





































-
地下水中磷富集已成为湖泊富营养化的重要来源之一[1]。地下水中的无机磷主要以正磷酸盐阴离子(H2PO4−和HPO42-)的形式存在于地下水环境中[2]。近年来,许多研究表明,全球洪泛区地下水含水层中磷含量达0.05~1.2 mg·g−1[3]。然而,对于地下水中磷的污染过程尚不清晰[4]。
胶体是一种颗粒粒径在1~1 000 nm的分散体,在天然水域的地球化学循环中起着至关重要的作用[5]。课题组前期工作中研究了江汉平原地下水中铁锰胶体与磷的含量分布情况,发现了地下水中铁锰胶体与磷之间存在较强的耦合控制作用[6]。ELEANER等[7]研究铁胶体在深层地下水系统中形成和迁移,同样发现三价铁(氢)氧化物胶体能使无机盐发生迁移。土壤/沉积物中常见的铁锰矿物有水铁矿、针铁矿、二氧化锰、水钠锰矿[8],易从沉积物表面分离而下渗到地下水中,并以胶体的形式参与到地下水中磷的吸附、释放、迁移及转化[9]。然而,地下水环境中铁锰胶体对无机磷的吸附-脱附机制鲜有报道。
本研究对比探讨了常见的4种铁锰胶体(水铁矿、针铁矿、二氧化锰及水钠锰矿)对地下水中无机磷的吸附-脱附行为,考察了胶体质量浓度、时间、pH、离子强度及有机质对磷在铁锰胶体上吸/脱附影响,探究了其吸/脱附机制,为认识地下水中磷污染过程提供基础。
-
矿物制备:水铁矿和针铁矿制备参考GOLDBERG等[10]和ZHANG等[11]的方法。二氧化锰和水钠锰矿制备根据LI等[12]和ATKINS等[13]的方法。胶体悬浮液制备:分别称取0.05 g铁锰矿物于50 mL 离心管中,并向其中加入50 mL 1.0 mmol·L−1氯化钠溶液,然后将离心管水浴超声1 h后转入高速离心机中于6 200 r·min−1下离心54 min(基于斯托克斯沉降定律),以去除大于1 μm的颗粒。最后,将上清液转移至干净的烧杯中以备用,并根据差重法测得胶体母液质量浓度约为0.8 mg·L−1。
胶体表征:胶体粒径及Zeta电位使用纳米粒度电位仪(Malvern Zetasizer Nano ZS90,英国)测定;比表面积使用多通道全自动比表面积及孔隙度分析仪(Tristar II 3020 Plus,中国)测定;胶体吸附磷前后官能团的变化使用傅里叶变换红外光谱仪(Nicolet iS50,美国)分析;胶体的晶面结构采用X射线衍射仪(Bruker D8 Advance,德国)进行分析。
-
所有实验所用溶液均通入高纯N2(纯度99.999%)中30 min以排除水里的溶解氧,并调节pH近中性用以模拟地下水环境。
吸附动力学实验。先移取20 mL 100 mg·L−1铁锰胶体悬浮液于500 mL厌氧瓶中,然后加入400 mL 15 mg·L−1的NaH2PO4(以磷计)溶液,避光密封后,置于220 r·min−1恒温振荡器中振荡,每间隔一段时间取出2 mL样品过0.1 μm滤膜,采用钼蓝比色法测定上清液中磷的质量浓度,根据式(1)计算吸附量。
式中:qt为t时刻对磷的吸附量,mg·g−1;t为吸附反应时间,min;Ce和C0分别为反应前后磷的质量浓度,mg·L−1;v为吸附溶液体积,mL;m为胶体分散体质量,等于0.2 mg。
等温吸附实验:根据上述实验,改变磷溶液初始质量浓度,样品经振荡4 h后过膜,测定上清液中磷的质量浓度,并计算平衡时的吸附量(qe)。
影响因素实验:所有影响因素实验条件均与上述实验条件一致,不同的是,移取2 mL 100 mg·L−1铁锰胶体悬浮液于20 mL 15 mg·L−1的NaH2PO4溶液中。pH影响实验控制体系的pH 5~8;离子强度影响实验控制背景电解质浓度分别为1.0、10.0、100.0、100 0.0 mmol·L−1;在有机质影响实验中,分别向初始磷溶液中加入10、25、50 mg·L−1的腐殖酸和富里酸。
脱附实验:采用含有不同质量浓度的KCl和CaCl2的溶液作为洗脱液。制备磷饱和的4种铁锰胶体样品,将其过0.1 μm滤膜,用超纯水冲洗滤膜,收集滤膜上的固体,经冷冻干燥后即得磷饱和铁锰胶体分散体粉末。取1 g所得粉末加入到装有200 mL质量浓度为25、50、100 mg·L−1洗脱液的厌氧瓶中,避光密封后将其置于220 r·min−1恒温振荡器中振荡。计算吸附量,每间隔一段时间将取出的悬浮液样品过膜,测定上清液中磷的质量浓度,根据式(2)计算其脱附量。根据式(3)计算其解吸率。所有实验在常温下进行,每组实验设置平行实验。
式中:q1为磷在胶体上的吸附量,mg·g−1;q2为磷在胶体上的脱附量,mg·g−1;C为脱附后磷的质量浓度,mg·L−1;V为脱附溶液体积,mL;M为磷饱和胶体分散体质量,为1 g;η为磷在胶体上的解吸率,%。
-
所制备的水铁矿、针铁矿、二氧化锰、水钠锰矿胶体,其胶体特性如表1所示。由表1可知,2种铁胶体表面带正电荷,锰胶体表面带负电荷;铁胶体的粒径高于锰胶体,但锰胶体的比表面积高于铁胶体。
-
磷在不同铁锰胶体上的吸附动力学曲线如图1(a)所示。磷在4种铁锰胶体上的吸附可以分为2个过程:50 min内的快速吸附以及50 min后的缓慢吸附并在180 min内达到平衡的过程。磷在铁锰胶体上的饱和吸附量遵循二氧化锰胶体>水钠锰矿胶体>水铁矿胶体>针铁矿胶体,对应的最大吸附量分别为32.09、25.52、71.10、46.17 mg·g−1。远高于研究[14~15]报道的水铁矿、针铁矿、二氧化锰矿物上的磷最大吸附量(12.6、3.6、12.8 mg·g−1)。
采用伪一级吸附动力学模型和伪二级吸附动力学模型对动力学曲线进行拟合,结果如表2所示。由表2可知,磷在铁锰胶体表面的吸附过程同时存在物理吸附和化学吸附,但伪二级动力学比伪一级动力学模型更接近实验数据。且水铁矿胶体、针铁矿胶体、二氧化锰胶体和水钠锰矿胶体上的吸附速率分别为0.04、0.07、0.06、0.09 mg·(g·min)−1,表明锰胶体吸附量和吸附速率均高于铁胶体。
采用Langmuir和Freundlich吸附模型对等温吸附结果进行拟合,结果如图1(b)及表3所示。由表3可知,Langmuir模型能更好拟合实验数据,表明铁锰胶体吸附过程主要为单分子层吸附。4种铁锰胶体对磷的吸附量均随着初始磷浓度的增加而增加。这可能是因为高浓度的磷可提供必要的驱动力,从而克服了其在固液相之间的传质阻力[16]。
-
pH对四种铁锰胶体吸附磷的影响如图2所示。由图2(a)可知,针铁矿胶体、二氧化锰胶体、水钠锰矿胶体在pH=7时对磷的吸附能力最好,而水铁矿胶体对磷的吸附随着pH的增高而降低。由图2(b)可知,2种锰胶体在pH=5~8内的电位值恒为负。在相同pH下水钠锰矿胶体表面电荷密度高于二氧化锰胶体,二氧化锰胶体表面所吸附的阳离子(H+、Na+)较水钠锰矿胶体少,与磷酸根之间的静电阻力弱,因此,相同pH下二氧化锰胶体对磷的吸附量比水钠锰矿胶体对磷的吸附量大。而水铁矿和针铁矿的Zeta电位值随着pH的升高而逐渐降低,其等电点分别在7.9和9.2[17]左右。水铁矿胶体在较低pH下表现出较高的吸附性能,这是因为当pH小于8.0时,水铁矿胶体表面带正电荷,与磷酸根之间存在静电引力,随着pH的升高,表面电荷量较小且发生了电荷性质的逆转,从而造成了吸附量的降低。针铁矿胶体在pH=5~10内对磷的吸附量变化趋势与Zeta电位的变化趋势一致。
离子强度对4种铁锰胶体吸附磷的影响结果如图3所示。由图3可知,当离子强度从0.001 mol·L−1增加到1.000 mol·L−1时,水铁矿胶体、针铁矿胶体、二氧化锰胶体和水钠锰矿胶体对磷的吸附量分别上升了25.24%、27.61%、18.73%、41.54%。说明离子强度的增加能够促进铁锰胶体对磷的吸附,这是因为离子强度的增加减弱了磷酸根与胶体之间的静电斥力,从而有助于其吸附,MUSTUFA等[18]也观察到同样的实验现象。另外,离子强度的增加能够压缩胶体的双电层厚度,使胶体表面的活性位点更多的暴露在环境中,从而导致与更多的磷结合,促进了磷在铁锰胶体上的吸附[19]。
有机质对铁锰胶体吸附磷的影响如图4所示。由图4可知,磷在4种胶体表面的吸附量随有机质浓度的升高而下降,这与一些研究结果类似[20]。这可能是因为有机质会占据铁锰胶体表面的吸附位点从而降低了磷在铁锰胶体上的吸附[21]。水铁矿胶体、针铁矿胶体、二氧化锰胶体、水钠锰矿胶体在添加50 mg·L−1腐殖酸后对磷的吸附量分别由32.09、25.51、71.10和46.17 mg·g−1下降到10.53、6.30、21.19和17.78 mg·g−1;在添加50 mg·L−1富里酸后,4种铁锰胶体对P的吸附量分别从32.09、6.30、21.19和17.78 mg·g−1下降到18.5、6.64、22.29和18.72 mg·g−1。上述结果表明腐殖酸对磷在铁锰胶体上的吸附抑制作用高于富里酸,这可能是由于2种有机质所含基团的种类及数量不同所致。
-
不同电解质对磷从水铁矿胶体、针铁矿胶体、二氧化锰胶体、水钠锰矿胶体上的最大脱附效率如图5所示。由图5可知,磷从铁锰胶体上的脱附效率随着电解质浓度的升高而增加。当氯化钾和氯化钙质量浓度达到100 mg·L−1时,磷从水铁矿胶体、针铁矿胶体、二氧化锰胶体和水钠锰矿胶体上的脱附效率分别可达到72.47%和92.36%、59.11%和63.09%、67.38%和74.61%、82.91%和89.24%。以上结果表明铁胶体的脱附效率高于锰胶体,氯化钙对磷从铁锰胶体上的脱附影响高于氯化钠。这可能是因为钙离子更易夺取胶体表面吸附的磷酸根形成难溶解的磷酸钙沉淀(Ksp=2.0×10−29),从而使得磷酸根解吸,尤其在负电性较大的锰胶体表面,高价态阳离子易发生配位交换,从而更易导致磷的解吸。
-
为了进一步探究铁锰胶体吸附-脱附磷机理,用傅里叶变换红外光谱仪分析铁锰胶体吸附无机磷前后官能团的变化,其红外光谱图如图6所示。由图6可知,吸附无机磷后的水铁矿胶体、针铁矿胶体、二氧化锰胶体、水钠锰矿胶体分别在106 2.44、106 2.08和109 5.43、106 2.44、1064.96和109 5.43 cm−1处出现信号强烈的P—O键伸缩振动峰。水铁矿胶体、针铁矿胶体、二氧化锰胶体、水钠锰矿胶体分别在163 2.31、163 9.82、162 9.46、162 7.45 cm−1处对应的—OH弯曲振动峰发生横移。同时,水铁矿胶体和针铁矿胶体在138 4.02、135 7.10 cm−1处对应的—OH弯曲振动峰的强度在吸附后发生明显降低,峰值的减少说明—OH遭到破坏。这些现象均表明磷在水铁矿胶体、针铁矿胶体、二氧化锰胶体、水钠锰矿胶体上的吸附是通过与-OH进行结合。此外,有研究表明,磷酸基团的氧原子可以直接与表面Fe配位,使磷酸盐与水铁矿强结合形成双角双齿配合物[22]。由pH和浓度对磷酸盐吸附的影响可知,锰胶体吸附磷酸盐离子的过程是与固体表面带正电的基团络合,这与其他研究结果一致[18]。
pH发生变化时,磷酸根在实验设置组中以H2PO4−和HPO42-两种形式同时存在。已有报道证明,在pH=3~5(磷酸根的形态主要以H2PO4−存在)时,固体优先吸附双电荷的HPO42-,随着pH的升高,胶体表面对单电荷磷酸根吸附增多[15],不同电荷数的磷酸根都能被有效吸附。本研究条件下铁胶体表面电荷恒为正,锰胶体表面电荷恒为负。铁胶体表面正电荷促进了磷酸基团以静电吸引的方式吸附。但由于锰胶体比铁胶体拥有更大的比表面积从而暴露出更多的羟基吸附位点,对无机磷进行化学吸附作用,进而导致磷在锰胶体上的吸附量高于铁胶体。
综上所述,铁锰胶体对磷的吸附/脱附机理图如图7所示。尽管铁锰胶体性质存在较大差异,但在近中性地下水环境中,铁锰胶体对磷的吸附主要通过:1)占据羟基活性位点的化学吸附作用;2)静电吸引作用;3)其他弱的吸附力作用(范德华力、氢键等)。磷从铁锰胶体表面的脱附主要受地下水电解质种类和浓度的影响,且高价阳离子更易与吸附的磷酸根发生配位交换,从而导致地下水磷的释放。
-
1) 水铁矿胶体、针铁矿胶体、二氧化锰胶体和水钠锰矿胶体对磷的吸附是一个快速的单分子层吸附过程,120 min内可达到吸附平衡,符合伪二级动力学模型和Langmuir等温吸附模型,其最大吸附量分别为磷32.09、25.52、71.10、46.17 mg·g−1。
2) pH值通过改变铁锰胶体表面的Zeta电位进一步影响磷在铁锰胶体上的吸附。当pH =7时有利于磷的吸附。由于离子强度的增加减弱了磷酸根与胶体之间的静电斥力,溶液离子强度升高可以促进四种铁锰胶体对磷的吸附。相反,有机质会竞争磷在铁锰胶体上的吸附位点从而抑制铁锰胶体对磷的吸附。
3) 磷从铁锰胶体的脱附随着电解质浓度的升高而增强,铁胶体的脱附效率高于锰胶体,且氯化钙的影响高于氯化钠。当氯化钙浓度达到100 mg·L−1时,磷从水铁矿胶体、针铁矿胶体、二氧化锰胶体和水钠锰矿胶体上的脱附率分别可达92.36%、63.09%、74.61%和89.24%。
铁锰胶体对地下水中无机磷的吸附-脱附特性
Adsorption and desorption properties of iron and manganese colloids to inorganic phosphorus in groundwater
-
摘要: 地下水中磷富集已成为水体富营养化的重要污染来源之一,但对于地下水中磷的污染过程认识不清。本研究对比了地下水中常见4种铁锰胶体(水铁矿胶体、针铁矿胶体、二氧化锰胶体和水钠锰矿胶体)对无机磷的吸附和脱附性能的差异。结果表明:铁锰胶体对磷的吸附主要是单分子层的化学吸附,锰胶体吸附量与吸附速率均大于铁胶体,中性pH和较高离子强度有利于铁锰胶体对磷的吸附,但有机质的存在会大大降低对磷的吸附量,尤其是针铁矿胶体受有机质的影响程度最大,其中腐殖酸影响比富里酸更大。同时,钙离子对磷从铁锰胶体上的脱附影响高于钠离子,且铁胶体的脱附效率高于锰胶体。以上研究结果为理解地下水中磷污染过程提供数据支撑。Abstract: Recent studies have shown that phosphorus enrichment in groundwater has become one of the important pollution sources of water eutrophication. However, the process of phosphorus pollution in groundwater is not clear. The differences of adsorption-desorption characteristics among four common iron and manganese colloids (ferrihydrite colloids, goethite colloids, manganese dioxide colloids and birnessite colloids) toward inorganic phosphoruswere compared. The results showed that phosphorus adsorption onto iron and manganese colloids was dominated by a monolayer and chemical process. The adsorption capacities and rates of manganese colloids were greater than those of iron colloids. The conditions of neutral pH and high ionic strength were favorable for the adsorption of phosphorus by iron and manganese colloids, while the presence of organic matter greatly decreased phosphorus adsorption capacity, especially the highest suppression effect occurred onto goethite colloids, and humic acid presented a stronger suppression than fulvic acid. Moreover, the phosphorus desorption were effected more seriously by Ca2+ ions than that by Na+ ones, and the desorption rate by iron colloids was higher than that by manganese ones. Therefore, above results can provide the data support for phosphorus pollution process in groundwater .
-
Key words:
- iron and manganese colloid /
- phosphorus /
- adsorption /
- desorption /
- underground water
-
提高人工快速渗滤(constructed rapid infiltration,CRI)系统的脱氮除磷性能是确保该工艺高效处理城镇生活污水的关键[1-2]。在诸多研究中,强化CRI系统中基于亚硝化的全程自养脱氮(completely autotrophic nitrogen removal over nitrite,CANON)作用被认为是提高该工艺脱氮效果的重要途径,此技术随之用于城镇生活污水处理[3]。截至目前,有学者相继开展了常温和低
NH+4 NH4+ 作为另一种备受关注的生物脱氮除磷新技术,反硝化除磷(denitrifying phosphorus removal,DPR)工艺可消耗有机碳源,并可发挥“一碳两用”的功能,使除磷和反硝化在缺氧环境下同时完成[9]。有研究结果初步证实,反硝化除磷耦合ANAMMOX作用的生物同步脱氮除磷工艺具备高效处理市政污水的潜力[10-11],如能在CRI系统中实现CANON作用与DPR作用的耦合,则CANON型CRI工艺的上述缺陷便有可能得到弥补。
本研究尝试构建了基于同步短程硝化、ANAMMOX、反硝化和反硝化除磷(simultaneous partial nitrification,ANAMMOX,denitrification and denitrifying phosphorus removal,SNADPR)作用的复合式人工快速渗滤(hybrid constructed rapid infiltration,H-CRI)系统,考察和探究了该系统的运行效能及微生物特性,而后对其中氮磷元素的归趋进行了解析。期望通过此研究,可探寻有效措施以弥补CANON型CRI工艺在处理城镇生活污水时的缺陷,提高其脱氮除磷性能及其稳定性,进而推动新型生物同步脱氮除磷工艺的研发及应用。
1. 材料与方法
1.1 实验装置
SNADPR型H-CRI装置构型见图1。
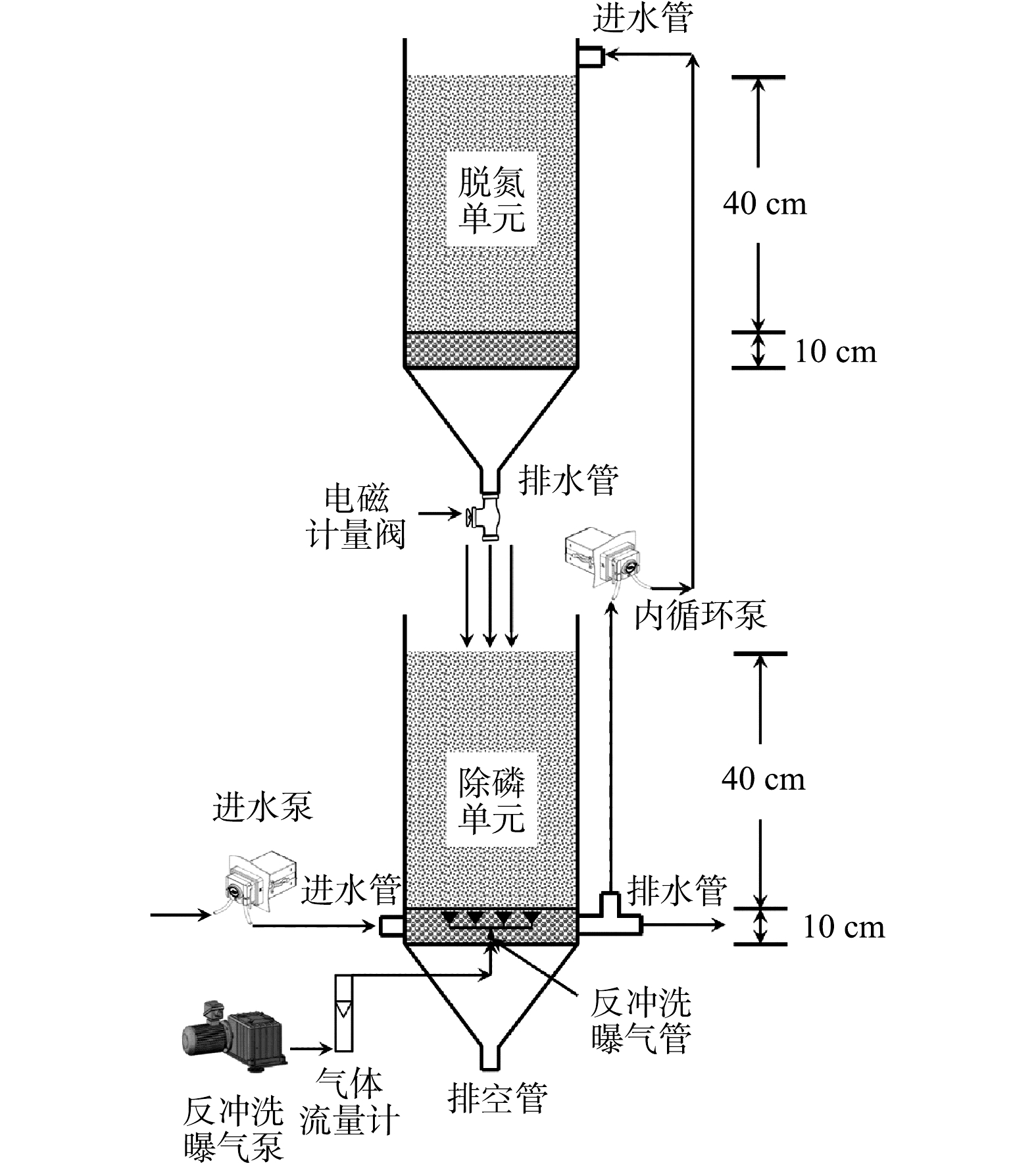 图 1 SNADPR型H-CRI装置构型Figure 1. Schematic of a H-CRI system utilizing the SNADPR process
图 1 SNADPR型H-CRI装置构型Figure 1. Schematic of a H-CRI system utilizing the SNADPR process前期实验分别构建了CANON型CRI系统和DPR型CRI系统[12-13]。其中:CANON型CRI系统由PVC管制成,表面积约314 cm2(φ=20 cm),其填料层厚度为50 cm且孔隙率约为37%:底部为5 cm厚砾石承托层,粒径为1~3 cm;上部为45 cm厚沸石层,粒径为5~10 mm。装置顶部和底部分别设有进水管和出水管,出水管附有计量阀控制装置的排水速率(vd)。前期研究中,当vd为0.5 L·min−1时,系统中的CANON作用得以强化,其TN和
NH+4 NO−3 NO−x SNADPR型H-CRI系统由CANON型CRI装置(标记为脱氮单元)和DPR型CRI装置(标记为除磷单元)耦合而成(图1),即两单元上下堆叠,并设置3台泵控制其运行。其中,1台为进水泵,与除磷单元的进水管相连;1台为内循环泵,分别与除磷单元的排水管和脱氮单元的进水管相连,可将前者出水泵入后者;1台为反冲洗曝气泵,与除磷单元的反冲洗曝气管相连。此外,本研究另设置1套CANON型CRI系统作为对照组,用于和SNADPR型H-CRI系统进行对比。
1.2 运行方式
考虑到好氧氨氧化菌(AOB)、厌氧氨氧化菌(AnAOB)、反硝化菌和反硝化聚磷菌(DPAOs)等功能微生物各自的生理生化特性,需对SNADPR型H-CRI系统进行空间或时间上的分割与交替,才能确保上述功能微生物的高效协同。为此,将该系统按照内循环潮汐流模式连续运行150个周期(图2),每个周期时长为12 h。运行方式为:将5 L进水泵入除磷单元中,使其进入释磷反应期,期间脱氮单元处于闲置期;随后,内循环泵将污水自除磷单元泵入脱氮单元中,使后者进入脱氮反应期,期间除磷单元处于中间闲置期;待脱氮单元的脱氮反应期结束后,此单元中的污水通过排水管以0.5 L·min−1的排水速率跌入除磷单元中,则除磷单元进入吸磷反应期,而脱氮单元则进入二次闲置期;最后,除磷单元将处理后的污水排出系统。按照时间顺序,上述典型周期内除磷单元的运行过程依次标记为进水期(时长为15 min)、释磷反应期(时长为150 min)、中间排水期(时长为15 min)、中间闲置期(时长为270 min)、中间进水期(时长为15 min)、吸磷反应期(时长为240 min)和排水期(时长为15 min)7个阶段;脱氮单元的运行过程依次为闲置期(时长为165 min)、进水期(时长为15 min)、脱氮反应期(时长为270 min)、排水期(时长为15 min)和二次闲置期(时长为255 min)5个阶段。H-CRI系统的污水处理量为10 L·d−1,水力负荷(HLR)为0.18 m3·(m2·d)−1。
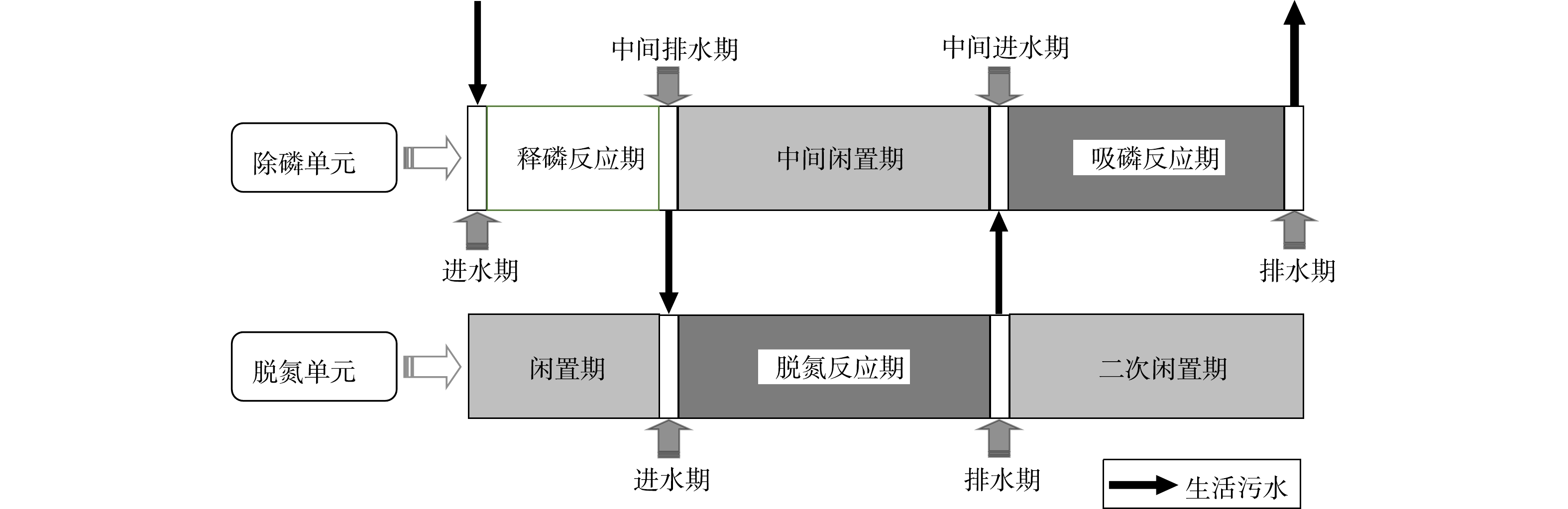 图 2 SNADPR型H-CRI系统的运行模式Figure 2. Operation mode of a H-CRI system utilizing the SNADPR process
图 2 SNADPR型H-CRI系统的运行模式Figure 2. Operation mode of a H-CRI system utilizing the SNADPR process对照组在实验阶段以潮汐流模式亦连续运行了150个周期,每个周期时长为12 h,包括进水期(时长为15 min)、淹水期(时长为270 min)、排水期(时长为15 min,vd=0.5 L·min−1)和闲置期(时长为420 min)4个阶段。该系统的污水处理量为10 L·d−1,HLR为0.32 m3·(m2·d)−1。
1.3 反冲洗操作
每周对H-CRI系统中的除磷单元进行反冲洗,以去除其中老化的生物膜,维持DPAOs活性并确保填料层的孔隙率。采用气-水联合反冲洗方式对此单元进行反冲洗:先对其单独气洗4 min,而后气-水联合反冲洗5 min,最后漂洗9 min。此过程中水洗和气洗的强度分别为6 L·(m2·s)−1和12 L·(m2·s)−1。清洗液和脱落的生物膜通过该单元的排空管进行收集。
1.4 进水水质
实验用水为安徽农业大学园区内生活污水,将其初沉后的上清液作为2组装置进水,其耗氧有机物(以COD计)、TN、
NH+4 NO−3 NO−2 1.5 分析方法
1)水样采集及分析方法。每天采集装置进出水水样;当各系统稳定运行时,在其典型周期内实时采集填料层中的水样。水样中COD、TN、
NH+4 NO−3 NO−2 2)填料样品采集。在实验阶段末采集2组实验装置中的填料样品,样品编号分别标记S1(取自对照组)、S2(取自H-CRI系统的脱氮单元)和S3(取自H-CRI系统的除磷单元)。
3)脱氮除磷性能测定。对于S1和S2,其亚硝酸化活性(PPNA)、硝酸化活性(PNA)、反硝化活性(PDA)、短程反硝化活性(PBDA)及厌氧氨氧化比活性(SAA)分别参照文献[15-16]中方法进行测定;对于S3,其生物膜中PHA、PHB、PHV、PH2MV和糖原(Gly)含量的测定参照文献[17],DPAOs占PAOs比例(即DPAOs/PAOs)的测定参照文献[18]。填料样品中全氮和全磷含量的测定方法均参照文献[19]。
4)基于16S rDNA的Illumina平台高通量测序。获取上述填料样品表面的生物膜,而后将其送至上海美吉生物科技医药公司进行高通量分析测序。测序分析后,根据Barcode序列区分各个样本的数据,进行嵌合体过滤,得到可用于后续分析的有效数据,即Clean reads。为了研究样品的物种组成多样性,对所有样品的Clean reads进行聚类,以97%的一致性(identity)将序列聚类成OTUs (operational taxonomic units),然后对OTUs的代表序列进行物种注释。
1.6 数据处理
采用SPSS 22.0对试验数据进行统计分析;采用one-way ANOVA进行方差分析;采用Origin 2018作图,图中相关数据为平均值±标准差;文中污染物去除(转化)率(量)、累积率(量)等的计算方法均参照文献[20];实验装置中氮磷去除途径的解析方法参考文献[21]。
2. 结果与讨论
2.1 运行性能
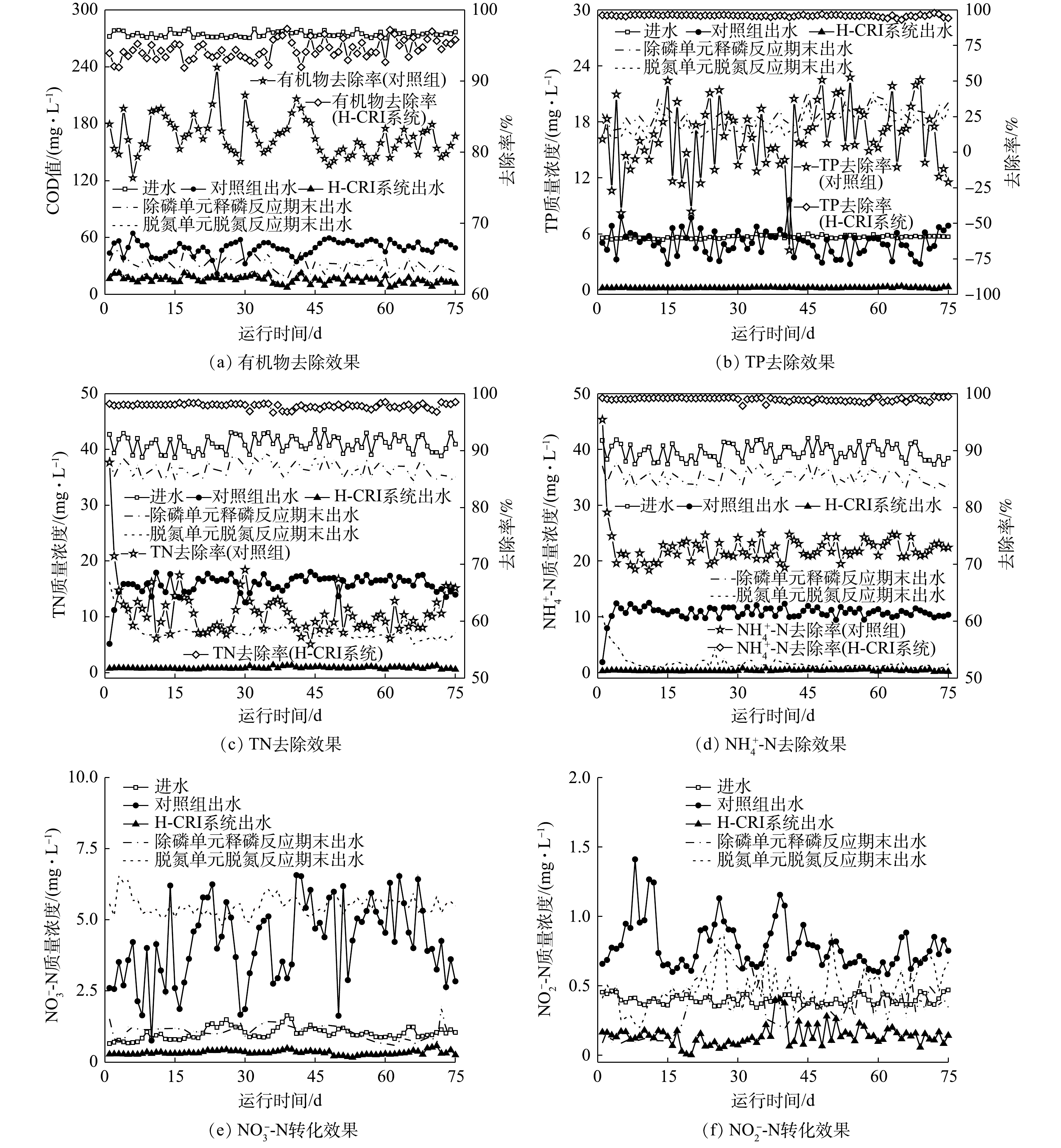
由图3可知,作为前期已成功启动的CANON型CRI系统,对照组具有良好的有机物去除性能[η≈(82.10±0.12)%],其出水的COD值<50 mg·L−1。有研究[4]表明,由于生物膜结构及其内部微环境的复杂性,某些CANON工艺中仍存在相当数量的异养型微生物,其可获得高效的有机物去除效果。然而,进水中的有机物却使对照组的脱氮性能出现下降,其TN和
NH+4 在稳定运行期间,SNADPR型H-CRI系统对进水中有机物、TN、
NH+4 NH+4 NO−3 NO−2 NH+4 NH+4 NO−3 NOx NO−3 NH+4 NO−3 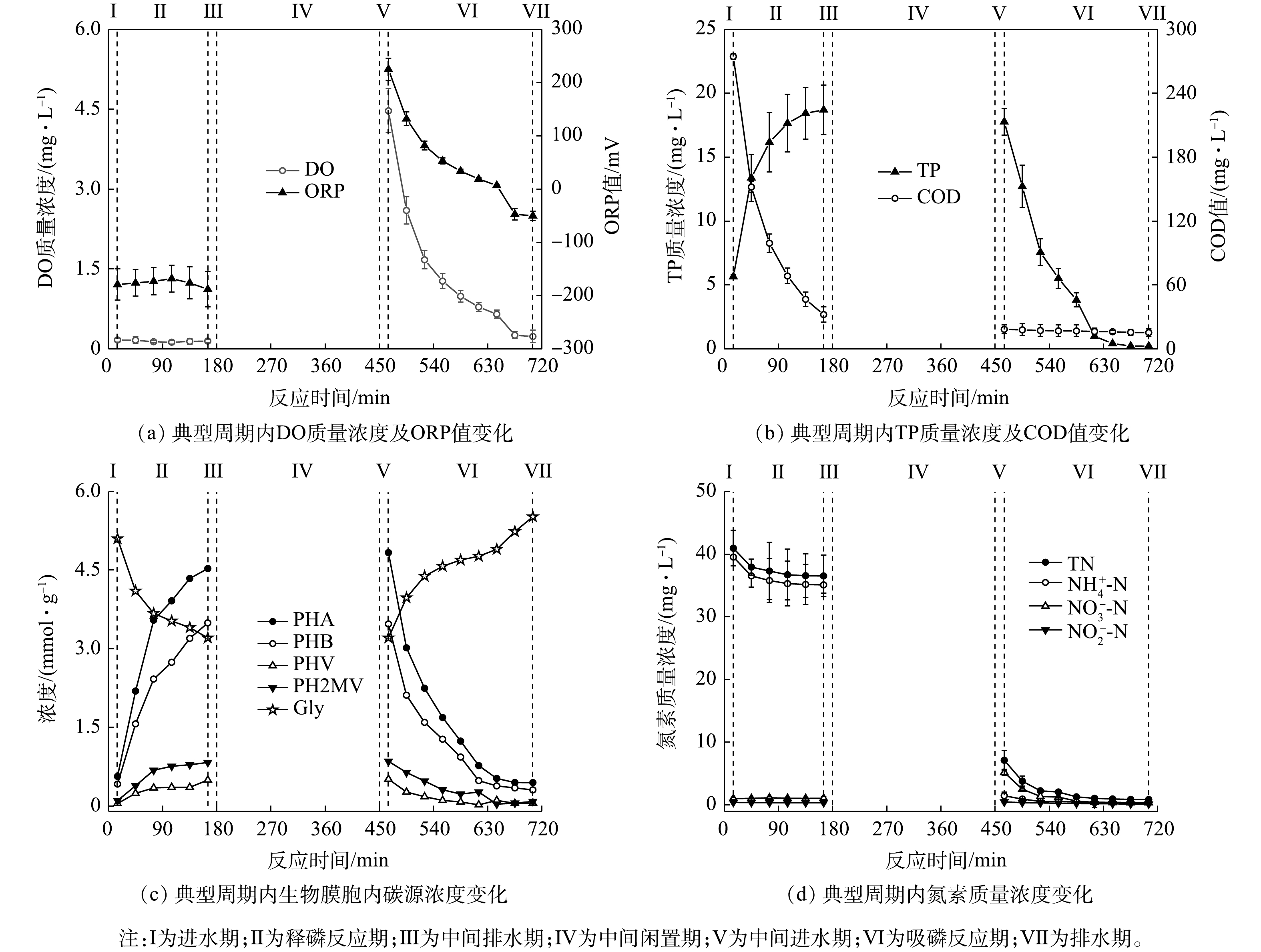 图 4 典型周期内H-CRI系统除磷单元中污染物质量浓度变化Figure 4. Pollutant concentration variation of contaminants in phosphorus removal unit of the H-CRI system during a typical cycle
图 4 典型周期内H-CRI系统除磷单元中污染物质量浓度变化Figure 4. Pollutant concentration variation of contaminants in phosphorus removal unit of the H-CRI system during a typical cycle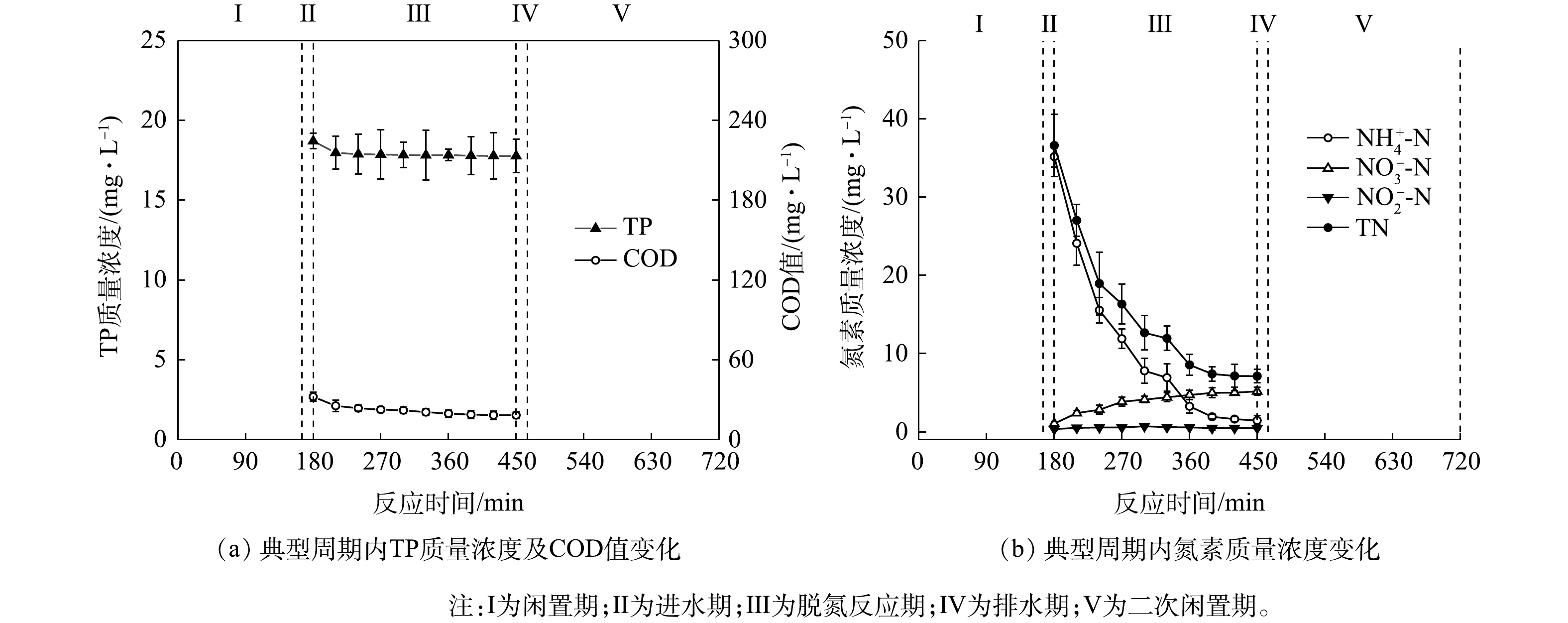 图 5 典型周期内H-CRI系统脱氮单元中污染物质量浓度变化Figure 5. Pollutant concentration variation of in nitrogen removal unit of the H-CRI system during a typical cycle
图 5 典型周期内H-CRI系统脱氮单元中污染物质量浓度变化Figure 5. Pollutant concentration variation of in nitrogen removal unit of the H-CRI system during a typical cycle上述结果表明,相较于对照组,H-CRI系统具备更佳的有机物及氮磷去除性能,可实现对生活污水的高效处理。对于该耦合装置,内循环潮汐流运行模式的设置可使其中的脱氮单元和除磷单元高效协作,且2单元中CANON作用和DPR作用的强度及稳定性均可得到充分保障。
2.2 微生物群落组成
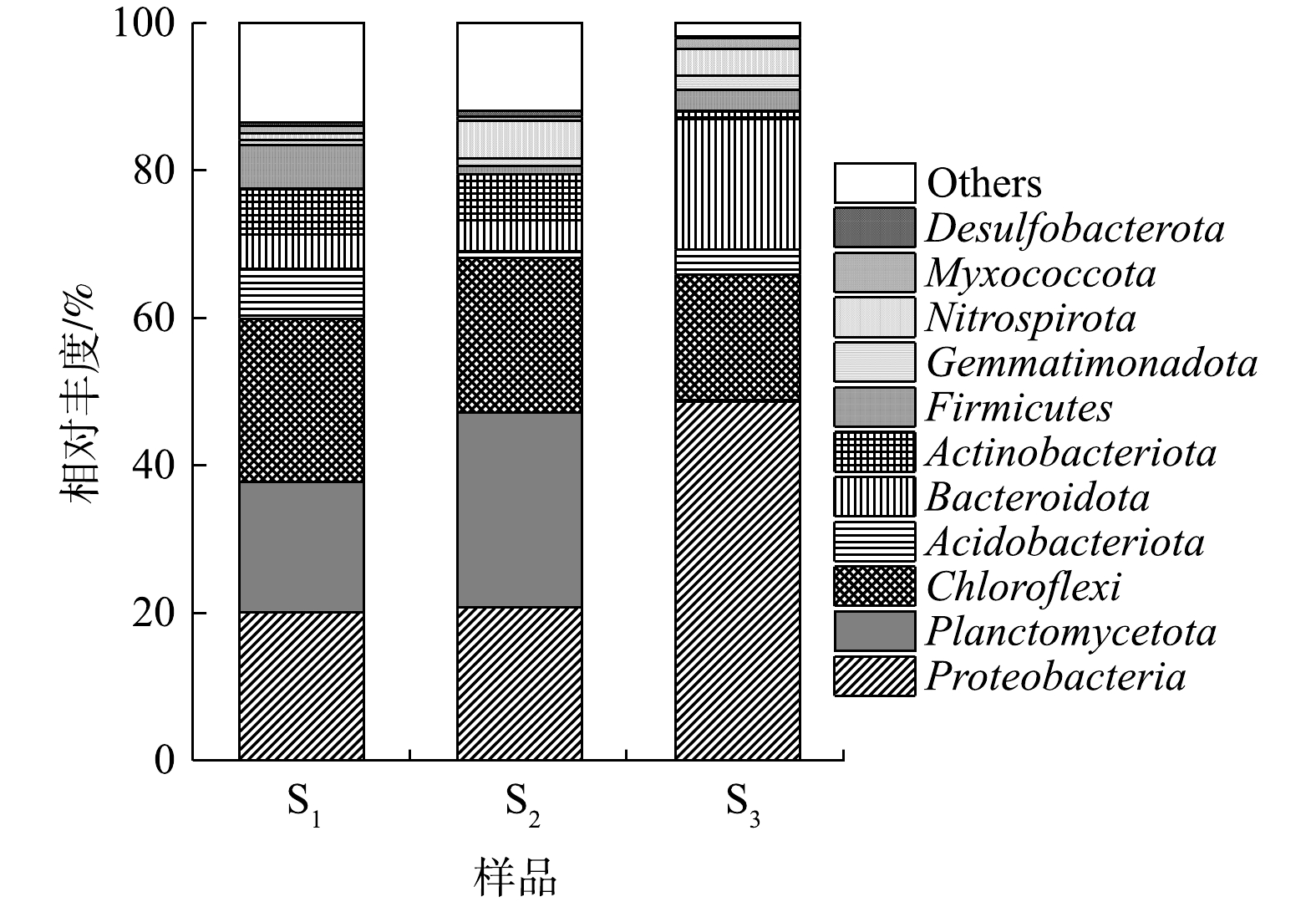
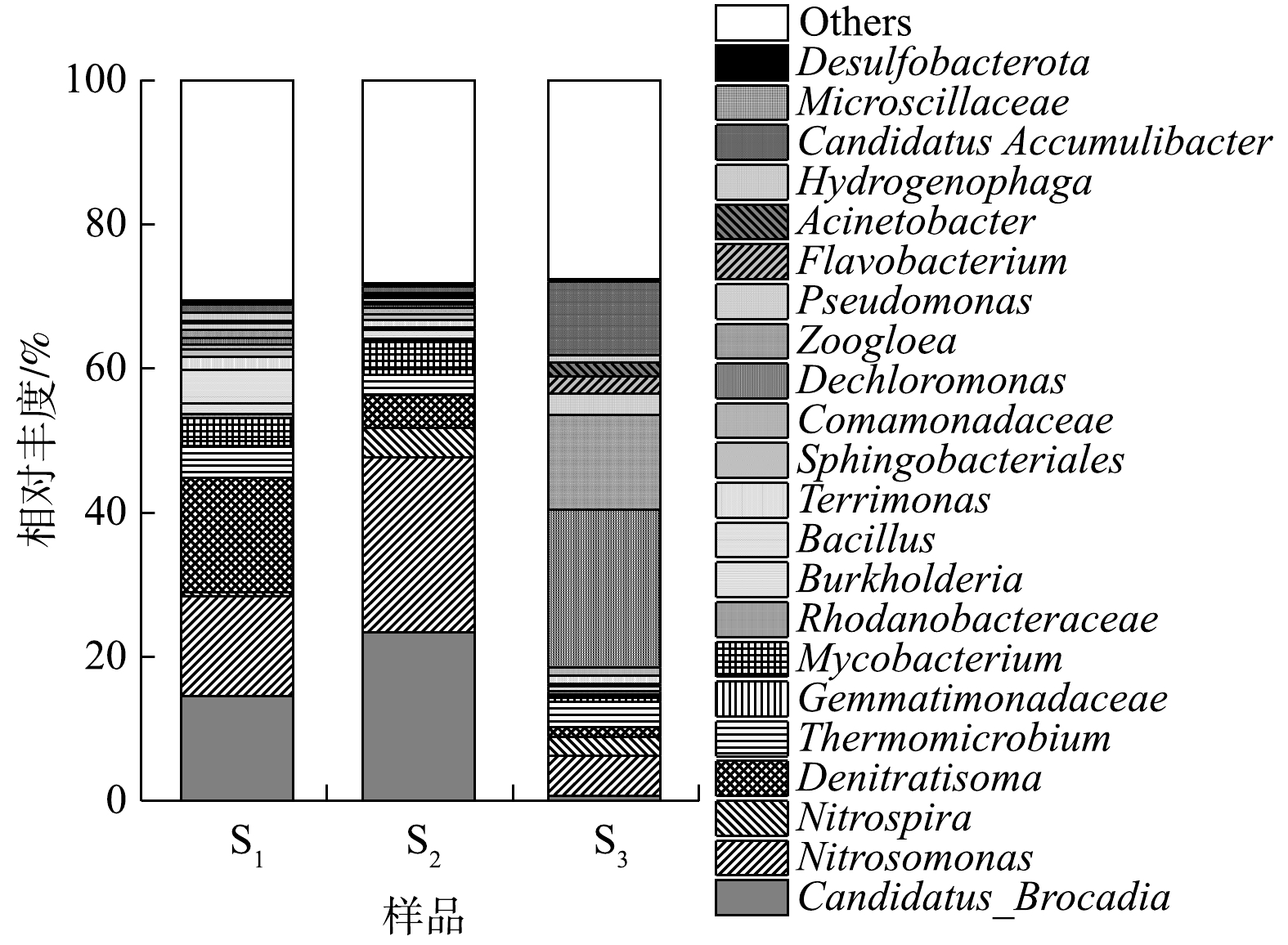
由图6可知,变形菌门(Proteobacteria)、浮霉菌门(Planctomycetota)和绿弯菌门(Chloroflexi)是S1和S2中相对丰度较高的3个菌门。其中,Proteobacteria在2组样本中的占比均>20%;Planctomycetota在S1中的相对丰度为17.66%,但其在S2中的含量却可达26.40%;此外,S1中硝化螺旋菌门(Nitrospirota)的相对丰度较S2(5.16%)显著下降至0.85%,但其中酸杆菌门(Acidobacteriota)的含量却增至6.72%,高于S2(0.91%)。有研究者指出,多数AOB(如Nitrosococcus、Nitrosomonas等)属Proteobacteria;AnAOB共包括5个属9个菌种,均属于Planctomycetota;亚硝酸盐氧化菌(NOB)属于Nitrospirota;反硝化菌则主要存在于Proteobacteria;另外,Acidobacteriota中的部分功能菌也具备还原
NO−x 由图7可知,S1和S2中的优势菌属均包括Candidatus Brocadia和Nitrosomonas,2种菌属与CANON反应相关[24]。其中,Candidatus Brocadia是上述样本中检出的唯一AnAOB,此结果与HU等[26]和GONZALEZ等[27]的研究结论相符。Nitrosomonas是各样本中唯一检出的AOB,相较于Nitrospira与Nitrospina,该菌属被证实更易在CANON系统中生长[28]。与图6相呼应,Candidatus Brocadia和Nitrosomonas在S1中的相对丰度(14.56%和13.80%)低于S2(23.38%和24.25%);硝化螺旋菌属(Nitrospira)在S1中的含量也明显低于S2。然而,属红环菌科(Rhodocyclaceae)的反硝化菌属——Denitratisoma此时却是S1中的优势菌属,其相对丰度可达15.89%,这表明,对照组的反硝化性能较H-CRI系统的脱氮单元有所增强。上述结果进一步证实:对照组和H-CRI系统的脱氮单元中均存在CANON反应体系;对照组由于受到进水中较高浓度有机物的影响,导致其中的异养菌(包括反硝化菌)滋生,随之影响了AOB与AnAOB的丰度及活性,进而影响了CANON作用的强度。而对于H-CRI系统的脱氮单元,由于除磷单元与之耦合后可缓解有机物对其微生物群落结构的冲击,则脱氮单元中CANON作用的强度及稳定性得到保障。S3中的第1大优势菌属为Dechloromonas(21.83%)。在侧流EBPR工艺中,Dechloromonas是常见的优势微生物,其具备缺氧吸磷的能力[29]。此结果表明H-CRI系统的除磷单元具备较理想的反硝化除磷性能。Zoogloea(13.11%)是S3中的第2大优势菌属,其能够合成胞内碳源进行内源反硝化作用[30],此样品中较高丰度的Zoogloea预示着H-CRI系统的除磷单元还具有一定的内源反硝化性能。值得注意的是,S3中还存在一定含量的Candidatus Accumulibacter (10.23%)和Pseudomonas (3.01%),此2种微生物均可进行反硝化除磷作用[31-32],则两者均对除磷单元的反硝化除磷效果有促进作用。有研究表明,除了
NO−3 2.3 功能微生物活性
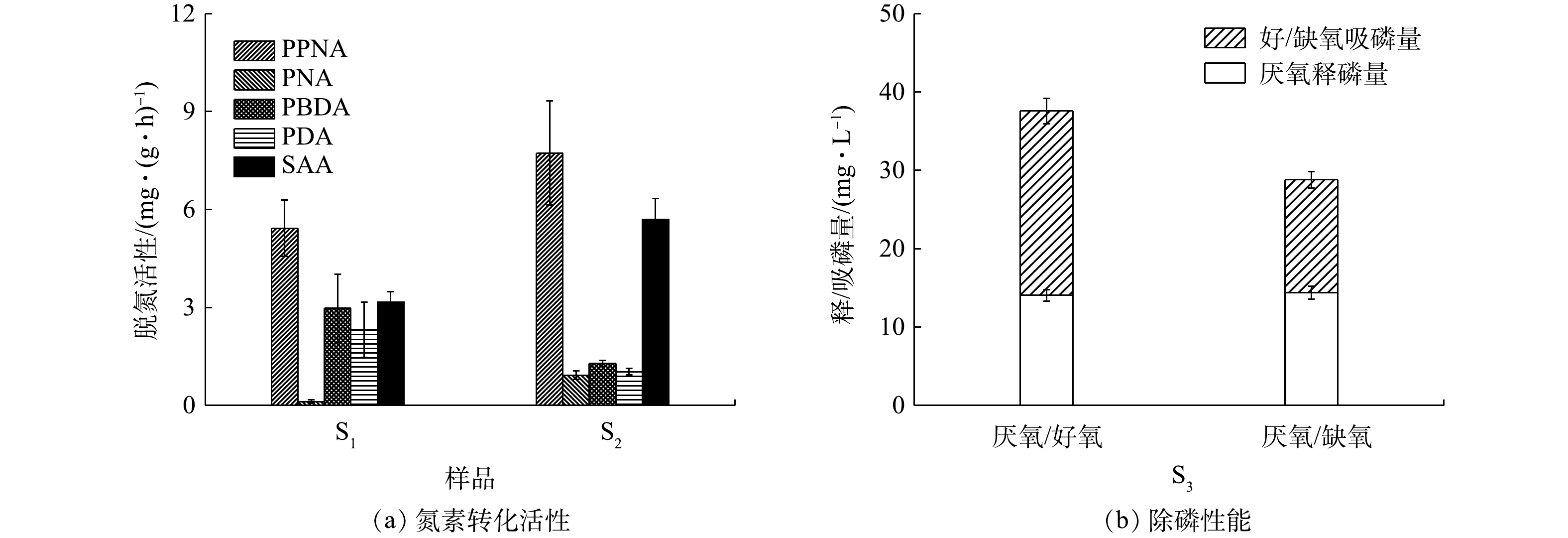
由图8可知,对照组的PPNA和SAA分别为(5.43±0.86) mg·(g·h)−1和(3.16±0.32) mg·(g·h)−1,而其PBDA和PDA则分别为(2.98±1.04) mg·(g·h)−1和(2.32±0.85) mg·(g·h)−1。与同类型研究[4]相比,此系统中CANON作用的强度偏低,但其反硝化性能却有较大幅度的提升。相较于对照组,H-CRI系统中脱氮单元的PPNA和SAA分别增至(7.73±1.60) mg·(g·h)−1和(5.70±0.64) mg·(g·h)−1,但该单元的PBDA和PDA有所下降,分别稳定在(1.29±0.08) mg·(g·h)−1和(1.03±0.10) mg·(g·h)−1。据此判断,脱氮单元中的CANON作用此时具备较高的强度,其反硝化性能也得到一定程度的强化。进水C/N会影响CANON系统中各功能微生物之间的竞争和协同关系[24]:当进水中有机物浓度适量时,各类脱氮功能微生物可共存并互相促进,即系统中的AOB和AnAOB协作完成CANON反应,反硝化菌则以有机碳源为电子供体,将体系中剩余的
NO−2 NO−3 上述结果表明,与对照组相比,内循环潮汐流运行模式下的H-CRI系统凭借其2个子单元的高效耦合分别为AOB、AnAOB、异养反硝化菌和PAOs(包括DPAOs)提供了其各自适宜的生态位,确保了此4类功能微生物对底物的合理竞争,进而实现了对生活污水的高效处理。其中:除磷单元中富集的PAOs(包括DPAOs)可在释磷反应期内利用有机物合成胞内碳源,因而削弱乃至消除了有机物对脱氮单元运行性能的影响,确保了其中CANON作用的稳定;除磷单元中的PAOs在吸磷反应期通过消耗内碳源实现了对脱氮单元出水中TP的超量吸收;脱氮单元出水中的
NOx− 2.4 氮磷去除途径解析
由图9可知,H-CRI系统在实验阶段的TN去除量为(1 082.39±27.82) g。其中,除磷单元的
NH+4 NO−3 NO−2 NH+4 NO−3 NO−2 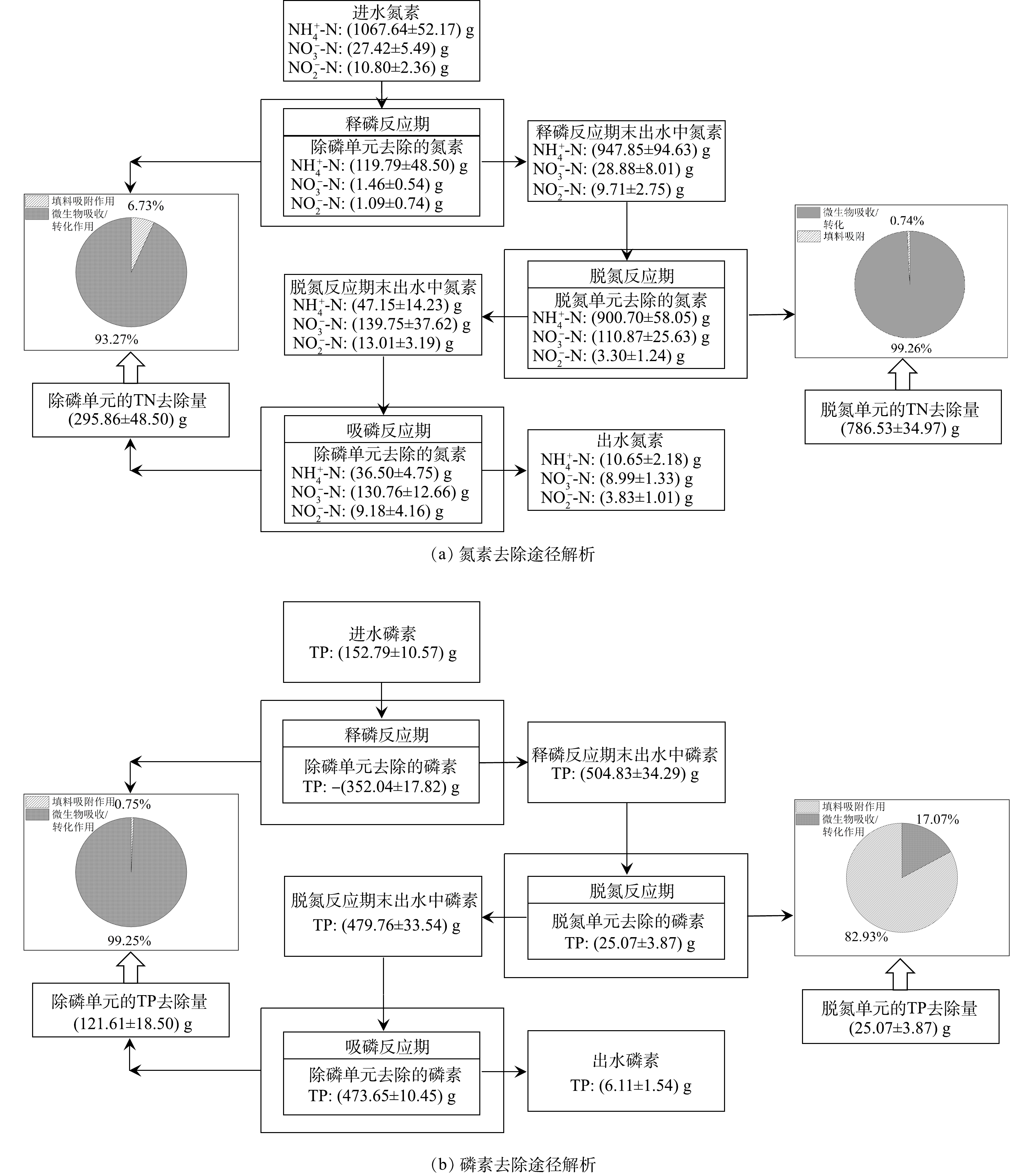 图 9 H-CRI系统中氮磷去除途径解析Figure 9. Nitrogen and Phosphorus removal pathways in the H-CRI system
图 9 H-CRI系统中氮磷去除途径解析Figure 9. Nitrogen and Phosphorus removal pathways in the H-CRI system综上所述,内循环潮汐流运行模式可将DPR型CRI装置与CANON型CRI装置成功耦合为SNADPR型H-CRI系统,此工艺在处理生活污水时可摆脱低
NH4+ 3. 结论
1)对于由CANON型CRI装置和DPR型CRI装置耦合而成的H-CRI系统,当其按照内循环潮汐流模式连续运行时,有助于其中SNADPR作用的发生和强化。
2)当HLR为0.18 m3·(m2·d)−1时,SNADPR型H-CRI系统可实现对生活污水中有机物及氮磷的高效同步去除,其对有机物、TN、
NH+4 3)脱氮单元中的CANON作用是SANDPR型H-CRI系统脱氮的主要途径,而系统中磷素的去除主要依赖于除磷单元中PAOs的过量吸磷作用,两者去除的氮磷量分别占H-CRI系统脱氮除磷总量的(72.13±6.12)%和(82.29±5.58)%。
-
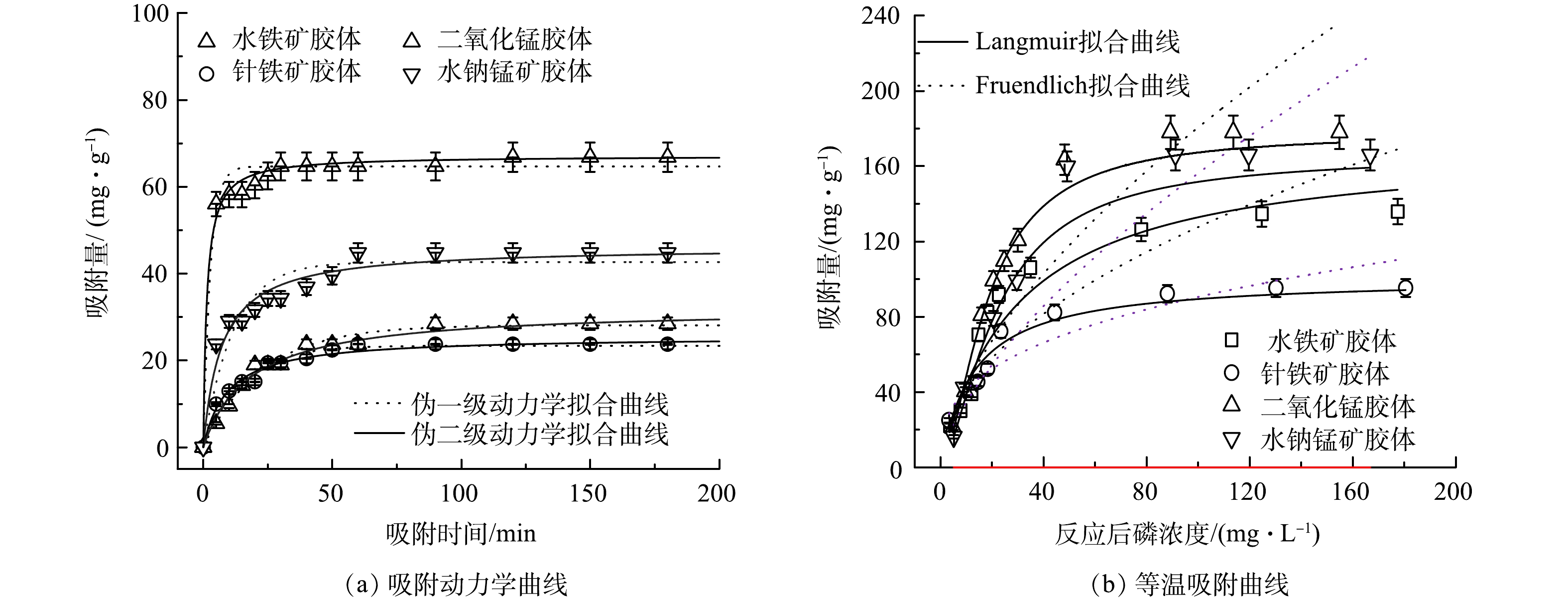
图 1 铁锰胶体对磷的吸附动力学曲线图和等温吸附曲线图
Figure 1. Adsorption kinetics of phosphorus on iron and manganese colloids and desorption isotherm of phosphorus on iron and manganese colloids
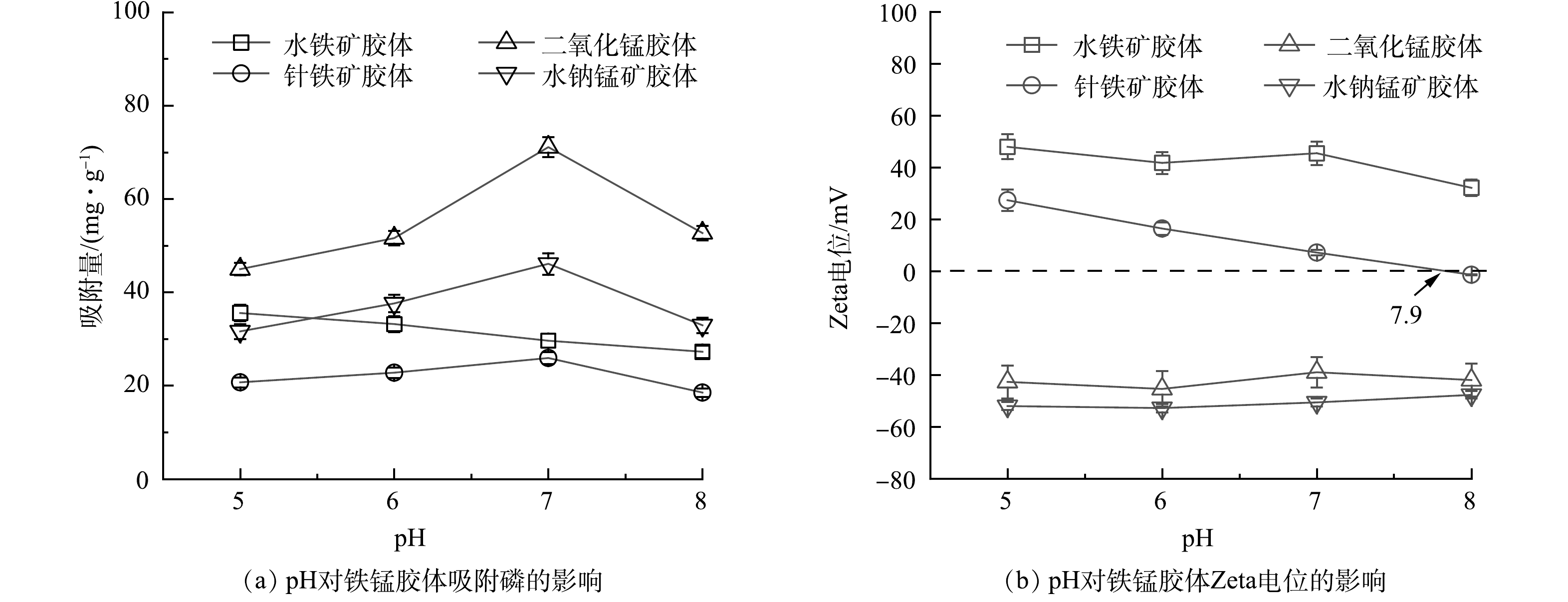
图 2 pH对铁锰胶体对磷的吸附量及Zeta电位的影响
Figure 2. Effect of pH on phosphorus adsorption onto iron and manganese colloid and its Zeta potential
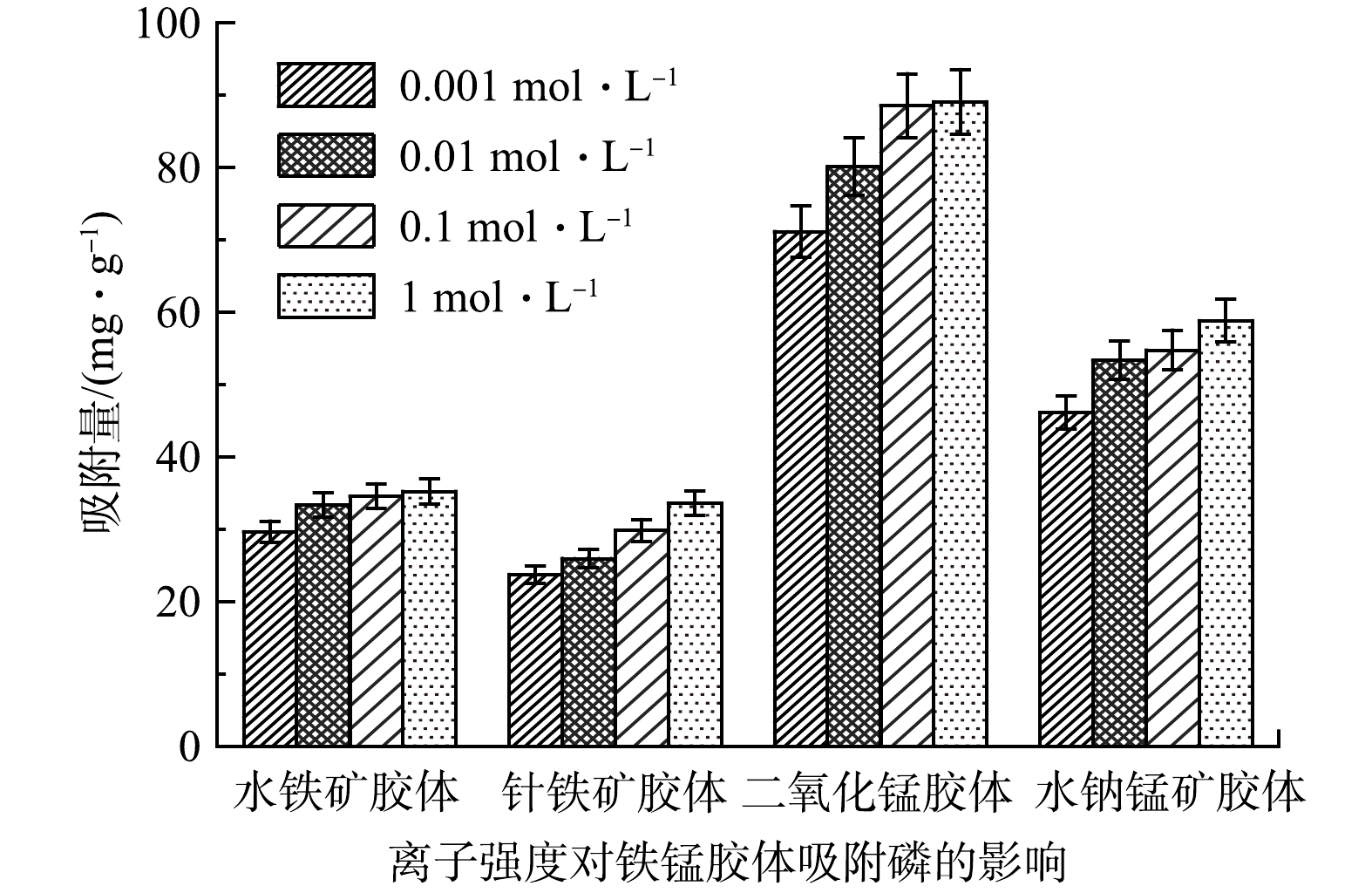
图 3 离子强度对铁锰胶体吸附磷的影响
Figure 3. Effect of ionic strength on P adsorption onto iron and manganese colloids

图 4 不同有机质对磷在水铁矿胶体、针铁矿胶体、二氧化锰胶体和水钠锰矿胶体上吸附的影响
Figure 4. Effect of different organic matter on adsorption of phosphorus onto ferrihydrite colloids, goethite colloids, manganese dioxide colloids and birnessite-manganese colloids

图 5 在不同浓度氯化钙和氯化钾中磷在不同胶体体系中的解吸效率
Figure 5. Desorption efficiency of phosphorus in different colloid systems with different concentrations of calcium chloride or potassium chloride

图 6 水铁矿胶体,针铁矿胶体,二氧化锰胶体和水钠锰矿胶体吸附磷前后的FT-IR图
Figure 6. FT-IR spectra of ferrihydrite colloid, goethite colloid, manganese dioxide colloid and birnessite colloid before and after adsorption of phosphorus
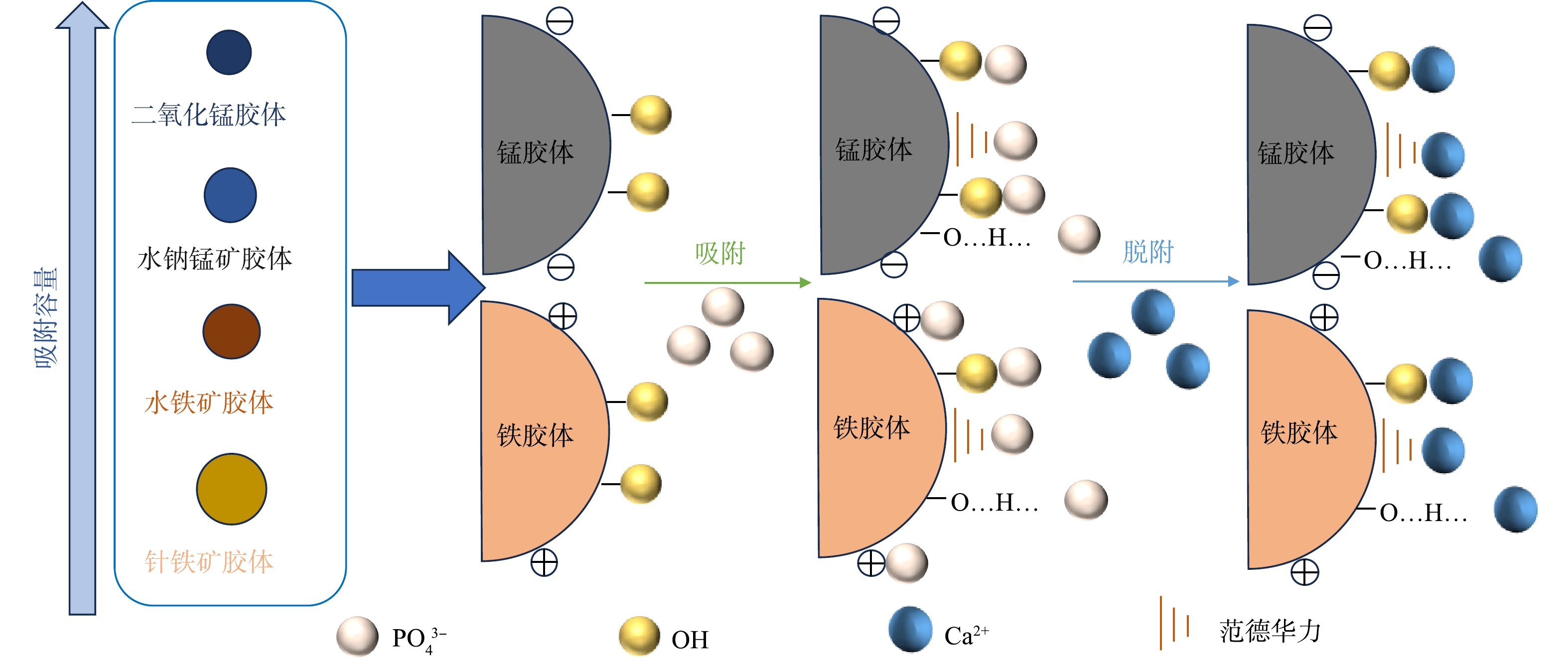
图 7 磷在铁锰胶体上的吸附-脱附机理的示意图
Figure 7. Schematic diagram of adsorption/desorption mechanism of phosphorus onto iron and manganese colloids
表 1 实验中不同铁锰胶体物理化学特征
Table 1. Physical and chemical characteristics of different iron and manganese colloids in the experiment
胶体 Zeta电位/mV 粒径/nm 比表面积/(m2·g−1) 水铁矿胶体 45.5 287.5 135.3 针铁矿胶体 7.2 452.8 124.9 二氧化锰胶体 -38.9 161.5 180.2 水钠锰矿胶体 -50.5 150.4 142.4  下载: 导出CSV
下载: 导出CSV
表 2 铁锰胶体对磷的吸附动力学曲线拟合参数
Table 2. Fitting parameters of phosphorus adsorption kinetics on iron and manganese colloids
胶体 伪一级吸附动力学模型 伪二级吸附动力学模型 K1/min−1 qe/(mg·g−1) R12 K2/(mg·(g·min)−1) qe /(mg·g−1) R22 水铁矿胶体 0.02 28.12 0.982 0.04 32.09 0.984 针铁矿胶体 0.01 23.37 0.967 0.07 25.52 0.981 二氧化锰胶体 0.05 64.63 0.957 0.06 71.10 0.981 水钠锰矿胶体 0.04 42.68 0.964 0.09 46.17 0.989
下载: 导出CSV
表 3 铁锰胶体对磷的等温吸附线的拟合参数
Table 3. Fitting parameters of phosphorus adsorption isotherm on iron and manganese colloids
胶体 Langmuir模型 Freundlich模型 KL/(L·mg−1) Qm/(mg·g−1) RL2 KF n RF2 水铁矿胶体 0.02 141.86 0.960 10.58 3.23 0.840 针铁矿胶体 0.02 100.79 0.967 9.71 2.76 0.926 二氧化锰胶体 0.02 177.96 0.990 10.19 9.71 0.853 水钠锰矿胶体 0.02 165.96 0.970 12.27 7.80 0.856
下载: 导出CSV
-
[1] LIU L, OUYANG W, LIU H, et al. Drainage optimization of paddy field watershed for diffuse phosphorus pollution control and sustainable agricultural development[J]. Agriculture, Ecosystems & Environment, 2021, 308: 107238. [2] BORCH T, KRETZSCHMAR R, KAPPLER A, et al. Biogeochemical redox processes and their impact on contaminant dynamics[J]. Environmental Science & Technology, 2010, 44(1): 15-23. [3] LI Y, YU C, ZHAO B, et al. Spatial variation in dissolved phosphorus and interactions with arsenic in response to changing redox conditions in floodplain aquifers of the Hetao Basin, Inner Mongolia[J]. Water Research, 2022, 209: 117930. doi: 10.1016/j.watres.2021.117930 [4] 张嘉雯, 魏健, 刘利, et al. 衡水湖沉积物营养盐形态分布特征及污染评价[J]. 环境科学, 2020, 41(12): 5389-5399. doi: 10.13227/j.hjkx.202004237 [5] ZHANG J, LIANG X, JIN M, et al. Identifying the groundwater flow systems in a condensed river-network interfluve between the Han River and Yangtze River (China) using hydrogeochemical indicators[J]. Hydrogeology Journal, 2019, 27(7): 2415-2430. doi: 10.1007/s10040-019-01994-1 [6] HUANG S, CHEN L, Li J, et al. The effects of colloidal Fe and Mn on P distribution in groundwater system of Jianghan Plain, China[J]. Science of The Total Environment, 2023, 854: 158739. doi: 10.1016/j.scitotenv.2022.158739 [7] SPIELMAN-SUN E, BLAND G, WIELINSKI J, et al. Environmental impact of solution pH on the formation and migration of iron colloids in deep subsurface energy systems[J]. Science of the Total Environment, 2023: 166409. [8] JIANMIN Z. Ferrihydrite: Surface structure and its effects on phase transformation[J]. Clays and Clay Minerals, 1994, 42(6): 737-746. doi: 10.1346/CCMN.1994.0420610 [9] DEGUELDRE C, BENEDICTO A. Colloid generation during water flow transients[J]. Applied Geochemistry, 2012, 27(6): 1220-1225. doi: 10.1016/j.apgeochem.2012.01.017 [10] GOLDDBERG S, JOHNSTO C T. Mechanisms of arsenic adsorption on amorphous oxides evaluated using macroscopic measurements, vibrational spectroscopy, and surface complexation modeling[J]. Journal of Colloid and Interface Science, 2001, 234(1): 204-216. doi: 10.1006/jcis.2000.7295 [11] ZHANG G, QU J, LIU H, et al. Preparation and evaluation of a novel Fe–Mn binary oxide adsorbent for effective arsenite removal[J]. Water Research, 2007, 41(9): 1921-1928. doi: 10.1016/j.watres.2007.02.009 [12] LI Z, DING Y, XIONG Y, et al. Rational growth of various α-MnO 2 Hierarchical Structures and β-MnO 2 nanorods via a homogeneous catalytic route[J]. Crystal Growth & Design, 2005, 5(5): 1953-1958. [13] ATKINS A L, SHAW S, PEACOCK C L. Release of Ni from birnessite during transformation of birnessite to todorokite: Implications for Ni cycling in marine sediments[J]. Geochimica et Cosmochimica Acta, 2016, 189: 158-183. doi: 10.1016/j.gca.2016.06.007 [14] 邵兴华, 章永松, 林咸永, 等. 三种铁氧化物的磷吸附解吸特性以及与磷吸附饱和度的关系[J]. 植物营养与肥料学报, 2006(2): 2208-2212. [15] CHEN P, ZHOU Y, XIE Q, et al. Phosphate adsorption kinetics and equilibria on natural iron and manganese oxide composites[J]. Journal of Environmental Management, 2022, 323: 116222. doi: 10.1016/j.jenvman.2022.116222 [16] SCHOTTING R J, MOSER H, HASSANIZADE S M. High-concentration-gradient dispersion in porous media: experiments, analysis and approximations[J]. Advances in Water Resources, 1999, 22(7): 665-680. doi: 10.1016/S0309-1708(98)00052-9 [17] 李政辉. 针铁矿/水铁矿-有机质复合体对有机磷的吸附特征研究[D]. 武汉: 华中农业大学, 2022. [18] MUSTAFA S, ZAMAN M I, Khan S. pH effect on phosphate sorption by crystalline MnO2[J]. Journal of Colloid and Interface Science, 2006, 301(2): 370-375. doi: 10.1016/j.jcis.2006.05.020 [19] 李惠. 铝胶体和腐殖酸对铀在饱和石英砂中迁移影响研究[D]. 衡阳: 南华大学, 2021. [20] BORGGAARD O K, RABEN-LANGE B, Gimsing A L, et al. Influence of humic substances on phosphate adsorption by aluminium and iron oxides[J]. Geoderma, 2005, 127(3/4): 270-279. [21] 宋嘉慧, 皇甫小留, 何强, 等. t al0–2对重金属Tl(Ⅰ)的吸附效能及影响因素[J]. 中国给水排水, 2019, 35(9): 53-57. [22] HIEMTRA T, VAN RIEMSDIJK W H. A surface structural model for ferrihydrite I: Sites related to primary charge, molar mass, and mass density[J]. Geochimica et Cosmochimica Acta, 2009, 73(15): 4423-4436. doi: 10.1016/j.gca.2009.04.032 -

 点击查看大图
点击查看大图
计量
- 文章访问数: 2109
- HTML全文浏览数: 2109
- PDF下载数: 124
- 施引文献: 0
