHEN S, FANG N, TANG L. Research progress in reduction of nitrate nitrogen in water by photocatalysis [J]. Environment and Development, 2018, 30(9): 105-106(in Chinese).
[2] DING Y J, SUN W Z, YANG W Y, et al. Formic acid as the in situ hydrogen source for catalytic reduction of nitrate in water by PdAg alloy nanoparticles supported on amine-functionalized SiO2 [J]. Applied Catalysis B:Environmental, 2017, 203: 372-380. doi: 10.1016/j.apcatb.2016.10.048 [3] MARTÍNEZ J, ORTIZ A, ORTIZ I. State-of-the-art and perspectives of the catalytic and electrocatalytic reduction of aqueous nitrates [J]. Applied Catalysis B:Environmental, 2017, 207: 42-59. doi: 10.1016/j.apcatb.2017.02.016 [4] LEI X H, LIU F, LI M, et al. Fabrication and characterization of a Cu-Pd-TNPs polymetallic nanoelectrode for electrochemically removing nitrate from groundwater [J]. Chemosphere, 2018, 212: 237-244. doi: 10.1016/j.chemosphere.2018.08.082 [5] 余静, 蒲生彦, 王晓科, 等. 磁性壳聚糖凝胶微球固定反硝化菌去除地下水中硝酸盐氮 [J]. 环境化学, 2020, 39(2): 416-425. doi: 10.7524/j.issn.0254-6108.2019032002 YU J, PU S Y, WANG X K, et al. Removal of nitrate nitrogen in groundwater by denitrifying bacteria immobilized on magnetic chitosan gel microspheres [J]. Environmental Chemistry, 2020, 39(2): 416-425(in Chinese). doi: 10.7524/j.issn.0254-6108.2019032002
[6] CHABANI M, AMRANE A, BENSMAILI A. Kinetics of nitrates adsorption on Amberlite IRA 400 resin [J]. Desalination, 2007, 206(1/2/3): 560-567. [7] LUBPHOO Y, CHYAN J M, GRISDANURAK N, et al. Influence of Pd-Cu on nanoscale zero-valent iron supported for selective reduction of nitrate [J]. Journal of the Taiwan Institute of Chemical Engineers, 2016, 59: 285-294. doi: 10.1016/j.jtice.2015.08.005 [8] HAO S Z, ZHANG H W. High catalytic performance of nitrate reduction by synergistic effect of zero-valent iron (Fe0) and bimetallic composite carrier catalyst [J]. Journal of Cleaner Production, 2017, 167: 192-200. doi: 10.1016/j.jclepro.2017.07.255 [9] ZHANG Q, DING L, CUI H, et al. Electrodeposition of Cu-Pd alloys onto electrophoretic deposited carbon nanotubes for nitrate electroreduction [J]. Applied Surface Science, 2014, 308: 113-120. doi: 10.1016/j.apsusc.2014.04.119 [10] DUAN W J, LI G, LEI Z C, et al. Highly active and durable carbon electrocatalyst for nitrate reduction reaction [J]. Water Research, 2019, 161: 126-135. doi: 10.1016/j.watres.2019.05.104 [11] SONG Q N, LI M, WANG L L, et al. Mechanism and optimization of electrochemical system for simultaneous removal of nitrate and ammonia [J]. Journal of Hazardous Materials, 2019, 363: 119-126. doi: 10.1016/j.jhazmat.2018.09.046 [12] HORÁNYI G, RIZMAYER E M. Electrocatalytic reduction of NO2− and NO3− ions at a platinized platinum electrode in alkaline medium [J]. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 1985, 188(1/2): 265-272. [13] FAN N W, LI Z K, ZHAO L, et al. Electrochemical denitrification and kinetics study using Ti/IrO2-TiO2-RuO2 as the anode and Cu/Zn as the cathode [J]. Chemical Engineering Journal, 2013, 214: 83-90. doi: 10.1016/j.cej.2012.10.026 [14] 陈西亮, 刘国, 高阳阳, 等. 零价纳米铁炭微电解体系去除水中硝酸盐 [J]. 环境化学, 2016, 35(8): 1670-1675. doi: 10.7524/j.issn.0254-6108.2016.08.2015123003 CHEN X L, LIU G, GAO Y Y, et al. Removal of nitrate from water by nano-zero-valent iron-carbon microelectrolysis system [J]. Environmental Chemistry, 2016, 35(8): 1670-1675(in Chinese). doi: 10.7524/j.issn.0254-6108.2016.08.2015123003
[15] GAO W C, GAO L L, LI D, et al. Removal of nitrate from water by the electrocatalytic denitrification on the Cu-Bi electrode [J]. Journal of Electroanalytical Chemistry, 2018, 817: 202-209. doi: 10.1016/j.jelechem.2018.04.006 [16] URETA-ZAÑARTU S, YÁÑEZ C. Electroreduction of nitrate ion on Pt, Ir and on 70: 30 Pt: Ir alloy [J]. Electrochimica Acta, 1997, 42(11): 1725-1731. doi: 10.1016/S0013-4686(96)00372-6 [17] LI M, FENG C P, ZHANG Z Y, et al. Efficient electrochemical reduction of nitrate to nitrogen using Ti/IrO2-Pt anode and different cathodes [J]. Electrochimica Acta, 2009, 54(20): 4600-4606. doi: 10.1016/j.electacta.2009.03.064 [18] LI A Z, ZHAO X, HOU Y N, et al. The electrocatalytic dechlorination of chloroacetic acids at electrodeposited Pd/Fe-modified carbon paper electrode [J]. Applied Catalysis B:Environmental, 2012, 111/112: 628-635. doi: 10.1016/j.apcatb.2011.11.016 [19] GARCIA-SEGURA S, LANZARINI-LOPES M, HRISTOVSKI K, et al. Electrocatalytic reduction of nitrate: Fundamentals to full-scale water treatment applications [J]. Applied Catalysis B:Environmental, 2018, 236: 546-568. doi: 10.1016/j.apcatb.2018.05.041 [20] JONOUSH Z A, REZAEE A, GHAFFARINEJAD A. Electrocatalytic nitrate reduction using Fe0/Fe3O4 nanoparticles immobilized on nickel foam: Selectivity and energy consumption studies [J]. Journal of Cleaner Production, 2020, 242: 118569. doi: 10.1016/j.jclepro.2019.118569 [21] ZHOU C H, BAI J, ZHANG Y, et al. Novel 3D Pd-Cu(OH)2/CF cathode for rapid reduction of nitrate-N and simultaneous total nitrogen removal from wastewater [J]. Journal of Hazardous Materials, 2021, 401: 123232. doi: 10.1016/j.jhazmat.2020.123232 [22] PIEROZYNSKI B, MIKOLAJCZYK T, LUBA M, et al. Kinetics of oxygen evolution reaction on nickel foam and platinum-modified nickel foam materials in alkaline solution [J]. Journal of Electroanalytical Chemistry, 2019, 847: 113194. doi: 10.1016/j.jelechem.2019.113194 [23] 中华人民共和国国家环境保护总局. 中华人民共和国环保行业标准: 水质 硝酸盐氮的测定 紫外分光光度法(试行) HJ/T 346—2007[S]. 北京: 中国环境科学出版社, 2007. State Environmental Protection Administration of the People's Republic of China. Environmental Protection Standard of the People's Republic of China: Water quality-Determination of nitrate-nitrogen-Ultraviolet spectrophotometry. HJ/T 346—2007[S]. Beijing: China Environment Science Press, 2007(in Chinese).
[24] 中国国家环境保护局. 水质 亚硝酸盐氮的测定 分光光度法: GB/T7493—1987[S]. 北京: 中国标准出版社, 1987. State Environmental Protection Administration of China. Water quality-Determination of nitrogen(nitrite)-Spectrophotometric method: GB/T7493—1987[S]. China Standard Press, 1987(in Chinese).
[25] 环境保护部. 中华人民共和国环保行业标准: 水质 氨氮的测定 纳氏试剂分光光度法 HJ 535—2009[S]. 北京: 中国环境科学出版社, 2010. Ministry of Environmental Protection of the People's Republic of China. Environmental Protection Standard of the People's Republic of China: Water quality-Determinatiion of ammonia nitrogen-Nesslers reagent spectrophotometry. HJ 535—2009[S]. Beijing: China Environment Science Press, 2010(in Chinese).
[26] LIU F, LIU K W, LI M, et al. Fabrication and characterization of a Ni-TNTA bimetallic nanoelectrode to electrochemically remove nitrate from groundwater [J]. Chemosphere, 2019, 223: 560-568. doi: 10.1016/j.chemosphere.2019.02.028 [27] DING J, LI W, ZHAO Q L, et al. Electroreduction of nitrate in water: Role of cathode and cell configuration [J]. Chemical Engineering Journal, 2015, 271: 252-259. doi: 10.1016/j.cej.2015.03.001 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 5062
- HTML全文浏览数: 5062
- PDF下载数: 152
- 施引文献: 0






















